

Abstract
Purpose:
Black men die from prostate cancer twice as often as White men, a disparity likely due to inherited genetics, modifiable cancer risk factors, and healthcare access. It is incompletely understood how and why tumor genomes differ by self-reported race and genetic ancestry.
Methods:
Among 2,069 men with prostate cancer (1,841 self-reported White, 63 Asian, 165 Black) with access to clinical-grade sequencing at the same cancer center, prevalence of tumor and germline alterations was assessed in cancer driver genes reported to have different alteration prevalence by race.
Results:
Clinical characteristics such as prostate-specific antigen and age at diagnosis as well as cancer stage at sample procurement differed by self-reported race. However, most genomic differences persisted when adjusting for clinical characteristics. Tumors from Black men harbored fewer PTEN mutations and more AR alterations than those from White men. Tumors from Asian men had more FOXA1 mutations and more ZFHX3 alterations than White men. Despite fewer TP53 mutations, tumors from Black men had more aneuploidy, particularly chromosome arm 8q gains, an adverse prognostic factor. Genetic ancestry was associated with similar tumor alterations as self-reported race, but also with modifiable cancer risk factors. Community-level average income was associated with chr8q gains after adjusting for race and ancestry.
Conclusions:
Tumor genomics differed by race even after accounting for clinical characteristics. Equalizing access to care may not fully eliminate such differences. Therapies for alterations more common in racial minorities are needed. Tumor genomic differences should not be assumed to be entirely due to germline genetics.
Keywords: prostate cancer, tumor genomics, race, ancestry, disparities
Introduction
Each year in the United States, there are 38 deaths from prostate cancer per 100,000 Black men, compared to 18 per 100,000 White men (1), a disparity that is greater than for any other cancer. It is critical to understand the contributions of the potential underlying causes, including differences in inherited (germline) genetics, modifiable risk factors, and healthcare access. Prostate cancer is highly heritable (2), and germline risk loci for prostate cancer differ by ancestry (3). Additionally, the burden of modifiable risk factors for prostate cancer death, such as smoking (4), is higher among Black men (5). Racial and ethnic groups have unequal healthcare access to cancer screening, diagnostics, and treatment. Consequently, prostate cancer among Black men may be diagnosed and treated at later stages with shorter survival after diagnosis.
Another potential contributor to racial differences is underlying differences in molecular tumor features that may indicate different cancer etiologies as well as higher or lower sensitivity of the tumor to targeted therapies. Prostate tumors differ by race in key somatic DNA alterations (6–10). For example, in the largest study to date (8), metastatic prostate tumors from Black men, compared to those from White men, were reported to have more DNA mutations in the androgen receptor AR, in BRAF, in DNA repair genes, and in genes considered “actionable,” and fewer TP53 alterations than tumors from Asian men. These differences suggest that for some therapies, treatment benefits may be different by race. However, previous genomic studies had limited or no data on differences in healthcare access or cancer risk factors, and they typically relied on either race or genetic ancestry.
A first question is whether differences in tumor genomics are a consequence of disparities in healthcare access. For example, if Black men had tumor sequencing later in their disease course than White men, finding more AR alterations in their tumors could merely reflect barriers to healthcare access. Conversely, if genomic differences persisted once consequences of healthcare access were accounted for, then race may need to be considered for precision prevention and treatment selection (11,12).
A second question is the interpretation of “race” and to what extent genetic ancestry is associated with genomic tumor differences associated with self-reported race. Race is a self-ascribed or socially-ascribed identity, whereas genetic ancestry describes components of an individual’s genetic origin (13). Yet both concepts are highly correlated and must be carefully disentangled.
Finally, a third question is to what extent differences in tumor genomics are driven by germline genetics or could be reduced by acting on racial disparities in cancer risk factors. We addressed these questions in one of the largest real-world, clinical-grade sequencing cohorts of prostate cancer.
Methods
Study population
This study included men with histologically-confirmed prostate cancer who had access to care at a comprehensive cancer center and who reported race on intake forms (Supplementary Fig. 1). Patients gave written informed consent for this institutional review board-approved study that was conducted in accordance with the U.S. Common Rule. Freely available definitions of data elements for a clinical-genomic research database, their validation, and the analysis pipeline were described elsewhere (14). Area-level median household income per 5-digit zip code was from the U.S. Census 2015–2019 American Community Survey.
Genomics
Tumor profiling used archival or fresh formalin-fixed paraffin-embedded tumor samples. Germline DNA from peripheral blood was only analyzed with specific consent. An FDA-cleared targeted hybridization capture-based gene panel, MSK-IMPACT, including 341 to 468 genes depending on its version, was used to capture single-nucleotide variants, small insertions and deletions, copy number alterations, and structural rearrangements (15).
TMPRSS2 and ERG alterations, more common in European-ancestry men (16), were not included in analyses given the suboptimal sensitivity of DNA panel sequencing for gene fusions like TMPRSS2:ERG and the analytical focus on absolute differences. Oncogenic or likely oncogenic function was annotated using OncoKB 2.8 (Sep 17, 2020) (17).
For copy number gains and deletions, absolute log2 copy number deviation >0.2 were required. The fraction of copy number-altered genome was defined as the proportion of the genome with such a copy number change relative to the size of the genome with copy number profiled. Estimates were corrected for pathologist-assessed tumor cellularity; there were no differences in tumor cellularity by race (Supplementary Fig. 7). CNTools v1.46, copynumber v1.30, and, for calling arm-level events, ASCETS (18) with breadth of coverage >0.5 were used.
To infer genetic ancestry, we used the 1000 Genomes Project (1KGP) with potentially admixed individuals excluded as reference. We selected 5,072 bi-allelic autosomal SNP markers from 1KGP with minor allele frequency >1% and within MSK-IMPACT468 bait intervals. For each patient, we genotyped these markers on the matched normal BAM files using GATK v4.0 Pileup, merged with 1KGP reference data and pruned with PLINK v1.9 (parameters --indep-pairwise 500kb 50 0.2) to keep only those markers that were in linkage equilibrium. We then performed supervised ADMIXTURE v1.3 (19) to estimate proportions of AFR, EUR, EAS, NAM (Native American; not included in analyses), and SAS ancestries.
Statistical analyses
Prevalence differences for genomic alterations by race (categorical) or ancestry (each ancestry modeled as a continuous variable) were estimated using marginal standardization of logistic regression, with bias-corrected accelerated (BCa) 95% CIs from parametric bootstrapping with 10,000 repeats (20). Regression models were used for differences in age at diagnosis and time to sequencing (quantile regression for the median), counts of actionable alterations (Poisson model), arithmetic mean ratios of fraction genome altered (generalized linear model with Gaussian distribution and log link), prevalence differences in chr8q gain by continuously modeled income (binomial model with identity link), and survival from sequencing to death or last contact (Cox regression with separate baseline hazards by disease extent at sequencing).
Results
Clinical characteristics and cancer risk factors
We first compared clinical features of 2,069 men with prostate cancer by self-reported race including 63 Asian men, 165 Black men, and 1,841 White men (Fig. 1A; Table 1). Despite being 3.0 years younger at diagnosis (95% CI, 0.6 to 5.4), Black men had median serum prostate-specific antigen (PSA) levels at diagnosis of 17 ng/ml, higher than White men (9 ng/ml; difference, 8.2 ng/ml; 95% CI, 0.1 to 16.3). Asian men had a more advanced cancer stage at sample procurement (32% castration-resistant) than White men (19%; difference, 13 percentage points; 95% CI, 1 to 24). Time to tumor sequencing was similar by race in our study (Table 1).
Figure 1.
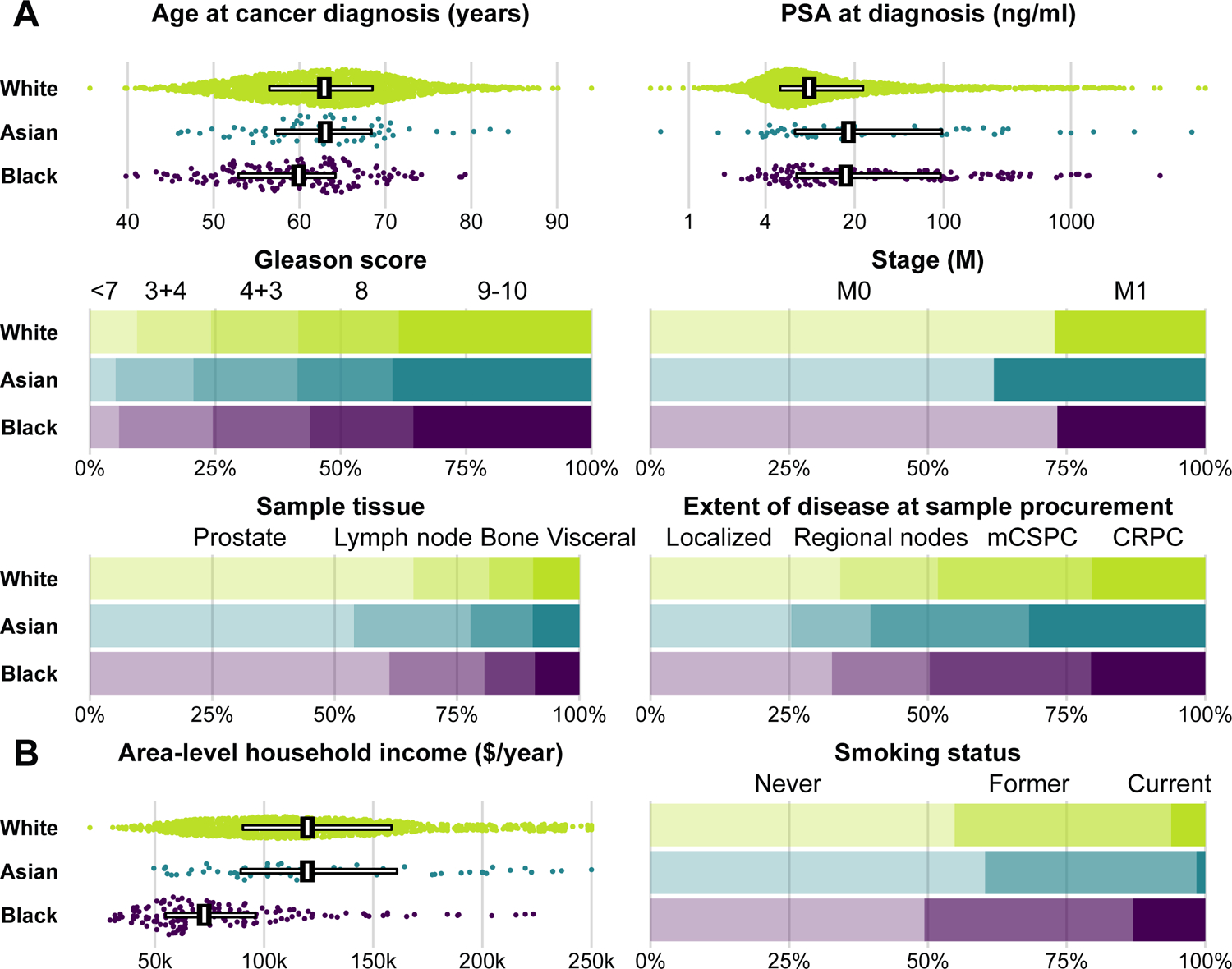
A, Clinical factors and B, cancer risk factors by self-reported race. For age, prostate-specific antigen, and area-level household income, individual data points are shown, with horizontal bars covering the 25th to 75th percentile and vertical bars indicating the median. For other characteristics, bars indicate proportions. PSA, prostate-specific antigen; CRPC, castration-resistant prostate cancer.
Table 1.
Characteristics of men and tumors with MSK-IMPACT sequencing.1
| By self-reported race | ||||
|---|---|---|---|---|
| Overall | White | Asian | Black | |
| 2,069 | 1,841 | 63 | 165 | |
| Age at diagnosis (years) | 63 (56, 68) | 63 (56, 68) | 63 (57, 68) | 60 (53, 64) |
| Age at sample (years) | 66 (59, 72) | 66 (59, 72) | 66 (60, 70) | 62 (56, 69) |
| Diagnosis to sample (months) | 3 (0, 43) | 3 (0, 42) | 4 (0, 29) | 3 (0, 50) |
| Diagnosis to sequencing (months) | 26 (6, 83) | 27 (6, 83) | 18 (7, 60) | 24 (6, 84) |
| Smoking status | ||||
| Never | 1,099 (55%) | 981 (55%) | 38 (60%) | 80 (49%) |
| Former | 783 (39%) | 698 (39%) | 24 (38%) | 61 (38%) |
| Current | 132 (7%) | 110 (6%) | 1 (2%) | 21 (13%) |
| Unknown | 55 | 52 | 0 | 3 |
| Area-level household income ($/year) | 115,844 (84,643, 156,558) | 119,884 (90,293, 158,420) | 119,780 (89,288, 160,935) | 72,568 (54,886, 96,086) |
| Unknown | 40 | 39 | 0 | 1 |
| PSA at diagnosis (ng/ml) | 9.2 (5.3, 26.8) | 8.8 (5.2, 23.3) | 17.9 (6.8, 97.0) | 17.0 (7.0, 94.5) |
| Unknown | 116 | 104 | 2 | 10 |
| Gleason grade | ||||
| <7 | 171 (9%) | 159 (9%) | 3 (5%) | 9 (6%) |
| 3+4 | 287 (15%) | 249 (15%) | 9 (16%) | 29 (19%) |
| 4+3 | 336 (18%) | 294 (17%) | 12 (21%) | 30 (19%) |
| 8 | 381 (20%) | 338 (20%) | 11 (19%) | 32 (21%) |
| 9–10 | 726 (38%) | 648 (38%) | 23 (40%) | 55 (35%) |
| Unknown | 168 | 153 | 5 | 10 |
| Stage (M) | ||||
| M0 | 1,501 (73%) | 1,341 (73%) | 39 (62%) | 121 (73%) |
| M1 | 568 (27%) | 500 (27%) | 24 (38%) | 44 (27%) |
| Sample tissue | ||||
| Prostate | 1,352 (65%) | 1,217 (66%) | 34 (54%) | 101 (61%) |
| Lymph node | 331 (16%) | 284 (15%) | 15 (24%) | 32 (19%) |
| Bone | 191 (9%) | 166 (9%) | 8 (13%) | 17 (10%) |
| Visceral | 125 (6%) | 110 (6%) | 3 (5%) | 12 (7%) |
| Other soft tissue | 70 (3%) | 64 (3%) | 3 (5%) | 3 (2%) |
| Extent of disease at sample | ||||
| Localized | 700 (34%) | 630 (34%) | 16 (25%) | 54 (33%) |
| Regional nodes | 362 (17%) | 324 (18%) | 9 (14%) | 29 (18%) |
| Metastatic hormone-sensitive | 578 (28%) | 512 (28%) | 18 (29%) | 48 (29%) |
| Non-metastatic castration-resistant | 17 (1%) | 16 (1%) | 0 (0%) | 1 (1%) |
| Metastatic castration-resistant | 405 (20%) | 353 (19%) | 20 (32%) | 32 (19%) |
| Metastatic, variant histology | 7 (0%) | 6 (0%) | 0 (0%) | 1 (1%) |
| Tumor cellularity by pathologist (%) | 40 (20, 50) | 40 (20, 50) | 40 (20, 50) | 40 (20, 50) |
| Unknown | 11 | 9 | 1 | 1 |
| Mean sequencing coverage (X) | 611 (461, 754) | 612 (461, 753) | 648 (539, 793) | 581 (453, 737) |
| Consent for germline analyses | 1,781 (86%) | 1,583 (86%) | 54 (86%) | 144 (87%) |
Values are count (percent) or median (quartile 1, quartile 3)
These differences in clinical cancer features prompted us to investigate two examples of potential upstream factors (Fig. 1B). Socioeconomic status has profound influences on cancer risk and survival (21) through cancer risk factors and healthcare use and access. Black men lived in zip code areas with an average household income 36% lower than that of White men (95% CI, 30 to 42%). There were no noticeable differences between Asian and White men. Twice as many Black men (13%) as White men (6%) were current smokers at the time of cancer diagnosis (difference, 7 percentage points; 95% CI, 2 to 12), a strong risk factor for prostate cancer death (4).
Tumor genomics by self-reported race before and after accounting for clinical features
We quantified alteration prevalence in oncogenes and tumor suppressors in tumor DNA (Fig. 2; Supplementary Fig. 2). Our focus was on genes that putatively differed by race in previous studies (7–10). Unlike previous studies, we considered all types of DNA alterations (mutations, copy number alterations, and fusions) but limited analyses to those classified as oncogenic or likely oncogenic based on a curated precision oncology database (17). We first compared expected tumor genomics by self-reported race because genetic ancestry is typically unknown in clinical practice until after a sequencing test is performed.
Figure 2.
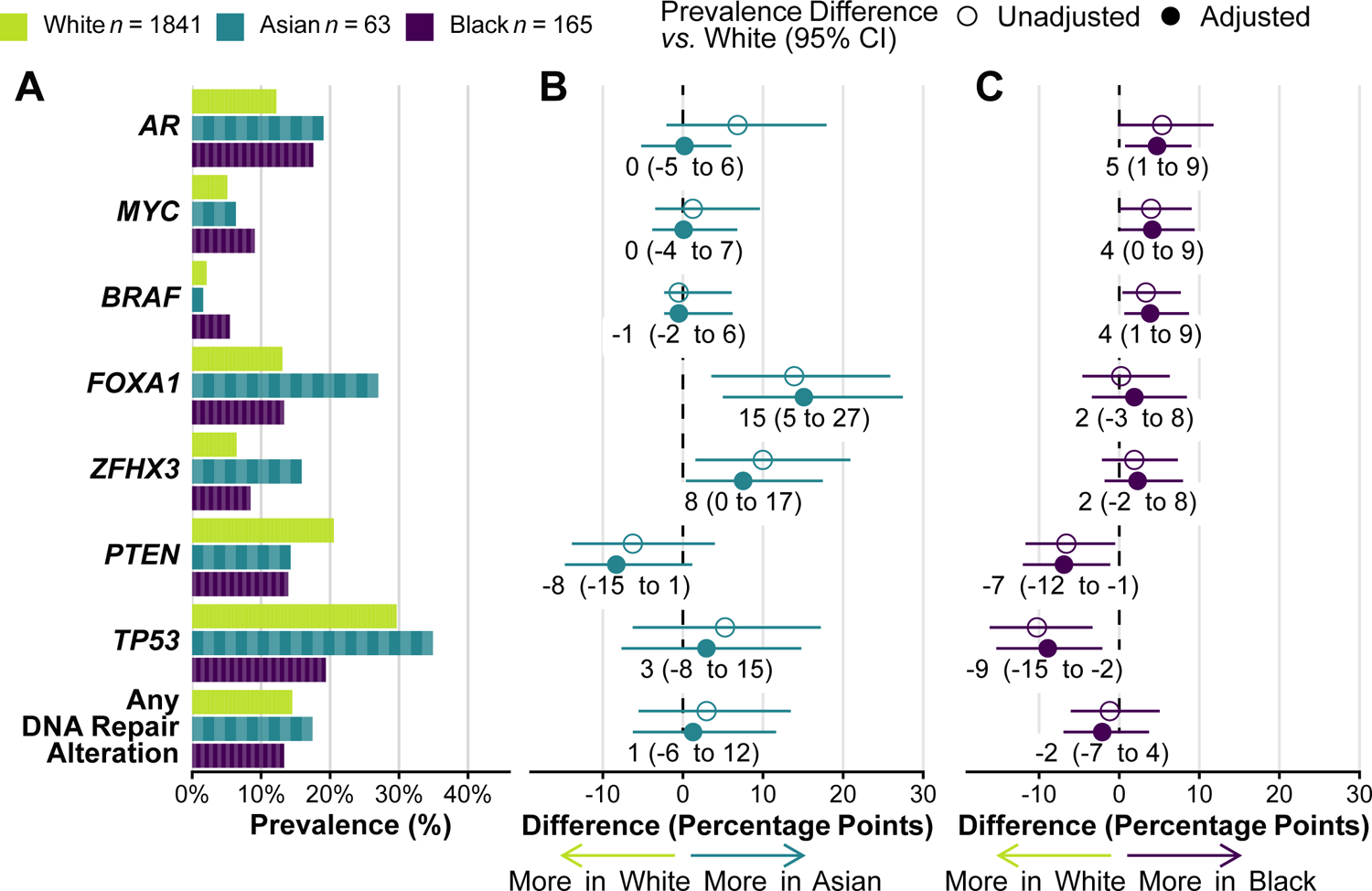
Self-reported race and tumor alterations. A, Prevalence of oncogenic alterations by self-reported race. Prevalence is a proportion; each “brick” represents one patient with an alteration (not visible for White men because of the high number). B, Differences in alteration prevalence, comparing Asian men vs. White men. C, Differences in alteration prevalence, comparing Black men vs. White men. Panels B and C show absolute differences in percentage points. Adjusted models include age at sample (linear), disease extent (localized, regional nodes, metastatic hormone-sensitive, or castration-resistant), prostate-specific antigen (PSA) levels at diagnosis (loge-transformed, mean-imputed), stage at diagnosis (de novo metastatic vs. non-metastatic), and sample type (prostate, lymph node, bone, or visceral). Individual genes are ordered by prevalence difference in Black men vs. White men. Only 1,924 samples (93%) were profiled for ZFHX3 due to its inclusion in a later panel version. For comparability with (8), DNA repair alterations encompass ERCC5, MRE11, TP53BP1, POLE, RAD21, MSH2, MSH6, BRCA1, BRCA2, ATR, and ATM.
Alterations in the androgen receptor gene (AR) occurred in 19% of tumors from Asian men and 18% among Black men, as compared to 12% among White men. Potential differences in AR alteration prevalence between tumors from Asian and White men were completely attenuated (prevalence difference, 0 percentage points; 95% CI, −5 to 6) after adjusting for clinical features (age, disease extent, PSA, stage, sample type) and thus accounting for the more advanced disease state when samples had been procured from Asian men. In contrast, AR alterations were slightly more common in tumors from Black men than White men even after adjusting for clinical features (difference, 5 percentage points; 95% CI, 1 to 8).
Other differences in tumor genomics by race persisted when adjusting for clinical features (Fig. 2), suggesting that the observed differences in tumor genomics were not driven exclusively by differences in disease extent. The strongest differences were in FOXA1, ZFHX3, TP53, and PTEN, with a smaller difference in MYC (copy number gains) and BRAF alterations (Fig. 2).
In FOXA1, 27% of tumors from Asian men had alterations compared 13% in White men (adjusted difference, 15 percentage points; 95% CI, 6 to 26), a difference driven by class-1 FOXA1 mutations in the forkhead domain (22), which occurred in 21% of tumors from Asian men compared to 8% from White men (Supplementary Fig. 3A). ZFHX3 alterations, both mutations and copy number alterations, were enriched in tumors from Asian men (17%) compared to White men (7%; adjusted difference, 8 percentage points; 95% CI, 1 to 17; Supplementary Fig. 2).
TP53 alterations were less common in tumors from Black men (19%) than White men (30%; adjusted difference, −10 percentage points; 95% CI, −3 to −17). This difference was likely driven by fewer TP53 missense mutations among tumors from Black men (11%) than White men (18%; adjusted difference, −4 percentage points; 95% CI, −9 to 1; Supplementary Fig. 3B). Differences were not explained by frameshift or truncating mutations or single hotspots, such as the most common mutation at amino acid R273 (prevalence among White men, 2%).
Alterations in the tumor suppressor PTEN were less common in tumors from Asian and Black men (14%) compared to White men (20%; adjusted difference Black vs. White, −7 percentage points; 95% CI, −12 to −1), particularly due to fewer PTEN mutations (8% in Black vs. 3% in White; adjusted difference, −4 percentage points; 95% CI, −7 to 0; Supplementary Fig. 3C).
In contrast to a previous study (8), we did not observe differences in the prevalence of DNA repair alterations (Fig. 2) or other potentially actionable alterations, nor did we find the (low) prevalence of microsatellite instability and the tumor mutation burden to differ by race (Supplementary Table 1). When combining gene-level alterations in oncogenic signaling pathways (23), differences in PI3K, RAS/RAF/MAPK, and MYC signaling pathways (Supplementary Fig. 4) were consistent with the observed differences in PTEN, BRAF, and MYC, respectively. In addition to these confirmatory analyses, we conducted an exploratory analysis of frequently altered genes (Supplementary Fig. 5), without additional candidates.
Tumor genomics and cancer risk factors by genetic ancestry
Single-nucleotide polymorphisms (SNPs) captured by the sequencing panels allowed us to infer genetic ancestry, which was strongly associated with but, as expected, not perfectly predictive of self-reported race (Fig. 3A). We then assessed how higher proportions of specific genetic ancestries were associated with tumor alterations that differed by self-reported race. We qualitatively observed the same associations between genetic ancestry and tumor alterations as between self-reported race and tumor alterations (Fig. 3B). For example, TP53 alteration prevalence was −13 percentage points lower (95% CI, −19 to −5) in tumors from men with 100% AFR (African) genetic ancestry markers than with 0% AFR ancestry.
Figure 3.
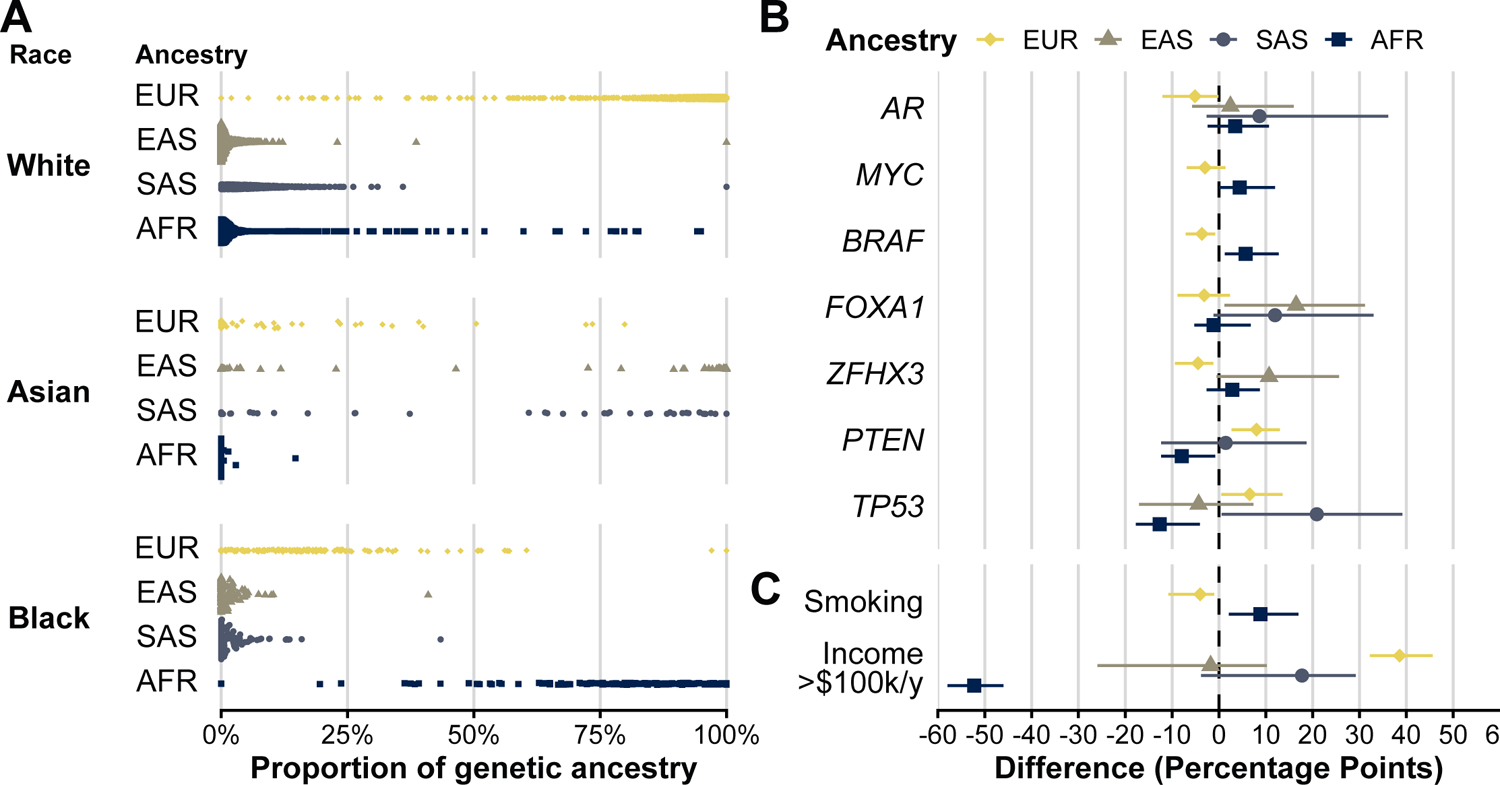
Genetic ancestry and tumor alterations. A, Self-reported race (left-most) and proportion of inferred genetic ancestry (x axis), by genetic superpopulation (EUR, European; EAS, East Asian; SAS, South Asian; AFR, African). B and C, Genetic ancestry and prevalence differences in tumors alterations (B) and cancer risk factors (C: current smoking vs. others; annual area-level household income >$100,000 vs. others). Each genetic ancestry was modeled continuously, and estimates are expressed as percentage-point differences in the outcome (with 95% CIs) for an extreme comparison of men with 100% of the respective genetic ancestry to those with 0%. No estimates for a specific ancestry are shown if <3 tumor alterations (or current smokers) were observed when categorizing men by predominant genetic ancestry.
We then probed to what extent ancestry can be interpreted causally as the influence of germline genetics or whether ancestry was also associated with non-genetic factors. We again encountered differences in smoking prevalence and in community-wide median household income, a unidimensional and imprecise measure of socioeconomic status, when comparing men by genetic ancestry (Fig. 3C). Among men with 100% AFR ancestry, 15% were predicted to live in a community with a median household income of at least $100,000 annually, compared to 68% among men with 0% AFR ancestry (difference, 52 percentage points; 95% CI, 46 to 58).
We assessed to what extent differences in tumor alteration prevalence could directly result from alterations present in the germline, which were included in all analyses. 1,781 men (86%) gave consent for germline genetic testing for alterations in genes associated with heritable cancer risk, without differences in consent by self-reported race. While germline alterations were included in all analyses, none of the genes with prevalence differences by race and ancestry had more than a single germline mutation among the 1,781 patients (Supplementary Table 2).
TP53 alterations and genomic instability
Tumor aneuploidy is strongly tied to TP53 alterations and genomic instability (24). Despite fewer TP53 alterations, tumors from Black men did not have a lower proportion of the tumor genome affected by copy number alterations (Fig. 4A). When adjusting for TP53 alteration status, tumors from Black men, but not Asian men, had slightly more copy number alterations than those from White men.
Figure 4.
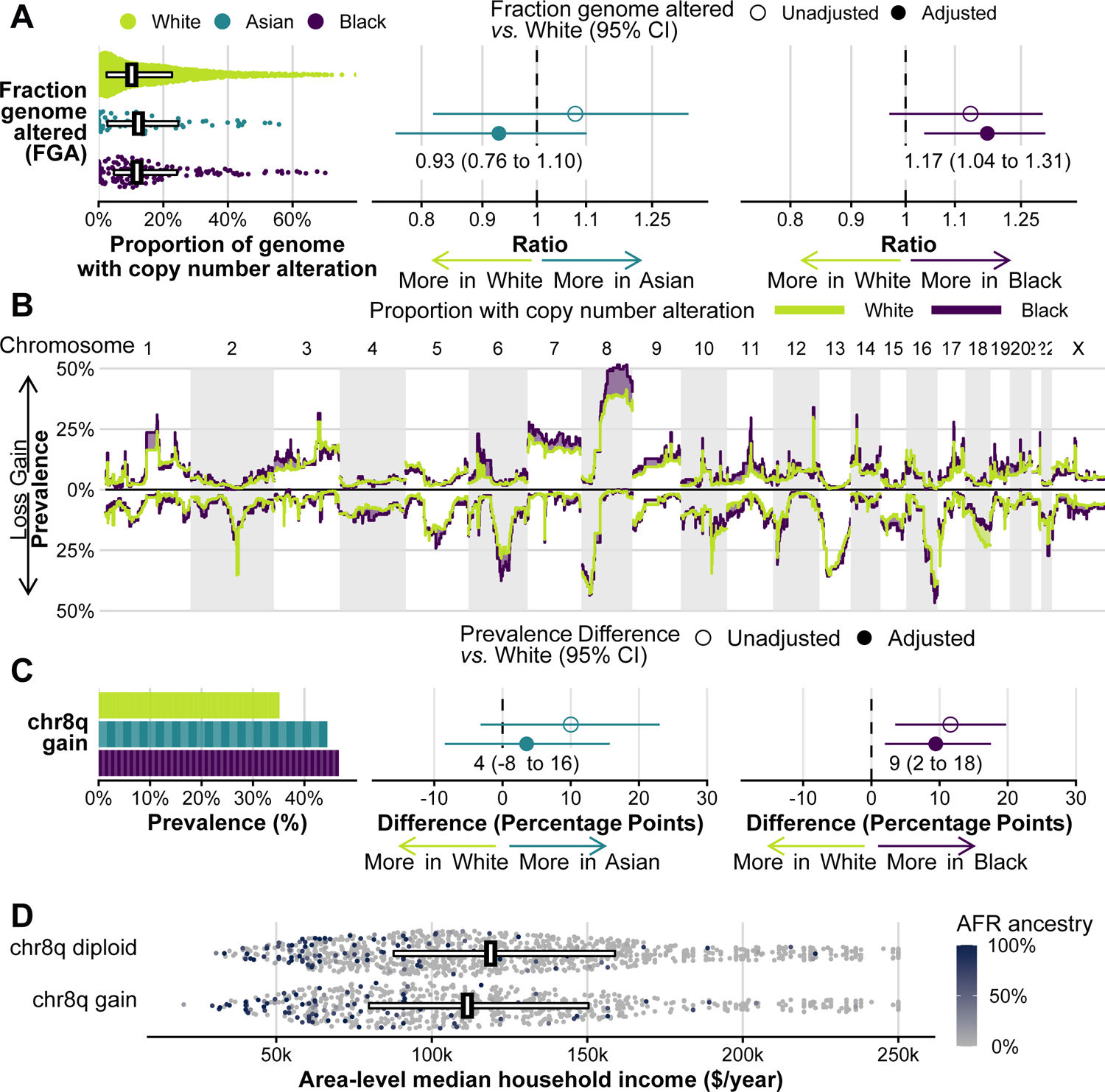
Tumor genomic instability. A, Fraction genome altered, the proportion of the genome affected by copy number alterations, by self-reported race. B, Segmental copy number profiles for White men and Black men, showing the proportion of tumors with copy number gains (upwards) or losses (downwards). Purple-colored areas indicate areas where copy number alterations are more common among tumors from Black men. C, Chromosome arm 8q gain by self-reported race. In A and C, the left panels show absolute values; one “brick” is one patient. The middle panels compare tumors from Asian men with tumors from White men; the right panel compares tumors from Black men with tumors from White men, giving the relative fraction genome altered (ratio, with 95% CI, in A) and the absolute difference in chr8q gain prevalence (with 95% CI, in C) from unadjusted comparisons and from adjusted comparisons that include the clinical factors as in Fig. 2 plus TP53 alteration status. D, Annual area-level household income, by chr8q status. One point is one individual; color indicates the proportion of AFR (African) ancestry markers; horizontal bars cover the 25th to 75th percentile and vertical bars indicate the median.
Patterns of copy number alterations appeared generally similar by race, except for chromosome arm 8q gains (Fig. 4B, Supplementary Fig. 6). 49% of tumors from Black men and 47% of tumors from Asian men had chr8q gains, as compared to 37% of tumors from White men. Adjusting for clinical factors attenuated the difference in chr8q gain prevalence for Asian men (difference to White, 5 percentage points; 95% CI, −6 to 17) but not for Black men (difference to White, 11 percentage points; 95% CI, 4 to 18; Fig. 4C).
As expected, chr8q gains were an adverse prognostic factor. Over a median of 30 months, 441 deaths occurred among 1690 White men with follow-up data after sequencing, 15 among 58 Asian men (precluding survival analyses), and 45 among 156 Black men. These data were not indicative of a generally worse prognosis among Black men in our study population, but we cannot rule out a difference (hazard ratio compared to White men, 1.15; 95% CI, 0.85 to 1.57). Tumors with chr8q gains, compared to tumors without chr8q gain but the same disease extent at sequencing, had a worse prognosis both among White men (hazard ratio, 1.61; 95% CI, 1.33 to 1.96) and among Black men (hazard ratio, 2.00; 95% CI, 1.00 to 4.01).
The difference in chr8q gain prevalence by race was mirrored by genetic ancestry. Tumors from men with 100% AFR ancestry had a 13 percentage-point higher prevalence of chr8q gains compared to 0% AFR ancestry (95% CI, 4 to 23). Yet even after adjusting for both self-reported race and genetic ancestry on an individual level, area-level median household income was associated with chr8q gain (Fig. 4D). For each $50,000/year increase, men of the same self-reported race and genetic ancestry were 2 percentage points less likely to have tumors with chr8q gain (95% CI, −4 to 0). Conversely, adjusting for community-level income reduced the above-noted difference in chr8q gains from 13 to 10 percentage points (95% CI, 0 to 20).
Discussion
Even within a group of men that ultimately had access to healthcare and tumor sequencing at the same cancer center, clinical factors and cancer risk factors differed noticeably by self-reported race and ancestry. These features are an essential part of the perspective on differences in tumor genomics. Tumor genomics still differed between men with similar prostate cancer clinical features but different self-reported race or genetic ancestry, in particular, prevalence of AR and BRAF alterations, class-1 FOXA1 mutations, ZFHX3 alterations, mutations of PTEN, missense mutations in TP53, and gains of chromosome arm 8q. Despite fewer TP53 alterations, tumors from Black men and those with greater AFR ancestry had more aneuploidy at chr8q, which may contribute to their aggressiveness. Our results suggest that self-reported race carries some information on what tumor genomic profiles to expect in clinic before a sequencing test is obtained. They also highlight the need for targeted treatments for genomic alterations more common in non-White men.
Our findings on differences in alteration prevalence of commonly altered genes in prostate cancer are generally consistent with previous studies (8–10,25). Some men in this study were included in a previous study (8) through Project GENIE (26). Our data are thus not an independent validation that alteration prevalence differs. Instead, the importance of our findings is the demonstration that most differences persist when restricting to oncogenic driver alterations, when accounting for different clinical features between men with different race, and when considering genetic ancestry.
The higher prevalence of AR and BRAF alterations in tumors from Black men is directly relevant in light of currently available targeted therapies (8). FOXA1, in which class-1 mutations in the DNA-binding forkhead domain were more common among tumors from Asian men, has been described functionally (22) but is not yet therapeutically actionable. In contrast to the class-1 FOXA1 mutations that occur early during carcinogenesis (22), a previously noted higher prevalence of (generally late-occurring) AR alterations in tumors from Asian men (8) was explained by more advanced disease among Asian men and thus should not influence clinical decision making. A lower prevalence of PTEN alterations in non-White men is relevant for PI3K inhibitors, which have reached phase 3 testing in prostate cancer (27).
Ancestry differences in aneuploidy, albeit in varying directions, have been observed in other cancer types (28). In our study, tumors with fewer TP53 alterations in Black men, specifically missense mutations, would be expected to have fewer chromosome arm losses and gains, given the strong association between TP53 alterations and aneuploidy burden (24). Surprisingly, tumors from Black men and those with higher AFR ancestry were no less aneuploid and instead harbored more chr8q gains, which are a poor prognostic indicator in primary prostate cancer (29) as well as in Black and White men in our study. chr8q gains explain differences in MYC alterations. Therapeutic targets on chr8q beyond MYC might be particularly relevant for Black men.
Many germline risk loci for prostate cancer are located at chr8q24, and some differ by ancestry (30). In our study, genetic ancestry had similar associations with chr8q gain and the other somatic alterations as did self-reported race. Germline genetics are clearly a contributor to tumor genomics. However, chr8q gains were also associated with a crude measure of socioeconomic status of neighborhoods, which differed strongly between Black and White men and by genetic ancestry. This crude, ecological measure does not allow pinpointing causes, which may include limited education opportunities and social services, neighborhood violence, environmental pollution, food insecurity, and many others (12), in addition to individual-level risk factors. Yet that modifiable risk factors have a strong impact is evidenced by prostate cancer rates among Asian-American men, which are lower than among Whites but substantially higher than for men of Asian ancestry living in Asia (31)—without changes in germline genetics upon immigration to the U.S. Our findings illustrate that studies on genetic ancestry as a potential cause of differences in tumor genomics need to consider confounding by non-genetic factors.
The underrepresentation of Asian and Black men in our study partially reflects disparities in access to healthcare. Increasing representation of racial and ethnic minorities will be important to allow for assessing differences by Hispanic ethnicity, between EAS and SAS ancestries, and by ancestry within each of the ancestry superpopulations, particularly between Northern, Eastern, and Western African ancestry that have been subsumed under AFR (11,32). Other facets of tumor genomics, and a richer set of cancer risk factors, need to be assessed. Even more important are population-based, rather than hospital-based study designs, without which we cannot know the denominator of men at risk of developing cancers with certain genomic features—a prerequisite for causal attributions and thus interventions that address causes, rather than just correlates of racial differences.
Taken together, tumor genomes differed by self-reported race even after accounting for differences in clinical features between men of different race. Cautiously considering race and tumor genomics jointly has potential to contribute to reducing the significant disparities in prostate cancer death rates that exist in the U.S. today (13). Differences in tumor genomics by race and ancestry should not be assumed to be completely genetically determined and thus immutable in the future.
Supplementary Material
Translational Relevance.
Prostate cancer has the greatest racial disparities in death rates in the United States of all cancers. As one contributor, tumor genomes have been suggested to differ by self-reported race. This study demonstrates that the same differences in tumor genomics as by self-reported race are observed if comparing men by genetic ancestry. There were notable differences in clinical factors by self-reported race among the men who underwent tumor–normal sequencing, but differences in tumor genomics persisted after adjusting for clinical characteristics. Tumors from Black men had more poor-prognosis chr8q gains, which beyond race and ancestry were also associated with a crude, area-level measure of socioeconomic status. These results underscore that therapies for genomic alterations more common in racial minorities are needed. Differences in tumor genomes should neither be assumed to result solely from access to care nor solely from germline genetics.
Acknowledgments
We are grateful for contributions of the Molecular Diagnostics Service and Marc Ladanyi, MD, to the MSK-IMPACT platform.
Financial support
This work was funded in part by the National Cancer Institute (1P01CA228696, to P.W. Kantoff; P30CA008748, Cancer Center Support Grant; P50CA092629, Prostate Cancer SPORE), the Department of Defense (Early Investigator Research Award W81XWH-18-1-0330, to K.H. Stopsack; Physician Research Award W81XWH-17-1-0124, to W. Abida), and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology. K.H. Stopsack, S.M. McBride, D.E. Rathkopf, L.A. Mucci, N. Schultz, and W. Abida are Prostate Cancer Foundation Young Investigators.
Conflicts of interest
S.M. McBride has received honoraria from AstraZeneca and research funding from AstraZeneca, Janssen, and Roche.
M.J. Morris is an uncompensated consultant to Bayer, Advanced Accelerator Applications, Johnson & Johnson, and Lantheus; a compensated consultant to Exelixis, NCCN, Athenex, Curium, and ORIC; and received institutional funding for the conduct of clinical trials for Bayer, Sanofi, Endoctye, Progenics, Corcept, Roche/Genentech, Janssen, and BMS.
D.E. Rathkopf is a consultant for Janssen, Genentech, AstraZeneca, Bayer, and Myovant Sciences, and has received research funding through her institution from Janssen Oncology, Medivation, Celgene, Tekeda, Millennium, Ferring, Novartis, Taiho Pharmaceutical, AstraZeneca, Genentech/Roche, TRACON Pharma, Bayer, and Phosplatin Therapeutics.
S.F. Slovin has received research support from Sanofi-Aventis, Novartis, Poseida, and the Prostate Cancer Foundation, and honoraria for advisory boards from Clovis, Janssen, Sanofi-Aventis, and PER.
D.C. Danila has received research support from the U.S. Department of Defense, American Society of Clinical Oncology, Prostate Cancer Foundation, Stand Up 2 Cancer, Janssen Research & Development, Astellas, Medivation, Agensys, Genentech, and CreaTV; he is a consultant for Angle LLT, Axiom LLT, Janssen Research & Development, Astellas, Medivation, Pfizer, Genzyme, and Agensys.
L.A. Mucci has provided expert testimony for Bayer and receives research funding from Janssen and AstraZeneca.
H. I. Scher reports the following support: compensated board of directors member of Asterias Biotherapeutics; compensated consultant/advisor to Ambry Genetics Corp, Konica Minolta, Bayer, Pfizer, Sun Pharmaceuticals, WCG Oncology; uncompensated consultant/advisory to Amgen, ESSA Pharma, Janssen Research & Development, LLC, Janssen Biotech, Sanofi Aventis; research funding to his institution from Epic Sciences, Illumina, Janssen, Menarini Silicon Biosystems, Prostate Cancer Foundation and ThermoFisher; intellectual property rights at BioNTech, Elucida Oncology, MaBVAX, Y-mAbs Therapeutics, non-financial support from Amgen, Asterias Biotherapeutics, Bayer, ESSA Pharma, Menarini Silicon Biosystems, Phosplatin, Pfizer, Prostate Cancer Foundation, and WCG Oncology.
D.B. Solit has consulted for/received honoraria from Pfizer, Loxo/Lilly Oncology, Vividion Therapeutics, Scorpion Therapeutics, and BridgeBio.
Y. Chen has interest in and research licencing with ORIC Pharmaceuticals.
M.F. Berger has received consulting fees from Roche and reseach support from Grail.
W. Abida has received honoraria from CARET, Roche, Medscape, and Aptitude Health; is a consultant for Clovis Oncology, Janssen, MORE Health, ORIC Pharmaceuticals, and Daiichi Sankyo; has received research funding through his institution from AstraZeneca, Zenith Epigenetics, Clovis Oncology, GlaxoSmithKline, ORIC Pharmaceuticals, and Epizyme; and he has had travel/accommodations/expenses paid by GlaxoSmithKline, Clovis Oncology, and ORIC Pharmaceuticals.
P.W. Kantoff reports the following disclosures for the last 24-month period: he has investment interest in Context Therapeutics LLC, DRGT, Placon, and Seer Biosciences; he is a company board member for Context Therapeutics LLC; he is a consultant/scientific advisory board member for Bavarian Nordic Immunotherapeutics, DRGT, GE Healthcare, Janssen, OncoCellMDX, Progenity, Seer Biosciences, and Tarveda Therapeutics; and he serves on data safety monitoring boards for Genentech/Roche and Merck.
K.H. Stopsack, S. Nandakumar, K. Arora, B. Nguyen, S.E. Vasselman, B. Nweji, K.A. Autio, M. Gönen, and N. Schultz have no disclosures.
Data availability
Clinical and genomic data from this study are available via cBioPortal: https://www.cbioportal.org/study/summary?id=prad_msk_stopsack_2021
References
- 1.Howlader N, Noone AM, Krapcho M, Miller D, Brest A, Yu M, et al. 2020 Feb 8, 2021. SEER Cancer Statistics Review, 1975–2017. National Cancer Institute <https://seer.cancer.gov/csr/1975_2017/>. Feb 8, 2021. [Google Scholar]
- 2.Hjelmborg JB, Scheike T, Holst K, Skytthe A, Penney KL, Graff RE, et al. The heritability of prostate cancer in the Nordic Twin Study of Cancer. Cancer Epidemiol Biomarkers Prev 2014;23(11):2303–10 doi 10.1158/1055-9965.EPI-13-0568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Conti DV, Darst BF, Moss LC, Saunders EJ, Sheng X, Chou A, et al. Trans-ancestry genome-wide association meta-analysis of prostate cancer identifies new susceptibility loci and informs genetic risk prediction. Nat Genet 2021;53(1):65–75 doi 10.1038/s41588-020-00748-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kenfield SA, Stampfer MJ, Chan JM, Giovannucci E. Smoking and prostate cancer survival and recurrence. JAMA 2011;305(24):2548–55 doi 10.1001/jama.2011.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cornelius ME, Wang TW, Jamal A, Loretan CG, Neff LJ. Tobacco Product Use Among Adults - United States, 2019. MMWR Morb Mortal Wkly Rep 2020;69(46):1736–42 doi 10.15585/mmwr.mm6946a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Awasthi S, Berglund A, Abraham-Miranda J, Rounbehler RJ, Kensler K, Serna A, et al. Comparative Genomics Reveals Distinct Immune-oncologic Pathways in African American Men with Prostate Cancer. Clin Cancer Res 2021;27(1):320–9 doi 10.1158/1078-0432.CCR-20-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li J, Xu C, Lee HJ, Ren S, Zi X, Zhang Z, et al. A genomic and epigenomic atlas of prostate cancer in Asian populations. Nature 2020;580(7801):93–9 doi 10.1038/s41586-020-2135-x. [DOI] [PubMed] [Google Scholar]
- 8.Mahal BA, Alshalalfa M, Kensler KH, Chowdhury-Paulino I, Kantoff P, Mucci LA, et al. Racial Differences in Genomic Profiling of Prostate Cancer. N Engl J Med 2020;383(11):1083–5 doi 10.1056/NEJMc2000069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan J, Kensler KH, Hu Z, Zhang Y, Zhang T, Jiang J, et al. Integrative comparison of the genomic and transcriptomic landscape between prostate cancer patients of predominantly African or European genetic ancestry. PLoS genetics 2020;16(2):e1008641 doi 10.1371/journal.pgen.1008641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koga Y, Song H, Chalmers ZR, Newberg J, Kim E, Carrot-Zhang J, et al. Genomic Profiling of Prostate Cancers from Men with African and European Ancestry. Clin Cancer Res 2020;26(17):4651–60 doi 10.1158/1078-0432.CCR-19-4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davis MB. Genomics and Cancer Disparities: The Justice and Power of Inclusion. Cancer Discov 2021;11(4):805–9 doi 10.1158/2159-8290.Cd-21-0225. [DOI] [PubMed] [Google Scholar]
- 12.Rebbeck TR. Prostate Cancer Disparities by Race and Ethnicity: From Nucleotide to Neighborhood. Cold Spring Harb Perspect Med 2018;8(9) doi 10.1101/cshperspect.a030387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borrell LN, Elhawary JR, Fuentes-Afflick E, Witonsky J, Bhakta N, Wu AHB, et al. Race and Genetic Ancestry in Medicine - A Time for Reckoning with Racism. N Engl J Med 2021;384(5):474–80 doi 10.1056/NEJMms2029562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keegan NM, Vasselman SE, Barnett ES, Nweji B, Carbone EA, Blum A, et al. Clinical Annotations for Prostate Cancer Research: Defining Data Elements, Creating a Reproducible Analytical Pipeline, and Assessing Data Quality. medRxiv 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn 2015;17(3):251–64 doi 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pettersson A, Graff RE, Bauer SR, Pitt MJ, Lis RT, Stack EC, et al. The TMPRSS2:ERG rearrangement, ERG expression, and prostate cancer outcomes: a cohort study and meta-analysis. Cancer Epidemiol Biomarkers Prev 2012;21(9):1497–509 doi 10.1158/1055-9965.EPI-12-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis Oncol 2017;2017 doi 10.1200/PO.17.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spurr LF, Touat M, Taylor AM, Dubuc AM, Shih J, Meredith DM, et al. Quantification of aneuploidy in targeted sequencing data using ASCETS. Bioinformatics 2020. doi 10.1093/bioinformatics/btaa980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res 2009;19(9):1655–64 doi 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stopsack KH. risks: Estimating risk ratios and risk differences using regression. https://stopsack.github.io/risks/.
- 21.Ward E, Jemal A, Cokkinides V, Singh GK, Cardinez C, Ghafoor A, et al. Cancer disparities by race/ethnicity and socioeconomic status. CA Cancer J Clin 2004;54(2):78–93 doi 10.3322/canjclin.54.2.78. [DOI] [PubMed] [Google Scholar]
- 22.Parolia A, Cieslik M, Chu SC, Xiao L, Ouchi T, Zhang Y, et al. Distinct structural classes of activating FOXA1 alterations in advanced prostate cancer. Nature 2019;571(7765):413–8 doi 10.1038/s41586-019-1347-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018;173(2):321–37 e10 doi 10.1016/j.cell.2018.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taylor AM, Shih J, Ha G, Gao GF, Zhang X, Berger AC, et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 2018;33(4):676–89 e3 doi 10.1016/j.ccell.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petrovics G, Li H, Stumpel T, Tan SH, Young D, Katta S, et al. A novel genomic alteration of LSAMP associates with aggressive prostate cancer in African American men. EBioMedicine 2015;2(12):1957–64 doi 10.1016/j.ebiom.2015.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.AACR Project GENIE Consortium. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov 2017;7(8):818–31 doi 10.1158/2159-8290.CD-17-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Bono JS, Bracarda S, Sternberg CN, Chi KN, Olmos D, Sandhu S, et al. LBA4 IPATential150: Phase III study of ipatasertib (ipat) plus abiraterone (abi) vs placebo (pbo) plus abi in metastatic castration-resistant prostate cancer (mCRPC). Ann Oncol 2020;31:S1153–S4 doi 10.1016/j.annonc.2020.08.2250. [DOI] [Google Scholar]
- 28.Yuan J, Hu Z, Mahal BA, Zhao SD, Kensler KH, Pi J, et al. Integrated Analysis of Genetic Ancestry and Genomic Alterations across Cancers. Cancer Cell 2018;34(4):549–60 e9 doi 10.1016/j.ccell.2018.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stopsack KH, Whittaker CA, Gerke TA, Loda M, Kantoff PW, Mucci LA, et al. Aneuploidy drives lethal progression in prostate cancer. Proc Natl Acad Sci U S A 2019;116(23):11390–5 doi 10.1073/pnas.1902645116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Troutman SM, Sissung TM, Cropp CD, Venzon DJ, Spencer SD, Adesunloye BA, et al. Racial disparities in the association between variants on 8q24 and prostate cancer: a systematic review and meta-analysis. Oncologist 2012;17(3):312–20 doi 10.1634/theoncologist.2011-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu H, Harris RE, Gao YT, Gao R, Wynder EL. Comparative epidemiology of cancers of the colon, rectum, prostate and breast in Shanghai, China versus the United States. Int J Epidemiol 1991;20(1):76–81 doi 10.1093/ije/20.1.76. [DOI] [PubMed] [Google Scholar]
- 32.Oni-Orisan A, Mavura Y, Banda Y, Thornton TA, Sebro R. Embracing Genetic Diversity to Improve Black Health. N Engl J Med 2021;384(12):1163–7 doi 10.1056/NEJMms2031080. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Clinical and genomic data from this study are available via cBioPortal: https://www.cbioportal.org/study/summary?id=prad_msk_stopsack_2021