

Abstract
Background:
Impairment of cellular cholesterol trafficking is at the heart of atherosclerotic lesions formation. This involves egress of cholesterol from the lysosomes and two lysosomal proteins, the Niemann–Pick C1 (NPC1) and NPC2 that promotes cholesterol trafficking. However, movement of cholesterol out the lysosome and how disrupted cholesterol trafficking leads to atherosclerosis is unclear. As the Wnt ligand, Wnt5a inhibits the intracellular accumulation of cholesterol in multiple cell types, we tested whether Wnt5a interacts with the lysosomal cholesterol export machinery and studied its role in atherosclerotic lesions formation.
Methods:
We generated mice deleted for the Wnt5a gene in vascular smooth muscle cells (VSMCs). To establish whether Wnt5a also protects against cholesterol accumulation in human VSMCs, we used a CRISPR/Cas9 guided nuclease approach to generate human VSMCs knockout for Wnt5a.
Results:
We show that Wnt5a is a crucial component of the lysosomal cholesterol export machinery. By increasing lysosomal acid lipase expression, decreasing metabolic signaling by the mTORC1 kinase, and through binding to NPC1 and NPC2, Wnt5a senses changes in dietary cholesterol supply and promotes lysosomal cholesterol egress to the endoplasmic reticulum (ER). Consequently, loss of Wnt5a decoupled mTORC1 from variations in lysosomal sterol levels, disrupted lysosomal function, decreased cholesterol content in the ER, and promoted atherosclerosis.
Conclusions:
These results reveal an unexpected function of the Wnt5a pathway as essential for maintaining cholesterol homeostasis in vivo.
Keywords: Atherosclerosis, Cell signaling/Signal transduction, Lipids and cholesterol, Metabolism, Physiology
INTRODUCTION
Wnt signaling has pleiotropic effects in diverse tissues. It plays an important role in cell differentiation, proliferation and migration during embryogenesis1. It has been associated with diverse human diseases1, and has been recently recognized as signaling involved in hyperlipemia syndrome and atherosclerosis. In mice, the mutation R611C of the Wnt co-receptor LRP6, a member of the LDLR gene family causes elevated plasma LDL and TG levels and fatty liver2. In humans, a missense mutation in LRP6 causes autosomal-dominant early onset cardiovascular disease and metabolic syndrome, including hyperlipidemia, diabetes, osteoporosis, and hypertension3. LRP6R611C mutation carriers exhibit impaired LDL clearance and internalization with elevated serum LDL cholesterol, implicating LRP6 and Wnt signaling as drivers of cholesterol trafficking3. More recently, Wnt signaling was also reported to promote resolution of atherosclerosis in mice and human4. However, how Wnt signaling mechanistically causes hyperlipidemia and impaired cholesterol trafficking remains an enigma.
Cholesterol uptake begins when LDL-derived cholesteryl esters are endocytosed through clathrin-coated pits into late endosomes/lysosomes (LELs) where they are hydrolyzed into free cholesterol and fatty acids by lysosomal acid lipase (LAL)5. Free cholesterol then binds two lysosomal proteins essential for cholesterol egress, the soluble Niemann–Pick C2 (NPC2) which is concentrated in LELs, and the polytopic membrane-bound Niemann–Pick C1 (NPC1)5. Upon binding to NPC2 in the lumen of the lysosome, cholesterol is handed off to NPC1 and then delivered to other cellular compartments including the plasma membrane and eventually the endoplasmic reticulum (ER) where regulation of cholesterol biosynthesis occurs. Heritable mutations in the NPC1 genes lead to massive cholesterol accumulation within LELs of many organs, including the brain. In macrophages, it causes impaired cholesterol efflux and atherosclerosis6. Whereas the functions of NPC1 and NPC2 are well understood at the molecular levels7, there are other crucial pieces of cholesterol export machinery we know very little about. For instance, in vitro binding experiments showed that whereas cholesterol binds rapidly to NPC2, handing it over to NPC1 is a slow process, especially at 4°C suggesting that other components beside NPC proteins are needed to perform efficient LELs cholesterol egress7.
The nutrient/energy/stress sensor mTORC1 was also suggested to participate in cholesterol trafficking. Accumulation of cholesterol activates the phosphatidylinositol 3-kinase (PI3K) and AKT which promotes translocation of mTORC1 from the cytosol to the lysosomal limiting membrane. Active mTORC1 then suppresses autophagy, steering the cell toward cholesterol accumulation8. Conversely, when mTORC1 activity is low, lysosomal exit of cholesterol raises ER cholesterol and suppresses activity of the sterol regulatory element-binding protein-2 (SREBP-2), a main transcriptional driver of cholesterol biosynthesis9. In NPC1-defective cells, mTORC1 signaling is elevated suggesting that NPCs, possibly in complex with other proteins function as sensors, regulating the mTORC1-scaffolding complex via cholesterol-dependent physical interaction.
We reported that mice deficient in the transmembrane receptor, the low density lipoprotein (LDL) receptor-related protein 1 (LRP1) in vascular smooth muscle cells (VSMCs) rapidly develop atherosclerosis10. LRP1 protects against atherosclerosis by integrating TGFβ and PDGF signaling and by stimulating the expression of a Wnt signaling protein, Wnt5a, an LRP6 ligand which we identified as an inhibitor of intracellular cholesterol accumulation11,12. LRP1 deficient mouse embryonic fibroblasts (MEFs) expressed very low endogenous Wnt5a11. When treated with a standard adipogenic mixture containing insulin, dexamethasone, and IBMX11, these cells accumulated large amounts of cholesterol within perinuclear cytosolic vesicles11, whereas their treatments with recombinant Wnt5a inhibited cholesterol accumulation. Similarly, mice overexpressing Wnt5a in adipose tissue display decreased cholesterol biosynthesis and accumulation in adipocytes12.
Wnt5a is also expressed in murine and human atherosclerotic lesions. Its expression positively corelates with the severity of the lesions13–15. Wnt5a upregulates the reverse cholesterol transport in fibroblasts, macrophages, and vascular smooth muscle cells11,15. However, it also stimulates endothelial cell proliferation and decreased endothelial NO production in vitro16, both of which being proatherogenic. Thus, its role in atherosclerosis lacks a solid mechanistic understanding. Here, we investigated whether, as previously reported with LRP1, augmenting Wnt5a in VSMCs protects against atherosclerosis in mice. We uncovered that lysosomal composition and function are profoundly disrupted in the absence of Wnt5a. We show that Wnt5a interacts with NPCs to down regulate mTORC1 signaling and lysosomal cholesterol egress. This mechanism is essential for maintaining cholesterol homeostasis and to protect against atherosclerosis.
METHODS
Data Availability.
The data and study material related to this study are available to other researchers on reasonable request. Methods are expanded in the Online Data Supplement.
RESULTS
Accelerated atherosclerosis in mice deleted for Wnt5a in VSMCs.
We achieved smooth muscle-specific Wnt5a inactivation by crossing SM22Cre transgenic mice with Wnt5aflox/flox animals. To increase susceptibility to spontaneous atherosclerosis these mice were crossed to LDL receptor knockout animals to generate SM22Cre+/Wnt5aflox/flox/LDLR−/− mice (smWnt5a−) and littermate controls SM22Cre-/Wnt5aflox/flox/LDLR−/− mice (smWnt5a+) (Fig. S1A). Only 20% of the smWnt5a− mice were born alive (Fig. S1B). Death was most likely due to heart defect as suggested by histology analysis showing cardiac tissue heavily disrupted and disorganized in dead mutants (data not shown). Surviving animals looked healthy. They were subjected to a cholesterol-rich (Paigen) diet for 5 months. As evaluated by p-NFκβ protein levels in the liver, we did not observe any toxicity of the diet (Fig. S1E). No significant difference was found in plasma cholesterol levels between mutants and controls (Fig. S1C). As shown by “en face” analysis (Fig. 1A) and quantification of the total size of the lesions in male and female animals (Fig. 1C), mutant mice developed twice as much atherosclerosis as controls. Interestingly, Oil-Red-O staining and histological analysis of thoracic and abdominal aortas showed lipid accumulation within lesions and in non-lesion areas in mutant mice, whereas in controls, lipid accumulated within lesions only (Fig. 1B, Fig. S2B), indicating that lysosomal function is impaired in the absence of Wnt5a. Thus, Wnt5a in VSMCs not only limits cholesterol accumulation, but also protects against atherosclerosis in mice, and in its absence lysosomal function is impaired.
Fig. 1. Accelerated formation of atherosclerotic lesions in smWnt5a− mice.
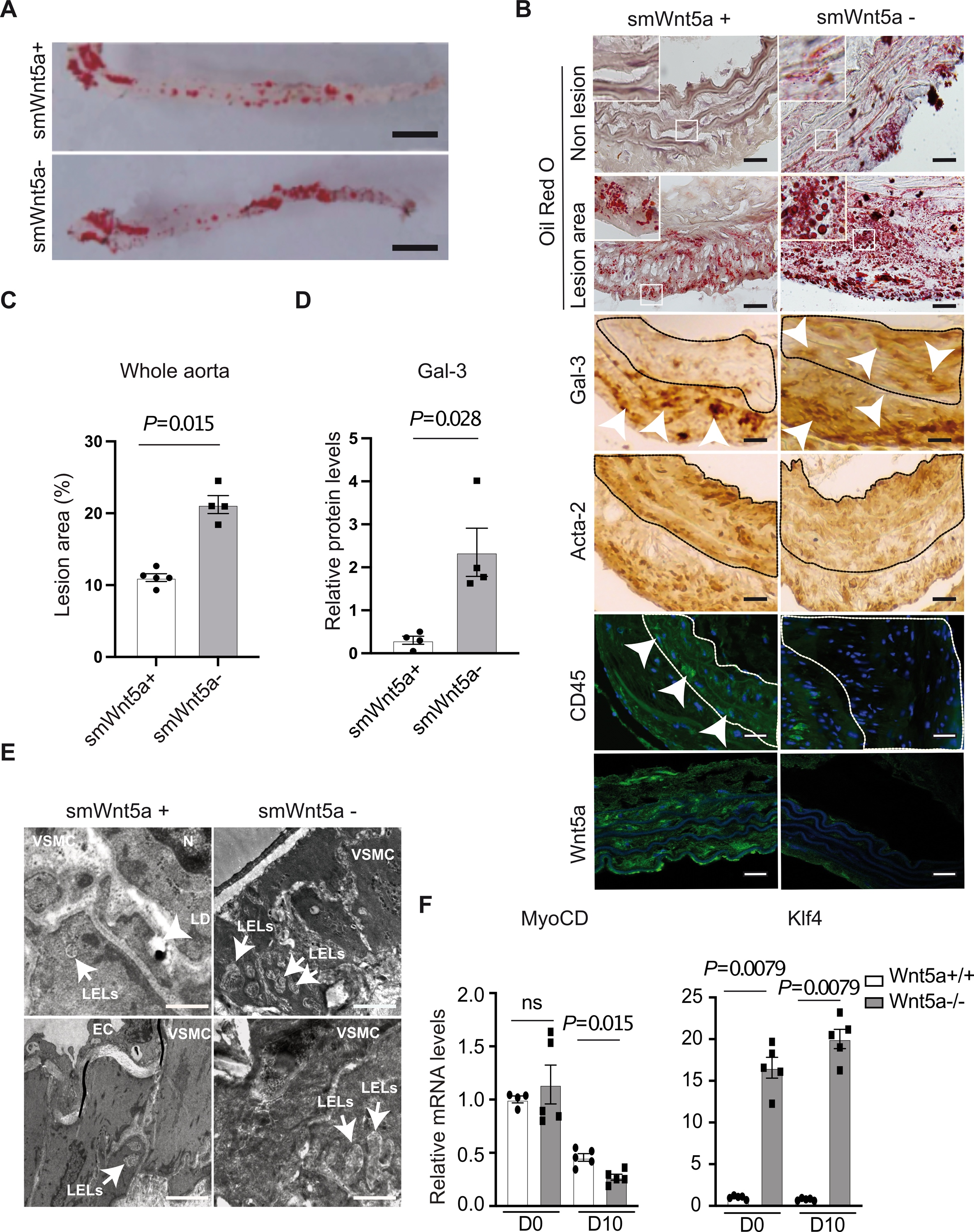
A, Opened and Sudan IV-stained whole aortas from cholesterol-fed LDL receptor deficient mice that express (smWnt5a+) or lack Wnt5a (smWnt5a−) in VSMCs. Scale bar 0,5 cm (n=6 mice/group). B, Oil Red O and immunohistochemical staining of Galectin-3, Acta-2, CD45, and Wnt5a in thoracic aortas from mice lacking Wnt5a in VSMCs or controls fed a cholesterol rich diet. The subpanels are higher magnification images (2.5X) of the areas outlined in white. The dashed line demarcates the media. Arrow heads show macrophages staining (Gal-3 and CD45). Scale bars 50μm (n=6 mice/group). C, Quantitative analysis of atherosclerotic lesion size in whole aortas from cholesterol-fed mice. The plot shows individual values with mean ± SEM (n=4 mice/group). D, Relative protein levels of Gal-3 in mutant and control mice. The plot shows individual scatter with mean ± SEM (n=4 mice/group). E, Representative electron micrographs show enlarged late endosomes/lysosomes (LELs) in VSMCs from thoracic aortas of mutants vs controls. Endothelial cells, nuclei, and lipid droplets are indicated (EC, N, LD). Scale bar top panels: 5μm, bottom panels 2μm (n=6 mice/group). F, Relative transcript levels of indicated genes in human VSMCs Wnt5a−/− and Wnt5a+/+ untreated (D0) and upon cholesterol loading (D10) taking GAPDH as internal control. The plot shows individual scatter with mean±SEM (MyoCD n=5 and Klf4 n=5). Statistical significance was assessed by Shapiro-wilk to test normality and Mann-Whitney test for C, D and F. C, P=0.015; D, P=0.028 indicate significance relative to smWnt5a+; F, P=0.015 and P=0.0079 indicate significance relative to Wnt5a+/+. ns indicates not significant.
Macrophage phenotypic switching in the absence of Wnt5a.
Because in advanced atherosclerosis 40–50% of VSMCs express macrophage markers17, we next evaluated whether the loss of Wnt5a affected VSMCs plasticity. In control mice, we found low expression of the macrophage marker Galectin-3 in the media, whereas the VSMCs marker Acta-2 is expressed in the media and at the periphery of plaques. By contrast in mutant mice, we found higher expression of Galectin-3 (Fig. 1B, D) and decreased expression of Acta-2 (Fig. 1B) within lesions and in the media of the aortas. The endothelial cell markers CD31 expression was not affected (Fig. S2A). In addition, CD45 a leukocyte/macrophage specific marker never expressed by VSMCs is expressed in the media in control mice, but not in the absence of Wnt5a indicating that these Galectin-3-positive cells are VSMCs in origin (Fig. 1B). Together, these data suggested that Wnt5a protects against macrophage phenotypic switching of VSMCs. To confirm this in human VSMCs, we used a CRISPR/Cas9 guided nuclease approach to generate human VSMCs knockout for Wnt5a (Wnt5a−/− VSMCs) and non-targeted controls (Wnt5a+/+ VSMCs). In human, Wnt5a gene produces two protein isoforms18. Both isoforms were deleted in human VSMCs (Fig. S3A, B). Cells were treated for 10 days with the cholesterol accumulation cocktail we previously reported, that consisted of a standard adipogenic mixture containing insulin, dexamethasone, IBMX, and the PPARγ agonist rosiglitazone11. In human Wnt5a−/− VSMCs western blot analysis revealed increased expression of the macrophage marker Galectin-3 (Fig. S1D) and decreased expression of the VSMCs marker MyoCD (Fig. 1F) upon cholesterol loading. In addition, mRNA expressions of the Kruppel-like transcription factor 4 (KLF4), a transcription factor known to silence VSMC marker genes17, and to promote formation of cholesterol-induced VSMC-derived foam cells strongly increased in untreated cells and upon cholesterol loading in the absence of Wnt5a (Fig. 1F). Thus, Wnt5 contribute to protect against macrophage phenotypic switching of VSMCs in vitro and during vascular injury.
Lipid accumulation in Wnt5a−/− human VSMCs.
We next tested whether Wnt5a protects against cholesterol accumulation in human VSMCs. When cultured in normal conditions, total cholesterol increased slightly but non significantly in human Wnt5a−/− VSMCs when compared to controls (Fig. 2A). However, cholesteryl-ester (CE) concentrations were significantly higher in Wnt5a−/− VSMCs (Fig. 2B). Upon cholesterol loading, Wnt5a protein levels increased over time in wild type human VSMCs (Fig. S3C) and neutral lipids accumulated modestly as shown by Oil-Red-O staining and microscopy analysis (Fig. S3E). By contrast, neutral lipids strongly accumulated in Wnt5a−/− VSMCs compared to controls, whereas their treatment with human recombinant Wnt5a rescued neutral lipids accumulation back to control levels (Fig. 2D). At day 10, CE level was strongly increased in Wnt5a−/− VSMCs (Fig 2B), and total cholesterol concentration was elevated as well (Fig. 2A). In both treated and untreated cells, triglyceride levels (Fig. S3D) as well as transcript levels of the acyl CoA:diacylglycerol acyltransferase 2 (DGAT2) enzyme that is required for triglyceride synthesis in adipocytes19, were similar in Wnt5a−/− VSMCs and controls (Fig. S3G). To confirm cholesterol accumulation in Wnt5a−/− VSMCs we used Ox-LDL as a lipoprotein vehicle for cholesterol loading. Previous reports indicated that upon ox-LDL treatments, RAW264.7 macrophages and VSMCs knockdown for Wnt5a accumulated cholesterol15. In agreement, we now show that when treated with ox-LDL for 12 days, Wnt5a−/− human VSMCs accumulated more neutral lipids than controls as evaluated by Oil Red O staining and microscopic analysis (Fig. S4A).
Fig. 2. Cholesterol accumulation and altered lysosomal function in human VSMCs Wnt5a−/−.
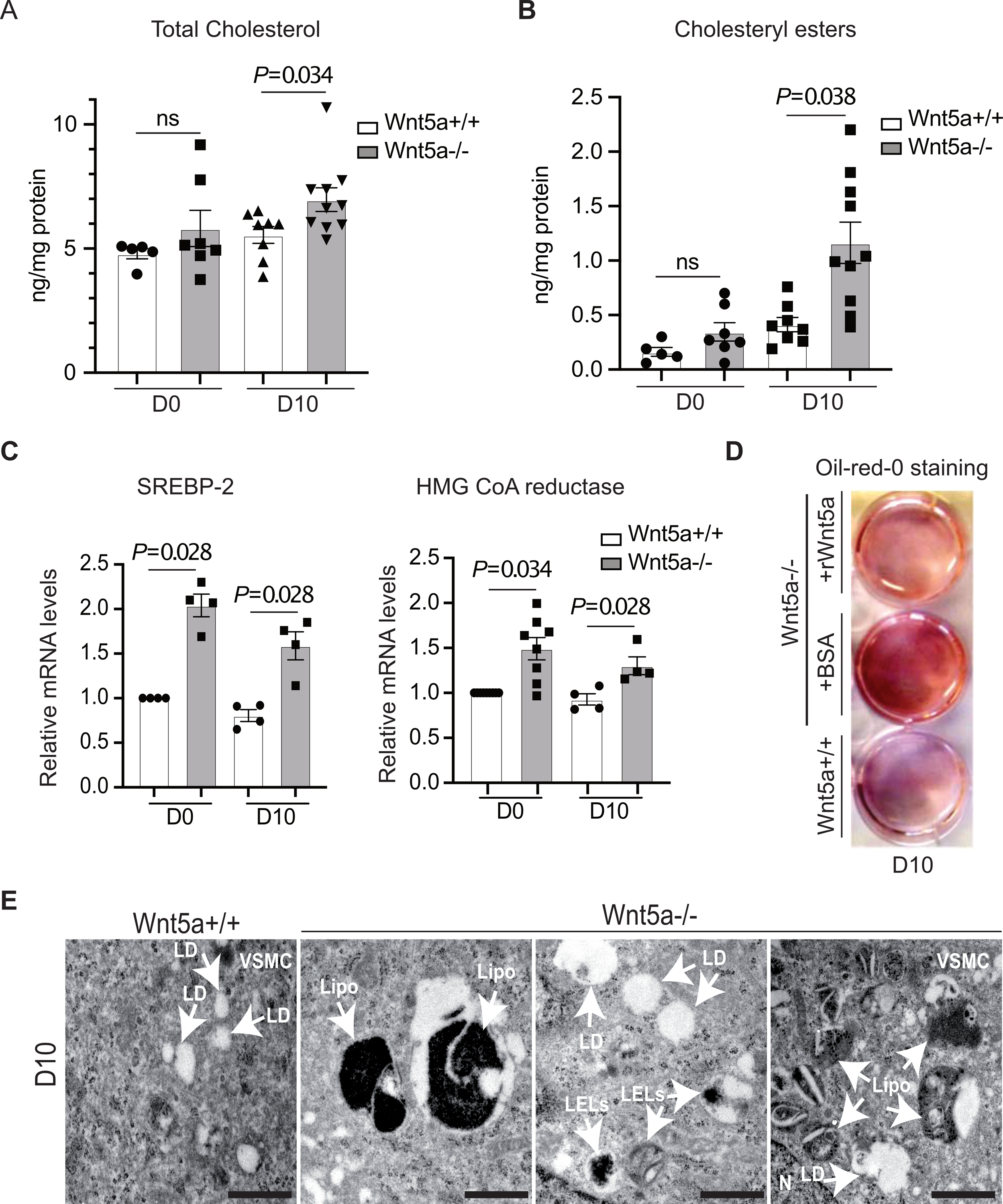
Human VSMCs Wnt5a−/− and Wnt5a+/+ were analyzed untreated (D0) or upon treatment with a cholesterol accumulation protocol during 10 days (D10). A, Quantification of total cholesterol n=5 D0 Wnt5a+/+, n=7 D0 Wnt5a−/−, n=8 D10 Wnt5a+/+, n=10 D10 Wnt5a−/−). B, Quantification of cholesteryl esters (n=5 D0 Wnt5a+/+, n=7 D0 Wnt5a−/−, n=8 D10 Wnt5a+/+, n=10 D10 Wnt5a−/−). C, Relative transcript levels of SREBP-2 (n=4 D0 and D10) and HMG CoA reductase (n=9 D0 Wnt5a+/+, n=8 D10 Wnt5a−/−, n=4 D10 Wnt5a +/+ and n=4 D10 Wnt5a −/−) taking GAPDH 18S as internal control. D, Oil Red O staining upon treatment for cholesterol accumulation during 10 days in presence of human recombinant Wnt5a (+rWnt5a) or BSA (+BSA) in human Wnt5a−/− VSMCs (n=3). E, Representative electron micrographs show presence of large lipofuscin containing vesicles (Lipo), lipid droplets (LD), and large LELs in human Wnt5a−/− VSMCs vs controls. Scale bar 1μm (n=3). A, B, and C show individual value along with mean±SEM. Statistical significance was assessed by Shapiro-wilk to test normality, unpaired t test for A and B, and Mann-Whitney test for C. A, P=0.034; B, P=0.038; C, P=0.028 and P=0.028 for SREBP-2 and P=0.034 and P=0.028 for HMG CoA reductase indicate significance relative to Wnt5a+/+. ns indicates not significant.
We next tested whether loss of Wnt5a activates cholesterol biosynthesis genes in human VSMCs. We found that mRNA levels of SREBP-2, and mRNA and protein levels of its target gene HMG CoA reductase were increased in the absence of Wnt5a in untreated and treated human VSMCs (Fig. 2C, Fig. S3F). Thus, loss of Wnt5a in human VSMCs constitutively increased the SREBP-2 pathway. These data are consistent with our previous report indicating that overexpression of Wnt5a in adipose tissue of mice decreased SREBP-2 activity and cholesterol biosynthesis12.
Absence of Wnt5a is associated with cholesterol accumulation in late endosomes.
We next analyzed in which compartment cholesterol accumulated in the absence of Wnt5a. Electron microscopic analysis of treated human Wnt5a−/− VSMCs revealed accumulation of lipid droplets, accumulation of numerous very large endosome/lysosome (LELs) containing cholesterol crystals, and accumulation of very large electron-dense cytoplasmic lipofuscin containing vesicles (Fig. 2E, Fig. S4B) that were comparable in shape and size to those seen in the heart and in the aortas of smWnt5a− mice (Fig. S2B). To determinate whether lipofuscin accumulated in endosomes, we used confocal imaging and the late endosomes marker (lysosome-associated membrane protein 1) Lamp1. We found that the lipofuscin-enriched large cytosolic vesicles that accumulate in Wnt5a−/− VSMCs are Lamp1 positive LELs (Fig. S4C).
The increased in cholesterol and CE levels upon treatment with the cholesterol accumulation cocktail, together with the alteration of the endosomal compartments suggested that in the absence of Wnt5a, cholesterol does not reach the ER. To test this, we quantified cholesterol inside cells with a specific fluorescent probe: the D4 fragment of perfringolysin O fused to GFP (GFP-D4). This probe can be efficiently used to label sterols in the PM or in subcellular structures such as endosomes20. We found more cholesterol-enriched cytosolic vesicles in Wnt5a−/− VSMCs as shown by immunofluorescence (Fig. 3A) and by quantification of GFP-D4 positive vesicles (Fig. 3B). Moreover, co-labelling between Lamp1 and the GFP-D4 probe showed an overlap of both signals indicating that cholesterol accumulates in endosomal compartments (Fig. 3A). Quantification of co-localization between GFP-D4 and Lamp1 show that there is three times more Lamp1+/GFP-D4+ overlapping vesicles in VSMCs lacking Wnt5a compared to controls upon cholesterol accumulation (Fig. 3C). In addition, immunofluorescence analysis of the LELs marker Lamp1 indicated that Lamp1 protein levels are substantially higher in cholesterol-fed mutant cells compared to controls (Fig. 3A). A time course experiment revealed that, whereas in control cells Lamp1 protein expression increased modestly during the course of cholesterol accumulation protocol, its expression reached higher levels in VSMCs lacking Wnt5a (Fig. 3D, Fig. S4D). Analysis of acidic compartments using LysoTracker reagent showed numerous and bigger acidic compartments in the absence of Wnt5a (Fig. 3E, F). Similarly, upon treatments with the Ox-LDL protocol immunohistochemistry experiments revealed numerous and bigger Lamp1 positive compartments in the absence of Wnt5a (Fig. S4A). Thus, in the absence of Wnt5a cholesterol accumulated in multivesicular inclusions and enlarged LELs.
Fig. 3. Cholesterol accumulated in Lamp1 positive cytosolic vesicles in human Wnt5a−/− VSMCs.
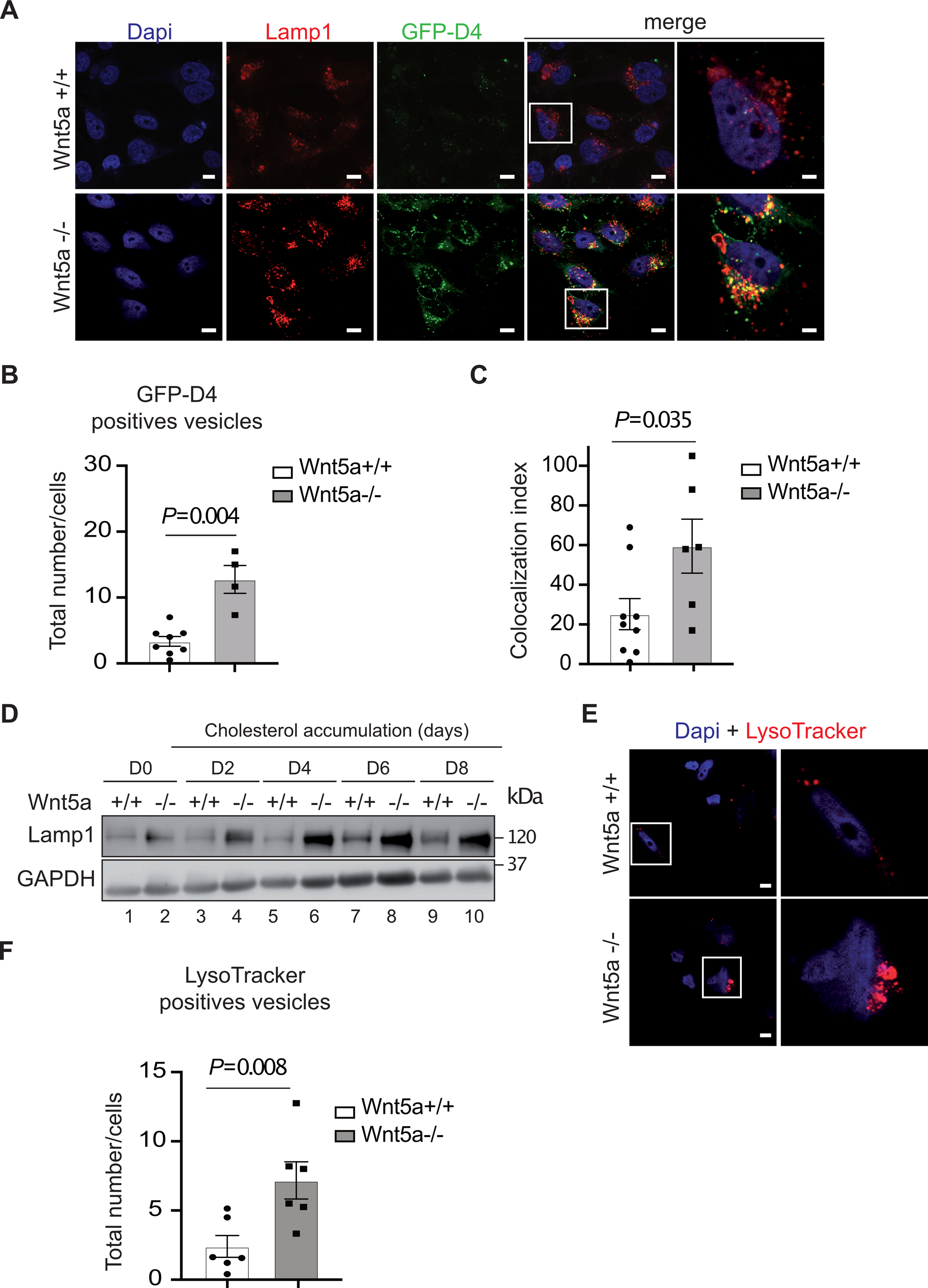
A, Cholesterol accumulation in human VSMCs Wnt5a−/− and controls labeled with anti-Lamp1 antibodies (red) a LELs marker, and with the fluorescent cholesterol probe GFP-D4 (green). Nuclei were stained in blue with Dapi. Higher magnification (2.5X) images of the areas outlined in white are shown in the right panels. Scale bar 5μm (n=4). B, Quantification of GFP-D4 positive vesicles/cells in human VSMCs Wnt5a−/− and controls (n=8 Wnt5a+/+, n=4 Wnt5a−/−). C, Quantification of confocal analysis shows increased colocalization between GFP-D4 (cholesterol) and Lamp1 (LELs) in human VSMCs Wnt5a−/− compared to controls (n=9 Wnt5a+/+, n=6 Wnt5a−/−). D, A representative immunoblot shows the protein expression of Lamp1 during the course of cholesterol accumulation (n=3). E, Human VSMCs Wnt5a−/− and controls were treated with 50 nM lysosome-tracker red. Higher magnification (2.5X) images of the area outlined in white are shown on the right. Scale bar 6μm (n=4). F, Quantification of lysosome-tracker red positive vesicles (n=6 Wnt5a+/+ and Wnt5a−/−). B, C, and F show individual value along with mean±SEM. For C, statistical significance was assessed by Shapiro-wilk to test normality followed by an unpaired t-test. For B and F statistical significance was assessed by Shapiro-wilk to test normality followed by a Mann-Whitney test. B, P=0.004; C, P=0.035 and F, P=0.008 indicate significance relative to Wnt5a+/+.
Absence of Wnt5a blocks cholesterol egress from LELs.
Because lysosome-derived cholesterol correlates closely with ER cholesterol, we indirectly assessed ER cholesterol levels by measuring ACAT1 (acyl-CoA cholesterolacyltransferase 1) expression and activity. ACAT1 is an ER resident enzyme that converts free cholesterol to cholesteryl ester. Its activity is controlled by cholesterol availability in the ER membrane21. In untreated cells, ACAT1 protein levels were markedly decreased in human Wnt5a−/− VSMCs compared to controls (Fig. 4A, B), indicative of low ER cholesterol. In addition, ACAT1 activity (Fig. 4C) was almost undetectable in human Wnt5a−/− VSMCs untreated or treated for cholesterol accumulation (Fig. 4C). LAL is the enzyme that hydrolyzes CE in lysosomes. Its activity in human VSMCs is markedly lower when compared to macrophages and this represent a mechanism for VSMCs foam cell formation22. To rule out the possibility that the CE accumulation observed in Wnt5a−/− VSMCs is due to decreased lysosomal hydrolysis of CE we measured its expression. We found the LAL protein levels almost undetectable in the absence of Wnt5a (Fig. 4D). Thus, the loss of Wnt5a may reduce its capacity to catabolize atherogenic lipoproteins and CE. This results in the accumulation of cholesterol and CE in Lamp1 positive compartments at the expense of the ER.
Fig. 4. Decreased concentrations of ER membrane cholesterol in human VSMCs Wnt5a -/−.
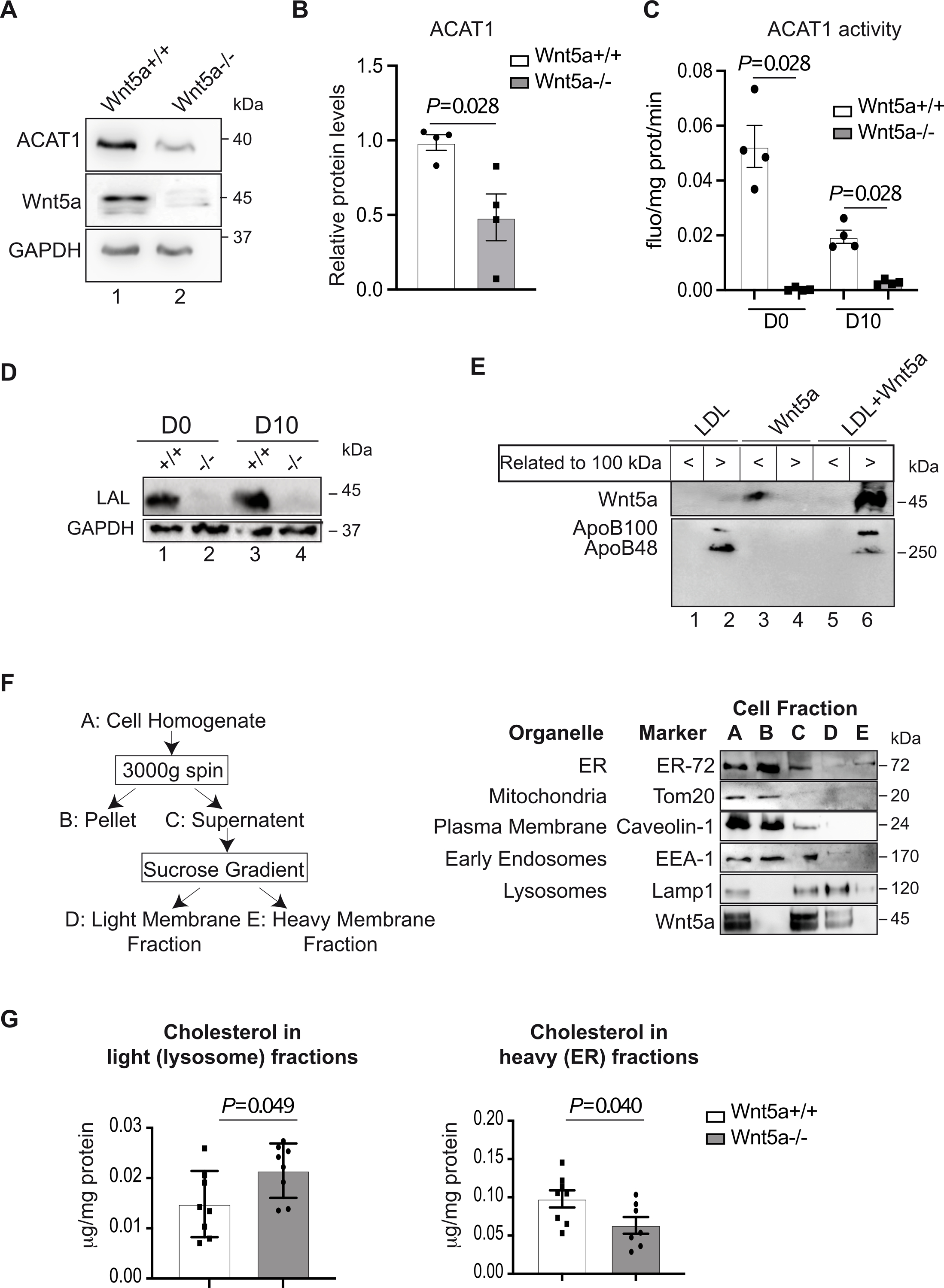
A, A representative immunoblot shows the protein expression of ACAT1 and B, its quantification in human Wnt5a−/− VSMCs vs controls upon cholesterol accumulation protocol calculated as relative to GAPDH expression (n=3). C, ACAT1 activity in human Wnt5a−/− VSMCs vs controls before (D0) and upon (D10) cholesterol accumulation treatments during 10 days (n=4). D, A representative immunoblot shows the protein expression of the Lysosomal acid lipase (LAL) (n=4). E, Wnt5a was incubated with human LDL particles as described in the method section. A representative immunoblot shows the presence of Wnt5a in the <100 kDa fraction when incubated with CHAPS, or in >100 kDa apoB positive fraction when incubated with human LDL (n=3). F, Purification of ER and LELs, purification of mitochondria, plasma and early endosomal membranes. Left panel is showing the diagram of LELs membrane fractionation scheme. A-E denote major fractions recovered and analyzed by western blot. Right panels, VSMCs were treated according to the fractionation scheme and as described in the method section. Aliquots representing equal volumes of each fraction (A-E) were subjected to immunoblot analysis for the indicated organelles markers and Wnt5a. G, Quantification of cholesterol concentrations in heavy (E, ER) and light (D, lysosomes) membrane fractions from human Wnt5a−/− VSMCs and Wnt5a+/+ (n=8 and n=7 Wnt5a−/− heavy fractions). B, C and G show individual data along with mean±SEM. For B, C and G, data were analyzed using a Shapiro-Wilk test to assess normality followed by Mann-Whitney test. For B, P=0.028; C, P=0.028 and G, P=0.049 and P=0.040 indicate significance relative to Wnt5a+/+.
Localization of Wnt5a in LEL particles.
To determine how Wnt5a enter the endocytic compartment we postulate that, as previously described in the cerebrospinal fluid23, Wnt5a associates with LDL particles. For this, recombinant Wnt5a was incubated with either 0.6% CHAPS or human LDL lipoprotein fraction for 4 h at 37 °C and filtered through 100 kDa cut-off protein concentrator. When recombinant Wnt5a was incubated with LDL, western blot confirmed presence of Wnt5a only in >100 kDa fraction but not in the <100 kDa fraction indicating that recombinant Wnt5a localized on LDL particles and forms a high-molecular weight complex (Fig. 4E).
Next, we hypothesize that Wnt5a might be internalized together with LDLs in LELs. For this, we used a subcellular fractionation approach to isolate LELs from human VSMCs as outlined in Fig. 4F. The cell homogenate (Fraction A) was subjected to two centrifugation steps in order to separate LELs membranes from other organelles (Fig. 4F, left panel). Centrifugation of the homogenate at 3000xg separates unbroken cells and the heaviest membranes. As assessed by immunoblot analysis of the fractions (Fig. 4F, right panel), a plasma membrane protein marker (Caveolin-1) and an ER membrane protein marker (ER-72) are mostly retained in the 3000xg pellet (Fraction B), whereas significant amounts of all other membrane markers, including the LEL protein marker Lamp1 were present in the 3000xg supernatant (Fraction C). Fraction C was then subjected to centrifugation through a discontinuous sucrose gradient and yielded two distinct bands of membranes (Fractions D, E). Fraction E contained ER membrane protein marker, whereas the LEL membrane protein Lamp1 was essentially present in Fraction D (Fig. 4F, right panel). Interestingly, Wnt5a was present in Fractions C and D, indicative of its localization in LELs. In addition, cholesterol in the fraction enriched in ER membranes (the heavy Fraction E) is decreased in mutant cells compared to controls, whereas cholesterol in the fraction enriched in LEL membranes (the light Fraction D) is increased (Fig. 4G). Thus, Wnt5a is directly associated with the LEL compartments, and in the absence of Wnt5a cholesterol is retained in LEL fractions.
Wnt5a down regulates the mTORC1 pathway.
LELs have emerged as the cellular site where the master growth regulator and nutrient sensor, mechanistic target of rapamycin complex 1 (mTORC1) is activated. Given that high mTORC1 activity coincides with low ER cholesterol, high ACAT-1 expression, and activation of SREBP-29, we assessed mTORC1 activation in Wnt5a−/− VSMCs. We found a robust increase of p-mTORC1 and its downstream targets, p-P70S6 and p-4EBP-1, and of p-Akt in the absence of Wnt5a in human VSMCs (Fig. 5A). During the course of cholesterol accumulation, p-mTORC1 remains highly activated in the absence of Wnt5a (Fig. S5A). Interestingly, rescue experiments show that treatment of human Wnt5a−/− VSMCs with conditioned media enriched in Wnt5a (Fig. S5B) decreased the p-mTORC1 target gene p-P70S6 to control levels, whereas mock media had no effect (Fig. 5B). Similarly, treatment with recombinant human Wnt5a decreased p-mTORC1 in human Wnt5a−/− VSMCs back to control levels (Fig. 5D). Treatment with Everolimus®, an mTORC1 inhibitor, decreased p-mTORC1 and p-Akt in human Wnt5a−/− VSMCs to control levels (Fig. S5C). Everolimus® treatment decreased cholesterol accumulation in the CD63 positive compartments in human Wnt5a−/− VSMCs (Fig. 5E). Similarly, Oil-red-o staining showed that neural lipids and cholesterol are reduced upon Everolimus treatments (Fig. S5D), and GC-MS quantification showed that Everolimus treatments decreased total cholesterol and cholesteryl esters in Wnt5a−/− VSMCs, however without reaching significance (Fig. S5E). Thus, loss of Wnt5a coincides with high mTORC1 activity, and cholesterol accumulation.
Fig. 5. Wnt5a decreases mTORC1 activity.
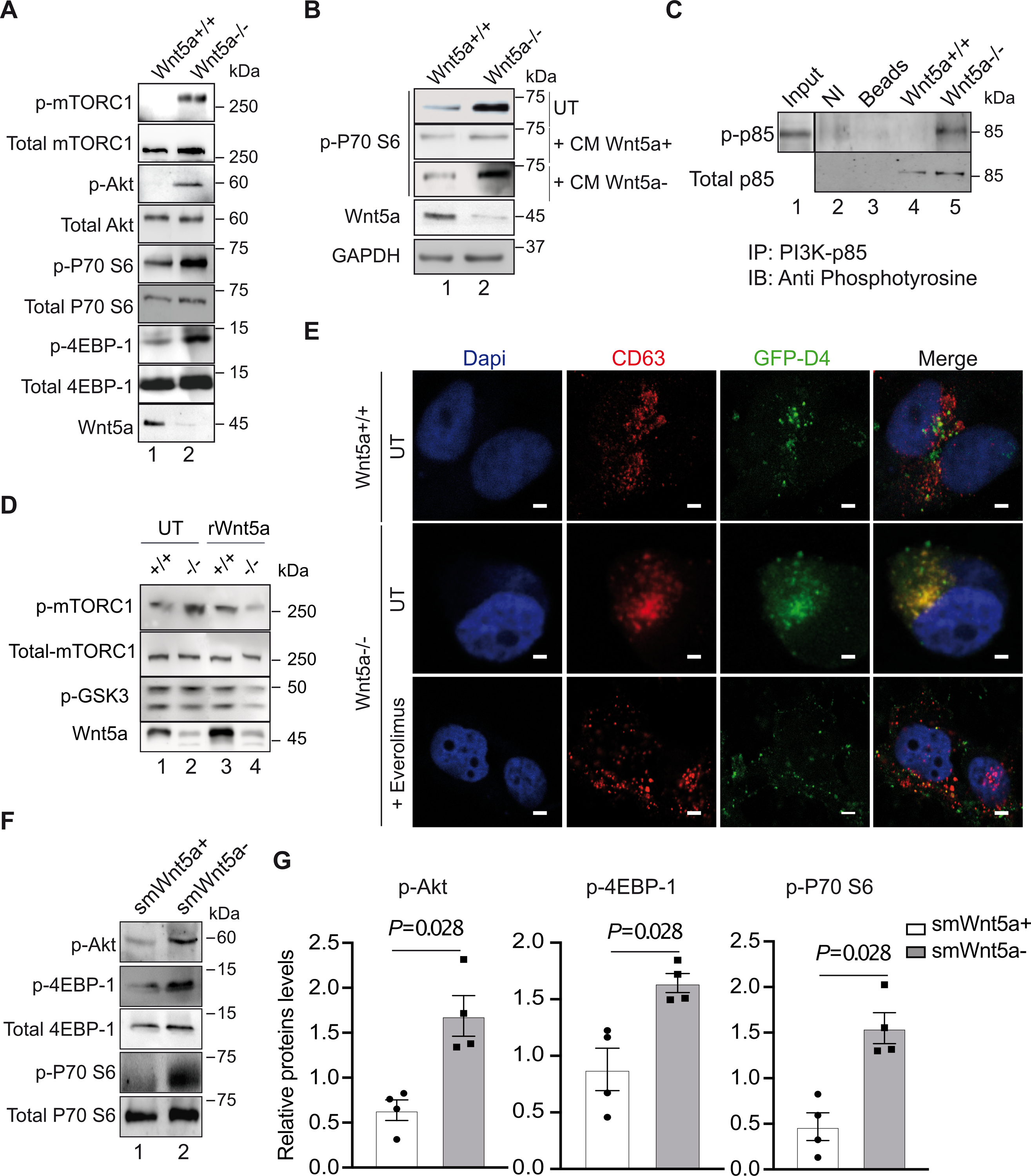
A, shows a representative immunoblot of the indicated genes in human Wnt5a−/− and control VSMCs upon cholesterol accumulation (n=3). B, Western blot analysis showing p-P70 S6 (Thr389) expressions in Wnt5a−/− VSMCs untreated (UT) or treated with conditioned medium enriched in Wnt5a (CM Wnt5a+) or mock medium (CM Wnt5a−) as described in the method section (n=3). C, Immunoprecipitation in Wnt5a−/− VSMCs and controls of the PI3K-p85 subunit followed with immunoblotting with anti-phosphotyrosine (4G10) antibodies show the tyrosine-phosphorylated form of p85 (p-p85) in the absence of Wnt5a. NI=non-immune antibodies, Beads = empty beads. Input is from whole cell lysate (n=3). D, A representative immunoblot shows p-mTORC1, total mTORC1, p-GSK3, and Wnt5a expressions in Wnt5a−/− VSMCs (−/−) and control cells (+/+) untreated (UT) or treated (rWnt5a) with human recombinant Wnt5a (n=3). E, Confocal analysis showing colocalization (yellow) between GFP-D4 positive vesicle (cholesterol, green) and CD63 (LELs, red) in untreated (UT) and Everolimus® (30nM) treated Wnt5a−/− VSMCs. Scale bars are 5μm (n=3). F, Western blot analysis and G, quantification of relative protein levels of the indicated genes in aortas from control (smWnt5a+) and mutant (smWnt5a−) mice (n=4 mice/group). G shows individual data along with mean±SEM. Data were analyzed using Shapiro-wilk followed by Mann-Whitney test. G, P=0.028 indicate significance relative to smWnt5a+ mice.
Changes in cholesterol levels can alter the activity of signaling pathways originating at the plasma membrane involving phosphinositide-3 kinase (PI3K) that can activate mTORC124. To test whether Wnt5a acts upstream of mTORC1 or directly on mTORC1 we measured PI3K activation in human VSMCs. We found the p85 subunit of PI3K to be highly tyrosine phosphorylated in human Wnt5a−/− VSMCs compared to controls (Fig. 5C) and treatment with the PI3K inhibitor Wortmannin inhibited mTORC1 activation in human Wnt5a−/− VSMCs (Fig. S5F). Marked activation of the Akt/mTORC1 pathway in the absence of Wnt5a was also observed in vivo with increased protein levels of p-Akt, p-P70 S6, and p-4EBP1 in aortas from smWnt5a− mice compared to controls (Fig. 5F, G). Overall, these data suggest that Wnt5a promotes LEL cholesterol egress by down regulating the PI3K/Akt/mTORC1 pathway in human VSMCs.
Absence of Wnt5a activated the calcineurin-dependent TFEB nuclear re-localization.
The MiT/TFE transcription factor family member, TFEB drives the expansion of the lysosomal compartment and lysosomal biogenesis25. To assess whether Wnt5a modulates TFEB activity, we measured mRNA levels of its target genes in human Wnt5a−/− VSMCs and controls. Results show an increase in SGSH, CTSF, and TPP1 mRNA levels in untreated mutant cells, and an increase in ATP6V mRNA levels in cholesterol-fed mutant cells (Fig. S4E, F) suggesting that TFEB is activated in the absence of Wnt5a. In agreement, we found that upon cholesterol accumulation TFEB translocated to the nucleus in the absence of Wnt5a (Fig. 6A, B). Conditions that promote TFEB nuclear translocation, such as starvation and lysosomal stress, dephosphorylated TFEB, and one phosphatase involved in TFEB dephosphorylation has been identified as calcineurin26. Conversely, mTORC1-mediated phosphorylation of TFEB at serine residues Ser142 and Ser211 promotes its cytoplasmic localization27. To test whether in the absence of Wnt5a, TFEB dephosphorylation is driven by calcineurin, human Wnt5a−/− VSMCs were treated with the calcineurin inhibitor FK506 upon cholesterol accumulation. FK506 treatments re-localized TFEB to the cytosol (Fig. 6A, B). In addition, using the Fura-8 fluorescent calcium marker, we found that basal intracellular calcium levels are increased in human Wnt5a−/− VSMCs compared to controls (Fig. 6C), an event known to activate calcineurin26. Collectively, these data suggest that in the absence of Wnt5a, activation of calcineurin by Ca2+ release from intracellular stores overrides the mTORC1 signal and causes TFEB to go to the nucleus.
Fig. 6. Calcineurin-dependent nuclear re-localization of TFEB in Wnt5a−/− VSMCs.
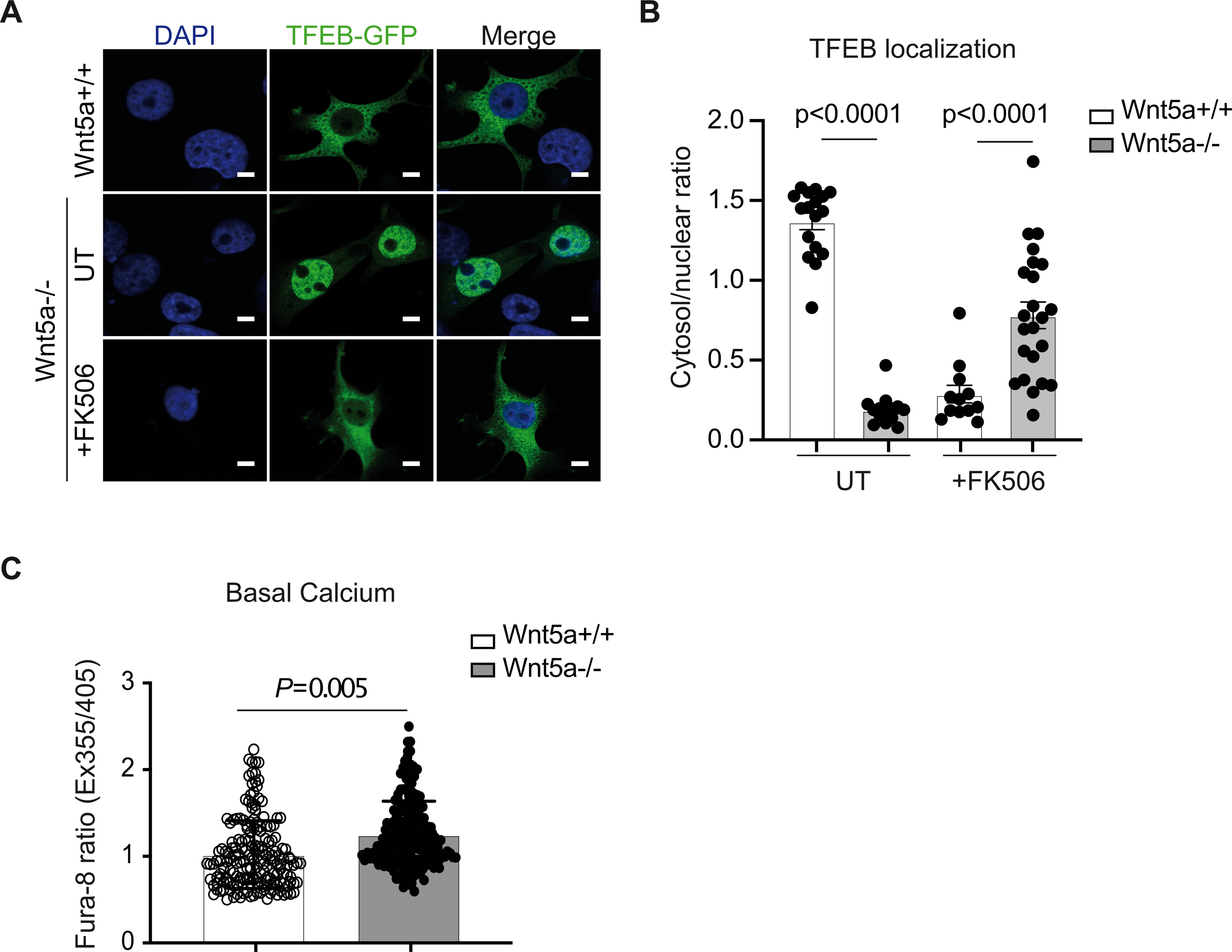
A, Nuclear localization of TEFB-GFP and its quantification B, in human VSMC Wnt5a−/− and controls untreated (UT) or treated with the calcineurin inhibitor FK506 (+FK506). Scale bars are 5μm (n=17 Wnt5a+/+ UT, n=14 Wnt5a−/− UT, n=12 Wnt5a+/+ +FK506 and n=23 Wnt5a+/+ +FK506). C, Intracellular calcium quantification in human VSMC Wnt5a−/− and controls (n=184 Wnt5a+/+ and n=187 Wnt5a−/−). B and C show individual data with mean ± SEM. Statistical analysis was done using Shapiro-Wilk test followed by Mann-Whitney test. B and C P<0.0001 indicate significance relative to Wnt5a+/+.
Wnt5a physically interacts with NPC proteins.
On the basis of our results indicating that absence of Wnt5a interferes with cholesterol trafficking in LELs, we examined whether the Wnt5a sequence contains cholesterol-binding motifs. We identified 5 putative cholesterol recognition amino acid consensus (CARC) motifs that we named D1–5 (Fig. 7A). The consensus CARC sequence (L/V)-X1–5-Y-X1–5-(K/R) has been described in transmembrane domains of cholesterol-regulated proteins such as caveolins, the Ca2+ channel Orai l, and SLC38A9 within the mTORC1 scaffolding complex28,29. However, cholesterol-binding domains have also been identified outside the membrane-spanning regions of soluble proteins such as α-synuclein30. Using the 3D structure of Wnt8 another Wnt ligand that is 33 % homologous to Wnt5a as a template31, we modeled in silico the 3D structure of Wnt5a and the subsequent localization of CARC motifs (Fig. 7A). CARC motifs D2, D3, and D4 were found at the periphery of the 3D structure of Wnt5a, accessible to cholesterol, and thus in a biologically relevant position (Fig. 7A). We next investigated the ability of Wnt5a to bind cholesterol in liposomes reconstituted with phosphatidylcholine, enriched with cholesterol (PC+Chol) or not (PC). Co-sedimentation assays revealed that Wnt5a did not pellet with PC-containing liposomes (Fig. 7B, lane 4). However, the inclusion of cholesterol in PC-containing liposomes enhanced their association with Wnt5a (Fig. 7B, lane 2) indicating that Wnt5a binds to cholesterol-enriched membranes.
Fig. 7. Wnt5a interacts with NPC proteins and cholesterol-enriched liposomes.
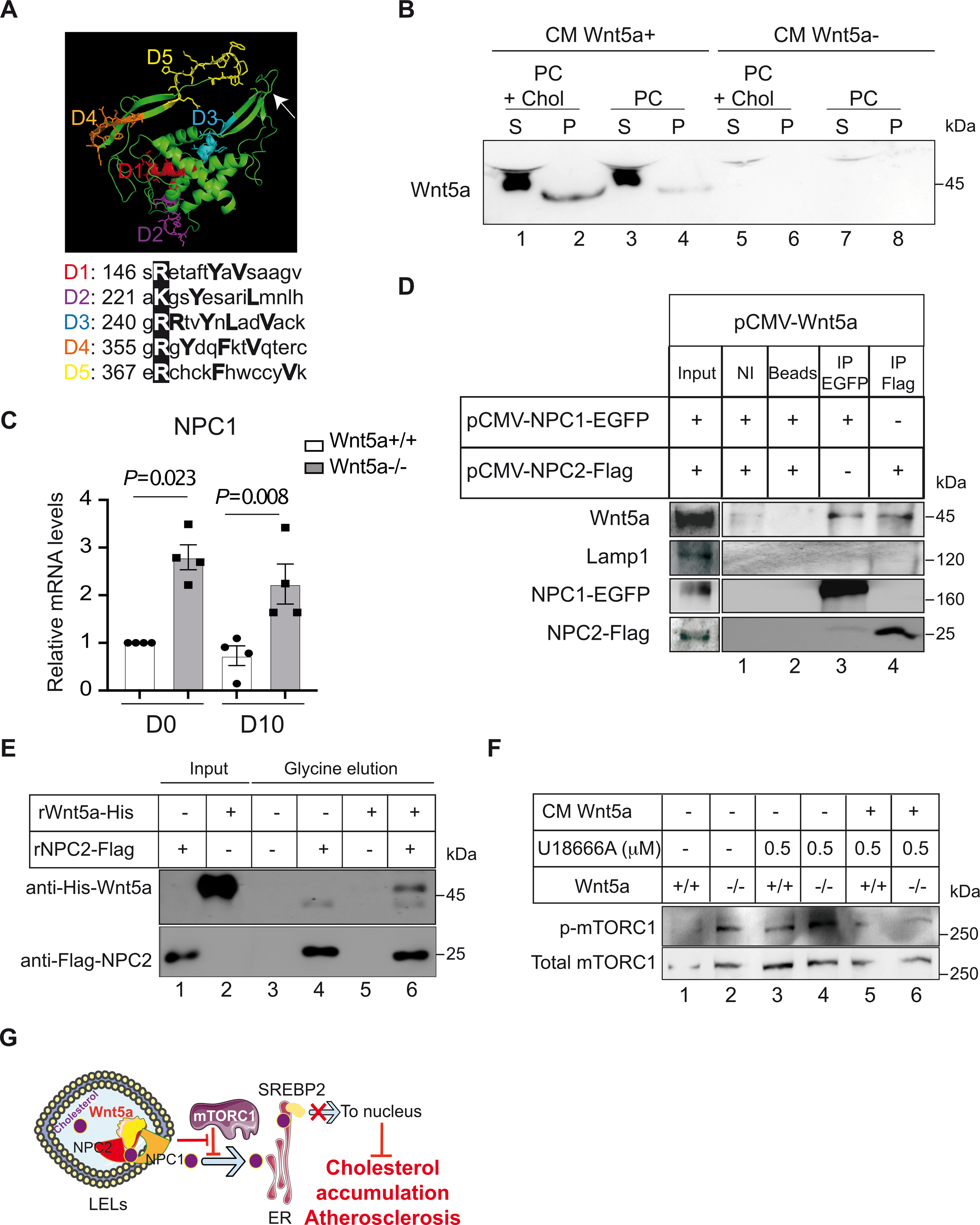
A, Predictive 3D structure of Wnt5a and localization of the CARC motifs. The arrow is showing localization for the palmitoleic acid lipid group. The essential Arg/Lys within the CARC motifs are boxed in dark. B, Conditioned medium enriched in Wnt5a (CM Wnt5a+) or mock medium (CM Wnt5a−) were incubated with liposomes containing 80–85% phosphatidylcholine (PC) or 80–85% phosphatidylcholine + 5% cholesterol (PC + Chol). Liposomes were sedimented, washed and analyzed by immunoblotting for the presence of Wnt5a. Supernatant (S) and pellets (P) were loaded (n=3). C, Relative transcript levels of NPC1 in human VSMC Wnt5a−/− and controls untreated (D0) or treated for cholesterol accumulation during 10 days (D10). GAPDH is taken as internal control. The plot shows individual values with mean±SEM (n=4). D, HEK293 cells were co-transfected with either pCMV-Wnt5a and pCMV-NPC1-EGFP or pCMV-Wnt5a and pCMV-NPC2-Tag expression plasmids. Immunoprecipitations of NPC1 (lane 3) or with NPC2 (lane 4), followed with immunoblotting with Wnt5a show that Wnt5a interacts with both NPC proteins. NI=non-immune antibodies, Beads=empty beads (n=3). E, In vitro direct binding between His-Wnt5a and Flag-NPC2. Recombinant Flag-NPC2 was incubated with Flag-NTA agarose beads allowing NPC2 to bind to the beads. Then 10 μg of human recombinant Wnt5a were added and incubated at room temperature. Complex of proteins was then eluted with a glycine buffer and analyzed by western blotting (n=4). F, Effects of the U18666A compound on the phosphorylation of mTORC1 in presence of control medium (CM Wnt5a−) or Wnt5a enriched condition medium (CM Wnt5a+) in human VSMC Wnt5a−/− and controls (n=3). G, Wnt5a is required for proper lysosomal functions. It promotes cholesterol egress from late endosomes to ER through inhibition of p-mTORC1. In LELs, upon binding to NPC2 and cholesterol, Wnt5a might facilitate cholesterol transfer to NPC1 and to the ER membrane. This suppresses SREBP-2 activity, limits cholesterol accumulation in VSMCs, and protect against atherosclerosis. Statistical analysis was done using a Shapiro-Wilk test followed by one-Way ANOVA and Tukey post hoc analysis for C. C, P=0.023 and P=0.088 indicate significance relative to Wnt5a+/+.
We next tested whether Wnt5a interacts with NPC1 and NPC2, and measured their mRNA levels in the absence of Wnt5a. We found that mRNA levels of NPC1 increased by 2–3X in Wnt5a−/− VSMCs compared to controls (Fig. 7C), whereas NPC2 levels remain unchanged (not shown). Moreover, when co-expressed in HEK293T cells, we found that Wnt5a co-immunoprecipitated with NPC1 and NPC2 (Fig. 7D). The immunoprecipitate does not contain the late endosome marker, Lamp1 suggesting that the interaction between Wnt5a and NPC proteins (Fig. 7D) is not a mere co-precipitation of proteins present in the same membrane complex. We also found that Wnt5a co-immunoprecipitated with NPC1 and NPC2 using mass spectrometry analysis of the interactome (Table S4). We further tested the specific interaction between Wnt5a and NPC2 using in vitro binding experiments and recombinant proteins. We found that purified recombinant Wnt5a physically interacts with recombinant NPC2 (Fig. 7E). This in vitro interaction demonstrates a direct binding between these two proteins and indicates that Wnt5a interacts with the lysosomal cholesterol export machinery.
The U18666A compound is a cholesterol transport-inhibiting agent, used to mimic NPC1 deficiency, which causes the accumulation of cholesterol in cells. Interestingly, we found that in human Wnt5a−/− VSMCs, Wnt5a− enriched conditioned medium decreased mTORC1 activity induced by U18666A treatments, whereas mock medium had no significant effect (Fig. 7F). Thus, loss of Wnt5a or NPC1 inhibition have similar effects on mTORC1 activation and cholesterol trafficking, and Wnt5a counteracts the activation of mTORC1 mediated by U18666A-induced NPC1 inhibition.
DISCUSSION
How cholesterol moves between cellular membranes in mammalian cells is not understood. Our study identifies Wnt5a as essential for lysosomal cholesterol egress.
Wnt5a act as intercellular signaling molecules but is lipid modified. Therefore, its intercellular transport requires extracellular carriers1. Here, we report that Wnt5a enter the endocytic compartment through binding to the LDL lipoparticles. In human or murine VSMCs Wnt5a−/− the LAL expression is very low and the cholesterol hydrolysis processes are overwhelmed. Cholesterol and CE accumulated in enlarged LELs and this coincide with high mTORC1 activity. Within LELs, Wnt5a interacts with NPC proteins, down regulates mTORC1, and promotes cholesterol egress to the ER and lysosomal function. Thus, Wnt5a appears to sense sterol abundance and translates cholesterol levels directly into mTORC1 activity, in parallel with the NPC proteins (Fig. 7G).
We previously reported that pre-adipocytes and mouse embryonic fibroblasts (MEFs) LRP1−/−, that expressed low endogenous Wnt5a, did not form adipocytes when submitted to an adipogenic cocktail. Instead, they accumulated cholesterol and CE11,12. Fatty acid synthesis is impaired and triglycerides levels remained unchanged11,12. Here, we found essentially the same changes in human Wnt5a−/− VSMCs.
In agreement with our data, previous reports showed that siRNA-mediated silencing of Wnt5a in both macrophages and VSMC significantly increased cholesterol content upon exposure to oxLDL15. In addition, Wnt signaling enhanced the macrophage response to inflammation resolving factors4. However, it was also reported that siRNA-mediated silencing of Wnt5a attenuated atherosclerosis potentially through inhibiting the proinflammatory NF-κB and mitogen-activated protein kinase (MAPK) signaling pathways32 suggesting that immune function and/or endothelial cell function are affected by the loss of VSMC Wnt5a. Here, we did not find any significant difference in the expression of the endothelial cell markers CD31 in the absence of Wnt5a in VSMC. In addition, an alteration of the immune or endothelial cell function would be against the atheroprotective effects of LRP6 observed in human. These apparent discrepancies may be related to the receptor availability of Wnt5a that can both activate canonical and non-canonical pathways. For instance, the non-canonical Wnt5a/CaMKII pathway contributes to the inflammatory response of human macrophages16, whereas the Wnt5a/beta-catenin or the LRP6 pathway prevent against cholesterol accumulation and atherosclerosis3,11.
As cholesterol accumulated, Wnt5a protein levels increased in human Wnt5a+/+ VSMCs (Fig. S3C) and, as described by others and we, also increased in the aortic wall during atherosclerosis13–15. However, the correlation between its expression and the accumulation of cholesterol in the arterial wall does not mean there is a causal effect. Here, by deleting Wnt5a in the VSMCs of mice we directly addressed its protective role against cholesterol-induced atherosclerosis. In addition, the absence of Wnt5a activated the PI3K/mTORC1 pathway. PI3K integrates signal from cellular growth and functions as a checkpoint for slowing or preventing degenerative disorders of the vascular wall. For instance, LRP1 mutant mice containing in VSMCs a mutated PDGF receptor-b kinase domain that disrupted the PI3K activation, are protected against atherosclerosis33.
Under nutrient-rich conditions, through mTORC1-mediated phosphorylation, TFEB is retained inactive in the cytoplasm. Upon starvation or under conditions of lysosomal dysfunction, TFEB is dephosphorylated by calcineurin and rapidly translocates to the nucleus to activate the transcription of its target genes. In human Wnt5a−/− VSMCs, we found TFEB in the nucleus and increased intracellular Ca2+ levels, an event known to activate calcineurin26. In the absence of Wnt5a, activated calcineurin overrides the mTORC1 signal and causes TFEB to go to the nucleus where it drives the expansion of the lysosomal compartment allowing cholesterol to accumulate.
In conclusion, the work presented here identifies a previously unrecognized crucial role for Wnt5a in controlling cholesterol homeostasis. The absence of Wnt5a down regulates LAL and this leads to the accumulation of CE in enlarged LELs. Accumulation of cholesterol activates the nutrient-dependent mTORC1 axis at the surface of LELs, whereas Wnt5a interacts with NPC proteins to communicate lysosomal sterol abundance to the mTORC1 complex. This is required for cholesterol transport across the lysosomal membrane and its trafficking to the ER for subsequent redistribution and export via ABC transporters. Through this mechanism, Wnt5a limits cholesterol accumulation in VSMCs, and protects against atherosclerosis in mice.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Disrupted cholesterol trafficking leads to atherosclerosis.
In humans, missense mutation in the Wnt co-receptor LRP6 causes autosomal-dominant early onset cardiovascular disease.
The LRP6 ligand, Wnt5a is expressed in murine and human atherosclerotic lesions.
Wnt5a deficiency in mouse embryonic fibroblasts (MEFs) promotes cholesterol accumulation within perinuclear cytosolic vesicles.
Wnt5a increases cholesterol export and decreases cholesterol bio-synthesis.
What New Information Does This Article Contribute?
Mice deficient for Wnt5a in vascular smooth muscle cells (VSMCs) developed twice as much atherosclerosis as controls.
Wnt5a binds to cholesterol-rich membranes and to the lysosomal Niemann–Pick C1 (NPC1) and NPC2 proteins, and promotes lysosomal cholesterol egress.
The loss of Wnt5a in VSMCs reduces the Lysosomal Acid Lipase expression and its capacity to catabolize atherogenic lipoproteins.
Wnt5a promotes cholesterol trafficking by down regulating the PI3K/Akt/mTORC1 pathway in human VSMCs.
The present studies provide evidence that Wnt5a is essential for cholesterol homeostasis. It activates lysosomal function, promotes cholesterol trafficking, and protects against atherosclerosis.
ACKNOWLEDGMENTS
We are grateful to Toshihide Kobayashi, Nario Tomishige and Motohide Murate (UMR CNRS 7021, University of Strasbourg) for helpful suggestions and EM analysis, Russell Debose-Boyd (UT Southwestern Medical Center at Dallas) for antibodies against HMG CoA reductase, and the Mouse Clinical Institute of Strasbourg (UMR CNRS 7021) for technical assistance.
SOURCES OF FUNDING
This work was supported by grants from Fédération Française de Cardiologie, and the Fondation de France (N°00107044). RSR was funded by Fondation Recherche Médicale doctoral fellowship (FRM, PLP20170939073). JH was supported by grants from the NHLBI (R37 HL063762), NIA (RF AG053391), the NINDS and NIA (R01 NS093382), BrightFocus A2016396S, the Bluefield Project to Cure FTD and a Harrington Scholar Innovator Award (2019).
Nonstandard Abbreviations and Acronyms:
- 4EBP1
Eukaryotic translation initiation factor 4E-binding protein 1
- ACAT1
Acyl-CoA cholesterolacyltransferase 1
- ACTA
Alpha-smooth luscle actin
- AKT
Protein kinase B
- ATP6V
V-type proton ATPase
- CARC
Cholesterol recognition amino acid consensus motifs
- CE
Cholesteryl-ester
- CTSF
Cathepsin F
- DGAT2
Acyl CoA:diacylglycerol acyltransferase 2
- ER
Endoplasmic reticulum
- GAL3
Galectin-3
- GFP-D4
D4 fragment of perfringolysin O fused to GFP
- LAL
Lysosomal acid lipase
- LAMP1
Lysosome-associated membrane protein 1
- LDLR
Low-density lipoprotein (LDL) receptor
- LELs
Late endosomes/lysosomes
- LRP1
Low-density lipoprotein (LDL) receptor-related protein 1
- LRP6
Low-density lipoprotein (LDL) receptor-related protein 6
- MAPK
Mitogen-activated protein kinase
- mTORC1
Mechanistic target of rapamycin complex 1
- MyoCD
Myocardin
- NF-kB
Nuclear factor kB
- NPC1/2
Niemann–Pick C1/2
- p-4EBP1
Phospho-Eukaryotic translation initiation factor 4E-binding protein 1
- P70 S6
Ribosomal protein S6 kinase beta-1
- p-Akt
Phospho-Protein kinase B
- PDGF
Platelet-derived growth factor
- PI3K
Phosphatidylinositol 3-kinase
- p-P70 S6
Phospho-Ribosomal protein S6 kinase beta-1
- SGSH
N-sulphoglucosamine sulphohydrolase
- SREBP-2
Sterol regulatory element-binding protein-2
- TGFβ
Transforming Growth Factor β
- TFEB
Transcription Factor EB
- TPP1
Tripeptidyl Peptidase 1
- KLF4
Kruppel-like transcription factor 4
- VSMCs
Vascular smooth muscle cells
- WT
Wild type
Footnotes
DISCLOSURES
The authors declare no competing interests.
SUPPLEMENTAL MATERIALS
REFERENCES
- 1.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. [DOI] [PubMed] [Google Scholar]
- 2.Go GW, Srivastava R, Hernandez-Ono A, Gang G, Smith SB, Booth CJ, Ginsberg HN, Mani A. The Combined Hyperlipidemia Caused by Impaired Wnt-LRP6 Signaling Is Reversed by Wnt3a Rescue. Cell Metab. 2014;19:209–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mani A, Radhakrishnan J, Wang H, Mani MA, Nelson-Williams C, Carew KS, Mane S, Najmabadi H, Wu D, Lifton RP. LRP6 mutation in a family with early coronary disease and metabolic risk factors. Science. 2007;315:1278–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weinstock A, Rahman K, Yaacov O, Nishi H, Menon P, Nikain CA, Garabedian ML, Pena S, Akbar N, Sansbury BE, Heffron SP, Liu J, Marecki G, Fernandez D, Brown EJ, Ruggles KV, Ramsey SA, Giannarelli C, Spite M, Choudhury RP, Loke P, Fisher EA. Wnt signaling enhances macrophage responses to IL-4 and promotes resolution of atherosclerosis. Elife. 2021;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maxfield FR, Tabas I. Role of cholesterol and lipid organization in disease. Nature. 2005;438:612–21. [DOI] [PubMed] [Google Scholar]
- 6.Zhang JR, Coleman T, Langmade SJ, Scherrer DE, Lane L, Lanier MH, Feng C, Sands MS, Schaffer JE, Semenkovich CF, Ory DS. Niemann-Pick C1 protects against atherosclerosis in mice via regulation of macrophage intracellular cholesterol trafficking. J Clin Invest. 2008;118:2281–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Infante RE, Wang ML, Radhakrishnan A, Kwon HJ, Brown MS, Goldstein JL. NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc Natl Acad Sci U S A. 2008;105:15287–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31:1095–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eid W, Dauner K, Courtney KC, Gagnon A, Parks RJ, Sorisky A, Zha X. mTORC1 activates SREBP-2 by suppressing cholesterol trafficking to lysosomes in mammalian cells. Proc Natl Acad Sci U S A. 2017;114:7999–8004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boucher P, Gotthardt M, Li WP, Anderson RG, Herz J. LRP: role in vascular wall integrity and protection from atherosclerosis. Science. 2003;300:329–32. [DOI] [PubMed] [Google Scholar]
- 11.Terrand J, Bruban V, Zhou L, Gong W, El Asmar Z, May P, Zurhove K, Haffner P, Philippe C, Woldt E, Matz RL, Gracia C, Metzger D, Auwerx J, Herz J, Boucher P. LRP1 controls intracellular cholesterol storage and fatty acid synthesis through modulation of Wnt signaling. J Biol Chem. 2009;284:381–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El Asmar Z, Terrand J, Jenty M, Host L, Mlih M, Zerr A, Justiniano H, Matz RL, Boudier C, Scholler E, Garnier JM, Bertaccini D, Thierse D, Schaeffer C, Van Dorsselaer A, Herz J, Bruban V, Boucher P. Convergent Signaling Pathways Controlled by LRP1 (Receptor-related Protein 1) Cytoplasmic and Extracellular Domains Limit Cellular Cholesterol Accumulation. J Biol Chem. 2016;291:5116–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woldt E, Terrand J, Mlih M, Matz RL, Bruban V, Coudane F, Foppolo S, El Asmar Z, Chollet ME, Ninio E, Bednarczyk A, Thierse D, Schaeffer C, Van Dorsselaer A, Boudier C, Wahli W, Chambon P, Metzger D, Herz J, Boucher P. The nuclear hormone receptor PPARgamma counteracts vascular calcification by inhibiting Wnt5a signalling in vascular smooth muscle cells. Nat Commun. 2012;3:1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christman MA, Goetz DJ, Dickerson E, McCall KD, Lewis CJ, Benencia F, Silver MJ, Kohn LD, Malgor R. Wnt5a is expressed in murine and human atherosclerotic lesions. Am J Physiol Heart Circ Physiol. 2008;294:H2864–70. [DOI] [PubMed] [Google Scholar]
- 15.Qin L, Hu R, Zhu N, Yao HL, Lei XY, Li SX, Liao DF, Zheng XL. The novel role and underlying mechanism of Wnt5a in regulating cellular cholesterol accumulation. Clin Exp Pharmacol Physiol. 2014;41:671–8. [DOI] [PubMed] [Google Scholar]
- 16.Bhatt PM, Malgor R. Wnt5a: a player in the pathogenesis of atherosclerosis and other inflammatory disorders. Atherosclerosis. 2014;237:155–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salmon M, Gomez D, Greene E, Shankman L, Owens GK. Cooperative binding of KLF4, pELK-1, and HDAC2 to a G/C repressor element in the SM22α promoter mediates transcriptional silencing during SMC phenotypic switching in vivo. Circ Res. 2012;111:685–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bauer M, Benard J, Gaasterland T, Willert K, Cappellen D. WNT5A encodes two isoforms with distinct functions in cancers. PLoS One. 2013;8:e80526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harris CA, Haas JT, Streeper RS, Stone SJ, Kumari M, Yang K, Han X, Brownell N, Gross RW, Zechner R, Farese RV. DGAT enzymes are required for triacylglycerol synthesis and lipid droplets in adipocytes. J Lipid Res. 2011;52:657–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ohno-Iwashita Y, Shimada Y, Waheed AA, Hayashi M, Inomata M, Nakamura M, Maruya M, Iwashita S. Perfringolysin O, a cholesterol-binding cytolysin, as a probe for lipid rafts. Anaerobe. 2004;10:125–134. [DOI] [PubMed] [Google Scholar]
- 21.Suckling KE, Stange EF. Role of acyl-CoA: cholesterol acyltransferase in cellular cholesterol metabolism. J Lipid Res. 1985;26:647–671. [PubMed] [Google Scholar]
- 22.Dubland JA, Allahverdian S, Besler KJ, Ortega C, Wang Y, Pryma CS, Boukais K, Chan T, Seidman MA, Francis GA. Low LAL (Lysosomal Acid Lipase) Expression by Smooth Muscle Cells Relative to Macrophages as a Mechanism for Arterial Foam Cell Formation. Arteriosclerosis, Thrombosis, and Vascular Biology. 2021;41:e354–e368. [DOI] [PubMed] [Google Scholar]
- 23.Kaiser K, Gyllborg D, Prochazka J, Salasova A, Kompanikova P, Molina FL, Laguna-Goya R, Radaszkiewicz T, Harnos J, Prochazkova M, Potesil D, Barker RA, Casado AG, Zdrahal Z, Sedlacek R, Arenas E, Villaescusa JC, Bryja V. WNT5A is transported via lipoprotein particles in the cerebrospinal fluid to regulate hindbrain morphogenesis. Nat Commun. 2019;10:1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perera RM, Zoncu R. The Lysosome as a Regulatory Hub. Annu Rev Cell Dev Biol. 2016;32:223–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E, Ballabio A. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–7. [DOI] [PubMed] [Google Scholar]
- 26.Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso C, Forrester A, Settembre C, Wang W, Gao Q, Xu H, Sandri M, Rizzuto R, De Matteis MA, Ballabio A. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015;17:288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31:1095–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Castellano BM, Thelen AM, Moldavski O, Feltes M, van der Welle RE, Mydock-McGrane L, Jiang X, van Eijkeren RJ, Davis OB, Louie SM, Perera RM, Covey DF, Nomura DK, Ory DS, Zoncu R. Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science. 2017;355:1306–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fantini J, Barrantes FJ. How cholesterol interacts with membrane proteins: an exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front Physiol. 2013;4:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fantini J, Carlus D, Yahi N. The fusogenic tilted peptide (67–78) of alpha-synuclein is a cholesterol binding domain. Biochim Biophys Acta. 2011;1808:2343–51. [DOI] [PubMed] [Google Scholar]
- 31.Janda CY, Waghray D, Levin AM, Thomas C, Garcia KC. Structural basis of Wnt recognition by Frizzled. Science. 2012;337:59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang L, Chu Y, Wang Y, Zhao X, Xu W, Zhang P, Liu X, Dong S, He W, Gao C. siRNA-mediated silencing of Wnt5a regulates inflammatory responses in atherosclerosis through the MAPK/NF-κB pathways. Int J Mol Med. 2014;34:1147–1152. [DOI] [PubMed] [Google Scholar]
- 33.Zhou L, Takayama Y, Boucher P, Tallquist MD, Herz J. LRP1 regulates architecture of the vascular wall by controlling PDGFRbeta-dependent phosphatidylinositol 3-kinase activation. PLoS One. 2009;4:e6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rohlmann A, Gotthardt M, Willnow TE, Hammer RE, Herz J. Sustained somatic gene inactivation by viral transfer of Cre recombinase. Nat Biotechnol. 1996;14:1562–1565. [DOI] [PubMed] [Google Scholar]
- 35.Student AK, Hsu RY, Lane MD. Induction of fatty acid synthetase synthesis in differentiating 3T3-L1 preadipocytes. Journal of Biological Chemistry. 1980;255:4745–4750. [PubMed] [Google Scholar]
- 36.Garratt AN, Voiculescu O, Topilko P, Charnay P, Birchmeier C. A dual role of erbB2 in myelination and in expansion of the schwann cell precursor pool. J Cell Biol. 2000;148:1035–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilhelm LP, Wendling C, Vedie B, Kobayashi T, Chenard MP, Tomasetto C, Drin G, Alpy F. STARD3 mediates endoplasmic reticulum-to-endosome cholesterol transport at membrane contact sites. EMBO J. 2017;36:1412–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–7. [DOI] [PubMed] [Google Scholar]
- 39.Zhang X, Julien-David D, Miesch M, Geoffroy P, Raul F, Roussi S, Aoude-Werner D, Marchioni E. Identification and quantitative analysis of beta-sitosterol oxides in vegetable oils by capillary gas chromatography-mass spectrometry. Steroids. 2005;70:896–906. [DOI] [PubMed] [Google Scholar]
- 40.Lada AT, Davis M, Kent C, Chapman J, Tomoda H, Omura S, Rudel LL. Identification of ACAT1- and ACAT2-specific inhibitors using a novel, cell-based fluorescence assay: individual ACAT uniqueness. J Lipid Res. 2004;45:378–386. [DOI] [PubMed] [Google Scholar]
- 41.Ortiz-Miranda S, Ji R, Jurczyk A, Aryee K-E, Mo S, Fletcher T, Shaffer SA, Greiner DL, Bortell R, Gregg RG, Cheng A, Hennings LJ, Rittenhouse AR. A novel transgenic mouse model of lysosomal storage disorder. Am J Physiol Gastrointest Liver Physiol. 2016;311:G903–G919. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data and study material related to this study are available to other researchers on reasonable request. Methods are expanded in the Online Data Supplement.