

Abstract
RG7388 (Idasanutlin) has been investigated as a small molecule inhibitor of oncoprotein murine double minute 2 (MDM2). Herein we investigated the feasibility of developing 18F-labeled RG7388 as a radiotracer for imaging MDM2 expression in tumors with positron emission tomography (PET). Two fluorinated analogues of RG7388, 6 and 7, were synthesized by attaching a fluoronicotinyl moiety to RG7388 via a polyethylene glycol (PEG3) or a propyl linker. The inhibitory potency (IC50) of 6 and 7 against MDM2 was determined by a fluorescence polarization (FP)-based assay. Next, compound 6 was labeled with 18F using a trimethylammonium triflate precursor to obtain [18F]FN-PEG3-RG7388 ([18F]6), and its properties were evaluated in MDM2 expressing wild-type p53 tumor cell lines (SJSA-1 and HepG2) in vitro and in tumor xenografts in vivo. The FP assays revealed an IC50 against MDM2 of 119 nM and 160 nM for 6 and 7, respectively. 18F-labeling of 6 was achieved in 50.3 ± 7.5% radiochemical yield. [18F]6 exhibited a high uptake (~70% of input dose) and specificity in SJSA-1 and HepG2 cell lines. Saturation binding assays revealed a binding affinity (Kd) of 128 nM for [18F]6 on SJSA-1 cells. In mice, [18F]6 showed fast clearance from blood with a maximum tumor uptake of 3.80 ± 0.85% injected dose per gram (ID/g) in HepG2 xenografts at 30 min post-injection (p.i.) and 1.32 ± 0.32% ID/g in SJSA-1 xenografts at 1 h p.i. Specificity of [18F]6 uptake in tumors was demonstrated by pre-treatment of mice with SJSA-xenografts with a blocking dose of RG7388 (35 mg/kg body weight, i.p.). In vivo stability studies in mice using HPLC showed ~60% and ~30% intact [18F]6 remaining in plasma at 30 min and 1 h p.i., respectively, with the remaining activity attributed to polar peaks. Our results suggest that RG7388 is a promising molecular scaffold for 18F-labeled probe development for MDM2. Additional labeling strategies and functionalizing locations on RG7388 are under development to improve binding affinity and in vivo stability of the 18F-labeled compound to make it more amenable for PET imaging of MDM2 in vivo.
Keywords: MDM2, fluorine-18, PET, RG7388, AMG232, SJSA-1
INTRODUCTION
Murine double minute 2 (MDM2, or the human homologue HDM2), has become a promising target for developing anti-cancer therapies. MDM2 is the most important negative regulator of the tumor suppressor protein p53 in cells1 and is frequently overexpressed in a wide variety of malignancies including breast cancer, liver cancer, glioblastoma and soft tissue sarcomas.2–5 To date, both small molecule- and peptide-based inhibitors of MDM2 have been developed, some of which have advanced to clinical trials including RG7112, RG7388, AMG232 and ALRN-6924.6–9 Results from these clinical trials suggest that MDM2 could serve as a predictive biomarker for selection of patients for MDM2 targeted therapies.6, 7
Currently, fluorescence in-situ hybridization (FISH) and immunohistochemical (IHC) staining of tumor biopsies are the main methods for assessing MDM2-amplification and/or protein overexpression in human cancers.10, 11 A promising alternative would be to exploit the noninvasive and real-time measurement features of positron emission tomography (PET) imaging. With the emergence of molecularly specific imaging agents, the concept of utilizing PET for guiding patient selection and monitoring therapeutic response for targeted therapeutics has become feasible and attractive.12 With a goal of developing a PET agent for imaging MDM2, we previously evaluated a fluorine-18 (18F, t1/2 = 109.8 min) labeled MDM2 inhibitor based on the well-studied MDM2 inhibitor SP-141.13, 14 The 18F-labeled SP-141 analogue showed high uptake and specific binding in MDM2 expressing wild-type p53 tumor cell lines but had suboptimal pharmacokinetics in mice bearing HepG2 tumor xenografts.13 In the present study, the potential utility of RG7388 (Idasanutlin) was evaluated for developing MDM2 targeted imaging agents based on its well-characterized nultin chemical scaffold.15 RG7388 is a potent, oral MDM2 inhibitor that has been extensively investigated in both preclinical and clinical studies.7, 16 RG7388 binds to a hydrophobic binding pocket on MDM2 by mimicking the three key amino acid residues (Phe19, Trp23 and Leu26) of p53 with its oxindole and spiro-pyrrolidine cores, thereby disrupting the MDM2-p53 interaction in cells with an IC50 of 6 nM.16 Furthermore, RG7388 has a higher binding affinity than SP-141 (Kd = 9.8 nM vs. 43.0 nM for SP-141)14, 16 and is among the most advanced MDM2 inhibitors to date.7, 17 In a recent clinical study, Reis et al. showed significant association between pretreatment MDM2 protein expression levels in leukemic blasts and clinical responses to RG7388 treatment in patients with acute myeloid leukemia.7 Thus, 18F-labeled analogue of RG7388 could serve as a companion imaging agent for MDM2 inhibitor therapy with RG7388 or other MDM2 antagonists.
In this study, the carboxyl group of RG7388 was functionalized with a fluoronicotinyl moiety via a PEG3 or a propyl linker. First, the nonradioactive fluorinated analogues of RG7388 were synthesized and evaluated for their ability to bind to MDM2 and inhibit the MDM2-p53 interaction using a fluorescence polarization (FP)-based assay. Based on these results, the RG7388-PEG3 analogue was labeled with 18F using a trimethylammonium triflate precursor and its uptake was evaluated in MDM2 expressing tumor cell lines in vitro. The in vivo characteristics of the labeled compound, referred to as [18F]FN-PEG3-RG7388, were investigated in athymic mice bearing SJSA-1 osteosarcoma and HepG2 hepatocellular carcinoma xenografts.
MATERIALS AND METHODS
All reagents were purchased from Sigma-Aldrich (St. Louis, MO) except where noted. o-(7-Azabenzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate (HATU) was obtained from Chem-Impex (Wood Dale, IL). tert-Butyl (2-(2-(2-(2-aminoethoxy)ethoxy)ethoxy)ethyl)carbamate (Boc-NH-PEG3-CH2CH2NH2) was purchased from AK Scientific (Union City, CA). RG7388 was purchased from Ontario Chemicals (Guelph, Ontario). Recombinant human MDM2 protein was purchased from Novus Biologicals (E3–204, 0.83 mg/mL; Bio-Techne Brands, Centennial, CO) and used as supplied. High-performance liquid chromatography (HPLC) analyses and purifications were performed using (1) a 1260 Infinity LC System equipped with a 1260 Infinity Multiple Wavelength Detector (Agilent Technologies, Santa Clara, CA) and a ExpressionL Compact Mass Spectrometer (Advion, Inc., Ithaca, NY) in series, or (2) a Beckman Gold system equipped with a Model 126 programmable solvent module coupled with a UV (Model 168 diode array) and a radiometric detector (Bioscan, Inc., Washington, D.C.). Nonradioactive reference fluoro analogues were purified by HPLC using a Zorbax SB-C18 reversed-phase semi-preparative column (9.4 mm I.D. × 250 mm, 5 μm, Agilent). The labeled compound [18F]FN-PEG3-RG7388 was purified by reversed-phase HPLC using an XBridge C18 column (4.6 × 150 mm, 5 μm, Waters Corporation, Milford, MA) with isocratic elution using 47% sodium acetate buffer (0.05 M, pH 5.5, A) and 53% ethanol (B) at a flow rate of 1.5 mL/min for 30 min. Quality control analysis of [18F]FN-PEG3-RG7388 was performed on an analytical HPLC system that consisted of an isocratic HPLC pump (Knauer, Germany) connected with an XBridge C18 column (3.5 μm, 3.0 × 100 mm, Waters) connected to a UV/VIS detector (Knauer) set at 254 nm and a radiometric detector (Carroll & Ramsey Associates, Berkeley, CA). Chromatograms were recorded using PeakSimple software (SRI Instruments, Torrance, CA). For metabolic studies, HPLC analysis of plasma and urine samples were performed using a Chromolith® Performance RP-18 end-capped 100–4.6 HPLC column (Merck KGaA, Darmstadt, Germany). Radioactivity in samples was measured using an automated gamma counter - either an LKB 1282 (Wallac, Finland) or a Wallac Wizard™ 1480 (PerkinElmer, Waltham, MA). Proton NMR spectra were obtained on a 400 MHz or a 500 MHz spectrometer (Varian/Agilent; Inova) and chemical shifts are reported in δ units using the residual solvent peaks as references. Mass spectra were recorded using an ExpressionL Compact Mass Spectrometer (Advion) by electrospray ionization (ESI) on the above described (System 1) LC/MS system or on a LC/MS-TOF system (ESI, Agilent Technologies); the latter being a high-resolution mass spectrometer.
Cell Culture Conditions
Cell culture reagents were purchased from Thermo Fisher Scientific (Waltham, MA) except where noted. SJSA-1 cells were purchased from American Type Culture Collection (ATCC, Manassas, VA), and HepG2 cells were obtained from the Duke University Cell Culture Facility. Cells were cultured in RPMI 1640 (SJSA-1) or MEM (HepG2) medium containing 10% fetal bovine serum and 1% Penicillin-Streptomycin, and maintained at 37°C in a CO2 (5%) humidified incubator.
Chemical Synthesis
tert-Butyl (1-(6-fluoropyridin-3-yl)-1-oxo-5,8,11-trioxa-2-azatridecan-13-yl)carbamate (2)
A mixture of 6-fluoropyridine-3-carboxylic acid (50 mg, 0.35 mmol), tert-butyl (2-(2-(2-(2-aminoethoxy)ethoxy)ethoxy)ethyl)carbamate (103 mg, 0.35 mmol), N,N-diisopropylethylamine (90.3 mg, 0.7 mmol) and HATU (200 mg, 0.53 mmol) in DMF (2 mL) was stirred at room temperature for 2 h. The crude mixture was purified by semi-preparative HPLC using a Zorbax SB-C18 column (9.4 × 250 mm, 5 μm, Agilent) eluted with a gradient of water (solvent A) and acetonitrile (solvent B), both containing 0.1% formic acid at a flow rate of 4 mL/min. The proportion of B was linearly increased from 15% to 35% over 15 min. The product eluted with a retention time (tR) of 11.1 min. Pooled HPLC fractions containing the product were lyophilized to obtain 81.2 mg (0.2 mmol, 55.9%) of compound 2 as a colorless oil: 1H-NMR (CD3OD, 500 MHz) δH = 1.45 (s, 9H), 3.22 (t, J = 5.57Hz, 2H), 3.51 (t, J = 5.57Hz, 2H), 3.55–3.75 (m, 12H), 7.19 (dd, J = 8.62, 2.52 Hz, 1H), 8.38 (td, J = 8.62, 2.52 Hz, 1H), 8.70 (d, J = 2.44Hz, 1H). LRMS (LC/MS-ESI) m/z: 416.3 (M+H)+. HRMS (ESI, m/z): calcd for C19H30FN3O6 (M+H)+: 416.2191; found: 416.2182.
tert-Butyl (3-(6-fluoronicotinamido)propyl)carbamate (3)
A mixture of 6-fluoropyridine-3-carboxylic acid (14 mg, 0.1 mmol), tert-butyl (3-aminopropyl)carbamate (17 mg, 0.1 mmol), N,N-diisopropylethylamine (25.8 mg, 0.2 mmol) and HATU (76 mg, 0.2 mmol) in DMF (1 mL) was stirred at RT for 2 h. The crude reaction mixture was purified by semi-preparative HPLC as described above except that the proportion of B was linearly increased from 20% to 40% over 15 min, at a flow rate of 4 mL/min; the product eluted with a retention time (tR) of 10.3 min. Pooled HPLC fractions containing the product were lyophilized to obtain 25.0 mg (0.08 mmol, 84.2%) of compound 3 as a colorless oil: 1H-NMR (CD3OD, 500 MHz) δH = 1.46 (s, 9H), 1.79 (t, J = 6.71, 2H), 3.16 (t, J = 6.64, 2H), 3.40–3.50 (m, 2H), 6.67 (s, 1H), 7.19 (dd, J = 8.54, 2.44 Hz, 1H), 8.37 (td, J = 8.05, 2.52 Hz, 1H), 8.69 (d, J = 2.14Hz, 1H). LRMS (LC/MS-ESI) m/z: 298.2 (M+H)+. HRMS (ESI, m/z): calcd for C14H20FN3O3 (M+H)+: 298.1565; found: 298.1573.
N-(2-(2-(2-(2-Aminoethoxy)ethoxy)ethoxy)ethyl)-6-fluoronicotinamide (4)
Compound 2 (78.1 mg, 0.19 mmol) was dissolved in 1 mL of TFA and stirred at RT for 30 min. Solvents were evaporated to yield 78.8 mg (97.8%, based on trifluoroacetate salt) of compound 4 as a colorless oil: 1H-NMR (D2O, 500 MHz) δH = 3.14 (t, J = 4.88 Hz, 2H), 3.55–3.75 (m, 16H), 7.19 (dd, J = 8.62, 2.52 Hz, 1H), 8.30–8.40 (m, 1H), 8.69 (d, J = 2.44Hz, 1H). LRMS (LC/MS-ESI) m/z: 316.2 (M+H)+. HRMS (ESI, m/z): calcd for C14H22FN3O4 (M+H)+: 316.1667; found: 316.1670.
N-(3-Aminopropyl)-6-fluoronicotinamide (5)
Compound 3 (25 mg, 0.08 mmol) was dissolved in 1 mL of TFA and stirred at RT for 30 min. Solvents were evaporated to yield 26.1 mg (99.9%, based on trifluoroacetate salt) of compound 5 as a colorless oil: 1H-NMR (CD3OD, 500 MHz) δH = 1.98 (quin, J = 6.98 Hz, 2 H), 2.85–2.90 (s, 1H), 2.95–3.10 (m, 2H), 3.52 (t, J = 6.64 Hz, 2H), 7.18 (dd, J = 8.55, 2.44 Hz, 1H), 7.95–8.05 (s, 1H), 8.35–8.45 (m, 1H), 8.69 (d, J = 2.14 Hz, 1 H). LRMS (LC/MS-ESI) m/z: 198.3 (M+H)+. HRMS (ESI, m/z): calcd for C9H12FN3O (M+H)+: 198.1044; found:198.1062.
FN-PEG3-RG7388 (6)
Compound 4 (17 mg, 54.0 μmol), RG7388 (33 mg, 54.0 μmol), HATU (31 mg, 82 μmol) and N,N-diisopropylethylamine (11.8 mg, 95 μmol) were stirred in 0.5 mL of DMF at RT for 2 h. The mixture was purified by semi-preparative HPLC as described above, but using an isocratic method employing 55% B (acetonitrile) for 20 min at a flow rate of 4 mL/min. Lyophilization of pooled HPLC fractions containing 6 (tR = 13.3 min) rendered 32.2 mg (35.3 μmol, 65.4%) of the compound as a colorless oil: 1H-NMR (CD3OD, 500 MHz) δH = 1.06 (s, 9H), 1.65–1.80 (m, 1H), 3.5–3.7 (m, 15H), 3.99 (s, 3H), 4.10 (d, J = 9.61 Hz, 1H), 4.65 (d, J = 8.39 Hz, 1H), 4.78 (d, J = 8.39 Hz, 1H), 7.13 (dd, J = 8.55, 2.44 Hz, 1H), 7.20–7.30 (m, 2H), 7.30–7.50 (m, 4H), 7.55 (d, J = 1.68 Hz, 1H), 7.74 (t, J = 6.79 Hz, 1H), 8.30–8.40 (m, 2H), 8.65 (d, J = 2.29 Hz, 1H). 19F-NMR (DMSO-d6, 471 MHz) δ ppm = −65.73 (s), −108.28 (s), −120.96 (s). LRMS (LC/MS-ESI) m/z: 913.2 (M+H)+. HRMS (ESI, m/z): calcd for C45H49Cl2F3N6O7 (M+H)+: 913.3065; found: 913.3035.
FN-3AP-RG7388 (7)
Compound 5 (3 mg, 10.0 μmol), RG7388 (6.1 mg, 10.0 μmol), HATU (6.5 mg, 17 μmol) and N,N-diisopropylethylamine (2.5 mg, 20 μmol) were taken in 0.2 mL of DMF and the resulting mixture was stirred at RT for 2 h. The crude mixture was subjected to semi-preparative HPLC purification as described above, but using an isocratic elution with 55% B (acetonitrile) for 20 min at a flow rate of 4 mL/min. Lyophilization of pooled HPLC fractions containing 7 (tR = 14.2 min) rendered 6.2 mg (7.8 μmol, 78.5%) of the compound as a colorless oil: 1H-NMR (CD3CN, 500 MHz) δH = 1.01 (s, 9H), 1.39 (d, J = 14.04 Hz, 1H), 1.6–1.7 (m, 1H), 1.82 (quin, J = 6.41 Hz, 2H), 3.4–3.5 (m, 5H), 3.96 (s, 3H), 4.05 (t, J = 10.91 Hz, 1H), 4.57 (t, J = 9.00 Hz, 1H), 4.67 (d, J = 8.39 Hz, 1H), 7.09 (dd, J = 8.54, 2.59 Hz, 1H), 7.20–7.45 (m, 7H), 7.50 (d, J = 1.68 Hz, 1H), 7.6–7.7 (m, 2H), 8.30 (td, J = 8.16, 2.44 Hz, 1H), 8.40 (d, J = 8.24 Hz, 1H), 8.66 (d, J = 2.29 Hz, 1H), 10.4 (s, 1H). 19F-NMR (DMSO-d6, 471 MHz) δ ppm = −65.85 (s), −108.29 (s), −120.96 (s). LRMS (LC/MS-ESI) m/z: 795.4 (M+H)+. HRMS (ESI, m/z): calcd for C40H39Cl2F3N6O4 (M+H)+: 795.2442; found: 795.2418.
5-((2,2-Dimethyl-4-oxo-3,8,11,14-tetraoxa-5-azahexadecan-16-yl)carbamoyl)-N,N,N-trimethylpyridin-2-aminium (9)
A mixture of N,N,N-trimethyl-5-((2,3,5,6-tetrafluorophenoxy)carbonyl)pyridin-2-aminium (compound 8, previously synthesized, 24 mg, 38.9 μmol), tert-butyl (2-(2-(2-(2-aminoethoxy)ethoxy)ethoxy)ethyl)carbamate (15 mg, 51.3 μmol) and N,N-diisopropylethylamine (11.1 mg, 85.8 μmol) in DMF (400 μL) was stirred at RT for 2 h. The product was isolated by semi-preparative HPLC as described above except that the proportion of B (acetonitrile) was linearly increased from 5% to 25% over 15 min at a flow rate of 4 mL/min; the product eluted with a retention time (tR) of 9.1 min. Pooled HPLC fractions containing the product were lyophilized to obtain 25.0 mg (41.4 μmol, 80.7%) of compound 9 as a colorless oil: 1H-NMR (CD3OD, 500 MHz) δH = 1.45 (s, 9H), 3.21 (t, J = 5.57 Hz, 2H), 3.51 (t, J = 5.65 Hz, 2H), 3.60–3.73 (m, 21H), 8.13 (d, J = 8.70 Hz, 1H), 8.57–8.59 (m, 1H), 9.07 (m, 1H). LRMS (LC/MS-ESI) m/z: 455.3 (M+). HRMS (ESI, m/z): calcd for C22H39N4O6 (M+): 455.2864; found: 455.2872.
5-((2-(2-(2-(2-Aminoethoxy)ethoxy)ethoxy)ethyl)carbamoyl)-N,N,N-trimethylpyridin-2-aminium (10)
Compound 9 (21 mg, 34.8 μmol) was dissolved in 0.3 mL of TFA and stirred at RT for 30 min. Solvents were evaporated to yield 21 mg (97.8%, based on trifluoroacetate salt) of compound 10 as a colorless oil: 1H-NMR (D2O, 500 MHz) δH = 3.19 (t, J = 4.65 Hz, 2H), 3.60–3.80 (m, 27H), 8.02 (d, J = 8.70 Hz, 1H), 8.45 (dt, J = 8.66, 1.93 Hz, 1H), 8.93–8.94 (m, 1H). LRMS (LC/MS-ESI) m/z: 355.3 (M+). HRMS (ESI, m/z): calcd for C17H31N4O4 (M+):355.2340; found: 355.2343.
5-((1-(4-((2R,3R,4R,5S)-3-(3-Chloro-2-fluorophenyl)-4-(4-chloro-2-fluorophenyl)-4-cyano-5-neopentylpyrrolidine-2-carboxamido)-3-methoxyphenyl)-1-oxo-5,8,11-trioxa-2-azatridecan-13-yl)carbamoyl)-N,N,N-trimethylpyridin-2-aminium (11)
A mixture of compound 10 (21 mg, 34.0 μmol), RG7388 (25 mg, 40.6 μmol), HATU (25.8 mg, 68.0 μmol) and N,N-diisopropylethylamine (8.8 mg, 68.0 μmol) in DMF (0.3 mL) was stirred at RT for 2 h. The crude mixture was purified by semi-preparative HPLC as described above but using a gradient wherein the proportion of solvent B (acetonitrile) was linearly increased from 30% to 70% over 15 min at a flow rate of 4 mL/min. Lyophilization of pooled HPLC fractions containing 11 (tR = 10.5 min) provided 23.0 mg (20.9 μmol, 61.5%) of the compound as a colorless oil: 1H-NMR (CD3CN, 500 MHz) δH = 1.01 (s, 9H), 1.39 (d, J = 14.35 Hz, 1 H), 3.40–3.70 (m, 29H), 3.94(s, 4H), 4.66 (d, J = 8.89 Hz, 1H), 7.00–7.50 (m, 8H), 7.85 (d, J = 8.20 Hz, 1H), 8.24 (s, 1H), 8.3–8.5 (m, 2H), 8.92 (d, J = 2.73 Hz, 1H), 10.40 (s, 1H). LRMS (LC/MS-ESI) m/z: 952.3 (M+). HRMS (ESI, m/z): calcd for C48H58Cl2F2N7O7 (M+): 952.3737; found: 952.3750.
Fluorescence Polarization (FP)-based Competitive Binding Assay
The inhibitory potency of nonradioactive FN-PEG3-RG7388 (6) and FN-3AP-RG7388 (7) fluoro analogues was determined by a fluorescence polarization-based competitive binding assay using recombinant human MDM2 protein and a 5-carboxyfluorescein-labeled p53-based peptide, 5-FAM-PMDM6 (AnaSpec Inc., Fremont, CA), as the fluorescent probe. First, 5-FAM-PMDM6 (1 nM) and increasing concentrations of MDM2 (0.23–500 nM, serial 3-fold dilutions, in triplicates) were mixed in 125 μL assay buffer (100 mM potassium phosphate, pH 7.5, 100 μg/mL bovine γ-globulin, 0.02% sodium azide, with 0.01% Triton X-100 and 4% DMSO) in a 96-well flat bottom, black polystyrene NBS™ microplate (Corning, NY). The plate was incubated at RT for 15 min, and the polarization values in millipolarization units (mP) were measured at an excitation wavelength of 485 nm and an emission wavelength of 530 nm on a CLARIOstar Microplate Reader (BMG LABTECH Inc., Cary, NC). From these experiments, the MDM2 concentration (6 nM) that provided 80% binding response to the PMDM6-F peptide was selected for subsequent experiments to determine the half-maximal inhibitory concentration (IC50) values of FN-PEG3-RG7388 and FN-3AP-RG7388. Briefly, 5 μL of the test compounds in DMSO (0.5–1000 nM, serial 3-fold dilutions, triplicates) were added to 120 μL of pre-incubated MDM2 (6 nM) / 5-FAM-PMDM6 (1 nM) complex in the assay buffer described above in a NBS™ microplate. After a 15-min incubation (RT), FP values were read in the microplate reader as described above. IC50 values were determined by nonlinear regression fitting of the sigmoidal dose-dependent decrease in FP as a function of compound concentration using Graphpad Prism®, v5.01.
Radiosynthesis of [18F]FN-PEG3-RG7388 ([18F]6)
Fluorine-18, trapped on a QMA cartridge (Waters Corp, Milford, MA), was obtained from PETNET Solutions (Durham, NC). The 18F activity was eluted from the cartridge using tetraethylammonium bicarbonate (3 mg/mL) in acetonitrile and water (80/20, v/v, 1 mL). Solvents were evaporated at 100°C and the residual water was removed by azeotroping with acetonitrile (3 × 0.3 mL) at 100°C. Trimethylammonium triflate precursor 11 (0.3 mg, 0.27 μmol) in 0.15 mL anhydrous acetonitrile was added to the dried 18F activity (2.0–3.1 GBq; 53.6–83.2 mCi), and the mixture was heated at 40°C for 15 min. The crude mixture containing [18F]6 was purified using the Beckman Gold HPLC system using an XBridge C18 column eluted with 53% ethanol in sodium acetate buffer (0.05 M, pH 5.5) at a flow rate of 1.5 mL/min (tR of [18F]6 = 16.4 min). For in vitro cell studies, [18F]6 isolated from the HPLC was added directly to the cell culture medium with the result that the ethanol concentration in the medium was <0.2%. For in vivo studies in mice, HPLC-purified [18F]6 was diluted with PBS to adjust the ethanol concentration to ≤10% before injection. For assessing the molar activity (Am) of [18F]6, the [18F]6 peak was concentrated using an Oasis® Light HLB cartridge (30 mg sorbent, Waters).
Determination of [18F]6 Lipophilicity
An aliquot of [18F]6 was added to a mixture of PBS (pH 7.4) and n-octanol (2 mL each, n=6). The mixture was vortexed for 1 min followed by centrifugation at 3000 rpm for 5 min. From each tube, 50 μL of n-octanol and 500 μL of the PBS phases were drawn into separate pre-weighed Eppendorf® tubes and the radioactivity was measured using an automated gamma counter. Sample weights were converted to volume by correcting for fluid density and the distribution constant (logD7.4) was calculated as the ratio of radioactivity concentration (CPM/mL) in the octanol phase to that in the aqueous phase.
Cell Uptake and Specificity Studies
Cell uptake studies were conducted on two MDM2 expressing, wild-type p53 tumor cell lines - HepG2 human hepatocellular carcinoma cells and SJSA-1 human osteosarcoma cells. These cell lines have high MDM2 expression levels and are frequently used for assessing MDM2 inhibitors, including RG7388.16, 18 A day before the assay, cells (2 × 105 cells per well in 0.5 mL medium) were seeded in 24-well plates and incubated overnight at 37°C. On the day of experiment, medium was replaced with fresh medium (0.5 mL, FBS free) containing [18F]6 (18.5 kBq) and the cells were incubated at 37°C. After 0.5, 1 or 2 h, supernatants were collected, cells were rinsed with cold PBS (3 × 0.5 mL) and lysed using Cell Culture Lysis Reagent (0.25 mL, Promega) followed by rinsing the well with 0.25 mL PBS. Radioactivity in supernatant, wash fractions and the cell lysate from triplicate wells were measured in an automated gamma counter. The protein concentration of the cell lysates was measured by Bradford assay (BIO-RAD, Hercules, CA), and cell uptake was normalized to protein content and expressed as % uptake per mg protein for each well. The specificity of [18F]6 uptake was determined as above but with the addition of 5–50 μM RG7388 to the labeled compound in the incubation medium using a 1 h incubation at 37°C. Additionally, the specificity of [18F]6 uptake in SJSA-1 cells was determined in blocking experiments with a MDM2 inhibiting stapled peptide, VIP116 (5–50 μM).
[18F]6 Saturation Binding Assay
SJSA-1 cells were plated in 24-well plates (2 × 105 cells per well) and incubated overnight at 37°C. On the day of experiment, supernatants were removed, cells were rinsed with PBS (0.5 mL) and incubated with increasing concentrations of [18/19F]6 in triplicate for 30 min. To obtain the desired [18/19F]6 concentrations, HPLC-purified [18F]6 was mixed with unlabeled 6 in serum-free medium; serial two-fold dilutions were made from 1000 nM to 7.8 nM [18/19F]6. At each concentration, non-specific binding was assessed in parallel by co-incubating the cells with 50 μM RG7388. After a 30 min incubation, supernatants were removed and the cells were processed as described above for the cell uptake studies. The data were corrected for non-specific binding and fit using GraphPad Prism® to calculate the equilibrium dissociation constant Kd for [18/19F]6 by plotting [18/19F]6 bound per mg protein vs. concentration.
In Vitro and In Vivo Stability Studies
In vitro stability was examined by incubating [18F]6 (7.4 MBq) in human serum (0.3 mL, Innovative Research Inc., Novi, MI) at 37 °C. After 1, 2, and 4 h, a 50 μL aliquot was analyzed by analytical HPLC using a Chromolith® Performance RP-18e column (4.6 × 100 mm, Merck KGaA) eluted with a gradient of ethanol and sodium acetate buffer (0.05 M, pH 5.5). The percentage of ethanol was kept constant at 5% for the first 5 min with a 0.5 mL/min flow rate, and subsequently increased to 90% over 15 min at a flow rate of 1 mL/min.
The in vivo stability of [18F]6 was investigated after intravenous injection (13.0–16.7 MBq) into BALB/c mice. At 0.5 and 1 h post-injection, blood and urine were collected and the mice were euthanized. Samples were centrifuged at 14,000 rpm for 5 min, and the plasma and urine supernatants were analyzed by HPLC as described above for the in vitro stability studies. For both analyses, the percentage of intact [18F]6 was determined by HPLC peak integration.
Tissue Distribution Studies
Animal experiments were conducted in accordance with the regulations of the Duke University Institutional Animal Care and Use Committee (IACUC) under an IACUC-approved protocol. Tissue distribution experiments were conducted in athymic male mice (20–25 g, Jackson Labs, Farmington, CT) bearing HepG2 or SJSA-1 xenografts implanted in the flank. For these experiments, 5 × 106 cells suspended in a mixture of Matrigel® (Corning, NY) and cell culture medium (1:1, 0.1 mL) were injected into the flank and tissue distribution studies were initiated 3–4 weeks later. Groups of five mice were injected via the tail vein with 740 kBq (20 μCi) of [18F]6 in 0.1 mL PBS (10% ethanol), and euthanized by isoflurane overdose at 0.5, 2 and 4 h post-injection for the HepG2 tumor model and 1 and 2 h for the SJSA-1 tumor model. Blood, urine, tumor and all major organs were collected, blot-dried, weighed and measured for radioactivity in an automated gamma counter. To determine the specificity of [18F]6 uptake in SJSA-1 tumors, an additional group (n=4) was injected intraperitoneally with RG7388 (35 mg/kg body weight) 1 h before receiving [18F]6, and tissue distribution was studied after 1 h. Biodistribution data for [18F]6 were expressed as percent injected dose per gram of tissue (% ID/g).
Statistical Analysis
Values are reported as mean ± SD. The statistical significance of differences in uptake with/without blocking in cells or in mice was determined by Student’s t-test using GraphPad QuickCalcs; a P value of ≤0.05 was considered significant.
RESULTS
Chemistry
As shown in Scheme 1, the fluorinated analogues FN-PEG3-RG7388 (6) and FN-3AP-RG7388 (7) were synthesized by conjugating commercially available 6-fluoropyridine-3-carboxylic acid 1 with RG7388 via a PEG3 or a propyl linker in three steps. The quaternary salt precursor 11 for 18F labeling was synthesized starting with the tetrafluorophenyl activated ester derivative 8 (Scheme 2), like the synthesis of nonradioactive reference compounds 6 and 7. In both cases, the pyridine reagent with the PEG3 or propyl linker was synthesized first (compounds 4, 5, 10), and then conjugated to RG7388 using a standard amide coupling reaction in 61–78% yield. The 1H-NMR (Figures S1–S9) and mass spectrometry data for compounds 2–11, and 19F-NMR data for compounds 6 and 7 (Figures S10 and S11) were consistent with their structures.
Scheme 1.
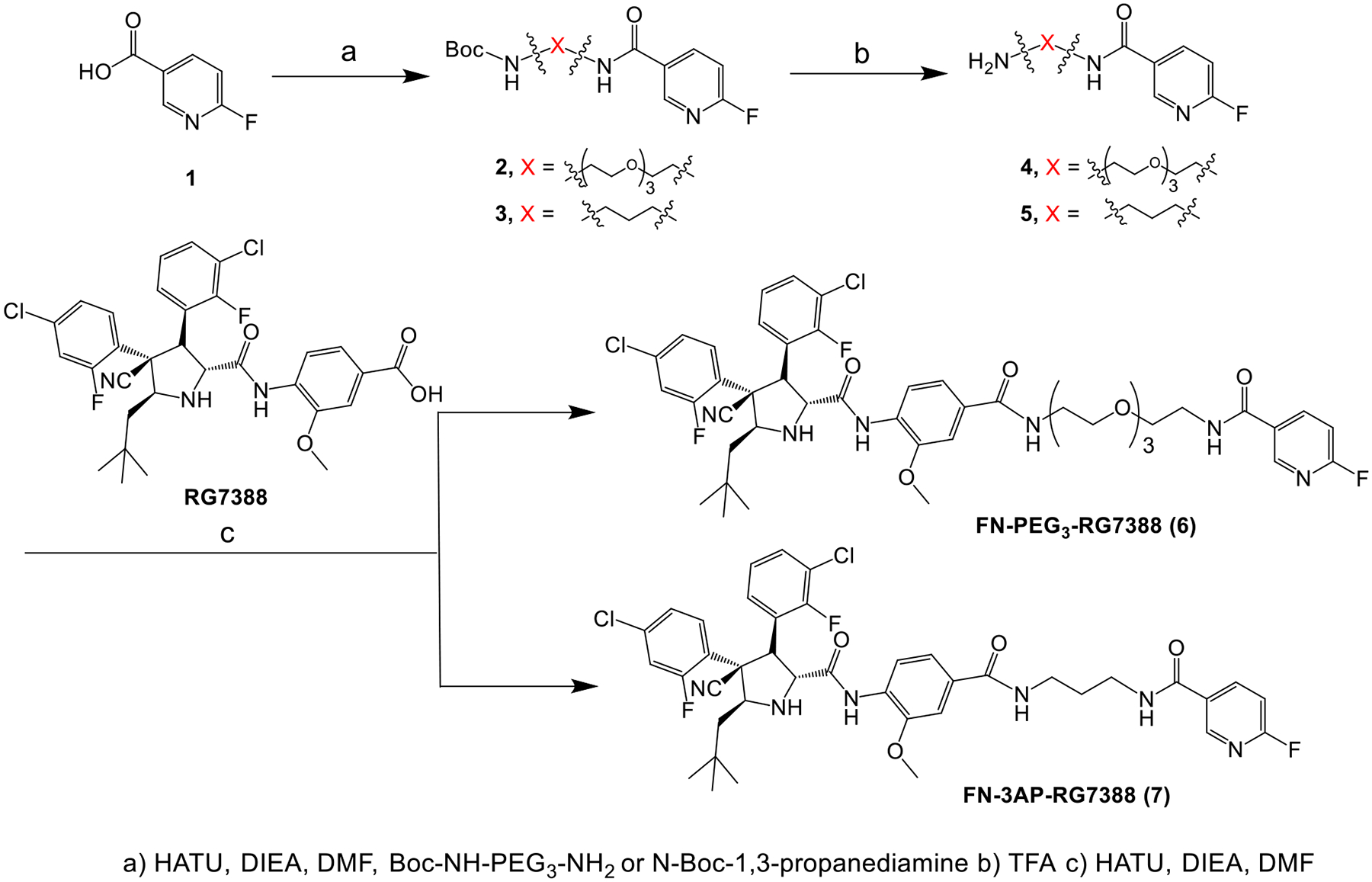
Synthesis of FN-X-RG7388
Scheme 2.
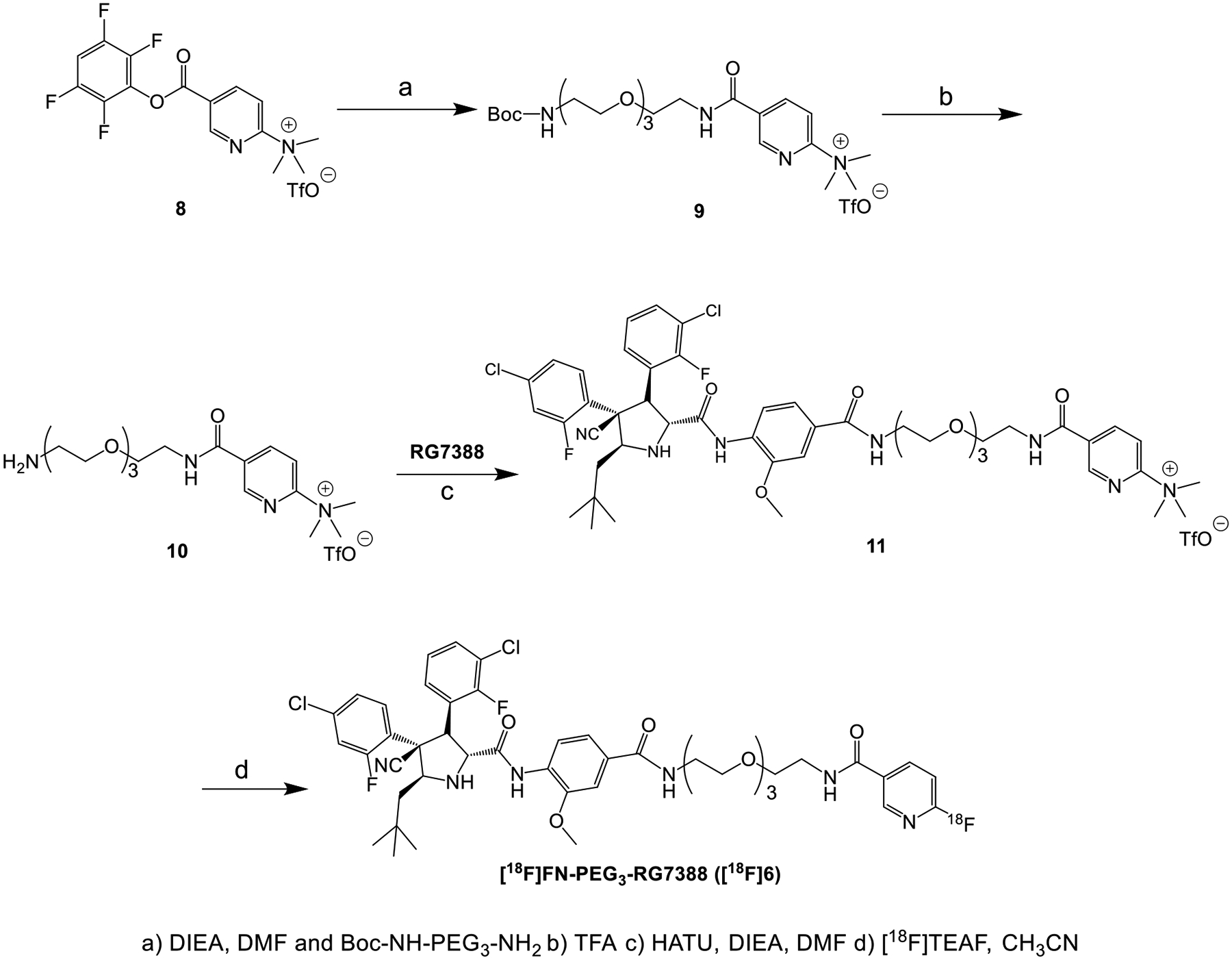
Synthesis of [18F]FN-PEG3-RG7388
Fluorescence Polarization (FP)-based Competitive Binding Studies
The ability of the nonradioactive fluoro analogues 6 and 7 to bind MDM2 and inhibit the MDM2-p53 interaction was evaluated by an FP-based assay. The binding affinity (Kd) of the FAM-labeled peptide towards MDM2, determined at 1 nM 5-FAM-PMDM6 with increasing concentrations of MDM2 protein (Figure S16), was 0.96 ± 0.2 nM, consistent with that reported in the literature (1.0 ± 0.1 nM) for this peptide by the same assay.19 Next, the IC50 (inhibitor concentration at which half of the bound 5-FAM-PMDM6 peptide was displaced from MDM2) of the test compounds was determined at fixed concentrations of 5-FAM-PMDM6 (1 nM) and MDM2 (6 nM) and 0.5–1000 nM of the fluoro analogues or the parent compound RG7388. The IC50 of 6 and 7 was 118.9 nM and 159.8 nM, respectively, compared to 8.7 nM for RG7388 (Figure 1).
Figure 1.
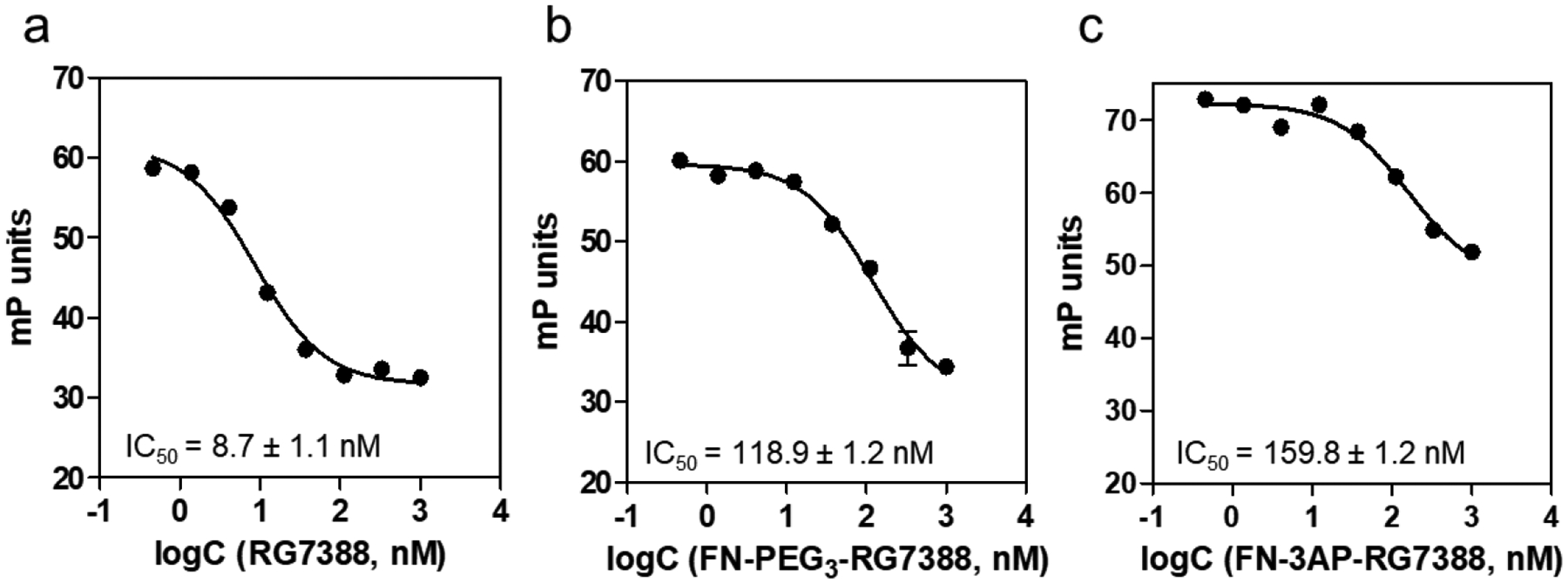
Determination of inhibitory potency of RG7388 (a), compound 6 (b) and 7 (c) through fluorescence polarization-based saturation experiments.
Radiochemistry
Based on its higher inhibitory potency, FN-PEG3-RG7388 (6) was labeled with 18F using the quaternary salt precursor 11 and [18F]tetraethylammonium fluoride ([18F]TEAF) via SNAr reaction (n=6; Scheme 2). About 0.74 GBq of the labeled product [18F]FN-PEG3-RG7388 ([18F]6) could be obtained starting with approximately 2.3 GBq of aqueous [18F]fluoride, resulting in a non-decay corrected radioactivity yield of about 32%. The decay-corrected radiochemical yield (RCY) was 50.3 ± 7.5% and the molar activity (Am) was 194.1 ± 19.1 GBq/μmol (n=3). The radiochemical purity was >98%, and the total synthesis time was about 65 min. The identity of [18F]6 was confirmed by comparing its retention time with that for unlabeled 6 on analytical HPLC (Figure S18).
Determination of Lipophilicity
The lipophilicity of [18F]6 was determined by its partitioning between PBS (pH 7.4) and n-octanol by the shake-flask method.13 [18F]6 exhibited a logD7.4 of 3.0 ± 0.1 (n=12).
Cell Uptake Studies
The ability of [18F]6 to bind to MDM2 expressing human tumor cells was assessed using the wild-type p53 SJSA-1 osteosarcoma and HepG2 hepatocellular carcinoma cell lines. The uptake and specificity of [18F]6 binding was assessed by co-incubating the cells with 5–50 μM RG7388 as a blocking agent for 1 h. RG7388 exerted a concentration-dependent inhibition of [18F]6 uptake on both cell lines (Figures 2a and 2b). Greater than 80% inhibition was observed with 50 μM RG7388, which decreased [18F]6 uptake from 780 ± 52 %/mg to 97 ± 16 %/mg protein in SJSA-1 cells, and from 361 ± 25 %/mg to 65 ± 5 %/mg in HepG2 cells. In a subsequent study investigating [18F]6 uptake kinetics from 30 to 120 min, maximal uptake occurred at 1 h with values of 1088 ± 84 %/mg and 559 ± 23 %/mg protein observed in SJSA-1 and HepG2 cells, respectively (Figures 2c and 2d). Blocking using 25 μM RG7388 significantly decreased [18F]6 uptake in both cell lines at all time points.
Figure 2.
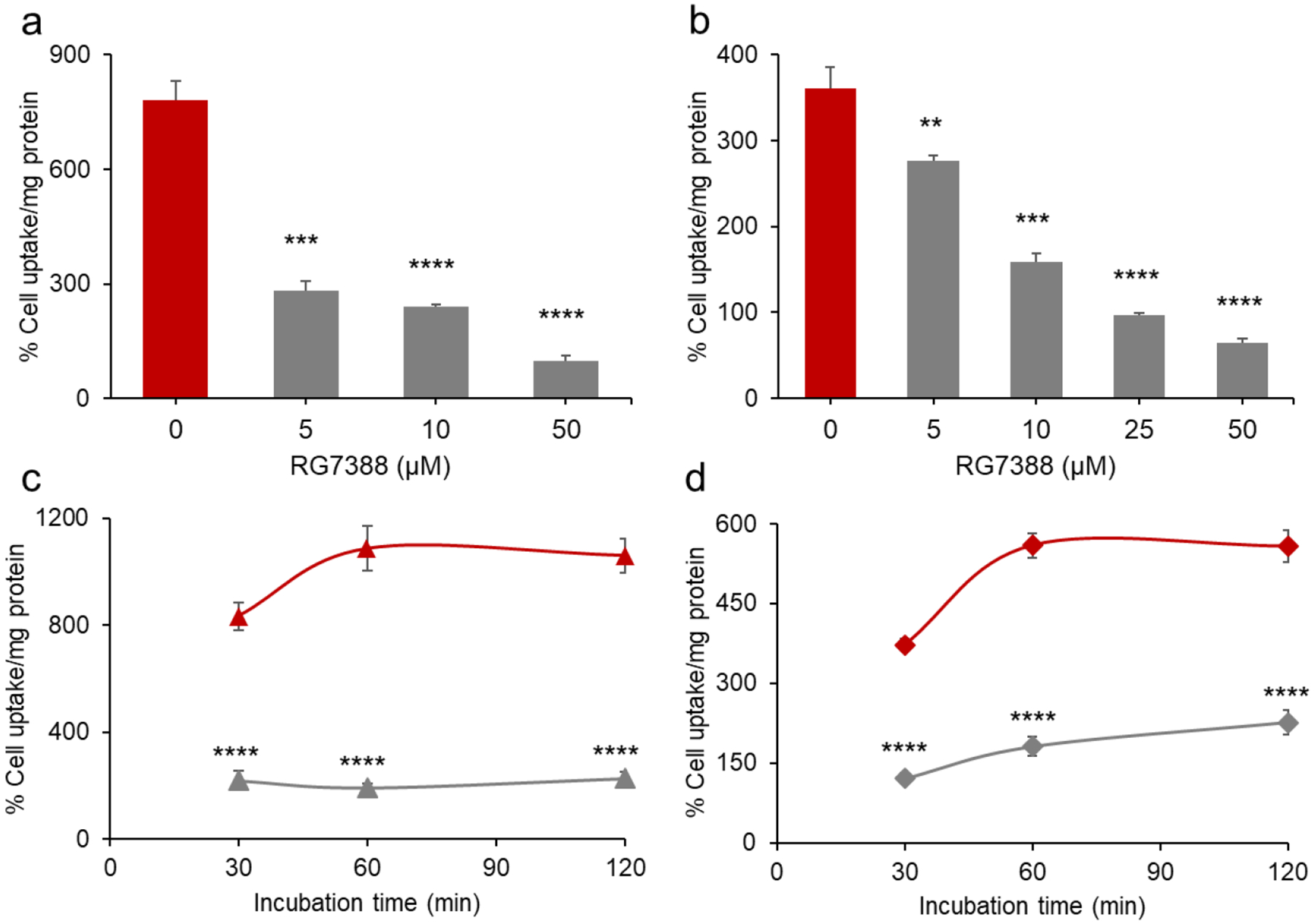
Uptake of [18F]6 in MDM2 expressing wild-type p53 tumor cell lines, (a,c) SJSA-1 and (b,d) HepG2. (a,b) Cells were incubated with [18F]6 and varying concentrations of RG7388 (0 – 50 μM) to assess [18F]6 uptake for 1 h. (c,d) Changes in [18F]6 uptake in tumor cells with (grey) or without (red) RG7388 (25 μM). Data are shown as mean ± SD for triplicates. **p ≤ 0.01, ***p ≤ 0.001, ***p ≤ 0.0001 vs. no RG7388 co-incubation (0 μM).
VIP116, a rationally designed stapled peptide and a potent inhibitor of the MDM2-p53 interaction (Kd = 15.1 nM),20, 21 was also tested as a blocking agent to confirm [18F]6 uptake specificity in SJSA-1 cells. As expected, co-incubation of SJSA-1 cells with [18F]6 and 5–50 μM VIP116 significantly decreased [18F]6 uptake, with >95% inhibition observed at 10 and 50 μM VIP116 (Figure 3a). Taken together, these studies confirmed the high and specific uptake of [18F]6 in MDM2 expressing tumor cells and suggest that, similar to RG7388, [18F]6 binds in the p53-binding pocket of the MDM2 protein.
Figure 3.
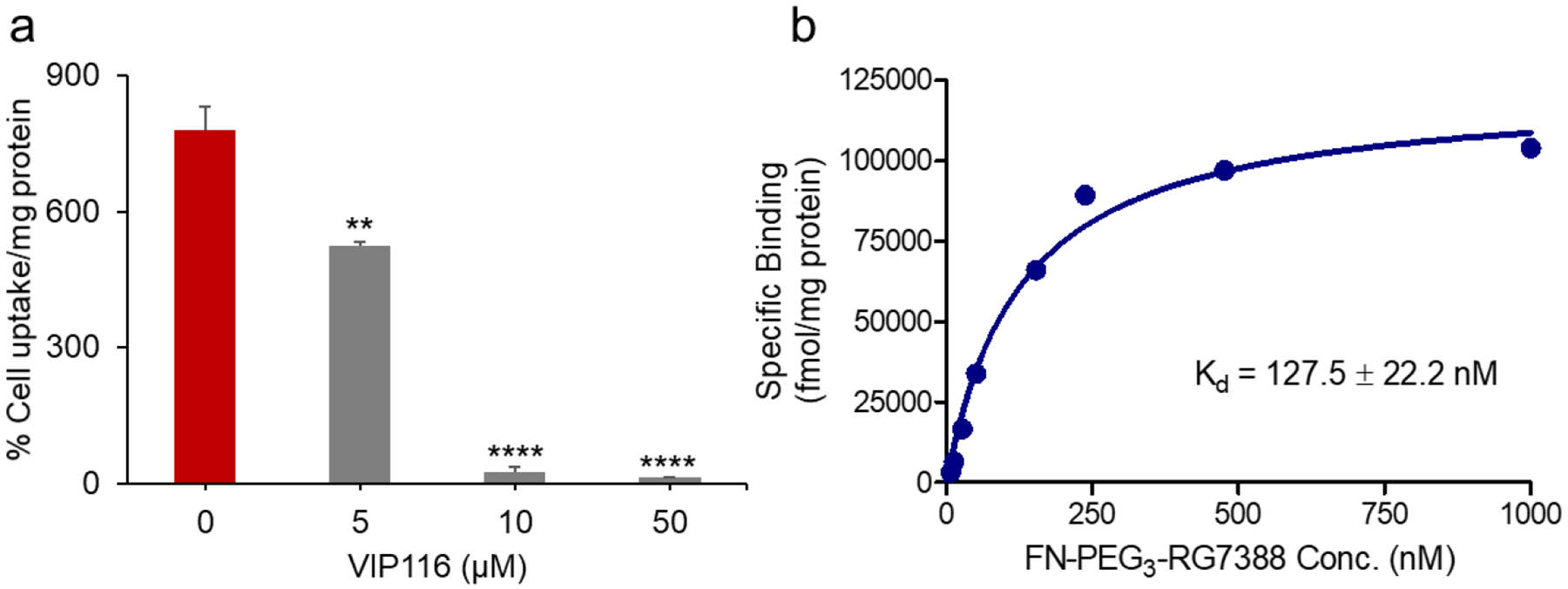
a) Uptake of [18F]6 in SJSA-1 tumor cells co-incubated with varying concentrations of the stapled peptide VIP116 (0 – 50 μM) was determined at 1 h post incubation, b) Binding affinity curve for [18F]6 on SJSA-1 cells. SJSA-1 cells were incubated with increasing concentrations of [18F]6, and the uptake was determined at 30 min post incubation. Nonspecific binding was assessed in parallel in cells that were exposed to RG7388 (50 μM). Data are shown as mean ± SD for triplicates. **p < 0.01, ***p < 0.0001 vs. no VIP116 co-incubation (0 μM).
Saturation Binding Assay
The binding affinity of [18F]6 on SJSA-1 cells was determined using a saturation binding assay. Because of the high molar activity of [18F]6 (~195 GBq/μmol), its nonradioactive counterpart 6 was added to obtain the desired concentration range (7.8–1000 nM). Nonspecific binding was determined in parallel by addition of 50 μM RG7388, a concentration that blocked >80% of [18F]6 cell uptake without significantly changing cell morphology during the 2 h experimental period. Nonlinear regression analysis of these binding data revealed a Kd of 127.5 ± 22.2 nM for [18F]6 on SJSA-1 cells (Figure 3b).
In vitro and in vivo stability studies
The in vitro stability of [18F]6 was examined by incubating the labeled compound with human serum at 37°C and analyzed by HPLC. After 1, 2 and 4 h, ~100% [18F]6 remained intact, indicating that the labeled compound is stable in human serum in vitro for up to 4 h (Figure 4a).
Figure 4.
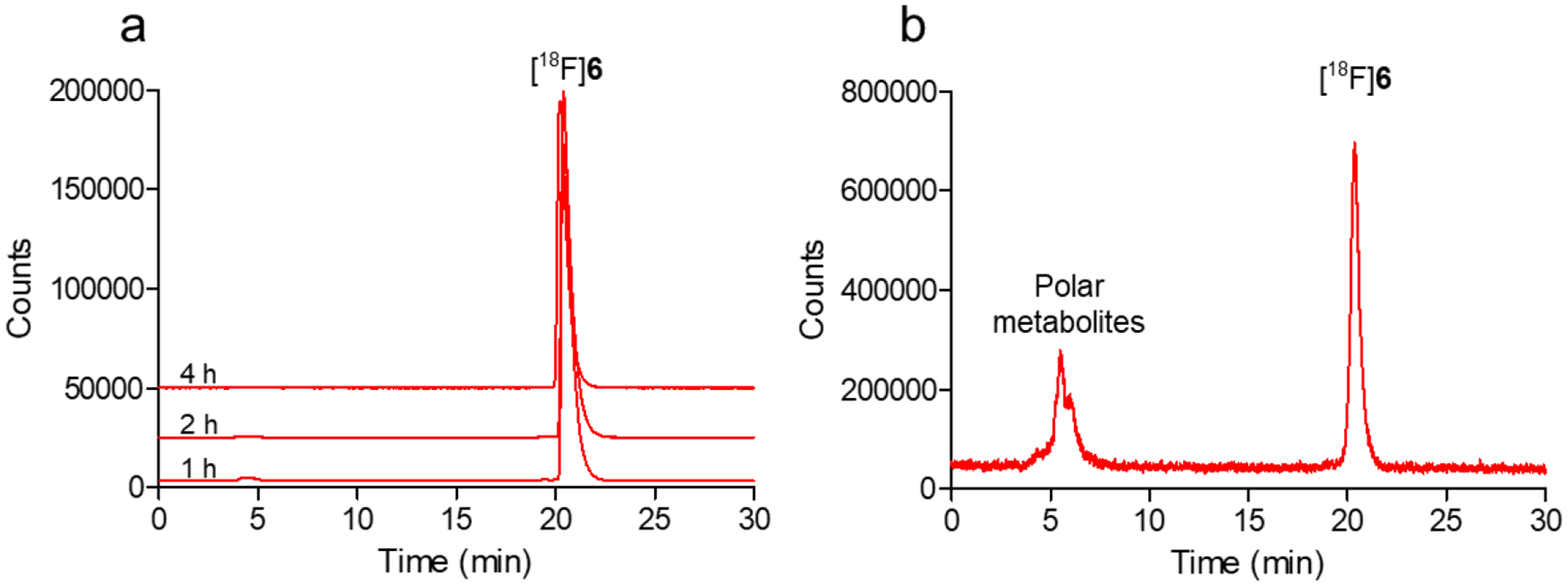
In vitro and in vivo stability of [18F]6. (a) Stability of [18F]6 in human serum after 1–4 h incubation in vitro; (b) Percentage of intact [18F]6 in mouse plasma at 30 min p.i. of the labeled compound in vivo
The stability of [18F]6 was also investigated after injection in BALB/c mice and analysis of plasma and urine samples by HPLC at 0.5 and 1 h post-injection. Figure 4b shows the HPLC profile of [18F]6 in mouse plasma at 30 min post-injection when about 59% of [18F]6 remained intact; this value decreased to 30% at 1 h. At both time points, the remaining 18F-activity was attributed to polar peaks, eluting at 5.5 min compared to a 20.4 min retention time for intact [18F]6. In urine, nearly all of the 18F-activity was associated with the aforementioned polar peaks (tR = ~5.5 min, Figure S20) at both time points, suggesting that unmetabolized [18F]6 is not excreted via kidneys, consistent with its lipophilicity (logD7.4 = 3.0).
Tissue Distribution Studies in HepG2 and SJSA-1 Xenografts
The normal tissue distribution and tumor uptake of [18F]6 were studied initially in athymic mice bearing HepG2 tumors followed by specificity studies in mice with SJSA-1 xenografts. Table 1 shows the tissue distribution of [18F]6, presented as % ID/g, in the HepG2 xenograft model. [18F]6 exhibited a fast clearance from the blood, with <1% ID/g remaining at 2 h compared to 3.90 ± 0.70% ID/g at 30 min. At 30 min, the highest radioactivity levels were observed in kidneys (15.04 ± 2.66%ID/g) followed by liver (10.24 ± 2.51%ID/g), suggesting both renal and hepatobiliary clearance of 18F-activity. No significant uptake was observed in the bone (0.65 ± 0.25%ID/g at 4 h), indicating minimal [18F]defluorination in vivo.
Table 1.
Biodistribution of [18F]6 in Mice Bearing HepG2 Xenografts1
| Organ | 30 min | 2 h | 4 h |
|---|---|---|---|
| Liver | 10.24 ± 2.51 | 2.14 ± 0.82 | 1.24 ± 0.82 |
| Spleen | 4.08 ± 1.04 | 0.79 ± 0.18 | 0.31 ± 0.04 |
| Lungs | 3.59 ± 0.90 | 0.81 ± 0.33 | 0.38 ± 0.12 |
| Heart | 3.52 ± 0.42 | 0.72 ± 0.16 | 0.26 ± 0.10 |
| Kidneys | 15.04 ± 2.66 | 2.54 ± 0.54 | 1.01 ± 0.33 |
| Bladder | 6.01 ± 5.14 | 2.55 ± 2.73 | 1.60 ± 1.96 |
| Stomach | 2.15 ± 0.80 | 1.72 ± 0.51 | 0.94 ± 0.26 |
| Sm. Int. | 9.46 ± 2.42 | 5.77 ± 2.59 | 1.89 ± 0.79 |
| Lg. Int. | 1.84 ± 0.43 | 16.28 ± 2.63 | 16.95 ± 1.45 |
| Bone | 1.58 ± 0.49 | 0.72 ± 0.13 | 0.65 ± 0.25 |
| Muscle | 2.43 ± 0.41 | 0.88 ± 0.18 | 0.39 ± 0.10 |
| Blood | 3.90 ± 0.70 | 0.83 ± 0.21 | 0.29 ± 0.12 |
| Brain | 0.84 ± 0.12 | 0.39 ± 0.15 | 0.16 ± 0.06 |
| Tumor | 3.80 ± 0.85 | 2.07 ± 0.41 | 0.98 ± 0.33 |
| tumor/muscle | 1.62 ± 0.55 | 2.46 ± 0.74 | 2.49 ± 0.58 |
| tumor/blood | 0.99 ± 0.22 | 2.66 ± 1.00 | 3.54 ± 1.25 |
| tumor/liver | 0.37 ± 0.03 | 1.09 ± 0.42 | 1.00 ± 0.52 |
Data presented as %ID/g
Comparison of the [18F]6 tissue distribution data between the two models at 2 h revealed somewhat faster clearance in SJSA-1 tumor-bearing mice than in HepG2 tumor-bearing mice, including in tumors (SJSA-1, 0.65 ± 0.12%ID/g; HepG2, 2.07 ± 0.41%ID/g). Additionally, the 18F-activity level in the large intestine was considerably lower in the SJSA-1 biodistribution study that likely reflected enhanced hepatobiliary clearance of the labeled compound through the feces. In HepG2 xenografts, the uptake of [18F]6 was highest at 30 min (3.80 ± 0.85%ID/g) but decreased to 0.98 ± 0.33%ID/g at 4 h post-injection. In SJSA-1 xenografts, [18F]6 exhibited peak tumor uptake (1.32 ± 0.32%ID/g) at 1 h, decreasing significantly to 0.57 ± 0.18% ID/g (P <0.01) when mice were pre-treated with a blocking dose of RG7388 (35 mg/kg, i.p.), indicating the specificity of [18F]6 tumor uptake in vivo. The higher tumor uptake of [18F]6 in HepG2 tumors resulted in more favorable tumor-to-blood and tumor-to-muscle ratios at 2 h than those observed in SJSA-1 xenografts.
DISCUSSION
p53 is a key tumor suppressor protein that is often inactivated in tumors due to overexpression of its negative regulatory protein, MDM2. Therefore, MDM2 is an important target for drug development, particularly for cancers that are MDM2-amplified or have high MDM2 protein expression levels.22 To date, several small molecule MDM2 inhibitors have been evaluated in clinical trials including RG7112, RG7388, and AMG232.6, 7, 9 The ability to noninvasively image MDM2 could facilitate the clinical development of MDM2-targeted agents and the assessment of treatment response;7 however, to the best of our knowledge, there are no methods currently available for noninvasive imaging of MDM2 at the protein level. With the goal of developing 18F-labeled agents for imaging MDM2 with PET, we have previously evaluated an 18F-labeled analogue of the well-studied small molecule MDM2 inhibitor SP-141 for this purpose. While the labeled SP-141 derivative had high uptake and specificity in MDM2 expressing tumor cell lines, it exhibited suboptimal biodistribution characteristics in mice, with poor clearance of 18F-activity from normal tissues.13
Encouraged by the promising in vitro characteristics of the 18F-labeled SP-141 analogue, in the present study, we evaluated two clinically-validated small molecule inhibitors, RG7388 and AMG232, as potential molecular scaffolds for developing 18F-labeled tracers for MDM2. Similar to RG7388, AMG232 is a potent inhibitor of MDM2 and restores p53 function in cancer cells by inhibiting the MDM2-p53 interaction with an IC50 of 0.6 nM.23 Besides having high inhibitory potency against MDM2, both RG7388 and AMG232 have a carboxyl group that can serve as a site for functionalizing, which was done herein with a fluoronicotinyl moiety for the purpose of 18F-labeling. This is an attractive strategy because this carboxyl group does not participate in the binding of these molecules with the hydrophobic Phe19, Trp23 and Leu26 binding pocket on MDM2.16, 24 However, co-crystal structure of a close structural analogue of AMG232 bound to MDM2 showed that the carboxyl group of AMG232 may form a hydrogen bond interaction with the adjacent His96 side chain of MDM2.23 Nonetheless, in the present study, we used a PEG3 or a propyl spacer via amide bonds to extend the [18F]fluoronicotinyl moiety away from the binding pocket to minimize its potential interference in the binding interaction of the molecules with MDM2 (Schemes 1 and S1).
Unexpectedly both modifications significantly impacted the inhibitory potency of the new RG7388 and AMG232 derivatives against MDM2 in FP assays. For RG7388 analogues 6 and 7, IC50 values increased by >10-fold (IC50, 6 = 118.9 nM, 7 = 159.8 nM) compared to RG7388 (IC50 = 8.7 nM) in the same assay (Figure 1). Likewise, IC50 values for the AMG232 analogues also increased by ≥ 6-fold, 12.0 nM for the PEG3 derivative 13 and 37.9 nM for 14 with the propyl linker compared to 2.0 nM for AMG232 (Figure S17). Our results for both RG7388 and AMG232 indicate better retention of inhibitory potency for compounds with the PEG3 linker (6 and 13) compared with those with the shorter chain length propyl linker (7 and 14),.
Based on the acceptable inhibitory potency exhibited by the two AMG232 derivatives, we labeled FN-3AP-AMG232 (14) with 18F via the same labeling approach as that for [18F]6 (Scheme S2) to assess the potential utility of AMG232 as a template for developing 18F-labeled compounds for PET imaging. Surprisingly, despite its nanomolar inhibitory potency against MDM2 (IC50 = 37.9 nM), [18F]14 showed negligible uptake (<2%) in SJSA-1 cells. However, co-incubation of cells with [18F]14 and 50 μM AMG232 resulted in a tenfold increase in [18F]14 uptake by SJSA-1 cells (20.1 ± 0.3% input dose, p < 0.0001, Figure S19). Furthermore, similarly enhanced cell [18F]14 uptake was observed when SJSA-1 cells were co-incubated with 50 μM verapamil or cyclosporine A, suggesting a potential role of efflux transporters (e.g., P-glycoprotein, organic anion transporting polypeptides, CYP3A4)25, 26 in limiting the cellular uptake of [18F]14 at tracer levels. Of note, it has been shown previously that RG7388 is not a good substrate of P-glycoprotein in neuroblastoma cells,27 which might explain the differences in the results obtained with [18F]6 vs. [18F]14. In view of these results and the limited uptake of [18F]14 in SJSA-1 cells, no additional experiments were conducted with the AMG232 derivatives, and [18F]FN-PEG3-RG7388 ([18F]6) was selected for further evaluation.
The equilibrium dissociation constant of [18F]6 (Kd, 127.5 nM) in the saturation binding assay with SJSA-1 cells was in line with its IC50 value (118.9 nM) measured in the FP assay using its nonradioactive counterpart 6, confirming that modification of the carboxyl group in RG7388 had a detrimental effect on its binding affinity toward MDM2. Although [18F]6 showed excellent stability in human serum for up to 4 h in vitro, we observed significant metabolism of [18F]6 in mice, with ~41% of the 18F activity in plasma corresponding to more polar species at 30 min post-injection. The mechanism(s) responsible for the instability of [18F]6 in vivo is not known at this time but likely reflects its metabolism by the liver or other metabolic organs in vivo. Although RG7388 has been shown to be fully stable in humans in vivo,17 Lai et al. reported the formation of polar radiometabolites in mice from a small molecule labeled via the same [18F]fluoronicotinyl moiety. They hypothesized that these species were derived from metabolic conversion of the [18F]fluoronicotinic acid functionality to [18F]fluoronicotinate mononucleotide and/or its derivatives via the NAD biosynthesis pathways in vivo.28, 29 Thus, it is possible that the polar labeled metabolites observed for [18F]6 in mice arise from metabolic breakdown of the [18F]fluoronicotinyl moiety; however, additional studies are needed to confirm the identity of these labeled metabolites. Additionally, future studies should include investigation of alternative 18F-labeling strategies in order to provide a more stable 18F-labeled RG7388 derivative for PET imaging.
[18F]6 showed a good uptake in HepG2 tumor xenografts (3.80 ± 0.85% ID/g) at 30 min post-injection, which should be compatible for PET imaging with 110-min half-life 18F. However, contrary to the high uptake observed in cell uptake studies, [18F]6 showed only a modest uptake in SJSA-1 xenografts in vivo (1.32 ± 0.32%ID/g at 1 h). Nonetheless, blocking studies conducted with the parent compound RG7388 demonstrated a more than twofold, significant decrease in [18F]6 uptake in SJSA-1 tumors and significantly lower tumor-to-muscle and tumor-to-blood ratios (Table 2), confirming the specificity of tumor [18F]6 uptake. Although specific, the overall tumor uptake levels and tumor-to-background ratios for [18F]6 need to be improved for successful translation of the labeled compound to PET imaging in vivo.
Table 2.
Biodistribution of [18F]6 in Mice Bearing SJSA-1 Xenografts1
| Organ | 1 h | 1 h Block2 | 2 h |
|---|---|---|---|
| Liver | 1.63 ± 0.35 | 1.27 ± 0.66 | 0.99 ± 0.47 |
| Spleen | 1.02 ± 0.29 | 0.79 ± 0.20 | 0.42 ± 0.14 |
| Lungs | 1.17 ± 0.40 | 1.38 ± 0.43 | 0.49 ± 0.15 |
| Heart | 1.16 ± 0.36 | 1.23 ± 0.26 | 0.48 ± 0.11 |
| Kidneys | 3.95 ± 1.60 | 2.93 ± 1.30 | 1.73 ± 0.74 |
| Bladder | 7.28 ± 9.23 | 7.39 ± 12.16 | 1.15 ± 0.63 |
| Stomach | 0.75 ± 0.33 | 0.50 ± 0.36 | 0.49 ± 0.14 |
| Sm. Int. | 2.85 ± 0.24 | 2.37 ± 1.48 | 1.39 ± 0.83 |
| Lg. Int. | 0.55 ± 0.36 | 0.92 ± 0.34 | 2.13 ± 0.83 |
| Bone | 0.61 ± 0.31 | 0.60 ± 0.18 | 0.37 ± 0.07 |
| Muscle | 0.85 ± 0.36 | 0.94 ± 0.28 | 0.53 ± 0.13 |
| Blood | 1.08 ± 0.25 | 1.03 ± 0.28 | 0.57 ± 0.11 |
| Brain | 0.45 ± 0.06 | 0.53 ± 0.08 | 0.19 ± 0.06 |
| Tumor | 1.32 ± 0.32 | 0.57 ± 0.18* | 0.65 ± 0.12 |
| tumor/muscle | 1.64 ± 0.26 | 0.66 ± 0.29* | 1.29 ± 0.45 |
| tumor/blood | 1.22 ± 0.02 | 0.60 ± 0.23* | 1.15 ± 0.13 |
| tumor/liver | 0.82 ± 0.08 | 0.52 ± 0.22* | 0.73 ± 0.21 |
Data presented as%ID/g;
Mice received RG7388 (1 mg) i.p. 1 h before i.v. [18F]6.
p<0.05 vs 1 h nonblocking group (n =4)
Decreased binding affinity and in vivo metabolism are likely factors contributing to the low tumor uptake of [18F]6 in vivo. While the carboxyl of RG7388 has been successfully modified by conjugation of other small molecules (e.g., BET inhibitor, Bcl-2 inhibitor) via a PEG3 or propyl linker, without a major loss in inhibitory potency,30, 31 this was not the case in our study with RG7388 derivatives 6 and 7. In a recent study investigating the potential utility of RG7388 as a clickable probe for cellular MDM2 imaging, Lebraud et al. modified the methoxy function to attach a trans-cyclo-octene (TCO) moiety via a propyl linker, with only a minor loss in inhibitory potency.32 Thus, modification the methoxy group of RG7388 to introduce an 18F-labeled function may address the current limitations of [18F]6, with minimal perturbation in MDM2 binding affinity. We speculate that this could lead to more favorable tumor uptake of the labeled compound in vivo, a strategy that we are currently pursuing.
CONCLUSION
In summary, we synthesized two fluorinated analogues of the clinically validated MDM2 inhibitor RG7388 and successfully radiolabeled one with 18F using a 6-[18F]fluoronicotinyl moiety. The labeled compound [18F]6 showed high uptake and specific binding in MDM2 expressing wild-type p53 SJSA-1 and HepG2 tumor cell lines. Specific tumor uptake of [18F]6 was demonstrated by pre-treatment with RG7388 in mice bearing SJSA-1 tumors. However, low in vivo stability and reduced binding affinity compromised the current design and resulted in low tumor uptake and tumor-to-background ratios for [18F]6 in vivo. Nonetheless, our results suggest that RG7388 is a promising molecular scaffold for future development of 18F-labeled MDM2-targeted probes. Labeling strategies utilizing different functional groups on RG7388 such as the methoxy group are currently being evaluated to improve binding affinity and in vivo stability with the goal of providing a more suitable tracer for PET imaging of MDM2.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Xiao-Guang Zhao for outstanding technical support with the animal studies, Dr. So Young Kim for helpful discussions on the FP assays, Dr. Fernando Ferrer, A*STAR, Singapore, for providing the VIP116 peptide and the Duke University School of Medicine for funding our use of Functional Genomics Core Facility for the FP assays. This research was supported by Grant CA241823 from the National Cancer Institute.
Footnotes
SUPPORTING INFORMATION
Synthesis of FN-PEG3-AMG232 (13) and FN-3AP-AMG232 (14); 18F-labeling of FN-3APAMG232 ([18F]14); NMR spectra; binding affinity of 5-FAM-PMDM6 towards MDM2; IC50 curves of AMG232 and its fluorinated analogues 13 and 14; identity confirmation of [18F]6; cell uptake data of [18F]FN-3AP-AMG232 ([18F]14); urine analysis of [18F]6 in mice.
DISCLOSURE
The authors declare no competing financial interest.
References
- 1.Piette J; Neel H; Marechal V MDM2: Keeping p53 Under Control. Oncogene 1997, 15, 1001–10. [DOI] [PubMed] [Google Scholar]
- 2.Leach FS; Tokino T; Meltzer P, et al. p53 Mutation and MDM2 Amplification in Human Soft Tissue Sarcomas. Cancer Res. 1993, 53, 2231–4. [PubMed] [Google Scholar]
- 3.Reifenberger G; Liu L; Ichimura K, et al. Amplification and Overexpression of the MDM2 Gene in a Subset of Human Malignant Gliomas without p53 Mutations. Cancer Res. 1993, 53, 2736–9. [PubMed] [Google Scholar]
- 4.Meng X; Franklin DA; Dong J, et al. MDM2–p53 Pathway in Hepatocellular Carcinoma. Cancer Res. 2014, 74, 7161–7167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Araki S; Eitel JA; Batuello CN, et al. TGF-beta1-Induced Expression of Human MDM2 Correlates with Late-Stage Metastatic Breast Cancer. J. Clin. Invest 2010, 120, 290–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andreeff M; Kelly KR; Yee K, et al. Results of the Phase I Trial of RG7112, a Small-Molecule MDM2 Antagonist in Leukemia. Clin. Cancer Res 2016, 22, 868–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reis B; Jukofsky L; Chen G, et al. Acute Myeloid Leukemia Patients’ Clinical Response to Idasanutlin (RG7388) Is Associated with Pre-Treatment MDM2 Protein Expression in Leukemic Blasts. Haematologica 2016, 101, e185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meric-Bernstam F. a.; Saleh MN; Infante JR, et al. Phase I Trial of a Novel Stapled Peptide ALRN-6924 Disrupting MDMX-and MDM2-Mediated Inhibition of WT p53 in Patients with Solid Tumors and Lymphomas. J. Clin. Oncol 2017, 35, S2505. [Google Scholar]
- 9.Gluck WL; Gounder MM; Frank R, et al. Phase 1 Study of the MDM2 Inhibitor AMG232 in Patients with Advanced p53 Wild-Type Solid Tumors or Multiple Myeloma. Invest. New Drug 2020, 38, 831–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sirvent N; Coindre J-M; Maire G, et al. Detection of MDM2-CDK4 Amplification by Fluorescence In Situ Hybridization in 200 Paraffin-Embedded Tumor Samples: Utility in Diagnosing Adipocytic Lesions and Comparison with Immunohistochemistry and RealTime Pcr. Am. J. Surg. Pathol 2007, 31, 1476–1489. [DOI] [PubMed] [Google Scholar]
- 11.Song MJ; Cho K-J; Lee J-S, et al. Application of MDM2 Fluorescence in Situ Hybridization and Immunohistochemistry in Distinguishing Dedifferentiated Liposarcoma from Other High-Grade Sarcomas. Appl. Immunohistochem. Mol. Morphol 2017, 25, 712–719. [DOI] [PubMed] [Google Scholar]
- 12.Pantel AR; Mankoff DA Molecular Imaging to Guide Systemic Cancer Therapy: Illustrative Examples of PET Imaging Cancer Biomarkers. Cancer Lett. 2017, 387, 25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chitneni SK; Zhou Z; Watts BE, et al. Feasibility of Developing Radiotracers for MDM2: Synthesis and Preliminary Evaluation of an 18F-Labeled Analogue of the MDM2 Inhibitor SP-141. Pharmaceuticals 2021, 14, 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang W; Qin J-J; Voruganti S, et al. The Pyrido[b]Indole MDM2 Inhibitor SP-141 Exerts Potent Therapeutic Effects in Breast Cancer Models. Nat. Commun 2014, 5, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vassilev LT; Vu BT; Graves B, et al. In Vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science 2004, 303, 844–848. [DOI] [PubMed] [Google Scholar]
- 16.Ding Q; Zhang Z; Liu J-J, et al. Discovery of RG7388, a Potent and Selective p53–MDM2 Inhibitor in Clinical Development. J. Med. Chem 2013, 56, 5979–5983. [DOI] [PubMed] [Google Scholar]
- 17.Pápai Z; Chen L-C; Da Costa D, et al. A Single-Center, Open-Label Study Investigating the Excretion Balance, Pharmacokinetics, Metabolism, and Absolute Bioavailability of a Single Oral Dose of [14C]-Labeled Idasanutlin and an Intravenous Tracer Dose of [13C]-Labeled Idasanutlin in a Single Cohort of Patients with Solid Tumors. Cancer Chemother. Pharmacol 2019, 84, 93–103. [DOI] [PubMed] [Google Scholar]
- 18.Wang W; Hu B; Qin J-J, et al. A Novel Inhibitor of MDM2 Oncogene Blocks Metastasis of Hepatocellular Carcinoma and Overcomes Chemoresistance. Genes Dis. 2019, 6, 419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding K; Lu Y; Nikolovska-Coleska Z, et al. Structure-Based Design of Spiro-Oxindoles as Potent, Specific Small-Molecule Inhibitors of the MDM2−p53 Interaction. J. Med. Chem 2006, 49, 3432–3435. [DOI] [PubMed] [Google Scholar]
- 20.Thean D; Ebo J; Luxton T, et al. Enhancing Specific Disruption of Intracellular Protein Complexes by Hydrocarbon Stapled Peptides Using Lipid Based Delivery. Sci. Rep 2017, 7, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yuen TY; Brown CJ; Xue YZ, et al. Stereoisomerism of Stapled Peptide Inhibitors of the p53-MDM2 Interaction: An Assessment of Synthetic Strategies and Activity Profiles. Chem. Sci 2019, 10, 6457–6466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rayburn E; Zhang R; He J, et al. MDM2 and Human Malignancies: Expression, Clinical Pathology, Prognostic Markers, and Implications for Chemotherapy. Curr. Cancer Drug Targets 2005, 5, 27–41. [DOI] [PubMed] [Google Scholar]
- 23.Sun D; Li Z; Rew Y, et al. Discovery of AMG232, a Potent, Selective, and Orally Bioavailable MDM2–p53 Inhibitor in Clinical Development. J. Med. Chem 2014, 57, 1454–1472. [DOI] [PubMed] [Google Scholar]
- 24.Rew Y; Sun DQ; Yan XL, et al. Discovery of AM-7209, a Potent and Selective 4Amidobenzoic Acid Inhibitor of the MDM2-p53 Interaction. J. Med. Chem 2014, 57, 10499–10511. [DOI] [PubMed] [Google Scholar]
- 25.Yang Y; Li P; Zhang Z, et al. Prediction of Cyclosporin-Mediated Drug Interaction Using Physiologically Based Pharmacokinetic Model Characterizing Interplay of Drug Transporters and Enzymes. Int. J. Mol. Sci 2020, 21, 7023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nanayakkara AK; Follit CA; Chen G, et al. Targeted Inhibitors of P-Glycoprotein Increase Chemotherapeutic-Induced Mortality of Multidrug Resistant Tumor Cells. Sci. Rep 2018, 8, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen L; Zhao Y; Halliday GC, et al. Structurally Diverse MDM2-p53 Antagonists Act as Modulators of MDR-1 Function in Neuroblastoma. Br. J. Cancer 2014, 111, 716–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lai TH; Toussaint M; Teodoro R, et al. Synthesis and Biological Evaluation of a Novel 18F-Labeled Radiotracer for PET Imaging of the Adenosine A2A Receptor. Int. J. Mol. Sci 2021, 22, 1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mori V; Amici A; Mazzola F, et al. Metabolic Profiling of Alternative NAD Biosynthetic Routes in Mouse Tissues. PLOS ONE 2014, 9, e113939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu D; Guo H; Chang Y, et al. Cell-and Tissue-Based Proteome Profiling and Dual Imaging of Apoptosis Markers with Probes Derived from Venetoclax and Idasanutlin. Angew. Chem. Int. Ed 2018, 57, 9284–9289. [DOI] [PubMed] [Google Scholar]
- 31.Hines J; Lartigue S; Dong H, et al. MDM2-Recruiting PROTAC Offers Superior, Synergistic Antiproliferative Activity via Simultaneous Degradation of BRD4 and Stabilization of p53. Cancer Res. 2019, 79, 251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lebraud H; Noble RA; Phillips N, et al. Highly Potent Clickable Probe for Cellular Imaging of MDM2 and Assessing Dynamic Responses to MDM2-p53 Inhibition. Bioconjugugate Chem. 2018, 29, 2100–2106. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.