

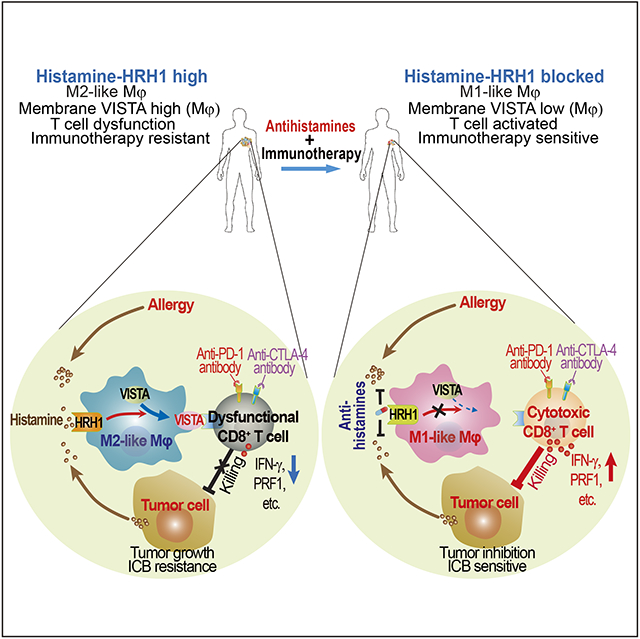
SUMMARY
Reinvigoration of anti-tumor immunity remains an unmet challenge. Our retrospective analyses revealed that cancer patients who took antihistamines during immunotherapy treatment had significantly improved survival. We uncovered that histamine and histamine receptor H1 (HRH1) are frequently increased in the tumor microenvironment and induce T cell dysfunction. Mechanistically, HRH1-activated macrophages polarize toward an M2-like immunosuppressive phenotype with increased expression of the immune checkpoint VISTA, rendering T cells dysfunctional. HRH1 knockout or antihistamine treatment reverted macrophages immunosuppression, revitalized T cell cytotoxic function, and restored immunotherapy response. Allergy, via the histamine-HRH1 axis, facilitated tumor growth and induced immunotherapy resistance in mice and humans. Importantly, cancer patients with low plasma histamine levels had more than tripled objective response rate to anti-PD-1 treatment compared to patients with high plasma histamine. Altogether, pre-existing allergy or high histamine levels in cancer patients can dampen immunotherapy responses and warrant prospectively exploring antihistamines as adjuvant agents for combinatorial immunotherapy.
Graphical Abstract
eTOC blurb
Li et al. investigate how cancer cells evade immune attack and resist immunotherapies. Cancer cell-derived or allergy-released histamine binds to HRH1 on tumor-associated macrophages that suppress CD8+ T cell function, accelerate tumor growth, and confer immunotherapy resistance. H1-antihistamines counteract histamine-mediated immunosuppression, reinforce anti-tumor immunity, and significantly enhance immunotherapy response.
INTRODUCTION
T cell-mediated anti-tumor immunity plays a central role in host’s defense against cancer. However, cancer cells can co-evolve with the tumor immune microenvironment and develop different strategies to evade T cell immune destruction. Tumor infiltrating T cells often manifest impaired effector function (i.e., dysfunction) and fail to eliminate cancer cells owing to various T cell inhibitory signals, e.g., cytotoxic lymphocyte antigen-4 (CTLA-4) and programmed cell death protein 1 (PD-1)/programmed death ligand 1 (PD-L1) (Jiang et al., 2018). Anti-CTLA-4 and anti-PD-1/PD-L1 antibodies, as immune checkpoint blockade (ICB) therapies, have yielded significant clinical benefits and durable responses in a subset of cancer patients. Yet, many cancer patients cannot benefit from these treatments, and it is highly challenging to reach immunotherapy’s full potential (Ribas and Wolchok, 2018; Sharma et al., 2017; Verma et al., 2019). To gain insights on what may impact on cancer patients’ immunotherapy response, we retrospectively analyzed patients who took different common pharmaceutical drugs during ICB treatments. Surprisingly, we found that antihistamines are associated with significantly improved clinical outcome and antihistamines may achieve this via reinforcing anti-tumor immunity, raising an interesting question: how antihistamines, which block histamine binding to histamine-receptors, influence anti-tumor immunity?
Histamine, a metabolite of histidine, is best known for its release from mast cells as a response to allergic reactions or tissue damage. Histamine exerts its effects primarily by binding to G protein-coupled receptors, designated histamine receptors H1 through H4 (HRH1/2/3/4). Among them, HRH1 is the major one involved in allergic response. During allergic reactions, mast cell-released histamines activate HRH1, which triggers contraction of smooth muscles and increases capillary permeability, resulting in classic allergy symptoms. HRH1 antagonists, mostly over-the-counter (OTC) drugs, are widely used to relieve allergy symptoms and to prevent nausea and vomiting in cancer treatment (Simons, 2004; Thurmond et al., 2008). Elevated levels of histamine have been detected in cancer patients’ blood and cancerous tissues although histamine has not been suggested in cancer etiology (Haak-Frendscho et al., 2000; Moriarty et al., 1988; Sieja et al., 2005; von Mach-Szczypinski et al., 2009). In addition, cancer cells frequently up-regulate a histamine-synthesizing enzyme, L-histidine decarboxylase (HDC), leading to increased histamine in cancer patients (Haak-Frendscho et al., 2000; Massari et al., 2018). Generally, the roles of histamine and histamine receptors in cancer development are unclear. Previous studies focus on HRHs’ expressions on various cancer cells, which lead to controversial reports that HRHs either promote or inhibit cancer growth (Faustino-Rocha et al., 2017). Moreover, allergic reaction releases lots of histamine and affects tens of millions of people every year, yet the potential impacts of allergy on cancer and cancer therapies have not been investigated.
RESULTS
Patients receiving antihistamines have better survival with ICB therapies
To assess the impact of taking other medications on therapeutic response to immunotherapy in cancer patients, we retrospectively evaluated the clinical outcomes of melanoma patients who took another medicine among forty charted common drugs (Table S1) while receiving immunotherapies (anti-PD-1/PD-L1) at The University of Texas MD Anderson Cancer Center. Our data showed that taking antibiotics (e.g., ampicillin) was associated with an increased death rate in immunotherapy-treated patients, consistent with a previous report (Elkrief et al., 2019); whereas taking aspirin was correlated with a reduced death rate in immunotherapy-treated patients, as found in mouse models (Figures 1A and S1A) (Zelenay et al., 2015). Among the forty common drugs examined, only HRH1-specific antihistamines (H1-antihistamines or second-generation antihistamines) significantly correlated with better survival of patients besides aspirin (Figure 1A). Clearly, melanoma patients who took H1-antihistamines during anti-PD-1/PD-L1 treatments had a highly significantly reduced death rate than did age-, sex-, or stage-matched patients without taking the H1-antihistamines (Figures 1B, 1C, S1B and Table S2). Among lung cancer patients receiving anti-PD-1/PD-L1 treatments, those taking H1-antihistamines also showed a statistically significant reduction of death rate than those without taking the H1-antihistamines (Figure 1B, and Table S2). Kaplan-Meier survival analysis also indicated significantly improved overall survival in melanoma and lung cancer patients who took H1-antibhistamins during anti-PD-1/PD-L1 treatment compared to control groups that didn’t (Figure 1D). Additionally, breast and colon cancer patients taking H1-antihistamines while receiving anti-PD-1/PD-L1 therapies also showed trends of reduced death rate (Figure S1C and Table S2), although no statistical significance due to relatively smaller numbers of patients taking the H1-antihistamines. These clinical data indicated that H1-antihistamines may augment immunotherapy. Notably, H1-antihistamines had minimal effect on the survival of chemotherapy-treated patients (Figures 1E and S1D), suggesting that H1-antihistamines may not target tumor cells directly.
Figure 1. Uptake of antihistamines correlated with better survival in ICB-treated patients and HRH1 expression is associated with T cell dysfunction.

(A) Scatter plot of the real numbers of deceased melanoma patients who took various commonly-used medicines (40 different drugs as listed in Table S1) during ICB treatment versus their estimated deaths (at 39% death rate based on 336 deceased patients out of total 865 ICB-treated patients at MDACC). Each dot represents a group of patients who took one type of medicine along with ICB.
(B) Percentages of deceased cancer patients taking H1-antihistamines during anti-PD-1/PD-L1 treatment versus those did not (Fisher exact test).
(C) Percentages of deceased melanoma patients who took H1-antihistamines during anti-PD-1/PD-L1 treatment or chemotherapy compared with sex- or stage-matched melanoma patients who did not.
(D) Kaplan-Meier overall survival analysis of cancer patients taking H1-antihistamines during anti-PD-1/PD-L1 treatment versus those who did not.
(E) Percentages of deceased cancer patients taking H1-antihistamines during chemotherapy treatment versus those did not (Fisher exact test).
(F) T cell dysfunction scores of HRH1-4 in indicated cancer types assessed by TIDE. T cell dysfunction score is defined as the z score of d/standard error (s.e.) following previous publication (Jiang et al., 2018).
(G) Kaplan-Meier overall survival analysis for CTL+ TNBC patients based on HRH1 level detected in tumors. The numbers at risk are the stratified HRH1 level high and low patient numbers of those who remained alive and uncensored after a certain time period.
(H) HRH1 mRNA expression in pre-treatment tumors of responder (n=15) versus non-responder (n=13) melanoma patients (t-test), and comparison of overall survival of melanoma patients who had high HRH1 vs. low HRH1 expressed in the tumors before anti-PD-1 treatment (GSE78220).
Mean±SEM, *P<0.05.
HRH1 correlates with T cell dysfunction in human cancers
The above clinical findings suggested that H1-antihistamines may enhance anti-tumor immunity. H1-antihistamines specifically block histamine binding to HRH1. Therefore, we examined whether HRH1 high-expressing tumors were associated with suppressed anti-tumor immunity in cancer patients. We adapted Tumor Immune Dysfunction and Exclusion computational framework (Jiang et al., 2018) to evaluate the impacts of HRH1 on T cell infiltration and T cell dysfunction in patient samples from The Cancer Genome Atlas (TCGA). Evidently, HRH1 expression yielded high tumor immune dysfunction scores in 9 of 12 TCGA cancer types analyzed, a higher proportion than CD274 (PD-L1) and SERPINB9 (Figure 1F), two genes well-known for inducing T cell dysfunction (Jiang et al., 2018). Notably, HRH1 expression was not associated with cytotoxic T lymphocyte (CTL) infiltration (Figure S1E), suggesting that HRH1 high expression may primarily induce T cell dysfunction. In contrast, other histamine-receptors (HRH/2/3/4) had much lower effect on T cell dysfunction compared with HRH1 (Figure 1F). The strong association between HRH1 expression and T cell dysfunction prompted us to examine whether high HRH1 expression correlates with poor clinical outcome in cancer patients, especially patients with CTL infiltrated (CTL+) tumors (Figure S1F). Indeed, high HRH1 expression was significantly associated with poor survival in patients with CTL+ triple negative breast cancer (TNBC) and lung adenocarcinoma as well as a strong trend of poor survival in melanoma patients (Figures 1G and S1G-S1I). Notably, HRH1 is among the top 20 genes that are strongly associated with poor survival in CTL+ TNBC patients (hazard ratio>2, Figure S1J).
Given that HRH1 is associated with T cell dysfunction in cancer patients and patients receiving H1-antihistamines along with ICB had better survival, we further explored whether high HRH1 expression is associated with immunotherapy resistance. Among melanoma patients treated with the anti-PD-1 drug pembrolizumab (GSE78220) (Hugo et al., 2016), non-responders had higher HRH1 mRNA expression in pre-treatment tumors than responders (Figure 1H, left). Anti-PD-1-treated patients with high HRH1-expressing tumors had devastatingly short overall survival compared with patients with low HRH1-expressing tumors (Figure 1H, right).
Histamine and HRH1 are up-regulated in tumor microenvironment
When deciphering how HRH1 induces T cell dysfunction, we surprisingly found that HRH1 expression was barely detectable in most of the tested human and mouse tumor cell lines (Figures S2A and S2B). Instead, using two deconvolution algorithms, i.e., Tumor Immune Estimation Resource (TIMER) and CIBERSORT (Li et al., 2017; Newman et al., 2015), we found that HRH1 expression was negatively correlated with tumor purity but positively correlated with tumor-associated macrophage (TAM) in the tumor microenvironment (TME) (Figures S2C and S2D), particularly in immunosuppressive M2-like macrophages among various cell types in human TME (Figure 2A). Furthermore, HRH1 was expressed mainly on M2-polarized (IL-4-treated) macrophages and TAMs in the TME, instead of naïve macrophages, M1-polarized (IFN-γ-treated) macrophages, or resident macrophages from normal mammary glands, in both humans (Figures 2B-2D and S2E top) and mice (Figures 2E-2G, S2E bottom, and S2F). In addition to HRH1 up-regulation on IL-4-induced M2-like macrophages (Orecchioni et al., 2019) (Figures 2B, 2E, and S2E), tumor-derived TGF-β also induced HRH1 expression on macrophages (Figure S2G). Additionally, we detected significantly increased levels of HRH1 ligand histamine in the blood of TNBC or colon cancer patients compared with that of healthy subjects (Figure 2H). Intriguingly, high histamine levels in TNBC patients’ blood was significantly correlated with low density of tumor-infiltrating GZMB+ cells (cytotoxic CD8+ T cell or NK cells) (Figure 2I), suggesting a potential link between histamine levels and immune cytotoxic cell infiltration. Increased histamine levels were also detected in tumor tissues (Figure S2H) and blood of tumor-bearing mice (Figure S2I) compared with corresponding normal tissues and blood from tumor-free mice, respectively, consistent with other reports (Moriarty et al., 1988; Sieja et al., 2005; von Mach-Szczypinski et al., 2009). Additionally, significantly increased histamine levels were detected in the tumor-conditioned-medium (TCM) derived from all the exanimated murine tumor cell lines and human breast cancer cell lines compared to normal culture medium and control medium from normal human breast epithelial cell line MCF-12A (Figure S2J), suggesting that cancer cells may be a major source of increased histamine detected in tumor-bearing mice and cancer patients (Figures 2H and S2H). Consistently, increased expression of HDC, the histamine-synthesizing enzyme, was also detected in patients’ breast cancer cells (Figure S2K). These data indicate that both histamine and HRH1 are up-regulated in the immunosuppressive TME.
Figure 2. Activated histamine-HRH1 axis in tumor microenvironment.

(A) Relative HRH1 mRNA levels in immune cell subsets assessed by CIBERSORT.
(B) Flow cytometry analysis of HRH1 expression on human peripheral blood monocytes (PBMC) - derived macrophages (n=3, one-way ANOVA).
(C) Representative images and quantification of HRH1+ macrophages in human breast tissues (n=9) and TNBC tumors (n=32, t-test). Blue, DAPI; red, CD68; green, HRH1. Scale bar, 25 μm.
(D) Percentage of CD68− or CD68+ cells (macrophages) in total HRH1+ cells in human TNBC tissues (n=32, t-test).
(E) Flow cytometry analysis of HRH1 expression on mouse bone marrow-derived (BMDM) naïve macrophages (M0) and polarized macrophages (M1 and M2-like) (n=3, one-way ANOVA).
(F) Mean florescent intensity (MFI) of HRH1 in indicated cell subsets of mouse mammary tumors (4T1 and EO771). Macrophages, CD45+CD11b+Grl−F4/80+; neutrophils, CD45+CD11b+Grl+; lymphocytes, CD45+CD11b−.
(G) Flow cytometry analysis of HRH1 expression on resident macrophages (nMφ) from mammary fat pad (MFP) of BALB/c mice and TAMs (4T1 and EMT6 tumors) (n=5, one-way ANOVA).
(H) Histamine levels in blood plasma from healthy subjects (n=20), patients with TNBC (n=50), colon cancer (n=28) detected by ELISA (one-way ANOVA).
(I) Pearson correlation analysis of the relationship between serum histamine level and GZMB+ cell density (%) in cancer tissues from TNBC patients (n=50). Mean ± SEM, **P<0.01, ****P<0.0001.
See also Figure S2.
Inhibition of HRH1 on macrophages restores T cell anti-tumor immunity
To investigate the specific function of the histamine-HRH1 axis in macrophages, we generated bone marrow-derived macrophages (BMDMs) from wild-type (WT) and HRH1-knockout (HRH1−/−) mice and treated them with TCM. Alternatively, we added an H1-antihistamine (fexofenadine, abbreviated as FEXO) to TCM-treated WT BMDMs. The expression ratio of major histocompatibility complex class II (MHCII, an M1 marker) versus CD206 (an M2 marker) were used to evaluate M1-M2 polarization status, which generally denotes anti-tumor versus pro-tumor activities of TAMs, although it does not fully reflect TAM’s complexity in the TME (Guerriero, 2018). Both HRH1−/− and FEXO treatment polarized macrophages toward an M1-like phenotype characterized by increased MHCII and decreased CD206 (Figure 3A). TAMs isolated from EO771 mammary tumors in HRH1−/− mice also had up-regulated M1-like pro-inflammatory molecules (Il1b, Il6, Il12b, and Nos2) and attenuated M2-like marker Arg1 compared with that in WT mice (Figure S3A). To examine the impact of macrophage HRH1 blockade on T cell activation, HRH1−/− or FEXO-treated macrophages were cultured with WT splenic T cells. HRH1−/− or FEXO treatment abrogated TAM-mediated T cell suppression, as signified by enhanced T cell proliferation, up-regulated cytotoxic and cytolytic effector molecules, including interferon gamma (IFN-γ) and perforin-1 (PRF1), and increased ovalbumin (OVA)-specific OT-I cell-mediated killing of OVA-transduced EO771 tumor cells (Figures 3B, and S3B-S3D). Notably, FEXO treatment of HRH1−/− macrophages didn’t increase MHCII:CD206 ratio (Figure 3A) or T cell activation/killing compared with vehicle-treated HRH1−/− macrophages (Figures 3B, S3C, and S3D), indicating that FEXO’s effects on macrophages are mediated by HRH1. Conversely, histamine (10 μM) treated mouse macrophages had increased M2-like marker CD206 and reduced M1-like marker MHCII compared to vehicle-treated ones (Figure S3E). Importantly, histamine-treated macrophages significantly suppressed T cell activation compared to vehicle-treated macrophages (Figures S3F and S3G). Next, we investigated the impact of histamine-HRH1 axis on TAMs and T cell immunity using two syngeneic tumor models in vivo. The EO771 mammary tumor cells or B16-GM (denotes B16-GM-CSF tumors with high tumor-infiltrating TAMs) melanoma cells (De Henau et al., 2016) were inoculated orthotopically into HRH1−/− mice and WT C57BL/6 mice. In separate experiments, WT mice were transplanted with these two cell lines and treated with vehicle or FEXO. Enhanced MHCII:CD206 ratio in the TAMs, increased numbers of IFN-γ+ and PRF1+ CD8+ T cells, and reduced tumor growth were found in HRH1−/− mice and FEXO treated mice compared to WT mice and vehicle treated mice, respectively (Figures 3C-3E, PRF1 data not shown). Furthermore, enzyme-linked immune absorbent spot (ELISPOT) assay revealed that tumor reactive T cells were increased in tumors from HRH1−/− mice compared to that from WT mice (Figure S3H). Similar changes were also detected in TNBC (4T1) or lung carcinoma tumors (LLC) in FEXO-treated mice compared with vehicle-treated mice (Figures S3I-S3K). The inhibition of B16-GM tumor growths in both HRH1−/− mice and FEXO-treated WT mice were blocked by depleting CD8+ T cells with anti-CD8 antibodies (Figures 3F and S3L), indicating that the enhanced anti-tumor activities by HRH1 blockade depend on CD8+ T cells. Although HRH1 was also expressed on endothelial cells (Lu et al., 2010), FEXO treatment did not show significant impact on angiogenesis in EO771 tumors (Figure S3M). To test whether HRH1 expressed on CD8+ T cells contributes to their biological functions, we compared HRH1−/− versus WT T cells and FEXO-treated versus vehicle-treated T cells, respectively, and found that they had similar proliferation rates and activities (Figure S3N), suggesting that increased T cell activation by blocking HRH1 in mice was unlikely resulted from direct inhibition of HRH1 on CD8+ T cells.
Figure 3. Inhibiting HRH1 on macrophages enhances T cell anti-tumor immunity.

(A) Flow cytometry analysis of M1-like (MHC II+) versus M2-like (CD206+) populations in bone marrow-derived macrophages (BMDMs) that were generated from wild-type (WT) or HRH1−/− mice and treated with vehicle or fexofenadine (FEXO) (10 μM) in the presence of EO771 tumor cell-conditioned medium (TCM) for 48 hours.
(B) Analysis of IFN-γ+CD8+ T cells in splenocytes co-cultured with vehicle- or FEXO (10 μM)-treated WT or HRH1−/− BMDMs (TCM-educated).
(C and D) Relative MHC II:CD206 MFI ratios of tumor-associated macrophages (TAMs) (C) and percentage of IFN-γ+CD8+ T cells (D) in EO771 tumors (left) or B16-GM tumors (right) growing in WT versus HRH1−/− mice, and vehicle-treated versus FEXO-treated WT mice (n=5-6, t-test).
(E) EO771 (left) and B16-GM (right) tumor growth in WT versus HRH1−/− mice, and vehicle-treated versus FEXO-treated WT mice (n= 5-8 mice/group, two-way ANOVA).
(F) B16-GM tumor growth with indicated treatment. CD8+ T cells were depleted by anti-CD8 antibodies (n=6-7 mice/group, two-way ANOVA).
(G) Growth of B16-GM tumor cells co-implanted with WT or HRH1−/− BMDMs in HRH1−/− or WT recipient mice, respectively (n=6-9 mice/group, two-way ANOVA).
(H) Percentages of IFN-γ+ and PRF1+ CD8+ T cells in primary tumors from WT and HRH1−/− mice transplanted with B16-GM tumor cells alone, or both tumor cells and BMDMs (HRH1−/− or WT respectively) (n=6, one-way ANOVA).
(I) t-distributed stochastic neighbor embedding (tSNE) plot of tumor-infiltrating leukocytes overlaid with color-coded clusters in EO771 tumors from WT or HRH1−/− mice. Dotted ellipses highlight clusters with significant differences between two groups.
Mean ± SEM, *P<0.05, **P<0.01, ***P<0.001.
See also Figures S3, S4 and S5.
To explore whether loss of HRH1 expression on non-immune cells in the TME may also contribute to tumor suppression in HRH1−/− mice, we generated chimeric mice by transplanting WT bone marrows into HRH1−/− mice (WT BM in HRH1−/−) or HRH1−/− bone marrows into WT mice (HRH1−/− BM in WT) following lethal irradiation. One month later, mice were orthotopically inoculated with EO771 tumor cells. HRH1−/− BM in WT mice showed decreased tumor growth along with increased MHCII:CD206 ratio in TAMs and increased IFN-γ+ CD8+ T cell infiltration compared to that of control WT mice reconstituted with WT bone marrow cells (WT BM in WT) (Figure S4A). Interestingly, WT BM in HRH1−/− mice seemed to have a minor tumor reduction although statistically insignificant (Figure S4A). To further determine the critical function of HRH1 on macrophages, we co-implanted WT or HRH1−/− macrophages with various types of cancer cells into recipient mice. Co-implantation of HRH1−/− BMDMs with B16-GM melanoma cells into WT mice significantly increased the activity of tumor-infiltrating CD8+ T cells and reduced tumor growth, which phenocopied the B16-GM tumors implanted in the HRH1−/− mice; whereas co-implanting WT BMDMs with B16-GM cells into HRH1−/− hosts enhanced tumor growth and suppressed CD8+ T cell activity, similar to the B16-GM tumors in WT mice (Figures 3G and 3H). Similarly, co-implanting HRH1−/− BMDMs with EO771 mammary tumor and LLC lung tumor cells into WT mice also significantly enhanced anti-tumor immunity and reduced tumor growths (Figure S4B, data not shown). Together, these data demonstrated that HRH1 activation on macrophages promotes CD8+ T cell activity suppression and tumor growth.
To evaluate the general impact of HRH1 blockade on the tumor immune microenvironment, we profiled CD45+ immune cells isolated from EO771 tumors grown in WT and HRH1−/− mice using mass cytometry (CyTOF), which revealed 12 distinct subsets, or clusters, of cells (Figures S4C-S4E). EO771 tumors from HRH1−/− mice had significantly fewer M2-like macrophages (cluster 7), whereas cytotoxic immune cells, including CD8+ T cells (cluster 2) were increased in tumors from HRH1−/− mice (Figures 3I and S4F), suggesting enhanced anti-tumor immunity. Moreover, the MHCII:CD206 ratio of TAMs was increased, along with granzyme B (GZMB)+ CD8+ T cells, in HRH1−/− mice compared with WT mice (Figure S4G).
HRH1 blockade also enhanced anti-tumor immunity in lung metastatic sites of two spontaneous lung metastasis models B16-GM and 4T1, as shown by increased M1-like polarization of resident alveolar macrophages (Misharin et al., 2013), increased cytotoxic CD8+ T cells, and reduced lung metastases (Figures S5A-S5C). B16-GM lung metastases in FEXO-treated mice were also inhibited compared to vehicle-treated mice after B16-GM primary melanomas were surgically removed upon growing to 100 mm3 of tumor size (Figure S5D), indicating that antihistamines enhanced anti-metastasis immune response.
HRH1 activation promotes VISTA membrane localization
To explore how HRH1 on macrophages suppresses T cell activities, EO771 TCM-treated WT or HRH1−/− macrophages were co-cultured with WT CD8+ T cells in direct contact or separately in a trans-well. Modulation of IFN-γ+PRF1+ CD8+ T cell by macrophage HRH1 was largely dependent on direct cell-cell contact (Figures 4A and S6A). Since macrophages and dendritic cells can regulate T cell function via engagement of co-stimulatory or inhibitory receptors on T cells (Guerriero, 2019; Ostuni et al., 2015), we investigated whether HRH1 on macrophages induces T cell dysfunction via regulating co-stimulatory or inhibitory receptors on T cells. Among the 13 ligands with co-stimulatory or inhibitory activities screened on EO771 or B16-GM TCM-treated macrophages, VISTA and TIM-3, known inhibitory molecules (Lines et al., 2014; Ocana-Guzman et al., 2016), were the most down-regulated molecules on HRH1−/− macrophages compared with WT macrophages (Figures 4B and S6B). Functionally, when WT macrophages in TCM were pre-treated with VISTA-blocking antibody and co-cultured with T cells, IFN-γ and PRF1+ CD8+ T cell levels and tumor cell killing activity increased to similar levels of the T cells co-cultured with HRH1−/− macrophages; a TIM-3-blocking antibody had a lesser effect (Figures 4C, S6C, and S6D), suggesting that VISTA is a major HRH1 downstream mediator of T cell dysfunction. In EO771 tumors from WT mice, HRH1 expression on TAMs was strongly correlated with VISTA expression (Figure S6E). Reduced VISTA expression was detected on TAMs from EO771 and B16-GM tumors in HRH1−/− mice or FEXO-treated WT mice compared to respective controls (Figure 4D). Decreased VISTA on TAMs was also observed in FEXO-treated other tumor models (e.g., 4T1 and LLC) in WT Balb/c or B6 mice compared to that of vehicle-treated ones (Figure S6F). Similarly, VISTA expression on alveolar macrophages from lung metastases of HRH1−/− mice bearing B16-GM tumor or of FEXO-treated WT mice bearing 4T1 tumor was also down-regulated compared with their controls (Figure S6G).
Figure 4. Histamine-HRH1 activation promotes VISTA membrane localization.

(A) Percentages of IFN-γ+ CD8+ T cells co-cultured with EO771 TCM-treated WT or HRH1−/− BMDMs in direct contact or separately in transwells (n=6, t-test).
(B) Heat-map depicting relative expression of co-stimulatory/inhibitory molecules on naïve or TCM-treated WT or HRH1−/− BMDMs measured by flow cytometry. The mean fluorescence intensity (MFI) of each molecule was normalized to the MFI of the naïve WT group.
(C) Percentages of IFN-γ+ CD8+ T cells co-cultured with EO771 TCM-treated WT or HRH1−/− BMDMs pretreated with IgG, anti-TIM-3 (10 μg/ml) and/or anti-VISTA (10 μg/ml) antibodies (n=3-4, one-way ANOVA).
(D) Flow cytometry analysis of VISTA+ TAMs (CD45+ CD11b+ F4/80+) from EO771 and B16-GM tumors growing in WT versus HRH1−/− mice, and vehicle-treated versus FEXO-treated WT mice (n=5-6, t-test).
(E) Western blot analysis of total VISTA and membrane VISTA expression on naïve or TCM-treated WT or HRH1−/− BMDMs. β-actin and CD11b were used as loading controls.
(F) Percentages of VISTA+ BMDMs after treatment with 10 μM BAPTA-AM (an intracellular calcium chelator) or 1 μg/ml ionomycin-Ca2+ (n=3, t-test).
All in vitro experiments were performed at least twice. Mean ± SEM, *P<0.05, **P<0.01, ***P<0.001, NS: not significant.
See also Figure S6.
Blocking HRH1 significantly reduced VISTA membrane expression on macrophages (Figures 4D and 4E) but VISTA mRNA and total protein expression didn’t change significantly (Figures 4E and S6H). Cell fractionation analysis confirmed that HRH1 blockade decreased cell membrane VISTA protein expression (Figure 4E). Calcium (Ca2+) facilitates protein trafficking to the plasma membrane and HRH1 activation induces Ca2+ release from the endoplasmic reticulum (Micaroni, 2010, 2012; Parsons and Ganellin, 2006). Thus, we explored whether HRH1 activation may foster VISTA membrane trafficking via releasing Ca2+. Indeed, after histamine or TCM treatment that induced activation of HRH1, intracellular free Ca2+ levels were higher in WT macrophages than that in HRH1−/− macrophages and FEXO-treated WT macrophages (Figure S6I). Blocking Ca2+ flux by BAPTA-AM, an intracellular calcium chelator, reduced HRH1-mediated membrane VISTA expression on WT TAMs, while the Ca2+ flux agonist ionomycin increased cell-surface VISTA expression on HRH1−/− TAMs (Figures 4F and S6I). These data suggest that HRH1-modulated Ca2+ release is critical for VISTA membrane localization.
HRH1 activation reshapes the transcriptomic landscape of macrophages
To gain deep insight into HRH1 downstream signaling that may contribute to the immunosuppressive phenotype of macrophages, we profiled the global transcriptome of TCM-treated WT and HRH1−/− macrophages by RNA sequencing. Compared to WT macrophages, HRH1−/− macrophages showed higher expression of genes associated with M1 polarization (e.g., CXCL10 and CD40), but lower expressions of many genes associated with M2-like phenotype (e.g., C1QB, C1QC, and MRC1) (Figure 5A). Gene set enrichment analysis (GSEA) identified key canonical pathways specifically up- or down-regulated in HRH1−/− macrophages compared to WT macrophages (Figure 5B). For example, TCM-treated HRH1−/− macrophages showed significantly upregulated TNF-α signaling, LPS and IFN-γ signaling (Figure 5B). Various pro-inflammatory cytokines and chemokines (e.g, Il6, Il1α, Cxcl10 and Cxcl11) are also significantly higher in HRH1−/− macrophages than in WT macrophages (Figure S6J). These upregulated signaling pathways and molecules in HRH1−/− macrophages are tightly associated with M1 polarization of macrophages and anti-tumor immune reactivity of macrophages (DeNardo and Ruffell, 2019), conceivably, they may contribute to the increased anti-tumor activities of HRH1−/− macrophages. On the other hand, reduced M2-polarized macrophage signature (Gerrick et al., 2018) was detected in HRH1−/− macrophages (Figure 5B), consistent with their M1-like polarization phenotype. Intriguingly, cholesterol biosynthesis and targets of sterol regulatory element binding transcription factors (SREBF1/2) are also among the inhibited signaling pathways in HRH1−/− macrophages compared to WT macrophages (Figure 5B).
Figure 5. HRH1 knockout reshapes the transcriptomic landscape of macrophages.

(A) Volcano plots of log2 fold change (FC) and log10 adjusted P -value of differentially expressed genes between TCM-treated WT and HRH1−/− macrophages. Red dots: genes up-regulated in WT macrophages; Blue dots: genes up-regulated in HRH1−/− macrophages.
(B) The pathway enrichment map of differentially expressed genes between WT and HRH1−/− macrophages.
(C) Dimensionality reduction by UMAP using all the single cells reveals the fine structure of CD45+ immune cells in mice. Manually inspected automatic annotation of cell identities are labeled in colors as their corresponding cell populations.
(D) Violin plot showing the expression-based score of M1-like (top) and M2-like macrophage (bottom), with their mean scores and P values labeled. The horizontal lines represent 25th percentile, median and 75th percentile of the scores. Significance levels are computed using the nonparametric Games-Howell Post-Hoc Test.
(E) Violin plot showing the expression-based score of exhausted CD8 T cells with their mean scores and P values labeled. Significance levels are computed using the nonparametric Games-Howell Post-Hoc Test.
(F) Scatter-plot results from the Pearson's correlation analysis of HRH1 and M2-like macrophage markers CD163 (left) and CD209 (right) at single-cell level in TAMs of human melanomas (GSE115978).
(G) Scatter-plot results from the Pearson's correlation analysis of HRH1 and M1-like macrophage markers IDO1 (left) and IRF1 (right) at single-cell level in TAMs of human melanomas (GSE115978).
See also Figure S6.
To further explore the broad impact of HRH1 blockade on macrophage phenotype and the landscape of tumor immune microenvironment in vivo, the CD45+ immune cells were isolated from EO771 tumors growing in WT versus HRH1−/− mice for single-cell RNA sequencing (scRNA-seq) analyses. Major immune cell types were predicted using built-in annotated human PBMC datasets as a reference and the automatic cell annotations were further calibrated by examining the most highly expressed marker genes between clusters (Figure 5C). The data showed that HRH1−/− primarily impacted on TAMs and T cells among CD45+ immune cells. Cell composition analysis showed reduced M2-like macrophages and slightly increased M1-like macrophages in tumors from HRH1−/− mice compared to that in WT mice (Figure S6K). We also calculated M1 and M2 gene signature-based scores at single-cell level in TAMs. Overall, macrophages isolated from EO771 tumors in HRH1−/− mice showed significantly higher M1-gene signature scores, but much lower M2-gene signature scores compared to macrophages from WT mice (Figure 5D). Since our above studies suggested correlation between HRH1 expression and T cell dysfunction (Figures 1F and S1F-S1H), we further evaluated exhausted CD8+ T cell gene signature score at single-cell level in CD8+ T cells isolated from the tumors. Indeed, CD8+ T cells isolated from EO771 tumors in HRH1−/− mice showed much lower exhausted CD8+ T cell gene signature scores compared to those from the WT mice (Figure 5E), indicating reduced T cell dysfunction by HRH1 blockade.
To validate the above findings in cancer patients, we further analyzed correlations between HRH1 and human M1- and M2- macrophage markers at single-cell level in TAMs collected from melanoma patients (GSE115978) (Jerby-Arnon et al., 2018). We found that HRH1 strongly and positively correlated with well-known human M2-macrophage markers (Martinez et al., 2006), e.g. CD163, CD209, C1QB/C, at the single-cell level (Figures 5F and S6M). On the other hand, HRH1 negatively correlated with human M1-macrophage markers, including IRF1 and IDO1, both of which are downstream of IFN-γ signaling (Figure 5G).
Taken together, blocking HRH1 reshaped the transcriptomic landscape of immune cells, among which reduced M2-like macrophage signatures and enhanced cytotoxic T cell functions mostly contribute to the alleviation of immunosuppression in TME.
HRH1 inhibition enhances therapeutic responses to ICB
HRH1 activation promotes VISTA membrane localization (Figure 4E) and patients with high HRH1-expressing tumors showed poor responses to anti-PD-1 immunotherapy (Figure 1G). VISTA and PD-1/PD-L1 suppress T cell activity non-redundantly, and up-regulation of VISTA has been linked with ICB resistance in cancer patients (Blando et al., 2019; Gao et al., 2017; Liu et al., 2015; Nowak et al., 2017). Therefore, we further investigated whether HRH1 high expression, via induction of VISTA membrane expression, would confer immunotherapy resistance, and HRH1 blockade could enhance response to immunotherapy. We found that among EO771 tumors in mice having heterogeneous responses to anti-PD-1 treatment, non-responding tumors had higher HRH1 and VISTA expression on TAMs than did partially responding tumors (Figure S7A). To test if inhibition of HRH1 would enhance anti-tumor activity of PD-L1 blockade, EO771 tumors in WT or PD-L1−/− mice were treated by vehicle or FEXO. FEXO-treated PD-L1−/− mice showed the most effective tumor inhibition and dramatically prolonged survival (Figure 6A) with 50% of mice remaining tumor free, whereas only 10% of vehicle-treated PD-L1−/− mice were tumor free. Similarly, growth of anti-PD-1-treated EO771 tumor in HRH1−/− mice was effectively inhibited accompanied by lower VISTA membrane expression on TAMs and more IFN-γ+ CD8+ T cells compared to that of anti-PD-1-treated WT mice (Figures 6B and S7B). Next, we examined whether inhibiting HRH1 activation with antihistamine could also enhance therapeutic efficacy of anti-CTLA-4 immune checkpoint inhibitor. FEXO or anti-CTLA-4 treatment both delayed tumor growth of CT26 murine colorectal carcinoma model in mice, and combinatorial treatment of FEXO plus anti-CTLA-4 more effectively inhibited CT26 tumor growth (Figure 6C, left). Remarkably, FEXO plus anti-CTLA-4 combinatorial treatment resulted in complete tumor remission and tumor-free survival in 40% of the mice while none of the mice in other groups survived by day 41 post-injection (Figure 6C, right). Furthermore, in ICB-resistant B16-GM melanoma model, FEXO treatment combined with ICB (anti-PD-1 plus anti-CTLA-4) achieved the highest therapeutic response and drastically inhibited both primary tumor growth and lung metastasis compared with FEXO or ICB alone (Figures 6D, 6E and S7C). Complete tumor remission was observed in 50% of the FEXO+ICB combination treatment group but in none of the other groups (Figure 6E), along with significantly down-regulated VISTA expression on TAMs and enhanced T cell function at both primary and metastatic tumor sites (Figures 6F and S7D). Next, the FEXO+ICB-treated B16-GM tumor-free mice were re-inoculated with B16-GM cells or EO771 cells. B16-GM cells, not EO771 cells, were rejected, indicating a persistent T cell memory for B16-GM tumor cells (Figure S7E). Together, HRH1 knockout or antihistamines combined with ICB greatly improved the therapeutic response of multiple tumor models, echoing our clinical findings that cancer patients who took H1-antihistamines during ICB treatments had better overall survival (Figure 1B).
Figure 6. HRH1 inhibition enhances ICB therapeutic efficacy.

(A) EO771 tumor growth and survival analysis of vehicle- or FEXO-treated WT and PD-L1−/− mice. n=15-23 mice/group for tumor volume analysis, n=9 mice/group for survival analysis, two-way ANOVA for tumor volume comparison, log-rank test for survival comparison.
(B) Flow cytometry analysis of VISTA+ TAMs and IFN-γ+ CD8+ T cells in EO771 tumors collected from WT and HRH1−/− mice receiving IgG or anti-PD-1 antibody treatment (n=6, one-way ANOVA).
(C) Primary tumor growth (left) and survival analysis (right) of CT26 tumor-bearing WT mice treated with FEXO alone, anti-CTLA-4 alone, or FEXO+anti-CTLA-4. n=9-10 mice/group for tumor volume and survival analysis.
(D and E) Tumor growth (D) and survival analysis (E) of B16-GM tumor-bearing WT mice with the indicated treatment (n=6-10 mice/group).
(F) Flow cytometry analysis of VISTA+ TAMs and IFN-γ+ CD8+ T cells in primary tumor tissues from B16-GM-bearing mice treated with indicated regimens (n=5, one-way ANOVA). (G) Flow cytometry analysis of MHCII:CD206 ratios of TAMs and IFN-γ+ CD8+ T cells in B16-GM primary tumor tissues from mice treated with indicated regimens (n=6, one-way ANOVA).
Mean ± SEM, two-way ANOVA for tumor volume comparison, log-rank test for survival comparison. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
See also Figure S7.
VISTA-blocking antibodies are currently being tested in clinical trials for anti-tumor efficacy (Nowak et al., 2017). We tested whether FEXO, a low-cost OTC drug, may have similar effects as anti-VISTA antibodies. FEXO monotherapy showed similar anti-tumor activity to that of VISTA antibodies in the B16-GM model (Figure 6D). When combined with ICB therapy, FEXO and the anti-VISTA antibody also had similar efficacy in controlling primary tumor growth (Figure 6D). Amazingly, FEXO+ICB was more effective than anti-VISTA+ICB for prolonging survival of mice because 50% of FEXO+ICB-treated mice had tumor-free survival but none of the anti-VISTA+ICB-treated mice survived (Figure 6E). FEXO+ICB was also more potent than anti-VISTA+ICB in promoting macrophage M1-like polarization and inhibiting lung metastasis (Figures 6G and S7F).
Allergies induce immunotherapy resistance which is mitigated by HRH1 blockade
The above studies indicate that histamine-HRH1 up-regulation in TME induces T cell dysfunction and immunotherapy resistance. Since allergic reactions release lots of histamine, we questioned whether allergy similarly impacts on anti-tumor immunity and immunotherapy response. To address the question, an OVA-induced allergic airway disease model (Nials and Uddin, 2008), in which BALB/c mice had two rounds of allergen (OVA) sensitization, was transplanted with tumor cells, followed by 1 week of airway OVA allergen exposure and treatment with FEXO, ICB, or FEXO+ICB (Figure 7A). To study allergy’s impact on tumor immunity and immunotherapy, we used two murine tumor models, EMT6 (mammary tumor) and CT26 (colon cancer), both of which were derived from BALB/c background that is susceptible to OVA-induced allergy (Kumar et al., 2008). High levels of histamine in plasma and tumor tissues were detected in mice after exposure to OVA (Figures 7B and S7G), indicating allergic reaction to OVA. Compared with the sham control group, mice with an OVA-induced allergic response had significantly accelerated EMT6 tumor growth, which was largely blocked by FEXO treatment (Figure 7C). Similarly, tumor growth of CT26 colon cancer cells in OVA-allergic BALB/c mice was significantly increased than that in sham control mice, and was largely blocked by FEXO treatment (Figure S7H). Both EMT6 or CT26 tumors in allergic mice also had increased VISTA expression on TAMs, a decreased MHCII:CD206 ratio, and reduced IFN-γ+ CD8+ T cells, all of which could be partially reversed by FEXO treatment (Figures 7D, S7I, and S7J).
Figure 7. HRH1 blockade rescues allergy-induced immunotherapy resistance.

(A) Experimental schematics of EMT6 tumor model with concurrent allergy.
(B) Serum histamine levels detected by ELISA in age-matched healthy mice, allergic mice, and tumor-bearing mice (10 days after EMT6 tumor cell inoculation) with or without induced allergy (n=6, t-test).
(C) EMT6 tumor growth in sham control group, allergy group, and allergy plus FEXO treatment group (n=6 mice per group).
(D) Flow cytometry analysis of VISTA+ TAMs and IFN-γ+ CD8+ T cells in EMT6 tumor tissues from sham control group, allergy induction group, and allergy plus FEXO treatment group (n=5, one-way ANOVA).
(E) EMT6 tumor growth in mice with or without concurrent allergic disease, treated with vehicle, ICB, or ICB+FEXO (n=6 mice per group).
(F) CT26 tumor growth in mice with or without concurrent allergic disease, treated with indicated therapies (n=7 mice per group).
(G) Comparison of deceased patient percentages according to patient allergy status before receiving anti-PD-1/PD-L1 treatment in melanoma and lung cancer patients.
(H) Comparison of plasma histamine levels in pre-treatment blood collected from cancer patient groups with different responses to anti-PD-1 treatment (one-way ANOVA). CR: complete response (100% remission), PR: partial response (≥30% remission), SD: stable disease (<30% remission to <20% increase of tumor size), PD: progressive disease (≥20% increase).
(I) A waterfall plot depicting the responses to anti-PD-1 treatment in cancer patients with low levels (<0.3 ng/ml), medium levels (>0.3 ng/ml to <0.6 ng/ml), and high levels (>0.6 ng/ml) of plasma histamine.
(J) Assessment of the objective response rate (ORR) and disease control rate (DCR) among cancer patients with different plasma histamine levels (low, medium, and high) (Fisher exact test).
(K) The distributions of age, sex, and tumor stage among anti-PD-1 treated lung cancer patients (n=48) with indicated histamine level (Fisher exact test).
Mean ± SEM, two-way ANOVA for tumor volume comparison. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, NS, no significant.
EMT6 and CT26 tumors are relatively sensitive to ICB treatment, as seen in sham control mice (Figures 7E and 7F) and as previously reported (Khononov et al., 2021; Mosely et al., 2017). However, both EMT6 and CT26 tumors became completely resistant to ICB therapy in allergic mice (Figures 7E and 7F). Remarkably, FEXO treatment largely restored sensitivity of EMT6 and CT26 tumors to ICB therapy in allergic mice (Figures 7E and 7F). The data indicate that allergic reaction promotes cancer immune evasion and immunotherapy resistance via the histamine-HRH1 axis, and this immune evasion could be mostly blocked by antihistamines.
Next, to examine the clinical impact of allergic response on immunotherapy efficacy in cancer patients, we retrospectively analyzed survival data of melanoma and lung cancer patients who reported allergic reactions before receiving anti-PD-1/PD-L1 treatment versus those who did not (Table S3). Indeed, cancer patients who experienced allergies had significantly worse outcomes compared with those patients who had no allergy (Figure 7G, melanoma patients: 51% versus 41% deceased; lung cancer patients: 64% versus 58% deceased).
Last, we directly examined whether plasma histamine levels are associated with patients’ response to immunotherapies. We measured pre-treatment histamine levels in plasma collected from a cohort of cancer patients (n=70) enrolled in a basket trial of anti-PD-1 treatment, which included lung cancer, breast cancer, and colon cancer patients. We found markedly lower levels of histamine in the blood of patients with complete response (CR) or partial response (PR) compared to that of patients with progressive disease (PD) (Figure 7H). Patients with stable disease (SD) had plasma histamine levels lower than PD patients but higher than CR/PR patients (Figure 7H). Next, we separated cancer patients into three groups based on their plasma histamine levels, patients with low levels of histamine (<0.3 ng/ml, which is the average level of histamine among healthy subjects, see Figure 2H), medium levels of histamine (0.3-0.6 ng/ml), and high levels of histamine (>0.6 ng/ml) (Tables S4-S6). Patients with low levels of plasma histamine had more than tripled overall response rate (ORR) and doubled disease control rate (DCR) (ORR: 55.6% vs 16%; DCR: 88.3% vs 44%) compared to patients with high levels of plasma histamine (Figures 7I and 7J). Notably, there were no significant differences in age, gender, and tumor stage among the three groups (Figure 7K and Tables S4-S6). These data from immunotherapy-treated cancer patients support the clinical relevance of our experimental findings from mouse tumor models with OVA-induced allergic airway disease (Figures 7E and 7F), suggesting that histamine release either from allergy response or by cancer cells attenuates response to immunotherapies, which can be mostly rescued by antihistamines.
DISCUSSION
In this study, we found that melanoma and lung cancer patients taking H1-antihistamines during immunotherapy treatment exhibited improved clinical outcomes with statistical significance. Similar trends were also observed in ICB-treated breast and colon cancer patients although didn’t achieve statistical significance which was likely due to the smaller patient numbers enrolled in the ICB treatment at the time compared to melanoma and lung cancer patients. These clinical data suggest that H1-antihistamines augment T cell-mediated anti-tumor immunity.
There were previous controversial reports on histamine modulation of myeloid-derived suppressive cells (MDSCs). Using different mouse and tumor models, some studies modulated histamine production and suggested that histamine reduced MDSCs and suppressed tumor growth (Grauers Wiktorin et al., 2019; Yang et al., 2011), while others found that histamine from mast cells increased MDSC proliferation and survival and promoted B16 melanoma metastasis (Martin et al., 2014). It is possible that different histamine concentrations and histamine receptors were inducing distinct effect on MDSCs. Antihistamines, when combined with chemotherapies, were reported to have either inhibitory or promoting effects on certain types of cancers, but antihistamines had not been tested for combination with any other therapies, especially not immunotherapy, for cancer treatment (Fritz et al., 2020). Our studies suggest that the histamine-HRH1 axis could serve as a potential biomarker of T cell dysfunction and immunotherapy response, as well as promising therapeutic targets for enhancing immunotherapy response. The strong correlation between low levels of plasma histamine and better response to ICB treatment in cancer patients infers that patients who have high levels of histamine in plasma thus respond poorly to immunotherapies may particularly benefit from antihistamine treatment. Based on our data, we consider that the low-cost OTC H1-antihistamines can be used as an adjuvant therapy in combination with immunotherapy to more effectively treat cancer patients.
HRH1 is also expressed in non-immune cells, including endothelial cells. Additionally, histamine may disintegrate endothelial barrier and induce vascular hyperpermeability (Ashina et al., 2015; Kugelmann et al., 2018). Our chimeric mice and macrophage co-implantation experiments indicated that HRH1 loss on TAMs is the major contributor of the enhanced immunity in HRH1−/− mice. However, HRH1−/− mice reconstituted with WT bone marrow (WT in HRH1−/−) also exhibited a slight tumor inhibition, suggesting that HRH1 loss in non-immune cells may also partly contribute to the tumor inhibition in HRH1−/− mice. Indeed, we found that compared to EO771 tumors in WT mice, tumors in HRH1−/− mice had reduced CD31+ blood vessel although they showed no significant difference in vascular permeability (data not shown). However, similar CD31+ blood vessel density was detected in FEXO-treated and vehicle-treated EO771 tumors. It is possible that HRH1−/− in endothelial cells induced impaired angiogenesis but FEXO treatment only temporally blocks binding of histamine to HRH1 without significant effects on endothelial cells as that in HRH1−/− mice. Some phenotypes observed in HRH1−/− mice reconstituted with WT bone marrow may also be associated with reduced angiogenesis in HRH1−/− mice.
A major downstream effector of the histamine-HRH1 axis is VISTA, which has been implicated in ICB resistance in patients (Blando et al., 2019; Gao et al., 2017; Liu et al., 2015). Knockout of HRH1 gene or antihistamine treatment reduced membrane VISTA on TAMs and boosted T cell anti-tumor immunity, similar to anti-VISTA antibody. Recently, VISTA was identified as an acidic pH-selective ligand for the co-inhibitory receptor P-selectin glycoprotein ligand-1 (PSGL-1) on T cells, thus suppresses T cell function (Johnston et al., 2019). It was suggested that acidic pH, which is frequently found in TME, is required for VISTA engaging with PSGL-1 and suppressing T cell immunity. Interestingly, we found that tumors growing in WT mice was more acidic compared to that in HRH1−/− mice (data not shown), which may favor binding of VISTA with PSGL-1 on T cells. Remarkably, when combined with ICB, antihistamines elicited a strong anti-tumor response superior to that of anti-VISTA antibody combined with ICB, suggesting that antihistamines also regulate other downstream effectors of immune stimulation/suppression, in addition to VISTA.
A most interesting finding from our studies is the potential impact of allergic reaction and histamine on anti-tumor immunity and immunotherapy response. Currently, studies regarding the relationship between allergy and cancer are controversial from epidemiological findings (Rittmeyer and Lorentz, 2012; Turner et al., 2006). Some studies suggested that allergies may reduce the risk of cancer by either increased immune surveillance after the immune hyperresponsiveness that may exert a protective effect against the development of cancer, or the physical effects of allergy symptoms that may inhibit cancer via removing potential carcinogens. In contrast, others suggested that the Th2 response and inflammation induced by allergy may facilitate development of cancer. The relationship between allergy and cancer was unclear since the potential impact of allergy on cancer hasn’t been experimentally investigated so far. Here, our experimental data from both mammary tumor and colon cancer models in mice clearly demonstrated that allergy fueled tumor growth and triggered resistance to immunotherapy through histamine-HRH1-mediated suppression of anti-tumor immunity, underscoring the previously unrecognized tumor-prone activity of allergy. Finally, our clinical data from ICB-treated cancer patients indicates that pre-existing allergy with high plasma histamine impairs cancer patients’ anti-tumor immune response and leads to their poor responses to immunotherapy.
Our clinical studies have a limitation that the numbers of patients with pre-existing allergies who received antihistamine treatment before ICB therapy were not recorded. Nevertheless, our finding that plasma histamine levels of, and uptake of antihistamines by, cancer patients are associated with their response to immunotherapies strongly supports using antihistamines to treat cancer patients who have allergy with high levels of plasma histamine. OTC H1-antihistamines can restore T cell function suppressed by cancer cell-secreted and/or allergy-released histamine and improve the efficacy of immunotherapies such as ICB therapies. Our findings necessitate further clinical studies to prospectively test the effect of H1-antihistamines as adjuvant therapies for enhancing immunotherapy responses in cancer patients.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and request for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Dihua Yu (dyu@mdanderson.org).
Materials Availability
The plasmids, antibodies, stable cell lines and mouse strains generated in this study have not been deposited to any repositories yet, however, these materials would be available upon request.
Data and Code Availability
RNA-seq data generated during this study have been deposited in the Gene Expression Omnibus (GEO) database under accession numbers GSE161484. Published datasets used in this study are available through GEO: GSE78220 (Hugo et al., 2016), GSE115978 (Jerby-Arnon et al., 2018) or cBioPortal database (http://www.cbioportal.org). Single-cell sequencing data has been submitted to Sequence Read Archive (SRA) and is available in SRA Run Selector with accession PRJNA756466.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6, BALB/c, and C57BL/6-background HRH1-knockout mice were purchased from The Jackson Laboratory. C57BL/6-background PD-L1-knockout mice were obtained and maintained as previously described (Chen et al., 2014). All mouse protocols and experiments were performed in accordance with National Institutes of Health guidelines and were approved by the MD Anderson Institutional Animal Care and Use Committee. All mice used in our experiments were between 6-8 weeks of age, and were housed under standard housing conditions at the MDACC animal facilities. Both male and female C57BL/6 or BALB/c mice were used for lung cancer model (LLC model), melanoma model (B16-GM model) and colon cancer model (CT26), and female BALB/c or C57BL/6 mice were used for breast cancer models (4T1, EMT6 and EO771 models). Animal numbers of each group were calculated by power analysis and animals are grouped randomly for each experiment.
Cell lines
Human mammary tumor cell lines including HCC1806, HS578T, BT-20, MDA-MB-435, MDA-MB- 436, MDA-MB-231 and BT-549, murine mammary tumor cell lines including 4T1, EMT6 and EO771, murine lung carcinoma cell line LLC1 (or LLC), colon cancer cell line CT26, L929 cell line and 293T cell line were obtained from American Type Culture Collection (ATCC) and cultured in endotoxin-free DMEM/F-12 medium supplemented with 10% fetal bovine serum (FBS) (HyClone). Murine melanoma cell line B16-BL6 obtained from ATCC and B16-GM described previously (De Henau et al., 2016) were cultured in endotoxin-free RPMI1640 medium supplemented with 10% FBS (HyClone). All the cells are not among commonly misidentified cell lines, and were tested for mycoplasma contamination annually using a Mycoplasma Detection Kit (Biotool #B3903). In order to prevent potential contamination, all the media were supplemented with Penicillin-Streptomycin (15-140-122, Thermo Fisher Scientific) according to the manufacturer’s instructions.
Human Samples
The untreated breast cancer samples including tumor tissues and blood plasma from TNBC patients who had undergone a mastectomy before therapy, normal breast tissues from women undergoing cosmetic breast surgery, and blood plasma from colon cancer patients and healthy subjects were collected at the First Affiliated Hospital of Chongqing Medical University. The blood plasma from patients with advanced lung (n=48), colon (n=12) and breast (n=10) cancers were collected pre-anti-PD1 treatment (patients were treated with Camrelizumab between Dec 24, 2019 and Feb 27, 2021) at the Beijing Friendship Hospital of Capital Medical. Best percentage change in the sum of the diameters for the selected target lesion is defined by Response Evaluation Criteria in Solid Tumors (RECIST version 1.1) on minimum 2 computed tomographic scans before treatment and 1 computed tomographic scan during treatment. The use of pathological specimens, as well as the review of all pertinent patient records, were approved by the Research Ethics Committee of the First Affiliated Hospital of Chongqing Medical University (project approval number 1005367 2017-012) and the Research Ethics Committee of Beijing Friendship Hospital of Capital Medical University (project approval number 2017-P2-141-01), and written informed consent were obtained from each participant and/or their legal representative, as appropriate. The information about the samples used is summarized in Table S7.
METHOD DETAILS
Survival analysis using The Cancer Genome Atlas (TCGA) data
TCGA breast cancer gene expression data and clinical data were downloaded from TCGA data portal (https://tcga-data.nci.nih.gov/tcga/dataAccessMatrix.htm). Triple-negative breast cancer (TNBC) cases were collected based on ESR1, PGR, and ERBB2 expression (RNAseq V2 RSEM) by using K-means 2 separation to define positive and negative groups and then combining triple-negative samples. CD8+ cytotoxic T lymphocyte (CTL) groups in TNBC samples were based on K-means 3 separation using a 15-gene signature (CD3E, CD8A, CCL2, CCL3, CCL4, CXCL9, CXCL10, GZMA, GZMK, HLA-DOA, HLA-DOB, HLA-DMB, ICOS, IRF1 and PRF1; Figure S1A). Survival analysis was performed for 77 CTL-high TNBC cases with survival data using the coxph function of R software using a resampling procedure (Yao et al., 2012). Candidate prognostic genes were collected from 100-round runs with a coxph P value of <0.02 in >75 rounds. Volcano plots were drawn using hazard ratios and P values from the coxph analysis on 16,975 genes after removal of low expressers with candidate genes associated with poor prognosis and favorable prognosis marked in both CTL-high and CTL-low groups. To test the association between HRH1 gene expression level and patient survival, Kaplan-Meier survival analysis was performed using the similar program described in the Human Protein Atlas (www.proteinatlas.org/humanpathology) (Uhlen et al., 2017). Briefly, survival analysis was performed using R package survival and survmine. Patients were stratified into gene expression high and low groups using Kmeans two separation of gene expression values (consensus from 20 iterations), which is an unbiased separation based on gene expression dispersion.
Epic SlicerDicer software analysis
Our retrospective study was conducted on melanoma patients encountered during 2016 to 2017 at The University of Texas MD Anderson Cancer Center, which draws a diverse range of local, regional, national, and international patients. The study population consisted of all melanoma patients with of International Classification of Diseases (ICD) code C43 (10th version). Anonymized aggregate-level data were collected using the SlicerDicer function within MD Anderson Epic electronic medical records. Institutional Review Board approval was therefore waived. Using the Epic SlicerDicer, we identified 9922 patients with a visit diagnosis, billing diagnosis, or active problem list with malignant skin of melanoma (ICD-10-CM:C43.*). EPIC SlicerDicer was used to further identify 878 of these patients who received anti-PD-1 antibodies (pembrolizumab, nivolumab, or cemiplimab) or anti-PD-L1 antibodies (durvalumab, atezolizumab, or avelumab) treatment during the study period. The comparison group was 1986 patients with the same diagnosis who received chemotherapies but no immunotherapeutic treatment. To analyze the impacts of 40 different common pharmaceutical drugs (Supplementary Table 1) on patients’ outcomes, the 878 patients receiving anti-PD-1/PD-L1 treatment were further divided into 40 patient subgroups based on the medications they took during immunotherapeutic treatment. The estimated patient deaths of each subgroup were calculated based on the patient number of each subgroup and the average patient death rate (39%, based on deceased patients out of total patients analyzed). The real patient deaths of each subgroup were compared to the estimated patient deaths in order to determine the overall impact of the medication on clinical outcome (Figure 1A). For further analysis of the H1-antihistamines, the 878 patients (received immunotherapy treatment) and 1986 patient (receiving chemotherapy treatment) were further subdivided into patients subgroups with or without uptake of H1-antihistamines that selectively target HRH1 (including fexofenadine, loratadine, desloratadine, cetirizine, levocetirizine, and azelastine) at the same time with anti-PD-1/PD-L1 antibody treatment or chemotherapies. Patient information, including age (<30 years, 30 to <50 years, 50-70 years, >70 years), sex (male or female), disease stage (0, I, II, III, IV, other, or unknown), survival (alive or dead), was extracted. Because many patients’ direct responses to therapy were not available in Epic SlicerDicer, the overall survival (up to June 15, 2021) was used as a surrogate indicator of patients’ therapeutic response. The Fisher exact test was used to identify any nonrandom association between antihistamine uptake and patients’ overall survival status. A similar analysis was performed in breast cancer patients (ICD code: C50) encountered at MD Anderson during 2016-2018, lung cancer patients (ICD code: C34) encountered at MD Anderson during 2016-2018, and colon cancer patients (ICD code: C18) encountered at MD Anderson during 2016-2018. To perform patient overall survival analysis, patient death date and the date receiving anti-PD-1 or PD-L1 treatment were pulled out from Epic. The survival time was calculated as the days between the two dates. To investigate the potential impact of melanoma patients’ allergy status on their response to ICB therapies, melanoma patients who received anti-PD-1/PD-L1 immunotherapy were divided into two groups based on their allergy status. Patients were considered to have had an allergic response if they had a specified diagnosis of allergy status to other drugs, medicaments, or biological substances (ICD code: Z88) documented as a visit diagnosis, billing diagnosis, or active problem list. To avoid conflating these allergic responses induced by anti-PD-1/PD-L1 antibodies, which may interfere with the treatment efficacy, we reserved the allergic response category for patients who reported an allergic response within 10 days before receiving anti-PD-1/PD-L1 treatment.
RNA sequencing and data analysis
WT and HRH1−/− BMDM were cultured with TCM or DMEM medium for two days and then total RNA was purified using Trizol (Invitrogen). After removing DNA by DNase, RNA was further purified using RNeasy MinElute Cleanup Kit (Qiagen). RNA samples were sent to UT health sequencing core for library construction and sequencing. After Raw fastq files from RNA sequencing were mapped to mm10 genome using STAR2, read counts for genes were prepared by htseq from which TPM were calculated. Principle component (PCA) analysis was done using R software. GSEA analysis was done using Java Web Start downloaded from https://www.gsea-msigdb.org/gsea/downloads.jsp. Differential expressed genes between the wild-type and HRH1-KO samples were obtained by using R package DESeq2 with filtering parameters of fold change above 3, adjusted P<0.01, and average log2(TPM) in the high expression group above 0. Volcano plots were drawn using log2 (fold change) and −log10 (adjusted p), ceiled at 5 and 100 respectively. We obtained a list of M1 and M2 macrophage signatures genes from previous publication (Gerrick et al., 2018) and generated two customized terms (M1_UP_SHORT and M2_UP_SHORT) using only genes with fold changes above 5. These terms were appended to the gmt file in mySigDB and used for GSEA analysis. For graphing top GSEA terms, we calculated a p value score using the average of −log10 (NOM p-val) and −log10 (FDR q-val) listed in the GSEA report where p values less than 1e-9 were set to 1e-9.
Single-cell transcriptomic profiling
0.2x106 EO771 tumor cells were transplanted into mammary fat pad of wide type or HRH1−/− mice. Three weeks later, tumors were harvested and 0.1 gram of tumor tissue of each tumor were collected. Three tumor tissues from the same group were randomly combined into one mixed sample and proceeded to digestion using 2mg/ml collagenase A. Single-cell suspension was generated following the method described below in “Isolation of tumor-infiltrating cells”. 106 live CD45+ immune cells were sorted by flowcytometry and submitted to UTHealth Cancer Genomics Core (CGC) for sequencing. Cells were labeled with multiplexing oligoes (#1000261, 10x Genomics, Pleasanton, CA) by following the cell multiplexing oligo labeling protocol (CG000391). The labeled samples were then pooled together. The single cell capture and library construction were performed by following the 10x Genomics Chromium Next GEM Single Cell 3′ Reagent Kits v3.1 protocol (CG000388). Briefly, the pooled samples were loaded onto Chromium Next GEM Chip G (PN-1000120, 10xGenomics, Pleasanton, CA) with partitioning oil and barcoded singe cell gel beads. The barcoded and full-length cDNA is produced after incubation of the gel beads-in-emulsion (GEMs) and amplified via PCR for library construction. The library preparation was performed by following the protocol of Chromium Single Cell 3′ GEM, Library & Gel Bead Kit v3 (PN-1000121, 10xGenomics, Pleasanton, CA). The quality of the final libraries was examined using Agilent High Sensitive DNA Kit (#5067-4626) by Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, USA), and the library concentrations were determined by qPCR using Collibri Library Quantification kit (#A38524500, Thermo Fisher Scientific) on a QuantStudio3 (ThermoFisher Scientific). The libraries were pooled evenly and underwent for the paired-end sequencing on an Illumina NextSeq 550 System (Illumina, Inc.) using High Output Kit v2.5 (#20024907, Illumina, Inc.).
Single-cell gene expression processing and analysis
CellRanger (10x Genomics; v6.0.1) subcommand multi was used to process the raw sequencing reads and generate count matrix per sample (Zheng et al., 2017). Reads were aligned to the mouse genome assembly (version mm10) which is pre-built by 10x Genomics. Raw count matrices were then merged and analyzed using the Seurat R package (v4.0.1) (Hao et al., 2021). Cell matrices were initially filtered by removing cells with barcodes lower than 20 UMIs, with lower than 200 expressed genes, or with more than 10% of reads mapping to the mouse mitochondrial genes. To avoid low-quality cells, empty droplets or multiplets, we further filtered cells based on the number of unique genes detected in each cell, which is capped in the range from 2.5th to 97.5th percentile. Counts for the remaining cells were normalized against library size and regressed for the unwanted cycling bias among proliferating cells, using S and G2M phase scores calculated by the CellCycleScoring function in Seurat package. Scaled and centered read counts were used as gene expression for further analysis.
Uniform Manifold Approximation and Projection for Dimension Reduction (UMAP) was applied to visualize inferred cell clusters (cite: arXiv:1802.03426v3) based on the top 30 principal components. Automatic immune cell annotation was performed using scPred package (v 1.9.0)(Alquicira-Hernandez et al., 2019). The built-in annotated human PBMC datasets were combined and used as a reference to predict the major immune cell types in our in-house mouse single-cell dataset. Manual inspections were carried out to calibrate the automatic cell annotations by examining the most highly expressed marker genes between clusters, as well as literature- and database-derived cell markers. Murine M1- and M2-like cell markers were extracted from prior publication (Jablonski et al., 2015), while exhausted CD8+ T cell markers were downloaded from CellMarker database (Zhang et al., 2019). To examine the phenotype shift between mouse wildtype and knockout groups, we calculated the M1-like and M2-like macrophages and the exhausted CD8+ T cell expression-based scores at single-cell level. Specifically, we overlapped cell type-specific markers with the top 50 highly expressed genes between clusters, and then took the average expression value as the score for each set of cell type-specific markers. All the statistical analyses were conducted using R software (v4.0.4).
To examine correlation of HRH1 gene expression with M1/M2 macrophage markers (Martinez et al., 2006) in human cancer, we obtained scRNA-seq data from GSE115978 (Jerby-Arnon et al., 2018) which included macrophages and other immune cells isolated from melanoma patients. To deal with missing data points in scRNA-seq, we focused on macrophage cells with normalized TPM above 0.5 for HRH1 and other macrophage marker genes of interest. Pearson correlation and scatter plots were done using log2 (1+TPM/10) values as describe in previous publication (Jerby-Arnon et al., 2018).
Generation of stable cells using lentiviral infection
Mouse TGF-β1-targeting shRNAs (shTGFB1-1: TRCN0000065993; shTGFB1-2: TRCN0000065994) were purchased from Sigma-Aldrich. For lentiviral production, the lentiviral expression vector was co-transfected with the third-generation lentivirus packing vectors into 293T cells using LipoD293 DNA in vitro Transfection Reagent (SignaGen Laboratories). Then, 48-72 hours after transfection, cancer cell lines were stably infected with viral particles.
Generation of naïve bone marrow-derived macrophages (BMDMs)
Bone marrow cells were collected from femurs obtained from 8- to 10-week-old C57BL/6 or BALB/c mice. After red blood cell lysis, bone marrow cells were seeded at a density of 5×106 cells/150×15 mm Petri dish and cultured at 37°C in complete Dulbecco modified Eagle medium (DMEM) containing 20% L929 cell-conditioned medium, providing macrophage colony-stimulating factor. Macrophages were ready for use on day 7 following a fresh medium change on day 4.
Isolation of murine peritoneal macrophages
2 ml of 3% Brewer thioglycollate medium per mouse was injected into the peritoneal cavity to trigger an inflammatory response. Allow inflammatory response to proceed for 4 days, and then collect peritoneal exudate macrophages. After red blood cell lysis, peritoneal macrophages were cultured at 37°C in complete DMEM medium, and ready for use.
In vitro co-culture with T cells
Spleens from wild-type C57BL/6 or BALB/c mice were harvested and filtered through a 40-μm cell strainer to generate a single-cell suspension. After red blood cell lysis, splenocytes were counted and plated in complete Roswell Park Memorial Institute 1640 medium supplemented with 50 μM β-mercaptoethanol and 10 mM HEPES onto 12-well plates coated with 2.5 μg/ml anti-CD3 (clone 145-2C11, BioLegend) and 3 μg/ml anti-CD28 (clone 37N, BioLegend) antibodies. Spleen T cells were activated for 48 hours before co-culture with macrophages. To educate the macrophages, we co-cultured naïve BMDMs with tumor cells at a ratio of 1:1 or with tumor cell-derived conditioned medium for 48 hours. Macrophages were seeded with activated T cells at a ratio of 5:1. After co-culture for another 24 or 48 hours, T cells were collected for flow cytometry analysis.
In vitro T cell killing assays
Ovalbumin (OVA)-expressing EO771 cells alone or co-cultured with macrophages at a ratio of 1:2 were plated into 12-well plates. The following day, OT-I T cells pre-activated by OVA257-264 were added to the plates at the indicated ratio. After co-culture for another 48 hours, the tumor cells were harvested and counted to determine T cell killing ability.
Macrophage polarization and stimulation
To polarize macrophages toward an M1-like phenotype, we stimulated PBMC-derived macrophages or BMDMs with IFN-γ (20 ng/ml, PeproTech) for 48 hours. To induce an M2-like phenotype, we treated THP-1-derived macrophages or BMDMs with IL-4 (20 ng/ml, PeproTech) for 48 hours. To stimulate macrophages with histamine, we cultured peritoneal macrophages with histamine (10 μM) for 48 hours. To stimulate macrophages with tumor cell-derived conditioned medium (TCM), we cultured BMDMs with complete medium containing 50% TCM (volume).
Tumor inductions and treatment experiments
For 4T1 and EO771 models, 2×105 tumor cells were orthotopically injected into female BALB/c or C57BL/6 mice, respectively. For LLC and B16-GM models, 2×105 tumor cells were subcutaneously injected into the back of C57BL/6 mice. For the CT26 model, 2×105 CT26 cells were subcutaneously injected into the back of BALB/c mice. The induction of allergic airway disease has been described previously. Briefly, BALB/c mice were sensitized using OVA (Sigma-Aldrich) at a dose of 0.01 mg/mouse in 0.2 ml incomplete Freund's adjuvant intraperitoneally on days 0 and 12. Control mice received the same volume of phosphate-buffered saline in incomplete Freund's adjuvant. All groups of mice were challenged daily with 5% OVA (aerosolized for 20 minutes) via the airways between days 19 and 24. EMT6 and CT26 models were constructed by orthotopic injection of 2×105 tumor cells on day 18 after the first OVA sensitization. Treatments were given as single agents or in combination, with the following regimen for each drug. HRH1 antagonist fexofenadine hydrochloride was administered by oral gavage once per day at 30 mg/kg. Treatments were initiated on day 5 after tumor inoculation for the entire duration of the experiment. Anti-PD-1 antibody (clone RMP1-14; the hybridoma RMP1-14 for αPD-1 production was provided by Dr. Hideo Yagita, 10 mg/kg) and anti-CTLA-4 antibody (clone 9D9, Bio X Cell, 5 mg/kg) were injected intraperitoneally on days 7, 10, 13, 16, and 19 after tumor inoculation. Anti-VISTA antibody (clone 13F3, Bio X Cell, 300 μg/mouse) was injected subcutaneously every day for 10 days starting on day 7, followed by continuous injection every 2 days for the rest of the duration of the experiment (Le Mercier et al., 2014). For in vivo CD8+ T cell depletion, mice were treated with 200 μg of anti-CD8 antibody every 3-4 days starting at 3 days before B16-GM tumor inoculation. For in vivo macrophage adoptive transfer experiments, B16-GM tumor cells mixed with primary wild-type or HRH1−/− BMDMs at a ratio of 1:5 were injected subcutaneously into HRH1−/− or wild-type host mice, respectively. For the re-challenge study, mice with complete responses were re-challenged with 2×105 B16-GM or EO771 tumor cells (on day 150 after tumor implant). Tumor size was measured by calipers every second or third day when tumors were palpable, and the volume was calculated using the formula V = as (width×width×length)/2.
Bone marrow transplantation
Bone marrow transplantation followed previous publication (Khononov et al., 2021). In brief, on day 1, recipient mice (8 weeks old) received 1,000 rads total body irradiation (137Cesium Gammacell source), and 4 hours later, they were transplanted with 5×106 bone marrow cells collected from femurs of the donor mice (5 weeks old) via tail vein injection. In a successful graft, the immunological reconstitution is expected complete within 4-5 weeks. 30 days after bone marrow transplantation, the recipient mice were ready for following experiments.
Isolation of tumor-infiltrating cells
Mouse tumor samples were chopped with scissors and then subjected to enzymatic digestion with 2 mg/ml collagenase A (Roche) in DMEM for 1 hour at 37°C. Next, tissues were filtered through 70-μm filters (BD Biosciences) to achieve single-cell suspensions. After treatment with red blood cell lysis buffer for 5 minutes at room temperature, all samples were washed and re-suspended in flow cytometry buffer (phosphate-buffered saline/0.5% albumin/2 mM EDTA) or DMEM depending on further use.
Flow cytometry staining and analysis
Live single cells were sub-gated by staining with Fixable Viability Dye eFluor 450 (eBioscience) for 15 minutes at 4°C. For blocking of Fc receptors, cells were then pre-incubated with purified anti-CD16/32 antibody (clone 93, BioLegend) for 10 minutes on ice before immunostaining. After one wash with flow cytometry buffer, cells were incubated with appropriate dilutions of various combinations of the following antibodies. Primary antibodies to cell surface markers directed against CD45 (30-F11), CD3 (145-2C11), CD8a (53-6.7), CD11b (M1/70), Gr-1 (RB6-8C5), F4/80 (BM8), CD206 (C068C2), I-A/I-E (M5/114.15.2), VISTA (MH5A), Tim-3 (B8.2C12), CD11c (N418), CD24 (M1/69), GITRL (YGL 386), ICOSL (HK5.3), 4-1BBL (TKS-1), CD276 (RTAA15), OX40L (RM134L), Galectin-9 (RG9-35), PD-L1 (10F.9G2), and PD-L2 (TY25) were from BioLegend; against HRH1 (480054), from R&D Systems; against HRH1 (AHR-001), from Alomone Labs; and against Siglec-F (E50-2440), from BD Biosciences. For intracellular staining, cells were fixed, permeabilized using Foxp3/Transcription Factor Staining Buffer Set (eBioscience), and then stained with fluorochrome-conjugated antibodies to Ki-67 (16A8) and PRF1 (S16009A) from BioLegend. For cytokine staining, cells were first stimulated with Cell Stimulation Cocktail (eBioscience) at 37°C for 4 hours, and then stained with anti-IFN-γ (XMG1.2) from BioLegend. The stained cells were acquired by a BD FACSCanto II Flow Cytometer using BD FACSDiva software (BD Biosciences), and data generated were processed using FlowJo software.
Mass cytometry and data analysis
Mouse tumor tissues were digested as described above. Then, for CyTOF analysis, cells were incubated with 25 μM cisplatin for 1 minute (viability staining) and subsequently stained with a metal-labeled monoclonal antibody cocktail against cell surface molecules. After treatment with the Fixation/Permeabilization Buffer (eBioscience), cells were further incubated with monoclonal antibody cocktails against intracellular proteins. Antibodies used in the mass cytometry analysis were purchased from Fluidigm. The samples were analyzed using the CyTOF 2 instrument (Fluidigm) in the Flow Cytometry and Cellular Imaging Core Facility at MD Anderson. All CyTOF files were normalized and manually gated in Cytobank software. Data were transformed using the cytofAsinh function before they were applied to the downstream analysis. Phenograph clustering analysis in the R cytofkit package was performed on pooled samples to automatically identify underlying immune subsets. Heat-maps were generated on the basis of the mean value for each marker in clusters. Cell frequency in each cluster was calculated as the assigned cell events divided by the total CD45+ cell events in the same sample.
Purification of myeloid cells or macrophages from tumors
Single-cell suspensions of mouse tumors were generated as described in the previous section. Single cells were stained with CD11b microbeads (Miltenyi Biotec) according to the manufacturer’s instructions to enrich the myeloid fractions. Cells were then stained with Fixable Viability Dye eFluor 450 (eBioscience) to exclude dead cells, and anti-Gr-1- phycoerythrin (PE) (clone RB6-8C5) and anti-F4/80- fluorescein isothiocyanate (FITC) (clone BM8) for flow sorting on a FACSAria II Cell Sorter (BD Biosciences).
qRT-PCR
cDNA was prepared using 1 μg of RNA with the iScript cDNA Synthesis Kit (Bio-Rad). SYBR green-based qRT-PCR was performed using mouse primers to Il1b, Il6, Il10, Il12b, Nos2, Arg1, Tgfb1, Vista, Hrh1, and 18s (Integrated DNA Technologies). mRNA levels were normalized to 18s (ΔCt=Ctgene of interest−Ct18s) and presented as relative mRNA expression (ΔΔCt=2−(ΔCtsample-ΔCtcontrol)) or fold change.
Western blotting
Western blotting was done as previously described (Zhang et al., 2015). The following primary antibodies were used: HRH1 (LS-C331459, LifeSpan BioSciences), VISTA (54979, Cell Signaling Technology) and CD11b (NB110-40766, Novus Biologicals).
Enzyme-linked immunosorbent assay (ELISA)
The levels of histamine in cell culture supernatant, serum/plasma, and tissues were detected by Histamine ELISA kits (ENZ-KIT140-0001, Enzo; ab213975, Abcam) according to the manufacturer’s instructions.
IFN-γ ELISPOT assay
The IFN-γ ELISPOT assay was done following the manufacturer’s protocol. Briefly, the CD45+ leukocytes were sorted from tumors by flow cytometry. The leukocytes were counted and seeded at 5x105 cells/well into pre-coated PVDF plates (ImmunoSpot® Kits, Cellular Technology Limited), stimulated with anti-CD3 antibody and IL-2 overnight, and secreted IFN-γ was quantified following standard protocol. Assay plates were scanned and analyzed using an automated ELISPOT reader system.
Immunohistochemistry (IHC) and immunofluorescence (IF) staining
Standard IHC and IF staining was performed as described previously (Zhang et al., 2020). The primary antibodies used for IHC staining include anti-GZMB (ab255598, Abcam), anti-HDC (ab37291, Abcam), and anti-CD31 (77699, Cell Signaling); used for IF staining include anti-CD68 (ab955, Abcam) and anti-HRH1 (ab75236, Abcam). DyLight 488- or DyLight 594-conjugated secondary antibodies against rabbit or mouse IgG were purchased from Thermo Fisher Scientific.
Intracellular calcium measurement
Intracellular Ca2+ was determined using the Fluo-Forte calcium assay kit (Enzo Life Sciences) according to the manufacturer’s instructions. Fluorescence was measured using a BD FACSCanto II Flow Cytometer.
Quantification and Statistical analysis
Prism 8.0 software (GraphPad) was used for statistical analysis. Analysis for significance was performed by one-way or two-way ANOVA when more than two groups were compared and by parametric or nonparametric Student t-test when only two groups were compared. Fisher exact test was used when percentages of cancer patients from different groups were compared. Chi-square test was performed to determine whether there was any significant difference of gender, age, and tumor stages between the patient groups that took antihistamines and that didn’t take antihistamines. P<0.05 was considered statistically significant (*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001). Survival was evaluated using the Kaplan-Meier method and analyzed by the Mantel-Cox log-rank test. All experiments were performed at least twice, and n refers to biological replicates.
Supplementary Material
Human samples used in the study, related to STAR Methods.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD45 PE/Cyanine5 | BioLegend | Cat# 103110; RRID: AB_312975 |
| Anti-mouse CD3e APC | BioLegend | Cat# 100312; RRID: AB_312677 |
| Anti-mouse CD8a PE/Cyanine7 | BioLegend | Cat# 100722; RRID: AB_312761 |
| Anti-mouse/human CD11b BV 510™ | BioLegend | Cat# 101263; RRID: AB_2629529 |
| Anti-mouse Ly-6G/Ly-6C (Gr-1) PE | BioLegend | Cat# 108408; RRID: AB_313373 |
| Anti-mouse F4/80 FITC | BioLegend | Cat# 123108; RRID: AB_893502 |
| Anti-mouse CD206 PE | BioLegend | Cat# 141706; RRID: AB_10895754 |
| Anti-mouse I-A/I-E Alexa Fluor® 488 | BioLegend | Cat# 107616; RRID: AB_493523 |
| Anti-mouse VISTA PE/Cyanine7 | BioLegend | Cat# 143714; RRID: AB_2632662 |
| Anti-mouse CD366 (Tim-3) PE | BioLegend | Cat# 134004; RRID: AB_1626177 |
| Anti-mouse CD11c APC | BioLegend | Cat# 117310; RRID: AB_313779 |
| Anti-mouse CD24 Alexa Fluor® 647 | BioLegend | Cat# 101818; RRID: AB_493484 |
| Anti-mouse GITR Ligand PE | BioLegend | Cat# 120306; RRID: AB_2207248 |
| Anti-mouse CD275 (ICOS Ligand) PE | BioLegend | Cat# 107405; RRID: AB_2248797 |
| Anti-mouse 4-1BB Ligand PE | BioLegend | Cat# 107105; RRID: AB_2256408 |
| Anti-mouse CD276 (B7-H3) PE | BioLegend | Cat# 124508; RRID: AB_1279206 |
| Anti-mouse CD252 Alexa Fluor® 647 | BioLegend | Cat# 108810; RRID: AB_2207379 |
| Anti-mouse Galectin-9 APC | BioLegend | Cat# 136110; RRID: AB_2561658 |
| Anti-mouse Ki-67 FITC | BioLegend | Cat# 652410; RRID: AB_2562141 |
| Anti-mouse PRF1 PE | BioLegend | Cat# 154306; RRID: AB_2721639 |
| Anti-mouse IFN-γ PE | BioLegend | Cat# 505810; RRID: AB_315404 |
| Anti-mouse CD274 PE/Cyanine7 | BioLegend | Cat# 124314; RRID: AB_10643573 |
| Anti-mouse CD3ε antibody | BioLegend | Cat# 100372; RRID: AB_2800556 |
| Anti-mouse CD28 antibody | BioLegend | Cat# 102132; RRID: AB_2810333 |
| Anti-mouse CD16/32 antibody | BioLegend | Cat# 101302; RRID: AB_312801 |
| Anti-human HRH1 Alexa Fluor® 647 | R&D Systems | Cat# FAB4726R |
| Anti-mouse Siglec-F PerCP-Cy™5.5 | BD Biosciences | Cat# 565526; RRID: AB_2739281 |
| HRH1 antibody | Alomone Labs | Cat# AHR-001; RRID: AB_2039915 |
| HRH1 antibody | LifeSpan BioSciences | Cat# LS-C331459 |
| HRH1 antibody | Abcam | Cat# ab75236; RRID: AB_2092479 |
| CD68 antibody | Abcam | Cat# ab955; RRID: AB_307338 |
| VISTA antibody | Cell Signaling Technology | Cat# 54979; RRID: AB_2799474 |
| CD11b/c antibody | Novus Biologicals | Cat# NB110-40766; RRID: AB_714950 |
| β-actin | Santa Cruz Biotechnology | Cat # SC47778; RRID: AB_2714189 |
| Anti-mouse IgG, DyLight 594 | Thermo Fisher Scientific | Cat # 35510; RRID: AB_1185569 |
| Anti-Rabbit IgG, DyLight 488 | Thermo Fisher Scientific | Cat # 35552; RRID: AB_844398 |
| Anti-mouse CTLA-4 (CD152) | Bio X Cell | Cat # BE0164; RRID: AB_10949609 |
| Anti-mouse VISTA | Bio X Cell | Cat # BE0310; RRID: AB_2736990 |
| Anti-mouse CD8α | Bio X Cell | Cat # BE0117; RRID: AB_10950145 |
| CD45, Label: 89Y | Fluidigm | Cat # 3089005B; RRID: AB_2651152 |
| FoxP3, Label: 165Ho | Fluidigm | Cat # 3165024A; RRID: AB_2687843 |
| Granzyme B, Label: 173Yb | Fluidigm | Cat # 3173006B; RRID: AB_2811095 |
| TIM-3, Label: 162Dy | Fluidigm | Cat # 3162029B; RRID: AB_2687841 |
| CD357, Label: 143Nd | Fluidigm | Cat # 3143019B |
| CD80, Label: 171Yb | Fluidigm | Cat # 3171008B |
| CD86, Label: 172Yb | Fluidigm | Cat # 3172016B |
| CD40, Label: 161Dy | Fluidigm | Cat # 3161020B |
| CD278, Label: 176Yb | Fluidigm | Cat # 3176014B |
| CD39, Label: 142Nd | Fluidigm | Cat # 3142005B |
| CD11b, Label: 148Nd | Fluidigm | Cat # 3148003B; RRID: AB_2814738 |
| CD11c, Label: 209Bi | Fluidigm | Cat # 3209005B; RRID: AB_2811244 |
| Ly-6C, Label: 150Nd | Fluidigm | Cat # 3150010B |
| Ly-6G, Label: 141Pr | Fluidigm | Cat # 3141008B; RRID: AB_2814678 |
| CD38, Label: 175Lu | Fluidigm | Cat # 3175014B |
| I-A/I-E, Label: 174Yb | Fluidigm | Cat # 3174003B |
| CD206, Label: 169Tm | Fluidigm | Cat # 3169021B; RRID: AB_2832249 |
| CD274, Label: 153Eu | Fluidigm | Cat # 3153016B; RRID: AB_2687837 |
| CD4, Label: 115In | BioLegend | Cat # 100506; RRID: AB_312709 |
| CD3e, Label: 152Sm | BioLegend | Cat # 100302; RRID: AB_312667 |
| CD8a, Label: 146Nd | BioLegend | Cat # 100702; RRID: AB_312741 |
| NK1.1, Label: 170Er | BioLegend | Cat # 108702; RRID: AB_313389 |
| CD19, Label: 149Sm | BioLegend | Cat # 115502; RRID: AB_313637 |
| T-bet, Label: 154Sm | BioLegend | Cat # 644825; RRID: AB_2563788 |
| IRF4, Label: 151Eu | BioLegend | Cat # 646402; RRID: AB_2280462 |
| CD152, Label: 163Dyv | BioLegend | Cat # 106202; RRID: AB_313247 |
| CD69, Label: 156Gd | BioLegend | Cat # 104533; RRID: AB_2563760 |
| CD14, Label: 158Gd | BioLegend | Cat # 123302; RRID: AB_940592 |
| F4/80, Label: 159Tb | BioLegend | Cat # 123102; RRID: AB_893506 |
| Ly-6G/C, Label: 139La | BioLegend | Cat # 108402; RRID: AB_313367 |
| CD103, Label: 147Sm | BioLegend | Cat # 121401; RRID: AB_535944 |
| GATA3, Label: 145Nd | Thermo Fisher Scientific | Cat # 14-9966-82; RRID: AB_1210519 |
| Ki67, Label: 168Er | BD Biosciences | Cat # 556003; RRID: AB_396287 |
| CCR7, Label: 155Gd | Thermo Fisher Scientific | Cat # 16-1971-85; RRID: AB_494123 |
| Bacterial and Virus Strains | ||
| DH5α | Thermo Scientific | Cat# 18265017 |
| Stbl3 | Thermo Scientific | Cat# C737303 |
| Biological Samples | ||
| Human samples (Normal breast tissues, breast and colon cancer tissues) | The First Affiliated Hospital of Chongqing Medical University, China. | Project approval number 1005367 2017-012 |
| The blood plasma from patients with advanced lung (n=48), colon (n=12) and breast (n=10) cancers collected pre-anti-PD1 treatment (patients were treated with Camrelizumab between Dec 24, 2019 and Feb 27, 2021) | Beijing Friendship Hospital of Capital Medical | Project approval number 2017-P2-141-01 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Fexofenadine HCl | Sigma-Aldrich | Cat # PHR1685; CAS: 153439-40-8 |
| Ovalbumin | Sigma-Aldrich | Cat # A5503; CAS: 9006-59-1 |
| Puromycin dihydrochloride | Sigma-Aldrich | Cat# P8833; CAS: 58-58-2 |
| Collagenase A | Roche | Cat # 11088793001 |
| 16% Formaldehyde, Methanol-free | Pierce | Cat # 28906 |
| Ionomycin, Calcium Salt | Cell Signaling Technology | Cat # 9995; CAS: 56092-82-1 |
| BAPTA-AM | Selleckchem | Cat # S7534; CAS: 126150-97-8 |
| Recombinant Murine IFN-γ | PeproTech | Cat # 315-05 |
| Recombinant Murine IL-4 | PeproTech | Cat # 214-14 |
| Fixable Viability Dye eFluor™ 450 | eBioscience | Cat # 65-0863-14 |
| CD11b MicroBeads | Miltenyi Biotec | Cat # 130-049-601 |
| Cell-ID™ Cisplatin | Fluidigm | Cat # 201064 |
| Critical Commercial Assays | ||
| Histamine ELISA kits | Enzo Life Sciences | Cat # ENZ-KIT140-0001 |
| iScript™ cDNA Synthesis Kit | Bio-rad | Cat # 1708891 |
| Intracellular Fixation & Permeabilization Buffer Set | eBioscience | Cat # 88-8824-00 |
| Maxima SYBR Green qPCR Master Mix | Thermo Fisher Scientific | Cat# K0253 |
| QIAGEN Plasmid Maxi Kit | QIAGEN | Cat# 12163 |
| ImmunoSpot® Kits | Cellular Technology Limited | Mouse IFN-γ Single-Color ELISPOT |
| Deposited Data | ||
| RNA-seq with mouse BMDM (Raw and analyzed data) | This paper | GEO: GSE161484 |
| RNA-seq with human melanomas | Hugo et al 2016 | GEO: GSE78220 |
| Single-cell RNA-seq of human melanoma | Livnat Jerby-Arnon et al 2018 | GEO: GSE115978 |
| Single-cell RNA-seq with CD45+ immune cells isolated from EO771 tumors | This paper | SRA Run Selector Accession: PRJNA756466 |
| Experimental Models: Cell Lines | ||
| 293T | ATCC | Cat# ACS-4500; RRID:CVCL_4V93 |
| 4T1 | ATCC | Cat# CRL-2539; RRID:CVCL_0125 |
| B16/BL6 | ATCC | Cat# CRL-6475; RRID:CVCL_0159 |
| BT20 | ATCC | Cat# HTB-19; RRID:CVCL_0178 |
| BT549 | ATCC | Cat# HTB-122; RRID:CVCL_1092 |
| CT26 | ATCC | Cat# CRL-2638; RRID:CVCL_7256 |
| EMT6 | ATCC | Cat# CRL-2755; RRID:CVCL_1923 |
| EO771 | ATCC | Cat# CRL-3461; RRID:CVCL_GR23 |
| HCC1806 | ATCC | Cat# CRL-2335; RRID:CVCL_1258 |
| HS578T | ATCC | Cat# HTB-126; RRID:CVCL_0332 |
| L929 | ATCC | Cat# CCL-1; RRID:CVCL_0462 |
| LLC1 | ATCC | Cat# CRL-1642; RRID:CVCL_4358 |
| MDA-MB-231 | ATCC | Cat# HTB-26; RRID:CVCL_0062 |
| MDA-MB-435 | ATCC | Cat# HTB-129; RRID:CVCL_0622 |
| MDA-MB-436 | ATCC | Cat# HTB-130; RRID:CVCL_0623 |
| THP-1 | ATCC | Cat# TIB-202; RRID:CVCL_0006 |
| B16-GMCSF | N/A | |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 | The Jackson Laboratory | N/A |
| Mouse: BALB/c | The Jackson Laboratory | N/A |
| Mouse: B6.129P2-Hrh1tm1Wtn/BrenJ | The Jackson Laboratory | Stock No: 029346 |
| Mouse: PD-L1−/− | Dr. Don L. Gibbons | Chen et al. (2014) |
| Oligonucleotides | ||
| See Table S2 for a detailed primer list | This paper | N/A |
| Recombinant DNA | ||
| pLKO.1-shTGFB1-1 | Sigma-Aldrich | TRCN0000065993 |
| pLKO.1-shTGFB1-2 | Sigma-Aldrich | TRCN0000065997 |
| Software and Algorithms | ||
| FlowJo | BD Biosciences | https://www.flowjo.com/solutions/flowjo/downloads |
| GraphPad Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| R Studio | N/A | https://www.rstudio.com |
| Cytofkit | Jinmiao Chen Lab | https://github.com/JinmiaoChenLab/cytofkit2 |
Highlights.
Histamine binding of HRH1 on macrophage induces an immunosuppressive phenotype;
H1-antihistamine treatment enhances immunotherapy response;
Allergic reaction promotes immune evasion and resistance to immunotherapy;
High histamine and HRH1 levels correlate with poor immunotherapy response in patients.
ACKNOWLEDGMENTS
We thank Dr. Jedd D. Wolchok for providing B16-GM cells; Dr. Mien-Chie Hung for providing EMT6 cells; Drs. Anil K. Sood, Chunru Lin, Lin Zhang, and Xiang H.-F. Zhang for reading the manuscript; Dr. Akosua A. Badu-Nkansah and Ms. Sarah Bronson for manuscript editing; and members of the Yu laboratory for helpful discussions. Dr. Weiya Xia helped to read and scored some IHC slides.
Funding:
This work was supported by the National Institutes of Health (NIH) grants R01CA112567 (D.Y.), R01CA184836 (D.Y.), R01CA208213 (D.Y.), the METAVivor grants 56675 and 58284 (D.Y.), and NIH Cancer Center Support Grant P30CA016672 to MD Anderson Cancer Center (Functional Genomics Core, Flow Cytometry and Cellular Imaging facility, Research Histology Core Laboratory, and Research Animal Support Facility - Houston). The sequencing data was generated by the UTHealth Cancer Genomics Core supported by the Cancer Prevention & Research Institute of Texas (CPRIT) grant RP180734. D.Y. is the Hubert L. & Olive Stringer Distinguished Chair in Basic Science at MDACC.
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list. The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Alquicira-Hernandez J, Sathe A, Ji HP, Nguyen Q, and Powell JE (2019). scPred: accurate supervised method for cell-type classification from single-cell RNA-seq data. Genome Biol 20, 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashina K, Tsubosaka Y, Nakamura T, Omori K, Kobayashi K, Hori M, Ozaki H, and Murata T (2015). Histamine Induces Vascular Hyperpermeability by Increasing Blood Flow and Endothelial Barrier Disruption In Vivo. PLoS One 10, e0132367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blando J, Sharma A, Higa MG, Zhao H, Vence L, Yadav SS, Kim J, Sepulveda AM, Sharp M, Maitra A, et al. (2019). Comparison of immune infiltrates in melanoma and pancreatic cancer highlights VISTA as a potential target in pancreatic cancer. Proc Natl Acad Sci U S A 116, 1692–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA, Zhang X, Yi X, Dwyer D, Lin W, et al. (2014). Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun 5, 5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, Budhu S, Ghosh A, Pink M, Tchaicha J, et al. (2016). Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature 539, 443–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNardo DG, and Ruffell B (2019). Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol 19, 369–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkrief A, Derosa L, Kroemer G, Zitvogel L, and Routy B (2019). The negative impact of antibiotics on outcomes in cancer patients treated with immunotherapy: a new independent prognostic factor? Ann Oncol 30, 1572–1579. [DOI] [PubMed] [Google Scholar]
- Faustino-Rocha AI, Ferreira R, Gama A, Oliveira PA, and Ginja M (2017). Antihistamines as promising drugs in cancer therapy. Life Sci 172, 27–41. [DOI] [PubMed] [Google Scholar]
- Fritz I, Wagner P, Bottai M, Eriksson H, Ingvar C, Krakowski I, Nielsen K, and Olsson H (2020). Desloratadine and loratadine use associated with improved melanoma survival. Allergy. [DOI] [PubMed] [Google Scholar]
- Gao J, Ward JF, Pettaway CA, Shi LZ, Subudhi SK, Vence LM, Zhao H, Chen J, Chen H, Efstathiou E, et al. (2017). VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat Med 23, 551–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrick KY, Gerrick ER, Gupta A, Wheelan SJ, Yegnasubramanian S, and Jaffee EM (2018). Transcriptional profiling identifies novel regulators of macrophage polarization. PLoS One 13, e0208602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grauers Wiktorin H, Nilsson MS, Kiffin R, Sander FE, Lenox B, Rydstrom A, Hellstrand K, and Martner A (2019). Histamine targets myeloid-derived suppressor cells and improves the anti-tumor efficacy of PD-1/PD-L1 checkpoint blockade. Cancer Immunol Immunother 68, 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerriero JL (2018). Macrophages: The Road Less Traveled, Changing Anticancer Therapy. Trends Mol Med 24, 472–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerriero JL (2019). Macrophages: Their Untold Story in T Cell Activation and Function. Int Rev Cell Mol Biol 342, 73–93. [DOI] [PubMed] [Google Scholar]
- Haak-Frendscho M, Darvas Z, Hegyesi H, Karpati S, Hoffman RL, Laszlo V, Bencsath M, Szalai C, Furesz J, Timar J, et al. (2000). Histidine decarboxylase expression in human melanoma. J Invest Dermatol 115, 345–352. [DOI] [PubMed] [Google Scholar]
- Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 e3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. (2016). Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 165, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonski KA, Amici SA, Webb LM, Ruiz-Rosado Jde D, Popovich PG, Partida-Sanchez S, and Guerau-de-Arellano M (2015). Novel Markers to Delineate Murine M1 and M2 Macrophages. PLoS One 10, e0145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, Leeson R, Kanodia A, Mei S, Lin JR, et al. (2018). A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 175, 984–997 e924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, Traugh N, Bu X, Li B, et al. (2018). Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med 24, 1550–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston RJ, Su LJ, Pinckney J, Critton D, Boyer E, Krishnakumar A, Corbett M, Rankin AL, Dibella R, Campbell L, et al. (2019). VISTA is an acidic pH-selective ligand for PSGL-1. Nature 574, 565–570. [DOI] [PubMed] [Google Scholar]
- Khononov I, Jacob E, Fremder E, Dahan N, Harel M, Raviv Z, Krastev B, and Shaked Y (2021). Host response to immune checkpoint inhibitors contributes to tumor aggressiveness. J Immunother Cancer 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugelmann D, Rotkopf LT, Radeva MY, Garcia-Ponce A, Walter E, and Waschke J (2018). Histamine causes endothelial barrier disruption via Ca(2+)-mediated RhoA activation and tension at adherens junctions. Sci Rep 8, 13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar RK, Herbert C, and Foster PS (2008). The "classical" ovalbumin challenge model of asthma in mice. Curr Drug Targets 9, 485–494. [DOI] [PubMed] [Google Scholar]
- Le Mercier I, Chen W, Lines JL, Day M, Li J, Sergent P, Noelle RJ, and Wang L (2014). VISTA Regulates the Development of Protective Antitumor Immunity. Cancer Res 74, 1933–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Fan J, Wang B, Traugh N, Chen Q, Liu JS, Li B, and Liu XS (2017). TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res 77, e108–e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lines JL, Pantazi E, Mak J, Sempere LF, Wang L, O'Connell S, Ceeraz S, Suriawinata AA, Yan S, Ernstoff MS, et al. (2014). VISTA is an immune checkpoint molecule for human T cells. Cancer Res 74, 1924–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yuan Y, Chen W, Putra J, Suriawinata AA, Schenk AD, Miller HE, Guleria I, Barth RJ, Huang YH, et al. (2015). Immune-checkpoint proteins VISTA and PD-1 nonredundantly regulate murine T-cell responses. Proc Natl Acad Sci U S A 112, 6682–6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Diehl SA, Noubade R, Ledoux J, Nelson MT, Spach K, Zachary JF, Blankenhorn EP, and Teuscher C (2010). Endothelial histamine H1 receptor signaling reduces blood-brain barrier permeability and susceptibility to autoimmune encephalomyelitis. Proc Natl Acad Sci U S A 107, 18967–18972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin RK, Saleem SJ, Folgosa L, Zellner HB, Damle SR, Nguyen GK, Ryan JJ, Bear HD, Irani AM, and Conrad DH (2014). Mast cell histamine promotes the immunoregulatory activity of myeloid-derived suppressor cells. J Leukoc Biol 96, 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FO, Gordon S, Locati M, and Mantovani A (2006). Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol 177, 7303–7311. [DOI] [PubMed] [Google Scholar]
- Massari NA, Nicoud MB, and Medina VA (2018). Histamine receptors and cancer pharmacology: an update. Br J Pharmacol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micaroni M (2010). The role of calcium in intracellular trafficking. Curr Mol Med 10, 763–773. [DOI] [PubMed] [Google Scholar]
- Micaroni M (2012). Calcium around the Golgi apparatus: implications for intracellular membrane trafficking. Adv Exp Med Biol 740, 439–460. [DOI] [PubMed] [Google Scholar]
- Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, and Perlman H (2013). Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol 49, 503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriarty CM, Stucky JL, Hamburger KW, Patil KD, Foley JF, and Koefoot RR (1988). Blood histamine and solid malignant tumors. J Cancer Res Clin Oncol 114, 588–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosely SI, Prime JE, Sainson RC, Koopmann JO, Wang DY, Greenawalt DM, Ahdesmaki MJ, Leyland R, Mullins S, Pacelli L, et al. (2017). Rational Selection of Syngeneic Preclinical Tumor Models for Immunotherapeutic Drug Discovery. Cancer Immunol Res 5, 29–41. [DOI] [PubMed] [Google Scholar]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, and Alizadeh AA (2015). Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 12, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nials AT, and Uddin S (2008). Mouse models of allergic asthma: acute and chronic allergen challenge. Dis Model Mech 1, 213–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak EC, Lines JL, Varn FS, Deng J, Sarde A, Mabaera R, Kuta A, Le Mercier I, Cheng C, and Noelle RJ (2017). Immunoregulatory functions of VISTA. Immunol Rev 276, 66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocana-Guzman R, Torre-Bouscoulet L, and Sada-Ovalle I (2016). TIM-3 Regulates Distinct Functions in Macrophages. Front Immunol 7, 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orecchioni M, Ghosheh Y, Pramod AB, and Ley K (2019). Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS−) vs. Alternatively Activated Macrophages. Front Immunol 10, 1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostuni R, Kratochvill F, Murray PJ, and Natoli G (2015). Macrophages and cancer: from mechanisms to therapeutic implications. Trends Immunol 36, 229–239. [DOI] [PubMed] [Google Scholar]
- Parsons ME, and Ganellin CR (2006). Histamine and its receptors. Br J Pharmacol 147 Suppl 1, S127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, and Wolchok JD (2018). Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittmeyer D, and Lorentz A (2012). Relationship between allergy and cancer: an overview. Int Arch Allergy Immunol 159, 216–225. [DOI] [PubMed] [Google Scholar]
- Sharma P, Hu-Lieskovan S, Wargo JA, and Ribas A (2017). Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 168, 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieja K, Stanosz S, von Mach-Szczypinski J, Olewniczak S, and Stanosz M (2005). Concentration of histamine in serum and tissues of the primary ductal breast cancers in women. Breast 14, 236–241. [DOI] [PubMed] [Google Scholar]
- Simons FE (2004). Advances in H1-antihistamines. N Engl J Med 351, 2203–2217. [DOI] [PubMed] [Google Scholar]
- Thurmond RL, Gelfand EW, and Dunford PJ (2008). The role of histamine H1 and H4 receptors in allergic inflammation: the search for new antihistamines. Nat Rev Drug Discov 7, 41–53. [DOI] [PubMed] [Google Scholar]
- Turner MC, Chen Y, Krewski D, and Ghadirian P (2006). An overview of the association between allergy and cancer. Int J Cancer 118, 3124–3132. [DOI] [PubMed] [Google Scholar]
- Uhlen M, Zhang C, Lee S, Sjostedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, et al. (2017). A pathology atlas of the human cancer transcriptome. Science 357. [DOI] [PubMed] [Google Scholar]
- Verma V, Shrimali RK, Ahmad S, Dai W, Wang H, Lu S, Nandre R, Gaur P, Lopez J, Sade-Feldman M, et al. (2019). PD-1 blockade in subprimed CD8 cells induces dysfunctional PD-1(+)CD38(hi) cells and anti-PD-1 resistance. Nat Immunol 20, 1231–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Mach-Szczypinski J, Stanosz S, Sieja K, and Stanosz M (2009). Metabolism of histamine in tissues of primary ductal breast cancer. Metabolism 58, 867–870. [DOI] [PubMed] [Google Scholar]
- Yang XD, Ai W, Asfaha S, Bhagat G, Friedman RA, Jin G, Park H, Shykind B, Diacovo TG, Falus A, et al. (2011). Histamine deficiency promotes inflammation-associated carcinogenesis through reduced myeloid maturation and accumulation of CD11b+Ly6G+ immature myeloid cells. Nat Med 17, 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Zhao Q, Yuan Y, Zhang L, Liu X, Yung WK, and Weinstein JN (2012). Identification of common prognostic gene expression signatures with biological meanings from microarray gene expression datasets. PLoS One 7, e45894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelenay S, van der Veen AG, Bottcher JP, Snelgrove KJ, Rogers N, Acton SE, Chakravarty P, Girotti MR, Marais R, Quezada SA, et al. (2015). Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 162, 1257–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Yao J, Wei Y, Zhou Z, Li P, Qu J, Badu-Nkansah A, Yuan X, Huang YW, Fukumura K, et al. (2020). Blocking immunosuppressive neutrophils deters pY696-EZH2-driven brain metastases. Sci Transl Med 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zhang S, Yao J, Lowery FJ, Zhang Q, Huang WC, Li P, Li M, Wang X, Zhang C, et al. (2015). Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 527, 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Lan Y, Xu J, Quan F, Zhao E, Deng C, Luo T, Xu L, Liao G, Yan M, et al. (2019). CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res 47, D721–D728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nat Commun 8, 14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Human samples used in the study, related to STAR Methods.
Data Availability Statement
RNA-seq data generated during this study have been deposited in the Gene Expression Omnibus (GEO) database under accession numbers GSE161484. Published datasets used in this study are available through GEO: GSE78220 (Hugo et al., 2016), GSE115978 (Jerby-Arnon et al., 2018) or cBioPortal database (http://www.cbioportal.org). Single-cell sequencing data has been submitted to Sequence Read Archive (SRA) and is available in SRA Run Selector with accession PRJNA756466.