

Abstract
Pear, belonging to the genus Pyrus, is one of the most economically important temperate fruit crops. Pyrus is an important genus of the Rosaceae family, subfamily Maloideae, and has at least 22 different species with over 5000 accessions maintained or identified worldwide. With the release of draft whole-genome sequences for Pyrus, opportunities for pursuing studies on the evolution, domestication, and molecular breeding of pear, as well as for conducting comparative genomics analyses within the Rosaceae family, have been greatly expanded. In this review, we highlight key advances in pear genetics, genomics, and breeding driven by the availability of whole-genome sequences, including whole-genome resequencing efforts, pear domestication, and evolution. We cover updates on new resources for undertaking gene identification and molecular breeding, as well as for pursuing functional validation of genes associated with desirable economic traits. We also explore future directions for “pear-omics”.
Introduction
As a member of the Rosaceae family and the subfamily Maloideae [1], pear has a wide range of germplasm resources and an ancient cultivation history. There are at least 22 known Pyrus species, with over 5000 accessions that are either cataloged or maintained around the world. These accessions have wide morphological and physiological variability and different ecological adaptations. Among these species, P. communis is mostly cultivated in Western countries, while P. pyrifolia, P. bretschneideri, P. ussuriensis, and P. × sinkiangensis are primarily cultivated in Asian countries. China is a major producer of pear, accounting for 71.40% of world pear production, and supplies approximately 17.60% of the export pear market (http://www.fao.org/faostat/en/#home, 2019). However, the current export price of Chinese pears is lower than that of the international average market price. This is because fruits of major Asian pear cultivars have higher stone cell contents, often lack an overall attractive appearance, particularly compared with those of red-colored European pears, and have relatively bland flavors [2–4]. Traditional pear breeding efforts are mainly dependent on sexual crosses between parental types and the selection of promising or opportunistic seedlings, mutation breeding and selection of improved mutants, and the selection of desirable spontaneous bud mutants. Overall, controlled sexual crosses are most often used in pear breeding.
Pears are often self-incompatible and have long juvenile periods (5–7 years), often leading to the independent segregation of traits of interest in the resultant hybrids from sexual crosses. Overall, traditional pear breeding is difficult, time-consuming, and costly, as it requires the long-term commitment of labor, materials, and land-space resources. Therefore, the availability and use of molecular markers and genomic selection can significantly contribute to overcoming some of these constraints of traditional breeding.
Prior to the release of the pear genome sequence, only a few genes and markers associated with important economic traits were identified. However, once genomic tools and new genetic approaches became available, a wide variety of valuable technologies and outcomes have been pursued and achieved in pear, including genetic transformation, genome sequencing, molecular markers, genetic and physical mapping, and comparative genomic analyses. Furthermore, the availability of tools and data resources has offered new opportunities for the efficient and robust discovery of genes controlling desirable fruit quality traits, fruit productivity traits, and postharvest storage life, as well as those that greatly shorten the breeding cycle in pear.
In 2013, the first genome sequence of Asian pear was released [5], thereby allowing delineation of chromosome evolution, genomic structure, and patterns of genetic variations. With the availability of a high-quality pear genome sequence, several gene families involved in controlling desirable and economic traits have now been identified [6–13]; the development of reliable and robust molecular markers [14], new genetic mapping initiatives [15], and analysis of genome evolution [5] have also occurred. This new knowledge and these resources will continue to have significant impacts on efforts for the genetic improvement of pears.
In this review, we will provide a summary of pear genome sequences and highlight the applications of high-throughput technologies that have contributed new knowledge about the biology of pear. These advances have provided new knowledge on the origins of pear and new insights into our understanding of the independent domestication history of Asian and European pears. Furthermore, the construction of high-density genetic linkage maps and the pursuit of genome-wide association studies (GWAS) have contributed to the identification of genomic loci for key genes regulating various economic and desirable agronomic traits. In addition, this review covers developments of multiple omics resources useful for the identification of candidate genes and evaluates the impacts of these findings on future efforts for pear breeding. This roadmap for the pear genome will serve as a useful guide for pursuing genetic improvement efforts toward developing new high-quality and well-adapted pear cultivars. This roadmap is also critical for addressing issues of the pear response to climate change, as well as of ever-changing consumer demands and preferences.
Overview of Asian and European pear genome sequences
Pear has a basic chromosome number of 17 (2n = 34). Different pear species have distinctly different genome sizes, ranging from 500 to 650 Mb, and possess high numbers of repeats and transposable elements (TEs), as well as high levels of heterozygosity.
The first draft genome sequence of the pear cv. ‘Dangshansuli’ (P. bretschneideri Rehd.), also known as Chinese white pear (Asian pear), was assembled based on a bacterial artificial chromosome (BAC)-by-BAC strategy, thereby alleviating issues of high rates of heterozygosity and the complexity of repeated reads [5]. A total of 2,103 scaffolds were assembled for this pear genome with an N50 of ~0.54 Mb, representing 97.1% (512.0 Mb) of the estimated genome size (527.0 Mb) with 194× genome coverage (Table 1). Based on a high-density genetic map, a total of 386.7 Mb sequences, corresponding to ~75.5% of the assembled genome, were anchored to all 17 chromosomes of the pear genome. Repetitive sequences accounted for 53.1% (271.9 Mb) of the assembled genome. A high long-terminal repeat (LTR) expansion rate suggested that the pear genome was in continuous expansion. Compared with the apple (Malus × domestica Borkh.) genome, another member of the subfamily Maloideae, it was proposed that the presence of large numbers of repeat sequences primarily contributed to the size differences between the pear and apple genomes [16]. Importantly, genes involved in the synthesis of stone cells, sugars, and volatile compounds, as well as those involved in disease resistance and self-incompatibility, were identified in the pear genome. However, haplotype features and allele-specific expression were difficult to determine owing to the high rates of heterozygosity, as well as the lack of available accessions with haplotype-derived homologous chromosomes in Asian pears. Subsequently, a haplotype-resolved genome for pear (P. bretschneideri) was developed using a new approach for protoplast isolation from pollen combined with single-cell DNA sequencing of 12 pollen cells and ‘barcode’ phasing of 38,304 BAC sequences [17]. Thus, the assembled genome sizes of the haploid genomes A and B were 546 Mb and 536 Mb, respectively. The haploid genome assembly also revealed that 8.12% of the genes identified in the first pear reference genome featured mosaic assemblies [17].
Table 1. Comparisons among different sequenced pear genomes.
| Contigs | ‘Dangshansuli’ | Bartlettv1.0 | BartlettDHv2.0 | ‘Shanxi Duli’ | ‘Zhongai 1’ |
|---|---|---|---|---|---|
| Number of contigs | 25,312 | 182,196 | 620 | 595 | 1241 |
| Total size of contigs (Mb) | 501.3 | 507.7 | 501 | 497 | 510.6 |
| N50 contig length (kb) | 35.7 | 6.6 | 5300 | 1571.5 | 1277.3 |
| Longest contig (Mb) | 0.3 | 0.1 | / | / | 6.5 |
| Scaffolds | |||||
| Number of scaffolds | 2,103 | 142083 | 592 | 139 | 784 |
| Total size of scaffolds (Mb) | 512 | 577.3 | 496.9 | 532.7 | 510.6 |
| N50 scaffold length (kb) | 540.8 | 88.1 | 6500 | 28122.4 | 23450 |
| Longest scaffold (Mb) | 4.1 | 1.2 | / | 45.5 | 31.9 |
| Anchored size to the chromosome (Mb) | 386.7 | 171.3 | 445.1 | 500 | 506.3 |
| Anchored rate to the chromosome (%) | 75.5 | 29.7 | 84.2 | 94 | 99.2 |
European pear, P. communis L., also an economically important species that is widely cultivated in Western countries, has distinct phenotypic and fruit quality characteristics that differ from those of Asian pear, including fruit shape, taste, lignin content, and aroma [2–4, 18, 19]. The whole genome sequence and annotation of European pear have aided in pursuing comparative genomic studies with Asian pear [20].
A draft genome sequence of the European pear ‘Bartlett’ version 1.0 was assembled and released using next-generation sequencing (NGS) technology (Roche 454) [20]. A total of 142,083 scaffolds were assembled, corresponding to 577.3 Mb, and represented 96.2% of the expected 600 Mb of the European pear genome (Table 1) [20]. The number of predicted genes in European pear was higher than that reported for most other plant species, but this number was similar to that identified in Asian pear. This result might be expected due to the incidence of whole genome duplication (WGD) events in members of the Maloideae subfamily [16]. In addition, the predicted coding region length (1,209 bp), exon length, and gene density in the European pear genome were found to be similar to those detected in Asian pear.
Recently, an updated version of the European pear genome using a double-haploid ‘Bartlett’ cultivar was published [21]. The quality and completeness of the European pear draft genome, designated BartlettDHv2.0, were greatly enhanced by integrating multiple technologies [21]. In this updated draft genome, a total of 496.9 Mb sequences were assembled, and 445.1 Mb were anchored and oriented across all 17 chromosomes of the pear genome using both Hi-C data and a high-density genetic map (Table 1). A total of 50% of the sequences (~247 Mb) in the European pear genome were found to be repetitive sequences, and 37,445 protein-coding genes were annotated, corresponding to a 13% reduction in predicted protein-coding genes from the previous two draft genomes of both P. communis and P. bretschneideri.
In addition to these three genome sequences of cultivated pears, genomes of the wild pear P. betulaefolia Bunge [22] and ‘Zhongai 1’ (P. ussuriensis × P. communis), a dwarfing hybrid rootstock [23], were recently sequenced and assembled in ongoing efforts to expand the pool of sequenced pear genomes. Most of the pear genome sequences and annotation-related datasets have been stored in the Genome Database of Rosaceae (GDR: https://www.rosaceae.org/tools/jbrowse) [24].
With the development of NGS, many resequencing studies are now underway. These expanded sequencing datasets have been widely used to explore the origin, domestication history, and evolutionary processes of various fruit crops, including those for peach [25], grape [26], and apple [27].
Recently, 113 representative Pyrus accessions worldwide were resequenced [28]. This study led to the proposal of a new paradigm for pear germplasm dispersion and domestication. In this paradigm, it was suggested that current Asian and European pears originated from the southeast region of China, spread over to central Asia, and were then subsequently dispersed across Asia and Europe. Thus, each of the cultivated Asian and European pears independently underwent domestication in local regions [28]. Due to such independent domestication events, Asian and European pears were subjected to different selection pressures to meet the fruit appearance and taste preferences of different human populations. Generally, Asian pear has a round-shaped fruit with notable features, including crisp flesh, high stone cell content, low acidity, minimal aroma, high sugar, and mild flavor, whereas European pear is characterized by a typical pyriform-shaped fruit with smooth flesh, few stone cells, soft flesh, strong aroma, and strong flavor [2–4, 29]. At the genomic level, a total of 9.29 Mb of the genome, containing 857 genes, was identified to carry selective signatures in Asian pears, whereas a total of 5.35 Mb of genomic regions, containing 248 genes, was identified to carry selective signatures regions in European pears. However, only 47 genes were found to be common between the two pear types, suggesting that these genes were selected upon during the domestication of both Asian and European pears. These findings supported the hypothesis that Asian pears and European pears were independently domesticated in different regions.
Pear domestication and improvement
Understanding the domestication and genetic improvement of a crop at the whole genome level can aid in future efforts to improve crop yield and other traits of interest.
By using resequencing and population genetic analysis of four different populations of pear (Asian wild, Asian cultivated, European wild, and European cultivated accessions), it was revealed that a weak domestication event occurred in pear [28]. The divergence time of Asian pears from European pears appeared to occur from 3.3 to 6.6 million years ago (MYA) compared to other plant species, including grape (Vitis vinifera), apple (M. × domestica), peach (Prunus persica), woodland strawberry (Fragaria vesca), poplar (Populus trichocarpa), papaya (Carica papaya), and the model plant Arabidopsis (Arabidopsis thaliana) (Fig. 1A). Furthermore, relationships between five domesticated pear populations and their corresponding wild relatives were delineated, with findings supporting an evolutionary model based on independent domestication of European and Asian pears (Fig. 1B). Specifically, it was demonstrated that S-RNase genes underwent rapid evolution and balancing selection. Furthermore, the relatively low level of overlap in signatures of selection between Asian and European pears suggested that the targets of selection differed between the two pear types. Moreover, the independent domestication model of Asian and European pears was also supported by the incidence of separate groups in the phylogenetic tree, which further demonstrated that the divergence time of Asian and European pears occurred much earlier and prior to any possible human intervention. Notably, genes associated with fruit size, sugars, organic acids, stone cells, and volatile compounds were present in regions with selective sweep signatures (Fig. 1C). To further explore genetic changes at the RNA level that occurred during domestication and improvement, Li et al. [30] used a transcriptome dataset of 41 pear (P. pyrifolia) genotypes, consisting of 14 wild, 12 landrace, and 15 improved genotypes, to explore the genetic changes related to the domestication and improvement of pear. It was found that 11.13 Mb of genome sequence carried selective signatures of pear domestication, while 4.04 Mb of genomic regions carried selective signatures of pear improvement (Fig. 1C). Of particular note, several genes related to sugar content, stone cell content and fruit size were located in these selected regions, with some of these genes mapping to previously reported QTLs [30].
Figure 1.

A schematic diagram of divergence time and an evolutionary model of multiple plant species, including Pyrus, as well as a model of the domestication of Asian and European pears. (A) Divergence time of nine species, including Vitis vinifera, Malus × domestica, Pyrus communis, Pyrus bretschneideri, Prunus persica, Fragaria vesca, Populus trichocarpa, Carica papaya, and Arabidopsis thaliana. The estimated divergence times (MYA) are inferred based on single-copy orthologous groups and shown at each node. WGD, whole genome duplication. (B) An evolutionary model of wild and cultivated pear species [28]. (C) A schematic representation of the independent domestication process of Asian and European pears along with subsequent modern breeding efforts for cultivar improvement. The domestication process of pear has experienced a weak bottleneck resulting in slightly reduced diversity, while the improvement process shows significantly reduced diversity; thus, new efforts for restoring diversity are extremely urgent in the genetic improvement of pear cultivars.
Genome-wide variations
A high degree of diversity can be expected at the genomic level in self-incompatible perennial plants, such as that reported for Pyrus [28]. Often, NGS and long-read sequencing analyses detect numerous types of DNA sequence variants, including SNPs, InDels, and structural variations (SVs).
Genome resequencing efforts offer new knowledge pertaining to taxonomic classifications, phylogenetic relationships, evolutionary history, domestication, and genetic resources for pursuing innovative molecular-based breeding efforts. A recent genome-wide variation study involving 113 pear accessions representing all known Pyrus species (cultivated and wild, collected from 26 countries) confirmed the delineation of Asian and European pears [28]. This study revealed that P.× sinkiangensis was derived from a hybridization event between a cultivated Asian pear and a cultivated European pear.
Although RNA-seq is often used to assess levels of genome-wide gene expression, it also allows for the identification of genomic variants within transcribed coding gene regions. RNA-seq was used for SNP mining of the fruit of five different pear cultivars, including ‘Hosui’, ‘Yali’, ‘Nanguoli’, ‘Kuerlexiangli’, and ‘Starkrimson’ [31]. Subsequently, fruits at the enlarged fruit stage were collected from a group of 41 accessions of P. pyrifolia consisting of 15 enhanced genotypes, 12 landraces, and 14 wild accessions and subjected to RNA-seq. This transcriptome analysis led to the identification of 875,319 high-quality SNPs [30]. Based on these SNP data, landrace and wild pears were found to be closely related to each other, while a low level of genetic diversity was observed in the improved cultivar group [30]. Moreover, using nucleotide diversity (π) and FST values, it was found that different selective sweeps occurred during both domestication and improvement. Moreover, the differential expression of selected genes showed a 20.89% decrease from the wild group to the landrace group, while a 23.13% increase was observed from the landrace group to the enhanced genotype group. This result indicated that diversifying selection might play an important role during the improvement of pear [30].
A reduced-representation genotyping-by-sequencing (GBS) strategy targeting regions flanking restriction enzyme (RE) sites was developed by Elshire et al. [32]. This genotyping approach focuses on DNA sequence polymorphisms around methylation-sensitive RE sites and yields genome-wide polymorphism data across numerous samples. Unlike SNP arrays, GBS involves the simultaneous detection and scoring of SNPs in a population of interest; thus, this approach is free from ascertainment bias. The availability of reference genome sequences for both Asian [5] and European [20] pears has facilitated the implementation of GBS in pear genetic studies. Kumar et al. [33] conducted a GBS study using a single RE (BamHI) to assess genetic diversity in Asian and European pears. Moreover, GBS was found to be useful for constructing a high-density linkage map, fine-mapping QTLs for red skin color [34], and pursuing genomic selection (GS) in interspecific populations of pear [35]. The restriction enzyme ApeKI was used to construct high-density linkage maps for Asian [36] and European pears [37]. Recently, a two-enzyme (EcoRI and NIaIII)-based GBS was used to construct a linkage map for Asian pear [38]. Regardless of the enzyme system, GBS poses technical uncertainties that can result in the uneven sequencing of samples, the nonuniform distribution of SNPs, and missing genotype data [39].
With the release of pear reference genome sequences along with the availability of large numbers of SNPs, it has become feasible to design high-density SNP arrays for pear. Recently, a high-density 200K SNP genotyping array was constructed [40]. This array was used to improve the genome assembly, genetic mapping, and GWAS in pear [40]. A 70K Axiom array was also released [41]. Owing to its efficiency, flexibility, high throughput, and low cost, such a SNP array will serve as an important reference tool for GWAS and will be highly useful in pursuing further germplasm improvement and breeding efforts.
Genetic linkage maps: Construction and mapping of trait-linked loci
Genetic linkage maps are powerful tools for use in investigating and understanding how agronomic traits are inherited from their parents. Prior to the assembly of the first Asian pear genome, studies focused on developing frameworks and encryption of linkage mapping [42–45]. It was not until Yamamoto et al. [46] used an F1 population of two European pear cultivars, ‘Bartlett’ and ‘La France’, that two independent maps for pear consisting of 17 linkage groups and corresponding to the basic chromosome number (n=17) were constructed. Subsequently, with the rapid development of pear genome sequences [5, 20], genome-wide DNA markers, including SNPs and simple sequence repeats (SSRs), were identified and developed. Wu et al. [15] used 3,143 SNPs from restriction-associated DNA sequencing (RAD-seq) to construct the first SNP-based high-density genetic map for pear and then used 98 SSR markers to anchor the corresponding linkage groups. The resulting map consisted of 3,241 markers, spanning 2,243.4 cM with an average distance of 0.70 cM between markers. This map has enhanced the development and analysis of pear genetic maps, along with the identification of QTLs for traits of interest that are useful in marker-assisted breeding (MAB) efforts [15].
Subsequently, linkage maps of different genetic backgrounds were constructed. However, these linkage maps lacked common markers, rendering it difficult to conduct comparative studies and to pursue analysis across different pear populations (Table 2). Li et al. [14] collected all genetic linkage maps of European pears and used common SSR markers across these different maps to merge them into a single integrated consensus map. This map allowed for anchoring a total of 291.5 Mb of the 'Bartlett' v1.0 sequence, exploring genetic structure patterns, conducting comparative studies among different maps, and identifying QTLs [14].
Table 2.
QTL mapping of different agronomic traits in pear
| Population | Population size | Marker type | Number of markers | Map length (cM) | Number of linkage groups (LGs) | Interval (cM) | Traits | LGs | Reference | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Female | Male | Female | Male | Female | Male | Female | Male | ||||||
| ‘Kinchaku’ × ‘Kosui’ | 82 | RAPD | 120 | 78 | 768 | 508 | 18 | 22 | 4.2 | Black spot | 11 | 42 | |
| ‘Bartlett’ × ‘Hosui’ | 63 | AFLP, SSR | 226 | 54 | 949 | 926 | 18 | 17 | 4.9 | Self-incompatibility | 17 | 43 | |
| ‘Passe Crassane’ × ‘Harrow Sweet’ | 99 | SSR, AFLP, MFLP, AFLP-RGA, RGA | 155 | 156 | 912 | 930 | 18 | 19 | 5.8 | 6 | Fire blight | 2a, 2b, 4, 9 | 45 |
| ‘Bartlett’ × ‘Hosui’ | 63 | AFLP, SSR | 256 | 180 | 1020 | 995 | 19 | 20 | 4 | 5.5 | Self-incompatibility | 17 | 44 |
| 'Bartlett' × 'Hosui' | 63 | AFLP, SSR | 447 | 1000 | 17 | 2.3 | - | - | 46 | ||||
| ‘Hosui’ × ‘La France’ | 55 | AFLP, SSR | 414 | 1156 | 17 | 2.8 | - | - | 46 | ||||
| ‘Abbè Fétel’× ‘Max Red Bartlett’ | 95 | MFLP, SSR | 123 | 110 | 908.1 | 879.8 | 18 | 19 | 7.4 | 8 | Pear scab | 3, 7 | 50 |
| ‘Abbè Fétel’ × ‘Max Red Bartlett’ | 95 | MFLP, SSR | 123 | 110 | 908.1 | 879.8 | 18 | 19 | 7.4 | 8 | Skin color | 4 | 48 |
| ‘Hosui' × ‘La France’ | - | AFLP, SSR | 484 | 1204 | 17 | 2.49 | Pear scab | 2,11,14 | 52 | ||||
| Self-incompatibility | 17 | ||||||||||||
| 'Bartlett' × 'Hosui' | - | AFLP, SSR | 504 | 356 | 1080 | 1180 | 17 | 16 | 2.1 | 3.31 | - | - | 53 |
| ‘Bartlett’ × ‘Hosui’ | 63 | AFLP, SSR | 335 | 1174 | 17 | 3.5 | - | - | 53 | ||||
| Leaf width | 10, 15 | ||||||||||||
| Leaf length/width | 5 | ||||||||||||
| ‘Yali’ × ‘Jingbaili’ | 145 | AFLP, SSR | 402 | 18 | 1395.9 | 3.8 | Leaf length | 8, 15, 16 | 54 | ||||
| Petiole length of leaf | 4, 15 | ||||||||||||
| ‘Abbè Fétel’ × ‘Max Red Bartlett’ | - | SSR | - | - | - | - | Red skin color | 9 | 55 | ||||
| ‘Niitaka’ × ‘Suhyangri’ | 94 | RAPD, AFLP, SSR | 106 | 122 | 1006 | 1168 | 19 | 19 | 9.5 | 9.6 | - | - | 56 |
| ‘Bayuehong’ × ‘Dangshansuli’ | 97 | AFLP, SRAP, and SSR | 214 | 122 | 1352.7 | 1044.3 | 17 | 17 | 6.3 | 8.6 | Fruit weight | 2, 7, 8, 10 | 47 |
| Fruit diameter | 10, 15 | ||||||||||||
| Fruit length | 7 (two years),8 | ||||||||||||
| Fruit shape index | 1, 2 (two years), 7, 8 | ||||||||||||
| SSC | 2, 5, 6 | ||||||||||||
| Fruit maturity date | 8 (two years) | ||||||||||||
| ‘Red Bartlett’ × ‘Nanguo pear’ | 74 | SRAP | 103 | 105 | 602.2 | 650 | 20 | 20 | 4.9 | 5.2 | - | - | 57 |
| ‘Bartlett’ × ‘Hosui’ | 63 | SSR, SNP | 485 | 965 | 17 | 2 | - | - | 58 | ||||
| ‘Housui’ × ‘La France’ | 55 | SSR, SNP | 370 | 415 | 1160 | 1177 | 17 | 20 | 3.1 | 2.8 | - | - | 58 |
| PEAR1 × PEAR2 | 143 | SSR, SNP | 250 | 314 | 1132.3 | 1136.8 | 17 | 17 | 4.53 | 3.62 | Pear scab | 2, 5, 7, 10, 17 | 59 |
| ‘Akiakari’ × ‘Taihaku’ | 93 | SSR, EST-SSR | 208 | 275 | 799.1 | 1039.1 | 17 | 17 | 3.84 | 3.78 | Fruit skin color | 8 (two years) | 60 |
| Fruit weight | 3, 11 | ||||||||||||
| Total soluble solids content | 4, 8 | ||||||||||||
| Firmness | 4 (two years) | ||||||||||||
| Preharvest fruit drop | 1, 15 (two years) | ||||||||||||
| Harvest time | 3 (two years), 15 (two years) | ||||||||||||
| ‘Bayuehong’ × ‘Dangshansuli’ | 56 | SSR | 734 | 1661.4 | 17 | 2.3 | - | - | 61 | ||||
| ‘Aihuali’ × ‘Chili’ | 215 | SSR, SLAF | 12 | - | 1 | - | Dwarf growth habit | 16 | 62 | ||||
| ‘Red Clapp’s Favorite’ × ‘Mansoo’ | 161 | SSR, SLAF | 4797 | 2703.6 | 17 | 0.56 | - | - | 63 | ||||
| Nine published maps | SSR, SNP | 5085 | 3266 | 17 | 0.6 | - | - | 14 | |||||
| ‘Spadona’ × ‘Harrow Sweet’ | 162 | SNP | 2036 | 1433 | 17 | 0.70 | Vegetative budbreak time | 5,8,9, 13,15,17 | 37 | ||||
| ‘Mantianhong’ × ‘Hongxiangsu’ | 345 | SNP | 2606 | 1847 | 17 | 0.71 | - | - | 64 | ||||
| Skin color | 4, 13, 16 (two years) | ||||||||||||
| Single fruit weight | 13, 17 | ||||||||||||
| Transverse diameter | 3, 11, 17 | ||||||||||||
| Vertical diameter | 11, 17 (two years) | ||||||||||||
| Soluble solid content | 5, 10, 14 | ||||||||||||
| ‘Bayuehong’ × ‘Dangshansuli’ | 102 | SSR, SNP | 3241 | 2243.4 | 17 | 0.7 | Flesh color | 9 (two years) | 15 | ||||
| Skin smooth | 2, 17 | ||||||||||||
| Length of pedicel | 2, 14, 17 | ||||||||||||
| Calyx status | 6 (two years) | ||||||||||||
| Juice content | 1, 5 | ||||||||||||
| Number of seeds | 5 (two years), 9, 14, 17 (two years) | ||||||||||||
| ‘Yuluxiang’ × ‘Mantianhong’ | 162 | SNP | 2489 | 1668 | 17 | 0.67 | - | - | 64 | ||||
| ‘E1 Dorado’ × ‘Potomac’ | 81 | SNP | 27052 | 1034.97 | 17 | 0.82 | Fire blight | 2 | 65 | ||||
| ‘Old Home’ × ‘Bartlett’ | 100 | SNP | 29703 | 1147.22 | 17 | 0.63 | Fire blight | 2 | 65 | ||||
| ‘NJA2R59T6’ × ‘Bartlett’ | 82 | SNP | 37182 | 1080.5 | 17 | 0.59 | Fire blight | 2 | 65 | ||||
Note: AFLP, amplified fragment length polymorphism; cM, centi-Morgan; EST, expressed sequenced tag; LGs, linkage group; MFLP, microsatellite-anchored fragment length polymorphism; RAPD, random amplified polymorphic DNA; RGA, resistance gene analog; SLAF, specific locus amplified fragment; SNP, single nucleotide polymorphism; SRAP, sequence-related amplified polymorphism; and SSR, simple sequence repeat.
Genetic linkage maps can also be used to localize positions or genetic regions controlling target agronomic traits. Several important pear fruit traits have been located on particular chromosomes. As most traits assessed in linkage mapping populations to date are quantitative in nature, QTLs related to these traits are distributed along several linkage groups (LGs) (Table 2) [15, 47]. For example, QTLs for single fruit weight were identified on LGs 2, 3, 7, 8, 10, 11, 13, and 17. Moreover, a QTL for fruit firmness was identified on LG4, while those for harvest time were identified on LG3 and LG15 in Japanese pear. Another important economic trait is fruit skin color, in particular red-color pigmentation, a QTL for which was first located on LG4 using an F1 population of ‘Max Red Bartlett’, a red color mutant of ‘Bartlett’ (Table 2) [48]. Subsequently, map-based cloning was used to identify a red skin-related QTL on the tail end of LG5 associated with the PyMYB114 gene, which regulates fruit anthocyanin biosynthesis [49]. Furthermore, QTLs for various disease resistance-related traits, such as fire blight [45], pear scab [50], and black spot [42], were identified along different linkage groups (Table 2) [51–65].
Genome-wide association studies (GWAS) in pear
Genome-wide association studies (GWAS) serve as effective approaches for exploring genome-level genetic architecture(s) and have been widely used to identify genetic variants associated with human-related diseases. These approaches are adopted for use in plant population studies to identify candidate loci associated with complex traits. Thus, GWAS provides opportunities for identifying candidate genetic loci associated with complex traits in pear and for developing robust molecular markers useful in pursuing MAB to enhance the rapid and accurate development of new cultivars.
The development of GWAS models varies with demands for degrees of accuracy, speed, and population size. A generalized linear model (GLM) can handle multiple variables and takes into account the population structure in the form of a covariance structure defined by the index Q [66]. Population structure can result in false-positive genotype-phenotype associations, and PCA is commonly used to quantify and correct for population structure in GWAS. Moreover, a mixed linear model (MLM) is also available for use in GWAS. An efficient mixed-model association (EMMA) [67] and a genome-wide efficient mixed-model association (GEMMA) [68] can serve as variance components and are exact methods, both belonging to MLM. EMMA eXpedited (EMMAX) [69, 70] and genome-wide rapid association using mixed model and regression (GRAMMAR) are approximate methods that allow for fast calculation of GWAS. However, GRAMMAR tends to underestimate associations [71]. Thus, a comprehensive overview of various factors must be taken into consideration, such as target species, quantitative and qualitative traits, population size, accuracy and speed, when different GWAS models are to be used.
For GWAS in perennial plants, such as fruit trees, natural germplasm populations are more readily available than controlled-cross (or hybridized) populations. GWAS has been conducted on various fruit trees, including apricots [72], peaches [73], and apples [27]. Recently, GWAS was conducted using 312 sand pear (P. pyrifolia) accessions and identified five loci associated with three fruit phenological traits, as well as 37 loci associated with eight fruit quality traits. Furthermore, a new gene, PbrSTONE, controlling stone cell conformation, was identified within these association loci [74].
Therefore, GWAS is a viable and useful approach for identifying QTLs/genes for various critical fruit quality traits and other desirable traits, as well as for contributing to the development of robust tools and resources for pursuing MAB in pear and other long-lived woody perennial trees, including self-incompatible fruit trees.
Marker-assisted selection (MAS), marker-assisted breeding (MAB), and genomic selection (GS) in pear
As pear has a long generation time (5 to 7 years), traditional pear breeding programs are expensive and time-consuming [75]. Marker-assisted selection (MAS) and marker-assisted breeding (MAB) are deemed viable approaches for the molecular mapping of genes, as well as for pursuing genetic improvement efforts, particularly of long-lived perennial fruit trees, as most economic traits are complex and controlled by QTLs [76].
Early use of molecular marker systems included SSRs, amplified-fragment length polymorphisms (AFLPs), sequence-characterized amplified regions (SCARs), random amplified polymorphic DNAs (RAPDs), and cleaved amplified polymorphic sequences (CAPS) [15, 77–79]. All these molecular markers relied on the use of gel electrophoresis methods for screening individuals, seedlings, selections, and germplasm accessions. The release of whole genome sequences and the availability of robust SNP markers have contributed to accelerated progress in breeding programs. For example, high-quality and high-density linkage maps combined with molecular markers have allowed for fine-mapping of the region around a gene for susceptibility to black spot disease, as well as for identifying QTLs for 11 pear fruit-related traits [80].
High-throughput and accurate SNP-based markers, SNP arrays, and GBS can assist in pursuing early trait-predictive assays [37, 81]. DNA markers flanking major causal loci, such as those for red pear fruit skin color, can be used for selection, but a traditional MAS scheme is not well suited for complex traits that are controlled by several loci. GS is a form of MAS that utilizes thousands of genome-wide markers simultaneously to calculate their associations with trait phenotypes in a training population and to predict the genomic estimated breeding values (GEBVs) of individuals in a tested population [82]. The GEBVs of selection candidates are estimated solely based on SNP genotypes and estimated SNP effects. Hence, outstanding candidates can be identified at very early stages of development prior to phenotyping, thereby reducing generation intervals and increasing breeding efficiency (Fig. 2). GS is best suited for polygenic traits, and high-density genotyping is essential for the application of GS to ensure that all QTLs are in population-wide linkage disequilibrium with SNP markers.
Figure 2.
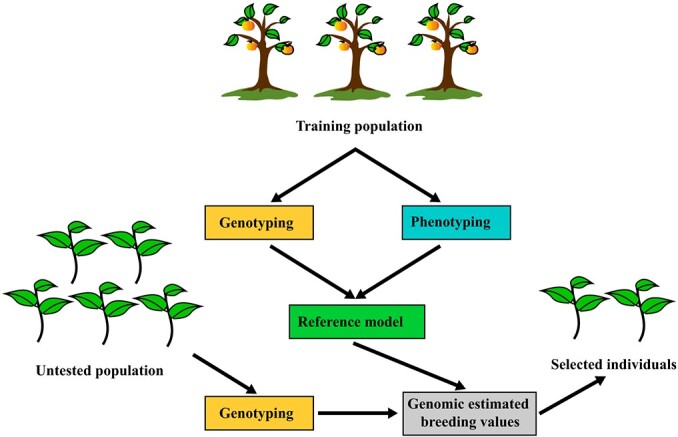
A pipeline for genomic selection
In a preliminary study to assess the utility of GS in a sand pear (P. pyrifolia) breeding program, Iwata et al. [83] screened 76 cultivars using 162 genome-wide markers and found that genome-wide predictions for GS were highly accurate (0.75) for harvest time and moderately accurate (0.38–0.61) for resistance to black spot, firmness of flesh, fruit shape (longitudinal section), fruit size, acid content, and number of spurs. Minamikawa et al. [84] used an Illumina Golden Gate genotyping assay comprising 1536 SNPs to conduct genomic predictions of fruit phenotypes in a P. pyrifolia population consisting of 86 cultivars and 765 seedlings. They found that the accuracy of genomic prediction was further enhanced when full-sib family data of a target family were available and suggested that phenotypic data collected in a breeding program were useful for pursuing GWAS and GS when these methods were combined with genome-wide marker data. Kumar et al. [35] evaluated the genetic architecture of 10 fruit phenotypes (including sensory traits) and used GBS to assess the potential of GS in 550 hybrid seedlings.
The above studies have demonstrated that the accuracy of genomic predictions in pear is moderate, particularly as most fruit quality traits are complex and polygenically controlled. Therefore, additional studies must be conducted to develop larger genotype-phenotype datasets to further improve the prediction accuracy of the fruit phenotypes of untested seedlings.
Multiple omics: Identifying genes related to important traits
Candidate gene mining at the whole-genome level
As pear genomes have become available, the ability to identify gene families associated with desirable traits or traits of interest has improved. Recently, studies have focused on identifying genes associated with fruit quality traits, such as those controlling skin color, sugar content, stone cells, and aromatic compounds. Members of the MADS-box gene family, MYB gene family, and lateral organ boundary domain (LBD) gene family were identified as candidate genes associated with anthocyanin biosynthesis in pear [8, 10, 85]. Sugar transporters, a SWEET gene family, the hexokinase gene family, and the phosphofructokinase gene family were identified as candidate genes associated with sugar biosynthesis [6, 9, 86, 87]. For the stone cell trait, MYB gene family, BZR gene family, and KNOX gene family were reported to be involved in stone cell conformation and related lignin biosynthesis in pear [8, 88, 89]. Flavor is a more complicated trait, and some candidate pear genes were identified as candidate genes contributing to aromatic compounds present in pear fruit [90, 91].
Transcriptomes
A transcriptome, usually generated by RNA-seq, corresponds to all transcripts within a single cell or a population of cells at either a particular developmental stage or a physiological state [92].
The release of large-scale transcriptomic data for pear, including those for different cultivated species, developmental stages, tissues, and treatments for specific trait investigations, has facilitated the exploration of functional genes. Transcriptomes of ‘Dangshansuli’, ‘Nanguoli’, ‘Yali’, ‘Hosui’, ‘Kuerlexiangli’, ‘Starkrimson’, and P. pyrifolia accessions at key developmental stages were released, and these provided insights into biological processes underlying fruit quality traits, including stone cell contents, sugars, and volatiles [31]. In addition, gene expression during four developmental stages of pear pollen provided opportunities for studying the growth and cessation of pear pollen tubes [93]. Furthermore, transcriptome data from seven tissues were exploited to track the expression patterns of gene pairs between two pear subgenomes, thus providing critical evidence for unbiased subgenome evolution following palaeopolyploidization in pear and serving as valuable resources for investigating tissue-specific gene expression in pear [94].
In pursuit of specific pear traits, transcriptome analysis studies have focused on color pigment development, dormancy, and biotic and abiotic stresses. Transcriptomes of ‘Starkrimson’ pear versus its green color mutant, bagged versus unbagged ‘Pingguoli’, color-fading ‘Red Bartlett’ versus non-color-fading ‘Starkrimson’, russet- versus green-pericarp individuals of the ‘Qingxiang’ × ‘Cuiguan’ F1 group, and the color mutant ‘Red Zaosu’ versus ‘Zaosu’ were compared. As a result, several key candidate genes, including LAR, ANR, Myb4-like1, myb4-like2, CCR, CAD, and PpBBX24, were identified as candidates for color pigmentation [95–99]. Bud dormancy is a key developmental process for perennial plants to survive under adverse environmental conditions. When the transcriptomes of endodormant and ecodormant Japanese pear (‘Kosui’) flower buds were subjected to RNA-seq, it was found that phytohormones, such as ethylene, were involved in endodormancy release [100]. Several studies were conducted to investigate cold, drought, salt stress, and black spot disease in pear, yielding critical datasets that would facilitate the identification and functional analysis of candidate genes [101–106].
Proteomics
Proteomics is a tool for investigating proteins associated with gene expression and for delineating biochemical networks. In pear, a total of 1810 proteins were identified to be involved during the three stages of pear fruit development [107]. Moreover, 35 differentially expressed proteins related to fruit quality were identified, including three proteins related to sugar formation, seven proteins related to aroma synthesis, and 16 proteins related to lignin formation [107]. In another study, a total of 2841 proteins were identified during seven different pear fruit development stages, and it was suggested that invertases do not play a major role in sugar conversion in developing pear fruit; rather, it was likely that sucrose was broken down by sucrose synthases [108]. Furthermore, several putative sugar transporters from diverse gene families demonstrated developmental regulation [108].
To delineate the mechanism of fruit maturity of an early-maturing bud sport of ‘Zaosu’ pear, 75 differentially expressed protein spots were identified between an early-maturing bud sport and its original cultivar ‘Zaosu’ using a combination of 2-DE and MALDI-TOF MS technology [109]. Most of these differentially expressed proteins were closely associated with maturation, thus elucidating the maturation process in these pear genotypes [109].
Metabolomics
Metabolomics deals with the analysis of large numbers of metabolites within a single cell, tissue, or organ in response to various conditions or treatments. In pear fruit, small molecule metabolites play important roles, such as those involved in basic metabolism; however, there are currently limited metabolomics studies on pear.
In early studies, the levels of glucose, fructose, sorbitol, and sucrose were determined in 10 pear accessions, while the levels of organic acids such as citric acid and malic acid were analyzed in 98 pear accessions using HPLC [3]. More than 100 volatile compounds could be identified in pear fruit [18, 19]. Recently, profiles of volatile compounds related to aroma were analyzed and compared in both ‘Dangshansuli’ and ‘Nanguo’ [110]. It was reported that fatty acid-related aroma volatiles were largely derived from metabolic precursors [110]. As wax is another important postharvest trait for pear, the metabolic profiles of wax compounds of 35 pear accessions from five pear species were evaluated. A total of 146 wax compounds were detected, including those composed of fatty acids, esters, primary alcohols, and terpenoids [111].
Small RNAs
MicroRNAs (miRNAs) are a class of 21- to 24-nucleotide noncoding RNAs that play important roles in growth and development [112]. As a result of NGS and high-throughput sequencing (HTS), many miRNAs associated with economic traits were detected in pear [113–115]. A particular focus was on pear fruit development and its regulation by miRNAs. Wu et al. [113] conducted deep sequencing of small RNAs from different developmental stages of ‘Dangshansuli’ fruit and found that miR160 acted as a regulator of an auxin response factor during fruit development [113]. Ma et al. [114] conducted an integrated analysis of RNA-seq and small RNA sequencing dataset from tissues of calyx abscission zones of ‘Korla fragrant pear’ and found that miRNAs miR858b and miR160a-3p were involved in calyx abscission [114]. In addition, miRNAs are also likely linked to pear fruit quality development. Using an integrated analysis of small RNAs and degradome sequencing of fruit skin tissues from ‘Meirensu’ pear combined with biochemical analysis, miR156 and its targeted SQUAMOSA-promoter binding like (SPL) gene family were found to participate in pear anthocyanin biosynthesis [115]. In another study, Wu et al. [113] identified nine sugar and acid metabolism-associated miRNAs (including miR1132, miR5077, and miR396b) and 11 lignin biosynthesis-related miRNAs (including miR397a, miR395a, and miR408b*) following small RNA sequencing of pear fruit collected at different developmental stages [113]. In a subsequent study, it was demonstrated that PbrmiR397a reduced the lignin content by silencing three PbrLAC genes [116]. Other miRNAs (pyr-miR1809 and pyr-miR144-3p) targeted LAC genes [117]. Several studies identified miRNAs associated with flavor and aroma in both ‘Suli’ and ‘Nanguoli’ pears, such as miR172, miR395, miR1132, and miR5077 [113, 118, 119]. Furthermore, biotic/abiotic stress-associated miRNAs were identified in pear through HTS, such as those involved in Apple Stem Grooving Virus (ASGV) defense, among other stress responses, including pbr-miR164, pbr-miR399, pbr-miR168, and pbr-miR171 [118, 120, 121]. Interestingly, a pear-specific miR6390 that might promote dormancy release by degrading DAM (dormancy-associated MADS-box) was identified using small RNA and degradome sequencing [122].
DNA methylation
DNA methylation is an important epigenetic modification that participates in diverse biological processes. Whole-genome bisulfite sequencing (WGBS or BS-Seq) or integrated analyses with RNA-seq are exploited in Rosaceae plants, and a Methylation Database for Rosaceae (MDR, http://mdr.xieslab.org/) has been established. As a result, various desirable traits associated with differentially methylated regions (DMRs) and DMR-regulated genes have been identified [123, 124]. DNA methylation plays essential roles in fruit size development and fruit ripening [125, 126]. Additionally, methylation plays an important role in regulating anthocyanin accumulation. It is well known that MYB TFs play important roles in anthocyanin biosynthesis in Rosaceous plants [127, 128] and that methylation levels of MYB promoters also influence anthocyanin biosynthesis. For example, methylation levels of the PcMYB10 promoter were associated with the green-skin sport of the pear cv. Max Red Bartlett, where it was observed that higher methylation of the PcMYB10 promoter resulted in lower expression levels of PcMYB10 and PcUFGT [128]. On the other hand, it was observed that the promoter PyMYB10 had lower levels of methylation in anthocyanin-rich tissues in the red sport of ‘Zaosu’ pear and its red-striped pigmentation patterns [127].
Stone cells
Stone cells are sclerenchyma cells that develop following cessation of cell expansion in pear fruit, wherein secondary cell walls are deposited along primary walls. These stone cells can result in a gritty texture and poor fruit taste and have a negative impact on consumer satisfaction. Moreover, lignin and cellulose are major components of the secondary cell walls of stone cells [129]. Lignin biosynthesis and deposition are highly correlated with the formation of stone cells within the flesh of pear fruit [130, 131]. The component of lignin in pear stone cells consists primarily of guaiacyl-lignin (G-lignin), along with small amounts of syringyl-lignin (S-lignin) and p-hydroxyphenyl lignin (H-lignin) [132, 133]. It is important to point out that the dominant monolignols are mainly bonded with β-O-4 linkages [133].
Currently, stone cell contents in pears are most often evaluated using a frozen-HCl protocol. This protocol was used to determine the stone cell contents of 236 accessions of sand pear (P. pyrifolia) at 50 days after bloom [134]. However, this method is limited due to the incomplete separation of stone cells from the flesh of pear fruits and it is both awkward and time-consuming to perform. Recently, a PearProcess software program was developed based on an imaging protocol using computer vision to digitize images, and image processing algorithms were applied [135].
In the lignin pathway (Fig. 3), a series of enzymes are involved in the synthesis of three monolignols (G-, S-, and H-type lignin) from phenylalanine, such as phenylalanine ammonia-lyase (PAL) and cinnamate-4-hydroxylase (C4H) [136, 137]. These monolignols are polymerized into lignin, which is catalyzed by either peroxidase (PRX) or laccase (LAC) enzymes [138]. Among these enzymes, members of the 4CL, CCR, CAD, OMT, PRX, and LAC gene families have been identified [5, 139–143]. The expression levels of Pb4CL1, PbCCR1, PbCCR2, PbCCR3, PbCAD2, PbCCOMT1, PbCCOMT3, five PbPRXs, and five PbLACs were found to be consistent with changes in the lignin content during pear fruit development [139–143]. Several transcription factors are predicted as potential regulators of the lignification of stone cells, such as PbWLIM1a, PbWLIM1b, PbKNOX1, PbIDD3, PbIDD5, PbMC1a/1b, PbMYB25, PbrMYB169, and PbMYB52 [89, 144–148]. PbrMYB169 was found to positively regulate lignification of stone cell formation via regulation of the expression of 4CL1, 4CL2, C3H1, HCT2, CCOMT2, CCR1, CAD, and LAC18 (Fig. 3) [144]. Furthermore, PbMC1a/1b, a member of the metacaspase gene family, was identified as an important gene that plays a key role in the lignification of the cell wall, probably by interacting with PbRD21 to regulate lignin synthesis-associated genes [148]. PbrmiR397a was found to be abundantly expressed during the early stages of pear fruit development [113]. It was revealed that PbrmiR397a was a key regulator of lignin synthesis in stone cells during fruit development by inhibiting three LAC genes coding for key enzymes in the lignin synthesis pathway [116]. Following analysis of the genome sequences of 60 pear cultivars, a SNP associated with low amounts of lignin in frutis was identified in the promoter of PbrmiR397a (Fig. 3) [116]. Thus, several SNP markers were developed for MAS to screen for pear stone cell contents.
Figure 3.
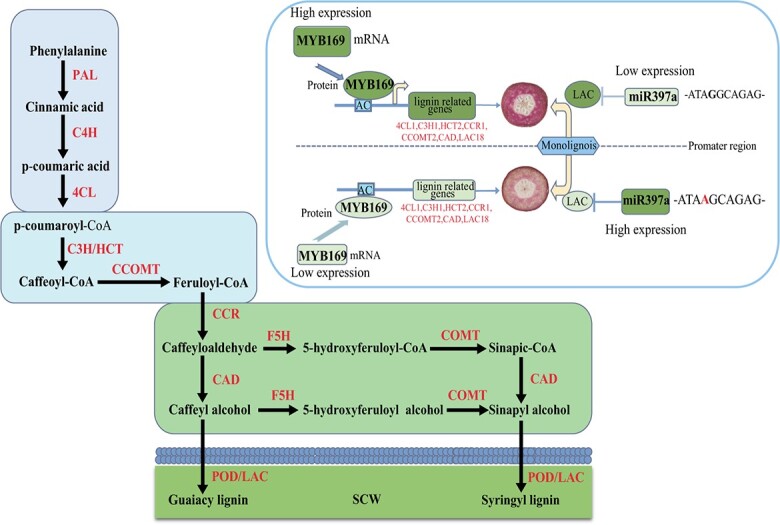
A proposed mechanism of stone cell development in pear fruit 4CL: 4-coumarate CoA ligase; C3H: p-coumaroyl shikimate 3’-hydroxylase; C4H: cinnamate 4-hydroxylase; CAD: cinnamyl alcohol dehydrogenase; CCOMT: caffeoyl CoA O-methyltransferase; CCR: cinnamoyl CoA reductase; COMT: caffeic acid O-methyltransferase; F5H: ferulic 5-hydroxylase; HCT: hydroxycinnamoyl transferase; LAC: laccase; PAL: phenylalanine ammonia lyase; POD: peroxidase; SCW: second cell wall.
Fruit color
Anthocyanins are secondary metabolites that play an important role in the color development of both flowers and fruits. Most red-skinned Asian pear fruits acquire anthocyanin-related coloration at the near-ripe stage, whereas the coloration of some European pears is observed at the beginning of fruit development; these fruits then lose this coloration but later regain red coloration upon approaching maturity.
In European pears, the red coloration gene was mapped to LG4 [48]. However, PyMYB10, a transcription factor (TF) controlling pear anthocyanin content, was located on LG9; thus, it was not directly responsible for red- versus yellow-colored pear cultivars [55]. Subsequently, QTLs for red skin color were detected on LG5 in a population of the Chinese white pear (P. bretschneideri) using linkage mapping [49]. There were only minor correlations between the expression patterns of anthocyanin biosynthetic genes and the MYB-bHLH-WD40 (MBW) regulatory complex (PyMYB10, PybHLH3, and PyWD40), suggesting that the regulatory pathway of anthocyanin biosynthesis in red pears was complex [149]. Furthermore, PyMYB10 was reported to regulate anthocyanin biosynthesis in some pear cultivars [150–152], and methylation levels of the PyMYB10 promoter were associated with the development of green-skinned sports in ‘Max Red Bartlett’ European pear [128]. Moreover, PbMYB10b and PbMYB9 were found to positively regulate flavonoid biosynthesis in pear fruit [153]. Subsequently, a map-based cloning strategy was used to identify a novel TF, PyMYB114, which was found to regulate fruit anthocyanin biosynthesis in pears [49]. In addition, PybHLH3, PyWRKY26, PybZIPa, and PyMADS18 were also found to be involved in anthocyanin biosynthesis in red pears [154–156].
PyMYB114 regulated anthocyanin biosynthesis by forming a complex with PybHLH3 and PyERF3 in Chinese red pears (Fig. 4) [49]. In ‘Hongzaosu’ pear, both PpERF24 and PpERF96 interacted with PpMYB114 to regulate blue light-modulated anthocyanin biosynthesis (Fig. 4) [157]. In addition, PyWRKY26 interacted with PybHLH3 to cotarget the promoter of PyMYB114 to regulate anthocyanin biosynthesis (Fig. 4) [156]. As light is essential for anthocyanin biosynthesis, Tao et al. [158] found that a blue-light signal transduction module, CRY-COP1-HY5, regulated anthocyanin biosynthesis in red pear [158]. Later, it was found that PybZIPa responded to light and promoted anthocyanin accumulation by activating PyUFGT in Asian pears [154]. Moreover, PyBBX16, PpBBX18 and PpBBX21 antagonistically regulated anthocyanin accumulation through competitive interactions with PpHY5 (Fig. 4) [159, 160].
Figure 4.
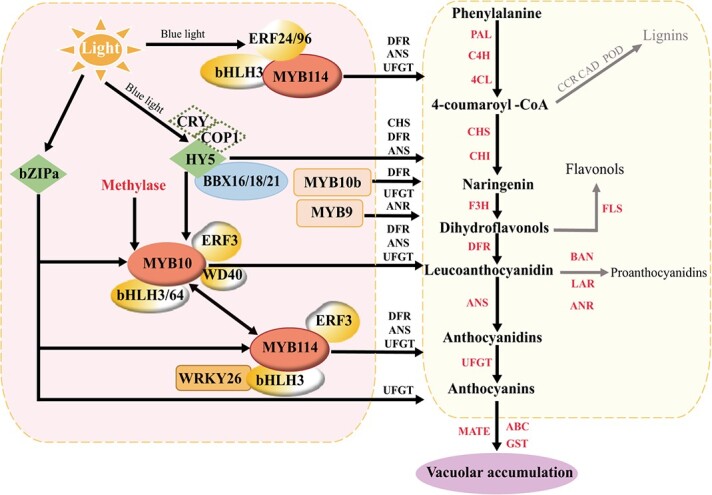
A proposed mechanism for fruit skin color development in pear ANS: anthocyanidin synthase; bZIP: basic region/leucine zipper; 4CL: 4-coumarate coenzyme A ligase; ABC: ATP-binding cassette transporter; ANR: anthocyanidin reductase; AP2/ERF: ethylene response factor/pathogenesis-related transcription factor; BAN: BANYULS; BBX: B-Box; bHLH: basic-helix-loop-helix; C4H: cinnamate 4-hydroxylase; CAD: cinnamyl alcohol dehydrogenase; CCR: cinnamoyl Co A reductase; CHI: chalcone isomerase; CHS: chalcone synthase; COP1: CONSTITUTIVE PHOTOMORPHOGENIC 1; CRY: cryptochrome; DFR: dihydroflavonol 4-reductase; F3H: flavanone 3-hydroxylase; FLS: flavonol synthase; GST: glutathione S- transferase; HY5: ELONGATED HYPOCOTYL 5; LAR: leucoanthocyanidin reductase; MATE: multidrug and toxic compound extrusion; MYB: v-myb avian myeloblastosis viral oncogene homolog; PAL: phenylalanine ammonialyase; POD: peroxidase; UFGT: UDP-glucose; flavonoid-3-O-glucosyltransferase; WD40: WD repeat protein; and WRKY: WRKYGQK domain.
Sugar and acid contents
Among the various factors influencing fruit quality, sugar is key in determining flavor. The major soluble sugars in ripe pear fruit are glucose, fructose, sucrose, and sorbitol [3]. Sorbitol, a major photosynthetic product in pear, is the predominant carbohydrate that is translocated in the phloem [161] and unloaded into the fruit via the apoplasm [162]. Monosaccharide transporters mediate the transport of a variety of sugars. Thus far, a total of 18 hexose transporters (STPs), six tonoplast monosaccharide transporters (TMTs or TSTs), six plastidic glucose transporters (pGlcTs), and six inositol transporters (INTs) have been detected in pear [6]. However, only PbTMT4 was experimentally demonstrated to participate in sugar accumulation in vacuoles (Fig. 5) [163].
Figure 5.
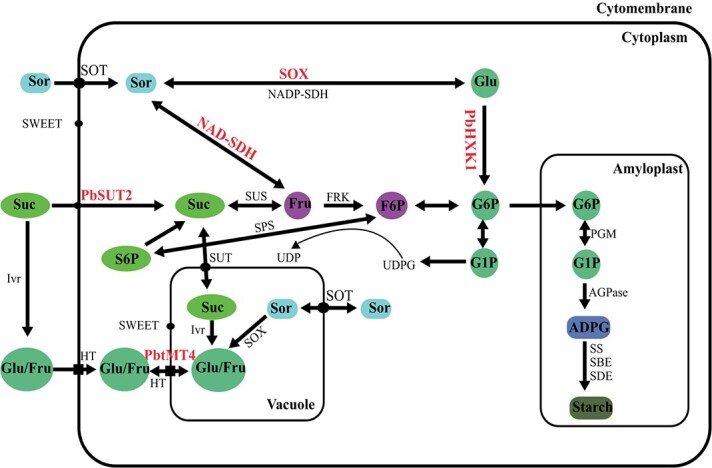
A proposed mechanism of sugar accumulation in pear fruit ADPase: ADP-glucose pyrophosphorylase; ADPG: ADP-glucose; FRK: fructokinase; Fru: fructose; G1P: glucose-1-phosphate; G6P: glucose-6-phosphate; Glu: glucose; HXK: hexokinase; HT: hexose transporter; Ivr: invertase; PGM: phosphoglucomutase; SBE: starch branching enzyme; SDE: starch debranching enzyme; Sor: sorbitol; SOT: sorbitol transporter; SOX: sorbitol oxidase; SPS: sucrose phosphate synthase; SUS: sucrose synthase; SS: starch synthase; Suc: sucrose; SUT: sucrose transporter; tMT: tonoplast monosaccharide transporter; UDP: uridine diphosphate; and UDPG: UDP-glucose.
Six sucrose transporters (SUTs) were identified in the pear genome [6]. Among these, PbSUT2 was found to influence the sucrose content in sinks, as well as the overall sugar content during flowering (Fig. 5) [164]. Oura et al. [165] reported that the rate of fruit growth depended on both the capability of the fruit to accumulate sorbitol and the rate of sorbitol conversion [165]. Sorbitol metabolic enzymes include NAD-sorbitol dehydrogenase (NAD-SDH), sorbitol-6-phosphate dehydrogenase (S6PDH), and sorbitol oxidase (SOX). Proteins and full-length cDNAs of both SD6PH and NAD-SDH in pears were purified and determined, respectively (Fig. 5) [165, 166].
Sucrose metabolic enzymes belong to the following two classes: sucrose phosphate synthase (SPS) and sucrose synthase (SS). It was previously reported that SPS and SS were involved in sucrose synthesis in pear, and with sucrose accumulation, the activities of both SPS and SS were found to increase [167]. Moreover, hexokinase (HXK) is a rate-limiting enzyme in glycolysis and controls cell survival by promoting metabolism and/or repressing apoptosis. HXK plays roles in sugar signaling and acts as a sugar sensor [168, 169]. Thus far, a total of 10 HXK protein sequences have been characterized in pear [87]. PbHXK1 was found to regulate the sugar content in pear fruit (Fig. 5) [170]. Furthermore, PbPFP1 was found to increase both fructose and sorbitol levels in pear fruit [9].
There are many different organic acids found in pear fruit that contribute to the unique flavors of different pear cultivars. The main organic acids are malic acid [171]. It was reported that cytosolic aconitase (ACO) and NADP-dependent isocitrate dehydrogenase (NADP-IDH) were involved in citric acid catabolism [172–174], while mitochondrial citrate synthase (CS) was involved in citric acid synthesis in various fruits [175]. The vacuolar H+-inorganic pyrophosphatase (V-PPase) [176] and V-ATPase [177] were purified and characterized, and expression of genes coding for these proteins during fruit development were investigated [178–180]. Additionally, a Py:vVpp gene encoding the V-PPase subunit, which is likely to be involved in the high citric acid content found in ‘Yandangxueli’, was characterized [181].
Aroma
Over 200 volatile compounds, primarily esters, alcohols, and aldehydes, have been identified in pear [110, 182, 183]. The fatty acid pathway is the main contributor of pear aroma components [110]. Major enzymes in this pathway include lipoxygenase (LOX), alcohol acyltransferase (AAT), alcohol dehydrogenase (ADH), and hydrogen peroxide lyase (HPL) [184, 185]. Based on RNA-seq, candidate genes for AAT, LOX, ADH, and HPL were identified [186–188]. In ‘Nanguo’ pear, PuADH3, PuLOX3, and PuAAT were found to be upregulated with increased amounts of aroma esters; thus, suggesting that these genes might regulate the synthesis of aroma [189]. Although ADH, AAT, LOX, and HPL are known to play important roles in both the formation and release of aroma in other species [190–192], TFs involved in aroma formation have not yet been reported in pear. Recently, several miRNAs were found to negatively regulate the expression of key aroma genes during cold storage of 'Nanguo' pear fruit, and they were also reported to play important roles in aroma formation [119].
Fruit maturity
It is likely that differential gene expression can either promote or delay fruit ripening. Thus, efforts are underway to delineate the mechanism of pear fruit maturity [193]. Some key genes regulating fruit maturity in pear have been investigated. Lelievre et al. [194] found that low-temperature treatment has a strong stimulating effect on 1-aminocyclopropane-1-carboxylic acid (ACC) oxidase activity, but it has a weak effect on ACC synthase activity, thus indicating that the expression of ACC synthase genes is regulated by low temperature and thereby contributes to fruit ripening [194].
The plant phytohormone indole acetic acid (IAA) is important in plant growth and development. IAA content can be modulated by conjugating IAA to amino acids using acyl acid amido synthetases of the Gretchen Hagen 3 (GH3) protein family. GH3 proteins can bind to IAA and IAA amide during pear fruit ripening; moreover, GH3.1 expression is activated by ERFs, thereby reducing the IAA content. Additionally, an actin-related protein, ARP4, is involved in ethylene-mediated fruit ripening, and it may work with pectin methylesterase 1 (PME1) to regulate the pear fruit ripening process [195]. Fruit softening serves as an important index for pear fruit ripening, with polygalacturaonase (PG)1 and PG2 playing critical roles in the rapid softening of ‘Starkrimson’ pear fruits [196].
In ‘La France’ pear, PcPG3, PcPME1, PcPME2, PcPME3, PcGAL1, and PcCel2 were proposed to be involved in fruit softening during storage [197, 198]. Moreover, after treatment of ‘Nanguo’ pear fruit with the chemical compound 1-methylcyclopropene (1-MCP), an ethylene inhibitor, ethylene biosynthesis (ACS1, ACS4, and ACO), reception (ERS1a), and response (PG1 and PG2) genes were inhibited by 1-MCP treatment. Furthermore, the -expansin-encoding genes PcExp2, PcExp3, PcExp5, and PcExp6 were also upregulated during ‘La France’ pear fruit ripening, and their expression patterns were consistent with the rates of softening [199].
Biotic and abiotic stress resistance
Biotic stress is one of the critical factors responsible for decreased fruit quality, yield gaps, tree losses, and increased production costs in pear. The major modes of mitigation for these biological stressors include chemical treatments, pruning, planting disease/pest-resistant cultivars, and physical barriers, such as netting. Often, pests and diseases that attack pear have a wide range of infection strategies and virulence mechanisms that are constantly evolving to adapt to their host. This renders effective and long-term control measures rather challenging to develop. The introduction of R genes, major resistance genes, into commercial cultivars is an effective strategy, but these genes can be overcome by the rapid adaption of a pathogenic strain due to mutations in individual effector genes [200]. Nevertheless, polygenic disease resistance consists of many additive quantitative loci that can confer durable resistance against rapidly evolving pathogen populations. Moreover, pyramiding of such quantitatively controlled disease and pest resistance loci into susceptible cultivated backgrounds can lead to the development of cultivars with wide-spectrum and durable resistance [201]. QTLs linked to disease and pest resistance have been identified in pear, and molecular markers are becoming widely available for use in breeding programs [202].
As mentioned above, genome sequences of both Asian and European pears provided opportunities to develop high-throughput genotyping platforms to construct dense genetic maps for QTL mapping, identify genes/QTLs for resistance genes, and identify robust molecular markers useful for early screening and genotyping of young seedlings. Due to their economic importance, the genetic basis for fire blight [45, 203, 204], pear scab [80, 205, 206], black spot [80], and pear psylla [201] resistance was of particular interest, and molecular markers linked to genetic resistance to some of these different biotic stressors were identified and became available for use in pear breeding programs.
Fire blight, caused by Erwinia amylovora, is a major threat to production in most of the countries where pears are grown. Different levels of susceptibility to fire blight were reported in European and Asian pear cultivars. Although no monogenic sources of fire blight resistance were reported, approximately 13 resistance loci of major and minor contributions to trait expression were identified for fire blight on LG2, LG4, LG9, LG10, LG11, and LG15 [45, 203, 204]. The genetic sources of these loci were derived from both European and Asian species, mainly P. communis and P. ussuriensis, respectively. Four QTLs linked to fire blight resistance were mapped onto LGs 2, 4, and 9 in the resistant European pear cultivar ‘Harrow Sweet’, and these QTLs had moderate/minor effects. A suite of AFLP-resistant gene analog (RGA) markers tightly linked to these QTLs were developed for use in MAB programs [45].
Abiotic stress is another important factor in the growth, development, productivity, and geographic adaptation of pear, including tolerance to drought, cold, and salt stresses. In recent years, a few key transcription factors associated with abiotic stress have been identified in pear, such as PbrWRKY53 [207], PbrMYB21 [208], PbrMYB5 [209], PbrbHLH1 [210], PbeNAC1 [211], and PbrNHX2 [212]. Among these, PbrWRKY53 was found to play a positive role in drought tolerance by binding to the W-box element in the promoter region of PbrNCED1 [207]. Moreover, transgenic tobacco expressing PbrMYB21 exhibited higher levels of arginine decarboxylase expression along with a higher accumulation of polyamine, thus contributing to enhanced tolerance to both dehydration and drought stresses [208]. Furthermore, a novel R2R3-type MYB transcription factor, designated PbrMYB5, could bind to the promoter of PbrDNAR2 and contribute to enhanced tolerance to chilling stress in pear [209].
Dwarf growth habit
The dwarf growth habit allows for the establishment of high-density plantings for pear production. Utilizing suitable dwarf rootstocks or dwarf growth habit cultivars is an important approach to achieve this goal. The French cultivar ‘Nain Vert’, a seedling mutant of P. communis exhibiting a dwarf growth habit architecture with distinctive features of a compact crown, short internodes, and short stature, is observed to carry a single dominant gene PcDw [62]. However, the molecular mechanism of this dwarf architecture trait remains unknown.
Based on available pear and apple genetic maps along with draft genome sequences [16, 20], several DNA molecular markers, including SSRs and SNPs, tightly linked to the dwarf growth habit trait of ‘Nain Vert’ were developed, and these could serve as useful tools for MAS [213]. To uncover the sequence information, structure, and functional regulation of PcDw, the region containing this locus was narrowed down by fine-mapping using these tightly linked DNA markers, and several candidate genes were inferred [213]. Recently, through comparative transcriptome analysis, the two most likely candidates, an arabinogalactan protein 7-like gene and a protein WVD2-like 7 gene, for the PcDw locus were identified, and a systemic overview of the complex regulatory network of the dwarf phenotype was provided [214].
As dwarf rootstocks are used for grafting scion cultivars to reduce the overall vigor of grafted trees, pear breeding efforts are underway to develop dwarf rootstocks that are graft-compatible with various scion cultivars, easily propagated, and highly resistant to both biotic and abiotic stresses. To accelerate the breeding scheme, it is necessary to elucidate the molecular mechanism and identify genetic determinants involved in the control of vigor in pear trees induced by rootstocks.
Two major QTLs, Dw1 and Dw2, were identified from the apple rootstock ‘M9’ (‘Malling 9’), accounting for most of the dwarfing effect conferred to the scion [215]. Recently, using an F1 segregating population of ‘Old Home’ × ‘Louise Bonne de Jersey’ (OH×LB), a QTL influencing scion vigor in pear was mapped to the syntenic position in the apple ‘M.9’ Dw1 locus, which is located at the upper end of LG5 [216]. Based on the high degree of synteny between apple and pear [217], it will be worthwhile to conduct comparative genome analysis to explore and delineate the genetic and molecular mechanisms of rootstock-induced dwarfing in pear.
Genome editing
Sequencing of the pear genome is important for identifying genes and desired traits using genome editing. Recently, CRISPR–Cas9 has been used for genome editing in diverse plant systems [218]. Owing to its high efficiency, simplicity, and versatility of multiplexing, Cas9, a Class 2 type II CRISPR system, was used extensively to manipulate different plant traits, such as disease resistance, crop yield, and quality [219–221]. Subsequently, Cas12a (formerly known as Cpf1), a Class 2 type V endonuclease, was used for editing several plant genomes, such as those for rice [222], tobacco [223], maize [224], and Arabidopsis [224], at high efficiencies.
Recently, genome editing studies have been undertaken in a number of horticultural crops. Among these, certain vegetable crops were subjected to more genome editing efforts (72% of all genome editing reports in horticultural crops) than other crops, particularly tomatoes (42% of all genome editing reports in horticultural crops) [225]. Fruit crops are very important commercial horticultural crops. Due to their time-consuming breeding cycle and complex genetic backgrounds, CRISPR–Cas systems have been successfully used in only a few fruit crops. Early efforts focused on using a mutation of the phytoene desaturase (PDS) gene that yielded an albino phenotype. This was demonstrated in banana [226], kiwifruit [227], grape [228], citrus [229], apple [230], and strawberry [231, 232]. Strawberry was used as a model fruit crop system for pursuing functional studies of particular genes of interest, such as the anthocyanin biosynthesis TF R2R3 MYB10, whereby CRISPR–Cas9-mediated gene editing was found to reduce fruit color [231]. In addition, the CRISPR–Cas9 system was designed to target the DIPM1, 2, and 4 genes encoding disease-specific (Dsp)A/E-interacting proteins and pathogenicity effectors of E. amylovora in apple to enhance genetic resistance against fire blight disease [233].
Efforts have been undertaken to generate compact plants that develop rapid terminal flowers and fruits to accelerate the breeding cycle in tree fruit crops by gene editing of floral repressor genes, such as CENTRORADIALIS (CEN)-like genes [234]. Recently, the CRISPR–Cas9 system was used for the first time in pear by targeting the TERMINAL FLOWER 1 (TFL1) gene, and early flowering phenotypes were observed in 9% of transgenic pear plants targeted for the PcTFL1.1 gene and 93% of transgenic apple lines targeted for the MdTFL1.1 gene [235].
Ongoing gene editing efforts are being pursued using the CRISPR–Cas9 system along with other novel gene editing systems, as these tools will be useful for the functional analysis of target genes and enhancing various desirable economic traits in pear.
Future research prospects
The first pear genome, reported in 2013, has served as a highly valuable resource for gaining sequence information, gene discovery, fine mapping, tool development, and variant detection. Over the last decade, WGS efforts for pear have had significant impacts on our knowledge of pear biology, gene function, inheritance, and trait physiology, leading to the availability of valuable resources and tools that will contribute to the genetic improvement of this highly valuable fruit crop. These resources will also support efforts to shorten the breeding cycle and increase breeding efficiency. For future pear research and breeding strategies, it is highly advisable to integrate different omics technologies into the discovery process of genes controlling traits of interest, the elucidation of gene function mechanisms, support for genotyping and phenotyping, and the acceleration of the breeding cycle. Additionally, advanced NGS methods and development, approaches to shorten the juvenility period, gene editing technology, and gene transfer technologies will all contribute to efforts to breed new pear cultivars with highly desirable economic characteristics.
Below are some specific areas of research interest that should be taken into consideration in future efforts.
Genome development
Although whole genome sequences of pear are available, each single reference genome does not represent the wide diversity within a species due to the presence of high levels of genomic variation. Recently, a pangenome concept has been proposed that involves comparing the genomes of multiple related individuals, wherein core genome segments are present in all individuals, while dispensable genome sequences are absent in some individuals. As pear has experienced independent domestication across Asian and European pears, major differences have been observed in different groups of wild and cultivated Asian and European pear accessions. Thus, pangenome sequencing will help us better characterize those subspecies of pear genomes and identify any unique genes that might be present in these genomes. Furthermore, it is necessary to construct a pear pangenome assembly to capture the wide genetic diversity present in Pyrus, as this will further support evolutionary studies and aid in breeding efforts in pear. Due to the high level of heterozygosity and large numbers of pear cultivars, short-read sequences and the uncertainty of the physical locations of genes will undoubtedly raise new challenges in constructing such a pear pangenome sequence assembly and sequence mapping.
It is also important to point out that the obtaining the haplotype genome of pear serves as another bottleneck due to the self-incompatibility and long generation time of pear fruit trees. However, long-read sequencing technology could help identify strand-specific sequences and overcome some of these problems.
Resequencing and phylogenetic studies
As NGS has become less expensive, long-read sequencing has become more popular. Earlier pear resequencing data reached tenfold coverage for NGS [28]; however, this is not sufficient for identifying structural variations (SVs), other than for SNP calling. Such SVs include deletions, insertions, duplications, inversions, and translocations, which can then be identified using NGS data and long-read sequencing combined with multiple algorithms, such as splitting reads, read pairs, and assembly methods, to discover and characterize SVs in the pear genome.
Whole-genome resequencing will also allow for a better understanding of the phylogenetics, domestication, improvement, and evolutionary history of a crop at the whole-genome level. This will provide unprecedented amounts of genomic datasets that will almost certainly allow for advances in modern pear molecular breeding programs.
Whole-genome association studies
For pear trait association studies, GWAS is an effective approach in natural populations, overcoming the long juvenile period and self-incompatibility issues in Pyrus. However, it is important to standardize parameters and units of measurements of various phenotypic data from multiple years and sites to uncover reliable trait associations in such GWAS. With the enhanced reliability and accuracy of trait associations, generated datasets will allow for investigating SVs across multiple genomes associated with target traits. Thus, in addition to SNPs and small InDels, these expanded datasets will allow for investigating genomes for copy number variants (CNVs), as these SVs confer larger effects on plant phenotypes. In recent years, several SV-GWAS have been undertaken in various plant species [236–238]; however, such studies have not yet been conducted in pear.
Integrated omics technologies
Currently, omics, which includes genetics, transcriptomics, epigenomics, proteomics, metabolomics, phenomics, and bioinformatics, offers various promising approaches that will accelerate breeding efforts. Thus, pursuing integrated omics efforts will allow for generating new knowledge of yet unknown genes, proteins, and metabolites. Such new knowledge will aid in genetic improvement and breeding efforts in pear. For future pear research, it is critical to exploit integrated omics approaches to enhance our knowledge not only of fruit development and quality traits but also of biotic and abiotic stresses and subsequently translate this knowledge into pear breeding efforts.
Gene editing
At present, gene editing offers a promising approach to improve new cultivars for traits of interest. Compared with other approaches, gene editing offers advantages in versatility, efficiency, and specificity. Currently, CRISPR-based gene editing can result in many types of mutations in target sequences, including precise base substitutions, small deletions/insertions, large fragment deletions, and gene replacement. Moreover, gene-edited plants can be free of exogenous DNA sequences either through genetic segregation or if CRISPR reagents are delivered as ribonucleoproteins to yield nontransgenic plants. This method may be a quick approach to developing new cultivars or improving existing cultivars.
As genetic transformation and genome editing technologies are limited for pear, it is important to undertake efforts to develop new protocols. This will be critical for pursuing the functional analysis of genes and for moving ahead with robust and efficient breeding of new and improved cultivars.
Supplementary Material
Acknowledgments
We thank the editor-in-chief, Prof. Zong-Ming (Max) Cheng, for initiating these review papers. We apologize to our colleagues whose work was not included here owing to space limitations. The authors are grateful to various funding agencies and projects that contributed to the present work, including the National Natural Science Foundation of China (31725024, 31820103012, 31801835, 31901978), National Key Research and Development Program (2018YFD1000200), National Natural Science Foundation of Jiangsu Province For Young Scholars (BK20180516, BK20180529), and China Agriculture Research System of MOF and MARA.
Author Contributions
J.W. and S.S.K. contributed to the manuscript outline and led editing of all sections of the manuscript; J.L. M.Z. and X.L. finalized the organization of the outline. The authors’ contributions to the different sections of the manuscript are as follows: “Overview of Asian and European pear genome sequences” and “Pear domestication and improvement” (X.L. S.Z.); “Genome-wide variations” (M.Z., R.W.); “Genetic linkage maps: Construction and trait linked loci mapping” (M.Q., and M.S.); “Marker-assisted selection, marker-assisted breeding, and genomic selection in pear” (A.C.A., K.L.W., and R.V.E); “Multiple omics: identifying genes related to important traits” (J.L., M.Z., X.L., A.K., C.W., R.W., C.X., G.Y., R.T., H.L., W.W., M.M, K.Z., B.B., J.N., and J.A.); and “Future research prospects” (J.L., M.Z. and X.L.). All authors reviewed the manuscript in the draft and final forms.
Conflict of interest statement
The authors declare that they have no conflicts of interest.
Supplementary data
Supplementary data is available at Horticulture Research online.
References
- 1. Potter D, Li J, Sun Jet al. Phylogeny and classification of Rosaceae. Plant Syst Evol. 2007;266:5–43. 10.1007/s00606-007-0539-9. [DOI] [Google Scholar]
- 2. Cao YF, Tian LM, Liu-Lin LIet al. Comparison studies on the stone cell content in flesh of pear cultivars. Acta Hortic Sinica. 2010;22:417–33. 10.16420/j.issn.0513-353x.2010.08.016. [DOI] [Google Scholar]
- 3. Yao GF, Zhang SL, Cao YFet al. Characteristics of components and contents of soluble sugars in pear fruits from different species. Scient Agric Sinica. 2010;43:4229–37. 10.3864/j.issn.0578-1752.2010.20.014. [DOI] [Google Scholar]
- 4. Quinet M, Wesel JP. Botany and taxonomy of pear. In: Korban SS, ed. The Pear Genome. Cham, Switzerland: Springer International Publishing, 2019,1–33. [Google Scholar]
- 5. Wu J, Wang Z, Shi Zet al. The genome of the pear (Pyrus bretschneideri Rehd.). Genome Res. 2013;23:396–408. 10.1101/gr.144311.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li JM, Zheng DM, Li LTet al. Genome-wide function, evolutionary characterization and expression analysis of sugar transporter family genes in pear (Pyrus bretschneideri Rehd). Plant Cell Physiol. 2015;56:1721–37. 10.1093/pcp/pcv090. [DOI] [PubMed] [Google Scholar]
- 7. Qiao X, Li M, Li et al. Genome-wide identification and comparative analysis of the heat shock transcription factor family in Chinese white pear (Pyrus bretschneideri) and five other Rosaceae species. BMC Plant Biol. 2015;15:12. 10.1186/s12870-014-0401-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li X, Xue C, Li Jet al. Genome-wide identification, evolution and functional divergence of MYB transcription factors in Chinese white pear (Pyrus bretschneideri). Plant Cell Physiol. 2016;57:824–47. 10.1093/pcp/pcw029. [DOI] [PubMed] [Google Scholar]
- 9. Lü H, Li J, Huang Yet al. Genome-wide identification, expression and functional analysis of the phosphofructokinase gene family in Chinese white pear (Pyrus bretschneideri). Gene. 2019;702:133–42. 10.1016/j.gene.2019.03.005. [DOI] [PubMed] [Google Scholar]
- 10. Song B, et al. Mining and evolution analysis of lateral organ boundaries domain (LBD) genes in Chinese white pear (Pyrus bretschneideri). BMC Genomics. 2020;21:644. 10.1186/s12864-020-06999-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sun M, Zhang M, Singh Jet al. Contrasting genetic variation and positive selection followed the divergence of NBS-encoding genes in Asian and European pears. BMC Genomics. 2020;21:809. 10.1186/s12864-020-07226-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hussain S, Niu Q, Qian Met al. Genome-wide identification, characterization, and expression analysis of the dehydrin gene family in Asian pear (Pyrus pyrifolia). Tree Genet Genomes. 2015;11:110. 10.1007/s11295-015-0938-y. [DOI] [Google Scholar]
- 13. Cao Y, Han Y, Meng Det al. B-BOX genes: genome-wide identification, evolution and their contribution to pollen growth in pear (Pyrus bretschneideri Rehd.). BMC Plant Biol. 2017;17:156. 10.1186/s12870-017-1105-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li L, Deng CH, Knabel Met al. Integrated high-density consensus genetic map of Pyrus and anchoring of the 'Bartlett' v1.0 (Pyrus communis) genome. DNA Res. 2017;24:dsw063–301. 10.1093/dnares/dsw063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wu J, Li LT, Li Met al. High-density genetic linkage map construction and identification of fruit-related QTLs in pear using SNP and SSR markers. J Exp Bot. 2014;65:5771–81. 10.1093/jxb/eru311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Velasco R, Zharkikh A, Affourtit Jet al. The genome of the domesticated apple (Malus × domestica Borkh.). Nat Genet. 2010;42:833–9. 10.1038/ng.654. [DOI] [PubMed] [Google Scholar]
- 17. Shi D, et al. Single-pollen-cell sequencing for gamete-based phased diploid genome assembly in plants. Genome Res. 2019;29:1889–99. 10.1101/gr.251033.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qin G, Tao S, Cao Yet al. Evaluation of the volatile profile of 33 Pyrus ussuriensis cultivars by HS-SPME with GC–MS. Food Chem. 2012;134:2367–82. 10.1016/j.foodchem.2012.04.053. [DOI] [PubMed] [Google Scholar]
- 19. Suwanagul A, Richardson DG. Identification of headspace volatile compounds from different pear (Pyrus communis L.) varieties. Acta Hortic. 1998;475:605–24. 10.17660/ActaHortic.1998.475.73. [DOI] [Google Scholar]
- 20. Chagne D, Crowhurst RN, Pindo Met al. The draft genome sequence of European pear (Pyrus communis L. 'Bartlett'). PLoS One. 2014;9:e92644. 10.1371/journal.pone.0092644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Linsmith G, Rombauts S, Montanari Set al. Pseudo-chromosome-length genome assembly of a double haploid "Bartlett" pear (Pyrus communis L.). Giga Science. 2019;8:giz138. 10.1093/gigascience/giz138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dong X, Wang Z, Tian Let al. De novo assembly of a wild pear (Pyrus betuleafolia) genome. Plant Biotechnol J. 2020;18:581–95. 10.1111/pbi.13226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ou C, Wang F, Wang Jet al. A de novo genome assembly of the dwarfing pear rootstock Zhongai 1. Scientific data. 2019;6:281. 10.1038/s41597-019-0291-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jung S, Lee T, Cheng CHet al. 15 years of GDR: new data and functionality in the genome database for Rosaceae. Nucleic Acids Res. 2019;47:D1137–45. 10.1093/nar/gky1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cao K, Zheng Z, Wang Let al. Comparative population genomics reveals the domestication history of the peach, Prunus persica, and human influences on perennial fruit crops. Genome Biol. 2014;15:415. 10.1186/s13059-014-0415-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhou Y, Massonnet M, Sanjak JSet al. Evolutionary genomics of grape (Vitis vinifera ssp. vinifera) domestication. Proc Natl Acad Sci U S A. 2017;114:11715–20. 10.1073/pnas.1709257114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Duan N, Bai Y, Sun Het al. Genome re-sequencing reveals the history of apple and supports a two-stage model for fruit enlargement. Nat Commun. 2017;8:249. 10.1038/s41467-017-00336-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wu J, Wang Y, Xu Jet al. Diversification and independent domestication of Asian and European pears. Genome Biol. 2018;19:77. 10.1186/s13059-018-1452-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Silva GJ, Souza TM, Barbieri RLet al. Origin, domestication, and dispersing of pear (Pyrus spp.). 82d Cong, 2d sess House of Representatives Report ; no 2356. 2014;2014:1. 10.1155/2014/541097. [DOI] [Google Scholar]
- 30. Li X, Li L, Ming Met al. Comparative transcriptomic analysis provides insight into the domestication and improvement of pear (P. pyrifolia) fruit. Plant Physiol. 2019;180:435–52. 10.1104/pp.18.01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang, M.Y, Xue, C, Xu, L.et al. Distinct transcriptome profiles reveal gene expression patterns during fruit development and maturation in five main cultivated species of pear (Pyrus L.). Sci Rep 6, ???, 10.1038/srep28130 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Elshire RJ, Glaubitz JC, Sun Qet al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One. 2011;6:e19379. 10.1371/journal.pone.0019379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kumar S, Kirk C, Deng Cet al. Genotyping-by-sequencing of pear (Pyrus spp.) accessions unravels novel patterns of genetic diversity and selection footprints. Hortic. Res. 2017;4:17015. 10.1038/hortres.2017.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kumar S, Kirk C, Deng CHet al. Fine-mapping and validation of the genomic region underpinning pear red skin colour. Hortic Res. 2019;6:29. 10.1038/s41438-018-0112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kumar S, Kirk C, Deng CHet al. Marker-trait associations and genomic predictions of interspecific pear (Pyrus) fruit characteristics. Sci Rep. 2019;9:9072. 10.1038/s41598-019-45618-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Han ML, Liu YL, Zheng XYet al. Construction of a genetic linkage map and QTL analysis for some fruit traits in pear. J Fruit Sci. 2010;27:496–503. [Google Scholar]
- 37. Gabay G, Dahan Y, Izhaki Yet al. High-resolution genetic linkage map of European pear (Pyrus communis) and QTL fine-mapping of vegetative budbreak time. BMC Plant Biol. 2018;18:175. 10.1186/s12870-018-1386-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xue L, Liu Q, Hu Het al. The southwestern origin and eastward dispersal of pear (Pyrus pyrifolia) in East Asia revealed by comprehensive genetic structure analysis with SSR markers. Tree Genet Genomes. 2018;14:1–12. 10.1007/s11295-018-1255-z. [DOI] [Google Scholar]
- 39. Beissinger TM, Hirsch CN, Sekhon RSet al. Marker density and read depth for genotyping populations using genotyping-by-sequencing. Genetics. 2013;193:1073–81. 10.1534/genetics.112.147710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li X, Singh J, Qin Met al. Development of an integrated 200K SNP genotyping array and application for genetic mapping, genome assembly improvement and genome wide association studies in pear (Pyrus). Plant Biotechnol J. 2019;17:1582–94. 10.1111/pbi.13085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Montanari S, Bianco L, Allen BJet al. Development of a highly efficient axiom 70 K SNP array for Pyrus and evaluation for high-density mapping and germplasm characterization. BMC Genomics. 2019;20:331. 10.1186/s12864-019-5712-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Iketani H, Abe K, Yamamoto Tet al. Mapping of disease-related genes in Japanese pear using a molecular linkage map with RAPD markers. Breed Sci. 2001;51:179–84. 10.1270/jsbbs.51.179. [DOI] [Google Scholar]
- 43. Yamamoto T, Kimura T, Shoda Met al. Genetic linkage maps constructed by using an interspecific cross between Japanese and European pears. Theor Appl Genet. 2002;106:9–18. 10.1007/s00122-002-0966-5. [DOI] [PubMed] [Google Scholar]
- 44. Yamamoto T, Kimura T, Saito Tet al. Genetic linkage maps of Japanese and European pears aligned to the apple consensus map. Acta Hortic. 2004;663:51–6. 10.17660/ActaHortic.2004.663.2. [DOI] [Google Scholar]
- 45. Dondini L, Pierantoni L, Gaiotti Fet al. Identifying QTLs for fire-blight resistance via a European pear (Pyrus communis L.) genetic linkage map. Mol Breed. 2005;14:407–18. 10.1007/s11032-005-0505-6. [DOI] [Google Scholar]
- 46. Yamamoto T, Kimura T, Tekakami Set al. Integrated reference genetic linkage maps of pear based on SSR and AFLP markers. Breed Sci. 2007;57:321–9. 10.1270/jsbbs.57.321. [DOI] [Google Scholar]
- 47. Zhang R, Wu J, Li XGet al. An AFLP, SRAP, and SSR genetic linkage map and identification of QTLs for fruit traits in pear (Pyrus L.). plant Mol. Plant Mol Biol Report. 2013;31:678–87. 10.1007/s11105-012-0544-1. [DOI] [Google Scholar]
- 48. Dondini L, Pierantoni L, Sansavini Set al. The inheritance of the red colour character in European pear (Pyrus communis) and its map position in the mutated cultivar ‘max red Bartlett’. Plant Breed. 2008;127:524–6. 10.1111/j.1439-0523.2008.01500.x. [DOI] [Google Scholar]
- 49. Yao G, Ming M, Allan ACet al. Map-based cloning of the pear gene MYB114 identifies an interaction with other transcription factors to coordinately regulate fruit anthocyanin biosynthesis. Plant J. 2017;92:437–51. 10.1111/tpj.13666. [DOI] [PubMed] [Google Scholar]
- 50. Pierantoni L, Dondini L, Cho KHet al. Pear scab resistance QTLs via a European pear (Pyrus communis) linkage map. Tree Genet Genomes. 2007;3:311–7. 10.1007/s11295-006-0070-0. [DOI] [Google Scholar]
- 51. Wu J, Qin M. Linkage mapping in pear. In: Korban SS, ed. The Pear Genome. Cham, Switzerland: Springer International Publishing, 2019,103–12. [Google Scholar]
- 52. Yamamoto T, Terakami S, Kimura Tet al. Reference genetic linkage maps of European and Japanese pears. Acta Hortic. 2009;814:599–602. 10.17660/actahortic.2009.814.101. [DOI] [Google Scholar]
- 53. Terakami S, Kimura T, Nishitani Cet al. Genetic linkage map of the Japanese pear 'Housui' identifying three homozygous genomic regions. Horticulture journal. 2009;78:417–24. 10.2503/jjshs1.78.417. [DOI] [Google Scholar]
- 54. Sun W, Zhang Y, Le Wet al. Construction of a genetic linkage map and QTL analysis for some leaf traits in pear (Pyrus L.). Front Agric China. 2009;3:67–74. 10.1007/s11703-009-0013-2. [DOI] [Google Scholar]
- 55. Pierantoni L, Dondini L, De Franceschi Pet al. Mapping of an anthocyanin-regulating MYB transcription factor and its expression in red and green pear, Pyrus communis. Plant Physiol Biochem. 2010;48:1020–6. 10.1016/j.plaphy.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 56. Choi JK, Na DY, Kim Det al. Genetic linkage mapping using interspecific hybrid population between Korean wild pear (Pyrus ussuriensis) and Japanese pear (P. pyrifolia). Hortic Environ Biotech. 2010;51:319–25. [Google Scholar]
- 57. Hao Y, Zhao Y, Guo Yet al. Genetic linkage maps of pear based on SRAP markers. Pak JBot. 2013;45:1265–71. [Google Scholar]
- 58. Yamamoto T, Terakami S, Moriya Set al. DNA markers developed from genome sequencing analysis in Japanese pear (Pyrus pyrifolia). Acta Hortic. 2013;976:477–83. 10.17660/ActaHortic.2013.976.67. [DOI] [Google Scholar]
- 59. Won K, Bastiaanse H, Song JHet al. Genetic mapping of polygenic scab (Venturia pirina) resistance in an interspecific pear family. Mol Breed. 2014;34:2179–89. 10.1007/s11032-014-0172-6. [DOI] [Google Scholar]
- 60. Yamamoto T, Terakami S, Takada Net al. Identification of QTLs controlling harvest time and fruit skin color in Japanese pear (Pyrus pyrifolia Nakai). Breed Sci. 2014;64:351–61. 10.1270/jsbbs.64.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chen H, Song Y, Li LTet al. Construction of a high-density simple sequence repeat consensus genetic map for pear (Pyrus spp.). Plant Mol. Biol. Rep. 2015;33:316–25. 10.1007/s11105-014-0745-x. [DOI] [Google Scholar]
- 62. Wang C, Tian Y, Buck EJet al. Genetic mapping of PcDw determining pear dwarf trait. J Am Soc Hortic Sci. 2011;136:48–53. [Google Scholar]
- 63. Wang L, Li X, Wang Let al. Construction of a high-density genetic linkage map in pear (Pyrus communis × Pyrus pyrifolia Nakai) using SSRs and SNPs developed by SLAF-seq. Sci Hortic. 2017;218:198–204. 10.1016/j.scienta.2017.02.015. [DOI] [Google Scholar]
- 64. Xue H, Wang S, Yao JLet al. Chromosome level high-density integrated genetic maps improve the Pyrus bretschneideri 'DangshanSuli' v1.0 genome. BMC Genomics. 2018;19:833. 10.1186/s12864-018-5224-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zurn JD, Norelli JL, Montanari Set al. Dissecting genetic resistance to fire blight in three pear populations. Phytopathology. 2020;110:1305–11. 10.1094/PHYTO-02-20-0051-R. [DOI] [PubMed] [Google Scholar]
- 66. Pritchard JK, Stephens MJ, Donnelly PJ. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59. 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kang HM, Zaitlen NA, Wade CMet al. Efficient control of population structure in model organism association mapping. Genetics. 2008;178:1709–23. 10.1534/genetics.107.080101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Aulchenko YS, Ripke S, Isaacs Aet al. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007;23:1294–6. 10.1093/bioinformatics/btm108. [DOI] [PubMed] [Google Scholar]
- 69. Abney M, Ober C, McPeek MS. Quantitative-trait homozygosity and association mapping and empirical genome-wide significance in large, complex pedigrees: fasting serum-insulin level in the hutterites. Am J Hum Genet. 2002;70:920–34. 10.1086/339705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Aulchenko YS, Koning DJ, Haley C. Genomewide rapid association using mixed model and regression: a fast and simple method for genome-wide pedigree-based quantitative trait loci association analysis. Genetics. 2007;177:577–85. 10.1534/genetics.107.075614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012;44:821–4. 10.1038/ng.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mariette S, Tai FWJ, Roch Get al. Genome-wide association links candidate genes to resistance to plum pox virus in apricot (Prunus armeniaca). New Phytol. 2016;209:773–84. 10.1111/nph.13627. [DOI] [PubMed] [Google Scholar]
- 73. Cao K, Zhou Z, Wang Qet al. Genome-wide association study of 12 agronomic traits in peach. Nat Commun. 2016;7:13246. 10.1038/ncomms13246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhang MY, Xue C, Hu Het al. Genome-wide association studies provide insights into the genetic determination of fruit traits of pear. Nat Commun. 2021;12:1144. 10.1038/s41467-021-21378-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Peace CP. DNA-informed breeding of rosaceous crops: promises, progress and prospects. Hortic. Res. 2017;4:17006. 10.1038/hortres.2017.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Collard BC, Mackill DJ. Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Philos Trans R Soc Lond Ser B Biol Sci. 2008;363:557–72. 10.1098/rstb.2007.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Itai A, Kotaki T, Tanabe Ket al. Rapid identification of 1-aminocyclopropane-1-carboxylate (ACC) synthase genotypes in cultivars of Japanese pear (Pyrus pyrifolia Nakai) using CAPS markers. Theor Appl Genet. 2003;106:1266–72. 10.1007/s00122-002-1186-8. [DOI] [PubMed] [Google Scholar]
- 78. Xu M, Huaracha E, Korban SS. Development of sequence-characterized amplified regions (SCARs) from amplified fragment length polymorphism (AFLP) markers tightly linked to the Vf gene in apple. Genome. 2001;44:63–70. 10.1139/gen-44-1-63. [DOI] [PubMed] [Google Scholar]
- 79. Moriya Y, Yamamoto K, Okada Ket al. Development of a CAPS marker system for genotyping European pear cultivars harboring 17 S alleles. Plant Cell Rep. 2007;26:345–54. 10.1007/s00299-006-0254-y. [DOI] [PubMed] [Google Scholar]
- 80. Terakami S, Moriya S, Adachi Yet al. Fine mapping of the gene for susceptibility to black spot disease in Japanese pear (Pyrus pyrifolia Nakai). Breed Sci. 2016;66:271–80. 10.1270/jsbbs.66.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Khan MA, Zhao YF, Korban SS. Identification of genetic loci associated with fire blight resistance in Malus through combined use of QTL and association mapping. Physiol Plant. 2013;148:344–53. 10.1111/ppl.12068. [DOI] [PubMed] [Google Scholar]
- 82. Meuwissen TH, Goddard ME. Prediction of identity by descent probabilities from marker-haplotypes. Genet Sel Evol. 2001;33:605–34. 10.1186/1297-9686-33-6-605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Iwata H, Hayashi T, Terakami Set al. Potential assessment of genome-wide association study and genomic selection in Japanese pear Pyrus pyrifolia. Breed Sci. 2013;63:125–40. 10.1270/jsbbs.63.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Minamikawa MF, Takada N, Terakami Set al. Genome-wide association study and genomic prediction using parental and breeding populations of Japanese pear (Pyrus pyrifolia Nakai). Sci Rep. 2018;8:11994. 10.1038/s41598-018-30154-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wang R, Ming M, Li Jet al. Genome-wide identification of the MADS-box transcription factor family in pear (Pyrus bretschneideri) reveals evolution and functional divergence. PeerJ. 2017;5:e3776. 10.7717/peerj.3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Li J, Qin M, Qiao Xet al. A new insight into the evolution and functional divergence of SWEET transporters in Chinese white pear (Pyrus bretschneideri). Plant Cell Physiol. 2017;58:839–50. 10.1093/pcp/pcx025. [DOI] [PubMed] [Google Scholar]
- 87. Yu L, Li J, Li Let al. Characterisation of the whole-genome wide hexokinase gene family unravels the functional divergence in pear (Pyrus bretschneideri Rehd.). J Hortic Sci Biotechnol. 2018;93:244–54. 10.1080/14620316.2017.1362961. [DOI] [Google Scholar]
- 88. Cao Y, Meng D, Li Xet al. A Chinese white pear (Pyrus bretschneideri) BZR gene PbBZR1 act as a transcriptional repressor of lignin biosynthetic genes in fruits. Front Plant Sci. 2020;11:1087. 10.3389/fpls.2020.01087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Cheng X, Li M, Abdullah Met al. In Silico genome-wide analysis of the pear (Pyrus bretschneideri) KNOX family and the functional characterization of PbKNOX1, an Arabidopsis BREVIPEDICELLUS orthologue gene, involved in cell wall and lignin biosynthesis. Front Genet. 2019;10:632. 10.3389/fgene.2019.00632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Liu C, Qiao X, Li Qet al. Genome-wide comparative analysis of the BAHD superfamily in seven Rosaceae species and expression analysis in pear (Pyrus bretschneideri). BMC Plant Biol. 2020;20:14. 10.1186/s12870-019-2230-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zeng W, Qiao X, Li Qet al. Genome-wide identification and comparative analysis of the ADH gene family in Chinese white pear (Pyrus bretschneideri) and other Rosaceae species. Genomics. 2020;112:3484–96. 10.1016/j.ygeno.2020.06.031. [DOI] [PubMed] [Google Scholar]
- 92. Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zhou H, Yin H, Chen Jet al. Gene-expression profile of developing pollen tube of Pyrus bretschneideri. Gene Expr Patterns. 2016;20:11–21. 10.1016/j.gep.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 94. Li QH, Qiao X, Yin Het al. Unbiased subgenome evolution following a recent whole-genome duplication in pear (Pyrus bretschneideri Rehd.). Hortic. Res. 2019;6:34. 10.1038/s41438-018-0110-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Yang YN, Yao GF, Yue WQet al. Transcriptome profiling reveals differential gene expression in proanthocyanidin biosynthesis associated with red/green skin color mutant of pear (Pyrus communis L.). Front Plant Sci. 2015;6:795. 10.3389/fpls.2015.00795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Liu BY, Wang L, Wang Set al. Transcriptomic analysis of bagging-treated 'Pingguo' pear shows that MYB4-like1, MYB4-like2, MYB1R1 and WDR involved in anthocyanin biosynthesis are up-regulated in fruit peels in response to light. Sci Hortic. 2019;244:428–34. 10.1016/j.scienta.2018.09.040. [DOI] [Google Scholar]
- 97. Wang ZG, Du H, Zhai Ret al. Transcriptome analysis reveals candidate genes related to color fading of 'Red Bartlett' (Pyrus communis L.). Front Plant Sci. 2017;8:455. 10.3389/fpls.2017.00455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Wang YZ, Dai MS, Zhang SJet al. Exploring candidate genes for pericarp russet pigmentation of sand pear (Pyrus pyrifolia) via RNA-seq data in two genotypes contrasting for pericarp color. PLoS One. 2014;9:e83675. 10.1371/journal.pone.0083675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Ou C, Zhang X, Wang Fet al. A 14 nucleotide deletion mutation in the coding region of the PpBBX24 gene is associated with the red skin of "Zaosu red" pear (Pyrus pyrifolia white pear group): a deletion in the PpBBX24 gene is associated with the red skin of pear. Hortic. Res. 2020;7:39. 10.1038/s41438-020-0259-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Bai SL, Saito T, Sakamoto Det al. Transcriptome analysis of Japanese pear (Pyrus pyrifolia Nakai) flower buds transitioning through endodormancy. Plant Cell Physiol. 2013;54:1132–51. 10.1093/pcp/pct067. [DOI] [PubMed] [Google Scholar]
- 101. Nham NT, Macnish AJ, Zakharov Fet al. 'Bartlett' pear fruit (Pyrus communis L.) ripening regulation by low temperatures involves genes associated with jasmonic acid, cold response, and transcription factors. Plant Sci. 2017;260:8–18. 10.1016/j.plantsci.2017.03.008. [DOI] [PubMed] [Google Scholar]
- 102. Yang TY, Huang XS. Deep sequencing-based characterization of transcriptome of Pyrus ussuriensis in response to cold stress. Gene. 2018;661:109–18. 10.1016/j.gene.2018.03.067. [DOI] [PubMed] [Google Scholar]
- 103. Li KQ, Xu XY, Huang XS. Identification of differentially expressed genes related to dehydration resistance in a highly drought-tolerant pear, Pyrus betulaefolia, as through RNA-Seq. PLoS One. 2016;11:e0149352. 10.1371/journal.pone.0149352. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 104. Li H, Lin J, Yang QSet al. Comprehensive analysis of differentially expressed genes under salt stress in pear (Pyrus betulaefolia) using RNA-Seq. Plant Growth Regul. 2017;82:409–20. 10.1007/s10725-017-0266-3. [DOI] [Google Scholar]
- 105. Kan JL, Liu T, Ma Net al. Transcriptome analysis of Callery pear (Pyrus calleryana) reveals a comprehensive signalling network in response to Alternaria alternata. PLoS One. 2017;12:e0184988. 10.1371/journal.pone.0184988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Yang XP, Hu H, Yu Det al. Candidate resistant genes of sand pear (Pyrus pyrifolia Nakai) to Alternaria alternata revealed by transcriptome sequencing. PLoS One. 2015;10:e0135046. 10.1371/journal.pone.0135046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Li JM, Huang X, S, Ting, L. T. et al. Proteome analysis of pear reveals key genes associated with fruit development and quality. Planta. 2015;241:1363–79. 10.1007/s00425-015-2263-y. [DOI] [PubMed] [Google Scholar]
- 108. Reuscher S, Fukao Y, Morimoto Ret al. Quantitative proteomics-based reconstruction and identification of metabolic pathways and membrane transport proteins related to sugar accumulation in developing fruits of pear (Pyrus communis). Plant Cell Physiol. 2016;57:505–18. 10.1093/pcp/pcw004. [DOI] [PubMed] [Google Scholar]
- 109. Liu X, Zhai R, Feng Wet al. Proteomic analysis of 'Zaosu' pear (Pyrus bretschneideri Rehd.) and its early-maturing bud sport. Plant Sci. 2014;224:120–35. 10.1016/j.plantsci.2014.04.012. [DOI] [PubMed] [Google Scholar]
- 110. Qin G, Tao S, Zhang Het al. Evolution of the aroma volatiles of pear fruits supplemented with fatty acid metabolic precursors. Molecules. 2014;19:20183–96. 10.3390/molecules191220183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Wu X, Yin H, Shi Zet al. Chemical composition and crystal morphology of epicuticular wax in mature fruits of 35 pear (Pyrus spp.) cultivars. Front Plant Sci. 2018;9:679. 10.3389/fpls.2018.00679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Kidner CA, Martienssen RA. The developmental role of microRNA in plants. Curr Opin Plant Biol. 2005;8:38–44. 10.1016/j.pbi.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 113. Wu J, Wang D, Liu Yet al. Identification of miRNAs involved in pear fruit development and quality. BMC Genomics. 2014;15:953. 10.1186/1471-2164-15-953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Ma L, Zhou L, Quan Set al. Integrated analysis of mRNA-seq and miRNA-seq in calyx abscission zone of Korla fragrant pear involved in calyx persistence. BMC Plant Biol. 2019;19:192. 10.1186/s12870-019-1792-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Qian M, Ni J, Niu Qet al. Response of miR156-SPL module during the red peel coloration of bagging-treated Chinese sand pear (Pyrus pyrifolia Nakai). Front Physiol. 2017;8:500. 10.3389/fphys.2017.00550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Xue C, Yao JL, Qin MFet al. PbrmiR397a regulates lignification during stone cell development in pear fruit. Plant Biotechnol J. 2019;17:103–17. 10.1111/pbi.12950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Cheng X, Yan C, Zhang Jet al. The effect of different pollination on the expression of Dangshan Su pear microRNA. Biomed Res Int. 2017;2017:1–18. 10.1155/2017/2794040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Niu Q, Qian M, Liu Get al. A genome-wide identification and characterization of mircoRNAs and their targets in ‘Suli’ pear (Pyrus pyrifolia white pear group). Planta. 2013;238:1095–112. 10.1007/s00425-013-1954-5. [DOI] [PubMed] [Google Scholar]
- 119. Shi F, Zhou X, Yao MMet al. miRNAs play important roles in aroma weakening during the shelf life of 'Nanguo' pear after cold storage. Food Res Int. 2019;116:942–52. 10.1016/j.foodres.2018.09.031. [DOI] [PubMed] [Google Scholar]
- 120. Liu J, Zhang X, Zhang FPet al. Identification and characterization of microRNAs from in vitro-grown pear shoots infected with apple stem grooving virus in response to high temperature using small RNA sequencing. BMC Genomics. 2015;16:945. 10.1186/s12864-015-2126-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Zhang Q, Zhang Y, Wang Set al. Characterization of genome-wide microRNAs and their roles in development and biotic stress in pear. Planta. 2019;249:693–707. 10.1007/s00425-018-3027-2. [DOI] [PubMed] [Google Scholar]
- 122. Niu Q, Li J, Cai Det al. Dormancy-associated MADS-box genes and microRNAs jointly control dormancy transition in pear (Pyrus pyrifolia white pear group) flower bud. J Exp Bot. 2016;67:239–57. 10.1093/jxb/erv454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Jiang SH, Sun QG, Chen Met al. Methylome and transcriptome analyses of apple fruit somatic mutations reveal the difference of red phenotype. BMC Genomics. 2019;20:117. 10.1186/s12864-019-5499-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Zhu YC, Zhang B, Allan ACet al. DNA demethylation is involved in the regulation of temperature-dependent anthocyanin accumulation in peach. Plant J. 2020;102:965–76. 10.1111/tpj.14680. [DOI] [PubMed] [Google Scholar]
- 125. Daccord N, Celton JM, Linsmith Get al. High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat Genet. 2017;49:1099–106. 10.1038/ng.3886. [DOI] [PubMed] [Google Scholar]
- 126. Cheng J, Niu Q, Zhang Bet al. Downregulation of RdDM during strawberry fruit ripening. Genome Biol. 2018;19:212. 10.1186/s13059-018-1587-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Qian M, Sun Y, Allan ACet al. The red sport of 'Zaosu' pear and its red-striped pigmentation pattern are associated with demethylation of the PyMYB10 promoter. Phytochemistry. 2014;107:16–23. 10.1016/j.phytochem.2014.08.001. [DOI] [PubMed] [Google Scholar]
- 128. Wang Z, Meng D, Wang Aet al. The methylation of the PcMYB10 promoter is associated with green-skinned sport in max red Bartlett pear. Plant Physiol. 2013;162:885–96. 10.1104/pp.113.214700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Smith WW. The course of stone cell formation in pear fruits. Plant Physiol. 1935;10:587–611. 10.1104/pp.10.4.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Martin-Cabrejas MA, Waldron KW, Selvendran RRet al. Ripening-related changes in the cell walls of Spanish pear (Pyrus communis). Physiol Plant. 1994;91:671–9. 10.1111/j.1399-3054.1994.tb03004.x. [DOI] [Google Scholar]
- 131. Tao S, Khanizadeh S, Zhang Het al. Anatomy, ultrastructure and lignin distribution of stone cells in two Pyrus species. Plant Sci. 2009;176:413–9. 10.1016/j.plantsci.2008.12.011. [DOI] [Google Scholar]
- 132. Cai Y, Li M, Li Det al. Study of the structure and biosynthetic pathway of lignin in stone cells of pear. Sci Hortic. 2010;125:374–9. 10.1016/j.scienta.2010.04.029. [DOI] [Google Scholar]
- 133. Jin Q, Yan C, Qiu Jet al. Structural characterization and deposition of stone cell lignin in Dangshan Su pear. Sci Hortic. 2013;155:123–30. 10.1016/j.scienta.2013.03.020. [DOI] [Google Scholar]
- 134. Zhang J, Li J, Xue Cet al. The variation of stone cell dontent in 236 germplasms of sand pear (Pyrus pyrifolia) and identification of related candidate genes. Hortic Plant J. 2021;7:108–16. 10.1016/j.hpj.2020.09.003. [DOI] [Google Scholar]
- 135. Xue Y-S, Xu S, Z, Xue, C. et al. Pearprocess: a new phenotypic tool for stone cell trait evaluation in pear fruit. J Integr Agric. 2020;19:1625–34. 10.1016/s2095-3119(20)63193-8. [DOI] [Google Scholar]
- 136. Vanholme R, Cesarino I, Rataj Ket al. Caffeoyl Shikimate esterase (CSE) is an enzyme in the lignin biosynthetic pathway in Arabidopsis. Science. 2013;341:1103–6. 10.1126/science.1241602. [DOI] [PubMed] [Google Scholar]
- 137. Zhao Q. Lignification: flexibility, biosynthesis and regulation. Trends Plant Sci. 2016;21:713–21. 10.1016/j.tplants.2016.04.006. [DOI] [PubMed] [Google Scholar]
- 138. Dean JF, Eriksson KEL. Laccase and the deposition of lignin in vascular plants. Holzforschung. 1994;48:21–33. 10.1515/hfsg.1994.48.s1.21. [DOI] [Google Scholar]
- 139. Cao Y, Han Y, Li Det al. Systematic analysis of the 4-coumarate: coenzyme a ligase (4CL) related genes and expression profiling during fruit development in the Chinese pear. Genes. 2016;7:89. 10.3390/genes7100089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Cao Y, Han Y, Meng Det al. Structural, evolutionary, and functional analysis of the class III peroxidase gene family in Chinese pear (Pyrus bretschneideri). Front Plant Sci. 2016;7:1874. 10.3389/fpls.2016.01874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Cheng X, Xiong Y, Li DHet al. Bioinformatic and expression analysis of the OMT gene family in Pyrus bretschneideri cv. GMR. 2016;15:15038664. 10.4238/gmr.15038664. [DOI] [PubMed] [Google Scholar]
- 142. Cheng X, et al. Characterization and analysis of CCR and CAD gene families at the whole-genome level for lignin synthesis of stone cells in pear (Pyrus bretschneideri) fruit. Biol Open. 2017;6:1602–13. 10.1242/bio.026997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Cheng X, Li G, Ma Cet al. Comprehensive genome-wide analysis of the pear (Pyrus bretschneideri) laccase gene (PbLAC) family and functional identification of PbLAC1 involved in lignin biosynthesis. PLoS One. 2019;14:e0210892. 10.1371/journal.pone.0210892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Xue C, Yao JL, Xue YSet al. PbrMYB169 positively regulates lignification of stone cells in pear fruit. J Exp Bot. 2019;70:1801–14. 10.1093/jxb/erz039. [DOI] [PubMed] [Google Scholar]
- 145. Cheng X, Li G, Muhammad Aet al. Molecular identification, phylogenomic characterization and expression patterns analysis of the LIM (LIN-11, Isl1 and MEC-3 domains) gene family in pear (Pyrus bretschneideri) reveal its potential role in lignin metabolism. Gene. 2019;686:237–49. 10.1016/j.gene.2018.11.064. [DOI] [PubMed] [Google Scholar]
- 146. Su X, Meng T, Zhao Yet al. Comparative genomic analysis of the IDD genes in five Rosaceae species and expression analysis in Chinese white pear (Pyrus bretschneideri). PeerJ. 2019;7:e6628. 10.7717/peerj.6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Cao Y, Han Y, Li Det al. MYB transcription factors in Chinese pear (Pyrus bretschneideri Rehd.): genome-wide identification, classification, and expression profiling during fruit development. Front Plant Sci. 2016;7:577. 10.3389/fpls.2016.00577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Gong X, Xie Z, Qi Ket al. PbMC1a/1b regulates lignification during stone cell development in pear (Pyrus bretschneideri) fruit. Hortic. Res. 2020;7:59. 10.1038/s41438-020-0280-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Yang YN, et al. Expression differences of anthocyanin biosynthesis genes reveal regulation patterns for red pear coloration. Plant Cell Rep. 2015;34:189–98. 10.1007/s00299-014-1698-0. [DOI] [PubMed] [Google Scholar]
- 150. Feng S, Wang Y, Yang Set al. Anthocyanin biosynthesis in pears is regulated by a R2R3-MYB transcription factor PyMYB10. Planta. 2010;232:245–55. 10.1007/s00425-010-1170-5. [DOI] [PubMed] [Google Scholar]
- 151. Zhang X, Allan AC, Yi Qet al. Differential gene expression analysis of Yunnan red pear, Pyrus pyrifolia, during fruit skin coloration. Plant Mol. Biol. Rep. 2011;29:305–14. 10.1007/s11105-010-0231-z. [DOI] [Google Scholar]
- 152. Yu B, Zhang D, Huang Cet al. Isolation of anthocyanin biosynthetic genes in red Chinese sand pear (Pyrus pyrifolia Nakai) and their expression as affected by organ/tissue, cultivar, bagging and fruit side. Sci Hortic. 2012;136:29–37. 10.1016/j.scienta.2011.12.026. [DOI] [Google Scholar]
- 153. Zhai R, Wang Z, Zhang Set al. Two MYB transcription factors regulate flavonoid biosynthesis in pear fruit (Pyrus bretschneideri Rehd.). J Exp Bot. 2016;67:1275–84. 10.1093/jxb/erv524. [DOI] [PubMed] [Google Scholar]
- 154. Liu H, Su J, Zhu Yet al. The involvement of PybZIPa in light-induced anthocyanin accumulation via the activation of PyUFGT through binding to tandem G-boxes in its promoter. Hortic. Res. 2019;6:134. 10.1016/j.na.2018.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Wu J, Zhao G, Yang Yet al. Identification of differentially expressed genes related to coloration in red/green mutant pear (Pyrus communis L.). Tree Genet Genomes. 2013;9:75–83. 10.1007/s11295-012-0534-3. [DOI] [Google Scholar]
- 156. Li C, Wu J, Hu KDet al. PyWRKY26 and PybHLH3 cotargeted the PyMYB114 promoter to regulate anthocyanin biosynthesis and transport in red-skinned pears. Hortic. Res. 2020;7:37. 10.1038/s41438-020-0254-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Ni J, Bai S, Zhao Yet al. Ethylene response factors Pp4ERF24 and Pp12ERF96 regulate blue light-induced anthocyanin biosynthesis in 'Red Zaosu' pear fruits by interacting with MYB114. Plant Mol Biol. 2019;99:67–78. 10.1007/s11103-018-0802-1. [DOI] [PubMed] [Google Scholar]
- 158. Tao R, Bai S, Ni Jet al. The blue light signal transduction pathway is involved in anthocyanin accumulation in 'Red Zaosu' pear. Planta. 2018;248:37–48. 10.1007/s00425-018-2877-y. [DOI] [PubMed] [Google Scholar]
- 159. Bai S, Tao R, Tang Yet al. BBX16, a B-box protein, positively regulates light-induced anthocyanin accumulation by activating MYB10 in red pear. Plant Biotechnol J. 2019;17:1985–97. 10.1111/pbi.13114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Bai S, Tao R, Yin Let al. Two B-box proteins, PpBBX18 and PpBBX21, antagonistically regulate anthocyanin biosynthesis via competitive association with Pyrus pyrifolia ELONGATED HYPOCOTYL 5 in the peel of pear fruit. Plant J. 2019;100:1208–23. 10.1111/tpj.14510. [DOI] [PubMed] [Google Scholar]
- 161. Bieleski RL, Redgwell RJ. Sorbitol metabolism in nectaries from flowers of Rosaceae. Funct Plant Biol. 1980;7:15–25. 10.1071/PP9800015. [DOI] [Google Scholar]
- 162. Zhang HP, Wu JY, Tao STet al. Evidence for apoplasmic phloem unloading in pear fruit. Plant Mol Biol Rep. 2014;32:931–9. 10.1007/s11105-013-0696-7. [DOI] [Google Scholar]
- 163. Cheng R, Cheng Y, Lu Jet al. The gene PbTMT4 from pear (Pyrus bretschneideri) mediates vacuolar sugar transport and strongly affects sugar accumulation in fruit. Physiol Plant. 2018;164:307–19. 10.1111/ppl.12742. [DOI] [PubMed] [Google Scholar]
- 164. Wang LF, Qi XX, Huang XSet al. Overexpression of sucrose transporter gene PbSUT2 from Pyrus bretschneideri, enhances sucrose content in Solanum lycopersicum fruit. Plant Physiol Biochem. 2016;105:150–61. 10.1016/j.plaphy.2016.04.019. [DOI] [PubMed] [Google Scholar]
- 165. Oura Y, Yamada K, Shiratake Ket al. Purification and characterization of a NAD(+)-dependent sorbitol dehydrogenase from Japanese pear fruit. Phytochemistry. 2000;54:567–72. 10.1016/S0031-9422(00)00158-8. [DOI] [PubMed] [Google Scholar]
- 166. Kim HY, Ahn JC, Choi JHet al. Expression and cloning of the full-length cDNA for sorbitol-6-phosphate dehydrogenase and NAD-dependent sorbitol dehydrogenase from pear (Pyrus pyrifolia N.). Sci Hortic. 2007;112:406–12. 10.1016/j.scienta.2007.01.015. [DOI] [Google Scholar]
- 167. Moriguchi T, Abe K, Sanada Tet al. Levels and role of sucrose synthase, sucrose-phosphate synthase, and acid invertase in sucrose accumulation in fruit of Asian pear. J Am Soc Hortic Sci. 1992;117:274–8. 10.21273/JASHS.117.2.274. [DOI] [Google Scholar]
- 168. Moore B, Zhou L, Rolland Fet al. Role of the Arabidopsis glucose sensor HXK1 in nutrient, light, and hormonal signaling. Science. 2003;300:332–6. 10.1126/science.1080585. [DOI] [PubMed] [Google Scholar]
- 169. Kim YM, Heinzel N, Giese JOet al. A dual role of tobacco hexokinase 1 in primary metabolism and sugar sensing. Plant Cell Environ. 2013;36:1311–27. 10.1111/pce.12060. [DOI] [PubMed] [Google Scholar]
- 170. Zhao B, Qi K, Yi Xet al. Identification of hexokinase family members in pear (Pyrus × bretschneideri) and functional exploration of PbHXK1 in modulating sugar content and plant growth. Gene. 2019;711:143932. 10.1016/j.gene.2019.06.022. [DOI] [PubMed] [Google Scholar]
- 171. Chen J, Wang Z, Wu Jet al. Chemical compositional characterization of eight pear cultivars grown in China. Food Chem. 2007;104:268–75. 10.1016/j.foodchem.2006.11.038. [DOI] [Google Scholar]
- 172. Popova TN, Pinheiro de Carvalho MA. Citrate and isocitrate in plant metabolism. Biochim Biophys Acta. 1998;1364:307–25. 10.1016/S0005-2728(98)00008-5. [DOI] [PubMed] [Google Scholar]
- 173. Sadka A, Dahan E, Or Eet al. NADP+-isocitrate dehydrogenase gene expression and isozyme activity during citrus fruit development. Plant Sci. 2000;158:173–81. 10.1016/S0168-9452(00)00328-9. [DOI] [PubMed] [Google Scholar]
- 174. Sadka A, Dahan E, Cohen Let al. Aconitase activity and expression during the development of lemon fruit. Physiol Plant. 2000;108:255–62. 10.1034/j.1399-3054.2000.108003255.x. [DOI] [Google Scholar]
- 175. Canel C, Bailey-Serres JN, Roose ML. Molecular characterization of the mitochondrial citrate synthase gene of an acidless pummelo (Citrus maxima). Plant Mol Biol. 1996;31:143–7. 10.1007/BF00020613. [DOI] [PubMed] [Google Scholar]
- 176. Suzuki Y, Kanayama Y, Shiratake Ket al. Vacuolar H(+)-pyrophosphatase purified from pear fruit. Phytochemistry. 1999;50:535–9. 10.1016/S0031-9422(98)00554-8. [DOI] [PubMed] [Google Scholar]
- 177. Hosaka M, Kanayama Y, Shiratake Ket al. Tonoplast H+-ATPase of mature pear fruit. Phytochemistry. 1994;36:565–7. 10.1016/S0031-9422(00)89775-7. [DOI] [Google Scholar]
- 178. Suzuki Y, Shiratake K, Yamaki S. Seasonal changes in the activities of vacuolar H+-pumps and their gene expression in the developing Japanese pear fruit. Engei Gakkai zasshi. 2000;69:15–21. 10.2503/jjshs.69.15. [DOI] [Google Scholar]
- 179. Hiratake K, Kanayama Y, Maeshima Met al. Changes in H(+)-pumps and a tonoplast intrinsic protein of vacuolar membranes during the development of pear fruit. Plant Cell Physiol. 1997;38:1039–45. 10.1093/oxfordjournals.pcp.a029269. [DOI] [PubMed] [Google Scholar]
- 180. Yasuo S, Masayoshi M, Shohei Y. Molecular cloning of vacuolar H+-pyrophosphatase and its expression during the development of pear fruit. Plant Cell Physiol. 1999;40:900–4. 10.1093/oxfordjournals.pcp.a029620. [DOI] [PubMed] [Google Scholar]
- 181. Lu XP, Liu YZ, Zhou GFet al. Identification of organic acid-related genes and their expression profiles in two pear (Pyrus pyrifolia) cultivars with difference in predominant acid type at fruit ripening stage. Sci Hortic. 2011;129:680–7. 10.1016/j.scienta.2011.05.014. [DOI] [Google Scholar]
- 182. Willner B, Granvogl M, Schieberle P. Characterization of the key aroma compounds in Bartlett pear brandies by means of the sensomics concept. J Agric Food Chem. 2013;61:9583–93. 10.1021/jf403024t. [DOI] [PubMed] [Google Scholar]
- 183. Wang C, Zhang W, Li Het al. Analysis of volatile compounds in pears by HS-SPME-GCxGC-TOFMS. Molecules. 2019;24:1795. 10.3390/molecules24091795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. El Hadi MA, Zhang FJ, Wu FFet al. Advances in fruit aroma volatile research. Molecules. 2013;18:8200–29. 10.3390/molecules18078200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Shi F, Zhou X, Yao MMet al. Low-temperature stress-induced aroma loss by regulating fatty acid metabolism pathway in 'Nanguo' pear. Food Chem. 2019;297:124927. 10.1016/j.foodchem.2019.05.201. [DOI] [PubMed] [Google Scholar]
- 186. Shi F, Zhou X, Zhou Qet al. Transcriptome analyses provide new possible mechanisms of aroma ester weakening of ‘Nanguo’ pear after cold storage. Sci Hortic. 2018;237:247–56. 10.1016/j.scienta.2018.04.013. [DOI] [Google Scholar]
- 187. Li M, Dunwell JM, Zhang Het al. Network analysis reveals the co-expression of sugar and aroma genes in the Chinese white pear (Pyrus bretschneideri). Gene. 2018;677:370–7. 10.1016/j.gene.2018.08.042. [DOI] [PubMed] [Google Scholar]
- 188. Wei S, Tao S, Qin Set al. Transcriptome profiling reveals the candidate genes associated with aroma metabolites and emission of pear (Pyrus ussuriensis cv.). Sci Hortic. 2016;206:33–42. 10.1016/j.scienta.2016.04.019. [DOI] [Google Scholar]
- 189. Luo M, Zhou X, Sun Het al. Insights into profiling of volatile ester and LOX-pathway related gene families accompanying post-harvest ripening of 'Nanguo' pears. Food Chem. 2021;335:127665. 10.1016/j.foodchem.2020.127665. [DOI] [PubMed] [Google Scholar]
- 190. Gonzalez M, Gaete-Eastman C, Valdenegro Met al. Aroma development during ripening of Fragaria chiloensis fruit and participation of an alcohol acyltransferase (FcAAT1) gene. J Agric Food Chem. 2009;57:9123–32. 10.1021/jf901693j. [DOI] [PubMed] [Google Scholar]
- 191. Zhou D, Sun Y, Li Met al. Postharvest hot air and UV-C treatments enhance aroma-related volatiles by simulating the lipoxygenase pathway in peaches during cold storage. Food Chem. 2019;292:294–303. 10.1016/j.foodchem.2019.04.049. [DOI] [PubMed] [Google Scholar]
- 192. Singh RK, Srivastava S, Chidley HGet al. Overexpression of mango alcohol dehydrogenase (MiADH1) mimics hypoxia in transgenic tomato and alters fruit flavor components. Agri gene. 2018;7:23–33. 10.1016/j.aggene.2017.10.003. [DOI] [Google Scholar]
- 193. Bapat VA, Trivedi PK, Ghosh Aet al. Ripening of fleshy fruit: molecular insight and the role of ethylene. Biotechnol Adv. 2010;28:94–107. 10.1016/j.biotechadv.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 194. Lelievre J-M, Tichit L, Dao Pet al. Effects of chilling on the expression of ethylene biosynthetic genes in Passe-Crassane pear (Pyrus communis L.) fruits. Plant Mol Biol. 1997;33:847–55. 10.1023/A:1005750324531. [DOI] [PubMed] [Google Scholar]
- 195. Yuan H, Zhang L, Jiang Zet al. Characterization of ripening-telated PuARP4 in pear (Pyrus ussuriensis). J Plant Growth Regul. 2017;36:766–72. 10.1007/s00344-017-9680-z. [DOI] [Google Scholar]
- 196. Song L, Wang Z, Wang Zet al. Screening of cell wall-related genes that are expressed differentially during ripening of pears with different softening characteristics. Postharvest Biol Technol. 2016;115:1–8. 10.1016/j.postharvbio.2015.12.012. [DOI] [Google Scholar]
- 197. Mercy WM, Mathooko FM, Matsuzaki Met al. Expression characteristics of seven members of the β-galactosidase gene family in ‘La France’ pear (Pyrus communis L.) fruit during growth and their regulation by 1-methylcyclopropene during postharvest ripening. Postharvest Biol Technol. 2005;36:253–63. 10.1016/j.postharvbio.2005.02.002. [DOI] [Google Scholar]
- 198. Sekine D, Munemura I, Gao Met al. Cloning of cDNAs encoding cell-wall hydrolases from pear (Pyrus communis) fruit and their involvement in fruit softening and development of melting texture. Physiol Plant. 2006;126:163–74. 10.1111/j.1399-3054.2006.00583.x. [DOI] [Google Scholar]
- 199. Hiwasa K, Rose JKC, Nakano Ret al. Differential expression of seven α-expansin genes during growth and ripening of pear fruit. Physiol Plant. 2003;117:564–72. 10.1034/j.1399-3054.2003.00064.x. [DOI] [PubMed] [Google Scholar]
- 200. Peil A. Inoculation of Malus × robusta 5 progeny with a strain breaking resistance to fire blight reveals a minor QTL on LG5. Acta Hortic. 2011;896:357–62. 10.17660/actahortic.2011.896.49. [DOI] [Google Scholar]
- 201. Perchepied L, Guerif P, Ravon Eet al. Polygenic inheritance of resistance to Cacopsylla pyri in a Pyrus communis × P. ussuriensis progeny is explained by three QTLs involving an epistatic interaction. Tree Genet Genomes. 2016;12:108. 10.1007/s11295-016-1072-1. [DOI] [Google Scholar]
- 202. De Franceschi, P. & Dondini, L.. Molecular mapping of major genes and QTLs in pear. In: Korban, S.S. (ed.) The Pear Genome, pp 113-131, Cham, Switzerland. 10.1007/978-3-030-11048-2_6 (2019). [DOI] [Google Scholar]
- 203. Bokszczanin K, Dondini L, Przybyla AA. First report on the presence of fire blight resistance in linkage group 11 of Pyrus ussuriensis maxim. J Appl Genet. 2009;50:99–104. 10.1007/bf03195660. [DOI] [PubMed] [Google Scholar]
- 204. Montanari S, Perchepied L, Renault Det al. A QTL detected in an interspecific pear population confers stable fire blight resistance across different environments and genetic backgrounds. Mol Breed. 2016;36:47. 10.1007/s11032-016-0473-z. [DOI] [Google Scholar]
- 205. Abe K, Saito T, Terai Oet al. Genotypic difference for the susceptibility of Japanese, Chinese and European pears to Venturia nashicola, the cause of scab on Asian pears. Plant Breed. 2008;127:407–12. 10.1111/j.1439-0523.2007.01482.x. [DOI] [Google Scholar]
- 206. Bouvier L, Bourcy M, Boulay Met al. A new pear scab resistance gene Rvp1 from the European pear cultivar 'Navara' maps in a genomic region syntenic to an apple scab resistance gene cluster on linkage group 2. Tree genet. Tree Genet Genomes. 2012;8:53–60. 10.1007/s11295-011-0419-x. [DOI] [Google Scholar]
- 207. Liu Y, Yang T, Lin Zet al. A WRKY transcription factor PbrWRKY53 from Pyrus betulaefolia is involved in drought tolerance and AsA accumulation. Plant Biotechnol J. 2019;17:1770–87. 10.1111/pbi.13099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Li K, Xing C, Yao Zet al. PbrMYB21, a novel MYB protein of Pyrus betulaefolia, functions in drought tolerance and modulates polyamine levels by regulating arginine decarboxylase gene. Plant Biotechnol J. 2017;15:1186–203. 10.1111/pbi.12708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Xing C, Liu Y, Zhao Let al. A novel MYB transcription factor regulates AsA synthesis and effects cold tolerance. Plant Cell Environ. 2019;42:832–45. 10.1111/pce.13387. [DOI] [PubMed] [Google Scholar]
- 210. Jin C, Huang XS, Li KQet al. Overexpression of a bHLH1 transcription factor of Pyrus ussuriensis confers enhanced cold tolerance and increases expression of stress-responsive genes. Front Plant Sci. 2016;7:441. 10.3389/fpls.2016.00441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Jin C, Li KQ, Xu Xet al. A novel NAC transcription factor, PbeNAC1, of Pyrus betulifolia confers cold and drought tolerance via interacting with PbeDREBs and activating the expression of stress-responsive genes. Front Plant Sci. 2017;8:1049. 10.3389/fpls.2017.01049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Dong H, Wang C, Xing Cet al. Overexpression of PbrNHX2 gene, a Na+/H+ antiporter gene isolated from Pyrus betulaefolia, confers enhanced tolerance to salt stress via modulating ROS levels. Plant Sci. 2019;285:14–25. 10.1016/j.plantsci.2019.04.021. [DOI] [PubMed] [Google Scholar]
- 213. Wang CH, Li W, Tian YKet al. Development of molecular markers for genetic and physical mapping of the PcDw locus in pear (Pyrus communis L.). J Hortic Sci Biotechnol. 2016;91:299–307. 10.1080/14620316.2016.1155319. [DOI] [Google Scholar]
- 214. Xiao Y, Wang C, Tian Yet al. Candidates responsible for dwarf pear phenotype as revealed by comparative transcriptome analysis. Mol Breed. 2019;39:1. 10.1007/s11032-018-0907-x. [DOI] [Google Scholar]
- 215. Foster TM, Celton JM, Chagne Det al. Two quantitative trait loci, Dw1 and Dw2, are primarily responsible for rootstock-induced dwarfing in apple. Hortic. Res. 2015;2:15001. 10.1038/hortres.2015.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Knäbel M, Friend AP, Palmer JWet al. Genetic control of pear rootstock-induced dwarfing and precocity is linked to a chromosomal region syntenic to the apple Dw1 loci. BMC Plant Biol. 2015;15:230. 10.1186/s12870-015-0620-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Celton J-M, Chagne D, Tustin SDet al. Update on comparative genome mapping between Malus and Pyrus. BMC Res Notes. 2009;2:182–2. 10.1186/1756-0500-2-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Nekrasov V, Staskawicz B, Weigel Det al. Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA-guided endonuclease. Nat Biotechnol. 2013;31:691–3. 10.1038/nbt.2655. [DOI] [PubMed] [Google Scholar]
- 219. Chandrasekaran J, Brumin M, Wolf Det al. Development of broad virus resistance in non-transgenic cucumber using CRISPR/Cas9 technology. Mol Plant Pathol. 2016;17:1140–53. 10.1111/mpp.12375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Peng A, Chen S, Lei Tet al. Engineering canker-resistant plants through CRISPR/Cas9-targeted editing of the susceptibility gene CsLOB1 promoter in citrus. Plant Biotechnol J. 2017;15:1509–19. 10.1111/pbi.12733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Tashkandi M, Ali Z, Aljedaani Fet al. Engineering resistance against tomato yellow leaf curl virus via the CRISPR/Cas9 system in tomato. Plant Signal Behav. 2018;13:e1525996. 10.1080/15592324.2018.1525996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Tang X, Ren Q, Yang Let al. Single transcript unit CRISPR 2.0 systems for robust Cas9 and Cas12a mediated plant genome editing. Plant Biotechnol J. 2019;17:1431–45. 10.1111/pbi.13068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Endo A, Masafumi M, Kaya Het al. Efficient targeted mutagenesis of rice and tobacco genomes using Cpf1 from Francisella novicida. Sci Rep. 2016;6:38169. 10.1038/srep38169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Malzahn AA, Tang X, Lee Ket al. Application of CRISPR-Cas12a temperature sensitivity for improved genome editing in rice, maize, and Arabidopsis. BMC Biol. 2019;17:9. 10.1186/s12915-019-0629-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Xu J, Hua K, Lang Z. Genome editing for horticultural crop improvement. Hortic. Res. 2019;6:113. 10.1038/s41438-019-0196-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Kaur N, Alok A, Kaur Net al. CRISPR/Cas9-mediated efficient editing in phytoene desaturase (PDS) demonstrates precise manipulation in banana cv. Rasthali genome. Funct. Integr. Genomics. 2018;18:89–99. 10.1007/s10142-017-0577-5. [DOI] [PubMed] [Google Scholar]
- 227. Wang Z, Wang S, Li Det al. Optimized paired-sgRNA/Cas9 cloning and expression cassette triggers high-efficiency multiplex genome editing in kiwifruit. Plant Biotechnol J. 2018;16:1424–33. 10.1111/pbi.12884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Wang X, Tu M, Wang Det al. CRISPR/Cas9-mediated efficient targeted mutagenesis in grape in the first generation. Plant Biotechnol J. 2018;16:844–55. 10.1111/pbi.12832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Zhang F, LeBlanc C, Irish VFet al. Rapid and efficient CRISPR/Cas9 gene editing in citrus using the YAO promoter. Plant Cell Rep. 2017;36:1883–7. 10.1007/s00299-017-2202-4. [DOI] [PubMed] [Google Scholar]
- 230. Nishitani C, Hirai N, Komori Set al. Efficient genome editing in apple using a CRISPR/Cas9 system. Sci Rep. 2016;6:31481. 10.1038/srep31481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Xing S, Jia M, Wei Let al. CRISPR/Cas9-introduced single and multiple mutagenesis in strawberry. J Genet Genomics. 2018;45:685–7. 10.1016/j.jgg.2018.04.006. [DOI] [PubMed] [Google Scholar]
- 232. Zhou J, Wang G, Liu Z. Efficient genome editing of wild strawberry genes, vector development and validation. Plant Biotechnol J. 2018;16:1868–77. 10.1111/pbi.12922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Malnoy M, Viola R, Jung MHet al. DNA-free genetically edited grapevine and apple protoplast using CRISPR/Cas9 ribonucleoproteins. Front Plant Sci. 2016;7:1904. 10.3389/fpls.2016.01904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Arkonyi-Gasic E, Wang T, Voogd Cet al. Mutagenesis of kiwifruit CENTRORADIALIS-like genes transforms a climbing woody perennial with long juvenility and axillary flowering into a compact plant with rapid terminal flowering. Plant Biotechnol J. 2019;17:869–80. 10.1111/pbi.13021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Charrier A, Vergne E, Dousset Net al. Efficient targeted mutagenesis in apple and first time edition of pear using the CRISPR-Cas9 system. Front Plant Sci. 2019;10:40. 10.3389/fpls.2019.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Wang X, Gao L, Jiao Cet al. Genome of Solanum pimpinellifolium provides insights into structural variants during tomato breeding. Nat Commun. 2020;11:5817. 10.1038/s41467-020-19682-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Guo J, Cao K, Deng Cet al. An integrated peach genome structural variation map uncovers genes associated with fruit traits. Genome Biol. 2020;21:258. 10.1186/s13059-020-02169-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Zhou Y, Minio A, Massonnet Met al. The population genetics of structural variants in grapevine domestication. Nat Plants. 2019;5:965–79. 10.1038/s41477-019-0507-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.