

Abstract
Background: In the past decade, several antibodies directed against the PD-1/PD-L1 interaction have been approved. However, therapeutic antibodies also exhibit some shortcomings. Using small molecules to regulate the PD-1/PD-L1 pathway may be another way to mobilize the immune system to fight cancer. Method: 52,765 marine natural products were screened against PD-L1(PDBID: 6R3K). To identify natural compounds, a structure-based pharmacophore model was generated, following by virtual screening and molecular docking. Then, the absorption, distribution, metabolism, and excretion (ADME) test was carried out to select the most suitable compounds. Finally, molecular dynamics simulation was also performed to validate the binding property of the top compound. Results: Initially, 12 small marine molecules were screened based on the pharmacophore model. Then, two compounds were selected for further evaluation based on the molecular docking scores. After ADME and toxicity studies, molecule 51320 was selected for further verification. By molecular dynamics analysis, molecule 51320 maintains a stable conformation with the target protein, so it has the chance to become an inhibitor of PD-L1. Conclusions: Through structure-based pharmacophore modeling, virtual screening, molecular docking, ADMET approaches, and molecular dynamics (MD) simulation, the marine natural compound 51320 can be used as a small molecule inhibitor of PD-L1.
Keywords: PD-L1, virtual screening, pharmacophore modeling, ADME, molecular dynamics
1. Introduction
Blocking the interaction of PD-1/PD-L1 and PD-1/PD-L1 pathway modulators has shown unprecedented clinical efficacy in a variety of tumor models [1,2,3,4]. Existing studies have shown that the programmed cell death protein 1 (PD-1)/programmed cell death ligand 1 (PD-L1) signaling pathway can induce tumor-specific T cell apoptosis by inhibiting T cell activation [5]. It plays a role in immune escape and immune suppression under pathological conditions such as inflammation [6,7,8,9,10,11].
Because PD-L1 is highly expressed in a variety of tumor cells, after PD-L1 binds to PD-1, T cell activation is inhibited, and T cells are in a state of immune tolerance [12]. At this time, the immune system cannot kill the cancer cells, and tumor immune escape occurs [13]. Therefore, this type of inhibitor has a wide range of tumors, especially for tumors with high PD-L1 expression, and the response rate is higher.
Therefore, a targeted inhibitor designed for PD-L1 can cut off the signal pathway and activate T cells [14]. Therefore, its immune tolerance is relieved, T cells are mobilized to kill tumors, and tumor treatment is realized. Anti-PD-L1 monoclonal antibody is one of the most important drugs for lung cancer immunotherapy [15]. However, therapeutic antibodies also show some disadvantages. For example, tumor penetration rate is low, it is difficult to overcome physiological barriers, and there is a lack of oral bioavailability, high manufacturing cost, inaccessibility to intracellular targets, and immune-related adverse events (irAE) [16]. Using small molecules to regulate the PD-1/PD-L1 pathway may be another way to mobilize the immune system to fight cancer. In 2015, Zak et al. reported the crystal structure of the hPD-1/hD-L1 complex, which is generally considered to provide important receptor-ligand interactions, and they are reasonable structure-based drugs on the surface of PD-L1 Design (SBDD), which provides several major active sites [17]. In recent years, several small molecule drugs that can bind to PD-L1 and inhibit the interaction of PD-1/L1 have been discovered [18,19]. Therefore, it is of great significance to generate simple, stable, and efficient PD-L1 small molecule inhibitors.
Due to its special ecological environment, the ocean contains rich natural products. With the development of terrestrial resources, the marine environment provides a new field for research. By the end of 2020, it is estimated that more than 29,000 marine natural products have been found, and marine natural products have also received increasing attention from scientists [20]. It has been found that marine natural products have different structural characteristics from terrestrial natural products with various biological activities such as antifungal, antiviral, anti-parasitic, anti-tumor, and anti-inflammatory [21,22]. Natural products are the best choice as a source of new drugs [23], and marine organisms are thus considered as the latest source of bioactive natural products related to terrestrial plants and non-marine microorganisms [24]. We have collected three marine natural product databases: Marine Natural Product Database (MNPD) [25], Seaweed Metabolite Database (SWMD) [26], and Comprehensive Marine Natural Product Database (CMNPD) [27]. In this study, 52,765 kinds of marine natural products were virtually screened by targeting PD-L1. In order to predict a variety of marine natural products that may inhibit PD-L1. We hope to provide new options for the development of new anti-tumor drugs.
Structure-based pharmacophore modeling, virtual screening, molecular docking, ADMET approaches, and molecular dynamics (MD) simulation were performed on a library of marine natural products to find the novel compounds against PD-L1, which are demonstrated as Figure 1.
Figure 1.

The virtual screening workflow (VSW) used in this work for the identification of hit molecules targeting PD-L1. A workflow overview of pharmacophore modeling, virtual screening, molecular docking, absorption, distribution, metabolism, elimination, and toxicity (ADMET) approaches, and MD simulation.
2. Results
2.1. Structure-Based Pharmacophore Modeling and Virtual Screening
2.1.1. Pharmacophore Model Establishment
Pharmacophore describes the three-dimensional arrangement of basic spatial and electronic characteristics to achieve the best combination of ligands and macromolecules. The main application area of the pharmacophore model is database search. By searching the compound database through the pharmacophore model, it is possible to find biologically active compounds with specific targets, and to find new chemical entities with similar biological activities and different skeleton structures. Depending on the available target or known ligand information, the design of the pharmacophore model can be structure-based or ligand-based [28,29,30]. In this study, the available PD-L1 information of its small molecule inhibitors was used to construct the structure-based pharmacophore model. Hence, the 7 pharmacophore models containing the feature set were generated (Table 1) [31,32,33,34,35]. Among the generated models, 6R3K composed of DHHHNP chemical characteristics with the highest selectivity score (16.25) was selected as the best model.
Table 1.
Ranking of pharmacophore models.
Therefore, the structure-based pharmacophore model was constructed based on PD-L1 (6R3K) and small molecule JQT (Figure 1). Ten pharmacophore models were generated. According to careful selection, we applied ligand 8 as the pharmacophore model for screening. DS software was used to generate key chemical features based on the pharmacophore model and co-crystalized ligand JQT and pharmacophore ligand 08 in Figure 2b. The total number of pharmacophores is 8. Two of them are hydrophobic, two hydrogen bond acceptors, two hydrogen bond donors, one positively charged ion center, and one negatively charged ion center (Figure 2c).
Figure 2.

The overall pharmacophore model generated during the study. (a) PD-L1 crystal (PDB ID:6R3K) with the co-crystalized ligand JQT and pharmacophore interaction map; (b) co-crystalized ligand JQT and pharmacophore ligand 08; (c) pharmacophore ligand 08.
2.1.2. Pharmacophore Model Validation
Verification is necessary to obtain true pharmacophore analysis and to evaluate the quality of molecular models [36]. Before database screening, the structure-based pharmacophore model established in this study was verified to verify whether the pharmacophore has a good ability to distinguish between active and inactive molecules. Receiver operating characteristic curve (ROC curve for short) is also known as sensitivity curve. The ROC curve graph is a curve reflecting the relationship between sensitivity and specificity. The X axis of the abscissa is 1-specificity, also known as false positive rate (false positive rate), the closer the X axis is to zero, the higher the accuracy rate; the Y axis of ordinate is called sensitivity, also known as true positive rate (sensitivity), the larger the Y-axis, the better the accuracy. According to the curve position, the whole graph is divided into two parts. The area under the curve is called AUC (area under curve), which is used to indicate the accuracy of prediction. The higher the AUC value, the larger the area under the curve, indicating the prediction, the higher the accuracy rate. The closer the curve is to the upper left corner (the smaller the X, the larger the Y), the higher the prediction accuracy rate [37]. In our verification process, the AUC (area under the ROC curve) at 1% threshold is 0.819 (Figure 3), which proves that our model has ability to distinguish between truly active substances and decoy compounds.
Figure 3.

Receiver operating characteristic (ROC) curve of Ligand_08.
2.1.3. Virtual Screening Based on Pharmacophore
A marine natural product library containing a total of 52,765 compounds was used for virtual screening based on pharmacophores against the generated pharmacophore models. A total of 12 compounds that meet the characteristics of all pharmacophores were generated. Compounds labeled HIT were retrieved and stored for further evaluation.
2.2. Molecular Docking
Molecular docking is an important part of the drug design process. This study aims to evaluate the binding ability of HITS compounds to the target PD-L1 protein. According to the previously obtained binding sites, a receptor grid with X = −7.1, Y = 59.3, and Z = −19.5 was prepared.
AutoDock was used to dock a specific number of drugs with PD-L1, and evaluate its binding ability, which is in line with the characteristics of the pharmacophore model. Among them, the binding affinity of compound 37080 and compound 51320 are −6.5 kcal/mol and −6.3 kcal/mol (Table 2), and their binding affinity is better than that of the PD-L1 inhibitor used in the process of generating the main pharmacophore model (−6.2 kcal/mol). The interaction of compound 37080 in the docking complex is shown in Figure 4a,b, and the interaction of compound 51320 is shown in the Figure 4c,d. In compound 51320 with good docking performance, it can be observed that the compound forms a hydrogen bond with Ala121, the oxygen atom interacts with the residue Asp122, and there is an ionic interaction with the residue ASP122 (Figure 4d). More importantly, the Pi–Pi interaction established between residue Ile54 and the compound and the Pi–Sigma interaction between residue Tyr123 and the compound also played a key role in ligand–receptor binding. Obviously, the rich interaction types between compound 513320 and the protein allow the best docking results between them. It can be seen from the interaction analysis that the docking result is reliable and the selected compound can be further analyzed.
Table 2.
Molecular docking results of JQT and 12 selected ligands from the library of marine natural products.
| Molecules | 2D Structure | Binding Affinity (kcal/mol) | Formula |
|---|---|---|---|
| 37080 |
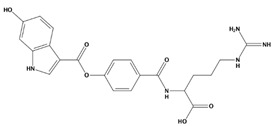
|
−6.5 | C22H23N5O6 |
| 51320 |
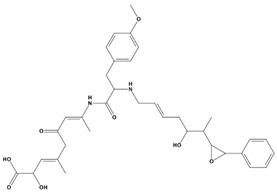
|
−6.3 | C35H44N2O8 |
| 37113 |
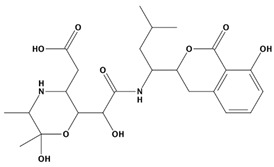
|
−6.2 | C24H34N2O9 |
| 38010 |
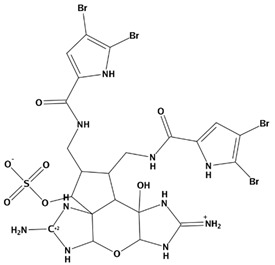
|
−5.7 | C22H25Br4N10O8S |
| 41160 |
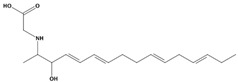
|
−5.2 | C18H29NO3 |
| 32979 |
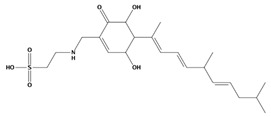
|
−5.1 | C22H35NO6S |
| 35432 |
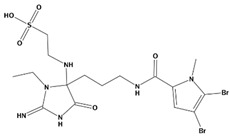
|
−4.7 | C16H24Br2N6O5S |
| 21793 |
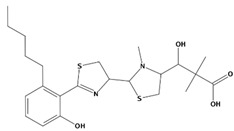
|
−4.6 | C23H34N2O4S2 |
| 23671 |
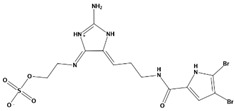
|
−4.6 | C13H16Br2N6O5S |
| 41159 |
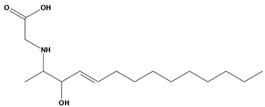
|
−4.5 | C16H31NO3 |
| 35433 |
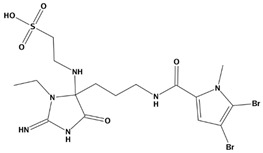
|
−4.4 | C16H24Br2N6O5S |
| 50094 |
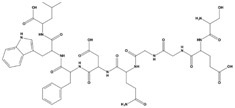
|
−3.9 | C47H63N11O16 |
| JQT |
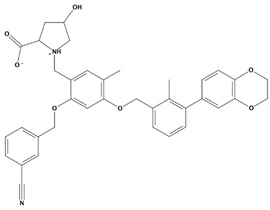
|
−6.2 | C36H33ClN2O7 |
Figure 4.

Interaction between the protein–ligand complex. (a) Three-dimensional binding mode of the 37080 and protein complex; (b) two-dimensional binding mode of the 37080 and protein complex; (c) three-dimensional binding mode of the 51320 and protein complex; (d) two-dimensional binding mode of the 51320 and protein complex.
2.3. Analysis of Pharmacophore Characteristics
The pharmacophore characteristics of compounds play an important role in the molecular recognition process of targeted biological macromolecules. The pharmacophore of a compound can be described according to the characteristics of H, AR, HBA or HBD, PI, and NI. This helps to identify and design new drugs for the treatment of selected diseases.
These features retain the necessary geometric arrangement of atoms required to produce a specific biological reaction. Therefore, the characteristics of the pharmacophore we generated were analyzed. As shown in Figure 5a, the overlap of the ligand and pharmacophore characteristics shows that the selected compound should be effective for our target protein. Superimposition of the top12 hit compounds on to the pharmacophore model was shown in Figure 5b and superimposition of JQT on to the pharmacophore model was shown in Figure 5c.
Figure 5.

Structure-based pharmacophore. (a) Aligned ligands. (b) Selected ligands superimposed to the simplified pharmacophore model. (c) Ligand JQT superimposed to the simplified pharmacophore model.
2.4. ADME and Toxicity Test ADME Properties Analysis
Swiss-ADME is a website (https://www.swissadme.ch, accessed on 9 October 2021) which allows the user to draw their respective ligand or drug molecule or include SMILES data from PubChem and provides parameters such as lipophilicity (iLOGP, XLOGP3, WLOGP, MLOGP, SILICOS-IT, Log P0/w), water solubility Log S (ESOL, Ali, SILICOS-IT), drug-likeness rules (Lipinski, Ghose, Veber, Egan, and Muegge), and Medicinal Chemistry (PAINS, Brenk, Lead-likeness, Synthetic accessibility) [38]. The ADME prediction study of the designed compounds demonstrated Table 3. The Swiss-ADME section gives a physicochemical property of possible oral drug candidates according to five different rules determined by Lipinski, Ghose, Veber, Egan, and Muegge [39,40,41,42]. The reference value of Log S for moderately soluble and highly soluble molecules ranged from −4 to −6 and −2 to −4, respectively. According to the results, all molecules are classified as moderately soluble and highly soluble. All these parameters infer that 51320 is close to a drug-like molecule.
Table 3.
ADME properties of JQT and selected ligands from the library of marine natural products.
| Molecule | MW | Rotatable Bonds | H-bond Acceptors | H-bond Donors | ESOL Log S | TPSA | WLOGP | GI Absorption | log Kp (cm/s) |
|---|---|---|---|---|---|---|---|---|---|
| 37080 | 453.45 | 12 | 7 | 7 | −3.18 | 190.62 | 1.54 | 0.78 | Low |
| 51320 | 620.73 | 19 | 9 | 5 | −3.82 | 157.72 | 3.33 | 0.99 | Low |
| 37113 | 494.53 | 9 | 10 | 6 | −1.91 | 174.65 | −0.09 | 0.04 | Low |
| 38010 | 909.18 | 10 | 8 | 11 | −5.32 | 271.15 | −2.38 | 0.43 | Low |
| 41160 | 307.43 | 12 | 4 | 3 | −1.6 | 69.56 | 3.22 | 0.35 | High |
| 32979 | 441.58 | 11 | 7 | 4 | −2.04 | 132.31 | 3.13 | 1.28 | High |
| 35432 | 572.27 | 11 | 7 | 5 | −2 | 165 | 0.94 | 0.31 | Low |
| 21793 | 466.66 | 9 | 6 | 3 | −3.74 | 143.96 | 3.07 | 2.18 | Low |
| 23671 | 528.18 | 9 | 6 | 5 | −3.99 | 166.6 | 1.06 | −0.33 | Low |
| 41159 | 285.42 | 13 | 4 | 3 | −2.01 | 69.56 | 3.11 | 0.14 | High |
| 35433 | 572.27 | 11 | 7 | 5 | −2 | 165 | 0.94 | 0.31 | Low |
| 50094 | 1038.07 | 40 | 17 | 15 | −0.68 | 449.83 | −3.85 | −4.93 | Low |
| JQT | 641.11 | 10 | 9 | 2 | −5.81 | 121.48 | 5.30 | 2.64 | Low |
2.5. Toxicity Analysis
In order to better select lead compounds, the measurement of toxicity within silicon is an important step before the candidate drug goes into clinical trials. Calculation-based electronic toxicity measurement is widely used because of its accuracy and accessibility. It can provide information on any synthetic or natural compound. In order to determine the toxicity and adverse effects of the two selected compounds, we used the freely available testing tool ProTox-II server [43]. The software evaluates several toxicological parameters, such as acute toxicity, liver toxicity, cytotoxicity, carcinogenicity, mutagenicity, and immunotoxicity, and is based on the predicted median lethal dose (LD50) (in mg/kg body weight) (Table 4). According to the ProTox-II server, compound 51320 belongs to 4 types of toxicity, with LD 50 of 300–2000 mg/kg, which is harmful when administered orally. Compound JQT belongs to grade 4 toxicity, and its LD 50 value is 800 mg/kg. However, it was active in cytotoxicity. Combined with the ADME results, 51320 is closer to drug-like molecules than JQT. Subsequently, MD simulations were performed on 51320 molecules and JQT.
Table 4.
List of toxicity properties (organ toxicity, toxicity endpoints, Tox21-nuclear receptor signaling pathways, Tox21-stress response pathway) of the selected 2 compounds.
| Endpoint | Target | JQT | 51320 |
|---|---|---|---|
| Organ toxicity | Hepatotoxicity | Inactive | Inactive |
| Toxicity end points | Carcinogenicity | Inactive | Inactive |
| Immunotoxicity | Inactive | Inactive | |
| Mutagenicity | Inactive | Inactive | |
| Cytotoxicity | Active | Inactive | |
| LD50 (mg/kg) | 800 | 765 | |
| Toxicity class | 4 | 4 | |
| Tox21-nuclear receptor signaling pathways | Aryl hydrocarbon receptor (AhR) | Inactive | Inactive |
| Androgen receptor (AR) | Inactive | Inactive | |
| Tox21-stress response pathways | Heat shock factor response element (HSE) | Inactive | Inactive |
2.6. Structure-Based Pharmacophore Modeling and Virtual Screening
MD simulations are used to explore the binding stability of the protein–ligand docking complex. MD simulations also provide information about molecular interactions within a reference time or provide valuable assessments in explaining drug resistance [44,45]. In this paper, MD simulation methods are used to analyze the docking file of a complex of a natural compound and PD-L1 protein to determine the stability and intermolecular interaction between the protein and the molecule within a 100 ns time interval. The trajectory of MD is extracted, and the simulation results of protein–ligand (P-L) interaction mapping based on RMSD and RMSF are described. The results show that compound 51320 is stable.
2.6.1. RMSD Analysis
In order to obtain the equilibrium time of each simulated protein ligand complex during the MD simulation, the root mean square deviation (RMSD) of the backbone was calculated. RMSD diagrams are commonly used to evaluate the time required for the system to reach structural equilibrium and estimate the duration of running simulations. RMSD is an important parameter for estimating changes or changes in molecular conformation. Due to the sudden change of structural conditions, the RMSD value of the analog complex including the reference suddenly increased, which is related to the protein crystallization method. The latter effect is expected, because in the crystal structure, the protein is rigid, and when it is solvated in the water tank, it resumes its dynamic motion.
The complex system with a time frame x should have an RMSD that can be calculated from the following Equation (1).
| (1) |
Here, the is the calculation of RMSD for the specific number of frames, is the number of selected atoms, is the reference or mentioned time, and is the selected atom in the frame after super imposing on the reference frame, is the recording intervals.
As shown in Figure 6a, the RMSD of selected compounds 51320 and JQT were analyzed to determine whether the system has been balanced. The system of compound 51320 was in equilibrium after 105 ns, and finally stabilized at 0.33 nm, which reflected the good stability of the whole system to some extent. In addition, the docking score of the compound was −6.3 kcal/mol. The JQT system was in equilibrium after 100 ns, but finally stabilized at 0.45 nm, and the score of the JQT complex was −6.2 kcal/mol. Overall, both systems could be in equilibrium after 150 ns simulation, and the stability of the two systems was good. Compound 51320 had better system stability.
Figure 6.

Root mean square deviation (RMSD) and root mean square fluctuation (RMSF) plots of JQT complex (black), 51320 (red). (a) RMSD values extracted from protein fit ligand of the protein–ligand docked complexes. RMSD plot of JQT complex (black), 51320 (red); (b) the RMSF graph of all complexes along with protein during 100 ns MD simulation. RMSF plot of JQT complex (black), 51320 (red).
2.6.2. RMSF Analysis
In order to determine the deviation of the ligand from the initial posture and the degree of movement of protein residues, the root mean square fluctuation (RMSF) values of all sampled conformations during the 30 ns simulation were also calculated. RMSF fluctuates greatly, indicating that the residue is unstable, otherwise the residue is stable. The RMSF for residue was calculated from the following Equation (2).
| (2) |
where is the overall trajectory time, is the residue location, is the reference time, is the location of atoms in residue after aligned on the reference, and the angle brackets are the average of the square distance.
The RMSF of two compounds was analyzed to measure the displacement of specific atoms during the simulation. In Figure 6b, the final image results of the two systems basically overlap between 0.05 and 1.25 nm, while RMSF values of docking pocket and residue generating interaction force are both lower than 0.25 nm, which to some extent indicates that the flexibility of the two systems is low, and the overall effect of compound binding is better.
2.7. MM/GBSA Analysis
Molecular mechanics Poisson–Boltzmann surface area (MM/PBSA) is an effective and reliable method for calculating the free energy of binding of small inhibitors to their protein targets. Another important indicator that considers the potential affinity of a ligand to its target is the free energy of binding calculated using MM-PBSA and MD. In general, complexes with lower binding free energy can be considered more stable, and their ligands are expected to have higher activity and potency. We summarize the interaction energy and binding free energy of the two complexes in Figure 7a,b, respectively. The MM/GBSA of the complex system is calculated from a single trajectory collected from the respective 100 ns simulation (Table 5). The analysis of the contribution of each energy term shows that the electrostatic interaction of 51320 (−179.032 kJ/mol) (Figure 8b) is much stronger than the corresponding term of JQT (Figure 8a) and PD-L1 (12.196 kJ/mol). Therefore, the compounds screened will be able to maintain a lasting interaction with the desired protein. The quantitative information on the contribution of each amino acid residue to energy is very helpful for a better understanding of the binding mechanism of inhibitor molecules. The analysis of the selected compounds in Figure 8 revealed that MET-115, TYR-56, and ILE-54 have high energy contributions in JQT and 51320. It can be seen from the above results that the selected compound can maintain long-term interaction with the binding site of the PD-L1 protein, resulting in the inhibition of the target protein.
Figure 7.

Residue wise decomposition of binding free energies obtained from the MMPBSA analyses. (a) JQT; (b) 51320.
Table 5.
Binding energy of binding for the protein complexed with ligands JQT, 51320.
| Criteria | JQT | 51320 |
|---|---|---|
| Van der Waal energy (kJ/mol) | −291.250 ± 1.797 | −227.131 ± 21.896 |
| Electrostatic energy (kJ/mol) | 12.196 ± 11.229 | −179.032 ± 18.056 |
| Polar solvation energy (kJ/mol) | 111.917 ± 12.103 | 271.823 ± 28.866 |
| SASA energy (kJ/mol) | −19.948 ± 0.176 | −20.535 ± 0.727 |
| Binding energy (kJ/mol) | −187.084 ± 0.748 | −154.875 ± 25.470 |
Figure 8.

Binding energy of binding for the protein complexed with ligands JQT, 51320. (a) JQT; (b) 51320.
3. Discussion
In recent years, anti PD-L1 monoclonal antibodies have also shown positive reactions in clinical trials of various malignant tumors. However, antibody drugs have some shortcomings, such as immunogenicity problems and poor tumor tissue permeability, resulting in a low overall response rate of PD-1/PD-L1 antibody drugs [29]. At present, small molecule inhibitors based on PD-1/PD-L1 are gradually recovering. Marine natural products are closely related to the fields of drug discovery and molecular biology, and have always attracted the attention of the scientific community. Therefore, our research aims to use marine natural products to perform virtual screening of PD-L1 detection sites.
In this study, we collected 7 structures of PD-L1 with low molecular mass inhibitor by reading the literature. A structure-based pharmacophore model was constructed by using DS 4.5, and 6R3K with the highest selection score was selected. The pharmacophore model constructed by the complex was used to screen the marine natural product database. The pharmacophore model is validated by the active compound, and the AUC under the ROC curve indicates that the model has good distinguishing ability. The validated pharmacophore model was used in the virtual screening process. A total of 12 compounds were retrieved for hits and further screened by molecular docking methods. According to the molecular docking score, the first two compounds with a better binding score than the original ligand JQT (−6.2 kcal/mol) were selected for further verification.
We conducted interaction analysis from the perspective of molecular docking. The rich interaction types between compound 513320 and protein can be seen, the docking result is reliable, and the selected conformation can be further analyzed. In addition, quantum mechanics/molecular mechanics (QM/MM) calculations can be performed on the complex to select conformations from docking simulations [46]. After years of development and calibration, QM/MM hybrid method has become an indispensable tool to study the dynamics of a variety of chemical and biochemical processes. For example, an article uses a molecular docking method combining quantum mechanics and molecular mechanics (QM/MM) to determine the resuscitation pathway of inhibited AChE [47]. Another article studied the reaction mechanism between oxime and MmAChE, using the sequential QM/docking (MM) method [48]. QM/MM is mostly used to characterize and study the transition state and activation energy of enzyme reactions. The conformation calculated by this method describes the surrounding environment in more detail. However, the calculation becomes more complicated and not easy to control. We selected the conformation by analyzing the interaction between molecules and combining scores, which not only pays attention to the binding mode but also has scores for reference. However, the influence of the environment is not considered, and some compounds will change the docking conformation due to environmental changes. All in all, it is better to use QM/MM for the selection of the molecular conformation of the enzyme’s active target. It is more visual and convenient to use molecular interaction force to choose the conformation of other molecules. In the future, further QM/MM research on PD-L1 and molecules can be carried out for better selection of molecular conformations.
The two selected compounds 51320 and 37080 have been evaluated based on ADME characteristics, and the 51320 showed good ADME characteristics. Compound 37080 violates Lipinski’s Five Rules, so this compound was skipped for further evaluation. Compounds with good ADME properties were further evaluated for toxicity properties to measure harmful effects on humans or animals. Toxicity analysis found that the selected compound 51320 has very low toxicity and JQT has cytotoxicity. Our selected compound 51320 has no cytotoxicity and is better than JQT.
The MD simulation method identifies the stability of the compound to the protein binding site. The 150 ns simulation trajectory was searched and analyzed based on RMSD and RMSF to confirm the stability of the compound and protein binding site. In addition, the MM/GBSA calculated from a single trajectory found a high ΔG binding value, indicating that the selected protein-ligand complex has long-term simulation stability.
All in all, marine natural products provide a lot of information for the discovery of new drugs. Through virtual screening, small-molecule inhibitors of PD-L1 were efficiently identified from more than 52,000 marine natural products. The expansion and further optimization of the screening range can finally identify useful immunomodulators to help improve public health.
4. Materials and Methods
4.1. Structure-Based Pharmacophore Modeling and Virtual Screening
4.1.1. Complex-Based Pharmacophore Modeling
Our first goal in this project was to collect as much information as possible about PD-L1 as an inhibitor target. Through a literature search, we downloaded 7 PD-L1 compounds and small molecule inhibitors from the PDB. Discovery Studio 4.5 was used to establish a pharmacophore model based on the compound (Table 1). The pharmacophore model with the highest score was used to screen the compound.
4.1.2. Pharmacophore Model Validation
In the modeling process, the receiver operating characteristic (ROC) curve analysis and verification method built in DS 4.5 was used. Pharmacophore validation helps to assess the potential properties of active and inactive compounds, usually derived from specific protein-ligand interactions. A total of 90 active antagonists obtained from patent and literature searches were used to validate the pharmacophore model, which is composed of 60 active compounds and 30 inactive compounds. Then, the generated pharmacophore was verified by using the verification option in the receptor ligand pharmacophore generation protocol implemented in DS 4.5. From the area under the ROC curve (AUC), we can judge whether a pharmacophore has successfully selected active ingredients and removed inactive ingredients [49,50]. The area under the ROC curve (AUC) is 0 ≤ A ≤ 1. When A > 0.5, the closer A is to 1, the higher the diagnostic accuracy. When A = 0.5, the diagnosis does not work at all. When A < 0.5, it does not meet the actual situation.
4.1.3. Virtual Screening Based on Pharmacophore
The marine small molecule databases (MND, SWMD, CMNPD) were screened according to the characteristics of the pharmacophore. DS 4.5 created and acquired 3D models in the case of protein-ligand interactions. These compounds were directly transferred to the database list for rapid virtual screening based on pharmacophore characteristics. According to the pharmacophore matching score, the fitted hit compounds were ranked and further verified. Of the 20 molecules obtained, a total of 12 hits were selected after careful visual inspection.
4.2. ADME and Toxicity Test
4.2.1. ADME
ADME is important to analyze the pharmacodynamics of the proposed molecule which could be used as a drug. The Swiss ADME server (http://www.swissadme.ch/, accessed on 9 October 2021) was used to evaluate the selected ligands which were harvested from PubChem, which was done on the basis canonical SMILES [38]. The ADME properties of the chosen compounds were calculated. The major ADME-associated parameters such as pharmacokinetic properties and the solubility of the drug were considered. The values of the observed properties are presented in Table 3.
4.2.2. Toxicity Test
Calculation-based methods have made it possible to obtain a safety profile of the desired compound to measure toxicity through computer methods. ProTox-II (http://tox-new.charite.de/protox_II/, accessed on 9 October 2021) server was used to determine the toxic effects of the two selected compounds [51]. The ProTox-II server predicts the median lethal dose (LD50), organ toxicity (hepatotoxicity), and toxicological endpoint (immunotoxicity and cytotoxicity) of the query molecule.
4.3. MD Simulation
In order to further verify the results obtained, the second docking program CDOCKER on DS 4.5 was adopted. The results were evaluated based on the interaction energy of CDOCKER, and a higher interaction energy of CDOCKER meant greater beneficial binding.
After docking, the compound with the highest binding energy for each target is simulated by MD simulation to check the stability of the compound in the binding pocket. Then, GROMACS 2019.1 software package [52], gromos54a7atb.ff force field and single point charge (SPC216) model was used for molecular dynamics simulation of the 150 ns. In order to ensure the total charge neutrality of the simulated system, a corresponding number of sodium ions was added to replace the water molecules in the three systems to produce a solvent box of appropriate size. Then, periodic boundary conditions (PBC) [53] were applied in the three directions of the system. Using the gromos54a7_atb force field, the force field parameters of the entire atom can be obtained from the ATB website (http://atb.uq.edu.au/, accessed on 9 October 2021). A first pass (EM) was conducted to minimize the energy of 50,000 steps of the entire system at 300 K, then through MD simulation with location constraints, through NVT collection (constant particle number, volume and temperature), and finally through NPT collection (constant particle number, pressure, and temperature) [53]. In addition, we balanced enzymes, ligand molecules, and solvents.
4.4. MM/GBSA
Improved MM-PBSA or Molecular Mechanics–Poisson Boltzmann Surface Area is an opensource software used to calculate the free energy of binding between the receptor and the inhibitor. As a scoring function, MM-PBSA has been used in the calculation method of drug design [54]. In this study, MM-PBSA was used to determine the binding free energy of JQT and molecule 51320, respectively.
The following Equation (3) describes the binding free energy:
| (3) |
The free energy of protein-inhibitor complex is represented by GComplex, the free energy of protein in solvent is represented by Gprotein, and the free energy of inhibitor in solvent is represented by Gligand.
5. Conclusions
PD-L1 has become a therapeutic target for many malignant tumors. In this study, a structure-based pharmacophore model was generated using the crystal structure of PD-L1 (6R3K) and the combined small molecule inhibitor JQT. These were used for virtual screening of a marine natural product database. Molecular docking, ADME analysis, and toxicity studies were performed on the obtained compounds. Subsequently, the molecule 51320 was selected for molecular dynamics simulation and MM/GBSA methods, revealing that 51320 is a potential small molecule inhibitor that helps inhibit PD-L1. The small molecule can be further evaluated through different laboratory-based experimental techniques to help determine the activity of the compound, thereby providing an alternative to immunotherapy.
Author Contributions
L.L. conceived and designed the study; A.Z., Q.W. and T.Z. contributed to the acquisition, analysis, and interpretation of data; L.L. and A.Z. wrote the manuscript; L.L. reviewed the paper and provided comments; and all authors reviewed the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This project was supported by Administration of Traditional Chinese Medicine of Guangdong Province (20201180); Administration of Traditional Chinese Medicine of Guangdong Province (20211223); Science and Technology Special Project of Zhanjiang (2019A01009); Basic and Applied Basic Research Program of Guangdong Province (2019A1515110201); Program of Department of Natural Resources of Guangdong Province (GDNRC [2020]038 and [2021]53); Discipline Construction Project of Guangdong Medical University (4SG21004G).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data used to support the findings of this study are included within the article.
Conflicts of Interest
The authors declare that they have no known competing financial interest or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Gong J., Chehrazi-Raffle A., Reddi S., Salgia R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: A comprehensive review of registration trials and future considerations. J. Immunother. Cancer. 2018;6:8. doi: 10.1186/s40425-018-0316-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Webb E.S., Peng L., Baleeiro R., Lemoine N.R., Ming Y., Wang Y.J. Immune checkpoint inhibitors in cancer therapy. J. Biomed. Res. 2018;32:317–326. doi: 10.7555/JBR.31.20160168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferrara R., Mezquita L., Texier M., Lahmar J., Audigier-Valette C., Tessonnier L., Mazieres J., Zalcman G., Brosseau S., Le Moulec S., et al. Hyperprogressive Disease in Patients with Advanced Non–Small Cell Lung Cancer Treated with PD-1/PD-L1 Inhibitors or with Single-Agent Chemotherapy. JAMA Oncol. 2018;4:1543–1552. doi: 10.1001/jamaoncol.2018.3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nishijima T.F., Shachar S.S., Nyrop K.A., Muss H.B. Safety and Tolerability of PD-1/PD-L1 Inhibitors Compared with Chemotherapy in Patients with Advanced Cancer: A Meta-Analysis. Oncologist. 2017;22:470–479. doi: 10.1634/theoncologist.2016-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mühlbauer M., Fleck M., Schütz C., Weiss T., Froh M., Blank C., Schölmerich J., Hellerbrand C. PD-L1 is induced in hepatocytes by viral infection and by interferon-α and -γ and mediates T cell apoptosis. J. Hepatol. 2006;45:520–528. doi: 10.1016/j.jhep.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 6.Alsaab H.O., Sau S., Alzhrani R., Tatiparti K., Bhise K., Kashaw S.K., Iyer A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017;8:561. doi: 10.3389/fphar.2017.00561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bari S., Muzaffar J., Chan A., Jain S.R., Haider A.M., Adams Curry M., Hostler C.J. Outcomes of Programmed Cell Death Protein 1 (PD-1) and Programmed Death-Ligand 1(PD-L1) Inhibitor Therapy in HIV Patients with Advanced Cancer. J. Oncol. 2019;2019:1–5. doi: 10.1155/2019/2989048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cimadamore A., Massari F., Santoni M., Lopez-Beltran A., Cheng L., Scarpelli M., Montironi R., Moch H. PD1 and PD-L1 Inhibitors for the Treatment of Kidney Cancer: The Role of PD-L1 Assay. Curr. Drug Targets. 2020;21:1664–1671. doi: 10.2174/1389450121666200324151056. [DOI] [PubMed] [Google Scholar]
- 9.Sun J., Zheng Y., Mamun M.A.A., Li X., Chen X., Gao Y. Research progress of PD-1/PD-L1 immunotherapy in gastrointestinal tumors. Biomed. Pharmacother. 2020;129:110504. doi: 10.1016/j.biopha.2020.110504. [DOI] [PubMed] [Google Scholar]
- 10.Seetharamu N., Preeshagul I., Sullivan K. New PD-L1 inhibitors in non-small cell lung cancer—Impact of atezolizumab. Lung Cancer Targets Ther. 2017;8:67–78. doi: 10.2147/LCTT.S113177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kumar R., Collins D., Dolly S., McDonald F., O’Brien M.E.R., Yap T.A. Targeting the PD-1/PD-L1 axis in non–small cell lung cancer. Curr. Probl. Cancer. 2017;41:111–124. doi: 10.1016/j.currproblcancer.2016.12.002. [DOI] [PubMed] [Google Scholar]
- 12.Cha J.H., Chan L.C., Li C.W., Hsu J.L., Hung M.C. Mechanisms Controlling PD-L1 Expression in Cancer. Mol. Cell. 2019;76:359–370. doi: 10.1016/j.molcel.2019.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang X., Wang J., Deng X., Xiong F., Ge J., Xiang B., Wu X., Ma J., Zhou M., Li X., et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer. 2019;18:10. doi: 10.1186/s12943-018-0928-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumagai S., Togashi Y., Kamada T., Sugiyama E., Nishinakamura H., Takeuchi Y., Vitaly K., Itahashi K., Maeda Y., Matsui S., et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat. Immunol. 2020;21:1346–1358. doi: 10.1038/s41590-020-0769-3. [DOI] [PubMed] [Google Scholar]
- 15.Park J.E., Kim S.E., Keam B., Park H.R., Kim S., Kim M., Kim T.M., Doh J., Kim D.W., Heo D.S. Anti-tumor effects of NK cells and anti-PD-L1 antibody with antibody-dependent cellular cytotoxicity in PD-L1-positive cancer cell lines. J. Immunother. Cancer. 2020;8:e000873. doi: 10.1136/jitc-2020-000873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Delanoy N., Michot J.-M., Comont T., Kramkimel N., Lazarovici J., Dupont R., Champiat S., Chahine C., Robert C., Herbaux C., et al. Haematological immune-related adverse events induced by anti-PD-1 or anti-PD-L1 immunotherapy: A descriptive observational study. Lancet Haematol. 2019;6:e48–e57. doi: 10.1016/S2352-3026(18)30175-3. [DOI] [PubMed] [Google Scholar]
- 17.Zak K.M., Grudnik P., Guzik K., Zieba B.J., Musielak B., Dömling A., Dubin G., Holak T.A. Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1) Oncotarget. 2016;7:30323. doi: 10.18632/oncotarget.8730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fang W., Zhang J., Hong S., Zhan J., Zhang L.J.O. EBV-driven LMP1 and IFN-γ up-regulate PD-L1 in nasopharyngeal carcinoma: Implications for oncotargeted therapy. Oncotarget. 2014;5:12189. doi: 10.18632/oncotarget.2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lim S.-O., Li C.-W., Xia W., Cha J.-H., Chan L.-C., Wu Y., Chang S.-S., Lin W.-C., Hsu J.-M., Hsu Y.-H., et al. Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell. 2016;30:925–939. doi: 10.1016/j.ccell.2016.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carroll A.R., Copp B.R., Davis R.A., Keyzers R.A., Prinsep M.R. Marine natural products. Nat. Prod. Rep. 2021;38:362–413. doi: 10.1039/D0NP00089B. [DOI] [PubMed] [Google Scholar]
- 21.Villa F.A., Gerwick L. Marine natural product drug discovery: Leads for treatment of inflammation, cancer, infections, and neurological disorders. Immunopharmacol. Immunotoxicol. 2010;32:228–237. doi: 10.3109/08923970903296136. [DOI] [PubMed] [Google Scholar]
- 22.Abdelmohsen U.R., Balasubramanian S., Oelschlaeger T.A., Grkovic T., Pham N.B., Quinn R.J., Hentschel U. Potential of marine natural products against drug-resistant fungal, viral, and parasitic infections. Lancet Infect. Dis. 2017;17:e30–e41. doi: 10.1016/S1473-3099(16)30323-1. [DOI] [PubMed] [Google Scholar]
- 23.Newman D.J., Cragg G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020;83:770–803. doi: 10.1021/acs.jnatprod.9b01285. [DOI] [PubMed] [Google Scholar]
- 24.Jiménez C. Marine Natural Products in Medicinal Chemistry. ACS Med. Chem. Lett. 2018;9:959–961. doi: 10.1021/acsmedchemlett.8b00368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blunt J.W., Copp B.R., Hu W.P., Munro M.H., Northcote P.T., Prinsep M.R. Marine natural products. Nat. Prod. Rep. 2008;25:35–94. doi: 10.1039/b701534h. [DOI] [PubMed] [Google Scholar]
- 26.Davis G.D.J., Vasanthi A.H.R. Seaweed metabolite database (SWMD): A database of natural compounds from marine algae. Bioinformation. 2011;5:361. doi: 10.6026/97320630005361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lyu C., Chen T., Qiang B., Liu N., Wang H., Zhang L., Liu Z. CMNPD: A comprehensive marine natural products database towards facilitating drug discovery from the ocean. Nucleic Acids Res. 2020;49:D509–D515. doi: 10.1093/nar/gkaa763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muneer I., ul Qamar M.T., Tusleem K., Abdul Rauf S., Hussain H.M.J., Siddiqi A.R. Discovery of selective inhibitors for cyclic AMP response element-binding protein: A combined ligand and structure-based resources pipeline. Anti-Cancer Drugs. 2019;30:363–373. doi: 10.1097/CAD.0000000000000727. [DOI] [PubMed] [Google Scholar]
- 29.Ul Qamar M.T., Kiran S., Ashfaq U.A., Javed M.R., Anwar F., Ali M.A., Gilani A.u.H. Discovery of novel dengue NS2B/NS3 protease inhibitors using pharmacophore modeling and molecular docking based virtual screening of the zinc database. Int. J. Pharmacol. 2016;12:621–632. [Google Scholar]
- 30.Kaserer T., Beck K.R., Akram M., Odermatt A., Schuster D.J.M. Pharmacophore models and pharmacophore-based virtual screening: Concepts and applications exemplified on hydroxysteroid dehydrogenases. Molecules. 2015;20:22799–22832. doi: 10.3390/molecules201219880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muszak D., Surmiak E., Plewska J., Magiera-Mularz K., Holak T. Terphenyl-based Small-Molecule Inhibitors of Programmed Cell Death-1/Programmed Death-Ligand 1 Protein—Protein Interaction. J. Med. Chem. 2021;64:15. doi: 10.1021/acs.jmedchem.1c00957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skalniak L., Zak K.M., Guzik K., Magiera K., Musielak B., Pachota M., Szelazek B., Kocik J., Grudnik P., Tomala M., et al. Small-molecule inhibitors of PD-1/PD-L1 immune checkpoint alleviate the PD-L1-induced exhaustion of T-cells. Oncotarget. 2017;8:72167–72181. doi: 10.18632/oncotarget.20050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guzik K., Zak K.M., Grudnik P., Magiera K., Musielak B., Törner R., Skalniak L., Dömling A., Dubin G., Holak T.A. Small-Molecule Inhibitors of the Programmed Cell Death-1/Programmed Death-Ligand 1 (PD-1/PD-L1) Interaction via Transiently Induced Protein States and Dimerization of PD-L1. J. Med. Chem. 2017;60:5857–5867. doi: 10.1021/acs.jmedchem.7b00293. [DOI] [PubMed] [Google Scholar]
- 34.Perry E., Mills J.J., Zhao B., Wang F., Sun Q., Christov P.P., Tarr J.C., Rietz T.A., Olejniczak E.T., Lee T., et al. Fragment-based screening of programmed death ligand 1 (PD-L1) Bioorganic Med. Chem. Lett. 2019;29:786–790. doi: 10.1016/j.bmcl.2019.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amaral M., Kokh D.B., Bomke J., Wegener A., Buchstaller H.P., Eggenweiler H.M., Matias P., Sirrenberg C., Wade R.C., Frech M.J.N.C. Protein conformational flexibility modulates kinetics and thermodynamics of drug binding. Nat. Commun. 2017;8:2276. doi: 10.1038/s41467-017-02258-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Temml V., Garscha U., Romp E., Schubert G., Gerstmeier J., Kutil Z., Matuszczak B., Waltenberger B., Stuppner H., Werz O.J.S.R. Discovery of the first dual inhibitor of the 5-lipoxygenase-activating protein and soluble epoxide hydrolase using pharmacophore-based virtual screening. Sci. Rep. 2017;7:42751. doi: 10.1038/srep42751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tai W., Tao L., Yuan H., Wang F., Liu H., Lu S., Leng Y., Zhang W., Jiang Y., Chen Y. Pharmacophore modeling and virtual screening studies to identify new c-Met inhibitors. J. Mol. Model. 2012;18:3087–3100. doi: 10.1007/s00894-011-1328-5. [DOI] [PubMed] [Google Scholar]
- 38.Antoine Daina O.M., Zoete V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baldwin J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000;43:3867–3877. doi: 10.1021/jm000292e. [DOI] [PubMed] [Google Scholar]
- 40.Ghose A.K., Viswanadhan V.N., Wendoloski J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999;1:55–68. doi: 10.1021/cc9800071. [DOI] [PubMed] [Google Scholar]
- 41.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997;23:3–25. doi: 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- 42.Muegge I., Heald S.L., Brittelli D. Simple Selection Criteria for Drug-like Chemical Matter. J. Med. Chem. 2001;44:1841–1846. doi: 10.1021/jm015507e. [DOI] [PubMed] [Google Scholar]
- 43.Banerjee P., Eckert A.O., Schrey A.K., Preissner R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018;46:W257–W263. doi: 10.1093/nar/gky318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.El-Hasab E.M., El-Bastawissy E.E., El-Moselhy T.F. Identification of potential inhibitors for HCV NS3 genotype 4a by combining protein–ligand interaction fingerprint, 3D pharmacophore, docking, and dynamic simulation. J. Biomol. Struct. Dyn. 2018;36:1713–1727. doi: 10.1080/07391102.2017.1332689. [DOI] [PubMed] [Google Scholar]
- 45.Alamri M.A., Qamar M.T.U., Mirza M.U., Bhadane R., Alqahtania S.M., Muneerf I., Froeyen M., Salo-Ahen O.M.H. Pharmacoinformatics and molecular dynamics simulation studies reveal potential covalent and FDA-approved inhibitors of SARS-CoV-2 main protease 3CLpro. J. Biomol. Struct. Dyn. 2020;39:4936–4948. doi: 10.1080/07391102.2020.1782768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ryde U. QM/MM Calculations on Proteins. Methods Enzym. 2016;577:119–158. doi: 10.1016/bs.mie.2016.05.014. [DOI] [PubMed] [Google Scholar]
- 47.Kuca K., Musilek K., Jun D., Zdarova-Karasova J., Nepovimova E., Soukup O., Hrabinova M., Mikler J., Franca T.C.C., Da Cunha E.F.F., et al. A newly developed oxime K203 is the most effective reactivator of tabun-inhibited acetylcholinesterase. BMC Pharm. Toxicol. 2018;19:8. doi: 10.1186/s40360-018-0196-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ramalho T.C., Cunha E.F.F.D., Castro A.A.D., Pereira A.F., de Lima W.E.A. Flexibility in the Molecular Design of Acetylcholinesterase Reactivators: Probing Representative Conformations by Chemometric Techniques and Docking/QM Calculations. Lett. Drug Des. Discov. 2016;13:360–371. [Google Scholar]
- 49.Kirchmair J., Markt P., Distinto S., Wolber G., Langer T. Evaluation of the performance of 3D virtual screening protocols: RMSD comparisons, enrichment assessments, and decoy selection—What can we learn from earlier mistakes? J. Comput.-Aided Mol. Des. 2008;22:213–228. doi: 10.1007/s10822-007-9163-6. [DOI] [PubMed] [Google Scholar]
- 50.Pin J.P., Bertrand O., Triballeau N., Brabet I., Acher F. Virtual screening workflow development guided by the “receiver operating characteristic” curve approach. Application to high-throughput docking on metabotropic glutamate receptor subtype 4. J. Med. Chem. 2005;48:2534–2547. doi: 10.1021/jm049092j. [DOI] [PubMed] [Google Scholar]
- 51.Drwal M.N., Priyanka B., Mathias D., Wettig M.R., Robert R. ProTox: A web server for the in silico prediction of rodent oral toxicity. Nucleic Acids Res. 2014;42:W53. doi: 10.1093/nar/gku401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., Lindahl E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- 53.Mark P., Nilsson L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A. 2001;105:9954–9960. doi: 10.1021/jp003020w. [DOI] [Google Scholar]
- 54.Kumari R., Kumar R., Lynn A., Open Source Drug Discovery Consortium g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014;54:1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are included within the article.