

Abstract
Introduction
Baricitinib, an oral Janus kinase (JAK)1/JAK2 inhibitor, is indicated in the European Union and Japan for treatment of moderate-to-severe atopic dermatitis (AD) in adults who are candidates for systemic therapy. In the ongoing, placebo-controlled, phase 3 trial BREEZE-AD5, once-daily oral baricitinib 2-mg monotherapy improved disease in moderate-to-severe AD patients who had an inadequate response or intolerance to topical corticosteroids. This post-hoc analysis aimed to identify responders to baricitinib 2 mg, using a proposed clinical tailoring approach based on baseline body surface area (BSA) affected and early clinical improvement, in BREEZE-AD5.
Methods
Classification and regression tree method was used to evaluate baseline predictors for the proportion of patients achieving ≥ 75% improvement in Eczema Area and Severity Index (EASI75) at week 16 among baricitinib 2-mg-treated patients. Two-by-two contingency tables evaluated the association between early response, defined as ≥ 50% improvement in BSA or ≥ 3-point improvement in Itch Numeric Rating Scale from baseline at weeks 4 or 8, and response at week 16 for the proportion of patients achieving EASI75, validated Investigator Global Assessment for AD (vIGA-AD) score of 0 or 1, or ≥ 4-point improvement in Itch (Itch ≥ 4), respectively. Missing data were imputed as non-responder.
Results
At week 16, EASI75 and vIGA-AD (0,1) were achieved by 37.5% and 31.7% of baricitinib 2-mg-treated patients with baseline BSA 10–50% compared with 9.5% and 4.8% with BSA > 50%. Early response in skin inflammation or itch at week 4 was associated with corresponding EASI75, vIGA-AD (0,1), and Itch ≥ 4 of 55.4%, 48.2%, and 39.3% at week 16, while early response at week 8 was associated with 66.7%, 56.1%, and 42.1% of patients achieving these endpoints.
Conclusion
Baseline BSA of 10–50% and early clinical improvement after 4 or 8 weeks of baricitinib 2-mg treatment may identify patients most likely to benefit from long-term baricitinib 2-mg therapy.
Clinical Trial Registration
Supplementary Information
The online version contains supplementary material available at 10.1007/s13555-021-00640-7.
Keywords: Atopic dermatitis, Baricitinib, Body surface area, Clinical tailoring, Clinical trial, Janus kinase inhibitor
Plain Language Summary
Baricitinib is a medication that helps a dysregulated immune system readjust. This leads to improvements in the inflammatory disease atopic dermatitis (AD). Baricitinib is approved for adults with moderate-to-severe AD in over 40 countries. In the ongoing study BREEZE-AD5, baricitinib 2 mg improved moderate-to-severe AD in patients who previously did not respond to or could not tolerate topical corticosteroids. Understanding which patients are likely to benefit most from a medication can improve patient experience with treatment. It can also ensure that only patients who are likely to benefit from a medication are exposed to it. This analysis aimed to identify patients who are most likely to benefit from baricitinib 2 mg in BREEZE-AD5, using an approach based on baseline body surface area (BSA) affected and early clinical improvement. We showed that patients with moderate-to-severe AD affecting between 10% and 50% of their BSA account for the majority of patients who respond to baricitinib 2 mg after 16 weeks of treatment. Clinical assessment of skin inflammation or itch in patients after 4–8 weeks of initiation of baricitinib 2-mg treatment further improved the ability to identify patients who are most likely to benefit from long-term therapy. This proposed clinical tailoring approach of baseline BSA of 10–50% and early clinical improvement after 4 or 8 weeks of baricitinib 2-mg treatment may allow for the treatment of patients who are most likely to respond to therapy, and rapid decision on discontinuation of treatment for those who are not likely to benefit from baricitinib 2 mg.
Supplementary Information
The online version contains supplementary material available at 10.1007/s13555-021-00640-7.
Video Abstract (MP4 25585 kb)
Key Summary Points
| Why carry out this study? |
| Baricitinib, an oral selective Janus kinase (JAK)1/JAK2 inhibitor, is indicated in Europe and Japan for the treatment of moderate-to-severe atopic dermatitis (AD) in adult patients who are candidates for systemic therapy. |
| Understanding which patients are likely to benefit most from therapy can improve patient experience with treatment, increase the cost-effectiveness of a therapy, and ensure that only patients who are likely to benefit from therapy are exposed to drug. |
| This post-hoc analysis aimed to identify patients who are likely to benefit from baricitinib 2 mg, using a clinical tailoring approach based on baseline body surface area (BSA) affected and early clinical improvement, in the phase 3 monotherapy trial BREEZE-AD5. |
| What was learned from the study? |
| Patients with moderate-to-severe AD affecting between 10% and 50% of their BSA account for the majority of week 16 responders to baricitinib 2 mg, and clinical assessment of patients after 4–8 weeks of initiation of baricitinib 2-mg treatment identified those who are likely to benefit from long-term baricitinib 2-mg therapy. |
| The proposed clinical tailoring approach for baricitinib 2 mg allows for the treatment of patients who are more likely to respond to therapy, and rapid decision on discontinuation of treatment for those who are not likely to benefit from baricitinib 2 mg. |
Digital Features
This article is published with digital features, including a video abstract, to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.16989175.
Introduction
Atopic dermatitis (AD) is a common, chronic, inflammatory skin disease characterized by complex pathophysiology and heterogeneous clinical phenotypes [1–3]. AD symptoms include intense itch, sleep disturbance, and skin pain, which significantly impact patient quality of life and work productivity [4–6]. Emollients and topical corticosteroids (TCS) are the mainstay of AD therapy [7–9]. In patients with moderate-to-severe AD for whom topical therapy does not adequately control the signs and symptoms of disease, addition of phototherapy and/or systemic treatment is recommended [10, 11]. Systemic treatments can be associated with variable efficacy and unnecessary cycling through ineffective medications resulting in prolonged burden and delayed improvements [10, 12]. Therefore, prediction of treatment efficacy is becoming increasingly important with the emergence of new therapies. An understanding of which patients are most likely to benefit from therapy can help tailor therapies to individual patient needs. Tailored therapeutic approaches can significantly refine the management of AD through improving patient experience with a treatment, increasing cost-effectiveness of a therapy, and ensuring that only patients who are likely to benefit from therapy are exposed to a given treatment.
Baricitinib, an oral selective Janus kinase (JAK) 1 and JAK2 inhibitor [13], is indicated in the European Union and Japan and being evaluated in the USA and other countries for treatment of moderate-to-severe AD in adult patients who are candidates for systemic therapy. Dependent upon country-specific approvals, the recommended starting dose of baricitinib for adults with moderate-to-severe AD is 2 mg or 4 mg [14–16]. In BREEZE-AD5, an ongoing, randomized, placebo-controlled, phase 3 trial of moderate-to-severe AD patients who had an inadequate response or intolerance to TCS, once-daily oral baricitinib 2-mg monotherapy improved several clinical signs and symptoms of AD at week 16 compared with placebo, with a safety profile consistent with previous studies of baricitinib 2 mg in AD [17, 18]. The objective of this post-hoc analysis was to identify patients who are most likely to benefit from baricitinib 2 mg, using a proposed clinical tailoring approach based on baseline body surface area (BSA) affected at drug initiation and early clinical improvement, in the phase 3 monotherapy trial BREEZE-AD5.
Methods
Study Design and Patients
BREEZE-AD5 (NCT03435081) is an ongoing phase 3 randomized, double-blind, placebo-controlled trial conducted in the USA and Canada [17]. Patients (N = 440) were randomized 1:1:1 to receive once-daily placebo, baricitinib 1 mg, or baricitinib 2 mg (Fig. S1). Eligible patients were ≥ 18 years of age and had a diagnosis of AD, as defined by the American Academy of Dermatology [19], ≥ 12 months prior to screening. Enrolled patients had moderate-to-severe disease, defined by baseline Eczema Area and Severity Index (EASI) score ≥ 16, validated Investigator Global Assessment for Atopic Dermatitis (vIGA-AD) score ≥ 3, and BSA involvement of ≥ 10%. Patients had a documented history of inadequate response or intolerance to topical therapies within 6 months before screening. Patients discontinued topical therapy 2 weeks and systemic therapy 4 weeks before randomization. This trial was conducted in accordance with the ethical principles of the Helsinki Declaration of 1964 and its later amendments and Good Clinical Practice guidelines and approved by the appropriate institutional review boards/ethics committees at each study site (Table S1), including the Quorum Review IRB (approval #33039). All patients provided written informed consent.
Outcomes
The primary endpoint of the trial was the proportion of patients who achieved 75% improvement in EASI score (EASI75) at week 16. Two secondary endpoints included the proportion of patients who achieved a vIGA-AD score of 0 (clear) or 1 (almost clear), with a ≥ 2-point improvement from baseline, and the proportion of patients who achieved a ≥ 4-point improvement from baseline in the Itch Numeric Rating Scale (NRS) [17]. These endpoints were analyzed in this post-hoc analysis in the subgroup of patients most likely to benefit from baricitinib 2-mg treatment.
Statistical Analyses
In this post-hoc analysis, classification and regression tree algorithm (CART) was used to identify baseline predictors of response. A variety of baseline measures were assessed, including, but not limited to patient demographics, prior treatment history, baseline disease severity [such as vIGA-AD, EASI, BSA, Itch NRS, Atopic Dermatitis Sleep Scale, Dermatology Life Quality Index (DLQI), SCORing Atopic Dermatitis, Hospital Anxiety Depression Scale], and laboratory parameters. Response was defined as the proportion of patients achieving EASI75 at week 16 among those treated with baricitinib 2 mg in the intent-to-treat (ITT) population. Two-by-two contingency tables with associated sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV) were generated between CART-identified baseline predictors and EASI75. As a sensitivity analysis, the same method was applied to identify baseline predictors for the proportion of patients who achieved vIGA-AD (0,1) at week 16. Sensitivity and NPV were examined closely. Sensitivity represents the proportion of patients with certain baseline characteristics, among those who were responders at week 16. A high sensitivity value indicates that most responders were identified by characteristics through the algorithm. NPV represents the proportion of patients who were not responders at week 16, among those without the same baseline characteristics. A high NPV indicates that patients in this group would likely not benefit from therapy. After baseline subgroups were identified using the CART method, response rate for EASI75, vIGA-AD (0,1), and ≥ 4-point improvement in Itch NRS by visits were analyzed with logistics regression models. Baseline disease severity (vIGA-AD), continuous version of endpoint at baseline, treatment group, baseline subgroup, and treatment-by-baseline subgroup interaction were included as model terms.
After identification of a baseline predictor of response, further analyses were conducted to determine if early response could be used to further refine the patient selection. The goal of this second analysis was to quickly identify any additional patients who are less likely to benefit from therapy and allow for quick clinical decision on whether to transition such patients to alternative therapies. Two-by-two contingency tables were used to evaluate the association between early response in skin inflammation (≥ 50% improvement in BSA) or itch (≥ 3-point improvement in Itch NRS) at week 4 or week 8, and response at week 16 for EASI75, vIGA-AD (0,1), and ≥ 4-point improvement in Itch NRS. Early measures of improvement (≥ 50% improvement in BSA and ≥ 3-point improvement in Itch NRS) were selected for being considered a meaningful improvement, easily noticeable by patients and physicians, and applicable in clinical practice [20]. EASI75, vIGA-AD (0,1), and ≥ 4-point improvement in Itch NRS are accepted regulatory endpoints in AD, which allowed for comparison between the ITT population results and this subgroup analysis. Sensitivity, specificity, PPV, and NPV were used to evaluate the performance of the two-by-two classification table.
Data collected after any rescue or treatment discontinuation were considered as missing. All missing data were imputed as non-responder. To use the same ITT population for evaluating skin inflammation and itch simultaneously in association with early improvement and week 16 response, a conservative approach to define improver and responder was used: any patient with a baseline Itch NRS score < 3 was automatically classified as a non-improver for ≥ 3-point improvement in Itch NRS at week 4 or 8, and any patient with a baseline Itch NRS score < 4 was automatically classified as a non-responder for ≥ 4-point improvement in Itch NRS at week 16.
Results
Classification and Regression Tree Analysis
Baseline BSA was identified as the strongest predictor of treatment outcome to optimize accuracy, with a cutoff of 50% being optimal for sensitivity and NPV. Application of baseline BSA 10–50% to predict response to baricitinib 2 mg resulted in selection of 90.7% (NPV 90.5%) and 94.3% (NPV 95.2%) of EASI75 and vIGA-AD (0,1) responders at week 16, respectively, in the overall study population.
Baseline Demographics and Disease Severity
Baseline demographics were balanced among the overall ITT population and baseline BSA 10–50% and BSA > 50% subgroups across treatment arms. EASI scores were lower in the BSA 10–50% subgroup compared with the ITT population; however, the overall burden of disease symptoms, including itch, sleep, skin pain, and impact on quality of life, was comparable between the two groups (Table 1).
Table 1.
Baseline demographics and disease severity by baseline BSA category
| BSA 10–50% | ITT population | BSA > 50% | ||||
|---|---|---|---|---|---|---|
| Placebo (N = 101) | Baricitinib 2 mg (N = 104) | Placebo (N = 147) | Baricitinib 2 mg (N = 146) | Placebo (N = 46) | Baricitinib 2 mg (N = 42) | |
| Age (years) | 40.2 (17.2) | 41.2 (14.9) | 39.0 (16.5) | 39.7 (15.0) | 36.4 (14.6) | 36.0 (14.8) |
| Female, n (%) | 47 (46.5) | 65 (62.5) | 67 (45.6) | 77 (52.7) | 20 (43.5) | 12 (28.6) |
| Race, n (%) | ||||||
| White | 58 (57.4) | 59 (56.7) | 80 (54.8) | 85 (58.2) | 22 (47.8) | 26 (61.9) |
| African American | 17 (16.8) | 20 (19.2) | 24 (16.4) | 30 (20.5) | 7 (15.2) | 10 (23.8) |
| Asian | 20 (19.8) | 17 (16.3) | 33 (22.6) | 22 (15.1) | 13 (28.3) | 5 (11.9) |
| Other | 6 (5.9) | 8 (7.7) | 9 (6.2) | 9 (6.2) | 3 (6.5) | 1 (2.4) |
| Duration since AD diagnosis (years) | 20.2 (16.4) | 23.9 (16.1) | 22.8 (16.8) | 23.9 (15.9) | 28.6 (16.6) | 24.0 (15.6) |
| vIGA-AD score of 4a, n (%) | 30 (29.7) | 30 (28.8) | 61 (41.5) | 61 (41.8) | 31 (67.4) | 31 (73.8) |
| EASIb | 21.5 (5.7) | 20.9 (4.7) | 27.0 (10.8) | 26.6 (11.4) | 39.0 (9.6) | 40.8 (10.6) |
| Body surface area affected by AD | 27.9 (10.9) | 27.7 (10.7) | 41.5 (23.1) | 39.7 (22.0) | 71.5 (11.6) | 69.5 (12.5) |
| Itch NRSc | 6.7 (2.5) | 7.1 (2.3) | 7.0 (2.4) | 7.3 (2.1) | 7.8 (1.8) | 7.8 (1.6) |
| Skin Pain NRSd | 6.3 (2.8) | 5.9 (2.5) | 6.5 (2.7) | 6.7 (2.6) | 7.1 (2.6) | 7.4 (2.2) |
| ADSS Item 2e | 1.9 (2.0) | 2.6 (3.3) | 2.0 (1.9) | 2.8 (3.2) | 2.3 (1.7) | 3.2 (2.8) |
| DLQIf | 13.4 (7.2) | 14.4 (7.4) | 14.7 (7.1) | 15.0 (7.6) | 17.6 (5.9) | 16.3 (7.8) |
| POEMg | 19.2 (6.9) | 21.1 (5.4) | 20.5 (6.5) | 21.7 (5.4) | 23.3 (4.2) | 23.1 (5.1) |
Data are mean (SD) unless otherwise indicated
AD atopic dermatitis, ADSS Atopic Dermatitis Sleep Scale, BSA body surface area, DLQI Dermatology Life Quality Index, EASI Eczema Area and Severity Index, ITT Intent-to-Treat, N number of subjects in the analysis population, n number of subjects in the specified category, NRS Numeric Rating Scale, POEM Patient Oriented Eczema Measures, SD standard deviation, vIGA-AD validated Investigator Global Assessment for Atopic Dermatitis
avIGA-AD measures the investigator global assessment of disease severity based on a static 5-point scale from 0 (clear skin) to 4 (severe disease)
bEASI scores range from 0 to 72, with higher scores indicating greater severity
cItch NRS ranges from 0 (no itch) to 10 (worst itch imaginable)
dSkin Pain NRS ranges from 0 (no pain) to 10 (worst pain imaginable)
eADSS Item 2 assesses the frequency of nighttime awakenings due to itch the previous night on a scale of 0–29
fDLQI evaluates health-related quality of life on a scale of 0–30
gPOEM is a composite measure of patient-reported symptoms, including the effect of symptoms on sleep, and evaluates the frequency of symptoms (including itch) and the effect of atopic dermatitis on sleep on a scale of 0–28
Clinical Response among Patients with Baseline BSA 10–50%
We evaluated the impact of baseline BSA on clinical responses to baricitinib 2-mg monotherapy in the ITT population over time. Baricitinib 2 mg resulted in a significantly higher proportion of patients achieving EASI75 and vIGA-AD (0,1) at week 16 in those with baseline BSA 10–50%, but not in BSA > 50% (Fig. 1A and B). Among patients with baseline BSA 10–50%, EASI75 and vIGA-AD (0,1) were achieved at week 16 in 37.5% and 31.7% with baricitinib 2 mg, compared with 9.9% and 6.9% with placebo (p ≤ 0.001 for both). In contrast, in patients with baseline BSA > 50%, EASI75 and IGA (0,1) responses at week 16 were only achieved in 9.5% and 4.8% with baricitinib 2 mg, compared with 4.3% and 2.2% with placebo (p = not significant for both) (Fig. 1A and B). Response rates for itch were also slightly higher in patients with baseline BSA 10–50% treated with baricitinib 2 mg (Fig. 1C).
Fig. 1.
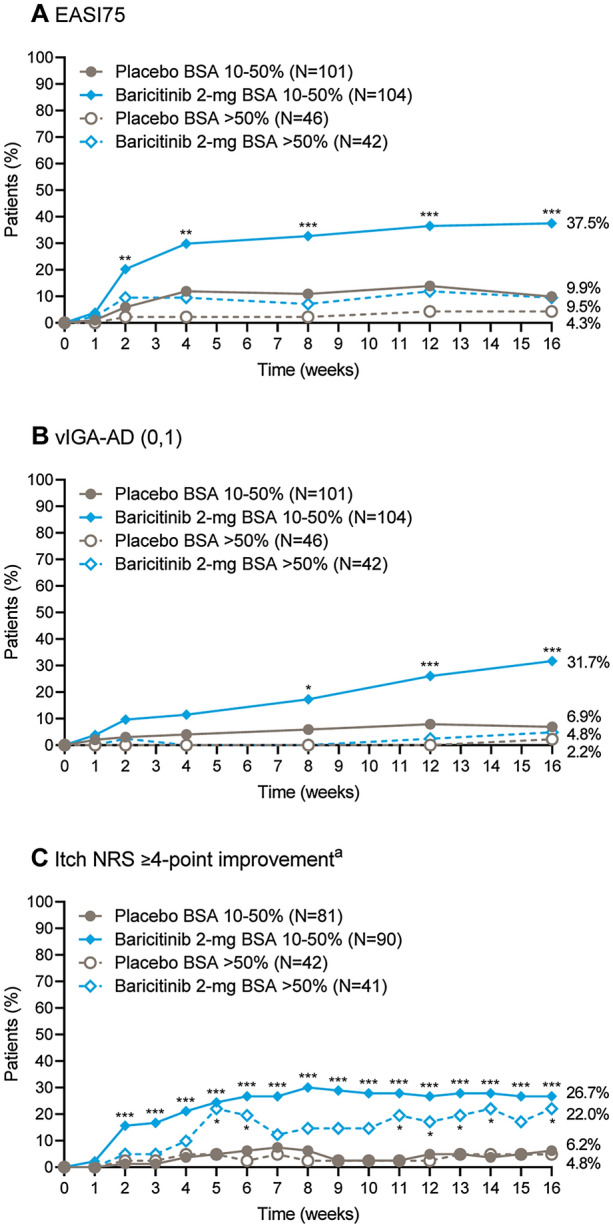
Clinical response by baseline BSA category. Proportion of patients achieving A a 75% improvement in total EASI score, B a vIGA-AD score of 0 or 1, or C a ≥ 4-point improvement in the Itch NRS response over time, among patients who had a baseline BSA of 10–50% or > 50%. aAssessed for patients with a baseline Itch NRS score ≥ 4. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 for baricitinib 2 mg compared with placebo. BSA body surface area, EASI75 75% improvement in Eczema Area and Severity Index score, NRS Numeric Rating Scale, vIGA-AD (0,1) validated Investigator Global Assessment for Atopic Dermatitis score of 0 or 1
Early meaningful response in skin inflammation (≥ 50% improvement in BSA) or itch (≥ 3-point improvement in Itch NRS) at week 8 or week 4, was able to further refine which patients were most likely to benefit from baricitinib 2-mg therapy longer term (Fig. 2). Among baricitinib 2-mg-treated patients with baseline BSA 10–50%, early response in skin inflammation or itch at week 8 was associated with EASI75, vIGA-AD (0,1), and ≥ 4-point improvement in Itch NRS of 66.7%, 56.1%, and 42.1% at week 16, compared with 2.1%, 2.1%, and 0% for those who did not have an early response at week 8 (Fig. 2A, C, and E). Assessment of early response in skin inflammation or itch at week 4 was associated with corresponding EASI75, vIGA-AD (0,1), and ≥ 4-point improvement in Itch NRS of 55.4%, 48.2%, and 39.3% at week 16, compared with 16.7%, 12.5%, and 4.2% for those who did not have an early response at week 4 (Fig. 2B, D, and F). High NPVs showed that, without early improvement in skin inflammation or itch, there were almost no EASI75, vIGA-AD (0,1), and ≥ 4-point improvement in Itch NRS responders at week 16 with baricitinib 2-mg treatment (Table 2). Among baricitinib 2-mg-treated patients with baseline BSA 10–50%, early response in skin inflammation or itch was associated with improved clinical response at week 16.
Fig. 2.

Clinical response over time by early response in skin inflammation or itch at week 4 or week 8 among patients with baseline BSA 10–50%. Proportion of patients achieving (A, B) a 75% improvement in total EASI score, (C, D) a vIGA-AD score of 0 or 1, or (E, F) a ≥ 4-point improvement in the Itch NRS response over time, among patients with a baseline BSA of 10–50% by early response at week 8 (A, C, E) or week 4 (B, D, F). Early response in skin inflammation or itch was defined as ≥ 50% improvement in BSA or ≥ 3-point improvement in Itch NRS, respectively. aAssessed for patients with a baseline Itch NRS score ≥ 4. BSA body surface area, EASI75 75% improvement in Eczema Area and Severity Index score, NRS Numeric Rating Scale, vIGA-AD (0,1) validated Investigator Global Assessment for Atopic Dermatitis score of 0 or 1
Table 2.
Two-by-two contingency tables for clinical response at week 16 by early response in skin inflammation or itch at week 4 or week 8
| Week 16 endpoint | Early-response assessment week | Sensitivity | Specificity | Positive predictive value | Negative predictive value |
|---|---|---|---|---|---|
| EASI75 | Week 4 | 0.795 | 0.615 | 0.554 | 0.833 |
| Week 8 | 0.974 | 0.708 | 0.667 | 0.979 | |
| vIGA-AD (0,1) | Week 4 | 0.818 | 0.592 | 0.482 | 0.875 |
| Week 8 | 0.970 | 0.648 | 0.561 | 0.979 | |
| Itch NRS ≥ 4-point improvement | Week 4 | 0.917 | 0.575 | 0.393 | 0.958 |
| Week 8 | 1.000 | 0.588 | 0.421 | 1.000 |
Early response in skin inflammation or itch was defined as ≥ 50% improvement in BSA or ≥ 3-point improvement in Itch NRS, respectively
BSA body surface area, EASI Eczema Area and Severity Index, NRS Numeric Rating Scale, vIGA-AD (0,1) validated Investigator Global Assessment for Atopic Dermatitis score of 0 or 1
Discussion
Personalized medicine has long been a goal in clinical practice [21, 22]. Providers seek to identify which patients are most likely to benefit from treatment in order to increase the probability of success with a given therapy and minimize risk of exposure to an ineffective treatment. In this study, we demonstrated that baricitinib 2 mg provides enhanced efficacy in patients with a baseline BSA ranging from 10% to 50%, with higher response rates for EASI75 and vIGA-AD (0,1), compared with patients with a baseline BSA > 50%.
Early assessment of clinical response was defined as an improvement of at least 50% in BSA or an improvement of at least 3 points in Itch NRS from baseline at week 4 or week 8. Early response in skin inflammation or itch resulted in an additional significant increase in patient response at week 16 among baricitinib 2-mg-treated patients with baseline BSA 10–50%, with as many as two-thirds of patients achieving an EASI75 at week 16 and over half of the patients achieving a vIGA-AD (0,1) at week 16. This is in contrast to 37.5% and 31.7% of responders observed for EASI75 and vIGA-AD (0,1), respectively, in the overall baseline BSA 10–50% group, and 29.5% and 24.0%, respectively, in the ITT population [17]. These high efficacy results were observed while maintaining a very high NPV for the analyses, showing that an early decision for discontinuation of therapy is possible in patients with a BSA of 10–50% at baseline. Patients who respond to baricitinib 2 mg experience clinical benefit early in their course of treatment. Patients who do not respond to baricitinib 2-mg treatment within the first 4–8 weeks are unlikely to respond to treatment in the long term.
Although BSA and associated EASI scores were lower in patients with BSA 10–50% at baseline, overall burden of disease by symptoms was comparable between the groups. Patients’ baseline itch, sleep disturbance due to itch, skin pain, Patient Oriented Eczema Measures scores, and quality of life (DLQI) were comparable between the groups, confirming the significant burden of disease in patients with baseline BSA 10–50%. Although the proposed clinical tailoring approach uses baseline BSA, a small increase in itch response was also observed in patients with baseline BSA 10–50% compared with patients with baseline BSA > 50%. Importantly, 100% of the patients who responded to baricitinib 2 mg with a ≥ 4-point improvement in Itch NRS at week 16 had achieved a ≥ 3-point improvement by week 8 (Table 2, sensitivity = 1.0, NPV = 1.0).
A limitation to this study is that these are post-hoc subgroup analyses as opposed to prespecified subgroup analyses [23]. The analyses focused on the 2-mg dose of baricitinib, and the lack of data from the 4-mg dose is a further limitation. In addition, patients discontinued topical and systemic treatments 2 and 4 weeks prior to randomization, respectively. The discontinuation of topical and systemic therapies weeks prior to the initiation of the study drug may lead to rescue earlier than if therapies were discontinued closer to the initiation of the clinical trial. In addition, this was a monotherapy trial. Although this study helped to better assess the impact of treatment in AD severity without the potential confounding effect of topical corticosteroids, topical calcineurin inhibitors, or topical phosphodiesterase-4 inhibitors, some of these treatments may be maintained in clinical practice during treatment. Other limitations are the lack of standardization in the assessment of BSA [24], as well as the inconsistent performance of a full-body skin examination in clinical practice. The BSA affords a rapid assessment on physical examination that may help to guide clinical decision-making, particularly with the advent of new therapies in the treatment armamentarium for AD.
In conclusion, patients with moderate-to-severe AD affecting between 10% and 50% of their BSA accounted for the majority of responders to baricitinib 2 mg. The clinical assessment of patients after 4–8 weeks of initiation of baricitinib 2-mg treatment predicted which patients are likely to benefit from long-term therapy. This analysis may allow for a precision-medicine approach to therapy in moderate-to-severe AD.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors would like to thank the patients and study investigators who participated in the study.
Funding
The study was sponsored by Eli Lilly and Company, under license from Incyte Corporation. The sponsor funded the journal’s Rapid Service fee.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
Study conception and design: Fabio P. Nunes. Acquisition of data: Jamie Weisman. Analysis and interpretation of data: Mark Boguniewicz, Yuxin Ding, Meghan Feely, Fabio P. Nunes, Jonathan I. Silverberg, Eric L. Simpson, Lindsay Strowd, Luna Sun, and Jill Waibel.
Medical Writing, Editorial, and Other Assistance
Medical writing assistance in preparation of this article was provided by Nicole Byers of Eli Lilly and Company. Support for this assistance was funded by Eli Lilly and Company.
Disclosures
Jonathan I. Silverberg served as a consultant and/or advisory board member for AbbVie, Arena, Asana, Bluefin, Boehringer-Ingelheim, Dermavant, Dermira, Eli Lilly and Company, Galderma, GlaxoSmithKline, Glenmark, Incyte, Kiniksa, Leo, Luna, Novartis, Pfizer, RAPT, Regeneron, Sanofi, receiving honoraria; served as a speaker for Regeneron-Sanofi; and received research grants from Galderma. Mark Boguniewicz served as an investigator for Incyte and Regeneron and a consultant and/or advisory board member for AbbVie, Eli Lilly and Company, Janssen, LEO Pharma, Pfizer, Regeneron, and Sanofi-Genzyme. Jill Waibel served as a consultant and/or investigator for, and/or received personal fees from AbbVie, Allergan, Almirall, AstraZeneca, Avita Medical, Biofrontera, Candela, Cytrellis, Dermira, Dominion Aesthetics, Eli Lilly and Company, Michaelson Diagnostics, Novartis, Pfizer, and Sciton. Jamie Weisman served as a speaker and/or investigator and/or has received grants and/or honoraria from AbbVie, Amgen, Biogen, Boehringer Ingelheim, Celgene, Eli Lilly and Company, Janssen, LEO Pharma, Merck, Novartis, Pfizer, Regeneron, Stiefel, and Valeant Pharmaceuticals. Lindsay Strowd has received support from Actelion, Eli Lilly and Company, Galderma, Pfizer, Regeneron, and Sanofi. Luna Sun, Yuxin Ding, and Meghan Feely are employees and shareholders of Eli Lilly and Company. Meghan Feely is also a clinical instructor at Mount Sinai. Fabio P. Nunes is a former employee of Eli Lilly and Company and is a current employee and shareholder of Janssen Pharmaceuticals Companies of Johnson and Johnson. Eric L. Simpson received grants and fees for participation as a consultant and principal investigator from Eli Lilly and Company, LEO Pharma, Pfizer, and Regeneron; grants for participation as a principal investigator from Galderma and Merck & Co.; and fees for consultant services from AbbVie, Boehringer Ingelheim, Dermavant Incyte, Forte Bio, Pierre Fabre Dermo, and Sanofi Genzyme.
Compliance with Ethics Guidelines
This trial was conducted in accordance with the ethical principles of the Helsinki Declaration of 1964 and its later amendments and Good Clinical Practice guidelines and approved by the appropriate institutional review boards/ethics committees at each study site (Table S1), including the Quorum Review IRB (approval #33039). All patients provided written informed consent.
Data Availability
Lilly provides access to all individual participant data collected during the trial, after anonymization, with the exception of pharmacokinetic or genetic data. Data are available to request 6 months after the indication studied has been approved in the US and EU and after primary publication acceptance, whichever is later. No expiration date of data requests is currently set once data are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, blank or annotated case report forms, will be provided in a secure data sharing environment. For details on submitting a request, see the instructions provided at https://www.vivli.org.
Prior Presentation
This work was presented as an oral presentation at the 2020 Revolutionizing Atopic Dermatitis Virtual Congress.
References
- 1.Brunner PM, Guttman-Yassky E, Leung DY. The immunology of atopic dermatitis and its reversibility with broad-spectrum and targeted therapies. J Allergy Clin Immunol. 2017;139(4s):S65–s76. doi: 10.1016/j.jaci.2017.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boguniewicz M, Fonacier L, Guttman-Yassky E, Ong PY, Silverberg J, Farrar JR. Atopic dermatitis yardstick: practical recommendations for an evolving therapeutic landscape. Ann Allergy Asthma Immunol. 2018;120(1):10–22.e2. doi: 10.1016/j.anai.2017.10.039. [DOI] [PubMed] [Google Scholar]
- 3.Czarnowicki T, He H, Krueger JG, Guttman-Yassky E. Atopic dermatitis endotypes and implications for targeted therapeutics. J Allergy Clin Immunol. 2019;143(1):1–11. doi: 10.1016/j.jaci.2018.10.032. [DOI] [PubMed] [Google Scholar]
- 4.Silverberg JI, Gelfand JM, Margolis DJ, Boguniewicz M, Fonacier L, Grayson MH, et al. Patient burden and quality of life in atopic dermatitis in US adults: a population-based cross-sectional study. Ann Allergy Asthma Immunol. 2018;121(3):340–347. doi: 10.1016/j.anai.2018.07.006. [DOI] [PubMed] [Google Scholar]
- 5.Vakharia PP, Chopra R, Sacotte R, Patel KR, Singam V, Patel N, et al. Burden of skin pain in atopic dermatitis. Ann Allergy Asthma Immunol. 2017;119(6):548–52.e3. doi: 10.1016/j.anai.2017.09.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andersen L, Nyeland ME, Nyberg F. Increasing severity of atopic dermatitis is associated with a negative impact on work productivity among adults with atopic dermatitis in France, Germany, the U.K. and the U.S.A. Br J Dermatol. 2020;182(4):1007–1016. doi: 10.1111/bjd.18296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eichenfield LF, Tom WL, Berger TG, Krol A, Paller AS, Schwarzenberger K, et al. Guidelines of care for the management of atopic dermatitis: section 2. Management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71(1):116–132. doi: 10.1016/j.jaad.2014.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ring J, Alomar A, Bieber T, Deleuran M, Fink-Wagner A, Gelmetti C, et al. Guidelines for treatment of atopic eczema (atopic dermatitis) part I. J Eur Acad Dermatol Venereol. 2012;26(8):1045–1060. doi: 10.1111/j.1468-3083.2012.04635.x. [DOI] [PubMed] [Google Scholar]
- 9.Katayama I, Aihara M, Ohya Y, Saeki H, Shimojo N, Shoji S, et al. Japanese guidelines for atopic dermatitis 2017. Allergol Int. 2017;66(2):230–247. doi: 10.1016/j.alit.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 10.Sidbury R, Davis DM, Cohen DE, Cordoro KM, Berger TG, Bergman JN, et al. Guidelines of care for the management of atopic dermatitis: section 3. Management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014;71(2):327–349. doi: 10.1016/j.jaad.2014.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ring J, Alomar A, Bieber T, Deleuran M, Fink-Wagner A, Gelmetti C, et al. Guidelines for treatment of atopic eczema (atopic dermatitis) Part II. J Eur Acad Dermatol Venereol. 2012;26(9):1176–1193. doi: 10.1111/j.1468-3083.2012.04636.x. [DOI] [PubMed] [Google Scholar]
- 12.Renert-Yuval Y, Guttman-Yassky E. New treatments for atopic dermatitis targeting beyond IL-4/IL-13 cytokines. Ann Allergy Asthma Immunol. 2020;124(1):28–35. doi: 10.1016/j.anai.2019.10.005. [DOI] [PubMed] [Google Scholar]
- 13.Fridman JS, Scherle PA, Collins R, Burn TC, Li Y, Li J, et al. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: preclinical characterization of INCB028050. J Immunol. 2010;184(9):5298–5307. doi: 10.4049/jimmunol.0902819. [DOI] [PubMed] [Google Scholar]
- 14.Olumiant. Australian Product Information. Eli Lilly Australia Pty Ltd. OLUMIANT vA6 Feb_2021. ARTG ID 277905. 2021. https://www.ebs.tga.gov.au/ebs/picmi/picmirepository.nsf/pdf?OpenAgent&id=CP-2018-PI-01225-1. Accessed 25 Mar 2021.
- 15.Olumiant. European Union Summary of Product Characteristics. Eli Lilly and Company. 2021. https://www.ema.europa.eu/en/documents/product-information/olumiant-epar-product-information_en.pdf. Accessed 23 Mar 2021.
- 16.Olumiant. Japan Product Information. Eli Lilly and Company. 2021. https://www.info.pmda.go.jp/go/pdf/530471_3999043F1020_1_08. Accessed 21 Sept 2021.
- 17.Simpson EL, Forman S, Silverberg JI, Zirwas M, Maverakis E, Han G, et al. Baricitinib in patients with moderate-to-severe atopicdermatitis: results from a randomized monotherapy phase 3 trial in the United States and Canada (BREEZE-AD5) J Am Acad Dermatol. 2021;85(1):62–70. doi: 10.1016/j.jaad.2021.02.028. [DOI] [PubMed] [Google Scholar]
- 18.King B, Maari C, Lain E, Silverberg JI, Issa M, Holzwarth K, et al. Extended safety analysis of baricitinib 2 mg in adult patients with atopic dermatitis: an integrated analysis from eight randomized clinical trials. Am J Clin Dermatol. 2021;22(3):395–405. doi: 10.1007/s40257-021-00602-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eichenfield LF, Tom WL, Chamlin SL, Feldman SR, Hanifin JM, Simpson EL, et al. Guidelines of care for the management of atopic dermatitis: section 1. Diagnosis and assessment of atopic dermatitis. J Am Acad Dermatol. 2014;70(2):338–351. doi: 10.1016/j.jaad.2013.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yosipovitch G, Reaney M, Mastey V, Eckert L, Abbé A, Nelson L, et al. Peak Pruritus Numerical Rating Scale: psychometric validation and responder definition for assessing itch in moderate-to-severe atopic dermatitis. Br J Dermatol. 2019;181(4):761–769. doi: 10.1111/bjd.17744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bieber T, D'Erme AM, Akdis CA, Traidl-Hoffmann C, Lauener R, Schappi G, et al. Clinical phenotypes and endophenotypes of atopic dermatitis: where are we, and where should we go? J Allergy Clin Immunol. 2017;139(4s):S58–s64. doi: 10.1016/j.jaci.2017.01.008. [DOI] [PubMed] [Google Scholar]
- 22.Renert-Yuval Y, Thyssen JP, Bissonnette R, Bieber T, Kabashima K, Hijnen D, et al. Biomarkers in atopic dermatitis-a review on behalf of the International Eczema Council. J Allergy Clin Immunol. 2021;147(4):1174–90.e1. doi: 10.1016/j.jaci.2021.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Curran-Everett D, Milgrom H. Post-hoc data analysis: benefits and limitations. Curr Opin Allergy Clin Immunol. 2013;13(3):223–224. doi: 10.1097/ACI.0b013e3283609831. [DOI] [PubMed] [Google Scholar]
- 24.Chopra R, Silverberg JI. Assessing the severity of atopic dermatitis in clinical trials and practice. Clin Dermatol. 2018;36(5):606–615. doi: 10.1016/j.clindermatol.2018.05.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Lilly provides access to all individual participant data collected during the trial, after anonymization, with the exception of pharmacokinetic or genetic data. Data are available to request 6 months after the indication studied has been approved in the US and EU and after primary publication acceptance, whichever is later. No expiration date of data requests is currently set once data are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, blank or annotated case report forms, will be provided in a secure data sharing environment. For details on submitting a request, see the instructions provided at https://www.vivli.org.