

Abstract
Computational methods to predict molecular properties regarding safety and toxicology represent alternative approaches to expedite drug development, screen environmental chemicals, and thus significantly reduce associated time and costs. There is a strong need and interest in the development of computational methods that yield reliable predictions of toxicity, and many approaches, including the recently introduced deep neural networks, have been leveraged towards this goal. Herein, we report on the collection, curation, and integration of data from the public datasets that were the source of the ChemIDplus database for systemic acute toxicity. These efforts generated the largest publicly available such dataset comprising > 80,000 compounds measured against a total of 59 acute systemic toxicity endpoints. This data was used for developing multiple single- and multi-task models utilizing Random Forest, deep neural networks, convolutional and graph convolutional neural network approaches. For the first time, we also reported the consensus models based on different multi-task approaches. To the best of our knowledge, prediction models for 36 out of the 59 endpoints have never been published before. Furthermore, our results demonstrated a significantly better performance of the consensus model obtained from three multi-task learning approaches that particularly predicted the 29 smaller tasks (less than 300 compounds) better than other models developed in the study. The curated dataset and the developed models have been made publicly available at https://github.com/ncats/ld50-multitask, https://predictor.ncats.io/ and https://cactus.nci.nih.gov/download/acute-toxicity-db (dataset only) to support regulatory and research applications.
Graphical Abstract
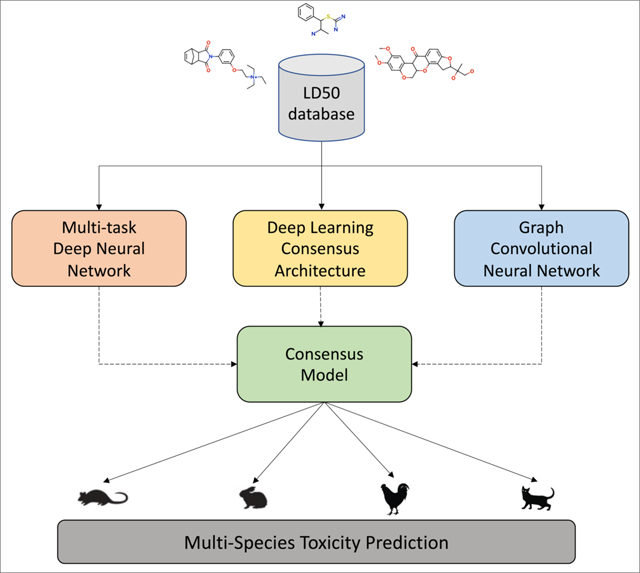
INTRODUCTION.
Early in silico assessment of small molecule toxicity is an indispensable step in drug discovery and development that helps reduce costs and labor, can inform on regulatory decision making, and has parallel applications in environmental chemical screening and prioritization1–6. The advent of big data in chemistry and biology, complemented by advances in screening technologies, has enabled the development of large-scale toxicity prediction models4,7–10. While the ChEMBL database11 serves as a major public resource of compound bioactivity data, there are other open-access databases that provide information on the toxicity of small molecules, such as TOXNET12 (www.nlm.nih.gov/toxnet/index.html) and DSSTox13. Dedicated resources with information related to specific toxicity endpoints are also becoming increasingly available14–16. RTECS® (Registry of Toxic Effects of Chemical Substances) is currently made available as a proprietary database that provides in vivo data for more than 180,000 chemical substances with a major focus on acute toxicity17. ChemIDplus18 on the other hand is a publicly available database that contains more that 150,000 compounds having acute systemic toxicity outcome records (e.g., lethal dose, 50% or LD50) in different species and multiple routes of administration.
A wide range of quantitative structure-activity relationship (QSAR) methods has been employed for computational toxicity prediction19–29. Machine learning methods such as Random Forest and Support Vector Machines have served as popular tools for building cheminformatics models30,31, and more recently, neural networks have emerged as robust methods that perform exceedingly well on large datasets and provide better extrapolation in comparison to traditional QSAR models4,32–36. However, models trained on small datasets often result in poor predictive performance on unseen external data37,38, and could potentially benefit from learning on biologically related endpoints. In this context, multi-task learning facilitates simultaneous modeling of multiple endpoints to develop better models, particularly when the endpoints are mechanistically correlated with one other36,39–41. Erhan et al.42 were the earliest to report single-task and multi-task predictive models for a family of biological targets in 2006. Later, a series of unified QSAR models were developed to predict antimicrobial activity of drugs against multiple fungal and bacterial species43–45. In 2008, Varnek et al.46 applied multi-task learning for modeling 11 types of tissue-air partition coefficients along with another inductive knowledge transfer technique known as Feature Net. In Feature Net approach, additional tasks are used to build models, predictions from which are used as descriptors for modeling the main task. Here, in the case of acute systemic toxicity, there are multiple specific endpoints in ChemIDplus18 that have a limited number of data points (e.g., human, cat, and rabbit lethal doses), but the expectation is that there is a finite number of mechanisms by which chemicals cause lethality and that these would be fairly consistent across species. Compounds that have data in one endpoint would then inform predictions on structurally similar compounds for related endpoints.
In 2018, at the CATMOS Meeting, Zakharov et al. proposed multi-task deep learning approach to model toxicity across 9 different endpoints47,48. Later, Sosnin et al.49 extended this approach for a total of 29 toxicity endpoints using data from RTECS® to report that these models outperformed single-task models based on other machine learning methods. More recently, Zakharov et al.4 proposed a novel deep learning consensus architecture (DLCA) to model more than 1000 endpoints using bioactivity data available from publicly accessible resources such as ChEMBL and Tox21 (https://tripod.nih.gov/tox21). Different types of graph convolutional neural networks (GCNN) were proposed and validated on benchmark datasets that include several multi-task classification and regression tasks50–52. Other studies showed that the implementation of transfer learning53–55 led to improved model performance40,41,56. In the toxicity domain, studies have used Tox2157, ToxCast58, SIDER59, and the recently introduced ClinTox dataset for benchmarking different machine learning methods19,50,57. While most of these datasets comprise multiple endpoints, consensus of multi-task approaches has not been extensively investigated. However, consensus modeling approaches have been reported to outperform simple QSAR models60–62. Thus, in this study, we consider consensus approaches that combined predictions from different multi-task learning approaches to predict acute toxicity.
Considering the chemical space coverage of the dataset and the sparsity of the measurements against 59 different endpoints, we thought that the acute toxicity data from ChemIDplus18 could be an ideal case study, both to improve existing models using multi-task learning and to implement and test new multi-task algorithms. Although several groups24,25,49,63–65 previously reported QSAR models for acute toxicity endpoints, most of which are publicly accessible, much of the modeling data were not made publicly available and therefore the quality could not be assessed. Here, we formulated the primary goals of the study as follows: i) collect, curate, and integrate all available data for systemic acute toxicity into the largest publicly available multi-species toxicity dataset; ii) use this dataset for benchmarking and comparing state-of-the-art single- and multi-task machine learning methods; iii) use the most recent advances in multi-task modeling to improve existing, or develop novel, models for a total of 59 acute toxicity endpoints spanning multiple species and routes of administration. Of the 59 endpoints, prediction models for 36 endpoints have not been previously reported in literature to the best of our knowledge.
MATERIAL AND METHODS.
Dataset.
The quality of experimental data is a crucial part of building machine learning models. In this study, we used data which are publicly available from ChemIDplus18. A set of 165,182 measurements related to 456 endpoints, representing 25 dosing routes across 28 species and expressed as LD50, lethal dose low (LDLo) and toxic dose low (TDLo) were extracted. The dataset consists of toxicity measurements in different units (mg/kg, mL/kg, gm/m3 and many others). In order to have a harmonized dataset, we considered the data from three measurement units: mg/kg, μg/kg, and ng/kg. This led us to a dataset of 159,968 measurements for 91,642 compounds tested against 437 endpoints.
Data curation.
The initial dataset containing 91,642 compounds was curated following a protocol previously developed by Fourches et al.66–68. Briefly, salts and solvents were stripped from all compounds followed by removal of counterions, large organic compounds (Da >= 2,000), mixtures, and inorganic compounds. Specific chemotypes such as aromatic, nitro groups, sulfo groups, tautomers, and protonation state were standardized using the ChemAxon Standardizer software (https://chemaxon.com/)69. If duplicates presented discordant potencies (i.e., > 0.2 −log units), both entries were excluded; if the reported potencies were similar, an average of the values was calculated, and one entry was retained in the dataset. Stereocenters were kept and enantiomers analyzed. After curation, 85,848 compounds and 255 endpoints were retrieved. In order to generate reliable prediction models, we removed the endpoints that had less than 100 reported measurements. This led us to a dataset of 80,081 unique compounds with 122,594 measurements against at least one of the 59 endpoints. Table S1 in the Supporting Information provides information on the number of measurements across each endpoint.
Molecular Descriptors.
Although the use of public descriptors in combination with commercial descriptors to model acute toxicity was previously reported49, we intended to stick to descriptors available in the open source domain. In 2018, Zakharov47 reported the superior performance of multi-task deep learning models for acute toxicity endpoints with Avalon fingerprints in comparison to Morgan fingerprints and RDKit descriptors. Therefore, we decided to use only Avalon fingerprints (1024 bits)70,71 in this study and calculated them using the RDKit Fingerprints node72 available in the KNIME analytics platform73.
Machine Learning Methods.
We used deep neural networks (DNNs) to build both multi-task (MT-DNN) and single-task (ST-DNN) models. In addition, we used Random Forest to build single-task (ST-RF) baseline models due to their widespread application and robust performance in cheminformatics and machine learning74–77. These methods are briefly explained below.
Deep Neural Networks (DNN)
DNNs have been reported to outperform most other machine learning methods for the prediction of molecular properties4,49,78,79. A DNN is an alteration of an artificial neural network (ANN) that consists of several sequential hidden layers. Each layer in a DNN is represented by a linear vector transformation Wx+b where W is a matrix of tunable weights and b is a bias vector, followed by a nonlinear transformation function (i.e., sigmoid). In our study, we developed multi-task DNN models utilizing the multi-layer feedforward neural networks implemented in Keras80 using the Tensorflow backend81. The loss function was minimized using the Adam algorithm82. In order to further identify the best hyperparameters for DNN, we used the grid search function available from the scikit-learn83 library. The grid search was performed for the following parameters: (i) number of epochs; (ii) batch size; (iii) activation function, (iv) learning rate of Adam optimizer, and (v) dense layer candidates, i.e., the number of neurons in each dense layer. The detailed list of hyperparameters optimized for the MT-DNN model can be found in Table S2 in the Supporting Information. While some parameters were fixed based on previous experience with the dataset, some were exhaustively searched to find the optimal performing hyperparameters. In the case of single-task DNN models, we used the best performing hyperparameters from the multi-task DNN since it would not be practical to evaluate an extensive list of hyperparameters over 59 different tasks individually. However, the learning rate for Adam optimizer was tuned for each task separately.
Random Forest
Random Forest (RF) is an ensemble of decision trees84. In this study, the single-task regression models (ST-RF) were built using the RF implementation in scikit-learn83. The number of trees was arbitrarily set to 100, since it has been shown that the optimal number of trees is usually 64 – 128, while further increasing the number of trees does not necessarily improve the model’s performance75,85. Due to the robust nature of RF86, no parameter optimization was performed.
Model Benchmarking
In addition to the single-task baseline models, we also benchmarked our DNN models with models reported in the literature. We first explored the ‘deep learning consensus architecture’ (DLCA) proposed by Zakharov et al.4. The approach averages the outputs of separate DNNs built using different descriptors inside a single neural net. This imposes a constraint on the learning algorithm to prevent propagation of corresponding errors, which leads to an improvement in the consensus results. In this study, we developed a DLCA model that combines descriptors-based and so-called descriptors-free models. The descriptors-based models were generated using three different types of fingerprints (Morgan, Avalon, and AtomPair), and RDKit descriptors. The descriptors-free model was created using SMILES notation and a convolutional neural net architecture based on 1D convolutional and GlobalMax pooling layers following by hidden dense and output layers (the architecture and training parameters are provided in Table S2 in the Supporting Information).87,88).
Next, we used the recently published graph convolutional neural networks (GCNN)52,89–93. In this study, we developed multi-task GCNN models by using a message-passing variant of GCNN as implemented in ChemProp51. These networks construct a learned molecular representation by operating on the graph structure of the molecule. Further, we also performed hyperparameter grid optimization and used the best settings to generate models for final validation. Optimization for the GCNN models was performed as proposed by Swanson et al.51,94,95.
Consensus Models
In this study, we developed two different consensus models from the best performing individual multi-task models. The first consensus model is based on the multi-task DNN model with hyperparameter grid optimization (MT-DNN), and multi-task GCNN model with grid optimization (GCNN). This is referred to as ‘consensus A’ in the rest of the study. The second is based on MT-DNN, GCNN, and multi-task DLCA models, referred as ‘consensus B’ in the rest of the study.
Model Validation and Statistical Performance.
To estimate the performance of the models developed in this study, we applied a 5-fold cross-validation procedure96. The dataset was randomly subdivided into five parts, where four parts were used as the training set for model building, and the remaining part was used as the test set for the assessment of predictive accuracy. As it was observed that the selection of hyperparameter plays a crucial role in the model performance97, in-order to have a fair and unbiased comparison, the best hyperparameters were selected based on grid search performed on the first fold of the dataset and applied on the remaining four folds. Further, in addition to random split, we also applied a scaffold-based splitting procedure as proposed by Yang et al.51
The performance of each model for 5-fold CV procedure was assessed on the basis of root mean squared error (RMSE) (Eq. 1), and determination coefficient R2 (Eq. 2),
| (1) |
| (2) |
is the predicted value for each particular compound; Yi is the observed value for each particular compound; is the mean value over all compounds; n is the number of compounds.
The difference between the model performance was evaluated using the Wilcoxon paired singed-rank non-parametric statistical test. For the given two methods, the predicted performance (RMSE and R2) for each of the 59 tasks was compared pairwise to identify the method that significantly outperforms the other. We defined the statistical significance as p-value less than 0.05.
Calculation of the Applicability Domain.
Applicability domain (AD) is a crucial part of the QSAR methodology that, if used correctly, may significantly improve the prediction results4,98. There are multiple ways to calculate the applicability of a QSAR model99–103. In this study, we used two different approaches for estimation of the model’s AD. In the first approach, we estimated the Tanimoto similarity104,105 between the test set compounds and nearest neighbor in the training set using Morgan fingerprints. For each fold, we filtered out those compounds that were below a certain similarity threshold and further calculated the RMSE (endpoint-wise) and the coverage of predictions as the percentage of compounds that fall within the model’s AD. In the second approach, since the DLCA model4 provides an integrated output from models based on different descriptors, we extracted the prediction output for each compound from individual descriptor models and calculated the standard deviation (SD) of prediction for each compound. Then, for each endpoint, we calculated the mean (μ) and standard deviation (σ) from the standard deviation of prediction for each compound. We then filtered out those compounds that were above the μ + 0.5 σ (t1), μ + σ (t2), μ + 2σ (t3) and μ + 3σ (t4), simultaneously and calculated the RMSE and coverage on the remaining.
RESULTS AND DISCUSSION
Data Overview.
After curation, 80,081 compounds remained in the dataset, with 122,594 measurements available for 59 endpoints. However, not all of these compounds were measured for all endpoints. The most frequently reported endpoints were for mouse, rat, and rabbit. For mouse, there were 12 different measurement types, i.e. combinations of dosing routes and acute systemic outcomes expressed as LD50, LDLo, and/or TDLo were reported (70,442 unique measurements). The second most reported species was rat (14,948 unique measurements) followed by rabbit (3,447 unique measurements) with 11 and 9 different measurement types, respectively. Oral, intravenous, and subcutaneous were the 3 most frequently studied routes of administration. The sparsity of the data matrix (80081×59) was found to be >97% (Table S3 in the Supporting Information). Next, PCA plots were generated based on Avalon fingerprints and the median acute systemic toxicity values across different endpoints for each molecule (Figure 1). Overall, the compounds span a fair extent of chemical space and potency. As can be seen, the majority of the overlapping chemical structures do not have distinct toxicity profiles. This indicates that the modelability106 of the dataset should be high, because ‘structurally similar molecules tend to exhibit similar properties’. This supports the idea of applying multi-task learning, in which the smaller tasks (endpoints with less than 300 measurements) are simultaneously learned with the larger tasks and the learner optimizes the performance across all tasks.
Figure 1.
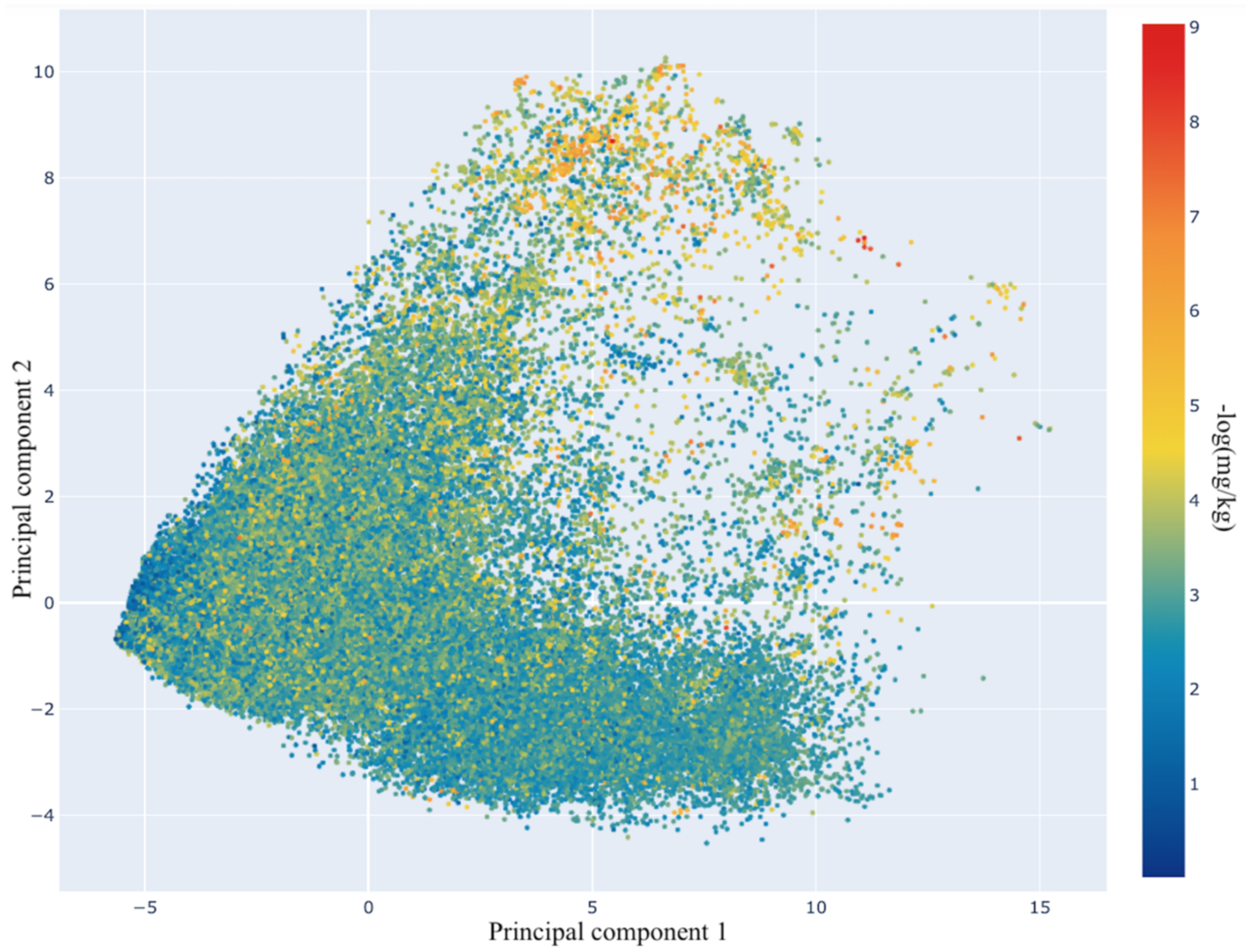
Two-dimensional PCA plot for the complete dataset based on Avalon fingerprints. The color scale represents the median toxicity value of the compounds against different endpoints in −log(mol/kg), i.e., the higher the values, the more toxic the compounds.
Modeling Results.
We evaluated the performance of multi-task regression models for different acute systemic toxicity endpoints, across species and dosing routes. We built multi-task DNN (MT-DNN), single-task models (DNN and Random Forest), multi-task DLCA (DLCA) and GCNN models. In order to avoid any bias that might occur due to the splitting schemes employed, all models were evaluated in a five-fold cross-validation scheme96,107. Figure 2 provides a comparison of the average performance (RMSE, R2) over 59 endpoints for different models generated in this study. The best results (average RMSE = 0.65; average R2 = 0.57) were obtained from the consensus B models, which is a consensus of the predictions from multi-task DNN, GCNN, and DLCA models. The DLCA models alone provided an average RMSE of 0.68. In general, our MT-DNN model performed slightly better than the GCNN model. The single-task DNN models on the other hand, performed the worst amongst all models in all folds. Though the single-task models based on Random Forest provided better performance than single-task DNNs, their performance was inferior compared to the multi-task models. The superior performance of the Random Forest model could be due to the robustness of algorithm108 as compared to the DNNs that require relatively large datasets in order to fit the hidden layers109,110. A similar performance trend was observed with the R2 values (Table S4 in the Supporting Information). Except for MT-DNN and GCNN, the difference in performance for any given pair of methods was found to be statistically significant (p < 0.05; Table S5 in the Supporting Information).
Figure 2.
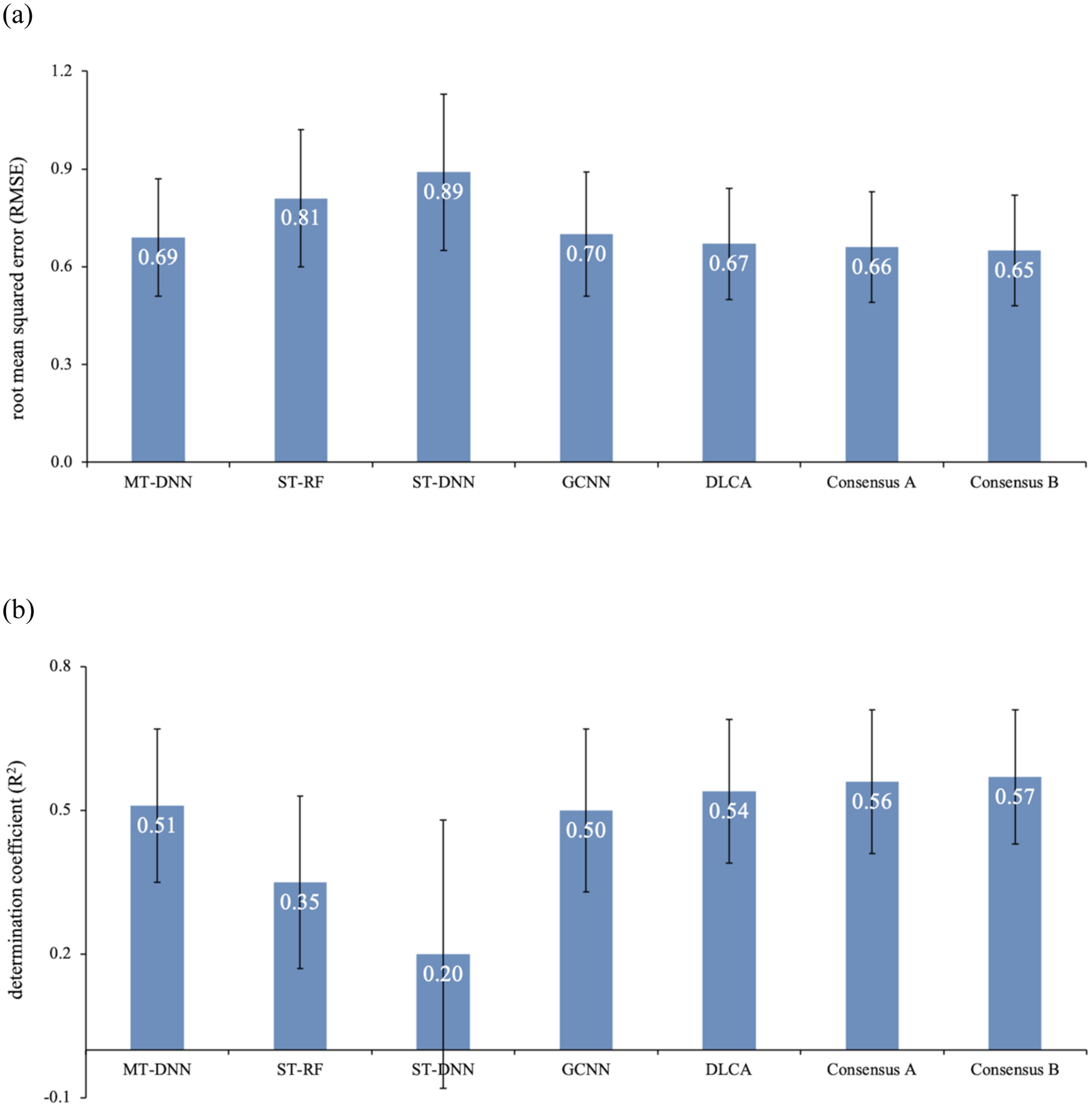
Average performance (a) RMSE, (b) R2 of all 59 endpoints for each approach over five-fold cross-validation based on training and test data generated using random splitting. The error bar represents the standard deviation of the average performance over five-folds.
With respect to specific endpoints, our best model (consensus B) predicted LD50 values fairly well for several species and several routes of administration, for example mouse oral, intravenous, intraperitoneal; rabbit skin; rat intraperitoneal, skin and others. The mouse oral LD50 had the best RMSE (RMSE = 0.43, R2 = 0.50), and was one of the most frequently measured endpoints with 23,373 values in the final dataset. The rabbit subcutaneous LDLo endpoint had the highest R2 value (R2 = 0.76, RMSE = 0.61) for our consensus B model, although it had one of the lowest incidences, with only 241 measurements. It should be noted that LDLo was predicted with lower accuracy than LD50 toxicity for all species and route of administration types, followed by TDLo. A possible reason could be that LD50 endpoints have comparatively higher numbers of measurements since it is more often evaluated as compared to TDLo and LDLo. Moreover, TDLo and LDLo are non-standard toxicity measurements and thus are less reliable due to lack of harmonized protocols causing variability in experimental conditions. The detailed model performance statistics can be found in Table S6 in the Supporting Information.
In addition to ‘random split,’ we also performed ‘scaffold split’, which is challenging, but a more realistic evaluation of the predictive power of the models4,51. ‘Scaffold split’ ensures that there is no molecular scaffold overlap between the train and test sets which indirectly mimics the evolution of new chemical space. As the ultimate goal of modeling is to predict properties of newly synthesized chemicals, performance assessment using ‘scaffold split’ can be considered a more realistic evaluation where new chemicals may not bear any resemblance to compounds in the training set111. In concordance with this, our results indicate superior performance of ‘random split’ in comparison to ‘scaffold split’. For the scaffold-split, DLCA model showed the best prediction results, followed by MT-DNN and GCNN models which provided similar performance. Detailed model statistics are provided in Table S4 in the Supporting Information.
Comparison to Previous Studies.
Outside of our own presentation at CATMOS meeting,47,48 only Sosnin et al.49 reported multi-task models for a total of 29 toxicity endpoints. They provided a comparison of both multi-task and single-task models using a wide range of molecular descriptors. It was shown that the best performance was obtained by averaging of the predictions of the top-five individual multi-task models (RMSE = 0.68; on 29 endpoints). In our study, the consensus B model combining three multi-task approaches provided the best performance with RMSE = 0.65 and R2 = 0.57 on 59 endpoints. Although we would like to benchmark the performance of our models against the results of Sosnin et al.,49 direct comparison is impossible because of the different numbers of compounds and endpoints. For some overlapping endpoints, we have fewer compounds in our dataset because of a more rigorous data curation procedure applied in this study. Furthermore, the raw data for our study were obtained from the ChemIDPlus portal, and therefore could have different numbers of compounds and measurements compared to the latest version of RTECS® dataset available from commercial vendors. These two reasons outlined above may explain the discrepancy in the number of measurements across different endpoints in Table S1 (in the Supporting Information). Ideally, future comparisons will be possible using newly obtained data. Despite the challenges in directly comparing the results, we checked for toxicity endpoint overlap with Sosnin et al.,49 and found that 23 were in common with the 59 endpoints addressed in this study. Of these 23 endpoints, we noticed that only 18 endpoints had a comparable number of measurements in both studies, considering a threshold of at least 300 measurements per endpoint (Table S1). We therefore drew parallels between both studies for these 18 relatively similar datasets. The best results from Sosnin et al.49 were achieved by a consensus model (RMSE = 0.54, R2 = 0.60). Our consensus B model provides an RMSE of 0.53 (R2 = 0.61) on the same 18 endpoints (Table S7 in the Supporting Information). Furthermore, we provide curated data and prediction models for 36 acute toxicity endpoints that to the best of our knowledge have never been published before. These include different combinations of species (dog, chicken, rabbit etc.), exposure route (oral, skin, intramuscular, etc.) and dose metric (LD50, LDLo, TDLo). While most of these represent non-standard endpoints in terms of internationally harmonized OECD guidelines, such studies are often performed and submitted to regulatory authorities as part of chemical toxicity evaluation packages. Ideally, the models presented here would substitute for future studies being performed using animals, saving considerable resources and providing reliable predictions using alternative approaches that have been trained on information from multiple species.
Multi-task Models versus Single-task Models.
It is clear from the results (Figure 3 and Table S6 in the Supporting Information) that our multi-task DNN models outperformed the single-task models on the smaller tasks (endpoints with fewer chemicals tested). This is expected according to the results from previous studies39,49,112–114 and due to the ability of multi-task methods to co-learn larger and smaller tasks115. Thus, multi-task models can learn from related tasks and thus tend to provide better performance on small (related) tasks compared to a single-task model trained using a smaller dataset. This emphasizes the advantage of using multi-task learning approaches for such understudied endpoints.
Figure 3.
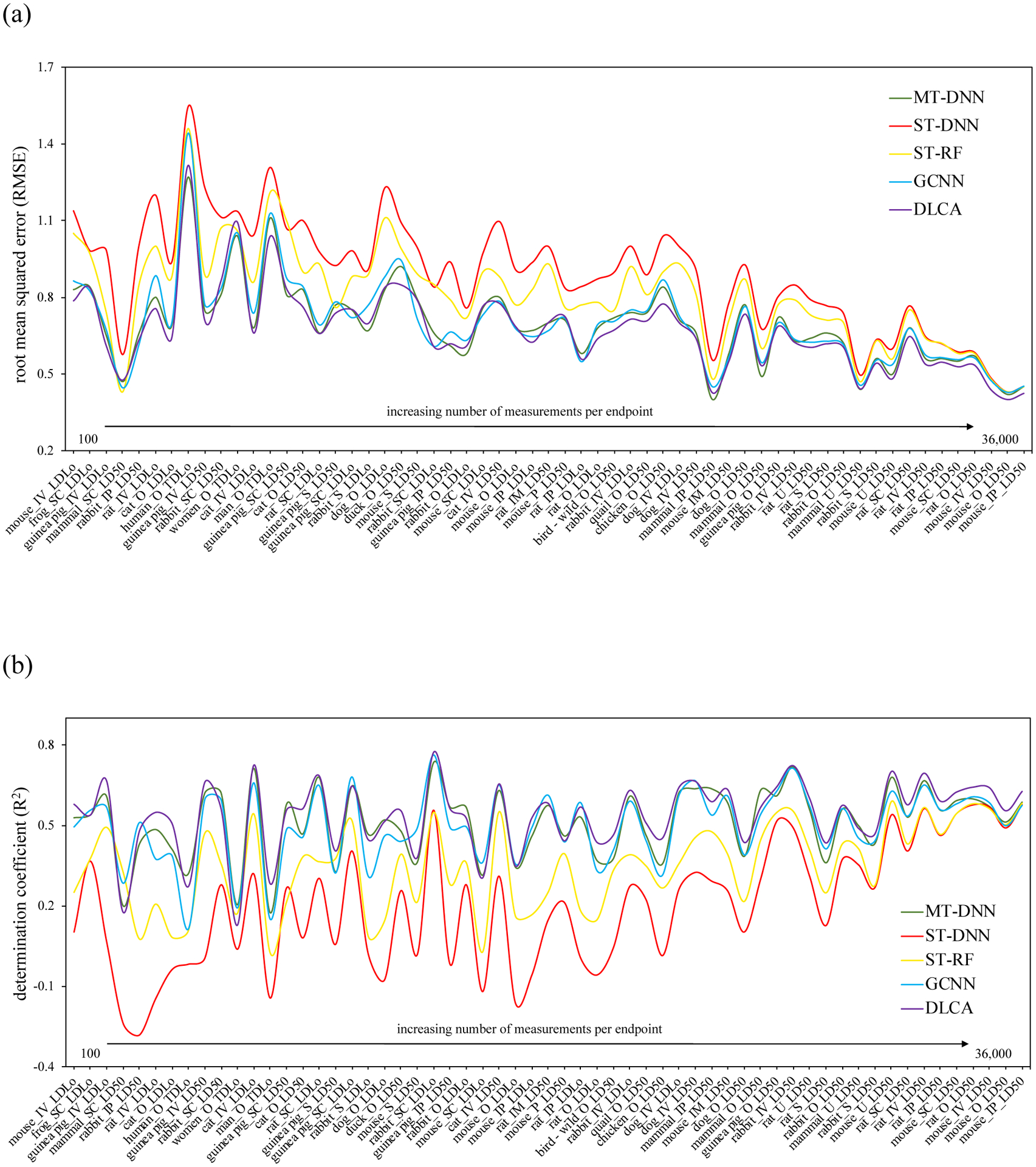
Performance (a) RMSE, (b) R2 of the best multi-task models obtained using different architectures and the single-task models for different endpoints ordered by the total number of measurements available in the dataset. (IM: intramuscular; IP: intraperitoneal; IV: intravenous; P: parenteral; S: skin; SC: subcutaneous; U: unreported; O: oral; mammal: mammal (species unspecified))
Applicability Domain Analysis.
Applicability domain (AD) of a QSAR model defines the limitations in its structural domain and response space. In this study, based on the two approaches (as presented in the ‘Materials and Methods’ section: ‘Calculation of the Applicability Domain’), the RMSE and the corresponding coverage (for the predictions from the DLCA model) were calculated and are presented in Figure 4 below with respect to the threshold values of AD (0.1–0.9) and SD (t1–t4) cut-offs. Figure S1 in the Supporting Information shows the R2, AD cut-off, SD cut-off and the corresponding coverage.
Figure 4.
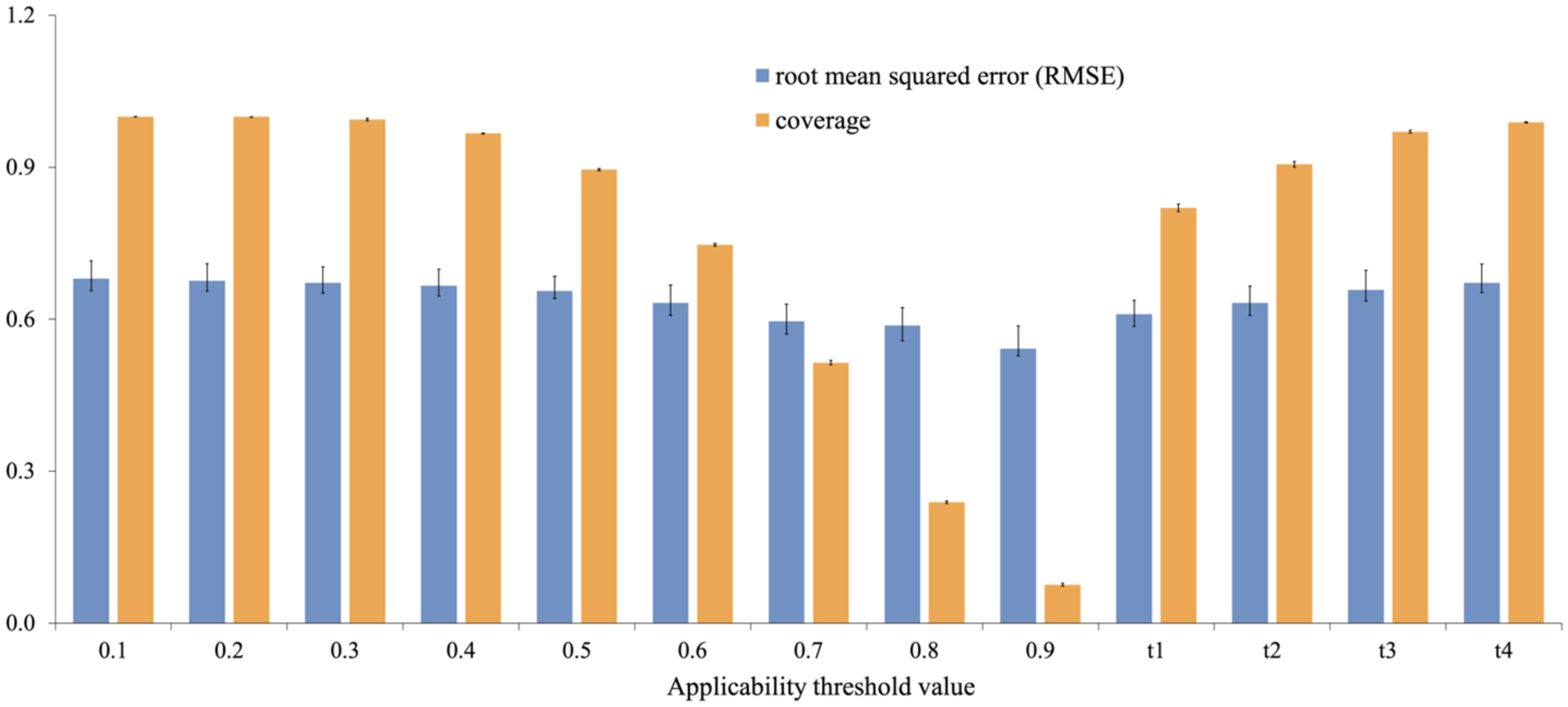
Distribution of the DLCA prediction results (RMSE) and coverage values over AD (0.1–0.9) and SD (t1–t4) cut-offs. The error bar represents the standard deviation of the average performance over five-folds.
Figure 4 shows an inverse correlation between the coverage and the accuracy of model prediction, meaning the higher the AD threshold, the better the accuracy of the model (lower RMSE) as expected. Based on the second approach, the higher the SD cut-off, the less accurate the model’s predictions (greater RMSE). The best results were obtained with AD = 0.9, which resulted in an RMSE value of 0.54 and 8% as the coverage of prediction. Considering both the coverage and the prediction accuracy, we found that t1 (mean + 0.5 SD) cut-off provides an optimal ratio between them, resulting in an RMSE value of 0.60 and coverage of 82%. Considering the Tanimoto similarity values, those predictions satisfying an AD threshold of 0.7 can be regarded as reasonable predictions (RMSE= 0.60; Coverage = 51%). Thus, both AD approaches could be used to select compounds with certain prediction confidence.
Online Service for Prediction of Acute Toxicity Profile of Chemical Compounds.
The MT-DNN model developed during the study are accessible via the NCATS Predictor (https://predictor.ncats.io/). Users can provide different molecular representations such as SMILES, SDF (structure data format) files or two-dimensional images of chemical structures as input. As an output, the online interface provides predictions for all 59 endpoints and reports the applicability domain assessment for each compound based on different models. The applicability domain calculation is based on the Tanimoto similarity to the nearest compound within the training set. A compound with a similarity value to the nearest neighbor falling in the region of (i) 1 to 0.7 is considered to be predicted with a high confidence; (ii) 0.7 to 0.5 is considered to be predicted with a medium confidence; (iii) less than 0.5 is considered to be predicted with a low confidence. This web service is provided to help researchers and regulators rapidly identify and prioritize compounds with toxic liabilities and gain additional insights based on the predicted profiles against the 59 multi-species acute-toxicity endpoints.
CONCLUSIONS.
Predicting molecular properties of small molecules is an essential step in modern drug discovery and environmental chemical assessment. Increasingly accurate computational methods for toxicity prediction are facilitated by data availability, novel algorithms, and computing power. Herein, we report on the collection, curation, and integration of all freely available data for systemic acute toxicity into the largest publicly available dataset (59 multi-species acute systemic toxicity endpoints and more than 8000 compounds). We used it for the development of deep-learning-based multi-task models and benchmarking them against state-of-the-art modeling techniques such as RF and recently proposed graph neural network architectures. We demonstrate that the MT-DNN approach offers a statistically significant advantage over single-task models, especially for endpoints with smaller number of compounds. Among multitask models, the DLCA model showed the best performance for both random and scaffold splitting procedures. Consensus predictors constructed from the results of MT-DNN, GCNN, and DLCA yielded the statistically highest predictive power. Both the curated acute toxicity dataset and the best performing models are made freely accessible to the research and regulatory community via the NCATS Predictor (https://predictor.ncats.io/), https://github.com/ncats/ld50-multitask as well as https://cactus.nci.nih.gov/download/acute-toxicity-db (dataset only) and can be readily used to predict and analyze acute toxicity of small molecules measured for different species and routes of administration.
Supplementary Material
Figure S1. Distribution of the DLCA prediction results (R2) over AD cut-offs and coverage values. The error bar represents the standard deviation of the average performance over five-folds. Figure S2. Average performance (a) RMSE, (b) R2 of all 59 endpoints for each approach over five-fold cross-validation based on training and test data generated using random splitting. The error bar represents the standard error mean of the average performance over five-folds.
S1. Provides information on the number of measurements across each endpoint extracted from the ChemIDPlus portal18. It also summarizes the extent of data overlap between the endpoints modeled in this study in comparison the study published by Sosnin et al.49 S2.The network architecture for MT-DNN, ST-DNN and DLCA and the list of hyperparameters optimized for the MT-DNN. S3. Provides information on the proportion of missing data across different endpoints. S4. Model statistics (5-fold cross-validation results) of multi-task regression models developed in this study. S5. Statistical analysis of prediction results using Wilcoxon paired singed-rank non-parametric statistical test. S6. Average RMSE of prediction across 5 folds for all endpoints by different models. S7. RMSE comparison of the overlapping endpoints for the models developed in this study and study published by Sosnin et al.49
ACKNOWLEDGEMENT
We would like to thank Dac-Trung Nguyen and Dr. Noel Southall for their valuable feedback and helpful discussions. This research was supported by the Intramural Research Program of the National Institutes of Health, National Center for Advancing Translational Sciences.
ABBREVIATIONS
- RTECS®
Registry of Toxic Effects of Chemical Substances
- QSAR
Quantitative Structure-Activity Relationship
- GCNN
Graph Convolutional Neural Networks
- LD50
Lethal Dose, 50%
- LDLo
Lethal Dose low
- TDLo
Toxic Dose low
- DNN
Deep Neural Network
- MT-DNN
Multi-Task Deep Neural Network
- ST-DNN
Single-Task Deep Neural Network
- ST-RF
Single-Task Random Forest
- RF
Random Forest
- DLCA
Deep Learning Consensus Architecture
- RMSE
Root Mean Squared Error
- AD
Applicability domain
- SD
Standard Deviation
- PCA
Principal Component Analysis
- CATMOS
Collaborative Modeling Project for Predicting Acute Oral Toxicity
- OECD
Organization for Economic Co-operation and Development
REFERENCES
- (1).Ting N Introduction and New Drug Development Process. In Dose Finding in Drug Development; Ting N, Ed.; Statistics for Biology and Health; Springer: New York, NY, 2006; pp 1–17. 10.1007/0-387-33706-7_1. [DOI] [Google Scholar]
- (2).Raies AB; Bajic VB In Silico Toxicology: Computational Methods for the Prediction of Chemical Toxicity. Wiley Interdiscip Rev Comput Mol Sci 2016, 6 (2), 147–172. 10.1002/wcms.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Tetko IV; Engkvist O; Koch U; Reymond J-L; Chen H BIGCHEM: Challenges and Opportunities for Big Data Analysis in Chemistry. Mol Inform 2016, 35 (11–12), 615–621. 10.1002/minf.201600073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Zakharov AV; Zhao T; Nguyen D-T; Peryea T; Sheils T; Yasgar A; Huang R; Southall N; Simeonov A Novel Consensus Architecture To Improve Performance of Large-Scale Multitask Deep Learning QSAR Models. J. Chem. Inf. Model 2019, 59 (11), 4613–4624. 10.1021/acs.jcim.9b00526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Lo Y-C; Rensi SE; Torng W; Altman RB Machine Learning in Chemoinformatics and Drug Discovery. Drug Discovery Today 2018, 23 (8), 1538–1546. 10.1016/j.drudis.2018.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Zhang L; McHale CM; Greene N; Snyder RD; Rich IN; Aardema MJ; Roy S; Pfuhler S; Venkatactahalam S Emerging Approaches in Predictive Toxicology. Environ. Mol. Mutagen 2014, 55 (9), 679–688. 10.1002/em.21885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Koutsoukas A; Lowe R; KalantarMotamedi Y; Mussa HY; Klaffke W; Mitchell JBO; Glen RC; Bender A In Silico Target Predictions: Defining a Benchmarking Data Set and Comparison of Performance of the Multiclass Naïve Bayes and Parzen-Rosenblatt Window. J. Chem. Inf. Model 2013, 53 (8), 1957–1966. 10.1021/ci300435j. [DOI] [PubMed] [Google Scholar]
- (8).Clark AM; Ekins S Open Source Bayesian Models. 2. Mining a “Big Dataset” To Create and Validate Models with ChEMBL. J. Chem. Inf. Model 2015, 55 (6), 1246–1260. 10.1021/acs.jcim.5b00144. [DOI] [PubMed] [Google Scholar]
- (9).Benigni R Predictive Toxicology Today: The Transition from Biological Knowledge to Practicable Models. Expert Opin Drug Metab Toxicol 2016, 12 (9), 989–992. 10.1080/17425255.2016.1206889. [DOI] [PubMed] [Google Scholar]
- (10).Blomme EAG; Will Y Toxicology Strategies for Drug Discovery: Present and Future. Chem. Res. Toxicol 2016, 29 (4), 473–504. 10.1021/acs.chemrestox.5b00407. [DOI] [PubMed] [Google Scholar]
- (11).Gaulton A; Bellis LJ; Bento AP; Chambers J; Davies M; Hersey A; Light Y; McGlinchey S; Michalovich D; Al-Lazikani B; Overington JP ChEMBL: A Large-Scale Bioactivity Database for Drug Discovery. Nucleic Acids Res 2012, 40. 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wexler P TOXNET: An Evolving Web Resource for Toxicology and Environmental Health Information. Toxicology 2001, 157 (1), 3–10. 10.1016/S0300-483X(00)00337-1. [DOI] [PubMed] [Google Scholar]
- (13).Richard AM; Williams CR Distributed Structure-Searchable Toxicity (DSSTox) Public Database Network: A Proposal. Mutat. Res 2002, 499 (1), 27–52. 10.1016/s0027-5107(01)00289-5. [DOI] [PubMed] [Google Scholar]
- (14).Hoofnagle JH; Serrano J; Knoben JE; Navarro VJ LiverTox. Hepatology 2013, 57 (3), 873–874. 10.1002/hep.26175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Montanari F; Knasmüller B; Kohlbacher S; Hillisch C; Baierová C; Grandits M; Ecker GF Vienna LiverTox Workspace—A Set of Machine Learning Models for Prediction of Interactions Profiles of Small Molecules With Transporters Relevant for Regulatory Agencies. Front Chem 2020, 7. 10.3389/fchem.2019.00899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Du F; Yu H; Zou B; Babcock J; Long S; Li M HERGCentral: A Large Database to Store, Retrieve, and Analyze Compound-Human Ether-à-Go-Go Related Gene Channel Interactions to Facilitate Cardiotoxicity Assessment in Drug Development. Assay Drug Dev Technol 2011, 9 (6), 580–588. 10.1089/adt.2011.0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).BIOVIA Databases | Bioactivity Databases: RTECS https://www.3dsbiovia.com/products/collaborative-science/databases/bioactivity-databases/rtecs.html (accessed Jan 29, 2020).
- (18).ChemIDplus: A Web-Based Chemical Search System, Mar–Apr 2000, NLM Technical Bulletin; https://www.nlm.nih.gov/pubs/techbull/ma00/ma00_chemid.html (accessed Jan 28, 2020). [Google Scholar]
- (19).Yang H; Sun L; Li W; Liu G; Tang Y In Silico Prediction of Chemical Toxicity for Drug Design Using Machine Learning Methods and Structural Alerts. Front. Chem 2018, 6. 10.3389/fchem.2018.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Cheng F; Shen J; Yu Y; Li W; Liu G; Lee PW; Tang Y In Silico Prediction of Tetrahymena Pyriformis Toxicity for Diverse Industrial Chemicals with Substructure Pattern Recognition and Machine Learning Methods. Chemosphere 2011, 82 (11), 1636–1643. 10.1016/j.chemosphere.2010.11.043. [DOI] [PubMed] [Google Scholar]
- (21).Cheng F; Yu Y; Zhou Y; Shen Z; Xiao W; Liu G; Li W; Lee PW; Tang Y Insights into Molecular Basis of Cytochrome P450 Inhibitory Promiscuity of Compounds. J Chem Inf Model 2011, 51 (10), 2482–2495. 10.1021/ci200317s. [DOI] [PubMed] [Google Scholar]
- (22).Pogodin PV; Lagunin AA; Filimonov DA; Poroikov VV PASS Targets: Ligand-Based Multi-Target Computational System Based on a Public Data and Naïve Bayes Approach. SAR QSAR Environ Res 2015, 26 (10), 783–793. 10.1080/1062936X.2015.1078407. [DOI] [PubMed] [Google Scholar]
- (23).Ji C; Svensson F; Zoufir A; Bender A EMolTox: Prediction of Molecular Toxicity with Confidence. Bioinformatics 2018, 34 (14), 2508–2509. 10.1093/bioinformatics/bty135. [DOI] [PubMed] [Google Scholar]
- (24).Drwal MN; Banerjee P; Dunkel M; Wettig MR; Preissner R ProTox: A Web Server for the in Silico Prediction of Rodent Oral Toxicity. Nucleic Acids Res. 2014, 42. 10.1093/nar/gku401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Banerjee P; Eckert AO; Schrey AK; Preissner R ProTox-II: A Webserver for the Prediction of Toxicity of Chemicals. Nucleic Acids Res 2018, 46. 10.1093/nar/gky318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Asadollahi-Baboli M Exploring QSTR Analysis of the Toxicity of Phenols and Thiophenols Using Machine Learning Methods. Environ. Toxicol. Pharmacol 2012, 34 (3), 826–831. 10.1016/j.etap.2012.09.003. [DOI] [PubMed] [Google Scholar]
- (27).Auerbach SS; Shah RR; Mav D; Smith CS; Walker NJ; Vallant MK; Boorman GA; Irwin RD Predicting the Hepatocarcinogenic Potential of Alkenylbenzene Flavoring Agents Using Toxicogenomics and Machine Learning. Toxicol. Appl. Pharmacol 2010, 243 (3), 300–314. 10.1016/j.taap.2009.11.021. [DOI] [PubMed] [Google Scholar]
- (28).Liu R; Zhang HY; Ji ZX; Rallo R; Xia T; Chang CH; Nel A; Cohen Y Development of Structure-Activity Relationship for Metal Oxide Nanoparticles. Nanoscale 2013, 5 (12), 5644–5653. 10.1039/c3nr01533e. [DOI] [PubMed] [Google Scholar]
- (29).Wang Y; Zheng M; Xiao J; Lu Y; Wang F; Lu J; Luo X; Zhu W; Jianga H; Chen K Using Support Vector Regression Coupled with the Genetic Algorithm for Predicting Acute Toxicity to the Fathead Minnow. SAR QSAR Environ Res 2010, 21 (5–6), 559–570. 10.1080/1062936X.2010.502300. [DOI] [PubMed] [Google Scholar]
- (30).Svetnik V; Liaw A; Tong C; Culberson JC; Sheridan RP; Feuston BP Random Forest: A Classification and Regression Tool for Compound Classification and QSAR Modeling. J Chem Inf Comput Sci 2003, 43 (6), 1947–1958. 10.1021/ci034160g. [DOI] [PubMed] [Google Scholar]
- (31).Varnek A; Baskin I Machine Learning Methods for Property Prediction in Chemoinformatics: Quo Vadis? J Chem Inf Model 2012, 52 (6), 1413–1437. 10.1021/ci200409x. [DOI] [PubMed] [Google Scholar]
- (32).Baskin II; Winkler D; Tetko IV A Renaissance of Neural Networks in Drug Discovery. Expert Opin Drug Discov 2016, 11 (8), 785–795. 10.1080/17460441.2016.1201262. [DOI] [PubMed] [Google Scholar]
- (33).Sosnin S; Vashurina M; Withnall M; Karpov P; Fedorov M; Tetko IV A Survey of Multi-Task Learning Methods in Chemoinformatics. Molecular Informatics 2019, 38 (4), 1800108. 10.1002/minf.201800108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Sze V; Chen Y-H; Yang T-J; Emer J Efficient Processing of Deep Neural Networks: A Tutorial and Survey. arXiv:1703.09039 [cs] 2017. [Google Scholar]
- (35).Lusci A; Pollastri G; Baldi P Deep Architectures and Deep Learning in Chemoinformatics: The Prediction of Aqueous Solubility for Drug-Like Molecules. J. Chem. Inf. Model 2013, 53 (7), 1563–1575. 10.1021/ci400187y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Ma J; Sheridan RP; Liaw A; Dahl GE; Svetnik V Deep Neural Nets as a Method for Quantitative Structure–Activity Relationships. J. Chem. Inf. Model 2015, 55 (2), 263–274. 10.1021/ci500747n. [DOI] [PubMed] [Google Scholar]
- (37).Gajewicz A How to Judge Whether QSAR/Read-across Predictions Can Be Trusted: A Novel Approach for Establishing a Model’s Applicability Domain. Environ. Sci.: Nano 2018, 5 (2), 408–421. 10.1039/C7EN00774D. [DOI] [Google Scholar]
- (38).Taskinen J; Norinder U 5.26 - In Silico Predictions of Solubility. In Comprehensive Medicinal Chemistry II; Taylor JB, Triggle DJ, Eds.; Elsevier: Oxford, 2007; pp 627–648. 10.1016/B0-08-045044-X/00279-0. [DOI] [Google Scholar]
- (39).Xu Y; Ma J; Liaw A; Sheridan RP; Svetnik V Demystifying Multitask Deep Neural Networks for Quantitative Structure–Activity Relationships. J. Chem. Inf. Model 2017, 57 (10), 2490–2504. 10.1021/acs.jcim.7b00087. [DOI] [PubMed] [Google Scholar]
- (40).Weiss K; Khoshgoftaar TM; Wang D A Survey of Transfer Learning. Journal of Big Data 2016, 3 (1), 9. 10.1186/s40537-016-0043-6. [DOI] [Google Scholar]
- (41).Wu P; Dietterich TG Improving SVM Accuracy by Training on Auxiliary Data Sources. In Proceedings of the twenty-first international conference on Machine learning; ICML ‘04; Association for Computing Machinery: Banff, Alberta, Canada, 2004; p 110. 10.1145/1015330.1015436. [DOI] [Google Scholar]
- (42).Erhan D; L’Heureux P-J; Yue SY; Bengio Y Collaborative Filtering on a Family of Biological Targets. J. Chem. Inf. Model 2006, 46 (2), 626–635. 10.1021/ci050367t. [DOI] [PubMed] [Google Scholar]
- (43).González-Díaz H; Prado-Prado FJ; Santana L; Uriarte E Unify QSAR Approach to Antimicrobials. Part 1: Predicting Antifungal Activity against Different Species. Bioorg Med Chem 2006, 14 (17), 5973–5980. 10.1016/j.bmc.2006.05.018. [DOI] [PubMed] [Google Scholar]
- (44).Prado-Prado FJ; González-Díaz H; Santana L; Uriarte E Unified QSAR Approach to Antimicrobials. Part 2: Predicting Activity against More than 90 Different Species in Order to Halt Antibacterial Resistance. Bioorganic & Medicinal Chemistry 2007, 15 (2), 897–902. 10.1016/j.bmc.2006.10.039. [DOI] [PubMed] [Google Scholar]
- (45).Prado-Prado FJ; González-Díaz H; de la Vega OM; Ubeira FM; Chou K-C Unified QSAR Approach to Antimicrobials. Part 3: First Multi-Tasking QSAR Model for Input-Coded Prediction, Structural Back-Projection, and Complex Networks Clustering of Antiprotozoal Compounds. Bioorg Med Chem 2008, 16 (11), 5871–5880. 10.1016/j.bmc.2008.04.068. [DOI] [PubMed] [Google Scholar]
- (46).Varnek A; Gaudin C; Marcou G; Baskin I; Pandey AK; Tetko IV Inductive Transfer of Knowledge: Application of Multi-Task Learning and Feature Net Approaches to Model Tissue-Air Partition Coefficients. J Chem Inf Model 2009, 49 (1), 133–144. 10.1021/ci8002914. [DOI] [PubMed] [Google Scholar]
- (47).Zakharov A Multitask Deep Learning Modelling of Rodent Acute Toxicity; 2018.
- (48).Kleinstreuer NC; Karmaus AL; Mansouri K; Allen DG; Fitzpatrick JM; Patlewicz G Predictive Models for Acute Oral Systemic Toxicity: A Workshop to Bridge the Gap from Research to Regulation. Computational Toxicology 2018, 8, 21–24. 10.1016/j.comtox.2018.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Sosnin S; Karlov D; Tetko IV; Fedorov MV Comparative Study of Multitask Toxicity Modeling on a Broad Chemical Space. J. Chem. Inf. Model 2019, 59 (3), 1062–1072. 10.1021/acs.jcim.8b00685. [DOI] [PubMed] [Google Scholar]
- (50).Wu Z; Ramsundar B; Feinberg EN; Gomes J; Geniesse C; Pappu AS; Leswing K; Pande V MoleculeNet: A Benchmark for Molecular Machine Learning. Chem. Sci 2018, 9 (2), 513–530. 10.1039/C7SC02664A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Yang K; Swanson K; Jin W; Coley C; Eiden P; Gao H; Guzman-Perez A; Hopper T; Kelley B; Mathea M; Palmer A; Settels V; Jaakkola T; Jensen K; Barzilay R Analyzing Learned Molecular Representations for Property Prediction. J. Chem. Inf. Model 2019, 59 (8), 3370–3388. 10.1021/acs.jcim.9b00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Na GS; Kim HW; Chang H Costless Performance Improvement in Machine Learning for Graph-Based Molecular Analysis. J. Chem. Inf. Model 2020. 10.1021/acs.jcim.9b00816. [DOI] [PubMed] [Google Scholar]
- (53).Pan SJ; Yang Q A Survey on Transfer Learning. IEEE Transactions on Knowledge and Data Engineering 2010, 22 (10), 1345–1359. 10.1109/TKDE.2009.191. [DOI] [Google Scholar]
- (54).Li X; Fourches D Inductive Transfer Learning for Molecular Activity Prediction: Next-Gen QSAR Models with MolPMoFiT. 2019. 10.26434/chemrxiv.9978743.v1. [DOI] [PMC free article] [PubMed]
- (55).Caruana R Multitask Learning. In Learning to Learn; Thrun S, Pratt L, Eds.; Springer US: Boston, MA, 1998; pp 95–133. 10.1007/978-1-4615-5529-2_5. [DOI] [Google Scholar]
- (56).Jiang J; Wang R; Wang M; Gao K; Nguyen DD; Wei G-W Boosting Tree-Assisted Multitask Deep Learning for Small Scientific Datasets. J. Chem. Inf. Model 2020. 10.1021/acs.jcim.9b01184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Huang R; Xia M Editorial: Tox21 Challenge to Build Predictive Models of Nuclear Receptor and Stress Response Pathways As Mediated by Exposure to Environmental Toxicants and Drugs. Front. Environ. Sci 2017, 5. 10.3389/fenvs.2017.00003. [DOI] [Google Scholar]
- (58).Dix DJ; Houck KA; Martin MT; Richard AM; Setzer RW; Kavlock RJ The ToxCast Program for Prioritizing Toxicity Testing of Environmental Chemicals. Toxicol. Sci 2007, 95 (1), 5–12. 10.1093/toxsci/kfl103. [DOI] [PubMed] [Google Scholar]
- (59).Kuhn M; Letunic I; Jensen LJ; Bork P The SIDER Database of Drugs and Side Effects. Nucleic Acids Res. 2016, 44 (D1), D1075–1079. 10.1093/nar/gkv1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Xu Y; Pei J; Lai L Deep Learning Based Regression and Multiclass Models for Acute Oral Toxicity Prediction with Automatic Chemical Feature Extraction. J. Chem. Inf. Model 2017, 57 (11), 2672–2685. 10.1021/acs.jcim.7b00244. [DOI] [PubMed] [Google Scholar]
- (61).Zakharov AV; Peach ML; Sitzmann M; Nicklaus MC QSAR Modeling of Imbalanced High-Throughput Screening Data in PubChem. J. Chem. Inf. Model 2014, 54 (3), 705–712. 10.1021/ci400737s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Zhu H; Tropsha A; Fourches D; Varnek A; Papa E; Gramatica P; Öberg T; Dao P; Cherkasov A; Tetko IV Combinatorial QSAR Modeling of Chemical Toxicants Tested against Tetrahymena Pyriformis. J. Chem. Inf. Model 2008, 48 (4), 766–784. 10.1021/ci700443v. [DOI] [PubMed] [Google Scholar]
- (63).Zhu H; Martin TM; Ye L; Sedykh A; Young DM; Tropsha A Quantitative Structure-Activity Relationship Modeling of Rat Acute Toxicity by Oral Exposure. Chem. Res. Toxicol 2009, 22 (12), 1913–1921. 10.1021/tx900189p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Li X; Chen L; Cheng F; Wu Z; Bian H; Xu C; Li W; Liu G; Shen X; Tang Y In Silico Prediction of Chemical Acute Oral Toxicity Using Multi-Classification Methods. J Chem Inf Model 2014, 54 (4), 1061–1069. 10.1021/ci5000467. [DOI] [PubMed] [Google Scholar]
- (65).Lagunin A; Zakharov A; Filimonov D; Poroikov V QSAR Modelling of Rat Acute Toxicity on the Basis of PASS Prediction. Molecular Informatics 2011, 30 (2–3), 241–250. 10.1002/minf.201000151. [DOI] [PubMed] [Google Scholar]
- (66).Fourches D; Muratov E; Tropsha A Trust, but Verify: On the Importance of Chemical Structure Curation in Cheminformatics and QSAR Modeling Research. J Chem Inf Model 2010, 50 (7), 1189–1204. 10.1021/ci100176x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Fourches D; Muratov E; Tropsha A Trust, but Verify II: A Practical Guide to Chemogenomics Data Curation. J Chem Inf Model 2016, 56 (7), 1243–1252. 10.1021/acs.jcim.6b00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Fourches D; Muratov E; Tropsha A Curation of Chemogenomics Data. Nature Chemical Biology 2015, 11 (8), 535–535. 10.1038/nchembio.1881. [DOI] [PubMed] [Google Scholar]
- (69).Chemaxon. Standardizer, JChem 5.4 http://www.chemaxon.com (accessed Feb 16, 2011).
- (70).Gedeck P; Rohde B; Bartels C QSAR – How Good Is It in Practice? Comparison of Descriptor Sets on an Unbiased Cross Section of Corporate Data Sets. J. Chem. Inf. Model 2006, 46 (5), 1924–1936. 10.1021/ci050413p. [DOI] [PubMed] [Google Scholar]
- (71).Riniker S; Landrum GA Open-Source Platform to Benchmark Fingerprints for Ligand-Based Virtual Screening. J Cheminform 2013, 5, 26. 10.1186/1758-2946-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Landrum G RDKit: Open-Source Cheminformatics.
- (73).Berthold MR; Cebron N; Dill F; Gabriel TR; Kötter T; Meinl T; Ohl P; Thiel K; Wiswedel B KNIME - the Konstanz Information Miner: Version 2.0 and Beyond. SIGKDD Explor. Newsl 2009, 11 (1), 26–31. 10.1145/1656274.1656280. [DOI] [Google Scholar]
- (74).Lai K; Twine N; O’Brien A; Guo Y; Bauer D Artificial Intelligence and Machine Learning in Bioinformatics. In Encyclopedia of Bioinformatics and Computational Biology; Ranganathan S, Gribskov M, Nakai K, Schönbach C, Eds.; Academic Press: Oxford, 2019; pp 272–286. 10.1016/B978-0-12-809633-8.20325-7. [DOI] [Google Scholar]
- (75).Jain S; Kotsampasakou E; Ecker GF Comparing the Performance of Meta-Classifiers-a Case Study on Selected Imbalanced Data Sets Relevant for Prediction of Liver Toxicity. J. Comput. Aided Mol. Des 2018, 32 (5), 583–590. 10.1007/s10822-018-0116-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Barta G Identifying Biological Pathway Interrupting Toxins Using Multi-Tree Ensembles. Front. Environ. Sci 2016, 4. 10.3389/fenvs.2016.00052. [DOI] [Google Scholar]
- (77).Chen J; Tang YY; Fang B; Guo C In Silico Prediction of Toxic Action Mechanisms of Phenols for Imbalanced Data with Random Forest Learner. J. Mol. Graph. Model 2012, 35, 21–27. 10.1016/j.jmgm.2012.01.002. [DOI] [PubMed] [Google Scholar]
- (78).Korotcov A; Tkachenko V; Russo DP; Ekins S Comparison of Deep Learning With Multiple Machine Learning Methods and Metrics Using Diverse Drug Discovery Datasets. Mol Pharm 2017, 14 (12), 4462–4475. 10.1021/acs.molpharmaceut.7b00578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Mayr A; Klambauer G; Unterthiner T; Hochreiter S DeepTox: Toxicity Prediction Using Deep Learning. Front. Environ. Sci 2016, 3. 10.3389/fenvs.2015.00080. [DOI] [Google Scholar]
- (80).Home - Keras Documentation https://keras.io/ (accessed Feb 16, 2020).
- (81).TensorFlow https://www.tensorflow.org/ (accessed Feb 16, 2020).
- (82).Kingma DP; Ba J Adam: A Method for Stochastic Optimization. 2014.
- (83).Pedregosa F; Varoquaux G; Gramfort A; Michel V; Thirion B; Grisel O; Blondel M; Prettenhofer P; Weiss R; Dubourg V; Vanderplas J; Passos A; Cournapeau D; Brucher M; Perrot M; Duchesnay É Scikit-Learn: Machine Learning in Python. Journal of Machine Learning Research 2011, 12, 2825–2830. [Google Scholar]
- (84).Breiman L Random Forests. Machine Learning 2001, 45 (1), 5–32. 10.1023/A:1010933404324. [DOI] [Google Scholar]
- (85).Oshiro TM; Perez PS; Baranauskas JA How Many Trees in a Random Forest? In Machine Learning and Data Mining in Pattern Recognition; Lecture Notes in Computer Science; Springer, Berlin, Heidelberg, 2012; pp 154–168. 10.1007/978-3-642-31537-4_13. [DOI] [Google Scholar]
- (86).Huang BFF; Boutros PC The Parameter Sensitivity of Random Forests. BMC Bioinformatics 2016, 17 (1), 331. 10.1186/s12859-016-1228-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Hochreiter S; Schmidhuber J Long Short-Term Memory. Neural Computation 1997, 9 (8), 1735–1780. 10.1162/neco.1997.9.8.1735. [DOI] [PubMed] [Google Scholar]
- (88).Sherstinsky A Fundamentals of Recurrent Neural Network (RNN) and Long Short-Term Memory (LSTM) Network. 2018. 10.1016/j.physd.2019.132306. [DOI]
- (89).Wang X; Li Z; Jiang M; Wang S; Zhang S; Wei Z Molecule Property Prediction Based on Spatial Graph Embedding. J. Chem. Inf. Model 2019, 59 (9), 3817–3828. 10.1021/acs.jcim.9b00410. [DOI] [PubMed] [Google Scholar]
- (90).Korolev V; Mitrofanov A; Korotcov A; Tkachenko V Graph Convolutional Neural Networks as “General-Purpose” Property Predictors: The Universality and Limits of Applicability. J. Chem. Inf. Model 2020, 60 (1), 22–28. 10.1021/acs.jcim.9b00587. [DOI] [PubMed] [Google Scholar]
- (91).Torng W; Altman RB Graph Convolutional Neural Networks for Predicting Drug-Target Interactions. J. Chem. Inf. Model 2019, 59 (10), 4131–4149. 10.1021/acs.jcim.9b00628. [DOI] [PubMed] [Google Scholar]
- (92).Li X; Yan X; Gu Q; Zhou H; Wu D; Xu J DeepChemStable: Chemical Stability Prediction with an Attention-Based Graph Convolution Network. J. Chem. Inf. Model 2019, 59 (3), 1044–1049. 10.1021/acs.jcim.8b00672. [DOI] [PubMed] [Google Scholar]
- (93).Ishida S; Terayama K; Kojima R; Takasu K; Okuno Y Prediction and Interpretable Visualization of Retrosynthetic Reactions Using Graph Convolutional Networks. J. Chem. Inf. Model 2019, 59 (12), 5026–5033. 10.1021/acs.jcim.9b00538. [DOI] [PubMed] [Google Scholar]
- (94).Swanson K Chemprop/Chemprop; chemprop, 2020. [Google Scholar]
- (95).Swanson K Message Passing Neural Networks for Molecular Property Prediction, Massachusetts Institute of Technology, 2019.
- (96).Berrar D Cross-Validation. In Encyclopedia of Bioinformatics and Computational Biology; Ranganathan S, Gribskov M, Nakai K, Schönbach C, Eds.; Academic Press: Oxford, 2019; pp 542–545. 10.1016/B978-0-12-809633-8.20349-X. [DOI] [Google Scholar]
- (97).Koutsoukas A; Monaghan KJ; Li X; Huan J Deep-Learning: Investigating Deep Neural Networks Hyper-Parameters and Comparison of Performance to Shallow Methods for Modeling Bioactivity Data. J Cheminform 2017, 9. 10.1186/s13321-017-0226-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Luque Ruiz I; Gómez-Nieto MÁ Study of the Applicability Domain of the QSAR Classification Models by Means of the Rivality and Modelability Indexes. Molecules 2018, 23 (11). 10.3390/molecules23112756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Kaneko H A New Measure of Regression Model Accuracy That Considers Applicability Domains. Chemometrics and Intelligent Laboratory Systems 2017, 171, 1–8. 10.1016/j.chemolab.2017.09.018. [DOI] [Google Scholar]
- (100).Patel M; Chilton ML; Sartini A; Gibson L; Barber C; Covey-Crump L; Przybylak KR; Cronin MTD; Madden JC Assessment and Reproducibility of Quantitative Structure-Activity Relationship Models by the Nonexpert. J Chem Inf Model 2018, 58 (3), 673–682. 10.1021/acs.jcim.7b00523. [DOI] [PubMed] [Google Scholar]
- (101).Keefer CE; Kauffman GW; Gupta RR Interpretable, Probability-Based Confidence Metric for Continuous Quantitative Structure-Activity Relationship Models. J Chem Inf Model 2013, 53 (2), 368–383. 10.1021/ci300554t. [DOI] [PubMed] [Google Scholar]
- (102).Yun Y-H; Wu D-M; Li G-Y; Zhang Q-Y; Yang X; Li Q-F; Cao D-S; Xu Q-S A Strategy on the Definition of Applicability Domain of Model Based on Population Analysis. Chemometrics and Intelligent Laboratory Systems 2017, 170, 77–83. 10.1016/j.chemolab.2017.09.007. [DOI] [Google Scholar]
- (103).Sushko I; Novotarskyi S; Körner R; Pandey AK; Cherkasov A; Li J; Gramatica P; Hansen K; Schroeter T; Müller K-R; Xi L; Liu H; Yao X; Oberg T; Hormozdiari F; Dao P; Sahinalp C; Todeschini R; Polishchuk P; Artemenko A; Kuz’min V; Martin TM; Young DM; Fourches D; Muratov E; Tropsha A; Baskin I; Horvath D; Marcou G; Muller C; Varnek A; Prokopenko VV; Tetko IV Applicability Domains for Classification Problems: Benchmarking of Distance to Models for Ames Mutagenicity Set. J. Chem. Inf. Model 2010, 50 (12), 2094–2111. https://doi.org/doi: 10.1021/ci100253r. [DOI] [PubMed] [Google Scholar]
- (104).Tanimoto TT An Elementary Mathematical Theory of Classification and Prediction; International Business Machines Corporation: New York, 1958. [Google Scholar]
- (105).Bajusz D; Rácz A; Héberger K Why Is Tanimoto Index an Appropriate Choice for Fingerprint-Based Similarity Calculations? Journal of Cheminformatics 2015, 7 (1), 20. 10.1186/s13321-015-0069-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Golbraikh A; Muratov E; Fourches D; Tropsha A Data Set Modelability by QSAR. J. Chem. Inf. Model 2014, 54 (1), 1–4. 10.1021/ci400572x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Johansson E; Eriksson L; Sandberg M; Wold S QSAR Model Validation. In Molecular Modeling and Prediction of Bioactivity; Gundertofte K, Jørgensen FS, Eds.; Springer US: Boston, MA, 2000; pp 271–272. 10.1007/978-1-4615-4141-7_36. [DOI] [Google Scholar]
- (108).Roy M-H; Larocque D Robustness of Random Forests for Regression. Journal of Nonparametric Statistics 2012, 24 (4), 993–1006. 10.1080/10485252.2012.715161. [DOI] [Google Scholar]
- (109).Ching T; Himmelstein DS; Beaulieu-Jones BK; Kalinin AA; Do BT; Way GP; Ferrero E; Agapow P-M; Zietz M; Hoffman MM; Xie W; Rosen GL; Lengerich BJ; Israeli J; Lanchantin J; Woloszynek S; Carpenter AE; Shrikumar A; Xu J; Cofer EM; Lavender CA; Turaga SC; Alexandari AM; Lu Z; Harris DJ; DeCaprio D; Qi Y; Kundaje A; Peng Y; Wiley LK; Segler MHS; Boca SM; Swamidass SJ; Huang A; Gitter A; Greene CS Opportunities and Obstacles for Deep Learning in Biology and Medicine. J R Soc Interface 2018, 15 (141). 10.1098/rsif.2017.0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Cao C; Liu F; Tan H; Song D; Shu W; Li W; Zhou Y; Bo X; Xie Z Deep Learning and Its Applications in Biomedicine. Genomics Proteomics Bioinformatics 2018, 16 (1), 17–32. 10.1016/j.gpb.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Sheridan RP Time-Split Cross-Validation as a Method for Estimating the Goodness of Prospective Prediction. J. Chem. Inf. Model 2013, 53 (4), 783–790. 10.1021/ci400084k. [DOI] [PubMed] [Google Scholar]
- (112).Seltzer ML; Droppo J Multi-Task Learning in Deep Neural Networks for Improved Phoneme Recognition. In 2013 IEEE International Conference on Acoustics, Speech and Signal Processing; 2013; pp 6965–6969. 10.1109/ICASSP.2013.6639012. [DOI] [Google Scholar]
- (113).Zhang Y; Yang Q A Survey on Multi-Task Learning. 2017.
- (114).Yuan H; Paskov I; Paskov H; González AJ; Leslie CS Multitask Learning Improves Prediction of Cancer Drug Sensitivity. Sci Rep 2016, 6 (1), 1–11. 10.1038/srep31619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Cai C; Wang S; Xu Y; Zhang W; Tang K; Ouyang Q; Lai L; Pei J Transfer Learning for Drug Discovery. J. Med. Chem 2020, 63 (16), 8683–8694. 10.1021/acs.jmedchem.9b02147. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Distribution of the DLCA prediction results (R2) over AD cut-offs and coverage values. The error bar represents the standard deviation of the average performance over five-folds. Figure S2. Average performance (a) RMSE, (b) R2 of all 59 endpoints for each approach over five-fold cross-validation based on training and test data generated using random splitting. The error bar represents the standard error mean of the average performance over five-folds.
S1. Provides information on the number of measurements across each endpoint extracted from the ChemIDPlus portal18. It also summarizes the extent of data overlap between the endpoints modeled in this study in comparison the study published by Sosnin et al.49 S2.The network architecture for MT-DNN, ST-DNN and DLCA and the list of hyperparameters optimized for the MT-DNN. S3. Provides information on the proportion of missing data across different endpoints. S4. Model statistics (5-fold cross-validation results) of multi-task regression models developed in this study. S5. Statistical analysis of prediction results using Wilcoxon paired singed-rank non-parametric statistical test. S6. Average RMSE of prediction across 5 folds for all endpoints by different models. S7. RMSE comparison of the overlapping endpoints for the models developed in this study and study published by Sosnin et al.49