

Abstract
Background and Purpose:
The calcitonin (CT) receptor family is complex, comprising two receptors (the CT receptor [CTR], and the CTR-like receptor [CLR]), three accessory proteins (RAMPs), and multiple endogenous peptides. This family contains several important drug targets, including CGRP which is targeted by migraine therapeutics. The pharmacology of this receptor family is poorly characterised in species other than rats and humans. To facilitate understanding of translational and pre-clinical data we need to know the receptor pharmacology of this family in mice.
Experimental Approach:
Plasmids encoding mouse CLR/CTR and RAMPs were transiently transfected into Cos7 cells. cAMP production was measured in response to agonists in the absence or presence of antagonists.
Key Results:
We report the first synthesis and characterisation of mouse adrenomedullin, adrenomedullin 2 and βCGRP, and of mouse CTR without or with mouse RAMPs. Receptors containing m-CTR had subtly different pharmacology than human receptors; they were promiscuous in their pharmacology, both in absence and presence of RAMPs. Several peptides, including mouse αCGRP and mouse adrenomedullin 2 were potent agonists of the m-CTR:m-RAMP3 complex. The pharmacological profile of receptors comprising m-CLR:m-RAMPs were generally similar to the profile of their human counterparts, albeit with reduced specificity.
Conclusion and Implications:
Mouse receptor pharmacology differed compared to human, with mouse receptors displaying reduced discrimination between ligands. This creates challenges for interpreting which receptor may underlie an effect in pre-clinical models, and thus translation of findings from mice to humans. It also highlights the need for new ligands to differentiate between these complexes.
Keywords: Adrenomedullin, calcitonin, amylin, CGRP, receptor activity-modifying protein, calcitonin receptor, calcitonin receptor-like receptor
Introduction
Migraine is a debilitating condition characterised by moderate to severe headache and other symptoms such as visual aura, nausea, and sensitivity to light/sound (Edvinsson et al., 2018). Migraine is a leading cause of disability worldwide and is the leading cause of neurological disability (Steiner et al., 2013). Recently, six drugs which are designed to reduce the signalling of calcitonin gene-related peptide (CGRP) have been approved to treat this disorder. Two drugs are small molecule receptors antagonists (ubrogepant and rimegepant), the remaining four are monoclonal antibodies of which three target CGRP itself, while the fourth (erenumab) targets a receptor for CGRP (Ray et al., 2021).
CGRP is part of a broader peptide family which includes calcitonin (CT), amylin (Amy), adrenomedullin (AM), and adrenomedullin 2/intermedin (herein referred to as AM2). These peptides have low amino acid sequence identity. However they are united by key structural features, namely, an amidated C-terminus, and an N-terminal disulfide loop between two cysteine residues (Figure 1). Although CGRP is the most prominently linked to migraine, other peptides from this family are being investigated for their role in the condition. Recent studies have reported that an Amy analogue (pramlintide [Pram]) and AM can provoke migraine-like headache (Ghanizada et al., 2021a; Ghanizada et al., 2021b). These investigations are supported by recent reports showing that Amy-like immunoreactivity in plasma was elevated in chronic migraineurs (Irimia et al., 2021), and the finding that repeated administration of AM can cause headaches in humans (Kita et al., 2020). Thus, it may be therapeutically worthwhile to extend beyond CGRP.
Figure 1.
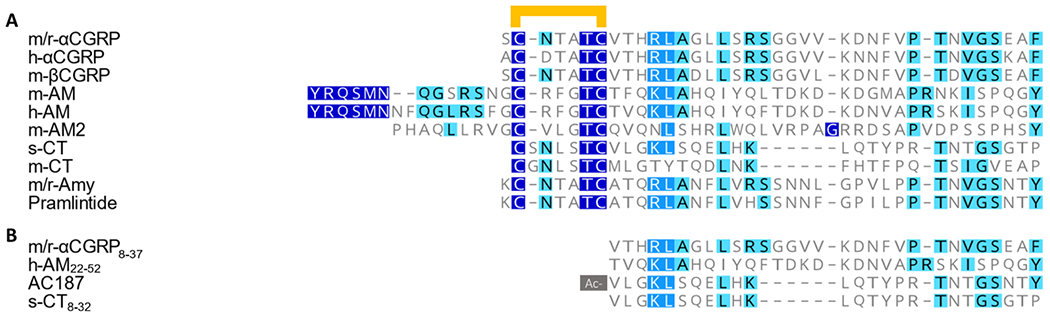
(a) Amino acid sequence alignment of agonists used in this study. In all cases there is a disulfide bond between the two conserved cysteine residues in the N-terminus (shown as a yellow bar). (b) Amino acid sequence alignment of antagonists used in this study. The N-terminal valine of AC187 is acetylated as indicated by the grey box. All agonists and antagonists are amidated on the C-terminal residue. All alignments were performed in Geneious Prime using the inbuilt ClustalW sequence alignment. A dark blue box with white text indicates an exact match, a medium blue box with white text indicates amino acids with >80% similarity, and a light blue box with black text indicates between 60% and 80% similarity as scored by Geneious.
The receptors for these peptides are formed from different protein subunits which combine to form different receptor subtypes. The CGRP receptor comprises a class B G protein-coupled receptor (GPCR), the calcitonin receptor-like receptor (CLR) in complex with the accessory protein receptor activity-modifying protein (RAMP) 1. However, when CLR combines with RAMP2 and RAMP3, it forms the AM1 and AM2 receptors, which have high affinity for AM and AM2. The human (h-) AM1 and AM2 receptors have relatively low affinity for CGRP. The calcitonin receptor (CTR) also forms complexes with the three RAMPs. These complexes are all activated by Amy, hence AMY1, AMY2 and AMY3 receptors. However, CGRP is also able to effectively activate the h-AMY1 receptor (Hay et al., 2018). The receptor nomenclature for these receptors implies that there is a clear distinction in ligand preference between the receptors, but the expanded experimental data now show that several of the receptor complexes are potently activated by several endogenous ligands. This appears to be more pronounced at rat (r-) CGRP family receptors. For example, CGRP gains potency compared to AM and Amy at the r-AM2 and r-AMY3 receptors, in addition to being a potent agonist of the r-CGRP and r-AMY1 receptors (Bailey et al., 2012; Hay et al., 2003).
Rodent models are commonly used to explore the physiological function of peptides, and to perform pre-clinical validation of drugs. Thus, knowledge of differences in receptor pharmacology between species is vital. Current migraine research includes a variety of mouse (m-) models in experiments (Vuralli et al., 2019), and includes models where h-RAMP1 overexpression in the central nervous system can be linked to sensitisation of migraine-like behaviours (Recober et al., 2009) in addition to metabolic effects (Zhang et al., 2011). The latter, then adds a mixed-species complexity. There are also ongoing drug discovery programmes for other peptides in this family, especially Amy for diabetes and obesity, which use rat and mouse models (Enebo et al., 2021; Henriksen et al., 2021; Zakariassen et al., 2020). Despite their common usage as models, we have only a rudimentary understanding of the pharmacology of CLR/CTR family receptors in mice. This is in contrast to the pharmacological profiles of h/r-CLR-based and h/r-CTR-based receptors which are better defined (Figure S 1, S 2 and S 3). This is an important gap in our knowledge because there are differences in the amino acid sequences of both the peptides and receptor components between species (Figure 1 a, Figure S 4 and S 5).
Early investigations into complexes of m-CLR with m-RAMP1, m-RAMP2, and m-RAMP3 showed that the profiles of agonists at mouse receptors were similar to their profile of human receptors, albeit with reduced selectivity. These initial reports did not investigate all endogenous agonists of the CT family, such as CT, AM2, Amy and βCGRP. Work with antagonists showed subtle differences between human and mouse receptors (Bailey et al., 2012; Husmann et al., 2003; Koller et al., 2004). m-CTR or mCTR:m-RAMP complexes have barely been studied and therefore it is not known whether these form receptors for Amy or CGRP in mice (Bohn et al., 2017). Without characterisation we are essentially blind when trying to interpret data from mouse models. Species such as mice, rats, and canines are vital for drug discovery, and thus information regarding how drug targets function across species is of high importance. In order to develop a framework for interpreting data for these receptors and peptides in mice, we set out to determine the pharmacology of m-CTR, m-CLR, and m-RAMPs in transfected cells.
Methods
DNA constructs
CTR can exist as one of multiple splice variants, with both tissue and species dependencies (Hay et al., 2018). Here, unless otherwise specified, CTR will refer to the mouse CT(a) receptor which we have used in all experiments. This splice variant is conserved between humans, rats, and mice, as opposed to some other variants which display differences between species. m-CT(a), m-CLR, m-RAMP1, m-RAMP2, and m-RAMP3 were purchased from Origene (Catalogue numbers MC216948, MC216307, MC206240, MC207508, and MC202180, respectively). These constructs were encoded in the pcmv6 expression vector and are Mus musculus sequences. All were cloned from C57BL6 sequences, except for m-RAMP1 which was originally cloned from the FVB/N strain. This DNA sequence is 99% identical to the m-RAMP1 sequence cloned from C57BL6, and the protein sequences are 100% identical. The m-RAMP2 used in this study has a threonine at position 10 within the signal sequence. This corresponds with GenBank Accession #AAH69992.1, while the sequence reported by UniProt has a proline in this position. This is likely to reflect a natural variation in the population, of which there may be more than one (Figure S 6). pcDNA3.1(+) was also used as an empty vector to explore the pharmacology of m-CLR and m-CTR in the absence of m-RAMPs. For full amino acid sequences, and comparison between human, rat and mouse sequences, see Figure S 4 and S 5. Constructs were confirmed by DNA sequencing, through the DNA Sequencing facility at the University of Auckland.
Peptides
The amino acid sequences for Amy and αCGRP are strictly conserved between rats and mice; this is indicated throughout by using the prefix m/r-. m/r-αCGRP was purchased from Bachem Pharmaceuticals (Cat# 4025897, Bubendorf, Switzerland), American peptide (Sunnyvale, CA, USA, supplier no longer available) or synthesised in-house; the three sources showed bioequivalence in cAMP assays (data not shown). Salmon [s]-CT and s-CT8-32, were purchased from American Peptide, m/r-Amy was purchased from American peptide. m/r-αCGRP8-37 and h-AM22-52 were purchased from Bachem (Cat# 4034544 and 4028071, respectively). m-βCGRP, h-αCGRP, Pram, AC187, m-AM, m-AM240, m-AM247, m-CT, and h-AM were synthesised in-house. Peptides were synthesised in-house using an Fmoc solid-phase synthesis approach. The synthesis of Pram, h-αCGRP, and h-AM has been reported previously (Bower et al., 2018; Garelja et al., 2020; Yule et al., 2016); detailed information regarding the synthesis of other peptides can be found in the supplemental information. Peptide sequences were in accordance with data from UniProt.
Where the peptide content was unknown, a peptide content of 80% was assumed. All peptides were reconstituted in UltraPure™ DNase/RNase-Free Distilled Water (Cat# 10977015, Thermofisher Scientific, New Zealand) and working stocks of either 10 mM or 1 mM were aliquoted into protein lo-bind tubes (Cat# EP0030108116, Eppendorf, Hamburg, Germany) and stored at −30° C. Peptides were further diluted as needed for cAMP assays.
Cell culture and transfection
Cell culture, plating, and transfection were conducted as previously described (Bailey & Hay, 2006; Bower et al., 2018). Cos-7 cells (originally from the American Type Culture Collection, RRID CVCL_0224) were used throughout this study. These cells were chosen as they do not endogenously express CTR, CLR, or RAMPs (Bailey & Hay, 2006), thus avoiding the confounding factor of background CT-family receptor expression. It would have been preferable to use a rodent-derived cell line but we are not aware of any that do not endogenously express the receptor components. Cells were tested for mycoplasma contamination using a MycoAlert kit according to the manufacturers protocol (Lonza, Basel, Switzerland; Cat# LT07-118) and found to be free from infection.
Cells were cultured in complete media, comprising Dulbecco’s modified Eagle media (DMEM; Cat# 11995065, ThermoFisher) supplemented with New Zealand origin heat-inactivated foetal bovine serum (Cat# 10372019, Gibco, MA, USA) to a final concentration of 8% v/v. Cells were grown in a 37 °C humidified incubator at 95% room air/5% CO2 to a maximum passage number of 30.
Cells were sub-cultured in either T-75 flasks (Cat#FAL353136, Corning, New York, USA) or T-175 cell culture flasks (Cat # CORN431080, Corning) until 80-90% confluent. On the day of passaging, complete media and TrypLE Express (Cat#12605-010, Gibco) were warmed to 37°C. For T-75 flasks, culture media was removed, and cells washed once with 5 mL room temperature Dulbecco’s phosphate-buffered saline (DPBS, Cat# 14190250, Gibco). DPBS was then removed and replaced with 5 mL TrypLE Express. Cells were then incubated at 37 °C for approximately five minutes. Cells were resuspended by agitation, then TrypLE was neutralised by adding 5 mL complete media. Either 1 or 2 mL of the resulting cell suspension was then transferred to a new T-75 flask containing 12 mL of complete media. These flasks would typically be ready for passaging after four or three days, respectively. For T-175 flasks, volumes were doubled.
Cells were seeded into 96 well SpectraPlates (Cat# 6005658, PerkinElmer, MA, USA) at a density range of 15,000-20,000 cells per well. Cell density was determined using a Countess II Cell Counter (Thermofisher). Following seeding, cells were returned to the incubator for 18-24 hours before being transfected with receptor components. Transfection was performed using polyethylenimine (Cat# 408727, Sigma-Aldrich, St. Louis, MO, US), as described previously (Bailey & Hay, 2006; Bower et al., 2018). Briefly, a transfection mix comprising 5% filter sterilised glucose (10% final volume; Cat# AJA783, ThermoFisher), 0.25 μg total DNA per well, polyethylenimine (ratio 10:1 to total DNA), and complete media was created. Media from plates were aspirated and replaced with 100 μL transfection mix per well. Constructs were transfected at a ratio of 1:1 (receptor:RAMP). When investigating the ability of m-CTR or m-CLR to signal in the absence of RAMPs, cells were transfected with m-CTR/m-CLR and pcDNA3.1(+) at a ratio of 1:1. Following transfection, cells were returned to the incubator for approximately 48 hours before being used in cAMP assays.
cAMP cell signalling assay
cAMP stimulation was performed using a method previously described (Woolley et al., 2017). All cAMP assays were performed using stimulation media comprising DMEM supplemented with 1 mM 3-isobutyl-1-methylxanthine (Cat# 15879, Sigma-Aldrich) and 0.1% w/v bovine serum albumin (Cat# ABRE-100g, MP Biomedicals, New Zealand). Forskolin, at a final concentration of 100 μM, was used as a positive control.
Prior to stimulation of Cos-7 cells, the transfection mix was aspirated and replaced with 50 μL of stimulation media and cells left to incubate for 30 minutes at 37 °C. Peptides were serially diluted in stimulation media to the appropriate concentrations. Following serum starvation, cells were stimulated with peptides or cAMP media alone for 15 minutes at 37 °C. For antagonism assays, the antagonist was added to cells followed immediately by the agonist. No pre-incubation was used. In these antagonist assays, we generally used the cognate agonist of each receptor, based on the known human pharmacology. For m-CTR:m-RAMPs we used both m-CT and m/r-Amy as agonists. Additionally, m/r-αCGRP was used as an agonist of m-CTR:m-RAMP1/3 (Bailey et al., 2012).
All media was then removed via aspiration and replaced with 50 μL of ice-cold absolute ethanol to stop cAMP production. Plates were then stored at −30 ° C for a minimum of 15 minutes and a maximum of two weeks before processing.
cAMP quantification using a LANCE protocol
cAMP production was quantified using a LANCE cAMP kit (Cat# AD0263, PerkinElmer) as previously described (Bower et al., 2018). Ethanol was evaporated in a fume hood. Cells were lysed using 50 μL lysis buffer (10 mM CaCl2, 50 mM HEPES, 0.35% TritonX-100, all in ddH2O, pH 7.4). After addition of lysis buffer, cells were gently shaken on an orbital shaker for 10-15 min to produce cell lysates.
Cell lysate (5 μL) was then transferred to a 384 well white OptiPlate (Cat# 6007290, PerkinElmer). In parallel, a standard curve was created according to kit instructions by serially diluting a stock standard of cAMP in lysis buffer. All lysate transfers, antibody and detection mix additions were performed manually or on a JANUS Automated Liquid Handling Station (PerkinElmer). The LANCE anti-cAMP antibody was diluted 1:200 in lysis buffer, and 5 μL added to each well. Following this, plates were pulse-centrifuged for 10-15 seconds, reaching 400 x g, then incubated for 30 minutes at room temperature. Detection mix (Europium-W8044 labelled streptavidin and biotin-cAMP diluted 1:4500 and 1:1500 in lysis buffer, respectively) was added to all wells (10 μL per well), and plates were pulse centrifuged for 10-15 seconds, reaching 400 x g. The plates were then incubated at room temperature for between 4 and 20 hrs. Plates were read on an Envision plate reader (PerkinElmer), with excitation at 340 nm and emissions detected at 615 and 665 nm. Molar quantities of cAMP were calculated using the standard curve created in each experiment.
Experimental design
For each transfection day, the order in which transfection mixes were added to plates was randomised. In cases where multiple transfections were being included on a single plate, these positions were randomised between days. On each stimulation day, the tested peptides were randomly assigned to positions on the plate. This was for both agonist assays (agonists randomly assigned positions), and antagonism assays (antagonists and media control randomly assigned to plate positions) to remove potential bias from plate positions.
In all cases, duplicate or triplicate technical replicates were performed within each biological replicate. A biological replicate consisted of plating cells from a distinct passage flask, transfection with separate transient transfection mixes, and stimulation with separate dilutions of peptides. Each biological replicate is referred to as an independent experiment and counted as one n. Blinding was not conducted in this study. This manuscript complies with the British Journal of Pharmacology’s recommendations and requirements relating to experimental design and analysis (Curtis et al., 2018).
Data analysis – Agonist assays
Experiments were biologically replicated a minimum of three or five times. Where three experiments did not produce quantifiable data sets, they were curtailed at n = 3 and no statistical analysis conducted. In all other cases, a minimum of five independent experiments were performed. There is variation in the sample sizes between some receptor/ligand combinations (range of 5-14 biological replicates). This is due to certain agonists being used as controls in early experiments. To avoid arbitrarily excluding data, these experiments were included in our analyses. Control curves from antagonist experiments were not included in agonist analyses because we did not want to further unbalance the size of data-sets.
Data were analysed using GraphPad PRISM 9 (GraphPad Software, La Jolla, CA). Curves were only fit to data when at least two data-points were above the basal cAMP production; otherwise the response was deemed unquantifiable and defined as not a curve. If a data set had sufficient points for a fit but did not reach a plateau, the curve-fit max was constrained using the mean response at the highest concentration of peptide (Garelja et al., 2020). When fitting agonist curves to individual experiments, F-tests were used to determine whether the Hill slope significantly differed from one (i.e. whether the data were better fit by three-parameter or four-parameter nonlinear regression). From each biological replicate we derived a pEC50 and Emax value; these values were then combined to generate mean data for analysis. pEC50 values were used in order to enable EC50 values to fit a Gaussian distribution. On rare occasions, there were some experiments for peptide:receptor combinations in which the Hill slope was significantly different from one, and others in which the Hill slope did not differ from one. In these cases, a “majority rules” approach was taken when presenting the combined data, however the individual values used in statistical analyses were derived from the curve of best-fit for each replicate.
Combined concentration-response curves were generated for presentation by combining the mean of data points from individual experiments. These data were then fit with either a three-parameter or four-parameter non-linear regression according to the “majority rules” approach dictated by the results of the individual experiments. pEC50 and Emax values were analysed using unpaired one-way analysis of variance (ANOVA). When this ANOVA reached a statistical threshold (p < 0.05) and there was no variance inhomogeneity, a post-hoc Tukey’s test was employed, comparing each peptide to every other peptide. All data are presented as the mean ± s.e.m. of n independent experiments. This unpaired, non-normalised approach was taken because the results from peptides were not linked in any way (unpaired analysis across peptides), nor was there a reference ligand on each plate to normalise to (non-normalised analysis). In all cases, a difference was considered significant when p < 0.05.
The purpose of this study was to develop parameters for receptor activation that can then be translated to in vivo mouse work. Thus, we have opted to focus on direct measures of receptor activation such as pEC50 in molar concentrations rather than binding.
Data analysis – Antagonist assays
For m-CLR:m-RAMP1, the agonist used was m/r-αCGRP and for m-CLR:m-RAMP2/3 this was m-AM. At m-CTR based receptors, we tested antagonists against m-CT and m/r-Amy. In select other cases we also used m/r-αCGRP as the agonist at m-CTR based receptors because we were interested in investigating agonist-dependent antagonism.
In order to determine whether antagonism was present in a given assay, we used a sum-of-squares F-test to compare whether the two datasets (agonist in the absence and presence of antagonist) could be fit with a shared EC50. In cases where a single EC50 could be fit to the two datasets, it was concluded that there was no detectable antagonism in the assay.
When no antagonism was detectable, we did not attempt to derive pA2 values, nor to perform statistical tests. A single experiment was performed using h-AM22-52 at m-CLR:m-RAMP1; this experiment detected no antagonism and was consistent with multiple previous reports. We did not repeat this experiment, and thus refer to this result as exploratory in text. In all other cases where antagonism was undetectable, we curtailed experiments after a minimum of three independent experiments. When this test showed that antagonism was detectable, a minimum of five independent experiments were performed. From each individual experiment we derived pA2 values using the Gaddum-Schild equation supplied in GraphPad PRISM 9. When examining the results of antagonist assays, parallelism of the curves was assessed by comparing the fits where the nH parameter was shared across the curve in the absence and presence of antagonist to fits in which each curve was allowed its own Hill slope factor. In the majority of tested data sets the Hill slope factor was equal to one in the absence and presence of antagonist, thus for all pA2 analyses the Hill slope was constrained to one. The antagonists used in this paper are fragments of the agonists, thus we assumed that antagonism would be competitive and surmountable. This assumption is consistent with results from previous studies using these antagonists (Bailey et al., 2012; Hay et al., 2003). Accordingly, the Schild slope was constrained to one in all cases.
pA2 values derived from these equations were then compared using either unpaired Student’s t-test or unpaired one-way ANOVA as dictated by the number of groups being compared. When this ANOVA reached a statistical threshold (p < 0.05) and there was no variance inhomogeneity, a post-hoc Tukey’s test was employed comparing the mean pA2 of each antagonist to all other antagonists. In all cases significance was accepted when p < 0.05.
Nomenclature of Targets and Ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019).
Results
Agonist pharmacology at m-CLR:m-RAMP complexes
We characterised mouse receptors using species-matched mouse peptides. In addition, we have also characterised receptor activation in response to other agonists that are commonly used in the study of this peptide family (h-AM and h-αCGRP, s-CT, and Pram, Figure 1). Two forms of m-AM2 were examined; a 47 amino acid peptide and a 40 amino acid peptide (which lacks the first seven amino acids of the 47 amino acid peptide). This approach was taken because both peptide lengths have been reported in humans, and there is evidence to suggest that these different lengths have different physiological effects in mice (Hong et al., 2012).
We first confirmed that m-CLR was unable to signal alone. Cells transfected with m-CLR:pcDNA did not respond to stimulation with m-AM or m/r-αCGRP at concentrations as high as 10 μM (Figure S 7). This is consistent with results from other mammalian species and suggests that without RAMP interaction, m-CLR does not translocate to the cell surface, nor bind peptide, though we did not test this (Hay et al., 2018).
We then characterised receptor complexes comprising m-CLR:m-RAMPs. For m-CLR:m-RAMP1, the most potent peptides were m/r-αCGRP, h-αCGRP, and m-βCGRP, which were all equipotent. The next most potent was m-AM240, which was approximately 14-fold less potent than m/r-αCGRP (Figure 2, Figure S 1). There were no significant differences in Emax between any peptides at this receptor.
Figure 2.
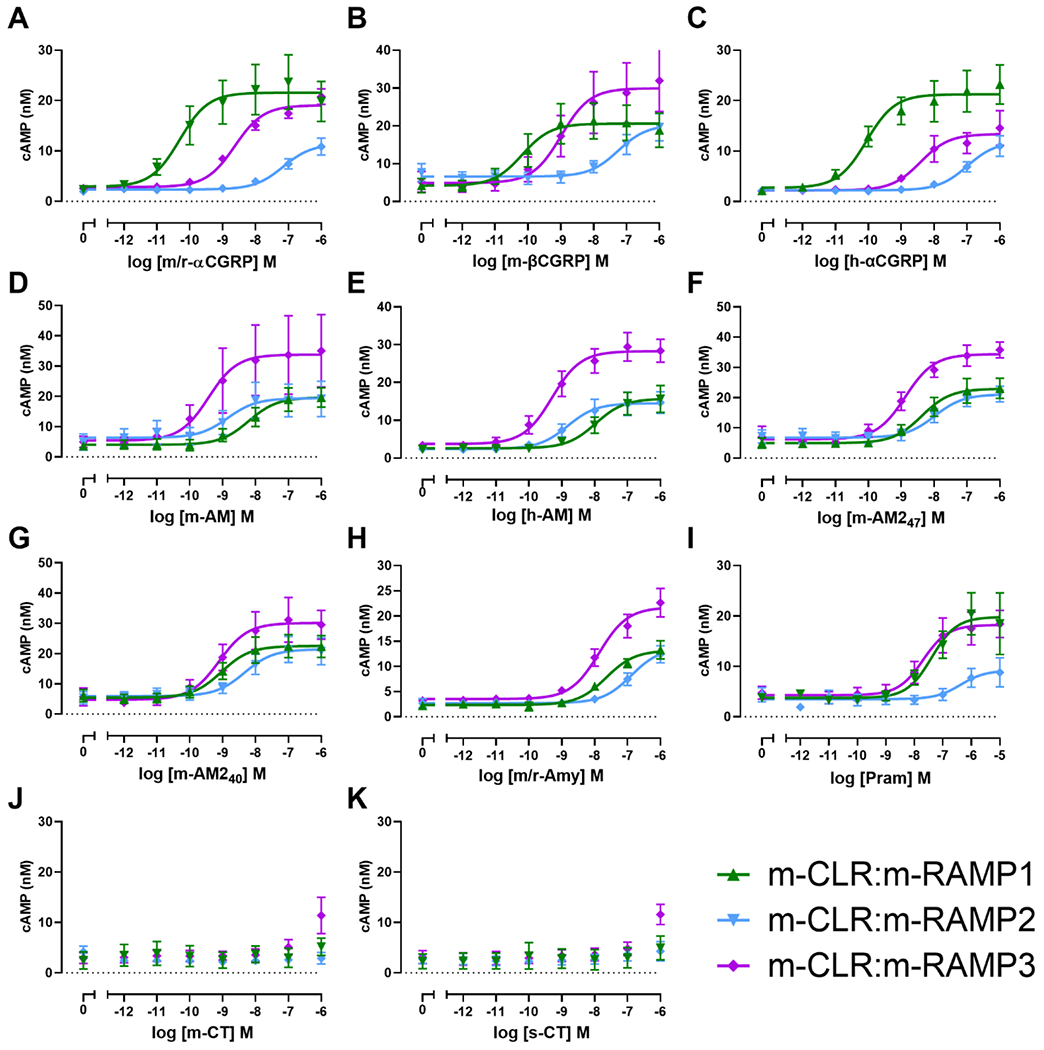
Concentration-response curves for m/r-αCGRP (A), m-βCGRP (B), h-αCGRP (C), m-AM (D), h-AM (E), m-AM247 (F), m-AM240 (G), m/r-Amy (H), Pram (I), m-CT (J), and s-CT (K) stimulating cAMP production in Cos7 cells transfected with m-CLR in the presence of m-RAMPs. Each point is the mean ± s.e.m. of at least three (flat-lines) or five (curves) independent experiments performed in duplicate or triplicate (see Table 1 for individual n numbers).
In cells transfected with m-CLR:m-RAMP2, the most potent peptides were m-AM, h-AM and m-AM240. These were followed by m-AM247, which was ~seven-fold less potent than m-AM (Figure 2, Figure S 1). There were no significant differences in Emax between any peptides at this receptor.
Receptors formed through the interaction of m-CLR and m-RAMP3 had less than a 10-fold difference in potency between m-AM (the most potent agonist) and the tested forms of CGRP, AM, and AM2 (Figure 2,). m/r-Amy and Pram were 32-fold and 55-fold less potent than m-AM, respectively. There were no significant differences in Emax between any peptides at this receptor. In all experiments using m-CLR, m-CT and s-CT were unable to stimulate quantifiable cAMP production within the tested concentration range (Figure 2).
Agonist pharmacology at m-CTR and m-CTR:m-RAMP complexes
In cells transfected with m-CTR:pcDNA (i.e. m-CTR alone) s-CT was the most potent peptide, m-βCGRP was the most potent non-CT peptide being ~10-fold more potent than m/r-Amy (Figure 3, Figure S 2). There were no significant differences in Emax between any peptides at this receptor.
Figure 3.
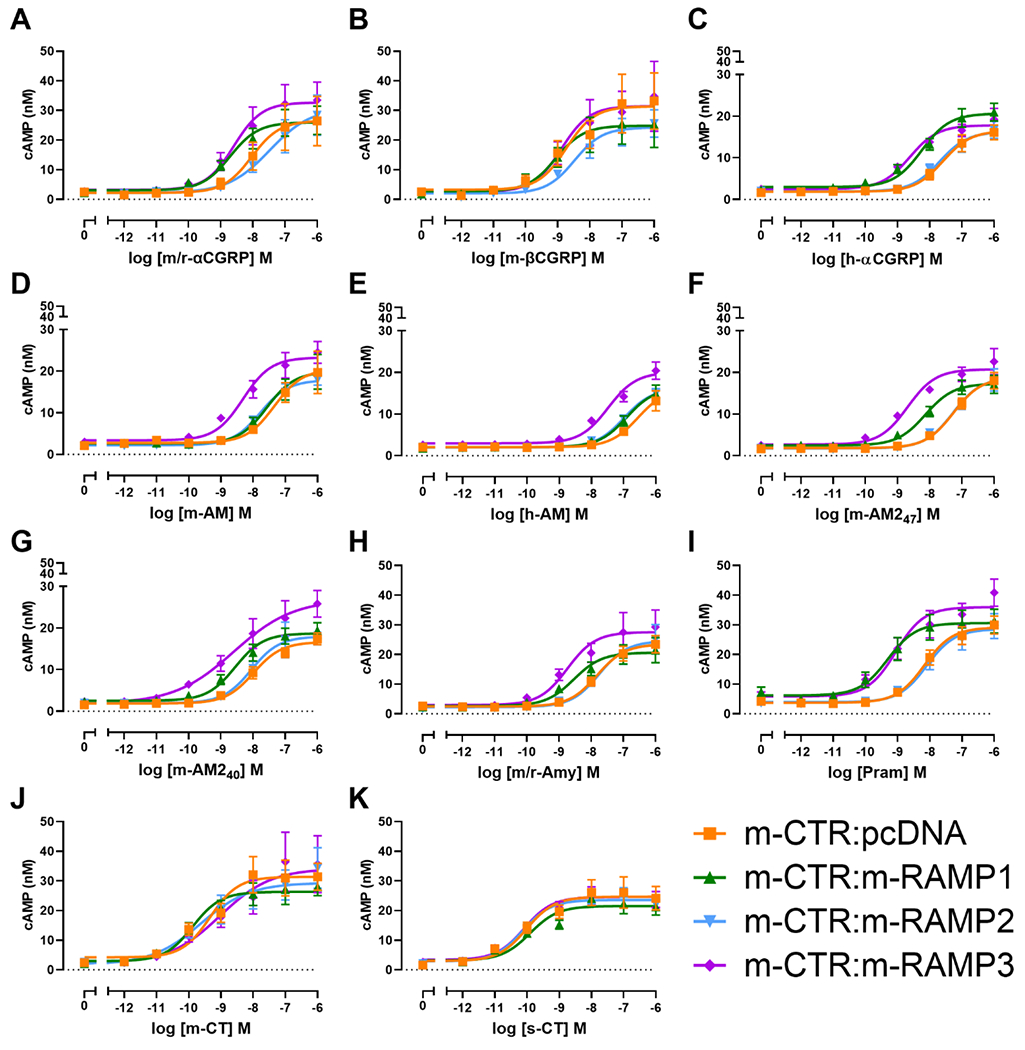
Concentration-response curves for m/r-αCGRP (A), m-βCGRP (B), h-αCGRP (C), m-AM (D), h-AM (E), m-AM247 (F), m-AM240 (G), m/r-Amy (H), Pram (I), m-CT (J), or s-CT (K) stimulating cAMP production in Cos7 cells transfected with m-CTR in the absence or presence of m-RAMPs as indicated. Each point is the mean ± s.e.m. of at least five independent experiments performed in duplicate or triplicate (see Table 1 for individual n numbers).
Broadly speaking, most non-CT peptides were more potent in cells transfected with m-CTR:m-RAMP1 than cells with m-CTR alone (Figure 3, Table 1). This was most pronounced for Pram (14-fold increase), m-AM247 (eight-fold increase), m/r-Amy and m/r-αCGRP (both five-fold increase; Figure S 3). βCGRP was also potent at this receptor (Figure S 2). There were no significant differences in Emax between any peptides at this receptor.
Table 1.
pEC50 and Emax values for CT-family peptides stimulating cAMP production in Cos7 cells transfected with combinations of m-CTR, m-CLR, and m-RAMPs.
| m-CLR:m-RAMP1 | m-CLR:m-RAMP2 | m-CLR:m-RAMP3 | m-CTR:pcDNA | m-CTR:m-RAMP1 | m-CTR:m-RAMP2 | m-CTR:m-RAMP3 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Agonist | pEC50 | Emax | n | pEC50 | Emax | n | pEC50 | Emax | n | pEC50 | Emax | n | pEC50 | Emax | n | pEC50 | Emax | n | pEC50 | Emax | n |
| m-CT | NC | NC | 3 | NC | NC | 3 | NC | NC | 3 | 9.42 ± 0.16 | 32.63 ± 5.25 | 5 | 9.83 ± 0.14# | 27.14 ± 3.40 | 5 | 9.45 ± 0.30# | 31.28 ± 6.56 | 5 | 9.04 ± 0.21 | 36.98 ± 9.91 | 5 |
| s-CT | NC | NC | 3 | NC | NC | 3 | NC | NC | 3 | 10.05 ± 0.14# | 24.80 ± 4.21 | 5 | 10.02 ± 0.18# | 22.45 ± 2.96 | 5 | 10.13 ± 0.11# | 24.59 ± 1.95 | 5 | 10.14 ± 0.11# | 25.07 ± 2.11 | 5 |
| m/r-Amy | 7.61 ± 0.19*# | 22.17 ± 2.76 | 5 | 6.88 ± 0.05*# | 13.71 ± 1.80 | 5 | 7.83 ± 0.15*# | 22.17 ± 2.76 | 5 | 7.83 ± 0.10*# | 23.77 ± 2.83 | 5 | 8.56 ± 0.11 | 21.56 ± 3.88 | 7 | 7.76 ± 0.10* | 28.81 ± 4.99 | 5 | 8.70 ± 0.17 | 29.89 ± 5.98 | 5 |
| Pram | 7.27 ± 0.13*# | 18.95 ± 2.80 | 5 | 6.49 ± 0.13*# | 8.18 ± 1.92 | 5 | 7.59 ± 0.18*# | 18.95 ± 2.79 | 5 | 8.11 ± 0.08*# | 29.57 ± 3.03 | 9 | 9.25 ± 0.12# | 30.83 ± 4.24 | 9 | 8.11 ± 0.09* | 28.71 ± 4.11 | 9 | 8.91 ± 0.20 | 37.3 ± 4.29 | 9 |
| m/r-αCGRP | 10.16 ± 0.12 | 19.11 ± 1.35 | 8 | 7.15 ± 0.20*# | 11.69 ± 1.81 | 5 | 8.66 ± 0.10*# | 19.11 ± 1.34 | 5 | 7.98 ± 0.07*# | 29.66 ± 7.24 | 5 | 8.64 ± 0.06* | 26.39 ± 4.34 | 14 | 7.53 ± 0.13* | 27.85 ± 6.30 | 6 | 8.59 ± 0.15* | 34.17 ± 6.51 | 5 |
| m-βCGRP | 10.05 ± 0.17 | 30.29 ± 8.14 | 5 | 7.33 ± 0.12*# | 20.89 ± 4.58 | 5 | 8.91 ± 0.16* | 30.29 ± 8.13 | 5 | 8.77 ± 0.10*# | 35.60 ± 10.90 | 5 | 9.07 ± 0.07*# | 25.47 ± 7.17 | 8 | 8.46 ± 0.13*# | 25.18 ± 4.31 | 6 | 8.95 ± 0.11* | 32.52 ± 9.70 | 6 |
| h-αCGRP | 10.08 ± 0.11 | 13.46 ± 2.94 | 6 | 7.03 ± 0.06*# | 11.89 ± 2.29 | 5 | 8.39 ± 0.08*# | 13.46 ± 2.93 | 5 | 7.54 ± 0.11*# | 16.69 ± 1.84 | 5 | 8.22 ± 0.08* | 20.58 ± 2.02 | 10 | 7.50 ± 0.08* | 17.26 ± 2.33 | 5 | 8.61 ± 0.20* | 18.21 ± 2.24 | 5 |
| m-AM | 8.18 ± 0.11*# | 30.86 ± 7.96 | 5 | 8.81 ± 0.07 | 19.43 ± 5.82 | 5 | 9.33 ± 0.16 | 30.86 ± 7.95 | 5 | 7.43 ± 0.12*# | 20.94 ± 5.71 | 5 | 7.56 ± 0.07*# | 16.65 ± 1.14 | 7 | 7.71 ± 0.14* | 18.65 ±1.87 | 5 | 8.45 ± 0.14 | 24.55 ± 2.82 | 5 |
| h-AM | 7.90 ± 0.13*# | 28.88 ± 3.08 | 5 | 8.80 ± 0.16 | 14.70 ± 3.02 | 7 | 9.26 ± 0.14 | 28.88 ± 3.07 | 5 | 6.72 ± 0.08*# | 14.67 ± 2.91 | 5 | 7.01 ± 0.12*# | 15.88 ± 2.79 | 5 | 7.00 ± 0.10*# | 15.87 ± 1.54 | 5 | 7.56 ± 0.10*# | 19.92 ± 2.24 | 5 |
| m-AM247 | 8.35 ± 0.10*# | 34.42 ± 2.84 | 5 | 7.98 ± 0.04*# | 21.10 ± 2.49 | 5 | 8.88 ± 0.10 | 34.42 ± 2.83 | 5 | 7.25 ± 0.07*# | 18.95 ± 2.11 | 6 | 8.16 ± 0.14* | 17.5 ± 2.07 | 6 | 7.37 ± 0.10* | 18.78 ± 3.13 | 6 | 8.67 ± 0.12 | 21.17 ± 1.95 | 6 |
| m-AM240 | 9.02 ± 0.06# | 30.30 ± 6.09 | 6 | 8.43 ± 0.13* | 21.47 ± 4.55 | 6 | 9.11 ± 0.18 | 30.29 ± 6.08 | 5 | 7.91 ± 0.17*# | 17.37 ± 0.92 | 5 | 8.52 ± 0.17 | 19.31 ± 1.88 | 5 | 8.09 ± 0.13* | 17.71 ± 2.77 | 5 | 8.78 ± 0.06 | 25.8 ± 3.19 | 5 |
Data are the mean ± s.e.m. of n independent experiments. Emax values are presented as nM values of cAMP. pEC50 and Emax values of individual peptides were compared across receptors using unpaired one-way ANOVA with Tukey’s post-hoc test comparing the results at a receptor to the results at each other receptor. In this table significance is shown only relative to a reference receptor (m-CLR:m-RAMP1 for m/r-αCGRP, m-βCGRP, and h-αCGRP, m-CLR:m-RAMP2 for m-AM and h-AM, and m-CLR:m-RAMP3 for m-AM240 and m-AM47, m-CTR:pcDNA for s-CT and m-CT, and m-CTR:m-RAMP1 for m/r-Amy and Pram), significance is indicated by an asterisk (*). pEC50 and Emax values were also compared within receptors using unpaired one-way ANOVA with Tukey’s post-hoc test, comparing the mean value of an individual peptide at a given receptor to each other peptide at the same receptor. In this table significance is shown only relative to a reference peptide at each receptor (m/r-αCGRP at m-CLR:m-RAMP1, m-AM at m-CLR:m-RAMP2/3, m-CT at m-CTR:pcDNA, and m/r-Amy at m-CTR:m-RAMP1/2/3), significance is indicated by a hash symbol (#). To view this data in graphical format, see Figure S 8.
Co-transfection of m-CTR with m-RAMP2 induced only minor changes in peptide potency relative to m-CTR alone, and did not affect the potency of m/r-Amy (Figure 3, Figure S 3). There were no significant differences in Emax between any peptides at this receptor.
Co-transfection of m-CTR with m-RAMP3 induced substantial increases in the potency of several peptides, relative to cells transfected with m-CTR alone. m/r-Amy was seven-fold more potent at m-CTR:m-RAMP3 over m-CTR (Figure 3, Figure S 3) but the increases were largest for m-AM247 (26-fold increase), h-αCGRP (12-fold increase) and m-AM (11-fold increase). The majority of tested peptides had a pEC50 within one log unit of m/r-Amy; the only exceptions were s-CT and h-AM (28-fold more potent, and 14-fold less potent than m/r-Amy, respectively; Figure S 2). There were no significant differences in Emax between any peptides at this receptor.
Note that m-CT responses could be acting through m-CTR alone in cells co-transfected with RAMPs. This has been discussed previously for human receptors and is a complication of CTR being able to traffic to the cell surface without RAMP (Hay et al., 2018). It is important to be aware of this when interpreting the data with m-CT.
Antagonist pharmacology
We additionally characterised these receptors using commonly cited antagonists (Figure 1 b). Our data above show that there is limited scope to differentiate receptors in vitro based on agonism alone, especially for m-CTR-based receptors which displayed little selectivity between agonists. Antagonists are useful tools when used in conjunction with agonists to better define receptors responsible for effects. Thus, we reasoned it will be important to combine agonists and antagonists in model systems to attribute discrete outcomes to defined receptors. We focused on peptide antagonists because the major class of small molecules targeting this receptor family (the -gepants) often have low affinity at receptors from non-primate species (Doods et al., 2000; Moore et al., 2020; Moore et al., 2010; Salvatore et al., 2008).
αCGRP8-37 is a commonly used antagonist of CGRP receptors. m/r-αCGRP8-37 was an effective antagonist of m-CLR:m-RAMP1 (pA2 of 8.48 ± 0.18, n = 5; Figure 4) and a significantly weaker antagonist of m-CLR:m-RAMP3 (pA2 of 6.20 ± 0.20, n = 5; Figure 4); m/r-αCGRP8-37 was unable to antagonise cAMP production in cells transfected with m-CLR:m-RAMP2 in four exploratory experiments (Figure 4). We compared this profile to the commonly used AM-receptor antagonist, h-AM22-52. In an initial exploratory experiment, 10 μM hAM22-52 was unable to cause a rightward shift at m-CLR:m-RAMP1, consistent with results from humans, rats, and previous mouse work (Booe et al., 2018; Husmann et al., 2003; Oliver et al., 2001). Concentrations higher than 10 μM were not practical to test, and therefore this experiment was not repeated (data not shown). In contrast, hAM22-52 was a weak antagonist at both m-CLR:m-RAMP2 and m-CLR:m-RAMP3 and (Figure 4). AM22-52 was significantly more effective at antagonising receptors comprising m-CLR:m-RAMP2 (pA2 of 5.71 ± 0.03, n = 5) over m-CLR:m-RAMP3 (pA2 of 5.19 ± 0.21, n = 5), although the difference was small (three-fold). Interestingly, at receptors comprising m-CLR:m-RAMP3, m/r-αCGRP8-37 was 10-fold more effective than h-AM22-52 at antagonising cAMP production; this difference was statistically significant.
Figure 4.
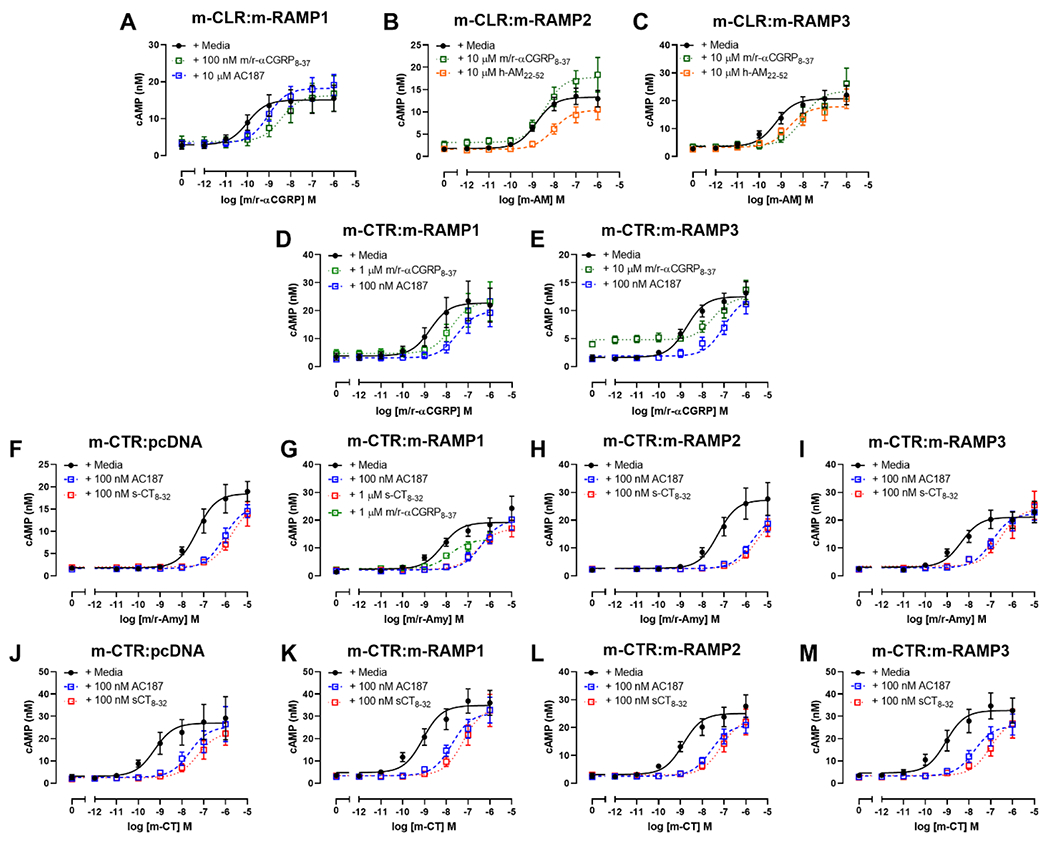
Concentration-response curves showing antagonism of m/r-αCGRP (A, D, E), m-AM (B and C), m/r-Amy (F, G, H, I), and m-CT (J, K, L, M) by various peptide antagonists in Cos7 cells. Cells were transfected with m-CLR:m-RAMP1 (A), m-CLR:m-RAMP2 (B), m-CLR:m-RAMP3 (C), m-CTR:m-RAMP1 (D, G, K), m-CTR:m-RAMP3 (E, I, M), m-CTR:pcDNA (F, J), or m-CTR:m-RAMP2 (H, L). Data are the mean ± s.e.m. of at least five independent experiments, except for m/r-αCGRP8-37 in panel B which is n = 4 (for individual n numbers, see Table 2).
Previous reports show that αCGRP8-37 can also act as an antagonist at the AMY1 receptor (Bailey et al., 2012; Hay et al., 2005; Walker et al., 2015). To explore the selectivity of this antagonist at mouse receptors, we characterised its ability to antagonise m/r-αCGRP stimulated cAMP production in cells transfected with m-CTR:m-RAMP1. m/r-αCGRP8-37 was 50-fold less effective at receptors comprising m-CTR:m-RAMP1 relative to m-CLR:m-RAMP1 (Table 2), this difference was statistically significant. Our work showed that m/r-αCGRP was also an effective agonist of m-CTR:m-RAMP3 (Figure 2), thus we included this receptor in our characterisation and found that m/r-αCGRP8-37 could antagonise m/r-αCGRP with a pA2 ~100-fold lower than its pA2 at m-CLR:m-RAMP1 (Table 2, Figure 4). We noted a partial agonist effect of m/r-αCGRP8-37 at m-CTR:m-RAMP3. This effect was statistically significant as determined by a paired Student’s t-test comparing the Emin of curves in the absence and presence of m/r-αCGRP8-37 (mean cAMP production of 1.78 ± 0.54 nM and 4.52 ± 0.57 nM, respectively, both n = 5; Figure 4), this is consistent with previous publications which have used high concentrations of this peptide (Walker et al., 2018). Small molecule antagonists (olcegepant and rimegepant) show differences in their ability to antagonise CGRP and Amy at the h-AMY1 receptor (Pan et al., 2020; Walker et al., 2018). We therefore investigated the ability of m/r-αCGRP8-37 to antagonise m/r-Amy at receptors comprising m-CTR:m-RAMP1 and found that m/r-αCGRP8-37 was equally effective against both agonists (Table 2, Figure 4).
Table 2.
pA2 values for antagonism at m-CTR-based receptors in transfected Cos7 cells.
| Antagonist | Agonist | m-CTR:pcDNA | m-CTR:m-RAMP1 | m-CTR:m-RAMP2 | m-CTR:m-RAMP3 | ||||
|---|---|---|---|---|---|---|---|---|---|
| pA2 | n | pA2 | n | pA2 | n | pA2 | n | ||
| sCT8-32 | m-CT | 9.17 ± 0.17 | 5 | 8.74 ± 0.27 | 5 | 8.79 ± 0.17 | 5 | 8.89 ± 0.27 | 5 |
| m/r-Amy | 8.44 ± 0.18^ | 5 | 7.47 ± 0.19*^ | 5 | 8.75 ± 0.05 | 5 | 8.41 ± 0.12 | 5 | |
| AC187 | m-CT | 8.59 ± 0.13 | 5 | 8.37 ± 0.19 | 6 | 8.34 ± 0.11 | 5 | 8.35 ± 0.26 | 5 |
| m/r-Amy | 8.07 ± 0.12*^ | 6 | 8.76 ± 0.12 | 6 | 8.57 ± 0.09 | 6 | 8.34 ± 0.10 | 6 | |
| m/r-αCGRP | - | - | 8.26 ± 0.14 | 5 | - | - | 8.65 ± 0.08* | 5 | |
| m/r-αCGRP8-37 | m/r-Amy | - | - | 6.88 ± 0.17 | 5 | - | - | - | - |
| m/r-αCGRP | - | - | 6.80 ± 0.08 | 5 | - | - | 6.49 ± 0.16 | 6 | |
Data are the mean ± s.e.m. of n independent experiments. An * (asterisk) indicates a significant difference between the pA2 of an antagonist across receptors (i.e. across a row). This explores whether a single antagonist is more effective at antagonising an individual agonist across different receptors. Analyses were performed using one-way ANOVA with Tukey’s post-hoc test, comparing the pA2 at each receptor to every other receptor, the exception being those with only two receptors, which were analysed by unpaired Student’s t-test. In this table, differences are shown relative to a reference receptor. The reference receptor for sCT8-32 was m-CTR:pcDNA, for AC187 and m/r-αCGRP8-37 the reference was m-CTR:m-RAMP1. A ^ (caret) indicates a significant difference between the pA2 of an antagonist between agonists (columns, within antagonists). Analyses were performed using unpaired Student’s t-tests, except for AC187 at m-CTR:m-RAMP1 and m-CTR:m-RAMP3, which were analysed by one-way ANOVA with Tukey’s post-hoc test comparing the pA2 for an agonist to each other agonist. For sCT8-32 and AC187, differences are shown compared to m-CT. There was no significant difference between the ability of m/r-αCGRP8-37 to antagonise m/r-Amy and m/r-αCGRP.
The N-terminally truncated analogue of sCT (sCT8-32), and the acetylated peptide AC187, are widely available and commonly cited peptide antagonists of CTR-based receptors. Thus, we sought to define the pharmacology of these peptides at m-CTR-based receptors. When using m/r-Amy as an agonist, sCT8-32 and AC187 were equivalent in all cases, except for receptors comprising m-CTR:m-RAMP1, where AC187 was 10-fold more effective than s-CT8-32. Additionally, AC187 was slightly more effective against m/r-Amy at receptors comprising m-CTR:m-RAMPs over m-CTR alone (Figure 4, Table 2).
We also investigated the selectivity of AC187 for m-CTR-based receptors over m-CLR:m-RAMP1 (Figure 4), where it was ~300-fold more effective at antagonising m/r-αCGRP at m-CTR:m-RAMP1 and m-CTR:m-RAMP3 (Table 2) compared to m-CLR:m-RAMP1 (pA2 of 5.84 ± 0.05, n = 5; Figure 4), these differences were statistically significant as determined by a one-way ANOVA with Dunnett’s post-hoc test comparing the pA2 of AC187 at m-CLR:m-RAMP1 to its pA2 at m-CLR:m-RAMP1 and m-CLR:m-RAMP3.
Discussion
Drugs which build upon the successes of CGRP-targeting therapeutics will be important in further reducing the burden of migraine. To facilitate the development of drugs targeting the CGRP/CT family, we need to understand their pharmacology in mice. We report here a reference framework which will be useful to investigators researching migraine, and other conditions such as cardiovascular and metabolic disorders (Hay et al., 2018).
To facilitate comparisons to human receptors, we have created two figures showing the relative potency of ligands at each receptor in humans and mice (Figure 5 and 6). Human data are the mean of values reported throughout the literature (see figure legends of Figure 5 and 6 for sources). The mouse receptor pharmacology for CLR-based receptors is a composite of our results in this study and previously published literature on these receptors. These figures are useful for comparing a single receptor complex between species; however they were not designed to compare across receptors. This is most relevant for CTR-based receptors, for which the fold-induction of peptide potency at CTR:RAMP complexes relative to CTR alone is important. For this comparison, we refer readers to Figure S 3.
Figure 5.
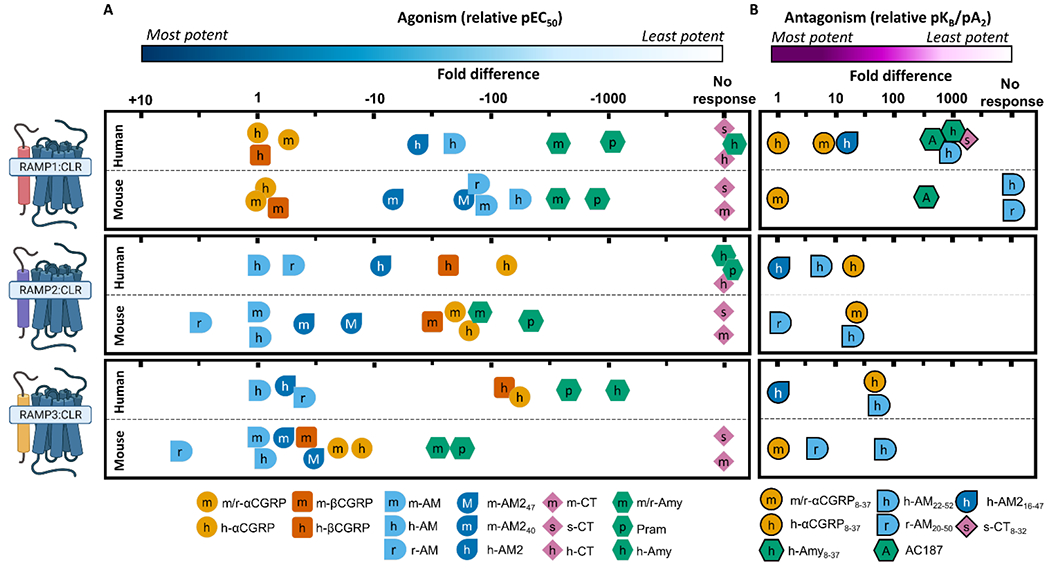
Relative potency of peptide agonists (A) or antagonists (B) at CLR-based receptors from human and mouse, to enable comparison of relative ligand profiles at each receptor between species. Agonist potency is pEC50 and antagonism is pKB/pA2 values. For agonism, the potency of each peptide is displayed relative to the most potent species matched peptide at each receptor, for antagonism the potency is displayed relative to the most potent antagonist. Each tick represents half a log unit. It is important to note that the absolute potencies are not captured in this figure. This presentation approach was taken because absolute agonist potency is system-dependent, and studies are more easily compared when using relative potency. Data for mouse receptors is from this paper, Husmann et al. (2003), and Koller et al. (2004); data for human receptors is collated from Hay et al. (2018) and Garelja et al. (2020), and updated to include Gingell et al. (2020), Ghanizada et al. (2021b), and Hendrikse et al. (2020). Receptor figures created with BioRender.com.
Figure 6.
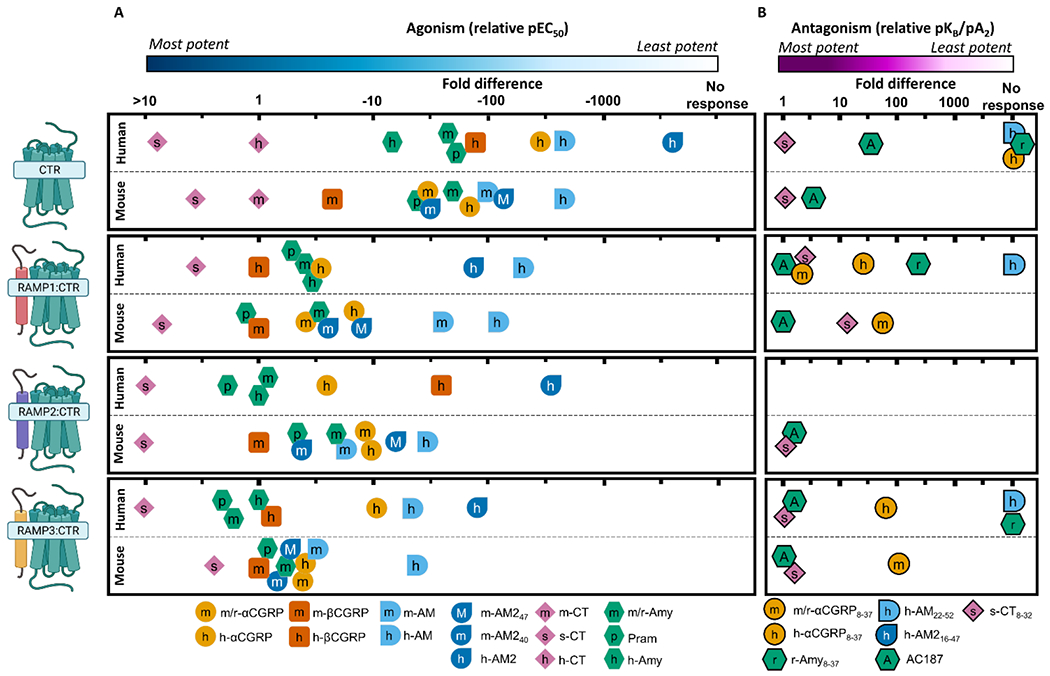
Relative potency of peptide agonists (A) or antagonists (B) at CTR-based receptors from human and mouse, to enable comparison of relative ligand profiles at each receptor between species. Agonist potency is pEC50 and antagonism is pKB/pA2. For agonism, the potency of each peptide is displayed relative to the most potent species matched peptide at each receptor, for antagonism the potency is displayed relative to the most potent antagonist. The antagonist data for CTR alone is antagonism of species matched CT, while the antagonist data for CTR:RAMP complexes is derived from antagonism of Amy or CGRP. Species matched CT was omitted from receptors comprising CTR in complex with RAMPs because these peptides exert their effect through free CTR, rather than RAMP complexed CTR (Hay et al., 2018). Each tick represents half a log unit. It is important to note that the absolute potencies are not captured in this figure. This presentation approach was taken because absolute agonist potency is system-dependent, and studies are more easily compared when using relative potency. Data for mouse receptors is from this paper; data from human receptors is collated from Hay et al., (2018), and updated to include Ghanizada et al., (2021b), Gingell et al., (2020), Hendrikse et al., (2020), and Yule et al. (2019). Receptor figures created with BioRender.com.
Consistent with receptors cloned from humans, our data show that m-CLR:m-RAMP1 is most potently activated by CGRP peptides and more weakly by AM peptides (Figure 5 A); this is congruous with rat receptors (Bailey et al., 2012). Likewise, our work with antagonists showed a consistent pharmacology across humans and mice, with α-CGRP8-37 being much more potent than AM-based antagonists (Figure 5 B), which is again congruous with rat receptors (Walker et al., 2015). Thus, the pharmacology of this receptor complex appears broadly consistent in all tested species.
m-CLR:m-RAMP2 had a strong preference for m-AM over m/r-αCGRP, which is consistent with human receptors (Figure 5 A) and rat receptors (Kuwasako et al., 2002). However, we report that m-AM and m-AM240 had similar pEC50 and Emax values at this receptor, while at receptors cloned from humans h-AM2 is a partial agonist that is approximately 10-fold less potent than h-AM (Garelja et al., 2020; Musa et al., 2019; Weston et al., 2016). The difference in potency between AM and AM2 is one of the hallmarks of a h-AM1 receptor; we now show that this does not appear to translate between species. We saw very weak antagonism with h-AM22-52 and no antagonism with m/r-αCGRP8-37. Previously, m/r-αCGRP8-37 has been reported as a very weak antagonist of m-CLR:m-RAMP2 (Husmann et al., 2003), thus it is likely that a higher concentration of antagonist would have induced measurable antagonism in our hands. Nevertheless, our antagonist findings seem to agree with human and rat receptors, where AM based antagonists are between two and 10-fold more effective than h-αCGRP8-37 and m/r-αCGRP8-37 (Figure 5 B; Hay et al., 2003).
m-CLR:m-RAMP3 showed only five-fold discrimination between m-AM and m/r-αCGRP (Figure 5 A). This is aligns with rat receptors and previous data from mouse receptors (Bailey et al., 2012; Hay et al., 2003; Husmann et al., 2003). In contrast, human AM2 receptors have a stronger preference for h-AM over CGRP peptides (Figure 5 A), highlighting a difference in pharmacology across species. However, both forms of m-AM2 were ~eight-fold more potent at m-CLR:m-RAMP3 compared to m-CLR:m-RAMP2 (Table 1), which is consistent with humans (Musa et al., 2019). Like rat receptors, our data showed that m/r-αCGRP8-37 was ~10-fold more effective than h-AM22-52 at m-CLR:m-RAMP3; in humans h-αCGRP8-37 and h-AM22-52 are equivalent (Figure 5 B), which again highlights a difference in receptor pharmacology across species. As with m-CLR:m-RAMP2, the differences in pharmacology between mice and humans preclude simple translation between a human AM2 receptor and a “mouse AM2 receptor”.
Consistent with cloned human, rat, and porcine receptors, m-CTR displayed a strong preference for m-CT and s-CT over m/r-Amy (Bailey et al., 2012; Kikumoto et al., 2003; Udawela et al., 2006) (Figure 6 A). However, while the human CTR has a preference for h-Amy over h-αCGRP and h-AM240, this was less apparent at the mouse CTR (Figure 6 A). We report m-βCGRP as a relatively potent agonist of m-CTR; this is unique to mice. βCGRP has previously been highlighted as a peptide which does not have conserved trends in pharmacology across species (Bailey et al., 2012); our data adds to this and highlights that further investigations into this underexplored peptide are warranted.
The “AMY receptor” nomenclature is defined by a significant enhancement of Amy binding and signalling upon co-expression of CTR with a RAMP (Poyner et al., 2002). Co-expression of m-CTR with m-RAMP1 or m-RAMP3 induced a five-fold and seven-fold increase in m/r-Amy potency, while co-expression with m-RAMP2 had no effect on m/r-Amy potency (Figure S 3, Table 1). On average, co-expression of h-CTR with h-RAMP1, h-RAMP2, or h-RAMP3 causes a 11-, two-, and six-fold increase in the potency of h-Amy (Figure S 3). Rat receptors have been defined only with r-RAMP1 and r-RAMP3; co-expression of r-CTR with r-RAMP1 or r-RAMP3 causes a nine-fold or 72-fold increase in the potency of m/r-Amy, respectively (Figure S 3). Our data suggest that the induction of m-AMY1/3 receptor phenotype is modest when compared to rat receptors, and slightly weaker than human receptors. It is worth highlighting that the pharmacology of h-CTR:h-RAMP2 is dependent on cell background, and this may also be the case for rat and mouse (Morfis et al., 2008; Tilakaratne et al., 2000).
AMY receptor nomenclature is also defined by Amy being more potent than other non-CT peptides. h-Amy is equipotent to h-αCGRP at the h-AMY1 receptor, and 10-fold more potent than h-αCGRP at the h-AMY3 receptor. Our data show that m/r-Amy and m/r-αCGRP are equipotent at receptors comprising m-CTR and either m-RAMP1 or m-RAMP3; this is consistent with rat receptors (Bailey et al., 2012). Additionally, our data show there is less discrimination between m/r-Amy and other peptides at m-CTR-based receptors relative to the human receptors. Thus, while there are some similarities across species, the pharmacology is clearly not conserved. These mixed profiles create significant challenges for nomenclature as some receptors do not have a clear endogenous agonist (Vanhoutte et al., 1996), especially when the most potent agonist of the “m-AMY receptors” appears to be m-βCGRP (Figure 6 A).
Despite these differences, we report a relatively conserved phenotype with regards to antagonist pharmacology (Figure 6 B). From these data, AC187 emerges as a potentially useful tool as it shows discrimination (>100-fold) between CTR and CLR-based receptors. CGRP8-37 had the opposite pharmacology, being more potent at CLR than CTR-based receptors, though the fold difference was lower than for AC187 (~50-fold discrimination). However, because both were antagonists of multiple receptors, it is important to use these peptides in conjunction with other pharmacological tools to accurately identify receptors.
These findings have implications for our understanding of physiological data. For instance, we show that m/r-αCGRP has nano-molar potency at four of the seven tested receptors. Thus, inferences drawn about receptors responsible for a given effect are complicated by the fact that effects could be occurring via m-CLR:m-RAMP1/3, or m-CTR:mRAMP1/3, or a combination of these receptors. m-βCGRP has an additional complication in that it is also a potent agonist of m-CTR alone. h-AM22-52 was a very weak antagonist of m-CLR:m-RAMP2/3, thus studies that use this to ascertain the contributions AM receptors make to a defined physiological outcome must use very high concentrations to see antagonism (Owji et al., 2008). Until now it has been unclear why the different lengths of human AM2 have distinct physiological effects in mice given the two lengths are equivalent in humans (Hong et al., 2012; Musa et al., 2019). Our data shows these two fragments have distinct pharmacology at mouse receptors, which could influence their physiological function. To the author’s knowledge, this is the first synthesis and characterisation of mouse AM2 in the literature, and given its apparently broad spectrum activity across several mouse receptors, it warrants further investigation.
After removing the signal sequences, h-CLR and m-CLR are 95% similar (BLOSUM90 matrix, threshold of 1). Using the same analysis technique, h-CTR and m-CTR are 88% similar. This could explain why the pharmacology of CLR-based receptors was more conserved across these two species. We explored this further using recently published structures of receptors from this family (Liang et al., 2020; Liang et al., 2018; Liang et al., 2017), and found that in addition to there being more differences across for species for CTR relative to CLR, the differences were also more distributed across the receptor. Differences for CLR were clustered in the extreme ECD, the linker region (which joins the ECD to transmembrane domain 1), and the intracellular portions of transmembrane domains 1/2. Conversely, for CTR the differences were found across the receptor including in the peptide binding pocket, the intracellular G protein binding pocket, and the extreme C-terminal tail (Figure 7). Cumulatively, these mutations may have a large impact on the ability of m-CTR to bind RAMP, peptides and interact with intracellular signalling proteins.
Figure 7.
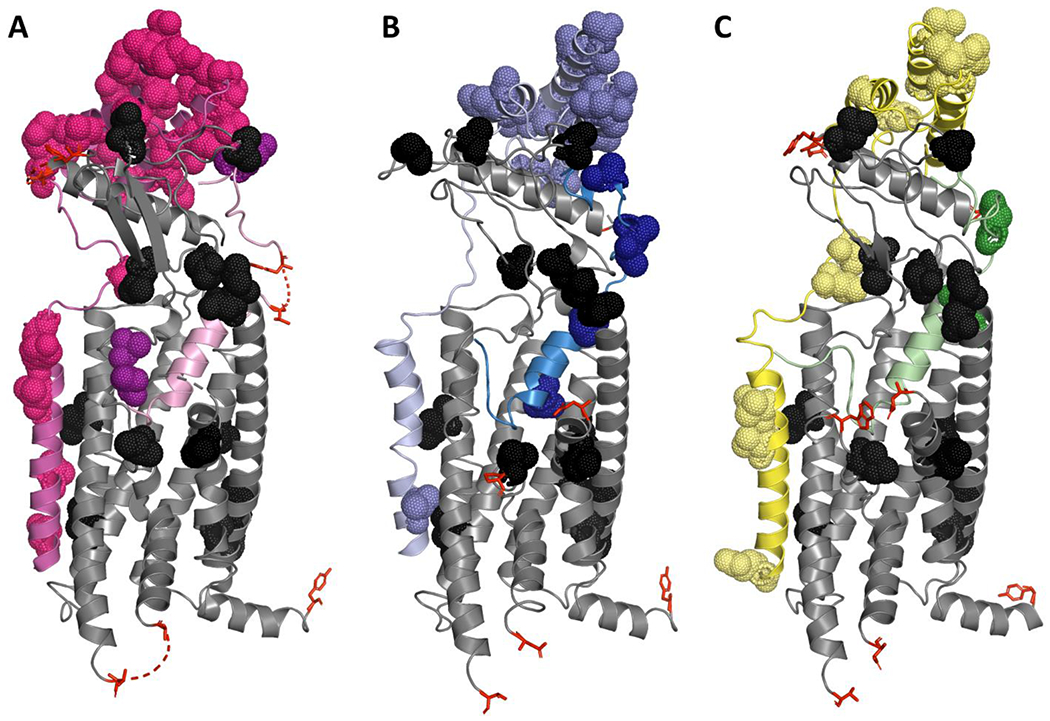
Structural models of (A) h-αCGRP (pink) bound to h-CLR:h-RAMP1 (grey:magenta, respectively), (B) h-AM (blue) bound to h-CLR:h-RAMP2 (grey:pale blue, respectively), and (C) h-AM2 (green) bound to h-CLR:h-RAMP3 (grey:yellow, respectively). Amino acids that differ between the human and mouse sequence are shown as black dots. Where there is an amino acid difference that is not captured in the deposited structure, the amino acids that flank the difference are shown as red sticks. The pdb codes for (A), (B), and (C) are 6E3Y, 6UUN, and 6UVA, respectively.
We used a similar approach to explore why m-RAMP3 might produce receptors with promiscuous profiles that differ from the defined profiles of h-RAMP3. This difference is surprising given the relative conservation of amino acid sequences between human and mouse RAMP3 (Figure S 5, 87% similarity between m-RAMP3 and h-RAMP3). RAMP1, which has a lower level of similarity between species (81% similarity between m-RAMP1 and h-RAMP1), has a more conserved effect on CTR and CLR when comparing across species. The differences between m-RAMP3 were not near the peptide binding pocket (Figure 7), suggesting that pharmacological differences were unlikely due to differential peptide contacts between species. The linker region of the RAMP has a strong influence on receptor phenotype by influencing conformational dynamics of the receptor complex (Liang et al., 2020). Examining the linker in m-RAMP3, there are two substantial differences between humans and mice, V109T, and L111W (changes presented relative to the human sequence). A structure captures these residues in the proximity of extracellular loop 2, specifically within 4 angstroms of CLR Y278/D280 (CTR F285/D287). These residues are known to influence the signalling of CLR and CTR (Pham et al., 2019; Watkins et al., 2016). Introducing side chains which change the polarity (V109T) and aromaticity (L111W) of the RAMP could influence receptor dynamics and therefore receptor pharmacology. While m-RAMP1 also has a change in the linker region, this change is less drastic, replacing a small hydrophobic residue with a slightly larger hydrophobic residue (V111L). This may explain why the m-RAMP1 pharmacology is more similar to h-RAMP1 than the m-RAMP3 is to h-RAMP3 however, this is speculative.
A limitation with this work is that the receptor sequences were all derived from the C57BL6 strain of mice. There are noted strain specific differences in CGRP induced migraine-like behaviours (Mason et al., 2017; Wattiez et al., 2019). There are numerous single nucleotide polymorphisms, insertions, and deletions in CLR, CTR, and RAMP genes between strains (Keane et al., 2011), which could manifest as differences in the sequence or expression of CGRP-responsive receptors. Future studies which explore the pharmacological effects of these genetic differences could shed light on the cause of physiological differences. Given the established use of mice genetically modified to over-express h-RAMP1 (Recober et al., 2009; Zhang et al., 2011), it would be interesting to further investigate receptors formed between mouse receptors and human RAMPs (Bohn et al., 2017). Previous work indicates that mixed species receptors do not necessarily replicate the pharmacology of receptors comprising subunits from individual species (Bohn et al., 2017; Hay et al., 2003; Husmann et al., 2000), thus further investigation would be invaluable to understanding these models. Mice which incorporate fully humanised receptors under endogenous promoters may also be useful for increasing the translatability of physiological findings, though issues relating to the differing splice variants across species must be considered. These mice would have the additional benefit of responding to small molecule antagonists such as olcegepant, which are less effective at rodent receptors than human receptors (Moore et al., 2020; Salvatore et al., 2008).
A further limitation to this work is that we have only measured cAMP production. There are reported differences in the ability of human receptors to stimulate alternative signalling pathways, for instance, the h-CGRP receptor can stimulate measurable IP1 production, while the h-AM1 receptor cannot (Garelja et al., 2020). Additionally, antagonists of these receptors can act in a pathway specific manner (Walker et al., 2018). Thus, future investigations into alternative signalling pathways regulated by mouse receptors may allow greater distinction between this family. Binding studies would also be valuable.
In conclusion, calcitonin family receptors cloned from mice are similar, but not directly comparable to, their human counterparts. Given the complicated pharmacology of this receptor family, a future research avenue might be to use genetically modified mice that express humanised receptors in place of mouse receptors to try improve the translatability of findings, thereby facilitating the discovery of novel therapeutics. In light of our findings, a cautious approach should be taken to assigning a particular calcitonin family receptor to a given function in mice.
Supplementary Material
What is already known
CGRP and related peptides are implicated in migraine pathophysiology
The pharmacology and physiology of this family is complex, involving multiple receptors and accessory proteins
What this study adds
Pharmacological characterisation of mouse receptors with endogenous ligands and commonly used pharmacological tools
Mouse receptors displayed reduced ligand specificity relative to their human counterparts
Clinical significance
Our findings are a framework for interpreting pre-clinical studies of this receptor family in mice
Acknowledgements
MAJ was supported by a Maurice Wilkins Centre PhD scholarship. DLH acknowledges receipt of research funding from the National Institutes of Health, U.S.A. (Grant NS113839); the contents of this article do not represent the views of the United States Government. CSW acknowledges receipt of a Sir Charles Hercus Health Research Fellowship from the Health Research Council (New Zealand).
Conflict of interest
D.L.H. has received research support from Living Cell Technologies and has acted as an advisor or consultant for Amgen, Merck and Intarcia.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design and Analysis, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Abbreviations
- AM
adrenomedullin
- Amy
Amylin
- CGRP
calcitonin gene-related peptide
- CLR
calcitonin receptor-like receptor
- CT
calcitonin
- CTR
calcitonin receptor
- DMEM
Dulbecco’s modified Eagle media
- DPBS
Dulbecco’s phosphate buffered saline
- GPCR
G protein-coupled receptor
- HEK293S
Human Embryonic Kidney cells
- Pram
Pramlintide
- RAMP
receptor activity-modifying proteins
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- Alexander Stephen P H, Christopoulos Arthur, Davenport Anthony P, Kelly Eamonn, Mathie Alistair, Peters John A, Veale Emma L, Armstrong Jane F, Faccenda Elena, Harding Simon D, Pawson Adam J, Sharman Joanna L, Southan Christopher, Davies Jamie A and CGTP Collaborators (2019) THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: G protein-coupled receptors. British Journal of Pharmacology, 176: S21–S141. 10.1111/bph.14748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey RJ, & Hay DL (2006, Jun). Pharmacology of the human CGRP1 receptor in Cos 7 cells. Peptides, 27(6), 1367–1375. 10.1016/j.peptides.2005.11.014 [DOI] [PubMed] [Google Scholar]
- Bailey RJ, Walker CS, Ferner AH, Loomes KM, Prijic G, Halim A, Whiting L, Phillips AR, & Hay DL (2012, May). Pharmacological characterization of rat amylin receptors: implications for the identification of amylin receptor subtypes. Br J Pharmacol, 166(1), 151–167. 10.1111/j.1476-5381.2011.01717.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn KJ, Li B, Huang X, Mason BN, Wattiez AS, Kuburas A, Walker CS, Yang P, Yu J, Heinz BA, Johnson KW, & Russo AF (2017, Jun). CGRP receptor activity in mice with global expression of human receptor activity modifying protein 1. Br J Pharmacol, 174(12), 1826–1840. 10.1111/bph.13783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booe JM, Warner ML, Roehrkasse AM, Hay DL, & Pioszak AA (2018, Apr). Probing the Mechanism of Receptor Activity-Modifying Protein Modulation of GPCR Ligand Selectivity through Rational Design of Potent Adrenomedullin and Calcitonin Gene-Related Peptide Antagonists. Mol Pharmacol, 93(4), 355–367. 10.1124/mol.117.110916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bower RL, Yule L, Rees TA, Deganutti G, Hendrikse ER, Harris PWR, Kowalczyk R, Ridgway Z, Wong AG, Swierkula K, Raleigh DP, Pioszak AA, Brimble MA, Reynolds CA, Walker CS, & Hay DL (2018, Sep 14). Molecular Signature for Receptor Engagement in the Metabolic Peptide Hormone Amylin. ACS Pharmacol Transl Sci, 1(1), 32–49. 10.1021/acsptsci.8b00002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Alexander S, Cirino G, Docherty JR, George CH, Giembycz MA, Hoyer D, Insel PA, Izzo AA, Ji Y, MacEwan DJ, Sobey CG, Stanford SC, Teixeira MM, Wonnacott S, & Ahluwalia A (2018, Apr). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. Br J Pharmacol, 175(7), 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doods H, Hallermayer G, Wu D, Entzeroth M, Rudolf K, Engel W, & Eberlein W (2000, Feb). Pharmacological profile of BIBN4096BS, the first selective small molecule CGRP antagonist. Br J Pharmacol, 129(3), 420–423. 10.1038/sj.bjp.0703110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvinsson L, Haanes KA, Warfvinge K, & Krause DN (2018, Jun). CGRP as the target of new migraine therapies - successful translation from bench to clinic. Nat Rev Neurol, 14(6), 338–350. 10.1038/s41582-018-0003-1 [DOI] [PubMed] [Google Scholar]
- Enebo LB, Berthelsen KK, Kankam M, Lund MT, Rubino DM, Satylganova A, & Lau DCW (2021, May 8). Safety, tolerability, pharmacokinetics, and pharmacodynamics of concomitant administration of multiple doses of cagrilintide with semaglutide 2.4 mg for weight management: a randomised, controlled, phase 1b trial. Lancet, 397(10286), 1736–1748. 10.1016/S0140-6736(21)00845-X [DOI] [PubMed] [Google Scholar]
- Garelja ML, Au M, Brimble MA, Gingell JJ, Hendrikse ER, Lovell A, Prodan N, Sexton PM, Siow A, Walker CS, Watkins HA, Williams GM, Wootten D, Yang SH, Harris PWR, & Hay DL (2020, Apr 10). Molecular Mechanisms of Class B GPCR Activation: Insights from Adrenomedullin Receptors. ACS Pharmacol Transl Sci, 3(2), 246–262. 10.1021/acsptsci.9b00083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanizada H, Al-Karagholi MA, Arngrim N, Morch-Rasmussen M, Walker CS, Hay DL, & Ashina M (2021a, May 18). Effect of Adrenomedullin on Migraine-Like Attacks in Patients With Migraine: A Randomized Crossover Study. Neurology, 96(20), e2488–e2499. 10.1212/WNL.0000000000011930 [DOI] [PubMed] [Google Scholar]
- Ghanizada H, Al-Karagholi MA, Walker CS, Arngrim N, Rees T, Petersen J, Siow A, Morch-Rasmussen M, Tan S, O’Carroll SJ, Harris P, Skovgaard LT, Jorgensen NR, Brimble M, Waite JS, Rea BJ, Sowers LP, Russo AF, Hay DL, & Ashina M (2021b, Jun). Amylin Analog Pramlintide Induces Migraine-like Attacks in Patients. Ann Neurol, 89(6), 1157–1171. 10.1002/ana.26072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingell JJ, Rees TA, Hendrikse ER, Siow A, Rennison D, Scotter J, Harris PWR, Brimble MA, Walker CS, & Hay DL (2020, Apr 10). Distinct Patterns of Internalization of Different Calcitonin Gene-Related Peptide Receptors. ACS Pharmacol Transl Sci, 3(2), 296–304. 10.1021/acsptsci.9b00089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay DL, Christopoulos G, Christopoulos A, Poyner DR, & Sexton PM (2005, May). Pharmacological discrimination of calcitonin receptor: receptor activity-modifying protein complexes. Mol Pharmacol, 67(5), 1655–1665. 10.1124/mol.104.008615 [DOI] [PubMed] [Google Scholar]
- Hay DL, Garelja ML, Poyner DR, & Walker CS (2018, Jan). Update on the pharmacology of calcitonin/CGRP family of peptides: IUPHAR Review 25. Br J Pharmacol, 175(1), 3–17. 10.1111/bph.14075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay DL, Howitt SG, Conner AC, Schindler M, Smith DM, & Poyner DR (2003, Oct). CL/RAMP2 and CL/RAMP3 produce pharmacologically distinct adrenomedullin receptors: a comparison of effects of adrenomedullin22-52, CGRP8-37 and BIBN4096BS. Br J Pharmacol, 140(3), 477–486. 10.1038/sj.bjp.0705472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrikse ER, Liew LP, Bower RL, Bonnet M, Jamaluddin MA, Prodan N, Richards KD, Walker CS, Pairaudeau G, Smith DM, Rujan RM, Sudra R, Reynolds CA, Booe JM, Pioszak AA, Flanagan JU, Hay MP, & Hay DL (2020, Apr 10). Identification of Small-Molecule Positive Modulators of Calcitonin-like Receptor-Based Receptors. ACS Pharmacol Transl Sci, 3(2), 305–320. 10.1021/acsptsci.9b00108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksen K, Broekhuizen K, de Boon WMI, Karsdal MA, Bihlet AR, Christiansen C, Dillingh MR, de Kam M, Kumar R, Burggraaf K, & Kamerling IMC (2021, May 21). Safety, tolerability and pharmacokinetic characterisation of DACRA KBP-042 in healthy male subjects. Br J Clin Pharmacol. 10.1111/bcp.14921 [DOI] [PubMed] [Google Scholar]
- Hong Y, Hay DL, Quirion R, & Poyner DR (2012, May). The pharmacology of adrenomedullin 2/intermedin. Br J Pharmacol, 166(1), 110–120. 10.1111/j.1476-5381.2011.01530.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husmann K, Born W, Fischer JA, & Muff R (2003, Dec 1). Three receptor-activity-modifying proteins define calcitonin gene-related peptide or adrenomedullin selectivity of the mouse calcitonin-like receptor in COS-7 cells. Biochem Pharmacol, 66(11), 2107–2115. 10.1016/j.bcp.2003.07.009 [DOI] [PubMed] [Google Scholar]
- Husmann K, Sexton PM, Fischer JA, & Born W (2000, Apr 25). Mouse receptor-activity-modifying proteins 1, -2 and -3: amino acid sequence, expression and function. Mol Cell Endocrinol, 162(1-2), 35–43. 10.1016/s0303-7207(00)00212-4 [DOI] [PubMed] [Google Scholar]
- Irimia P, Martinez-Valbuena I, Minguez-Olaondo A, Dominguez-Vivero C, Sanchez-Arias JA, Martinez-Vila E, Luquin MR, & Leira R (2021, Apr). Interictal amylin levels in chronic migraine patients: A case-control study. Cephalalgia, 41(5), 604–612. 10.1177/0333102420977106 [DOI] [PubMed] [Google Scholar]
- Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M, Furlotte NA, Eskin E, Nellaker C, Whitley H, Cleak J, Janowitz D, Hernandez-Pliego P, Edwards A, Belgard TG, Oliver PL, McIntyre RE, Bhomra A, Nicod J, Gan X, Yuan W, van der Weyden L, Steward CA, Bala S, Stalker J, Mott R, Durbin R, Jackson IJ, Czechanski A, Guerra-Assuncao JA, Donahue LR, Reinholdt LG, Payseur BA, Ponting CP, Birney E, Flint J, & Adams DJ (2011, Sep 14). Mouse genomic variation and its effect on phenotypes and gene regulation. Nature, 477(7364), 289–294. 10.1038/nature10413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikumoto K, Katafuchi T, & Minamino N (2003, Feb). Specificity of porcine calcitonin receptor and calcitonin receptor-like receptor in the presence of receptor-activity-modifying proteins. Hypertens Res, 26 Suppl, S15–23. 10.1291/hypres.26.s15 [DOI] [PubMed] [Google Scholar]
- Kita T, Kaji Y, & Kitamura K (2020). Safety, Tolerability, and Pharmacokinetics of Adrenomedullin in Healthy Males: A Randomized, Double-Blind, Phase 1 Clinical Trial. Drug Des Devel Ther, 14, 1–11. 10.2147/DDDT.S225220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koller D, Ittner LM, Muff R, Husmann K, Fischer JA, & Born W (2004, May 7). Selective inactivation of adrenomedullin over calcitonin gene-related peptide receptor function by the deletion of amino acids 14-20 of the mouse calcitonin-like receptor. J BiolChem, 279(19), 20387–20391. 10.1074/jbc.M313058200 [DOI] [PubMed] [Google Scholar]
- Kuwasako K, Kitamura K, Onitsuka H, Uemura T, Nagoshi Y, Kato J, & Eto T (2002, May 22). Rat RAMP domains involved in adrenomedullin binding specificity. FEBS Lett, 519(1-3), 113–116. 10.1016/s0014-5793(02)02721-7 [DOI] [PubMed] [Google Scholar]
- Liang YL, Belousoff MJ, Fletcher MM, Zhang X, Khoshouei M, Deganutti G, Koole C, Furness SGB, Miller LJ, Hay DL, Christopoulos A, Reynolds CA, Danev R, Wootten D, & Sexton PM (2020, Apr 10). Structure and Dynamics of Adrenomedullin Receptors AM1 and AM2 Reveal Key Mechanisms in the Control of Receptor Phenotype by Receptor Activity-Modifying Proteins. ACS Pharmacol Transl Sci, 3(2), 263–284. 10.1021/acsptsci.9b00080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang YL, Khoshouei M, Deganutti G, Glukhova A, Koole C, Peat TS, Radjainia M, Plitzko JM, Baumeister W, Miller LJ, Hay DL, Christopoulos A, Reynolds CA, Wootten D, & Sexton PM (2018, Sep). Cryo-EM structure of the active, Gs-protein complexed, human CGRP receptor. Nature, 561(7724), 492–497. 10.1038/s41586-018-0535-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang YL, Khoshouei M, Radjainia M, Zhang Y, Glukhova A, Tarrasch J, Thal DM, Furness SGB, Christopoulos G, Coudrat T, Danev R, Baumeister W, Miller LJ, Christopoulos A, Kobilka BK, Wootten D, Skiniotis G, & Sexton PM (2017, Jun 1). Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature, 546(7656), 118–123. 10.1038/nature22327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason BN, Kaiser EA, Kuburas A, Loomis MM, Latham JA, Garcia-Martinez LF, & Russo AF (2017, Jan 4). Induction of Migraine-Like Photophobic Behavior in Mice by Both Peripheral and Central CGRP Mechanisms. J Neurosci, 37(1), 204–216. 10.1523/JNEUROSCI.2967-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore E, Fraley ME, Bell IM, Burgey CS, White RB, Li CC, Regan CP, Danziger A, Stranieri Michener M, Hostetler E, Banerjee P, & Salvatore C (2020, Jan 28). Characterization of Ubrogepant: A Potent and Selective Antagonist of the Human Calcitonin Gene-Related Peptide Receptor. J Pharmacol Exp Ther, 373(1), 160–166. 10.1124/jpet.119.261065 [DOI] [PubMed] [Google Scholar]
- Moore EL, Gingell JJ, Kane SA, Hay DL, & Salvatore CA (2010, Mar 26). Mapping the CGRP receptor ligand binding domain: tryptophan-84 of RAMP1 is critical for agonist and antagonist binding. Biochem Biophys Res Commun, 394(1), 141–145. 10.1016/j.bbrc.2010.02.131 [DOI] [PubMed] [Google Scholar]
- Morfis M, Tilakaratne N, Furness SG, Christopoulos G, Werry TD, Christopoulos A, & Sexton PM (2008, Nov). Receptor activity-modifying proteins differentially modulate the G protein-coupling efficiency of amylin receptors. Endocrinology, 149(11), 5423–5431. 10.1210/en.2007-1735 [DOI] [PubMed] [Google Scholar]
- Musa H, Hendrikse ER, Brimble MA, Garelja ML, Watkins HA, Harris PWR, & Hay DL (2019, Aug 13). Pharmacological Characterization and Investigation of N-Terminal Loop Amino Acids of Adrenomedullin 2 That Are Important for Receptor Activation. Biochemistry, 58(32), 3468–3474. 10.1021/acs.biochem.9b00571 [DOI] [PubMed] [Google Scholar]
- Oliver KR, Kane SA, Salvatore CA, Mallee JJ, Kinsey AM, Koblan KS, Keyvan-Fouladi N, Heavens RP, Wainwright A, Jacobson M, Dickerson IM, & Hill RG (2001, Aug). Cloning, characterization and central nervous system distribution of receptor activity modifying proteins in the rat. Eur J Neurosci, 14(4), 618–628. 10.1046/j.0953-816x.2001.01688.x [DOI] [PubMed] [Google Scholar]
- Owji AA, Chabot JG, Dumont Y, & Quirion R (2008, Jul). Adrenomedullin-2/intermedin induces cAMP accumulation in dissociated rat spinal cord cells: evidence for the existence of a distinct class of sites of action. J Mol Neurosci, 35(3), 355–361. 10.1007/s12031-008-9062-x [DOI] [PubMed] [Google Scholar]
- Pan KS, Siow A, Hay DL, & Walker CS (2020). Antagonism of CGRP Signaling by Rimegepant at Two Receptors. Front Pharmacol, 11, 1240. 10.3389/fphar.2020.01240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham V, Zhu Y, Dal Maso E, Reynolds CA, Deganutti G, Atanasio S, Hick CA, Yang D, Christopoulos A, Hay DL, Furness SGB, Wang MW, Wootten D, & Sexton PM (2019, Jun 14). Deconvoluting the Molecular Control of Binding and Signaling at the Amylin 3 Receptor: RAMP3 Alters Signal Propagation through Extracellular Loops of the Calcitonin Receptor. ACS Pharmacol Transl Sci, 2(3), 183–197. 10.1021/acsptsci.9b00010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poyner DR, Sexton PM, Marshall I, Smith DM, Quirion R, Born W, Muff R, Fischer JA, & Foord SM (2002, Jun). International Union of Pharmacology. XXXII. The mammalian calcitonin gene-related peptides, adrenomedullin, amylin, and calcitonin receptors. Pharmacol Rev, 54(2), 233–246. 10.1124/pr.54.2.233 [DOI] [PubMed] [Google Scholar]
- Ray JC, Kapoor M, Stark RJ, Wang SJ, Bendtsen L, Matharu M, & Hutton EJ (2021, Jan 25). Calcitonin gene related peptide in migraine: current therapeutics, future implications and potential off-target effects. J Neurol Neurosurg Psychiatry. 10.1136/jnnp-2020-324674 [DOI] [PubMed] [Google Scholar]
- Recober A, Kuburas A, Zhang Z, Wemmie JA, Anderson MG, & Russo AF (2009, Jul 8). Role of calcitonin gene-related peptide in light-aversive behavior: implications for migraine. J Neurosci, 29(27), 8798–8804. 10.1523/JNEUROSCI.1727-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvatore CA, Hershey JC, Corcoran HA, Fay JF, Johnston VK, Moore EL, Mosser SD, Burgey CS, Paone DV, Shaw AW, Graham SL, Vacca JP, Williams TM, Koblan KS, & Kane SA (2008, Feb). Pharmacological characterization of MK-0974 [N-[(3R,6S)-6-(2,3-difluorophenyl)-2-oxo-1-(2,2,2-trifluoroethyl)azepan-3-yl]-4-(2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-1-yl)piperidine-1-carboxamide], a potent and orally active calcitonin gene-related peptide receptor antagonist for the treatment of migraine. J Pharmacol Exp Ther, 324(2), 416–421. 10.1124/jpet.107.130344 [DOI] [PubMed] [Google Scholar]
- Steiner TJ, Stovner LJ, & Birbeck GL (2013, Apr). Migraine: the seventh disabler. Cephalalgia, 33(5), 289–290. 10.1177/0333102412473843 [DOI] [PubMed] [Google Scholar]
- Tilakaratne N, Christopoulos G, Zumpe ET, Foord SM, & Sexton PM (2000, Jul). Amylin receptor phenotypes derived from human calcitonin receptor/RAMP coexpression exhibit pharmacological differences dependent on receptor isoform and host cell environment. J Pharmacol Exp Ther, 294(1), 61–72. https://www.ncbi.nlm.nih.gov/pubmed/10871296 [PubMed] [Google Scholar]
- Udawela M, Christopoulos G, Morfis M, Christopoulos A, Ye S, Tilakaratne N, & Sexton PM (2006, Nov). A critical role for the short intracellular C terminus in receptor activity-modifying protein function. Mol Pharmacol, 70(5), 1750–1760. 10.1124/mol.106.024257 [DOI] [PubMed] [Google Scholar]
- Vanhoutte PM, Humphrey PP, & Spedding M (1996, Mar). X. International Union of Pharmacology recommendations for nomenclature of new receptor subtypes. Pharmacol Rev, 48(1), 1–2. https://www.ncbi.nlm.nih.gov/pubmed/8685244 [PubMed] [Google Scholar]
- Vuralli D, Wattiez AS, Russo AF, & Bolay H (2019, Jan 31). Behavioral and cognitive animal models in headache research. J Headache Pain, 20(1), 11. 10.1186/s10194-019-0963-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker CS, Eftekhari S, Bower RL, Wilderman A, Insel PA, Edvinsson L, Waldvogel HJ, Jamaluddin MA, Russo AF, & Hay DL (2015, Jun). A second trigeminal CGRP receptor: function and expression of the AMY1 receptor. Ann Clin Transl Neurol, 2(6), 595–608. 10.1002/acn3.197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker CS, Raddant AC, Woolley MJ, Russo AF, & Hay DL (2018, Mar). CGRP receptor antagonist activity of olcegepant depends on the signalling pathway measured. Cephalalgia, 38(3), 437–451. 10.1177/0333102417691762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins HA, Chakravarthy M, Abhayawardana RS, Gingell JJ, Garelja M, Pardamwar M, McElhinney JM, Lathbridge A, Constantine A, Harris PW, Yuen TY, Brimble MA, Barwell J, Poyner DR, Woolley MJ, Conner AC, Pioszak AA, Reynolds CA, & Hay DL (2016, May 27). Receptor Activity-modifying Proteins 2 and 3 Generate Adrenomedullin Receptor Subtypes with Distinct Molecular Properties. J Biol Chem, 291(22), 11657–11675. 10.1074/jbc.M115.688218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wattiez AS, Wang M, & Russo AF (2019, 2020 Feb 9). CGRP in Animal Models of Migraine. Handbook of Experimental Pharmacology, 255, 85–107. 10.1007/164_2018_187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston C, Winfield I, Harris M, Hodgson R, Shah A, Dowell SJ, Mobarec JC, Woodlock DA, Reynolds CA, Poyner DR, Watkins HA, & Ladds G (2016, Oct 14). Receptor Activity-modifying Protein-directed G Protein Signaling Specificity for the Calcitonin Gene-related Peptide Family of Receptors. J Biol Chem, 291(42), 21925–21944. 10.1074/jbc.M116.751362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolley MJ, Reynolds CA, Simms J, Walker CS, Mobarec JC, Garelja ML, Conner AC, Poyner DR, & Hay DL (2017, Oct 15). Receptor activity-modifying protein dependent and independent activation mechanisms in the coupling of calcitonin gene-related peptide and adrenomedullin receptors to Gs. Biochem Pharmacol, 142, 96–110. 10.1016/j.bcp.2017.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yule LR, Bower RL, Kaur H, Kowalczyk R, Hay DL, & Brimble MA (2016, Jun 21). Synthesis and amylin receptor activity of glycomimetics of pramlintide using click chemistry. Org Biomol Chem, 14(23), 5238–5245. 10.1039/c6ob00850j [DOI] [PubMed] [Google Scholar]
- Yule LR, Garelja ML, Hendrikse ER, Gingell JJ, Poyner DR, Harris PWH, Brimble MA, & Hay DL (2019, Nov). A potent fluorescent calcitonin gene-related peptide analogue enables visualization of receptor internalization. Peptide Science, 111(6), e24126. https://doi.org/ARTN e24126 10.1002/pep2.24126 [DOI] [Google Scholar]
- Zakariassen HL, John LM, & Lutz TA (2020, Sep). Central control of energy balance by amylin and calcitonin receptor agonists and their potential for treatment of metabolic diseases. Basic Clin Pharmacol Toxicol, 127(3), 163–177. 10.1111/bcpt.13427 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Liu X, Morgan DA, Kuburas A, Thedens DR, Russo AF, & Rahmouni K (2011, Apr). Neuronal receptor activity-modifying protein 1 promotes energy expenditure in mice. Diabetes, 60(4), 1063–1071. 10.2337/db10-0692 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.