

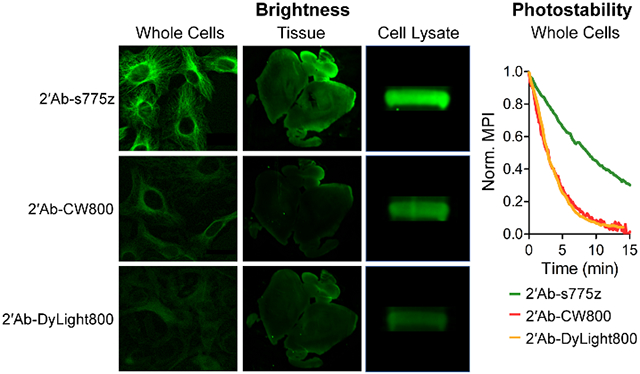
Abstract
A broad array of imaging and diagnostic technologies employ fluorophore-labeled antibodies for biomarker visualization, an experimental technique known as immunofluorescence. Significant performance advantages, such as higher signal-to-noise ratio, are gained if the appended fluorophore emits near-infrared light with a wavelength >700 nm. However, the currently available near-infrared fluorophore antibody conjugates are known to exhibit significant limitations including low chemical stability and photostability, weakened target specificity, and low fluorescence brightness. These fluorophore limitations are resolved by employing a near-infrared heptamethine cyanine dye called s775z whose chemical structure is very stable, charge-balanced, and sterically shielded. Using indirect immunofluorescence for imaging and visualization, a secondary IgG antibody labeled with s775z outperformed IgG analogues labeled with the commercially available near-infrared fluorophores, IRDye 800CW® and DyLight800®. Comparison experiments included three common techniques: immunocytochemistry, immunohistochemistry, and western blotting. Specifically, the secondary IgG labeled with s775z was 3-8 times brighter, 3-6 times more photostable, and still retained excellent target specificity when the degree of antibody labeling was high. The results demonstrate that antibodies labeled with s775z can emit total photon counts that are 1-2 orders of magnitude higher than currently possible, and thus, enable unsurpassed performance for near-infrared fluorescence imaging and diagnostics. They are especially well suited for analytical applications that require sensitive near-infrared fluorescence detection or use modern photon-intense methods that require high photostability.
Graphical Abstract
INTRODUCTION
Antibodies are widely used in therapeutics, diagnostics, and vaccine development due to their high avidity and specificity for a target antigen. Many imaging and diagnostic applications employ an antibody labeled with a reporter group that emits a detectable signal. When the appended reporter group is a fluorescent dye, the technique is called immunofluorescence.1 Fluorescent dyes that emit visible light are commonly used for assays that analyze thin and transparent samples. However, there is an emerging need for primary and secondary antibodies that are labeled with longer wavelength fluorophores. One reason for this situation is the development of new multiplex detection methods that employ multiple fluorescent antibodies in the same sample with distinguishable emission wavelengths. Another factor for long wavelength fluorophores is the increasing interest in imaging thick and opaque samples. Fluorophores that emit near-infrared (NIR) light with wavelengths >700 nm are especially attractive due to the relatively deep penetration of NIR light through biological tissue, low absorption and background autofluorescence by endogenous pigments in the tissue, and low scattering of the light.2,3
Cyanine heptamethine dyes are well-known NIR fluorophores, with emission wavelengths >740 nm, and several are commercially available as reagents for labeling antibodies, and in some cases the dye-labeled antibody can be purchased for immediate use. At present, two of the most popular commercial NIR cyanine heptamethine fluorophores for antibody conjugation are IRDye 800CW® (CW800) and DyLight800® (DyLight800). The most common way to fluorescently label an antibody is to use amine reactive NHS ester chemistry, where the dye’s activated carboxyl group reacts with an exposed lysine amine residue on the antibody surface to form an amide linkage (Scheme 1A).4 This bioconjugation method is attractive because the procedure is rapid, and experimentally straightforward. Moreover, antibodies typically have around 10-20 exposed lysine residues available for chemical reaction which raises the possibility of producing a high degree of labeling (DOL).5,6 While the currently available commercial conjugatable NIR dyes are undoubtedly useful for many types of immunofluorescence technologies,7,8,9,10,11 the resulting NIR dye-labeled antibodies sometimes exhibit performance limitations. In the case of CW800 and DyLight800, there are three inherent fluorophore concerns. (1) Common to both chemical structures is a meso-OAryl group connected directly to the heptamethine fluorochrome. This group is susceptible to nucleophilic displacement by biological amines and thiols,12,13 resulting in diminished chemical stability of the dye-antibody conjugates during synthesis, storage, or the time-course of an imaging experiment. In addition, the electron donating meso-OAryl group promotes high fluorochrome reactivity with electrophilic singlet oxygen, resulting in relatively poor dye photostability.14,15 (2) Both dyes are flat molecules with a hydrophobic core and a polyanionic charge periphery. Thus, chemical conversion of a small polar, cationic lysine residue on the antibody surface to a large hydrophobic, polyanionic dye derivative has the potential to produce substantial changes in antibody folding and physiochemical properties, leading to lower antibody stability and decreased target specificity.15-18 (3) When reactive versions of these hydrophobic dyes are conjugated to an antibody surface, they tend to attach at proximal lysine sites as stacked face-to-face dimers, which produces a diagnostic H-dimer peak in the absorbance spectra that is non-fluorescent (Scheme S1).19,20 Moreover, close stacking of proximal conjugated CW800 or DyLight800 on an antibody surface amplifies the potential for a deleterious effect on antibody targeting due to a localized patch of polyanionic charge and hydrophobicity.
Scheme 1.

(A) Amide bond coupling of NIR dyes, s775z (green), CW800 (red), and DyLight800 (orange) to secondary goat anti-rabbit IgG antibodies. Representative (B) normalized absorbance and (C) relative fluorescence emission spectra (ex: 745 nm) of aqueous solutions containing identical concentrations of dye-labeled antibodies.
Recently, we showed how all these dye performance problems could be resolved by developing a conceptually new class of NIR fluorophores called shielded heptamethine cyanine dyes.21 The prototype fluorophore is called s775z and addresses the three structural concerns of CW800 and Dylight800: (1) The reactive meso-OAryl group in CW800 and DyLight800 is replaced by a meso-Aryl group with a much less reactive C-C linkage, which greatly enhances chemical stability and photostability. (2) The periphery of s775z is charge-balanced with an equal number of anionic sulfonate and cationic ammonium residues; a structural feature that is known to reduce interactions with off-target biological surfaces.22,23 (3) The meso-Aryl group in s775z projects two shielding arms containing triethyleneglycol chains directly over both faces of the heptamethine fluorochrome. These shielding arms block any undesired bimolecular association processes, and thus, enhance fluorescence brightness (Scheme S1). In addition, a preliminary set of antibody labeling experiments indicated no stacking of multiple s775z fluorophores appended to the antibody surface.21
The promising initial results motivated us to conduct a more systematic assessment of bioconjugate properties, and here we report a direct comparison of immunoglobulin G (IgG) antibody labeled with s775z, CW800 or DyLight800. We evaluated the performance of the fluorescent conjugates in solution-state studies and as secondary antibodies for indirect immunofluorescence. Indirect immunofluorescence is a paired fluorescence detection system comprised of a primary antibody that targets the antigen and a secondary antibody with appended fluorophore that has selective avidity for the primary antibody, and thus, permits its visualization. Specifically, performance was evaluated in three common types of indirect immunofluorescence experiments; immunocytochemistry,24 immunohistochemistry,25 and western blotting.26-28 The results confirm our hypotheses that s775z is a superior NIR fluorophore and produces fluorescent antibody conjugates that are substantially brighter and more photostable; moreover, they retain excellent target specificity, even when the DOL is very high.
EXPERIMENTAL SECTION
Materials.
s775z NHS ester was synthesized following a previously published protocol with characterization shown in the Supporting Information. CW800 NHS ester was purchased from LI-COR (929-70020) and goat anti-rabbit IgG was purchased from Thermo Fischer Scientific (31210). Goat anti-rabbit IgG labeled with CW800 at DOL = 1.8 (2’Ab-CW800) was purchased from LI-COR (926-32211). Goat anti-rabbit IgG labeled with DyLight800 at DOL = 3.5 (2’Ab-DyLight800) was purchased from Thermo Fischer Scientific (SA5-10036). Both samples of commercial NIR dye-labeled IgG included bovine serum albumin as a storage stabilizer which enhanced the absorption peak at 280 nm. Donkey anti-mouse antibody labeled with FITC (2’Ab-FITC) was purchased from Thermo Fisher Scientific (A16012). All other reagents were purchased from Sigma Aldrich.
Cell Culture.
All cells were cultured and maintained in a humidified incubator at 37 °C under 5% CO2. The cell media (EMEM, F12-K) were purchased from American Type Culture Collection (ATCC). Fetal bovine serum was purchased from Atlanta Biologicals, and all other supplies were purchased from Sigma Aldrich. HT-1080 (ATCC CCL-121) human fibrosarcoma cells and U87-MG (ATCC HTB-14) glioblastoma cells were maintained in EMEM medium supplemented with 10% fetal bovine serum, 0.1 mM non-essential amino acids, 1.5 g/L sodium bicarbonate, 1 mM sodium pyruvate, and 1% penicillin/streptomycin. A549 (ATCC CCL-185) human non-small cell lung adenocarcinoma cells were maintained in F-12K medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin.
Antibody Conjugation.
Goat anti-rabbit IgG was concentrated to 12.85 mg/mL in Phosphate Buffered Saline and was further diluted to 5 mg/mL using 0.1 M Sodium Phosphate buffer pH 8.5. Separate stock solutions of CW800 NHS ester and s775z NHS ester were prepared in DMSO (10 mM). In each case, a 10 μL aliquot of dye NHS ester was added to 200 μL of goat anti-rabbit IgG (5 mg/mL) to give a molar stoichiometry of reactants (antibody: dye NHS ester) of 1:10. The reaction was shaken at room temperature for 2 hr and was filtered using a PD-10 desalting column (GE Healthcare Life Sciences) pre-equilibrated with 0.1 M Sodium Phosphate buffer pH 8.5. The eluent was collected and concentrated using an Amicon® ultra-4 centrifugal filter unit. Each antibody conjugate was washed once with Sodium Phosphate buffer, washed several times in Phosphate Buffered Saline, and concentrated to make a ~2 mg/mL solution in Phosphate Buffered Saline. The final DOL for purified 2’Ab-s775z was 4.2, and the final DOL for purified 2’Ab-CW800 was 4.4. A second labeling reaction used an antibody:CW800 NHS ester stoichiometry of 1:5 and produced 2’Ab-CW800 with a DOL of 2.5. This second reaction was shaken at room temperature for 1 hr, filtered using a PD-10 desalting column and concentrated using the same procedure as described above. Fluorescence emission spectra of goat anti-rabbit IgG conjugated to dyes were collected in Phosphate Buffered Saline using Horiba Fluoromax-4 Fluorometer with FluorEssence software (ex: 745 nm, slit width: 4 nm).
Immunocytochemistry.
HT-1080, A549, or U87-MG cells were seeded and grown to 80% confluency in an 8-well chambered coverglass. Cells were washed with Phosphate Buffered Saline and fixed with 4% paraformaldehyde for 20 min at room temperature. For the NIR microscopy experiments that imaged α/β-Tubulin alone (Figure 1), the cells were incubated with blocking buffer (Phosphate Buffered Saline containing 5% Bovine Serum Albumin and 0.3% TWEEN-20). For the multiplex microscopy experiments that imaged α/β-Tubulin and β-Actin (Figure 3), the cells were incubated with cold methanol at −20 °C for 10 minutes. The cells were washed twice with Phosphate Buffered Saline and incubated with blocking buffer (Phosphate Buffered Saline containing 10% Donkey Serum and 0.3% TWEEN-20) for 1 hr at room temperature. In all cases, the blocking buffer was removed, and the cells were co-incubated overnight at 4 °C with primary anti-Tubulin (1:100) (Cell Signaling, 2148) alone or mixed with primary anti-Actin (1:2500) (Cell Signaling, 3700) in Phosphate Buffered Saline containing 1% Bovine Serum Albumin and 0.3% TWEEN-20. The following day, the primary antibodies were removed, and the cells were washed twice with Phosphate Buffered Saline. Then the cells were incubated for 2 hr at room temperature with the appropriate dye-labeled secondary antibody (2 mg/mL in each case; 1:1000) in Phosphate Buffered Saline containing 2% Bovine Serum Albumin and 0.3% TWEEN-20. The secondary antibodies were removed, and the cells were washed three times with Phosphate Buffered Saline and co-stained with 3 μM Hoechst 33342 for 10 min at room temperature. Cells were washed two times with Phosphate Buffered Saline and imaged on a Zeiss Axiovert 100 TV epifluorescence microscope equipped with a UV filter (ex: 387/11, em: 447/60), FITC filter (ex: 485/20, em: 524/24), and ICG filter (ex: 769/41, em: 832/37).
Figure 1.

Comparison of secondary goat anti-rabbit IgG antibodies labeled with NIR dyes for targeting primary anti-Tubulin in fixed cells. HT-1080, U87-MG, and A549 cells were fixed, incubated with anti-Tubulin overnight at 4 °C, and incubated the following day with a fluorescent secondary antibody (2’Ab-s775z, 2’Ab-CW800, or 2’Ab-DyLight800) for 2 hr at room temperature. (A,B,C) Representative epifluorescence cell micrographs depict fluorescent secondary antibody without (left) or with primary anti-Tubulin (right). (D,E,F) Graphs of mean pixel intensity (MPI) for cell micrographs, where the cells were incubated with primary anti-Tubulin and a fluorescent secondary antibody. Scale bar = 30 μm. p-value < 0.05 (*), <0.01 (**), < 0.0001 (****)
Figure 3.

Multiplex imaging of HT-1080 cells. HT-1080 cells were fixed, co-incubated with primary rabbit anti-Tubulin and primary mouse anti-Actin overnight at 4 °C. Cells were co-incubated the following day with secondary goat anti-rabbit labeled with s775z (2’Ab-s775z) and secondary donkey anti-mouse labeled with FITC (2’Ab-FITC) for 2 hr at room temperature. Cells were co-stained with Hoechst 33342. Scale bar = 30 μm.
The following procedure was used to quantify the cell fluorescence intensity. With each micrograph, a background subtraction with a rolling ball radius of 200 pixels was applied using ImageJ2 software. A built-in ImageJ2 threshold was chosen and used to calculate the average mean pixel intensity (MPI) and SEM , which were plotted in GraphPad Prism. A total of 4 micrographs were evaluated per condition, and each experiment was conducted in triplicate.
For photostability analysis, time-lapse cell micrographs were captured at 10 or 20 sec time intervals over 15 minutes. Image processing for each micrograph was then conducted using ImageJ2 software with a 200-pixel rolling ball radius background subtraction. A photostability plot of the micrograph MPI over time was generated and fit with a one phase decay using GraphPad Prism.
Immunohistochemistry.
Mouse brain tissues were excised, fixed, and processed using a Leica TP 1020 Tissue Processor. The brain tissue was then embedded into paraffin, sectioned into 4 μm slices, and placed onto a glass slide. The tissues were sequentially placed in xylene, 100% ethanol, 95% ethanol, and water. Then the tissues were put in a water bath of 0.01 M Sodium Citrate buffer, pH 6.0 at 95 °C for 10 min; the tissues were removed from the water bath and cooled with deionized water for three, 5-min intervals. The tissues were incubated with blocking buffer (Phosphate Buffered Saline containing 5% Bovine Serum Albumin and 0.3% TWEEN-20) for 1 hr at room temperature. Blocking buffer was removed, and the tissues were incubated overnight at 4 °C with primary anti-Tubulin (1:100) (Cell Signaling, 2148) in Phosphate Buffered Saline containing 1% Bovine Serum Albumin and 0.3% TWEEN-20. The following day, the primary antibody was removed, and the tissues were washed twice with Phosphate Buffered Saline for 5 min each. Then the tissues were incubated for 2 hr at room temperature with the secondary antibody (2 mg/mL in each case; 1:250) in Phosphate Buffered Saline containing 2% Bovine Serum Albumin and 0.3% TWEEN-20. The secondary antibody was removed, and the tissues were washed three times with Phosphate Buffered Saline for 5 min each. Phosphate Buffered Saline was added, and a coverslip was placed onto the tissues. Tissues were imaged using a LI-COR Odyssey Infrared Imaging System equipped with an 800 nm detection channel. For magnified micrographs, tissues were imaged on a Zeiss Axiovert 100 TV epifluorescence microscope equipped with an ICG filter (ex: 769/41, em: 832/37). Each experiment was conducted in triplicate with three slides (one for each secondary antibody) to account for variability between experiments.
Western Blotting.
HT-1080 cells were grown to 80% confluency in a cell culture flask. Cells were washed with Phosphate Buffered Saline, SDS sample buffer was added (1x Tris buffer pH 6.8 containing 6% SDS, 30% glycerol and 56 mM DTT) and the cells transferred to a microcentrifuge tube. The cell lysate was sonicated for 15 sec and heated at 95 °C for 5 min. The cell lysate was cooled on ice and centrifuged at 10,000 g for 10 min. The total protein concentration was determined using the Bradford Protein Assay (Bio-Rad). Next, three 20 μL samples were loaded into a 4-15% Mini-PROTEAN TGX precast protein gel (Bio-Rad). The gel was run at 125 V for 60 min. Separated proteins were transferred to a polyvinylidene fluoride membrane (Azure) using a XCell II Blot Module (Thermo Fisher Scientific) at 25 V for 1.5 hr. The membrane was rinsed with Tris Buffered Saline and blocked for 1 hr at room temperature using Tris Buffered Saline plus Tween (TBST) containing 5% nonfat dried milk. The membrane was incubated overnight at 4 °C with primary anti-Tubulin (1:1000) (Cell Signaling, 2148). The following day, the membrane was washed with TBST three times for 5 min each and was cut into three pieces. The three pieces were placed in separate containers and incubated for 1 hr at room temperature with different secondary antibodies (2 mg/mL in each case; 1:20,000) in TBST containing 5% Bovine Serum Albumin and 0.01% SDS. The three pieces were rinsed with TBST three times for 5 min each and then placed in 1x Tris buffer. The pieces were imaged on a LI-COR Odyssey Infrared Imaging System equipped with an 800 nm detection channel. Experiment was conducted in triplicate.
RESULTS AND DISCUSSION
Antibody Labeling and Photophysical Characterization.
The project objective was to label goat anti-rabbit IgG antibody with three different NIR fluorophores and assess the capability of each conjugate for indirect NIR fluorescence detection. Shown in Scheme 1 are the three conjugate structures and identifiers: 2’Ab-s775z, 2’Ab-CW800, and 2’Ab-DyLight800. The conjugates were either procured commercially or synthesized by amine reactive NHS ester chemistry. The synthetic procedure involved mixing the antibody with a molar excess of s775z NHS ester or CW800 NHS ester to permit amide bond formation with multiple lysine amine groups on the antibody surface.29 The DOL was determined using a standard absorbance-based method, and gel electrophoresis was used to verify that each sample was a covalently bonded conjugate. In each case, a single NIR fluorescent band was observed at the expected IgG molecular weight of ~150 kDa with no measurable amount of unconjugated dye (Figure S4A). The chemical stabilities of the 2’Ab-s775z and 2’Ab-CW800 conjugates in Phosphate Buffered Saline at 4 °C were evaluated by monitoring the NIR absorbance. There was no spectral change for either conjugate over 10 days, indicating high chemical stability (Figure S2). But as shown in Scheme 1B, there are obvious differences in the NIR absorbance spectra for the antibody conjugates (expanded absorption spectra for all dye-labeled antibodies are provided in Figure S1). The sample of 2’Ab-s775z exhibited a single peak at 775 nm that matched the wavelength and peak shape exhibited by a solution of free s775z dye. Thus, the appended fluorochromes within 2’Ab-s775z behave like monomeric dye molecules and produce high fluorescence emission at 794 nm as seen in Scheme 1C. In contrast, the absorption spectra of 2’Ab-CW800 and 2’Ab-DyLight800 each exhibited a large blue-shifted “H-dimer” peak at ~710 nm, a common feature of antibodies labeled with heptamethine cyanine dyes.15,19 Low conjugate fluorescence due to stacking of proximal appended fluorochromes explains the antibody fluorescence trend in Scheme 1C and Figure S3 where the NIR fluorescence intensity for 2’Ab-CW800 and 2’Ab-DyLight800 (large fraction of stacked appended fluorochromes) is substantially lower than 2’Ab-s775z (no stacked appended fluorochromes).
Degree of Labeling for Each Dye-labeled Antibody.
An important early goal was to identify the near-optimal DOL for each dye-labeled antibody for subsequent comparative evaluations. In the case of 2’Ab-s775z, we found that NIR fluorescence intensity increased with DOL , up to the highest DOL tested, which was 17.5 (Figure S5).21 We decided to use a sample of 2’Ab-s775z with DOL = 4.2 for comparative testing, a compromise choice of DOL that ensured high brightness and negligible loss in antibody avidity/selectivity.
In the case of 2’Ab-CW800, we characterized three separate samples with DOL = 4.4, 2.5, or 1.8 and found that all exhibited the same NIR fluorescence intensity (solution-state measurements in Figure S3 and gel band measurements in Figure S4B). We attribute the invariance of 2’Ab-CW800 fluorescence brightness with DOL to a compensating effect caused by two factors, an increase in the fraction of non-fluorescent H-dimer with increasing DOL (Figure S1) and a general increase in fluorescence quenching due to non-radiative energy transfer between appended fluorochromes at higher DOL.30 We also evaluated the three samples of 2’Ab-CW800 (with different DOL) as secondary antibodies for immunocytochemistry. As described in the next section, the 2’Ab-CW800 antibodies were used in conjunction with primary anti-Tubulin to visualize α/β-Tubulin in NIR fluorescence cell micrographs. For 2’Ab-CW800 with DOL 1.8, moderately sharp fluorescence imaging of α/β-Tubulin was observed with negligible non-specific fluorescence staining of the cells when the primary anti-Tubulin was absent (Figure S6). In comparison, micrographs of cells treated with primary anti-Tubulin and 2’Ab-CW800 (DOL 2.5) displayed some background signal in the cytoplasm, and cells treated with primary anti-Tubulin and 2’Ab-CW800 (DOL 4.4) showed significant off-target fluorescence accumulation in the nucleus. When the primary antibody was absent, extensive non-specific accumulation of 2’Ab-CW800 (DOL 4.4) in the nucleus was still observed. The same differences in cell micrographs were observed using two other two cell lines (Figure S6). This loss of target specificity with increased CW800 labeling has been reported before by Choi and coworkers who observed higher background fluorescence with secondary antibodies labeled with CW800 (DOL 2.5) compared to versions with a lower DOL of 1.2.16 We speculate that the undesired nuclear accumulation of 2’Ab-CW800 (DOL 4.4) is due to strong electrostatic attraction of the anionic dye-labeled conjugate to cationic histones in the nucleus.31 Because 2’Ab-CW800 (DOL 1.8) exhibited the cleanest targeting of primary anti-Tubulin with minimal non-specific signal, it was chosen as a near-optimal 2’Ab-CW800 conjugate for comparison studies. The comparison studies also included a commercial sample of 2’Ab-DyLight800 (DOL 3.5) as an optimized secondary antibody from a reputable vendor.
The following sections describe comparative performance evaluations of 2’Ab-s775z (DOL 4.2), 2’Ab-CW800 (DOL 1.8), and 2’Ab-DyLight800 (DOL 3.5), as secondary antibodies for immunocytochemistry, immunohistochemistry, and western blotting. In all cases, the experiments using 2’Ab-s775z produced much higher immunofluorescence intensities and one obvious reason for this outcome is its higher DOL of 4.2. But it is very important to realize in the case of 2’Ab-CW800 (and undoubtedly also with 2’Ab-DyLight800) that NIR fluorescence brightness does not increase with DOL. Furthermore, 2’Ab-CW800 with DOL values higher than 1.8 exhibit non-specific targeting (as described above). Thus, the indirect immunofluorescence performance of 2’Ab-CW800 and 2’Ab-DyLight800 as secondary antibodies cannot be improved significantly beyond the results presented in the following sections.
Immunocytochemistry.
Fluorescence cell microscopy was used to compare the dye-labeled goat anti-rabbit IgG antibodies in two sets of common immunocytochemistry procedures. The first set of single-color imaging experiments focused on visualizing α/β-Tubulin, an abundant intracellular protein that is centrally important in cell biology.32,33 Three cell lines were chosen for evaluation: HT-1080 fibrosarcoma cells, U87-MG glioblastoma cells, and A549 lung adenocarcinoma cells. Cells were grown in an 8-well chambered coverglass, fixed with 4% paraformaldehyde, and incubated with blocking buffer (PBS, 5% BSA, 0.3% TWEEN-20) for 1 hr at room temperature. The blocking buffer was removed, and the cells were incubated overnight at 4 °C with rabbit anti-Tubulin. The following day, each well was incubated for 2 hr at room temperature with one of the dye-labeled goat anti-rabbit antibodies followed by cell imaging using NIR fluorescence microscopy. Shown in Figure 1 is a comparison of cell micrographs obtained using primary anti-Tubulin and 2’Ab-s775z, 2’Ab-CW800, or 2’Ab-DyLight800. Quantification of the cell fluorescence showed that treatment with 2’Ab-s775z produced fluorescence intensities that were 6, 4, and 8 times higher in HT-1080, U87-MG, and A549 cells, respectively, compared to cells treated with 2’Ab-CW800 or 2’Ab-DyLight800. When the primary antibody was absent, there was negligible cell staining by any of the dye-labeled secondary antibodies.
Our preliminary study of s775z as a free dye in solution showed that it was significantly more photostable than other benchmark NIR dyes.21 Chemically, the main reason for the enhanced photostability is lower fluorochrome reactivity with photogenerated singlet oxygen due to the electronic deactivation of the meso-Aryl group in s775z. To confirm that high photostability is maintained when 2’Ab-s775z is used as a secondary antibody for indirect immunofluorescence, a series of photostability experiments were conducted by photobleaching fluorescently labeled cells. Cells were incubated with primary anti-Tubulin and the secondary antibody conjugate followed by continuous irradiation with NIR light within a microscope. During the 15-min irradiation, time-lapse NIR fluorescence cell micrographs were captured at 10-20 sec time intervals, and the mean pixel intensity (MPI) was quantified for each time point. For all three cell lines stained with 2’Ab-s775z, the rate of fluorescence photobleaching was 3-6 times slower than the photobleaching of the cells stained using 2’Ab-CW800 or 2’Ab-DyLight800 (Figure 2 and Table S2). It is important to emphasize that the plots in Figure 2 show the change in relative fluorescence intensity for each separate sample. Since cells stained with 2’Ab-s775z start out 4-8 times brighter, they still exhibit equal or higher brightness after 15 min of continuous irradiation, than cells that were stained using 2’Ab-CW800 or 2’Ab- DyLight800 and kept in the dark (i.e., no microscope NIR irradiation) (Figure S9). The same photostability trend was observed with stained cells exposed to a long period of continuous sunlight. As shown in Figure S10, all three cell lines stained with 2’Ab-s775z and exposed to sunlight for 16 hr were significantly brighter than cell micrographs that had been stained with 2’Ab-CW800 or 2’Ab- DyLight800 and kept in the dark.34
Figure 2.

Photostability comparison of secondary goat anti-rabbit IgG antibodies labeled with NIR dyes. Cells were incubated with primary anti-Tubulin and fluorescent secondary antibody (2’Ab-s775z, 2’Ab-CW800, or 2’Ab-DyLight800) followed by continuous irradiation with NIR light within a microscope equipped with an ICG filter (ex: 769/41, em: 832/37). Plots show normalized mean pixel intensity (MPI) for time-lapse cell micrographs.
Described in the Supporting Information is a second set of independent immunocytochemistry experiments that focused on indirect fluorescence imaging of HSP72, a member of the 70-kDa heat shock protein family (HSP70) that is strongly expressed in HT-1080 fibrosarcoma cells.35-37 In short, the same trends were observed; cells stained with 2’Ab-s775z were 3 times brighter (Figure S7) and photobleached 5 times more slowly than cells stained with 2’Ab-CW800 (Table S2 and Figure S11).
The final set of immunocytochemistry experiments evaluated suitability of 2’Ab-s775z as a secondary antibody for multiplex immunofluorescence. The NIR fluorescent 2’Ab-s775z can easily be combined with other visible fluorescent probes to permit multiplex cell imaging with no spectral overlap. In principle, two-color indirect immunofluorescence can be achieved in four steps: (1) add primary antibody, (2) add complementary secondary antibody, (3) add different primary antibody, (4) add complementary secondary antibody. However, we focused on a shorter two-step method: (1) add both primary antibodies together, and (2) add both secondary antibodies together. The convenience of the second method is attractive; however, it requires each dye-labeled secondary antibody to be very specific for its respective primary antibody partner. We used the two-step multiplex indirect immunofluorescence imaging experiment to image α/β-Tubulin and β-Actin in the same cells. A population of HT-1080 cells were co-incubated with primary rabbit anti-Tubulin and primary mouse anti-Actin. The cells were then co-incubated with a NIR-emitting secondary goat anti-rabbit antibody (2’Ab-s775z) and a commercial green-emitting secondary donkey anti-mouse antibody (2’Ab-FITC). The multiplex image depicted in Figure 3 shows the nuclear stain, Hoechst 33342, as blue, β-Actin as red, and α/β-Tubulin as green. Furthermore, control experiments that incubated HT-1080 cells with the mismatched pair of primary mouse anti-Actin and secondary anti-rabbit 2’Ab-s775z produced minimal cell fluorescence, confirming no cross-reactivity (Figure S8a). Additional control experiments with commercial 2’Ab-FITC are described in Figure S8b.
Immunohistochemistry.
Immunohistochemistry typically employs a much larger field-of-view than immunocytochemistry and captures multi-dimensional data related to tissue architecture and spatial distribution of cell phenotypes.38 Immunohistochemistry identifies prognostic markers in cancer, infections, and neurodegenerative diseases,39 and is a common and valuable diagnostic method in clinical practice.40 NIR fluorescent immunohistochemistry is especially useful for analyzing antigen expression levels in thick and opaque histological sections of biopsy and surgical samples.38
The relative performance of 2’Ab-s775z, 2’Ab-CW800 and 2’Ab- DyLight800 as NIR-emitting secondary goat anti-rabbit antibodies for fluorescence immunohistochemistry was assessed by staining and imaging mouse brain tissue sections composed of neurons with high tubulin expression.41 Thin sections of normal mouse brain tissues were incubated with primary rabbit anti-Tubulin overnight at 4 °C. The following day, separate sections were incubated for 2 hr at room temperature with one of the three secondary antibody conjugates, and then imaged using a LI-COR Odyssey Infrared Imaging System equipped with an 800 nm fluorescence observation channel. As illustrated by the images in Figure 4, the NIR fluorescence intensity of brain tissue stained with 2’Ab-s775z was 2.5 or 2.6 times higher than corresponding tissues sections stained with equal amounts of 2’Ab-CW800 or 2’Ab-DyLight800, respectively. Control experiments showed minimal non-specific staining of the tissue when the primary anti-Tubulin was absent (Figure S12). Magnified images of the antibody treated tissue sections were collected using an epifluorescence microscope. The magnified images stained with 2’Ab-s775z revealed an α/β-Tubulin staining pattern that matched literature micrographs,41,42 whereas magnified images stained with 2’Ab-CW800 or 2’Ab-DyLight800 showed very low fluorescence (inset Figure 4). The low fluorescence intensity produced by 2’Ab-CW800 is consistent with the findings of a recently published study that was forced to use a primary antibody, an unlabeled secondary antibody as signal amplifier, and a tertiary antibody conjugated to CW800 to identify α-Methylacyl-CoA Racemase in human prostate cancer tissues.16 In comparison, the capability of 2’Ab-s775z to visualize α/β-Tubulin (a protein with lower expression level than α-Methylacyl-CoA Racemase in prostate cancer specimens)43 without the need for signal amplification is a good indication of its superior detection sensitivity.
Figure 4.

Comparison of secondary goat anti-rabbit IgG antibodies labeled with NIR dyes for targeting primary anti-Tubulin in mouse brain tissue. Mouse brain tissue slice was incubated with anti-Tubulin overnight at 4 °C and incubated the following day with a fluorescent secondary antibody (2’Ab-s775z, 2’Ab-CW800, or 2’Ab-DyLight800) for 2 hr at room temperature. Each NIR fluorescent image is a brain tissue slice with the inset showing a magnified micrograph. Scale bars on the brain tissue and micrograph inset are 1 mm and 30 μm, respectively.
Western Blotting.
Western blotting is a rapid and sensitive method to identify and quantify protein abundance, kinase activity, and protein–protein interactions.44 There are three standard types of optical signal output: colorimetric, chemiluminescence, and fluorescence.44,45 Fluorescence imaging of western blots has several attractive features including high sensitivity and the capability of simultaneous multiplex detection. For example, the popular Odyssey Infrared Imaging System from LI-COR is equipped with two detection channels (700 and 800 nm) to simultaneously record signals from separate fluorescent probes with appended pentamethine cyanine (700 nm) or heptamethine cyanine (800 nm) dyes.
A series of western blots were performed in order to compare the relative performance of 2’Ab-s775z, 2’Ab-CW800 and 2’Ab-DyLight800 as NIR-emitting secondary goat anti-rabbit antibodies for staining and imaging of α/β-Tubulin in cell lysate. The primary antibody was the rabbit anti-Tubulin used above. As explained by the workflow diagram in Scheme S2, HT-1080 cell lysate was aliquoted equally into three lanes on a polyacrylamide SDS gel, and after protein separation, the protein bands were transferred to a polyvinylidene fluoride membrane. The membrane was blocked and incubated overnight at 4 °C with primary rabbit anti-Tubulin. The following day, the membrane was cut into three pieces, each containing an identical cell lysate lane, and each piece was separately incubated with 2’Ab-s775z, 2’Ab-CW800 or 2’Ab-DyLight800. As shown in Figure 5, the α/β-Tubulin band stained with 2’Ab-s775z produced 2.6 or 3.5 times higher fluorescence intensity than the analogous bands stained with 2’Ab-CW800 or 2’Ab-DyLight800, respectively. The trend of higher fluorescence intensity with 2’Ab-s775z was maintained when the experiment was repeated using varying amounts of intracellular total protein (Figure S13). With the lowest amount of total protein (107 ug/mL), the α/β-Tubulin band stained with 2’Ab-CW800 or 2’Ab-DyLight800 was very faint, but the α/β-Tubulin band stained with 2’Ab-s775z was easily observed. We infer from the linear dependence that secondary antibodies labeled with s775z should permit visualization of protein bands within western blots at 2.6 or 3.5 times lower concentration compared to secondary antibodies labeled with CW800 or DyLight800.
Figure 5.

Comparison of secondary goat anti-rabbit IgG antibodies labeled with NIR dyes for targeting primary anti-Tubulin in western blots. HT-1080 cell lysate was run on a polyacrylamide SDS gel, and the separated proteins were transferred to a polyvinylidene fluoride membrane. The membrane was incubated with primary anti-Tubulin overnight at 4 °C and incubated with a fluorescent secondary antibody (2’Ab-s775z, 2’Ab-CW800 or 2’Ab- DyLight800), then a NIR fluorescence image of each α/β-Tubulin band was acquired.
CONCLUSION
Using indirect immunofluorescence for imaging and visualization, a goat anti-rabbit IgG antibody labeled with the new NIR fluorophore s775z (2’Ab-s775z) significantly outperformed analogous IgG conjugates labeled with the commercially available NIR fluorophores IRDye 800CW® (2’Ab-CW800) and DyLight800® (2’Ab-DyLight800). A series of comparative immunocytochemistry, immunohistochemistry, and western blotting experiments assessed performance as fluorescent secondary antibodies. The immunocytochemistry experiments imaged the intracellular proteins, α/β-Tubulin and HSP72, and found that 2’Ab-s775z produced micrographs that were 3-8 times brighter and 3-6 times more photostable. Moreover, multiplex microscopy imaging experiments showed excellent target specificity, even when the degree of antibody labeling was high. The immunohistochemistry experiments imaged tubulin expression in mouse brain tissue sections and found that equivalent tissue sections stained with 2’Ab-s775z were 3 times brighter. The western blotting experiments assessed immunofluorescence staining of α/β-Tubulin from cell lysate where staining with 2’Ab-s775z was about 3 times brighter. Thus, a specific tangible outcome of this study is identification of 2’Ab-s775z as a superior near-infrared fluorescent secondary antibody for indirect imaging and detection of the common and broadly useful diagnostic biomarkers α/β-Tubulin46,47,48,49 and HSP7250,51,52. In terms of long-term future impact, the results demonstrate that secondary IgG antibodies labeled with s775z have the capacity to emit total photon counts that are 1-2 orders of magnitude higher than currently possible using the commercial options. This is a major advance in NIR immunofluorescence and will enable researchers to substantially improve various types of indirect NIR immunofluorescence imaging and diagnostics applications that require high sensitivity, and also develop new photon-intense techniques that require high photostability. It is quite likely that antibodies labeled with s775z will also be very useful for direct immunofluorescence methods where high levels of NIR fluorescence brightness are essential. A particularly attractive clinical application is direct NIR fluorescence imaging of living subjects, especially intraoperative imaging53,54 and rapid pathology.55 These studies are underway and the results will be published in due course.
Supplementary Material
ACKNOWLEDGMENT
We are grateful for funding support from the US NIH (R35GM136212 and T32GM075762) and a Berry Family Foundation fellowship from the University of Notre Dame.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website: Antibody conjugate characterization, photophysical properties, imaging data and analysis (PDF)
Conflict of Interest
B.D.S and D.-H.L. have filed a provisional patent application involving shielded cyanine dyes, whose value might be affected by this publication.
REFERENCES
- (1).Im K; Mareninov S; Diaz MFP; Yong WH In Biobanking: Methods and Protocols; Yong WH, Ed.; Springer New York: New York, NY, 2019; pp 299–311. [Google Scholar]
- (2).Flanagan JH; Khan SH; Menchen S; Soper SA; Hammer RP Bioconjug. Chem 1997, 8 (5), 751–756. [DOI] [PubMed] [Google Scholar]
- (3).Li Y; Zhou Y; Yue X; Dai Z Adv. Healthc. Mater 2020, 9, 2001327. [DOI] [PubMed] [Google Scholar]
- (4).Berlier JE; Rothe A; Buller G; Bradford J; Gray DR; Filanoski BJ; Telford WG; Yue S; Liu J; Cheung CY; Chang W; Hirsch JD; Beechem JM; Haugland RP; Haugland RP J. Histochem. Cytochem 2003, 51 (12), 1699–1712. [DOI] [PubMed] [Google Scholar]
- (5).Jain N; Smith SW; Ghone S; Tomczuk B Pharm. Res 2015, 32 (11), 3526–3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Dennler P; Fischer E; Schibli R Antibodies 2015, 4 (3), 197–224. [Google Scholar]
- (7).Milovancev M; Hilgart-Martiszus I; McNamara MJ; Goodall CP; Seguin B; Bracha S; Wickramasekara SI BMC Vet. Res 2013, 9, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Liu WH; Zheng J; Feldman JL; Klein MA; Kuznetsov VI; Peterson CL; Griffin PR; Denu JM Nat. Commun 2020, 11 (1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Nowak K; Rosenthal F; Karlberg T; Bütepage M; Thorsell AG; Dreier B; Grossmann J; Sobek J; Imhof R; Lüscher B; Schüler H; Plückthun A; Leslie Pedrioli DM; Hottiger MO Nat. Commun 2020, 11 (1), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Varadaraj A; Jenkins LM; Singh P; Chanda A; Snider J; Lee NY; Amsalem-Zafran AR; Ehrlich M; Henis YI; Mythreye K Mol. Biol. Cell 2017, 28 (9), 1195–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Wu J; Bell OH; Copland DA; Young A; Pooley JR; Maswood R; Evans RS; Khaw PT; Ali RR; Dick AD; Chu CJ Mol. Ther 2020, 28 (3), 820–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Van Der Wal S; Kuil J; Valentijn ARPM; Van Leeuwen FWB Dye. Pigment 2016, 132, 7–19. [Google Scholar]
- (13).Zaheer A; Wheat TE; Frangioni JV Mol. Imaging 2002, 1 (4), 354–364. [DOI] [PubMed] [Google Scholar]
- (14).Tynan CJ; Clarke DT; Coles BC; Rolfe DJ; Martin-Fernandez ML; Webb SED PLoS One 2012, 7(4):, e36265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Luciano MP; Crooke SN; Nourian S; Dingle I; Nani RR; Kline G; Patel NL; Robinson CM; Difilippantonio S; Kalen JD; Finn MG; Schnermann MJ ACS Chem. Biol 2019, 14 (5), 934–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Choi HS; Gibbs SL; Lee JH; Kim SH; Ashitate Y; Liu F; Hyun H; Park G; Xie Y; Bae S; Henary M; Frangioni JV Nat. Biotechnol 2013, 31 (2), 148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Warram JM; de Boer E; Korb M; Hartman Y; Kovar J; Markert JM; Yancey Gillespie G; Rosenthal EL Br J Neurosurg 2015, 29 (6), 850–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Szabó Á; Szendi-Szatmári T; Ujlaky-Nagy L; Rádi I; Vereb G; Szöllősi J; Nagy P Biophys. J 2018, 114 (3), 688–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Gruber HJ; Hahn CD; Kada G; Riener CK; Harms GS; Ahrer W; Dax TG 2000, 696–704. [DOI] [PubMed] [Google Scholar]
- (20).Pauli J; Grabolle M; Brehm R; Spieles M; Hamann FM; Wenzel M; Hilger I; Resch-Genger U Bioconjug. Chem 2011, 22 (7), 1298–1308. [DOI] [PubMed] [Google Scholar]
- (21).Li D; Schreiber CL; Smith BD Angew. Chem. Int. Ed 2020, 12154–12161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Sato K; Gorka AP; Nagaya T; Michie MS; Nani RR; Nakamura Y; Coble VL; Vasalatiy OV; Swenson RE; Choyke PL; Schnermann MJ; Kobayashi H Bioconjug. Chem 2016, 27 (2), 404–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Erfani A; Seaberg J; Aichele CP; Ramsey JD Biomacromolecules 2020, 21 (7), 2557–2573. [DOI] [PubMed] [Google Scholar]
- (24).Tello-Lafoz M; Martínez-Martínez G; Rodríguez-Rodríguez C; Albar JP; Huse M; Gharbi S; Merida I Traffic 2017, 18 (8), 491–504. [DOI] [PubMed] [Google Scholar]
- (25).Dixon AR; Bathany C; Tsuei M; White J; Barald KF; Takayama S Expert Rev. Mol. Diagn 2015, 15 (9), 1171–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Ramos J; Han L; Li Y; Hagelskamp F; Kellner SM; Alkuraya FS; Phizicky EM; Fu D Mol. Cell. Biol. 2019, 39 (19). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Sachar M; Li F; Liu K; Wang P; Lu J; Ma X; Res C; Author T Chem. Res. Toxicol 2016, 29 (8), 1293–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Menz C; Parsi MK; Adams JRJ; Sideek MA; Kopecki Z; Cowin AJ; Gibson MA PLoS One 2015, 10 (8), e0135577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Shrestha D; Bagosi A; Szöllősi J; Jenei A Anal. Bioanal. Chem 2012, 404 (5), 1449–1463. [DOI] [PubMed] [Google Scholar]
- (30).Luchowski R; Matveeva EG; Gryczynski I; Terpetschnig EA; Patsenker L; Laczko G; Borejdo J; Gryczynski Z Curr. Pharm. Biotechnol 2008, 9 (5), 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Zinchenko A; Chen Q; Berezhnoy NV; Wang S; Nordenskiöld L Soft Matter 2020, 16 (18), 4366–4372. [DOI] [PubMed] [Google Scholar]
- (32).Brouhard GJ; Rice LM Nat. Rev. Mol. Cell Biol 2018, 19 (7), 451–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Goodson HV ; Jonasson EM Cold Spring Harb. Perspect. Biol 2018, 10 (6), a022608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).One way to increase photostability of NIR dyes is to add a standard antifade buffer, and preliminary experiments on fixed tissue showed that SlowFade™ Diamond Antifade (ThermoFisher) increased the photostability of all antibody conjugates as expected.
- (35).Albakova Z; Armeev GA; Kanevskiy LM; Kovalenko EI; Sapozhnikov AM Cells 2020, 9 (3), 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Multhoff G; Botzler C; Wiesnet M; Müller E; Meier T; Wilmanns W; Issels RD Int. J. Cancer 1995, 61 (2), 272–279. [DOI] [PubMed] [Google Scholar]
- (37).Yaglom JA; Gabai VL; Sherman MY Cancer Res. 2007, 67 (5), 2373–2381. [DOI] [PubMed] [Google Scholar]
- (38).Stack EC; Wang C; Roman KA; Hoyt CC Methods 2014, 70 (1), 46–58. [DOI] [PubMed] [Google Scholar]
- (39).Orakpoghenor O; Avazi DO; Markus TP; Olaolu OS Immunogentics 2018, 3 (1), 1000120. [Google Scholar]
- (40).De Matos LL; Trufelli DC; De Matos MGL; Da Silva Pinhal MA Biomark. Insights 2010, 5, 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Vu HT; Akatsu H; Hashizume Y; Setou M; Ikegami K Sci. Rep 2017, 7 (1), 40205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Mozes E; Hunya A; Toth A; Ayaydin F; Penke B; Datki ZL Brain Res. Bull 2011, 86 (3–4), 217–221. [DOI] [PubMed] [Google Scholar]
- (43).Kumar-Sinha C; Shah RB; Laxman B; Tomlins SA; Harwood J; Schmitz W; Conzelmann E; Sanda MG; Wei JT; Rubin MA; Chinnaiyan AM Am. J. Pathol 2004, 164 (3), 787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Bass JJ; Wilkinson DJ; Rankin D; Phillips BE; Szewczyk NJ; Smith K; Atherton PJ Scand. J. Med. Sci. Sports 2017, 27 (1), 4–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Gingrich JC; Davis DR; Nguyen Q Biotechniques 2000, 29 (3), 636–642. [DOI] [PubMed] [Google Scholar]
- (46).Nakamura Y; Ishigaki Y Oncol. Lett 2014, 8 (6), 2387–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Cuenca-Zamora EJ; Ferrer-Marín F; Rivera J; Teruel-Montoya R Int. J. Mol. Sci 2019, 20(14), 3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Duran GE; Wang YC; Moisan F; Francisco EB; Sikic BI Br. J. Cancer 2017, 116 (10), 1318–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Schwarzerová K; Bellinvia E; Martinek J; Sikorová L; Dostál V; Libusová L; Bokvaj P; Fischer L; Schmit AC; Nick P Sci. Rep 2019, 9 (1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Thakur SS; James JL; Cranna NJ; Chhen VL; Swiderski K; Ryall JG; Lynch GS Cell Stress Chaperones 2019, 24 (4), 749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Tang XM; Dai J; Sun HL Mol. Med. Rep 2019, 20 (1), 205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Hamamoto T; Suzuki K; Kodama S; Sasaki H; Abe K; Hayashi T; Watanabe M Int. J. Hyperth 2007, 23 (4), 363–370. [DOI] [PubMed] [Google Scholar]
- (53).Garner RC; Lappin G Br. J. Clin. Pharmacol 2006, 61 (4), 367–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Burt T; Vuong LT; Baker E; Young GC; McCartt AD; Bergstrom M; Sugiyama Y; Combes R Altern. to Lab. Anim 2018, 46 (6), 335–346. [DOI] [PubMed] [Google Scholar]
- (55).Schaefer JM; Barth CW; Davis SC; Gibbs SL J. Biomed. Opt 2019, 24 (02), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.