

Abstract
Methylation is one of the most common and considerable modifications in biological systems mediated by multiple enzymes. Recent studies have shown that methylation has been widely identified in different RNA molecules. RNA methylation modifications have various kinds, such as 5-methylcytosine (m5C). However, for individual methylation sites, their functions still remain to be elucidated. Testing of all methylation sites relies heavily on high-throughput sequencing technology, which is expensive and labor consuming. Thus, computational prediction approaches could serve as a substitute. In this study, multiple machine learning models were used to predict possible RNA m5C sites on the basis of mRNA sequences in human and mouse. Each site was represented by several features derived from k-mers of an RNA subsequence containing such site as center. The powerful max-relevance and min-redundancy (mRMR) feature selection method was employed to analyse these features. The outcome feature list was fed into incremental feature selection method, incorporating four classification algorithms, to build efficient models. Furthermore, the sites related to features used in the models were also investigated.
1. Introduction
Methylation is one of the most common and considerable modifications in biological systems mediated by multiple enzymes. The substrates of biological methylation are diverse, with DNA as the most common one. Previous studies on methylation mostly focused on DNA methylation, revealing its specific role in transcriptional activity regulation during development, aging, and pathogenesis [1]. However, recent studies have widely identified methylation among different RNA molecules, including mRNA, snoRNA, miRNA, and rRNA (not restricted to functional mRNAs) [2]. RNA methylation enables the posttranscriptional control of gene expression by changing how RNA interacts with other components of the cell as an important part of epitranscriptome [3]. RNA methylation is actively involved in posttranscriptional regulatory bioprocesses, like RNA splicing, transport, stability, and translatability, and it has strong relationships with mammalian development and diseases [4–6].
Among the various kinds of RNA methylation modifications, N6-methyladenosine (m6A), the methylation modification on the nitrogen at the sixth position of the adenosine base, is the most prevalent internal mRNA modification, accounting for 50% of the total methylated ribonucleotides [2, 7]. M6A broadly affects mRNA metabolism, and it is widely distributed in all kinds of RNA transcripts, including coding and noncoding regions. The deposition of m6A modification in the transcriptome has its unique pattern: the m6A modification sites have a typical consensus sequence DRACH (D = G, A, or U; R = G or A; H = A, C, or U), which is widely dispersed over coding sequence and untranslated region (UTR) and highly enriched near the stop codon area [8]. Recent evidence has proven that m6A RNA methylation plays a vital role in pre-mRNA splicing, mRNA stability regulation, mRNA export, mRNA degradation, translation regulation, and miRNA processing [9–11]. M6A modification is dynamic, it could be reversible, and it may vary between different genes and different tissues [12, 13]. With the increase in the number of m6A mapping studies, the list of specific genes containing a disproportionately high level of m6A was revealed. For example, Han et al. found a series of m6A methylated genes related to the presynaptic membrane, the postsynaptic membrane, and the synaptic growth in Alzheimer's disease (AD) mouse models, suggesting that m6A may be involved in the occurrence of AD [14]. While the function of m6A modification is context-dependent and dynamic, many m6A sites are evolutionally conserved among species. One-third of mammalian mRNAs share the same m6A modifications, and many of them are conserved with single-nucleotide specificity [15].
Another kind of RNA methylation modification, namely, 5-methylcytosine (m5C), which is the methylation of carbon 5 in cytosine, also acts as an important regulator in gene expression, including RNA localization, ribosome assembly, translation regulation, and mRNA stabilization. Among all the mRNA methylation sites, the proportion of m5C could be up to 20% in human cells [16]. The distribution of m5C sites in mRNA is not random; in HeLa and mouse cells, m5C methylation were found to be enriched in 5′ and 3′ UTRs rather than coding regions [16]. Like m6A, m5C acts its function in dynamic ways. M5C methylation occurs dynamically during testis development and helps maintain the stability of maternal mRNA in embryonic development [17].
Though RNA methylation plays a pivotal role in bioprocess and is of great importance to posttranscriptional regulation, their functions in individual methylation sites still remain to be elucidated. Testing of all the methylation sites relies heavily on high-throughput sequencing technology, which is expensive and labor consuming; thus, computational prediction approaches could serve as a substitute [18]. As mentioned above, the distribution of m5C in mRNA has its own enrichment pattern and is not random. With adequate datasets and statistic method, predicting accurate m5C RNA methylation sites and gaining an enhanced understanding of their functions are doable.
In this study, multiple machine learning models were applied to predict the possible m5C RNA methylation sites in mRNA sequences of human and mouse. For each m5C, a subsequence containing such site as center was extracted from the RNA sequence. The features of k-mers yielded by RNA2Vec [19] were refined to represent the subsequence. The powerful max-relevance and min-redundancy (mRMR) feature selection method [20] was employed to analyse all features. Obtained feature list was fed into incremental feature selection (IFS) [21] method, incorporating four classification algorithms, to build efficient models. In addition to prediction models, we also investigated the sites related to features used in the models, trying to discover special patterns around mouse and human m5C sites. Comparison of those prediction results may help obtain a dynamic RNA methylation profile and build relationships between the RNA methylation sites and human diseases.
2. Materials and Methods
2.1. Data
M5C is a common RNA modification in mammals. Human and mouse m5C data were downloaded from one previous study (iRNA-m5C, http://lin-group.cn/server/iRNA-m5C/download.html) [22]. In fact, the human m5C data was first used in [23], which was extracted from the original data retrieved from RMBase database [24]. The original data was processed by CD-HIT program [25] so that the sequence similarity of any remaining sequences was less than 0.7. As a result, 120 positive and 120 negative m5C sites were obtained. As for mouse m5C data, it was constructed in [22]. It was directly retrieved from RMBase database [24] and was not processed by CD-HIT program [25] because its size was so small. The mouse data consisted of 97 positive and 97 negative m5C sites. As the sites around the m5C sites have some special patterns, which can help to identify m5C sites in RNA sequence, 20 upstream sites and 20 downstream sites were picked up. These sites together with the m5C site at the center constructed a subsequence with 41 bp. Some features would be extracted from this subsequence to represent the m5C site.
2.2. Problem Description and Study Design
For a given RNA sequence, it is essential to identify m5C sites in it. The machine learning models can give a deep investigation on current known m5C sites and learn a special pattern to make prediction. The prediction procedure can be deemed as a function f, formulated by
| (1) |
where Ψ denoted the site set for human or mouse RNA sequences and +(−) represented whether the input site was an m5C site or not.
Generally, we want to discover an optimal function such that its loss was smallest. Because machine learning algorithms were employed to design such function, we adopted the following steps:
For any site in the human or mouse m5C data, sites around it were picked up to comprise a subsequence, which can indicate the surrounding information of the investigated site. This step was described in section “Feature Engineering”
Each subsequence was represented by a number of features, which can reflect its essential information. This step was described in section “Feature Engineering”
A feature selection method was adopted to analyse all features and produce a feature list. This step was described in section “Max-Relevance and Min-Redundancy (mRMR) Feature Selection”
The IFS method was applied on such feature list to find out which classification algorithm and which features can yield the best performance (smallest loss). This step was described in section “Incremental Feature Selection (IFS).” The descriptions of four classification algorithms used in IFS method can be found in section “Classification Algorithm.” The loss was determined by one measurement listed in section “Performance Measurement”
2.3. Feature Engineering
To build efficient models for identifying m5C site in RNA sequence, it is very important to extract essential features from the subsequence consisting of this site, 20 upstream sites and 20 downstream sites. This study adopted a natural language processing approach to extract features, which were further used to represent the subsequence containing m5C site.
RNA2Vec [19] was adopted to extract sequence features for each k-mers (subsequences of length k). In detail, this method employed the whole human genome as corpus. A sliding window technique was used to split the RNA sequence into several fix-length words. If an RNA sequence with length L was formulated by
| (2) |
it was split into L − k + 1 words, say R1R2 ⋯ Rk, R2R3 ⋯ Rk+1, ⋯, RL−k+1RL−k+2 ⋯ RL. All obtained words were fed into GloVe algorithm [26], a type of Word2vec method, to extract features of words, i.e., features of k-mers. Here, we selected k = 4. Features of 4-mers were directly retrieved from https://github.com/HsiaoYetGun/MiRLocator/blob/master/RNA2Vec/RNAVectors.txt. Each 4-mers was represented by 30 features.
Given a 41 bp long RNA subsequence SS, formulated by
| (3) |
where R21 was the m5C site, we extracted all 4-mers from this subsequence. Because the R21 was always same for all investigated subsequences, the 4-mers containing this site were discarded. 34 4-mers can be obtained from each RNA subsequence. Their 30 features obtained by RNA2Vec were collected together to represent the subsequence SS. Accordingly, 1020 (34 × 30) features were adopted to encode each subsequence with 41 bp.
2.4. Max-Relevance and Min-Redundancy (mRMR) Feature Selection
The mRMR is a powerful feature selection method [20, 27–30], which evaluates the importance of features from two aspects: (1) relevance to class labels and (2) redundancies to other features. The mutual information (MI) is used to quantify the relevance and redundancy. For two variables x and y, their MI is computed by
| (4) |
where p(x) and p(y) stand for the marginal probabilistic densities of x and y, respectively, and p(x, y) stands for the joint probabilistic density of x and y. Generally, a high MI indicates the strong relevance or high redundancy of two variables. The mRMR method tries to keep features with high relevance to class labels and low redundancies to other features. However, this is a NP-hard problem. The mRMR method employed a heuristic way to evaluate features, which sorts all investigated features in a list, namely, mRMR feature list. At the beginning, this list is empty. For each feature f that is not in this list, compute its relevance to class labels, measured by MI(f, c), where c is a variable representing class labels, and redundancies to features that are already in the list, measured by the average MI between f and features in the current list. The difference of these two values is computed. The feature with highest difference is selected and appended to the list. When all features have been in the list, the procedures stop. Feature ranks in this list indicate the importance of features. Generally, features with high ranks are more important than those with low ranks.
The mRMR program used in this study was downloaded from http://penglab.janelia.org/proj/mRMR/. For convenience, it was executed using its default parameters.
2.5. Incremental Feature Selection (IFS)
Although mRMR method produced a feature list, it is still a problem that which features should be selected to construct the model. In view of this, this study employed the IFS method [21], which can aid to choose proper features for any given classification algorithm. In detail, on the basis of the mRMR feature list, IFS produces several feature subsets with a step interval as one. For instance, the first feature subset has the top feature in the mRMR list, and the second feature subset has the first two features, and so on. Then, a model based on a certain classification algorithm can be constructed on the training data, where samples are represented by feature in each feature subset. All constructed models are assessed by one cross-validation method [31]. The model yielding the best performance is picked up and called the optimum model. The feature subset used in this model is termed as the optimum feature subset.
2.6. Classification Algorithm
As mentioned above, IFS method needs one classification algorithm. Here, four classification algorithms were used, including (1) random forest (RF) [32], (2) support vector machine (SVM) [33], (3) K-nearest neighbor (kNN) [34], and (4) decision tree (DT) [35]. These algorithms have been widely used to tackle various medical problems [36–48]. Their brief descriptions are as follows.
2.6.1. Random Forest
RF is a powerful and classic classification algorithm. In fact, it is an ensemble algorithm that contains several DTs. Each DT is built using two random selection procedures. The first procedure is to select samples, whereas the second procedure is for the selection of features. Given a query sample, each DT yields the prediction. RF integrates these predictions with majority voting. Although DT is a quite weak classification algorithm, RF is much more robust. Thus, it is always an important candidate for constructing prediction models.
2.6.2. Support Vector Machine
SVM is another powerful and classic classification algorithm. Its main idea is to find out a hyperplane for separating samples in two classes. However, such hyperplane does not exist in many cases. SVM maps the original data with nonlinear pattern in low-dimensional space to a new data with linear pattern in high-dimensional space. Then, the hyperplane is constructed in such new space by maximizing interval between samples in two classes. Finally, it predicts the class label of a new sample according to which side of hyperplane this new data point belongs to.
2.6.3. K-Nearest Neighbor
kNN is a simple but also efficient classification algorithm. It is not a strict machine learning algorithm because there is no training procedures. Several computational steps are conducted to determine the class of a test sample, such as computing the distance between the test sample and all training samples, ranking all training samples by those distances, selecting the k high-ranked training samples (i.e., nearest k neighbors), estimating the class label distribution of such k samples, and predicting the class label of the test sample as the one with the highest distribution frequency.
2.6.4. Decision Tree
It aims to learn the human understanding classification and regression models. It generally uses IF–TEHN format to describe individual features' roles and weights in classification or regression models, thereby providing interpretative rules in a white box model. To date, several types of DT have been proposed. In this work, the CART algorithm with the Gini index was adopted to build DT model.
To quickly implement above-mentioned four classification algorithms, we employed corresponding packages collected in Scikit-learn (https://scikit-learn.org/stable/). They were executed using their default parameters.
2.7. Performance Measurement
In this study, the MCC [49] within 10-fold cross-validation [31] was used to evaluate each model's performance. A two-class classification model was obviously built here; thus, the MCC for binary problem was used as follows:
| (5) |
where TP, TN, FP, and FN represent the sample numbers with true-positive, true-negative, false-positive, and false-negative predictions, respectively. The MCC value ranges from −1 to +1. When one classification model has the best performance, its MCC achieves +1.
Besides, we further computed other measurements to fully assess the performance of models, including sensitivity (SN) (same as recall), specificity (SP), accuracy (ACC), precision, and F1-measure. They can be calculated by
| (6) |
2.8. Feature Frequency Visualization
Each feature was related to four sites in the sequence to understand the biological meaning of the extracted sequence features. After the optimum features for one classification algorithm were obtained, the related sites of each feature were picked up, and the frequency of each site was counted and plotted as a bar illustration.
3. Results
In this study, we adopted the features of k-mers yielded by RNA2Vec to represent m5C sites. Some machine learning algorithms were employed to analyse these features and further build efficient models for identifying m5C site in RNA sequences. The whole procedures are shown in Figure 1. The detailed results were described in this section.
Figure 1.

Flow chart to construct models for the prediction of m5C sites. A subsequence with 41 bp is used to represent each m5C site. Features of k-mers obtained by RNA2Vec are adopted to constitute features of the subsequence. All features are analysed by max-relevance and min-redundancy method. The outcome feature list is fed into incremental feature selection, incorporating four classification algorithms and 10-fold cross-validation, to construct optimum models.
3.1. Selection of m5C Methylation-Associated Features for Mouse
For mouse m5C data, the mRMR method was first employed to analyse all 1020 features. An mRMR feature list was obtained. This list was fed into the IFS method that integrated one of four classification algorithms. On each feature subset, a model was built based on one classification algorithm and was further evaluated by 10-fold cross-validation. The performance of each model, including SN, SP, ACC, MCC, precision, and F1-measurem is provided in Supplementary file S1. MCC was selected as the key measurement. Accordingly, a curve is plotted in Figure 2 for each classification algorithm, which defined MCC as y-axis and number of features as the x-axis. For kNN, RF, and SVM, they can provide perfect performance with MCC = 1 when top 3, 10, and 3 features were adopted. The corresponding optimum kNN/RF/SVM model can be built with these features. The detailed performance of these models is listed in Table 1. All measurements reached the maximum of 1.000. For DT, the highest MCC was 0.990, which can be obtained by using top 195 features. Accordingly, the optimum DT model was set up with these features. Its detailed performance is listed in Table 1. It can be observed that all measurements were very high. All these indicated that the models with features yielded by RNA2Vec were quite efficient for identification of mouse m5C sites, also confirming the utility of these features to predict mouse m5C sites.
Figure 2.
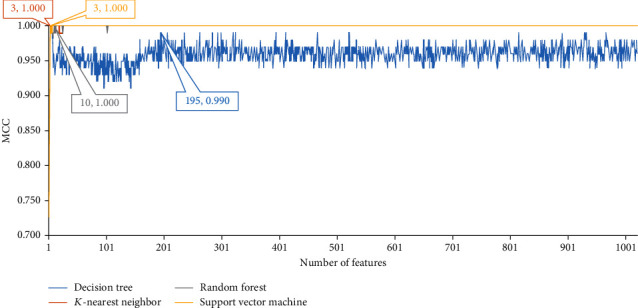
IFS curves with different classifiers on different numbers of sequence features on mouse m5C data.
Table 1.
Performance of models based on different classification algorithms for predicting mouse m5C sites.
| Classification algorithm | Number of features | SN | SP | ACC | MCC | Precision | F1-measure |
|---|---|---|---|---|---|---|---|
| Decision tree | 195 | 1.000 | 0.990 | 0.995 | 0.990 | 0.990 | 0.995 |
| K-nearest neighbor | 3 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 |
| Random forest | 10 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 |
| Support vector machine | 3 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 |
3.2. Selection of m5C Methylation-Associated Features for Human
For human m5C data, the same procedures were conducted. The performance of four classification algorithms on all possible feature subsets is provided in Supplementary file S2. Similarly, one curve was plotted for each classification algorithm (as shown in Figure 3). It can be observed that four classification algorithms yielded the highest MCC values of 0.576, 0.627, 0.742, and 0.790, respectively. Such performance was obtained by using top 15, 84, 543, and 114 features. Accordingly, optimum DT/kNN/RF/SVM model can be set up with these features. The detailed performance of these models is listed in Table 2. Evidently, the performance of these models was much lower than that of models for mouse.
Figure 3.
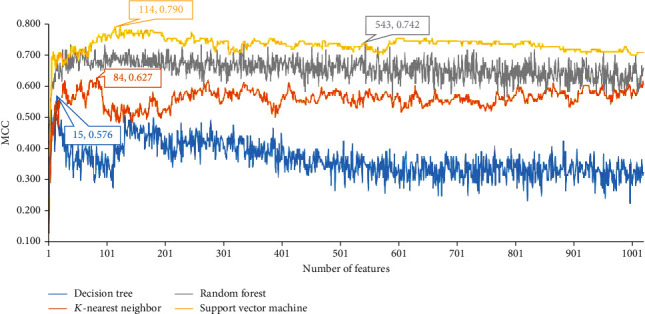
IFS curves with different classifiers on different numbers of sequence features on human m5C data.
Table 2.
Performance of models based on different classification algorithms for predicting human m5C sites.
| Classification algorithm | Number of features | SN | SP | ACC | MCC | Precision | F1-measure |
|---|---|---|---|---|---|---|---|
| Decision tree | 15 | 0.767 | 0.808 | 0.788 | 0.576 | 0.800 | 0.783 |
| K-nearest neighbor | 84 | 0.683 | 0.925 | 0.804 | 0.627 | 0.901 | 0.777 |
| Random forest | 543 | 0.875 | 0.867 | 0.871 | 0.742 | 0.868 | 0.871 |
| Support vector machine | 114 | 0.825 | 0.958 | 0.892 | 0.790 | 0.952 | 0.884 |
3.3. Feature Frequency Analysis
The purpose of this study was not only to set up efficient models for prediction of m5C sites but also to discover novel patterns around the m5C sites, thereby providing more biological insights. Thus, we conducted feature frequency analysis in this section.
For mouse m5C data, four optimum models were built, which adopted some top features in the list. For each model, the number of selected features related to each site was counted. A bar chart was plotted to display such number of each site (as shown in Figure 4). Detailed discussion would be given in section “m5C Methylation-Associated Features in Mouse.”
Figure 4.
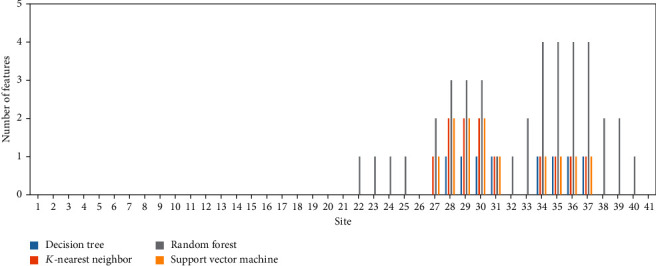
Frequency visualization for sequence features related to mouse m5C.
For human m5C data, we conducted the same operations. For each optimum model, the number of selected features related to each site is shown in Figure 5. Evidently, Figures 4 and 5 displayed quite different patterns, indicating the difference between mouse and human m5C sites. In section “m5C Methylation-Associated Features in Human,” a discussion would be given.
Figure 5.

Frequency visualization for sequence features related to human m5C.
3.4. Comparison with Previous Models
This study used the mouse and human m5C data reported in [22]. In that study, several models with different classification algorithms were built and evaluated by 10-fold cross-validation, including DT, RF, SVM, Naïve Bayes, Bayes net, and logistic regression. The performance of models with DT, RF, and SVM is listed in Tables 3 and 4. For easy comparison, the performance of our models with same classification algorithms is also provided in these two tables. For mouse m5C data, our model with DT was slightly superior to the model in [22] with the same classification algorithm. As for other two classification algorithms, all models with one of them gave perfect performance. For human m5C data, DT provided better performance in our model than the model in [22], whereas other two classification algorithms generated lower performance in our model than the model in [22]. However, the gap was not very big. As a whole, our models and those in [22] were almost at the same level.
Table 3.
Comparison with previous models on mouse m5C data.
| Classification algorithm | Model | SN | SP | ACC | MCC |
|---|---|---|---|---|---|
| Decision tree | Our model | 1.000 | 0.990 | 0.995 | 0.990 |
| Model in [22] | 1.000 | 0.835 | 0.918 | 0.847 | |
|
| |||||
| Random forest | Our model | 1.000 | 1.000 | 1.000 | 1.000 |
| Model in [22] | 1.000 | 1.000 | 1.000 | 1.000 | |
|
| |||||
| Support vector machine | Our model | 1.000 | 1.000 | 1.000 | 1.000 |
| Model in [22] | 1.000 | 1.000 | 1.000 | 1.000 | |
Table 4.
Comparison with previous models on human m5C data.
| Classification algorithm | Model | SN | SP | ACC | MCC |
|---|---|---|---|---|---|
| Decision tree | Our model | 0.767 | 0.808 | 0.788 | 0.576 |
| Model in [22] | 0.783 | 0.783 | 0.783 | 0.567 | |
|
| |||||
| Random forest | Our model | 0.875 | 0.867 | 0.871 | 0.742 |
| Model in [22] | 0.900 | 0.917 | 0.908 | 0.817 | |
|
| |||||
| Support vector machine | Our model | 0.825 | 0.958 | 0.892 | 0.790 |
| Model in [22] | 0.842 | 0.967 | 0.904 | 0.815 | |
As mentioned in the above section, the purpose of this study further included the discovery of special patterns around m5C sites. This was the exclusive contributions of this study compared with the previous study.
4. Discussion
Multiple machine learning models were used to distinguish samples/sites with or without a different kind of RNA methylation (human or mouse), focusing on the significant pattern of RNA methylation as m5C [50–52]. With the help of IFS, the optimal number of essential features was selected for RNA methylation prediction. The distribution of predicted features in the 41 nt sequence was summarized to evaluate the discriminative contributions of different RNA loci for RNA methylation [53]. The detailed analyses on the results of m5C methylation in mouse or human tissues could be seen below, along with their respective distribution patterns.
4.1. m5C Methylation-Associated Features in Mouse
Multiple physiochemical features were used to encode the 41 nt sequence [53] of RNA. For the evaluation of the differential contribution of RNA sites for m5C methylation, four machine learning models were applied (DT, KNN, RF, and SVM) to identify the optimal combination of features for m5C methylation prediction. The distribution of features' respective RNA loci is shown in Figure 4. As identified from the feature distribution, all the selected features belong to the back end of the selected sequence, from 23 nt to 41 nt, just behind the candidate m5C methylation site (21 nt). In particular, two regions (27–31 nt and 34–37 nt) were predicted by at least three machine learning models to be associated with m5C methylation. According to recent publications based on the biological functions of m5C, the two kinds of m5C sites in multiple subgroups of RNAs are (1) type I m5C, which is followed by a G-rich triplet motif, and (2) type II m5C, which is adjacent to a downstream UCCA motif; both have specific sequence characteristics in the following region of m5C methylation loci [54], which corresponded with the prediction results in the present study. Further studies have also confirmed that specific regions in the downstream of m5C loci may have different sequence contexts, indicating that the feature-enriched regions in the prediction list in the present study could definitely be associated with m5C methylation efficiency. In 2019, a systematic analyses on mRNA 5-methylcytosine in mammals identified that the sequence context at the downstream of the captured m5C loci was alternate with different m5C locus methylation status, regulated by a specific 5-methylcytosine methyltransferase called NSUN2 [55, 56]. For comparison, the sequence before the m5C loci in mouse did not considerably change with NSUN2 wild-type, knock-out, or rescue status, implying that the m5C loci and their downstream sequence, especially for the following 10 nt sequence [55, 56], which corresponded with the prediction distribution in the present study. In addition, another similar 5-methylcytosine methyltransferase NSUN6 in mouse functioned as an mRNA m5C methyltransferase [54]. As a methyltransferase of type II m5C, the m5C targets of such gene have a symbolic downstream UCCA tail located at the first ambiguous peak (only predicted via RF method) in the prediction result of the present study (1–4 nt following the methylation region) [54]. Furthermore, different from the biological regulatory effects of NSUN2, the flanking regions around 15 nt were found to have another low base-pairing regions, which include more variants, by using the same procedure that detects the sequences with methyltransferase knock-out, rescue, and wild-type statuses [54]. This finding indicated the importance of sequence around such region. All in all, the predicted distribution of m5C methylation-associated loci has been validated by recent publications.
4.2. m5C Methylation-Associated Features in Human
The m5C-associated feature distribution among 40 flanking sequences (20 downstream and 20 upstream) from human tissues was also identified. According to the same publications [54, 55], the following 1–4 nt (22–26 nt) and 13–15 nt (34–37 nt) were also associated with the efficacy of m5C methylation, which corresponded with the prediction of the present study. As seen in Figures 4 and 5, the feature peaks in the downstream region (21–41 nt) were quite similar between the human and mouse data, reflecting the similarity of m5C methylation-associated patterns among different species. However, obvious differences were also observed, implying the presence of biological differences in m5C methylation among different species. In human beings, recent publications revealed that the distribution of RBP (RNA-binding protein) target density, which reflects the binding efficacy of the related region, was significant at the m5C candidate site, and gradually, not suddenly going down in both directions [56, 57]. Therefore, the sequences around m5C in each direction may also be not randomized but with specific sequence characteristics. Further, in 2015, an analysis on the regulatory homologous proteins of yeast and human from the same protein family (Nop2/NSUN/NOL family) showed that specific binding domains (e.g., SAM-binding domain) may be located behind the m5C loci, and they may affect regulatory effects. Therefore, although they were not directly validated, some nucleotides located before the m5C loci may be essential for the prediction of methylation status [58].
4.3. Biological Significance of Identified m5C Methylation-Associated Features
As summarized above, we identified m5C-associated features in mouse and human. The biological significance of identified m5C methylation features can be clustered into two parts:
The specific and diverse distribution of m5C associated features in human or mouse. In this part, we identified that mouse m5C methylations are generally only associated with 28-31 nt and 34-37 nt regions in the 41 nt subsequence, while in human tissues, apart from 19-21 nt regions, most positions of the 41 nt sequence are associated with m5C methylation. These results identified key regulatory regions associated with m5C methylation and the differences between regulatory effects on m5C methylation in different species, reflecting the evolution conservation of m5C methylation regulatory mechanisms
The downstream regulatory network associated with m5C methylation is essential for gene transcription and translation. Generally, m5C methylation can help bind hydrogen with guanine to stabilize the complete RNA structures and fold into unique spatial conformation [59]. According to recent publications, m5C regulator NSUN2 has been shown to alter m5C capacity in certain RNA regions. Genes like p27 (KIPI), CDK1, p21, and ErbB2 have all been shown to be regulated by m5C methylation and further related to tumorigenesis [59, 60]. The sequence loci of m5C methylation have been shown to be specifically affects the downstream cell proliferation and inflammation associated pathway [61, 62], indicating the specific biological significance of m5C methylation. Therefore, the identification of different contribution of nucleotide from different sequence location can help demonstrate the specific regulatory effects for abnormal m5C methylation during different pathogenic conditions
Therefore, the identification of loci-related characters regulating m5C methylation between different species can not only help us reveal the consistence and evolution conservation of m5C methylation associated sequences but also connect specific sequence loci with significant m5C methylation-associated phenotypes or diseases.
5. Conclusions
All in all, as discussed above, the top optimal methylation sites in the prediction list have been supported by recent publications. The RNA methylation patterns were validated to be different in multiple species by comparing the results of m5C methylation-associated loci in human and mouse tissues. The discriminative feature distribution patterns for different methylation patterns were also detected by comparing the results of m5C distribution patterns. Therefore, the results not only evaluated the discriminative contribution of different loci for important RNA methylation patterns but also revealed the site distribution differences of m5C methylation types between species (human and mice).
Acknowledgments
This work was supported by the Strategic Priority Research Program of Chinese Academy of Sciences (XDA26040304 and XDB38050200), National Key R&D Program of China (2018YFC0910403), and the Fund of the Key Laboratory of Tissue Microenvironment and Tumor of Chinese Academy of Sciences (202002).
Contributor Information
Tao Huang, Email: tohuangtao@126.com.
Yu-Dong Cai, Email: cai_yud@126.com.
Data Availability
The original data used to support the findings of this study are available at iRNA-m5C (http://lin-group.cn/server/iRNA-m5C/download.html).
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.”
Authors' Contributions
Lei Chen, ZhanDong Li, and ShiQi Zhang contributed equally to this work.
Supplementary Materials
Supplementary Material S1: performance of IFS for mouse m5C sites. Supplementary Material S2: performance of IFS for human m5C sites.
References
- 1.Greenberg M. V., Bourc’his D. The diverse roles of DNA methylation in mammalian development and disease. Nature Reviews Molecular Cell Biology . 2019;20(10):590–607. doi: 10.1038/s41580-019-0159-6. [DOI] [PubMed] [Google Scholar]
- 2.Shi H., Wei J., He C. Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Molecular Cell . 2019;74(4):640–650. doi: 10.1016/j.molcel.2019.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li X., Xiong X., Yi C. Epitranscriptome sequencing technologies: decoding RNA modifications. Nature Methods . 2017;14(1):23–31. doi: 10.1038/nmeth.4110. [DOI] [PubMed] [Google Scholar]
- 4.du K., Zhang L., Lee T., Sun T. m6A RNA methylation controls neural development and is involved in human diseases. Molecular Neurobiology . 2019;56(3):1596–1606. doi: 10.1007/s12035-018-1138-1. [DOI] [PubMed] [Google Scholar]
- 5.Heck A. M., Wilusz C. J. Small changes, big implications: the impact of m6A RNA methylation on gene expression in pluripotency and development. Biochimica et Biophysica Acta (BBA)-Gene Regulatory Mechanisms . 2019;1862(9, article 194402) doi: 10.1016/j.bbagrm.2019.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chokkalla A. K., Mehta S. L., Vemuganti R. Epitranscriptomic regulation by m6A RNA methylation in brain development and diseases. Journal of Cerebral Blood Flow & Metabolism . 2020;40(12):2331–2349. doi: 10.1177/0271678X20960033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gu C., Shi X., Dai C., et al. RNA m6A modification in cancers: molecular mechanisms and potential clinical applications. The Innovation . 2020;1(3, article 100066) doi: 10.1016/j.xinn.2020.100066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zaccara S., Ries R. J., Jaffrey S. R. Reading, writing and erasing mRNA methylation. Nature Reviews Molecular Cell Biology . 2019;20(10):608–624. doi: 10.1038/s41580-019-0168-5. [DOI] [PubMed] [Google Scholar]
- 9.Min K. W., Zealy R. W., Davila S., et al. Profiling of m6A RNA modifications identified an age-associated regulation of AGO 2 mRNA stability. Aging Cell . 2018;17(3, article e12753) doi: 10.1111/acel.12753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee Y., Choe J., Park O. H., Kim Y. K. Molecular mechanisms driving mRNA degradation by m6A modification. Trends in Genetics . 2020;36(3):177–188. doi: 10.1016/j.tig.2019.12.007. [DOI] [PubMed] [Google Scholar]
- 11.Lesbirel S., Wilson S. A. The m6A‑methylase complex and mRNA export. Biochimica et Biophysica Acta (BBA)-Gene Regulatory Mechanisms . 2019;1862(3):319–328. doi: 10.1016/j.bbagrm.2018.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu J.’., Li K., Cai J., et al. Landscape and regulation of m6A and m6Am methylome across human and mouse tissues. Molecular Cell . 2020;77(2):426–440.e6. doi: 10.1016/j.molcel.2019.09.032. [DOI] [PubMed] [Google Scholar]
- 13.Roundtree I. A., Evans M. E., Pan T., He C. Dynamic RNA modifications in gene expression regulation. Cell . 2017;169(7):1187–1200. doi: 10.1016/j.cell.2017.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han M., Liu Z., Xu Y., et al. Abnormality of m6A mRNA methylation is involved in Alzheimer's disease. Frontiers in Neuroscience . 2020;14:p. 98. doi: 10.3389/fnins.2020.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Z., Chen L. Q., Zhao Y. L., et al. Single-base mapping of m6A by an antibody-independent method. Science Advances . 2019;5(7, article eaax0250) doi: 10.1126/sciadv.aax0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trixl L., Lusser A. The dynamic RNA modification 5-methylcytosine and its emerging role as an epitranscriptomic mark. Wiley Interdisciplinary Reviews: RNA . 2019;10(1, article e1510) doi: 10.1002/wrna.1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang X., Yang Y., Sun B. F., et al. 5-methylcytosine promotes mRNA export -- NSUN2 as the methyltransferase and ALYREF as an m5C reader. Cell Research . 2017;27(5):606–625. doi: 10.1038/cr.2017.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu X., Wei Z., Chen K., et al. m6Acomet: large-scale functional prediction of individual m6A RNA methylation sites from an RNA co-methylation network. BMC Bioinformatics . 2019;20(1):1–12. doi: 10.1186/s12859-019-2840-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiao Y., Cai J., Yang Y., Zhao H., Shen H. Prediction of MicroRNA Subcellular Localization by Using a Sequence-to-Sequence Model. 2018 IEEE International Conference on Data Mining (ICDM); 2018; Singapore. IEEE; [Google Scholar]
- 20.Peng H., Fulmi L., Ding C. Feature selection based on mutual information criteria of max-dependency, max-relevance, and min-redundancy. Ieee Transactions On Pattern Analysis And Machine Intelligence . 2005;27(8):1226–1238. doi: 10.1109/TPAMI.2005.159. [DOI] [PubMed] [Google Scholar]
- 21.Liu H. A., Setiono R. Incremental feature selection. Applied Intelligence . 1998;9(3):217–230. doi: 10.1023/A:1008363719778. [DOI] [Google Scholar]
- 22.Lv H., Zhang Z. M., Li S. H., Tan J. X., Chen W., Lin H. Evaluation of different computational methods on 5-methylcytosine sites identification. Briefings in Bioinformatics . 2020;21(3):982–995. doi: 10.1093/bib/bbz048. [DOI] [PubMed] [Google Scholar]
- 23.Feng P., Ding H., Chen W., Lin H. Identifying RNA 5-methylcytosine sites via pseudo nucleotide compositions. Molecular BioSystems . 2016;12(11):3307–3311. doi: 10.1039/C6MB00471G. [DOI] [PubMed] [Google Scholar]
- 24.Sun W. J., Li J. H., Liu S., et al. RMBase: a resource for decoding the landscape of RNA modifications from high-throughput sequencing data. Nucleic Acids Research . 2016;44(D1):D259–D265. doi: 10.1093/nar/gkv1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu L., Niu B., Zhu Z., Wu S., Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics . 2012;28(23):3150–3152. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pennington J., Socher R., Manning C. D. Glove: global vectors for word representation. Proceedings of the 2014 Conference on Empirical Methods in Natural Language Processing (EMNLP); 2014; Doha, Qatar. [Google Scholar]
- 27.Zhang S., Zeng T., Hu B., et al. Discriminating origin tissues of tumor cell lines by methylation signatures and Dys-methylated rules. Frontiers in Bioengineering and Biotechnology . 2020;8:p. 507. doi: 10.3389/fbioe.2020.00507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang S., Pan X. Y., Zeng T., et al. Copy number variation pattern for discriminating MACROD2 states of colorectal cancer subtypes. Frontiers in Bioengineering and Biotechnology . 2019;7:p. 407. doi: 10.3389/fbioe.2019.00407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen L., Zeng T., Pan X., Zhang Y. H., Huang T., Cai Y. D. Identifying methylation pattern and genes associated with breast cancer subtypes. International Journal of Molecular Sciences . 2019;20(17):p. 4269. doi: 10.3390/ijms20174269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao X., Chen L., Lu J. A similarity-based method for prediction of drug side effects with heterogeneous information. Mathematical Biosciences . 2018;306:136–144. doi: 10.1016/j.mbs.2018.09.010. [DOI] [PubMed] [Google Scholar]
- 31.Kohavi R. A study of cross-validation and bootstrap for accuracy estimation and model selection. International Joint Conference On Artificial Intelligence . 1995;14(2) [Google Scholar]
- 32.Breiman L. Random forests. Machine Learning . 2001;45(1):5–32. doi: 10.1023/A:1010933404324. [DOI] [Google Scholar]
- 33.Cortes C., Vapnik V. Support-vector networks. Machine Learning . 1995;20(3):273–297. doi: 10.1007/BF00994018. [DOI] [Google Scholar]
- 34.Cover T., Hart P. Nearest neighbor pattern classification. IEEE Transactions on Information Theory . 1967;13(1):21–27. doi: 10.1109/TIT.1967.1053964. [DOI] [Google Scholar]
- 35.Safavian S. R., Landgrebe D. A survey of decision tree classifier methodology. IEEE Transactions on Systems, Man, and Cybernetics . 1991;21(3):660–674. doi: 10.1109/21.97458. [DOI] [Google Scholar]
- 36.Zhang Y.-H., Li Z., Zeng T., et al. Detecting the multiomics signatures of factor-specific inflammatory effects on airway smooth muscles. Frontiers in Genetics . 2021;11, article 599970 doi: 10.3389/fgene.2020.599970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pan X., Li H., Zeng T., et al. Identification of protein subcellular localization with network and functional embeddings. Frontiers in Genetics . 2021;11, article 626500 doi: 10.3389/fgene.2020.626500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y.-H., Zeng T., Chen L., Huang T., Cai Y. D. Determining protein-protein functional associations by functional rules based on gene ontology and KEGG pathway. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics . 2021;1869(6, article 140621) doi: 10.1016/j.bbapap.2021.140621. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y.-H., Li H., Zeng T., et al. Identifying transcriptomic signatures and rules for SARS-CoV-2 infection. Frontiers in Cell and Developmental Biology . 2021;8, article 627302 doi: 10.3389/fcell.2020.627302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Onesime M., Yang Z., Dai Q. Genomic island prediction via chi-square test and random forest algorithm. Computational and Mathematical Methods in Medicine . 2021;2021:9. doi: 10.1155/2021/9969751.9969751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y., Xu Y., Yang Z., Liu X., Dai Q. Using recursive feature selection with random forest to improve protein structural class prediction for low-similarity sequences. Computational and Mathematical Methods in Medicine . 2021;2021:9. doi: 10.1155/2021/5529389.5529389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang Y., Chen L. Identification of drug–disease associations by using multiple drug and disease networks. Current Bioinformatics . 2021;16 doi: 10.2174/1574893616666210825115406. [DOI] [Google Scholar]
- 43.Zhou J.-P., Chen L., Wang T., Liu M. iATC-FRAKEL: a simple multi-label web server for recognizing anatomical therapeutic chemical classes of drugs with their fingerprints only. Bioinformatics . 2020;36(11):3568–3569. doi: 10.1093/bioinformatics/btaa166. [DOI] [PubMed] [Google Scholar]
- 44.Zhu Y., Hu B., Chen L., Dai Q. iMPTCE-Hnetwork: a multi-label classifier for identifying metabolic pathway types of chemicals and enzymes with a heterogeneous network. Computational and Mathematical Methods in Medicine . 2021;2021 doi: 10.1155/2021/6683051.6683051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jia Y., Zhao R., Chen L. Similarity-based machine learning model for predicting the metabolic pathways of compounds. IEEE Access . 2020;8:130687–130696. doi: 10.1109/ACCESS.2020.3009439. [DOI] [Google Scholar]
- 46.Chen L., Wang S., Zhang Y. H., et al. Identify key sequence features to improve CRISPR sgRNA efficacy. IEEE Access . 2017;5:26582–26590. doi: 10.1109/ACCESS.2017.2775703. [DOI] [Google Scholar]
- 47.Liu H., Hu B., Chen L., Lu L. Identifying protein subcellular location with embedding features learned from networks. Current Proteomics . 2021;18(5):646–660. doi: 10.2174/1570164617999201124142950. [DOI] [Google Scholar]
- 48.Chen W., Chen L., Dai Q. iMPT-FDNPL: identification of membrane protein types with functional domains and a natural language processing approach. Computational and Mathematical Methods in Medicine . 2021;2021 doi: 10.1155/2021/7681497.7681497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matthews B. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochimica et Biophysica Acta (BBA)-Protein Structure . 1975;405(2):442–451. doi: 10.1016/0005-2795(75)90109-9. [DOI] [PubMed] [Google Scholar]
- 50.Jia L., Chen J., Liu H., et al. Potential m6A and m5C methylations within the genome of a Chinese African swine fever virus strain. Virologica Sinica . 2021;36(2):321–324. doi: 10.1007/s12250-020-00217-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu Z.-X., Li L. M., Sun H. L., Liu S. M. Link between m6A modification and cancers. Frontiers in Bioengineering and Biotechnology . 2018;6:p. 89. doi: 10.3389/fbioe.2018.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sommer S., Salditt-Georgieff M., Bachenheimer S., et al. The methylation of adenovirus-specific nuclear and cytoplasmic RNA. Nucleic Acids Research . 1976;3(3):749–766. doi: 10.1093/nar/3.3.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dao F. Y., Lv H., Yang Y. H., Zulfiqar H., Gao H., Lin H. Computational identification of N6-methyladenosine sites in multiple tissues of mammals. Computational and Structural Biotechnology Journal . 2020;18:1084–1091. doi: 10.1016/j.csbj.2020.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu J., Huang T., Zhang Y., et al. Sequence-and structure-selective mRNA m5C methylation by NSUN6 in animals. National Science Review . 2021;8(6) doi: 10.1093/nsr/nwaa273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang T., Chen W., Liu J., Gu N., Zhang R. Genome-wide identification of mRNA 5-methylcytosine in mammals. Nature Structural & Molecular Biology . 2019;26(5):380–388. doi: 10.1038/s41594-019-0218-x. [DOI] [PubMed] [Google Scholar]
- 56.Li Q., Li X., Tang H., et al. NSUN2-mediated m5C methylation and METTL3/METTL14-mediated m6A methylation cooperatively enhance p21 translation. Journal of Cellular Biochemistry . 2017;118(9):2587–2598. doi: 10.1002/jcb.25957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Squires J. E., Patel H. R., Nousch M., et al. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Research . 2012;40(11):5023–5033. doi: 10.1093/nar/gks144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bourgeois G., Ney M., Gaspar I., et al. Eukaryotic rRNA modification by yeast 5-methylcytosine-methyltransferases and human proliferation-associated antigen p120. PLoS One . 2015;10(7, article e0133321) doi: 10.1371/journal.pone.0133321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bohnsack K. E., Höbartner C., Bohnsack M. T. Eukaryotic 5-methylcytosine (m5C) RNA methyltransferases: mechanisms, cellular functions, and links to disease. Genes . 2019;10(2):p. 102. doi: 10.3390/genes10020102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xiang S., Ma Y., Shen J., et al. m5C RNA methylation primarily affects the ErbB and PI3K–Akt signaling pathways in gastrointestinal cancer. Frontiers in Molecular Biosciences . 2020;7 doi: 10.3389/fmolb.2020.599340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee W.-H., Morton R. A., Epstein J. I., et al. Cytidine methylation of regulatory sequences near the pi-class glutathione S-transferase gene accompanies human prostatic carcinogenesis. Proceedings of the National Academy of Sciences . 1994;91(24):11733–11737. doi: 10.1073/pnas.91.24.11733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li Y., Li J., Luo M., et al. Novel long noncoding RNA NMR promotes tumor progression via NSUN2 and BPTF in esophageal squamous cell carcinoma. Cancer Letters . 2018;430:57–66. doi: 10.1016/j.canlet.2018.05.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material S1: performance of IFS for mouse m5C sites. Supplementary Material S2: performance of IFS for human m5C sites.
Data Availability Statement
The original data used to support the findings of this study are available at iRNA-m5C (http://lin-group.cn/server/iRNA-m5C/download.html).