

Abstract
Thienopyrimidines are widely represented in the literature, mainly due to their structural relationship with purine base such as adenine and guanine. This current review presents three isomers—thieno[2,3-d]pyrimidines, thieno[3,2-d]pyrimidines and thieno[3,4-d]pyrimidines—and their anti-infective properties. Broad-spectrum thienopyrimidines with biological properties such as antibacterial, antifungal, antiparasitic and antiviral inspired us to analyze and compile their structure–activity relationship (SAR) and classify their synthetic pathways. This review explains the main access route to synthesize thienopyrimidines from thiophene derivatives or from pyrimidine analogs. In addition, SAR study and promising anti-infective activity of these scaffolds are summarized in figures and explanatory diagrams. Ligand–receptor interactions were modeled when the biological target was identified and the crystal structure was solved.
Keywords: thienopyrimidine, antibacterial, antifungal, antiparasitic, antiviral
1. Introduction
In recent years, thieno-fused derivatives are of growing interest and are found in many original bioactive molecules [1,2], even if the thiophene ring is known to potentially generate reactive metabolites [3]. Among thieno-fused derivatives, thienopyrimidines have been widely studied in the literature, probably due to their structural relationship with purine bases and their easy synthetic access. Considering the fusion between pyrimidine and thiophene rings, three different thienopyrimidines can be obtained, namely thieno[2,3-d]pyrimidines, thieno[3,2-d]pyrimidines and thieno[3,4-d]pyrimidines (Figure 1). All three have been studied and many derivatives have shown a large range of biological activities, such as anticancer, antioxidant, and central nervous system (CNS) protection. Some of them are still in clinical trials [2], while others have even reached the market (e.g., Relugolix, a gonadotropin-releasing hormone (GnRH) receptor antagonist, Figure 1).
Figure 1.

Structure of thienopyrimidines and of Relugolix (the thienopyrimidine scaffold is highlighted in blue).
In a recent review, Ali et al. summed up the biological activities of the thieno[2,3-d]pyrimidine scaffold until the end of 2018, with a particular attention provided onto their anticancer activities [2]. Due to our interest in the development of new anti-infective compounds [4,5,6,7,8,9], the objective of the present review is to provide an overview of the access routes to thienopyrimidine derivatives and to discuss the significance of this scaffold for the discovery of anti-infective drugs. In this review, we have collected all references until September 2021 involving the three isomers presented above with anti-infective properties. Only compounds with quite similar or higher activities compared to the selected reference drugs are presented.
2. Synthesis of Thienopyrimidines
Different synthetic pathways involving the construction of the pyrimidine or the thiophene ring were reported in the literature to access polysubstituted thienopyrimidines. In these approaches, the synthetic strategies mostly involved the synthesis of a thienopyrimidin-4-one derivative, where position 4 could be modified via further functionalization.
2.1. Synthesis from Thiophene Derivatives
Due to the high diversity of supplies, the reaction between an aminothiophene derivative bearing an electrophilic center (ester or nitrile) and a carbonyl or an amine reactant is probably the easiest way for produce thienopyrimidin-4-one derivatives. The leading routes to afford thienopyrimidines from aminothiophene derivatives are described in Scheme 1.
Scheme 1.

Main synthetic pathways to produce thienopyrimidin-4-ones from thiophene derivatives.
2.1.1. Cyclization with Carbonyl Reactants
The most efficient chemical approach to access 2- and 3-unsubstituted thieno[2,3-d]pyrimidin-4(3H)-ones involved a condensation reaction between an aminothiophene substrate and formamide. Thus, compounds 1a–e treated with an excess of formamide at high temperature led to compounds 2a–e with good yields (76 to 97%), except for compound 1e for which the methoxy group in R3 decreased the reaction yields compared to the ethoxy group (1a) (Scheme 2) [10,11,12,13,14].
Scheme 2.

Access to 2- and 3-unsubstituted thieno[2,3-d]pyrimidin-4-one derivatives (Me = methyl, Et = ethyl, and Ph = phenyl).
In contrast, mild conditions were sufficient to perform cyclization reaction with formamide to synthesize the thieno[3,2-d]pyrimidin-4(3H)-one isomers 4a–b with good yields (60 to 65%, Scheme 3) [15].
Scheme 3.

Synthesis of 3-unsubstituted thieno[3,2-d]pyrimidines 4a–b.
Woodring et al. presented a variant of this process that also involved formamide in combination with ammonium formate [14]. Cyclization of the thiophene intermediate 5 at 150 °C led to the unsubstituted thieno[3,2-d]pyrimidin-4-one 6 with a 56% yield (Scheme 4).
Scheme 4.

Synthetic route to unsubstituted thieno[3,2-d]pyrimidin-4-one 6.
In addition, reaction of 2-amino-3-cyanothiophene derivatives with formic acid could also be considered to access 2- and 3-unsubstituted thieno[2,3-d]pyrimidin-4-ones [13]. In such approach, the cyano group is firstly converted into its corresponding primary amide, which could then be cyclized in the presence of formic acid. Kanawade et al. used such an approach to prepare thienopyrimidinone 8a from 2-amino-3,5-dicyanothiophene 7a (Scheme 5). Replacing formic acid by formamide led to the formation of the 4-amino analogue, as reported by Aly et al. [16]. Thus, cyclocondensation involving 7b and formamide occurred under reflux to afford the expected 8b with a 83% yield (Scheme 5).
Scheme 5.

Access to thieno[2,3-d]pyrimidine derivatives from 2-amino-3-cyanothiophene derivatives.
Cyclocondensation of thiophene carboxamide 9 in the presence of sodium hydroxide was used to synthesize thieno[3,4-d]pyrimidin-4(3H)-one 10 (Scheme 6). The expected molecule was isolated with a moderate yield (40%) after a 1 h reaction in refluxing methanol.
Scheme 6.

Synthesis of 2-methyl-thieno[3,4-d]pyrimidin-4(3H)-one 10.
Using a similar approach, but with a nitrile group as the precursor of the primary amide, Desroches et al. synthesized 2-methyl- and 2-trichloromethyl-thieno[2,3-d]pyrimidin-4(3H)-ones 12 and 13, respectively (Scheme 7) [17]. Thus, treatment of 3-cyanothiophene acetamide 11a with hydrogen peroxide in alkaline medium (NaOH) afforded 2-methyl-thieno[2,3-d]pyrimidin-4(3H)-one 12 with a 72% yield. Using 3-cyanothiophene trichloroacetamide as a substrate and phosphoric acid in polyphosphoric acid triggered the cyclocondensation reaction and the formation of the 2-trichloromethyl-thieno[2,3-d]pyrimidin-4(3H)-one 13 with good yields (90%).
Scheme 7.

Synthesis of thieno[2,3-d]pyrimidin-4(3H)-ones substituted in position 2.
2.1.2. Cyclization with Nitrile Reactants
Various pathways exploiting nitrile condensation were reported in the literature to produce thieno-fused analogues. De Schutter et al. used a synthetic route involving a thiophene amino ester treated in strongly acidic conditions by a cyanoalkyl derivative at 90 °C (Scheme 8) [18]. Thieno[2,3-d]pyrimidin-4(3H)-ones 16c, substituted in positions 2, 5, and 6 were then obtained in 1,4-dioxane in moderate to good yields (50 to 90%). In addition, Mavrora et al. used the same synthetic pathway and obtained chloroethyl derivatives 16a–b with good yields (Scheme 8) after nitrile cyclocondensation at room temperature [19]. Likewise, thieno[3,2-d]pyrimidinones 15 substituted at position 2 were prepared from cyclization of the starting thiophene with the appropriate cyanoalkyl in acidic conditions at 90 °C in 1,4-dioxane (Scheme 8) [18]. To introduce a trichloromethyl group at position 2 of the thieno[3,2-d]pyrimidine core, Desroches et al. used trichloroacetonitrile in acetic acid, saturated with HCl gas, to afford 2-trichloromethyl-thieno[3,2-d]pyrimidine 17 with a 63% yield (Scheme 8) [17].
Scheme 8.

Synthesis of 2-substituted thienopyrimidin-4-ones using nitrile reactants.
Using the same strategy, Kim et al. introduced a chloromethyl group at position 2 of thieno[3,2-d]pyrimidinones after slight modifications of the reaction conditions [20]. Formation of the thieno-fused core occurred with the cyclocondensation of malononitrile with 2-methyl-3-aminothiophene carboxylate under acidic conditions and mild heating to offer 18 with high yields (Scheme 9).
Scheme 9.

Synthesis of 2-chloromethyl-thieno[3,2-d]pyrimidinone 18.
Slavinski et al. presented another synthetic pathway to introduce a sulfonamide group at position 2, using sulfonyl cyanamide potassium salts 19 [21]. Acidification of the reaction with boiling glacial acetic acid led to cyclization and afforded 2-sulfonamide-thieno[3,2-d]pyrimidinone derivatives 20 with low yields (20–34%, Scheme 10).
Scheme 10.

Formation of 2-sulfonamide-thieno[3,2-d]pyrimidinones 20.
2.1.3. Synthesis from (Thio)urea Reagents, Iso(Thio)cyanate or (Thio)cyanate Derivatives
An easy way to access thienopyrimidin-2,4-dione or 2-thioxo-thienopyrimidin-4-one derivatives consisted of cyclocondensation of the appropriate ethyl aminothiophene-carboxylate with potassium (thio)cyanate in an acidic medium. Patel et al. obtained 2-thioxo-thieno[2,3-d]pyrimidin-4-one 22a with a 58% yield, using hydrochloric acid in refluxing 1,4-dioxane (Scheme 11) [22], whereas Temburkinar et al. and other groups [23,24,25] used potassium cyanate in acetic acid to obtain thieno[3,2-d]pyrimidin-2,4-dione 21a with 71 to a 88% yield.
Scheme 11.

Synthesis of 2-thioxo-thieno[2,3-d]pyrimidin-4-one 22a and thieno[3,2-d]pyrimidin-2,4-dione 21a.
Another way to access such compounds was to condensate the starting aminothiophene with urea or thiourea, followed by cyclization to afford thienopyrimidinone compounds 21 or 22. Ortikov and Prabhakar teams used such conditions to synthesize 2-thioxo-thieno[2,3-d]pyrimidin-4-one 22b and thieno[2,3-d]pyrimidine-2,4-diones 22c (Scheme 12) with good yields (72–91%) [11,26,27]. Condensation and cyclization only occurred at very high temperatures after 2 or 3 h of heating without solvent. Thieno[3,2-d]pyrimidin-2,4-one 21b could be synthesized under these conditions, whereas the synthesis of 2-thioxo-thieno[3,2-d]pyrimidin-4-ones 21c required the use of N,N-dimethylformamide (DMF) as a solvent (Scheme 12) [28,29].
Scheme 12.

Formation of 2-thioxo-thienopyrimidin-4-ones and thienopyrimidine-2,4-diones using (thio)urea.
Kankanala et al. used a common synthetic pathway to access 3-hydroxythieno[2,3-d]pyrimidin-2,4-diones and thieno[3,2-d]pyrimidin-2,4-diones [30] bearing various groups in α and β positions of the sulfur atom. Firstly, the aminothiophene reacted with 1,1′-carbonyldiimidazole (CDI) to afford the imidazole-carboxamide intermediate after 2 h in refluxing toluene (Scheme 13). Secondly, the substitution of the imidazole group by protected hydroxylamine generated the hydroxyurea intermediate. Then, a basic treatment deprotonated hydroxyurea to allow cyclization. Afterward, deprotection of the hydroxyurea led to the final compounds 23 with correct to good yields (40–85%).
Scheme 13.

Synthetic pathway to afford 3-hydroxythienopyrimidin-2,4-diones 23.
To introduce more chemical diversity at position 3, a convenient synthetic route described by Abu-Hashem et al. involved nucleophilic attack of an aminothiophene derivative on an isocyanate or thioisocyanate in the presence of a catalytic amount of triethylamine in refluxing 1,4-dioxane (Scheme 14A) [31]. The (thio)ureidothiophene intermediate 24 or 25 was then isolated on average with good yields (60 and 70%). Thereafter, basic treatment of 24 or 25 with sodium ethoxide in refluxing ethanol led to thieno-fused derivatives 26 and 27 with good yields (70% and 75%) after 8 h. Dewal et al. obtained similar results using sodium methoxide under refluxing methanol to prepare trisubstituted thieno[2,3-d]pyrimidin-2,4-dione derivatives 26 with 88–90% yields [32]. In addition, Abu-Hashem et al. reported a one-pot reaction with phenylisothiocyanate and sodium hydroxide as a base, in refluxing ethanol for 6 h [31]. Both the two-step procedure and the one-pot reaction offered 27a with a 70% yield (Scheme 14A). Furthermore, the use of potassium carbonate in refluxing acetonitrile led to the 2-mercapto-thieno[2,3-d]pyrimidin-4-one analogues 28b–c in even higher yields (78%) [12,33]. In a similar way, 3-ethyl-2-thioxo-thieno[3,2-d]pyrimidin-4-one 27b was also accessible via the cyclization of 2-methyl-3-aminothiophene carboxylate with ethylisothiocyanate in refluxing pyridine [34]. In addition, 6-bromothieno[3,2-d]pyrimidin-2,4-diol 30 was synthesized in milder conditions with potassium tert-butoxide in DMF at room temperature and obtained it with a quantitative yield (Scheme 14B) [35]. It was then possible to introduce further chemical diversity in positions 2, 4, and 6, starting from this bicyclic product.
Scheme 14.

(A). Synthesis of 3-substituted 2-thioxo-thienopyrimidin-4-ones or thienopyrimidine-2,4-diones 26–28. (B). Synthesis of 6-bromothieno[3,2-d]pyrimidine-2,4-diol 30.
Alternately, Cohen et al. suggested an original synthetic pathway to obtain thieno[3,2-d]pyrimidin-4(3H)-one derivatives 34, substituted in position 2 by an amino group [36]. This one-pot procedure involved first the condensation of the starting material with ethoxycarbonyl isothiocyanate in DMF to generate the thiourea carbamate intermediate 32, that was not isolated (Scheme 15). Afterward, a primary alkylamine reacted with this species, previously mixed with 1-ethyl-3-(3′-dimethylaminopropyl)carbodiimide (EDCI.HCl) and triethylamine. Guanidine intermediate 33 was observed but was not isolated. Then, this intermediate cyclized at 170 °C to afford thieno-fused derivatives 34 with 42 to 70% yields depending on the substituents.
Scheme 15.

Synthetic pathway purposed by Cohen et al. [36].
2.1.4. Synthesis via a Tetrazole Intermediate
To generate thieno[2,3-d]pyrimidines substituted in positions 2 and 3 by an amino group, Abu-Hashem et al. purposed an access route via a tetrazole intermediate (Scheme 16) [31]. Firstly, the tetrazole ring was formed by treating 35 with triethyl orthoformate and sodium azide to generate 36 with good yields (70%). Then, refluxing 36 in the presence of a large excess of hydrazine hydrate led to two consecutive hydrazide intermediates 37 and 38. Intramolecular cyclization of 38 afforded 39 with good yields (75%).
Scheme 16.

Synthesis of 2,3-diaminothieno[2,3-d]pyrimidine 39.
2.1.5. Cyclization with Amine/Hydrazine Derivatives
A more common way to access 3-amino-thieno[2,3-d]pyrimidin-4-ones consisted of the condensation and cyclization between a thiophene derivative and hydrazine monohydrate in refluxing ethanol. Using this strategy, several groups reported the synthesis of compounds 42a–b with moderate to good yields (Scheme 17) [12,37]. Aly et al. employed the same reaction conditions to generate 3-amino-thieno[2,3-d]pyrimidin-4-one 42c. Only the starting thiophene was different and achieved cyclocondensation with good yields (80%).
Scheme 17.

Synthesis of 3-amino-thienopyrimidin-4-ones 42.
To introduce chemical diversity at position 3, a similar route was followed by Habib et al. using various primary amines to synthesize a set of 3-substituted thieno[2,3-d]pyrimidinone derivatives 43 [12]. Firstly, the 2-aminothiophene 1c reacted with triethyl orthoformate under reflux to prepare the imino intermediate, which was not isolated (Scheme 18). Then, the appropriate amine was added to allow cyclization and obtain 3-substituted thienopyrimidinone derivatives 43 with good yields (79–85%).
Scheme 18.

Access route to synthesize 3-substituted thieno[2,3-d]pyrimidin-4-ones 43.
Finally, condensation of ammonia with N-acylaminothiophenes 44 allowed access to 3-unsubstituted thieno[2,3-d]pyrimidin-4-ones 45 [15,17]. The first synthetic route involved 25% ammonia heated at 105 °C in a sealed vial to obtain thieno[3,2-d]pyrimidin-4-one 46a after 3 h, with a 63% yield (Scheme 19). In contrast, using milder conditions with 30% ammonia at room temperature for 6 to 8 h led generally to lower yields (28–60%). Moreover, it has been observed by Desroches et al. that this method was not efficient when R = CCl3 (compound 45e) [17]. Indeed, with this substrate, cyclization in the presence of 25% ammonium hydroxide in a sealed vial failed.
Scheme 19.

Synthesis of 3-unsubstituted-thienopyrimidin-4-ones 45 (Pr = propyl).
2.2. Synthesis of Thienopyrimidines from Pyrimidine Derivatives
2.2.1. Synthesis from the Thorpe-Ziegler Reaction
One of the possibilities to shape the thieno-fused ring from pyrimidine derivatives is the Thorpe-Ziegler cyclization. A six-membered ring bearing a mercaptocarbonitrile group was the starting point to synthesize thienopyrimidines (Scheme 20). After substitution of alkyl chloroacetate by the sulfhydryl group (compound 47), and subsequent deprotonation, cyclization can occur in basic conditions. In such a way, Abdel Hamid et al. reported the synthesis of compound 48 with a 71% yield [38].
Scheme 20.

Synthesis of thienopyrimidin-4-one 48 via a Thorpe-Ziegler cyclization.
A variant of the previous approach was purposed by Ali and Saleh for the synthesis of 2-thioxo-1,2,3,4-tetrahydrothieno[3,4-d]pyrimidine 52 [39]. First, thiobarbituric acid 49 was deprotonated in α-position of the two carbonyl groups at room temperature (Scheme 21). Then, nucleophilic substitution on phenyl isothiocyanate led to the ketene aminothioacetal 50. Thereafter, the addition of alkyl bromoacetate allowed cyclocondensation of 51 in basic conditions. The final product 52 was obtained with good yields (74%).
Scheme 21.

Synthesis of 2-thioxo-1,2,3,4-tetrahydro thieno[3,4-d]pyrimidin-4-one 52.
2.2.2. Synthesis from the Gewald Reaction
The Gewald reaction is a versatile reaction to access 2-aminothiophene derivatives involving one-pot cyclocondensation of ketones or aldehydes with activated nitrile derivatives and elemental sulfur. Using thiobarbituric acid 49 as the starting ketone, 2-thioxo-6-aminothieno[3,2-d]pyrimidin-4-one derivatives could be easily accessible. Treatment of 49 with piperidine in the presence of the appropriate alkyl cyanide led to the aminothieno-fused derivatives 53a and 53b with good yields (Scheme 22) [39].
Scheme 22.

Synthesis of 2-thioxo-thieno[3,2-d]pyrimidines 53 by the Gewald reaction.
As shown in the previous examples, many access routes to these compounds are possible and allow to easily prepare a wide range of polysubstituted thienopyrimidines. Therefore, these compounds have been included in many biological studies. More particularly, their antiparasitic, antibacterial, antifungal and antiviral activities have been studied.
3. Antiparasitic Activity of Thienopyrimidines
3.1. Antimalarial Activity
Malaria is a parasitic disease caused by protozoan parasites belonging to the Plasmodium genus. Five species are known to infect humans, namely P. falciparum, P. vivax, P. malaria, P. ovale, and P. knowlesi. The female Anopheles mosquito acts as the transmission vector of the infection. P. falciparum is the most virulent species in humans while P. falciparum and P. vivax represent the greatest threat [40]. In 2019, P. falciparum generated most malaria cases in Africa, South-East Asia, Eastern Mediterranean, and Western Pacific while P. vivax was the most prevalent in the Americas. In 2019, the number of cases was estimated at 229 million in the world and 409,000 deaths due to malaria were identified by the World Health Organization (WHO) [41]. The resurgence of resistance to current antimalarial drugs such as artemisinin derivatives [42] represents a major health issue. Therefore, the development of novel antimalarial drugs remains an urgent need [43].
3.1.1. Thieno[2,3-d]pyrimidine Derivatives with Antiplasmodial Activity
Zhu et al. elaborated a small library of thieno[2,3-d]pyrimidine derivatives as falcipain-2 inhibitors [44]. The cysteine protease falcipain-2 (FP-2) of P. falciparum is a major cysteine protease and an essential hemoglobinase of erythrocytic trophozoites [45]. Inhibition of FP-2 blocks hemoglobin hydrolysis and stops the development of the parasite. Therefore, the FP-2 enzyme would represent an attractive target for antimalarial drug development [46]. Enzyme inhibition assays showed inhibitory potential for the whole series. The inhibition rate of these derivatives ranges between 53.0 and 94.3% at 10 μM (Table 1). Falcipain-2 inhibitors described in the literature are peptidic analogues that exhibit nanomolar IC50 values [47]. In contrast, Zhu et al. derivatives demonstrated moderate activity with micromolar IC50 values. IC50 values of these compounds against FP-2 showed that allyl, cyclohexyl, para- or meta-phenyl groups at position 3 were tolerated (54a to 54d, IC50 = 1.46 to 2.81 μM). Para-chloro-phenyl and benzyl groups led to a slight loss of potency (54e and 54f, IC50 = 4.30 and 5.74 μM, respectively). Replacing the phenyl group on the thiophene ring of 54a by a meta-substituted phenyl group led to a loss of potency (54h), whereas a para-substituted phenyl group maintained activity (54g). N-substitution of the amide at position 2 globally maintained the inhibitory activity of compounds 55a to 55f with IC50 values from 2.49 to 6.63 μM.
Table 1.
The inhibitory activity of thienopyrimidine derivatives 54a–f on FP-2.
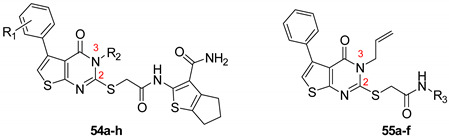
| |||||
|---|---|---|---|---|---|
| Cpd | R1 | R2 | R3 | Inhibitory Activity Against FP-2 | |
| Inhibition Rate at 10 μM (%) | IC50 (μM) | ||||
| 54a | H | Allyl | - | 88.7 | 2.81 |
| 54b | H | Cyclohexyl | - | 92.7 | 1.46 |
| 54c | H | 3-F-Ph | - | 79.0 | 2.05 |
| 54d | H | 4-NO2-Ph | - | 85.4 | 2.77 |
| 54e | H | 4-Cl-Ph | - | 84.7 | 4.30 |
| 54f | H | Benzyl | - | 90.6 | 5.74 |
| 54g | 4-Cl | Allyl | - | 85.7 | 2.95 |
| 54h | 3-NO2 | Allyl | - | 53.0 | 11.8 |
| 55a | - | - |
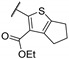
|
93.3 | 6.63 |
| 55b | - | - |
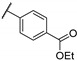
|
94.3 | 5.70 |
| 55c | - | - |
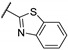
|
90.3 | 3.31 |
| 55d | - | - |
|
93.2 | 2.49 |
| 55e | - | - |
|
72.0 | 5.58 |
| 55f | - | - |
|
92.0 | 5.43 |
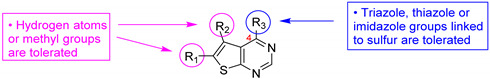
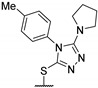
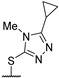
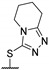
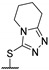
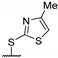
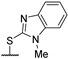
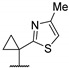
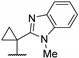
Then, a series of thieno[2,3-d]pyrimidines was discovered by Edlin and Barrows’ teams as potent antimalarial agents with micromolar or submicromolar activities [48,49]. All derivatives synthesized were evaluated in vitro against P. falciparum NF54 or P. falciparum 3D7 (clone of NF54 and chloroquine-sensitive) strains (Table 2). SAR study demonstrated that bulky groups supported by the triatrazole ring at position 4 increased antiplasmodial activity (compounds 56a and 56c compared to 56b). However, the introduction of methyl groups at R1 and R2 slightly decreased activity (56c vs. 56d). Thereafter, a thiazole group linked with thioether at position 4 led to strong antiplasmodial activity (56e, EC50 = 34 nM on P. falciparum 3D7 strains). In contrast, a benzimidazole group reduced activity (56f, EC50 = 0.191 µM). Bioisosteres of 56e and 56f (56g and 56h) were synthesized and evaluated to avoid the S-oxidation metabolism of the sulfur at position 4. Unfortunately, 56g and 56h were inactive against P. falciparum 3D7 strains. Only cytotoxicity of compounds 56a to 56d was determined (EC50 < 4 µM on HEK-293 mammalian cell lines). Afterward, 56c was assessed in vivo on mice infected by Plasmodium berghei at 50 mg/kg once a day for 4 days. At the end of the experiment, the parasitemia decreased by 34%, and no gain in survival days was observed compared to the untreated mice.
Table 2.
Antiplasmodial activity of thieno[2,3-d]pyrimidines 56a–h.
Additional studies carried out with compound 56e demonstrated that this family of compounds targets coenzyme A (CoA) synthetic pathway. CoA is involved in metabolic functions and is necessary for parasite survival. CoA operates during the asexual and sexual stages of P. falciparum [50,51,52]. Compound 56e exhibited strong antiplasmodial activity on both the asexual and the sexual stage of P. falciparum (EC50s = 0.06 to 0.120 µM). 56e could block parasite transmission. In addition, no cytotoxicity on HEK-293 cell lines was observed (EC50 = 40 µM) [50,51]. Then, Weidner et al. further studied this series of 4-thioether-thieno[2,3-d]pyrimidines [52]. To identify active compounds, a first screening was realized at 3 µM on the asexual erythrocytic stage of transgenic NF54-luc P. falciparum, using a luciferase-based viability assay. All derivatives presented in Table 3 decreased the viability of erythrocytic asexual stage P. falciparum NF54-luc parasites (compounds 56i to 56o). Then, the authors demonstrated that supplementation of CoA in the growing medium decreases the antiplasmodial activity of thienopyrimidines (Pf3D7 EC50 vs. Pf3D7 + 0.8 mM CoA EC50). These results corroborated the hypothesis that the inhibition of CoA synthesis was a mechanism associated with the antiplasmodial activity of this series. Lastly, all derivatives exhibited low to moderate cytotoxicity on THP-1 cells, a human monocytic cell line, and on HEK-293 cell line.
Table 3.
Biological evaluations of 4-thioether-thieno[2,3-d]pyrimidines 56i–o.
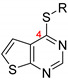
| ||||||
|---|---|---|---|---|---|---|
| Compound | R | % Inhibition, PfNF54-Luc at 3 µM |
Pf3D7 EC50 (µM) |
Pf3D7 + 0.8 mM CoA EC50 (µM) |
THP-1 EC50 (µM) |
HEK-293 EC50 (µM) |
| 56i |
|
80.8 ± 0.5 | 0.283 ± 0.073 | 4.61 ± 0.15 | 6.13 | >10 [48] |
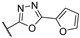
| 56j |
|
99.9 ± 0.0 | 0.0388 ± 0.0010 | 2.61 ± 0.42 | 27.5 | >10 |
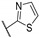
| 56k |
|
99.5 ± 0.5 | 0.0747 ± 0.021 | 2.58 ± 0.42 | 33.6 | >10 |
| 56l |
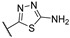
|
99.7 ± 0.1 | 0.0958 ± 0.0063 | 11.2 ± 2.6 | 11 | >4 |
| 56m |
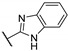
|
95.0 ± 1.1 | 0.0734 ± 0.0022 | 0.531 ± 0.007 | 16.5 | >10 |
| 56n |
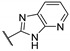
|
98.0 ± 0.2 | 0.149 ± 0.021 | 1.06 ± 0.03 | 28 | >20 |
| 56o |
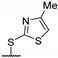
|
- | 0.0370 ± 0.0017 | 1.51 ± 0.19 | - | >40 |
| Chloroquine | - | - | 0.0195 ± 0.0034 | 0.0271 ± 0.0094 | - | >40 |
| Artemisinin | - | - | 0.00337 ± 0.00065 | 0.00490 ± 0.0011 | - | - |
3.1.2. Thieno[3,2-d]pyrimidine Derivatives with Antiplasmodial Activity
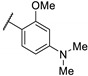
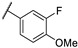
In 2006, Kikuchi et al. synthesized a bioisostere of febrifugine represented in Figure 2 [53]. Febrifugine is a quinazolinone alkaloid first isolated from the Chinese herb Dichroa febrifuga Lour. which has been used in traditional Chinese medicine for over 2000 years for the treatment of various diseases, including malaria. Its analogue exhibited a strong antiplasmodial activity against P. falciparum FCR-3 and K1 cell lines (chloroquine-sensitive or resistant strains). Unfortunately, this analogue was cytotoxic on mouse-L929 cells (EC50 = 0.563 μg/mL). In vivo, this thienopyrimidinone exhibited similar activity than chloroquine (ED50 = 2.95 and 2.53 mg/kg, respectively) and a moderate cytotoxicity (LD50 = 88 mg/kg).
Figure 2.

Structure and activity of a thienopyrimidinyl analogue of febrifugine.
A few years later, Cohen et al. reported the antimalarial activity of a series of thieno[3,2-d]pyrimidinone derivatives. A library of 120 derivatives was synthesized and tested in vitro on P. falciparum K1-resistant strain and HepG2 cell line [36]. SARs in this series revealed that a tert-butyl- or an isopropyl-amino group at position 2 was essential to obtain antimalarial activity (compared compounds 57a–b to compound 57c, Table 4), along with a phenyl ring at position 6 (compared compound 57d to 57e). Para-substitution of this phenyl ring increased activity on P. falciparum strains (compared compound 57a to 57d and 57f–g), but only with a methyl group or a chlorine atom (57a, 57h vs. 57i). To improve the aqueous solubility of these derivatives, various salts were synthesized. Hydrochloric salt 57j was identified as a lead compound with an antimalarial activity tenfold higher than chloroquine and with cytotoxicity like chloroquine. The advantage of compound 57j was its activity on the erythrocytic stage of P. falciparum K1 strain and the liver stage of P. yoelii strain (EC50 = 35 nM). A preliminary in vivo evaluation revealed that 57j reduced parasitemia by 45% compared to untreated infected mice, proving that its antiplasmodial activity was preserved in vivo. Bosson-Vanga et al. discovered 57j displayed activity on P. falciparum at the three stages of the parasite cycle (erythrocytic, hepatic and sexual stages) and reduced transmission of the parasite in a mouse model [9]. However, the original mechanism of action of these compounds remains to be elucidated.
Table 4.
SAR and antimalarial activity on P. falciparum resistant K1 strains and cytotoxicity on HepG2 cells of thieno[3,2-d]pyrimidin-4(3H)-one derivatives 57a–j.
| ||||
|---|---|---|---|---|
| Compound | R1 | R2 | Antiplasmodial Activity a on K1 EC50 (µM) |
Cytotoxicity a on HepG2 CC50 (µM) |
| 57a | 4-Me-Ph | NH-tBu | 0.2 ± 0.02 | 25.6 ± 3.1 |
| 57b | 4-Me-Ph | NH-iPr | 0.8 ± 0.5 | 49.4 ± 1 |
| 57c | 4-Me-Ph | NH-nPr | >5 b | >62.5 b |
| 57d | Ph | NH-tBu | 1 | 12.5 ± 2.5 |
| 57e | H | NH-tBu | >5 b | 8.4 ± 3.5 |
| 57f | 3-Me-Ph | NH-tBu | 3.6 | 14.1 ± 1.4 |
| 57g | 2-Me-Ph | NH-tBu | 1.7 | 4.0 ± 1.1 |
| 57h | 4-Cl-Ph | NH-tBu | 0.8 | 15.0 ± 2.7 |
| 57i | 4-F-Ph | NH-tBu | >5 b | 5.1 ± 1.0 |
| 57j | 4-Me-Ph | NH-tBu·HCl | 0.045 | 24.0 |
| Chloroquine c | - | - | 0.5 | 30 |
a The values are the means ± SD of three independent experiments. b No activity was observed at the highest concentration tested. c Antimalarial drug reference.
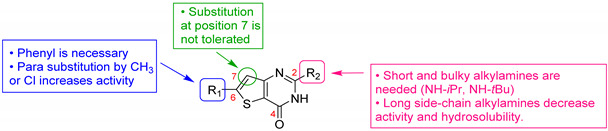
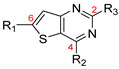
González Cabrera et al. were interested in similar derivatives but including an amine function at position 4 of the thienopyrimidine core [35,54]. A SAR study demonstrated the influence of various substituents at positions 2, 4, and 6 of the thieno[3,2-d]pyrimidine ring (Table 5). These derivatives displayed in vitro activity on both P. falciparum chloroquine-sensitive strains (NF54) and resistant strains to chloroquine, pyrimethamine, and proguanil (K1) with EC50 values in the submicromolar range (Table 5). At position 6, a phenyl group was essential for antimalarial activity (58a vs. 58b). Para- or meta-monosubstitution and meta-meta- or para-ortho-disubstitution of this phenyl ring by electron-withdrawing groups were tolerated (58c to 58h). Compounds 58c and 58f were 10-fold more potent than chloroquine, with EC50 = 19 nM and 17 nM vs. 194 nM, respectively. Morpholinophenyl derivative 58i displayed strong antimalarial activities against both strains (EC50 of 12 and 7 nM, respectively). Unfortunately, this compound showed poor in vitro microsomal stability after incubation with human liver microsomes (half-life = 15 min). The most stable compound 58d was chosen for in vivo studies on a P. berghei infected mouse model. Compound 58d reduced parasitemia by more than 99.8% when administered orally (50 mg/kg once daily for 4 days). This administration scheme led to a mean survival time of 23 days, which is comparable to chloroquine at 4 × 30 mg/kg. The major drawback of this series was the metabolic weakness due to the dealkylation of positions 2 and 4, along with the inhibition of hERG channels, which could cause cardiac toxicity [55]. The SAR study was extended and the metabolic stability of these thieno[3,2-d]pyrimidine derivatives was explored. Various aminoalkyl chains were introduced at position 4 (compounds 58j to 58p). These modifications led globally to a decrease in the activity, except when an aminopiperidine group or an alkylmorpholine group was introduced (compounds 58n to 58p). Interestingly, these latter compounds showed improved microsomal metabolic stability. Substitution of position 2 was also studied. Replacing the amino group with another substituent like a hydrogen or a chlorine atom, a methoxy, a morpholino or a phenylmethanesulfonyl group was not tolerated. In contrast, a benzylamino group (compound 58q) or a primary amine (compounds 58r to 58t) was well tolerated. Compound 58r presented good in vitro antimalarial activity and led to the N-methyl dealkylated as one of the main identified metabolites. Two new compounds, 58s and 58t, were identified as displaying a strong activity, 5- to 8-fold better than chloroquine on P. falciparum K1 and NF54 strains, together with a high in vitro microsomal stability. Unfortunately, these two thieno[3,2-d]pyrimidine derivatives exhibited significant affinity for hERG channels. The cytotoxicity of compounds 58n, 58o and 58t was measured against CHO and Vero cell lines and evidenced a strong in vitro cytotoxic effect (CC50s between 2.17 and 4.30 µM).
Table 5.
Antimalarial activity of thieno[3,2-d]pyrimidine derivatives 58a–t.
| Compound |
|
Activity on P. falciparum Strains EC50 (nM) a |
Degradation Half-Life (min) | Ref. | |||
|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | K1 | NF54 | |||
| 58a | Ph |
|
NHMe | 73 | 28 | 344.9 | [35] |
| 58b | Br |
|
NHMe | 973 | 636 | - | [35] |
| 58c | 4-CN-Ph |
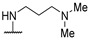
|
NHMe | 19 | 9 | 238 | [35] |
| 58d | 4-CF3-Ph |
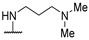
|
NHMe | 32 | 29 | 124 | [35] |
| 58e | 4-CF3O-Ph |
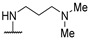
|
NHMe | 26 | 28 | >250 | [35] |
| 58f | 3-CN-Ph |
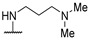
|
NHMe | 17 | 4 | - | [35] |
| 58g | 3-CF3-Ph |
|
NHMe | - | 13 | 31.1 | [35] |
| 58h | 2-Cl-4-CF3-Ph |
|
NHMe | 47 | 16 | 130 | [35] |
| 58i |
|
|
NHMe | 12 | 7 | 15 | [35] |
| 58j | 4-CF3-Ph |
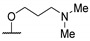
|
NHMe | >2436 | 1832 | - | [54] |
| 58k | 4-CF3-Ph |
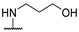
|
NHMe | 233 | 111 | - | [54] |
| 58l | 4-CF3-Ph |
|
NHMe | >2608 | >2608 | - | [54] |
| 58m | 4-CF3-Ph | NH2 | NHMe | - | 1158 | - | [54] |
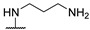
| 58n | 4-CF3-Ph |
|
NHMe | - | 42 | - | [54] |
| 58o | 4-CF3-Ph |
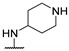
|
NHMe | 24 | 19 | - | [54] |
| 58p | 4-CF3-Ph |
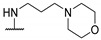
|
NHMe | - | 43 | 23.9 | [54] |
| 58q | 4-CF3-Ph |
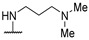
|
NH-benzyl | - | 42 | - | [54] |
| 58r | 4-CF3-Ph |
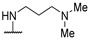
|
NH2 | - | 58 | 104 | [54] |
| 58s | 4-CF3-Ph |
|
NH2 | 24 | 25 | >150 | [54] |
| 58t | 4-CF3-Ph |
|
NH2 | 33 | 20 | >150 | [54] |
| Chloroquine b | 194 | 16 | - | [54] | |||
| Artesunate b | 3 | 4 | - | [54] | |||
a Mean from n values of ≥2 independent experiments with multi-drug-resistant (K1) and sensitive (NF54) strains of P. falciparum. b Antimalarial drug references.
Van der Watt et al. also highlighted the antiplasmodial activity of 2,4-diaminothienopyrimidine derivatives on the asexual blood stage of P. falciparum with nanomolar EC50s [56]. Twelve 2,4-diaminothienopyrimidines were studied on the asexual and sexual stages. Overall, these compounds presented a better activity on the asexual stage than on the sexual stage (gametocytes) of the parasite. These results are quite mitigated because the ultimate goal of antimalarial chemotherapy is to act on both the asexual and the sexual stage of the parasite, to eradicate its development in humans and block the transmission of the disease. Nevertheless, these thieno[3,2-d]pyrimidine derivatives were still slightly effective against early and late-stage gametocytes. SARs were inferred from the chemical features and biological activities in this series. A diaminothienopyrimidine scaffold, a N-methylation at position 2, and a para-phenyl substitution with lipophilic groups at position 6 were identified as important criteria for gametocidal activity (Figure 3).
Figure 3.

SARs of thieno[3,2-d]pyrimidine compounds according to González Cabrera and Van der Watt works.
Woodring et al. carried out a large repositioning campaign of compounds, by screening them against several protozoan parasites [14,57]. Some of them incorporating a thienopyrimidine core and a fluorobenzylaminophenoxy group were identified as potential antimalarial drugs. These derivatives presented EC50 values in the order of the micro- or submicromolar range on drug-sensitive D6 P. falciparum strains (Table 6). SAR studies revealed that morpholinophenyl (compounds 59a and 59b) and alkynyl substituents (59c and 59d) at position 2 led to very good antimalarial activity. A lead compound 59a was identified as displaying a strong in vitro antimalarial potency against P. falciparum sensitive D6 strains (EC50 = 27 nM).
Table 6.
SAR studies and inhibition profile of thienopyrimidine derivatives against P. falciparum D6.
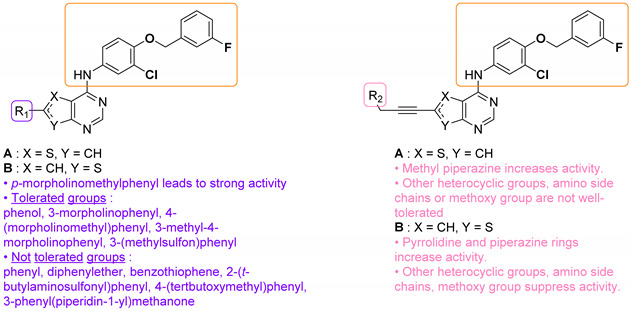
| |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | P. falciparum D6 EC50 (µM) (r2) a | Ref. | |
| 59a | A |
|
- | 0.027 (0.97) | [14] |
| 59b | B | 0.089 (0.81) | |||
| 59c | A | - |
|
0.64 (0.99) | [57] |
| 59d | B | 0.26 (0.94) | |||
a Compounds screened against P. falciparum (D6 strain) either in duplicate or quadruplicate and had r2 values >0.90 except for 59b (r2 = 0.81).
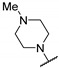
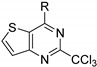
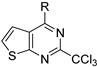
Further works, carried out by Desroches et al., also demonstrated that compounds with a thienopyrimidine core, in [3,2-d] or [2,3-d] series, exhibited antimalarial activities on K1 P. falciparum resistant strains with EC50 values in the submicromolar range [17]. Substitution of positions 2 and 4 was explored exclusively. SAR studies at position 2 were performed on the pyrimidine core. Various substituents were introduced such as a hydrogen atom, a methyl, or halogenated methyl groups. The antiplasmodial activity was maintained only in the presence of the trichloromethyl group. Despite the promising in vitro activity profile of these compounds, consequent cytotoxicity on HepG2 human hepatic cell line and low selectivity were the main drawbacks of these series (Table 7). Interestingly, the SAR study evidenced no significant difference between the two thienopyrimidine series.
Table 7.
SAR data from Desroches et al.’s studies.
| R |
|
|
|
|---|---|---|---|
|
K1 P. falciparum IC50 (µM) | 0.6 | 0.5 |
| HepG2 CC50 (µM) | 3.2 | 6.2 | |
| SIa | 5.3 | 12.4 | |
|
K1 P. falciparum IC50 (µM) | 0.9 | 0.6 |
| HepG2 CC50 (µM) | 0.7 | 6.2 | |
| SI a | 0.8 | 6.7 | |
|
K1 P. falciparum IC50 (µM) | 0.6 | - |
| HepG2 CC50 (µM) | 4.3 | - | |
| SI a | 7.2 | - | |
|
K1 P. falciparum IC50 (µM) | 0.4 | 0.8 |
| HepG2 CC50 (µM) | 6.9 | 6.2 | |
| SI a | 17.2 | 16.6 | |
| Doxycycline b | K1 P. falciparum IC50 (µM) | 6.0 | |
| HepG2 CC50 (µM) | 20 | ||
| SI a | 3.3 | ||
| Atovaquone b | K1 P. falciparum IC50 (µM) | 0.001 | |
| HepG2 CC50 (µM) | >15.6 | ||
| SIa | 15,600 | ||
| Chloroquine b | K1 P. falciparum IC50 (µM) | 0.6 | |
| HepG2 CC50 (µM) | 30 | ||
| SI a | 50 | ||
| Doxorubicine | HepG2 CC50 (µM) | 0.2 | |
a SI: Selectivity Index as a ratio of Hep G2 CC50/K1 EC50. b Doxycycline, atovaquone, and chloroquine were used as antimalarial reference drugs.
Through the various examples presented above, the thienopyrimidine core proved to be attractive to discover new antimalarial agents. In these different studies, thieno[2,3-d]pyrimidines and thieno[3,2-d]pyrimidines were considered and modulations at positions 2, 3, 4, 5, 6 and 7 of the thienopyrimidine core have been carried out to reach antiplasmodial activities in the order of the micro or submicromolar range. The most promising compounds seem to be thieno[3,2-d]pyrimidines substituted in positions 2, 4, and 6. Their weak point remains their significant cytotoxicity on HepG2 and CHO cells. In addition, for most of the examples presented, no target was identified except for compounds 54 and 55, which inhibited the cysteine protease falcipain-2 (FP-2). Further works need to be carried out to identify the plasmodial target(s) involved in the mechanism of other compounds.
3.2. Anti-Trypanosomatid Activity
Trypanosomatid parasites are the causative agents of several neglected tropical diseases [58]. Among them, Trypanosoma brucei, Trypanosoma cruzi, and Leishmania sp. are responsible for human African trypanosomiasis (HAT), Chagas disease, and leishmaniasis, respectively. Regarding leishmaniasis, L. donovani and L. infantum cause visceral leishmaniasis in humans, which is a mortal disease if untreated.
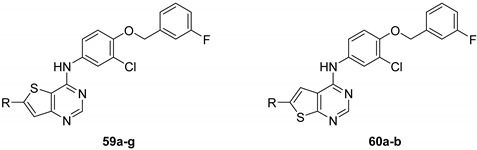
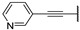
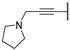
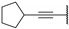
In 2015, after having tested thienopyrimidines on P. falciparum strains [14] (see previous section), Woodring et al. screened more than 35 of these compounds on L. major and identified several molecules with micromolar activity on the amastigote form and submicromolar activity on the promastigote form of the parasite (Table 8). To broaden the spectrum of anti-trypanosomatid activities, a new thienopyrimidine scaffold was designed. It was inspired by compound GW837016X [59] (compound 59a), which presented good activity on T. brucei strains with an EC50 = 0.26 μM. Compound 59b was the most potent molecule of this series against L. major promastigotes (EC50 = 0.22 µM) but displayed no activity on the amastigote form (EC50 > 15 µM), making it irrelevant for potential human use (Table 8). To explore their whole antitrypanosomatid potential, Woodring et al. also evaluated these compounds on T. brucei brucei strain 427 and T. cruzi amastigotes. A single compound, 59c, presented a strong activity against T. b. brucei, with submicromolar activity comparable to the reference drug suramin, and no cytotoxic effects toward the HepG2 cell line. 59d was also identified as a potent anti-T. cruzi hit compound, with the same in vitro activity as benznidazole. Three years later, new thieno[3,2-d]pyrimidine derivatives substituted by various alkynyl groups at position 6 were synthesized by the same team [57]. Among these compounds, 60b revealed a strong activity against T. cruzi amastigotes, like benznidazole. Only 59e presented a submicromolar EC50 value against L. major amastigote, but with an activity 10-fold lower than amphotericin B. Other compounds 59f, 59g and 60a displayed antitrypanosomal activity on T. b. brucei with submicromolar EC50s. Complementary experiments proved that 59a stopped trypanosome proliferation after G2 phase and before cytokinesis.
Table 8.
Antitrypanosomatid activity of thienopyrimidine derivatives 59a–g and 60a–b.
| |||||
|---|---|---|---|---|---|
| Cpd | R |
L. major Amastigotes EC50 (µM) (r2) a |
L. major Promastigotes EC50 (µM) (r2) a |
T. brucei brucei EC50 ± SEM (µM) |
T. cruzi Amastigotes EC50 ± SEM (µM) |
|
59a
(GW837016X) |
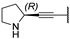
|
- | - | 0.26 | - |
| 59b |
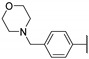
|
>15 | 0.22 (0.84) | 1.1 ± 0.0 | >50.0 |
| 59c |
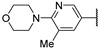
|
>3 | >3 | 0.084 ± 0.0 | 3.3 ± 1.2 |
| 59d |
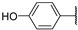
|
1.58 (0.83) | >20 | 2.2 ± 0.1 | 0.75 ± 0.02 |
| 59e |
|
0.38 (0.94) | - | 1.3 ± 0.3 | 12 ± 0.45 |
| 59f |
|
4.1 (0.77) | - | 0.28 ± 0.07 | 1.8 ± 0.17 |
| 59g |
|
>15 | - | 0.76 ± 0.07 | >50 |
| 60a |
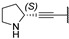
|
4.2 (0.94) | - | 0.22 ± 0.02 | 3.7 ± 0.23 |
| 60b |
|
9.1 (0.88) | - | 1.9 ± 0 | 0.61 ± 0.16 |
| Amphotericin B b | - | 0.035 (0.90) | - | - | - |
| Suramin b | - | - | - | 0.04 ± 0 | - |
| Benznidazole b | - | - | - | - | 0.79 ± 0.01 |
a Compounds screened against L. major amastigotes and promastigotes were tested in duplicate and had r2 values >0.75. b Drug references.
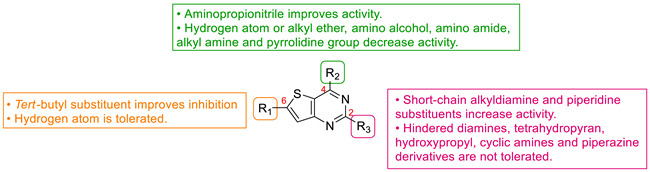
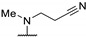
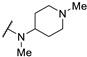
Five years later, Bell et al. developed novel thienopyrimidines as inhibitors of leishmanial N-myristoyltransferase (NMT) [60]. NMT plays a key role in the growth and development of eukaryotes by catalyzing the co-translational N-terminal myristoylation of several proteins. Bell et al. studied thienopyrimidine derivatives, substituted at positions 2, 4, and 6 (Table 9). Position 4 was functionalized by an aminopropionitrile group and modifications at other positions were explored. Compounds 61a and 61b were identified as potential selective inhibitors of NMT in L. donovani (IC50 = 0.34 µM and 0.15 µM, respectively) and L. major NMT (IC50 = 0.20 µM and 2.7 µM, respectively). Replacing the piperidine ring with a tetrahydropyran (61c), a hydroxypropyl (61d), a piperazine (61e–f), cyclic amines (61g), or a pyrrolidine-amine (61h) led to a loss of potency (IC50 from 7.7 µM to >100 µM). Slight modifications on this piperidine substituent at position 2 (61i–j) were tolerated, except when a ramified alkyl chain was introduced (compound 61k). Modifying the diamine chain of compound 61b caused a decrease in the inhibitory activity on L. donovani NMT (61l–n). Replacement of the aminopropionitrile group at position 4 by a hydrogen atom, alkyl ether (61p), amino alcohol (61q), amino amide (61r–s), alkyl amine (61t), and pyrrolidine group (61u) led to a drop in potency on L. donovani and L. major NMT. However, compound 61v, substituted by a pyrrolidine group at position 4 and an N-methylpiperidine group at position 2, exhibited a submicromolar activity against LdNMT and a strong selectivity for leishmanial NMT over the human isoform (IC50(HsNMT1)/IC50(LdNMT) > 660). Moreover, a tert-butyl group at position 6 (61w and 61x) improved the Leishmania NMT inhibition. Compound 61x exhibited the best IC50 value against L. donovani and L. major NMT (46 and 42 nM, respectively). However, the selectivity against the human form of the enzyme was lost. The co-crystallization of 12 derivatives complexed with L. major NMT and myristoyl-CoA highlighted the key inhibitor-enzyme interactions. Co-crystallization of compound 61x suggested that the aromatic system of the pyrimidine core was involved in π-π staking interactions with Tyr217. The lipophilicity of the rigid core and the specific geometry of the piperidine group at position 2 appeared to be crucial interaction parameters. Moreover, the basic center of this group established an ionic interaction with the carboxylate of the C-terminal Leu421 carboxylate. Finally, the nitrogen atom of the pyrrolidine at position 4 was also essential as it formed a hydrogen bond with the hydroxyl group of Tyr217 (Figure 4).
Table 9.
Thienopyrimidine derivatives as selective inhibitors of L. donovani and L. major NMT.
| ||||||
|---|---|---|---|---|---|---|
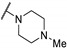
| Cpd | R1 | R2 | R3 | CMP Assay IC50 (μM) | ||
| LdNMT | LmNMT | HsNMT1 | ||||
| 61a | H |
|
|
0.34 | 0.20 | 5.7 |
| 61b | H |
|
0.15 | 2.7 | >100 | |
| 61c | H |
|
>100 | - | - | |
| 61d | H |
|
>100 | - | - | |
| 61e | H |
|
28 | - | - | |
| 61f | H |
|
30 | - | - | |
| 61g | H |
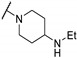
|
7.7 | - | 46 | |
| 61h | H |
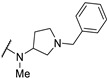
|
>100 | - | - | |
| 61i | H |
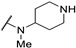
|
0.5 | - | 11 | |
| 61j | H |
|
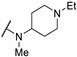
0.83 | 0.62 | 17 | |
| 61k | H |
|
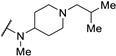
4.0 | - | >100 | |
| 61l | H |
|
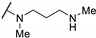
5.2 | - | - | |
| 61m | H |
|
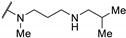
>100 | - | - | |
| 61n | H |
|
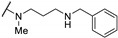
>100 | - | - | |
| 61o | H | H |
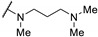
|
>100 | - | - |
| 61p | H |
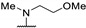
|
44 | - | - | |
| 61q | H |
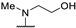
|
22 | 36 | - | |
| 61r | H |
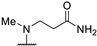
|
- | 13 | - | |
| 61s | H |
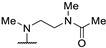
|
>100 | - | >100 | |
| 61t | H |
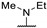
|
>100 | - | - | |
| 61u | H |
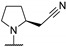
|
3.6 | - | 16 | |
| 61v | H |
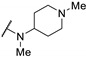
|
0.15 | - | >100 | |
| 61w | t-Bu |
|
|
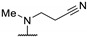
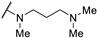
0.36 | 0.16 | - |
| 61x | t-Bu |
|
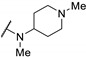
0.046 | 0.042 | 0.55 | |
Figure 4.

Co-crystallized structure of compound 61x and L. major NMT (PDB: 6QDD). (A)—Positioning of 61x and myristoyl-Co A in the pockets of NMT. The surface of the enzyme is represented in gray and the compounds are shown as stick representations. (B)—Key interactions of 61x into the active site of NMT. The enzyme is represented as pale cyan cartoon mode and the compounds are shown as stick representation. Key bonding interactions are indicated as yellow dotted lines.
3.3. Antihelminthic Activity
To our knowledge, only one study reported the activity of thienopyrimidine derivatives on helminths, and more specifically against Trichinella spiralis. Trichinellosis is caused by larva of these nematodes, which settles in the muscular tissues of the host. Humans get infected by this parasite after consumption of raw or inadequately cooked meat, containing encysted larvae.
Mavrova et al. synthesized and evaluated thieno[2,3-d]pyrimidine derivatives as antihelminthic agents against Trichinella spiralis [19]. The substitution of the alkyl chain in position 2 of the thienopyrimidine ring by a benzimidazole moiety was essential for the antihelminthic activity (Table 10). The most active compound 62a presented after 48h incubation a percentage of efficacy against T. spiralis larvae 5-fold better than albendazole, chosen by the authors as the reference drug. The addition of a sulfide group at position 2, as a link between the ethyl chain and the benzimidazole ring, was also tolerated (compounds 63a and 63c, 59.75% and 80.05% efficacy after 48 h incubation, respectively), except for 63c which was not active in vitro. Further experiments also demonstrated an in vivo antiprotozoal activity of these compounds against Lamblia muris.
Table 10.
Antihelminthic activity of thienopyrimidine derivatives against Trichinella spiralis.
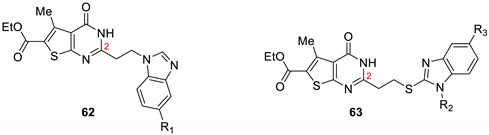
| |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | Efficacy (%) a after 24 h b 5 μg/mL |
Efficacy (%) after 48 h 5 μg/mL |
| 62a | H | - | - | 79.8 | 85.30 |
| 63a | - | Et | H | 39.07 | 59.75 |
| 63b | - | H | NO2 | 50.00 | 80.05 |
| 63c | - | H | Cl | 0.00 | 5.09 |
|
Albendazole
(20 μg/mL) |
- | - | - | 10.8 | 14.8 |
a Control—96 parasites. b p > 0.05.
4. Thienopyrimidines with Antituberculosis Activity
Tuberculosis (TB) is an infectious disease caused by the bacterium Mycobacterium tuberculosis (MTB). This mycobacterium spreads through the air and infects the lungs. The WHO reported that 10 million people contracted TB and approximately 1.5 million died from this infection in the world in 2020 [61]. The most vulnerable people to TB are those that already have a disease that weakens their immune system, such as human immunodeficiency viruses (HIV) infection. However, this infection remains curable, except for multi-drug-resistant TB (MDR-TB), which contaminated approximately 206,000 people in 2019 [61]. TB is the source of a public health crisis as a threat to health security. Nowadays, the major issue is to offer new antibacterial treatments, effective on MDR-TB, inexpensive and accessible to all.
In this context, Rashmi et al. discovered new potent antituberculosis agents with a thieno[2,3-d]pyrimidine core [62]. All synthesized compounds were evaluated against MTB H37Rv (AT27294) by determining minimum inhibitory concentration (MIC). Regarding the SAR study, electron-donating substituents of the phenyl group, in para- or ortho-position (compounds 64b–f) led to higher antituberculosis activity compared to compound 64a (MICs = 32 to 71 and 320 µM, respectively) and similar activity to pyrazinamide, used as the reference drug (MICs = 64 to 71 µM and 60.97 µM, respectively) (Table 11). Bulkier 3,4,5-trimethoxyphenyl group was also tolerated (compound 64g). The most interesting compounds demonstrated weak cytotoxicity against THP-1 human monocytic cell line.
Table 11.
Antituberculosis activity of thieno[2,3-d]pyrimidine derivatives 64–64g.
| |||
|---|---|---|---|
| Compound | R |
M. tuberculosis H37Rv MIC (µM) |
THP-1 IC50 (µM) |
| 64a | Ph | 320 | ND a |
| 64b | 4-((Me)2)N-Ph | 70 | 2492.90 |
| 64c | 2-Br-Ph | 64 | 2387.80 |
| 64d | 2-NO2-Ph | 70 | 2415.90 |
| 64e | 4-Cl-Ph | 71 | 2264.80 |
| 64f | 4-MeO-Ph | 66 | 2454.70 |
| 64g | 3,4,5-(MeO)3-Ph | 62 | 2019.90 |
| Pyrazinamide | - | 60.97 | ND |
a ND: not determined.
From a high-throughput screening of a 100,997 compound library, Ananthan et al. identified thienopyrimidinone derivatives with antituberculosis potential [63]. Five of these derivatives (compounds 65a–e) exhibited moderate to high antimycobacterial activity against MTB H37Rv (Table 12). However, the limited number of thienopyrimidines in the screened libraries as well as the lack of reported IC90 for reference drugs included in the assay do not allow any firm conclusion on SARs.
Table 12.
Thienopyrimidin-4-one derivatives with antimycobacterial activity.
| ||||
|---|---|---|---|---|
| Compound | R1 | R2 | R3 | TB IC90 (µg·mL−1) |
| 65a | H | 4-Me-Ph | CH2Ph-Cl-4 | 1.0 |
| 65b | H | 3,4-(Me)2-Ph | Cyclohexyl | 1.7 |
| 65c | CH3 | Me | Cyclohexyl | 1.8 |
| 65d | H | 4-Br-Ph | CH2Ph | 2.2 |
| 65e | H | 4-Et-Ph | Et | 6.6 |
Several years later, Harrison et al. reported the activity of a series of 4-amino substituted thieno[2,3-d]pyrimidine derivatives against Mycobacterium tuberculosis [64] (Table 13). Biological studies were carried out on M. tuberculosis strains to determine antibacterial activities via a Microplate Alamar Blue assay (MABA). Regarding SAR studies, some derivatives with a long aminoalkyl chain displayed significant antibacterial activities with IC50 values in the micromolar or submicromolar range (IC50 = 0.083 to 2.7 µM). A lead compound (66f) was identified with stronger antituberculosis activity compared to the reference drug thioridazine (IC50 = 0.083 and 11.2 µM, respectively) but with a similar activity than bedaquiline. Several bulky groups at R such as alkyl and alkylaryl chains were tolerated (66b to 66e and 66g). Various experiments carried out by Harrison et al. indicated that these 4-aminothieno[2,3-d]pyrimidines could target QcrB, a subunit of the electron transport chain (ETC) enzyme cytochrome bc1 oxidoreductase [64]. A recent study suggests that combination of QcrB inhibitors and current treatments tends to amplify the antimycobacterial activity of the treatments and presents a possible alternative to improve current antitubercular drugs [65]. However, the safety of such approach needs to be confirmed.
Table 13.
4-amino-substituted thienopyrimidine derivatives with antimycobacterial activity.
| ||
|---|---|---|
| Compound | R | MABA in M. tuberculosis IC50 (µM) |
| 66a |

|
2.7 |
| 66b |

|
0.11 |
| 66c |
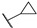
|
0.62 |
| 66d |
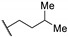
|
0.32 |
| 66e |
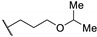
|
0.32 |
| 66f |
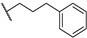
|
0.083 |
| 66g |
|
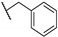
0.15 |
| Bedaquiline | - | <0.078 |
| Thioridazine | - | 11.2 |
5. Thienopyrimidines with Antibacterial Activities Other than Tuberculosis
Since the discovery of the first antibiotic, penicillin G by Alexander Fleming in 1928, antibiotics have been extensively used to treat all types of microbial infections. However, despite the existence of a wide range of antibiotics, the number of bacterial infections is constantly increasing with greater difficulties to cure them [66]. Even if the reasoned use of antibiotics has limited the development of resistance, this strategy is not sufficient to stop its progression and bacterial resistance becomes a growing scourge for humans. Therefore, the development of new antibiotics became an urgent concern, and the scientific community is thus mobilized to find new efficient antibacterial candidates. Among the different scaffolds under study, thienopyrimidine derivatives revealed to be attractive to discover new antibacterial compounds. However, the identification of active compounds in these series was mostly performed by phenotypic screening. To our knowledge, only one study reports an activity on an identified target, namely an amino-sugar acetyltransferase enzyme, named protein glycosylation D (PglD).
5.1. Inhibition of the Protein Glycosylation D (PglD) of Campylobacter Jejuni
Campylobacter jejuni is an intestinal Gram-negative bacterium. It most often causes severe diarrhea, which can be fatal to young children. C. jejuni can also be the cause of other serious infections such as hepatitis, pancreatitis and could provoke miscarriages, autoimmune diseases, or Guillain–Barré syndrome [67]. In recent years, the emergence of resistant strains toward front-line antibiotics against this bacterium was increasingly observed [68]. It has been reported that highly modified sugars, including 2,4-diacetamido-2,4,6-trideoxy-D-glucose (2,4-diacetylbacillosamine or diNAcBac) play a key role in host-cell interactions and can influence the virulence of Gram-negative bacteria. In addition, when certain enzymes involved in carbohydrate biosynthesis are suppressed, bacterial strains lose their activity. In this context, De Schutter team discovered a series of thienopyrimidine derivatives as inhibitors of an amino-sugar acetyltransferase enzyme, named protein glycosylation D (PglD), essential in the biosynthesis pathway of UDP-2,4-diacetamidobacillosamine of C. jejuni [18].
A wide range of compounds was synthesized and evaluated in vitro on C. jejuni (NCTC 11168) PglD acetyltransferase. Optimization of these thieno[2,3-d]pyrimidines activity was established from compound 67a (Table 14). Replacement of the methyl group by a bulkier group such as phenyl substituent doubled affinity for PglD (67a vs. 67b). Para-phenyl substitution or di-substitution in position 4 led to a strong PglD inhibition with submicromolar IC50s values (compounds 67c to 67f). In addition, ortho- or meta-substitutions were tolerated. Other bulky substituents such as pyridin-2-yl (67g) and benzo[d][1,3]dioxol-5-yl (67h) increased activity. Then, the replacement of the phenyl group at position 2 by a pyridin-3-yl ring led to a strong PglD inhibition (67i vs. 67a). Insertion of a 4-acetamidophenylethyl group (67j) in position 2 allowed good C. jejuni PglD inhibition, the same way as a 2-methoxy-2-phenylethyl group (67k to 67m).
Table 14.
Thieno[2,3-d]pyrimidine derivatives as potent PglD inhibitors.

| |||
|---|---|---|---|
| Compound | R1 | R2 |
C. jejuni PglD IC50 (µM) |
| 67a | Ph | Me | 2.2 ± 0.4 |
| 67b | Ph | PhCH2CH2 | 1.4 |
| 67c | Ph | 4-MeO-PhCH2CH2 | 0.54 |
| 67d | Ph | 4-F-PhCH2CH2 | 0.42 |
| 67e | Ph | 4-Me-PhCH2CH2 | 0.72 |
| 67f | Ph | 3,5-(MeO)2-PhCH2CH2 | 0.37 |
| 67g | Ph |
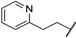
|
0.42 |
| 67h | Ph |
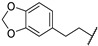
|
0.59 |
| 67i |
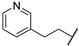
|
Me | 0.46 ± 0.05 |
| 67j | 4-AcNH-Ph |
|
0.28 |
| 67k | 4-AcNH-Ph |
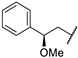
|
0.27 ± 0.09 |
| 67l | Benzyl |
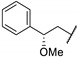
|
0.87 |
| 67m | Benzyl |
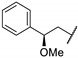
|
0.42 |
A co-crystallization of C. jejuni PglD with inhibitor 67a was obtained (Figure 5) and three ligand–receptor interactions were identified. A π-staking interaction between the thiophene ring and Phe155 of PglD was observed. In addition, the carboxylic acid of inhibitor 67a formed two hydrogen bonds with Ser139 and Ile158.
Figure 5.

Co-crystallized structure of compound 67a in the protein glycosylation D (PglD) active site (PDB: 5T2Y). The enzyme is represented as pale cyan cartoon mode and the inhibitor is shown as stick representation. Key bonding interactions are indicated as yellow dotted lines.
5.2. Compounds wih Broad-Spectrum Antibacterial Activity
In this section, the compounds were classified according to their chemical structure.
5.2.1. Thieno[2,3-d]pyrimidin-4-one and Pyrimidin-2,4-dione Derivatives
Abu-Hashem et al. identified six thieno[2,3-d]pyrimidinone derivatives as antibacterial agents [31]. All final compounds were evaluated in vitro against Escherichia coli, Staphylococcus aureus, and Bacillus cereus (Table 15). Compounds 68 and 69a–c displayed high activity against the three tested bacterial strains. Interestingly, their thienopyridine analogues 71a and 71b only showed moderate antibacterial activity, which could suggest that the carbonyl group at position 4 is important for the activity. Similar thienopyrimidin-4-ones 70a–b and thienopyrimidine-2,4-diones 69d–e were reported by Ortikov et al. Compounds were evaluated in vitro (Table 15) against the three previous bacterial strains and against P. aeruginosa [11]. Globally, this series showed lower antibacterial efficacy than the previous series, suggesting that the presence of a benzoyl group at R1 is important for the activity (compare 69c to 69e). Moreover, replacing the methyl group (70a) at R2 with a nitro group (70b) increased antibacterial activity against all tested strains. In addition, a sulfur atom in position 2 (69e) led to a better antibacterial activity than its oxo analogue 69d against S. aureus and B. cereus. In contrast, 69d strongly inhibited E. coli growth compared to 69e (16 vs. 6 mm, respectively). However, none of these compounds exhibited better antibacterial activity than levofloxacin, used as the reference drug.
Table 15.
Antibacterial activity of thieno[2,3-d]pyrimidin-4- (or 2,4-di)ones derivatives.
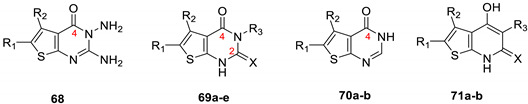
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | X | Inhibition Zone Diameter (mm) | Ref. | |||
| S. aureus | E. coli | B. cereus | P. aeruginosa | ||||||
| 68 | COPh | Me | - | - | 26 | 24 | 27 | NT a | [31] |
| 69a | COPh | Me | Ph | S | 24 | 23 | 26 | NT | [31] |
| 69b | COPh | Me | Ph | O | 22 | 21 | 23 | NT | [31] |
| 69c | COPh | Me | H | S | 21 | 20 | 22 | NT | [31] |
| 71a | COPh | Me | COMe | O | 9 | 10 | 12 | NT | [31] |
| 71b | COPh | Me | CN | O | 11 | 12 | 14 | NT | [31] |
| 70a | Me | Me | - | - | 6 | 6 | 6 | 6 | [11] |
| 70b | Me | NO2 | - | - | 12 | 15 | 16 | 10 | [11] |
| 69d | Me | Me | H | O | 8 | 16 | 8 | 6 | [11] |
| 69e | Me | Me | H | S | 12 | 6 | 12 | 8 | [11] |
| Levofloxacin | - | - | - | - | 26 | 30 | 28 | NT | [31] |
a NT: not tested.
De Candia et al. studied a series of spiro thienopyrimidin-4-one derivatives against several resistant bacterial strains to usual antibiotics [69] (Table 16). These compounds were screened in vitro to determine their minimum inhibitory concentration (MIC). Globally, all compounds displayed lower antibacterial activity than ampicillin against the three tested strains (S. agalactiae, E. faecalis and S. epidermidis). Regarding SAR studies, the modification of R1 did not affect the activity (compounds 72a to 72c). At R2, the introduction of a phenyl group was tolerated (compound 72b). However, the introduction of a para-methoxy (72h) or a para-nitro (72g) substituent on the phenyl ring decreased the activity, whereas a para-methoxy group maintained it (72h). Replacement of the methyl carboxylate (72d) by an acetyl group at position Y (72e) or replacement of the hydrogen atom (72d) by a methyl carboxylate group at position X (72f) were not tolerated. In addition, cytotoxicity of compounds 72b and 72d was determined on four cancer cell lines (Table 17). Compound 72b revealed a moderate to strong cytotoxicity (GI50 = 8 to 23 µM). A slightly lower cytotoxic effect on cancerous cell lines was observed for compound 72d (GI50 = 16 to 48 µM).
Table 16.
Antibacterial activity of spiro thieno[2,3-d]pyrimidine-4-one derivatives 72a–h.
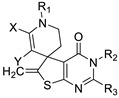
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cpd | X | Y | R1 | R2 | R3 | Antibacterial Activity MIC (µM) |
|||
| S. agalactiae (1) a | S. agalactiae (2) a | E. faecalis a | S. epidermidis a | ||||||
| 72a | H | CO2Me | Bn | H | CH2OMe | 25 | 25 | 25 | >250 |
| 72b | H | CO2Me | iPr | H | CH2Cl | 25 | 25 | 25 | >250 |
| 72c | H | CO2Me | Et | Ph | H | 25 | 25 | 25 | >250 |
| 72d | H | CO2Me | Et | 4-Cl-Ph | H | 125 | 25 | 25 | 25 |
| 72e | H | COMe | Et | 4-Cl-Ph | H | >250 | 250 | 250 | 250 |
| 72f | CO2Me | CO2Me | Et | 4-Cl-Ph | H | >250 | 250 | NT b | >250 |
| 72g | H | CO2Me | Et | 4-NO2-Ph | H | 250 | 250 | NT | >250 |
| 72h | H | CO2Me | Et | 4-MeO-Ph | H | 25 | 25 | 25 | 250 |
| Ampicillin | - | - | - | - | - | 1.5 | 1.5 | 25 | 25 |
a Resistance profiles were evaluated by antimicrobial susceptibility testing. S. agalactiae (1): tetracycline; S. agalactiae (2): clindamycin, erythromycin, tetracycline; E. faecalis: chloramphenicol, rifampicin, Synercid; S. epidermidis: fosfomycin, ampicillin, and penicillin G (β-lactamase positive). b NT: not tested.
Table 17.
Cytotoxic activity of spiro thieno[2,3-d]pyrimidine-4-one derivatives 72b–d.
| Compound | Cancer Cell Lines a | |||
|---|---|---|---|---|
| GI50 b (µM) | ||||
| MDA-MB-231 | OV2008 | HepG2 | C6 | |
| 72b | 8 | 7 | 21 | 23 |
| 72d | 44 | 16 | 40 | 48 |
a MDA-MB-231: human breast carcinoma cells, OV2008: human ovarian carcinoma cells, HepG2: human liver carcinoma cells, C6: rat glioma cell line. b GI50: concentration of half-maximal inhibition of cell proliferation.
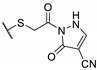
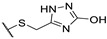
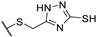
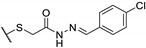
A wide range of thienopyrimidinone derivatives, substituted in positions 2, 3, 5, and 6, were synthesized by Shaaban et al. and Habib et al. [12,33]. Antibacterial activity was determined against a large panel of Gram-positive and Gram-negative bacteria. However, all tested compounds were revealed to have weaker antibacterial activities than the reference drugs (Table 18). In particular, compounds 73b, 73e, 73g–h displayed moderate antibacterial activity against B. subtilis, 2-fold lower than ampicillin (MIC = 25 vs. 12.5 µg/mL, respectively). Regarding inhibition of Gram-negative bacteria, compounds 73b–c, 73e, 73g–h were half as potent as levofloxacin against P. aeruginosa (MIC = 25 vs. 12.5 µg/mL, respectively). In the same way, P. vulgaris was modestly inhibited by compounds 73a, 73d, 73f, and 73i (MIC = 25 µg/mL).
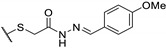
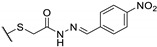
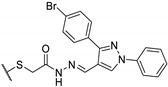
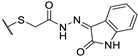
Table 18.
Antibacterial activity of thieno[2,3-d]pyrimidin-4-one derivatives 73a–i.
| ||||
|---|---|---|---|---|
| Compound | R | MIC (µg/mL) | ||
| Gram Positive | Gram Negative | |||
| B. subtitlis | P. aeruginosa | P. vulgaris | ||
| 73a |
|
50 | 50 | 25 |
| 73b |
|
25 | 25 | 100 |
| 73c |
|
50 | 25 | 100 |
| 73d |
|
100 | 100 | 25 |
| 73e |
|
25 | 25 | 100 |
| 73f |
|
100 | 100 | 25 |
| 73g |
|
25 | 25 | 100 |
| 73h |
|
25 | 25 | 100 |
| 73i |
|
100 | 100 | 25 |
| Ampicillin | - | 12.5 | 125 | - |
| Levofloxacin | - | - | 12.5 | 12.5 |
Chambhare et al. studied a series of 5-furyl-thienopyrimidone derivatives that were tested in vitro against Gram-positive (Staphylococcus aureus, Bacillus subtilis) and Gram-negative bacteria (Escherichia coli and Salmonella typhi) [37]. Among these molecules, twelve of them demonstrated strong antibacterial activity against the four tested strains. Para-substitution of the phenyl ring with electron-donating groups associated with a carboxamide or an NCH spacer was associated to good antibacterial activities (compounds 74a–b, 74e–f) (Table 19). Nonetheless, compounds 74c and 74g bearing a para-nitro group exhibited even stronger antibacterial activities (MIC = 8 to 12 µmol·L−1 and 4 to 7 µmol·L−1, respectively). Ortho-para-halogenation also increased antibacterial activity against all bacterial strains (74d and 74h compared to 74a and 74e). Overall, this series represented a broad-spectrum antibacterial potential.
Table 19.
Antibacterial activity of 5-furyl thieno[2,3-d]pyrimidin-4-ones derivatives.
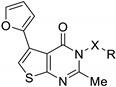
| ||||||
|---|---|---|---|---|---|---|
| Compound | X | R2 | Antibacterial Activity MIC (µmol·L−1) |
|||
| Gram-Positive Bacteria | Gram-Negative Bacteria | |||||
| S. aureus | B. subtilis | E. coli | S. typhi | |||
| 74a | NHCO | 4-F-Ph | 9 | 9 | 9 | 11 |
| 74b | NHCO | 4-MeO-Ph | 10 | 9 | 10 | 12 |
| 74c | NHCO | 4-NO2-Ph | 6 | 5 | 7 | 6 |
| 74d | NHCO | 2,4-F2-Ph | 4 | 5 | 4 | 5 |
| 74e | NCH | 4-F-Ph | 10 | 9 | 9 | 8 |
| 74f | NCH | 4-MeO-Ph | 11 | 11 | 12 | 12 |
| 74g | NCH | 4-NO2-Ph | 4 | 5 | 7 | 7 |
| 74h | NCH | 2,4-F2-Ph | 5 | 4 | 5 | 6 |
| Ampicillin | - | - | 50 | 4 | 4 | 49 |
| Penicillin-G | - | - | 6 | 22 | 5 | 5 |
| Chloramphenicol | - | - | 4 | 6 | 5 | 6 |
Two derivatives were identified as antibacterial agents by Dewal et al. [32] (Figure 6). Compound 75a displayed broad-spectrum antibacterial properties with MIC values comprised between 2 and 32 mg·L−1 against vancomycin-resistant S. aureus, S. pneumoniæ, E. faecium, P. aeruginosa, K. pneumoniæ, and E. aerogenes. In contrast, compound 75b only inhibited E. aerogenes with MIC equal to 8 mg·L−1. Compounds 75a–b showed a slight cytotoxic effect against NIH-3T3 mammalian cells (GI50 = 52 and 98 mg·L−1, respectively). In addition, compound 75a had no hemolytic activity.
Figure 6.
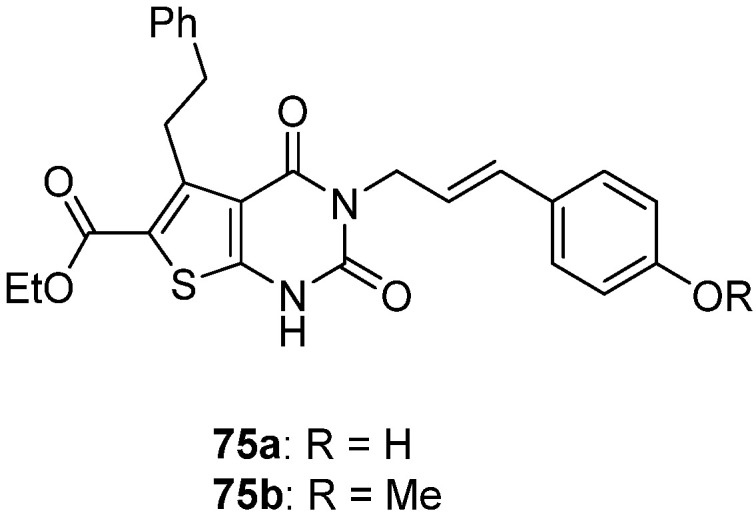
Structure of two thieno[2,3-d]pyrimidin-2,4-dione derivatives as antibacterial candidates identified by Dewal et al.
5.2.2. Other thieno[2,3-d]pyrimidine Derivatives
Tolba et al. discovered a novel series of thieno[2,3-d]pyrimidine derivatives with antibacterial potential [70]. Four compounds were synthesized and evaluated in vitro against Bacillus cereus, Staphylococcus aureus, Pseudomonas aeruginosa, and Escherichia coli (Table 20). All derivatives were active against these bacterial strains at low concentrations (MICs = 4.0 to 5.0 µg·mL−1), except compound 76d on E. coli (MIC = 8.0 µg·mL−1). All these compounds showed antibacterial activity in the same range as reference drugs.
Table 20.
Antibacterial activity of thieno[2,3-d]pyrimidine derivatives 76a–d.
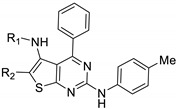
| ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | Antibacterial Activity MIC (µg·mL−1) |
|||
| Gram Positive | Gram Negative | |||||
| B. cereus | S. aureus | P. aeruginosa | E. coli | |||
| 76a | H |
|
5.0 | 5.0 | 4.0 | 5.0 |
| 76b | H |
|
4.0 | 5.0 | 5.0 | 4.0 |
| 76c | COCH2Cl |
|
4.0 | 5.0 | 4.0 | 5.0 |
| 76d | COCH2NHPh |
|
5.0 | 4.0 | 5.0 | 8.0 |
| Reference drugs | - | - | 5.0 Ofloxacin |
4.0 Levofloxacin |
4.0 Clindamycin |
5.0 Nitrofurantoin |
Saddik et al. identified new antibacterial candidates with a thienopyrimidine core substituted in positions 2, 4, 5, and 6 [71]. All compounds were evaluated against five bacterial strains (Table 21) and molecules 77g and 77i were the only derivatives showing moderate activity against Staphylococcus aureus and Escherichia coli, respectively. Regarding the SAR study, benzoyl (compound 77d) and carboxamide groups (compounds 77b, 77f–i) at position 6 (R) were not tolerated against Salmonella sp. In contrast, cyano (compound 77a), ethyl carboxylate (compound 77c), and acetyl groups (compound 77e) led to moderate activity against Salmonella sp. All derivatives were slightly active against Bacillus subtilis compared to the reference drug ampicillin, except compounds 77c, 77g, and 77i.
Table 21.
Antibacterial activity of thieno[2,3-d]pyrimidine derivatives 77a–i.
| |||||
|---|---|---|---|---|---|
| Compound | R | Zone of Bacterial Inhibition at 10 mg/mL (in mm) | |||
| Gram Positive | Gram Negative | ||||
| S. aureus | B. subtilis | Salmonella sp. | E. coli | ||
| 77a | CN | 0 | 14 | 14 | 0 |
| 77b | CONH2 | 0 | 16 | 0 | 0 |
| 77c | CO2Et | 0 | 0 | 14 | 0 |
| 77d | COPh | 0 | 15 | 0 | 0 |
| 77e | COMe | 0 | 15 | 14 | 0 |
| 77f | CONHPh | 0 | 15 | 0 | 0 |
| 77g | CONHPh-Cl-4 | 15 | 0 | 0 | 0 |
| 77h | CONHPh-OMe-4 | 0 | 15 | 0 | 0 |
| 77i | CONHPh-Br-4 | 0 | 0 | 0 | 20 |
| Ampicillin | - | 23 | 32 | - | - |
| Gentamycin | - | - | - | 17 | 19 |
Abdel Hamid et al. discovered a new thieno[2,3-d]pyrimidine series substituted in positions 2, 4, 5, and 6 [38]. Antibacterial activity of these derivatives was determined by measuring the inhibition zone diameter of Gram-positive and Gram-negative bacterial growth (Table 22). All derivatives showed moderate antibacterial activity against the tested strains. Regarding the SAR study, modification of ethyl carboxylate (78a) at R2 by carboxamide groups (78b and 78c) implied similar potency. In the same way, modification of the amine at R1 by N-heterocyclic ring (78d and 78e) and thiourea (78f) led to equivalent antibacterial activities. Only compound 78g demonstrated similar antibacterial activity against B. subtilis, E. coli, and P. aeruginosa compared to ampicillin.
Table 22.
Antibacterial activity of 2-furyl thieno[2,3-d]pyrimidine derivatives 78a–g.

| ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | Inhibition Zone Diameter (mm/mg Sample) | |||
| Gram Positive | Gram Negative | |||||
| B. subtilis | S. aureus | E. coli | P. aeruginosa | |||
| 78a | NH2 | CO2Et | 17 | 19 | 14 | 10 |
| 78b | NH2 | CONHNH2 | 18 | 18 | 10 | 14 |
| 78c | NH2 | CONHPh | 14 | 18 | 16 | 10 |
| 78d |
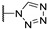
|
CO2Et | 15 | 13 | 16 | 15 |
| 78e |
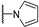
|
CO2Et | 11 | 15 | 10 | 17 |
| 78f |
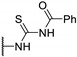
|
CO2Et | 13 | 17 | 19 | 19 |
| 78g |
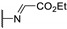
|
CO2Et | 20 | 16 | 22 | 21 |
| Ampicillin | - | - | 26 | 21 | 25 | 26 |
New thieno[2,3-d]pyrimidine derivatives incorporating an aminophenyl group or a pyrazole ring at position 4 were identified by Bhagchand et al. and Prabhakar et al. for their antibacterial potential [27,72]. Seven compounds were evaluated in vitro to determine the zone of growth inhibition of Escherichia coli, Bacillus sphaericus, Bacillus subtilis, Staphylococcus aureus, and Klebsiella pneumonia (Table 23). All derivatives exhibited a moderate activity against E. coli, whereas only compounds 79a and 79b were slightly active against B. sphaericus. However, the main drawback of this study was the absence of reference drugs. In contrast, other bacterial strains such as B. subtilis, S. aureus, and K. pneumonia were moderately inhibited by compounds 79d to 79g compared to amoxicillin.
Table 23.
Antibacterial activity of 4-aminophenyl- or 4-pyrazolyl thieno[2,3-d]pyrimidine derivatives.
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Cpd | R1 | R2 | Inhibition Zone (mm) | Ref. | ||||
| E. coli | B. sphaericus | B. subtilis | S. aureus | K. pneumonia | ||||
| 79a | Cl |
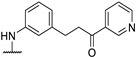
|
16 a | 8 a | - | - | - | [72] |
| 79b | Cl |
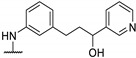
|
17 a | 10 a | - | - | - | [72] |
| 79c | Cl |
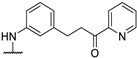
|
11 a | 0 a | - | - | - | [72] |
| 79d | 4-CF3-Ph |
|
15.5 b | - | 11.5 b | 12.5 b | 14.5 b | [27] |
| 79e |
|
|
16.5 b | - | 12.5 b | 14.5 b | 15 b | [27] |
| 79f |
|
|
17 b | - | 13 b | 15 b | 16.5 b | [27] |
| 79g |
|
|
13 b | - | 11 b | 11.5 b | 12.5 b | [27] |
| Amoxicillin | - | - | 19.6 b | - | 15.7 b | 17.4 b | 18 b | [27] |
a Experiments were realized at a concentration of 80 µL/mL. b Experiments were realized at a concentration of 100 µL/mL.
Aly et al. studied a thieno[2,3-d]pyrimidine derivative 80a including an aminopyrazole at position 5 [16], as well as its original thienothiadiazine analogue 80b. These two compounds were evaluated against Staphylococcus aureus, Bacillus subtilis, and Escherichia coli by determining a zone of growth inhibition of bacteria. Compounds 80a and 80b exhibited moderate antibacterial activity (Figure 7).
Figure 7.

Antibacterial agents proposed by Aly et al. [16].
Afterward, new antibacterial agents were suggested by Kanawade et al. [13]. A series of sixteen derivatives was synthesized and evaluated in vitro against Gram-negative (Escherichia coli and Pseudomonas aeruginosa) and Gram-positive bacteria (Staphylococcus aureus and Streptococcus pyogenes). Overall, these compounds demonstrated weak antibacterial activities compared to the reference drugs tested in the same conditions (Table 24).
Table 24.
Kanawade et al. derivatives as antibacterial agents.
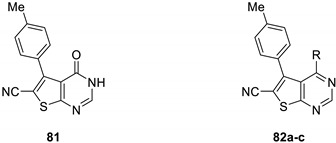
| |||||
|---|---|---|---|---|---|
| Compound | R | Antibacterial Activity MIC (µg/mL) |
|||
| Gram-Negative Bacteria | Gram-Positive Bacteria | ||||
| E. coli | P. aeruginosa | S. aureus | S. pyogenes | ||
| 81 | - | 100 | 62.5 | 200 | 250 |
| 82a | Piperazinyl | 125 | 250 | 62.5 | 100 |
| 82b | Piperazinyl-carboxylate | 62.5 | 100 | 250 | 250 |
| 82c | 4-Cl-Ph | 125 | 62.5 | 200 | 100 |
| Ampicillin | - | 100 | NA a | 250 | 100 |
| Chloramphenicol | - | 50 | 50 | 50 | 50 |
| Ciprofloxacin | - | 25 | 25 | 50 | 50 |
a NA: not active.
5.2.3. Thieno[3,2-d]pyrimidine Derivatives
Temburnikar et al. synthesized three 2-chlorothieno[3,2-d]pyrimidine derivatives [25] and evaluated them against several bacterial strains, including resistant strains (Escherichia coli, Bacillus subtilis, methicillin-resistant Staphylococcus aureus, vancomycin-resistant Enterococcus faecalis, and Pseudomonas aeruginosa), and also against several fungi (see Section 6). Among these three compounds, only 2,4-dichlorothieno[3,2-d]pyrimidine (Figure 8) showed a low antibacterial activity against one bacterial strain, B. subtilis with 43% inhibition at 100 µM.
Figure 8.
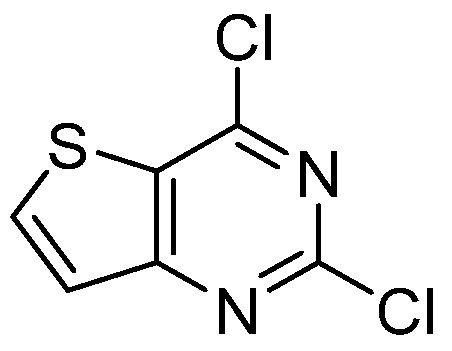
Structure of 2,4-dichlorothieno[3,2-d]pyrimidine.
Giri et al. identified new thieno[3,2-d]pyrimidine derivatives including an acyl hydrazone moiety as potential antibacterial agents [23]. All derivatives were evaluated in vitro against E. coli, Pseudomonas sp., S. aureus, and Bacillus sp. (Table 25). In this series, modulations were only studied on the hydrazone part of the molecule. Globally, these compounds showed similar activities against the tested strains, except compounds 83c and 83f, which were only active towards S. aureus and Pseudomonas, respectively. However, the exhibited antibacterial activities were twice lower than the reference drug streptomycin.
Table 25.
Thieno[3,2-d]pyrimidine derivatives incorporating an acylhydrazone motif as antibacterial agents.
| |||||
|---|---|---|---|---|---|
| Compound | R | Zone of Inhibition of the Bacteria (mm, C = 30 µL) | |||
| E. coli | Pseudomonas sp. | S. aureus | Bacillus sp. | ||
| 83a |
|
13 | 17 | 25 | 17 |
| 83b |
|
14 | 5 | 14 | NA a |
| 83c |
|
NA a | NA a | 17 | NA a |
| 83d |
|
15 | 14 | 16 | 15 |
| 83e |
|
15 | 14 | 19 | NA a |
| 83f |
|
NA a | 14 | NA a | NA a |
| Streptomycin | - | 35 | 32 | 34 | 37 |
a NA: not active.
Afterward, Hafez et al. undertook considerable work on spiro derivatives of thieno[3,2-d]pyrimidine-4-one [73]. Approximately forty compounds were synthesized and evaluated in vitro on six Gram-positive and Gram-negative bacterial strains (Table 26). Regarding SAR studies, the introduction of an oxygen atom in position 3 (compound 84a) caused a loss of potency compared to compounds 84b–j. Compounds 84b, 84e, 84h–j displayed better or similar inhibitory activity against all bacterial strains (MIC = 1 to 4 µmol·L−1) than the reference drug ciprofloxacin. In contrast, compounds 84c–g exhibited moderate antibacterial activity. Overall, all these compounds appeared to be broad-spectrum antibacterial molecules.
Table 26.
Antibacterial activity of thieno[3,2-d]pyrimidin-4-one derivatives 84a–j.
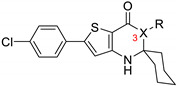
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Cpd | X | R | MIC (µmol·L−1) | |||||
| Gram-Negative Bacteria | Gram-Positive Bacteria | |||||||
| E. coli | K. pneumoniae | P. aeruginosa | S. lactis | S. aureus | E. faecalis | |||
| 84a | O | - | 8 | 8 | 7 | 6 | 5 | 8 |
| 84b | N |
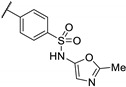
|
2 | 3 | 2 | 2 | 2 | 4 |
| 84c | N |
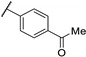
|
7 | 7 | 5 | 5 | 4 | 7 |
| 84d | N |
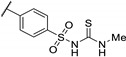
|
6 | 5 | 5 | 5 | 4 | 6 |
| 84e | N |
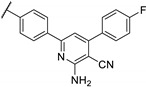
|
4 | 3 | 3 | 2 | 2 | 4 |
| 84f | N |
|
9 | 7 | 6 | 5 | 5 | 7 |
| 84g | N |
|
7 | 8 | 7 | 8 | 6 | 8 |
| 84h | N |
|
3 | 2 | 3 | 2 | 1 | 2 |
| 84i | N |
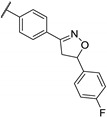
|
3 | 3 | 2 | 3 | 2 | 4 |
| 84j | N |
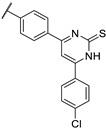
|
2 | 1 | 4 | 1 | 2 | 3 |
| Ciprofloxacin | - | - | 5 | 4 | 4 | 2 | 2 | 4 |
Shao et al. also reported the antibacterial activity of a series of thieno[3,2-d]pyrimidine-4-one [15], showing that such compounds may be effective against Clostridium difficile. This Gram-positive bacterium is naturally present in the intestinal flora of humans. In frail people, taking some antibiotics such as amoxicillin, clindamycin, and cephalosporins may cause C. difficile infection, which can be difficult to treat [74]. During this infection, bacterial toxins formation can lead to severe diarrhea and pseudomembranous colitis [75]. A wide range of 67 thieno[3,2-d]pyrimidin-4-one incorporating a nitro group at position 7 was synthesized and evaluated against two C. difficile strains (ATCC BAA 1870 and ATCC 43255). A dozen of these molecules inhibited C. difficile moderately, with MIC values between 1 and 16 µg·mL−1. Regarding the SAR study, positions 2, 3, and 4 were modified. Small groups such as a hydrogen atom or a methyl group at R1 led to moderate antibacterial activity (85a and 85b, Table 27). The introduction of a styryl group (85c), its bioisostere vinylthiophene (85d), or a meta-substituted phenyl ring (85g) kept the activity. In contrast, ortho- and para-phenyl substitution slightly increased antibacterial activity (85e–f, 85h–I compared to 85c) on C. difficile. Other positions were then modulated while keeping a methyl group in position 2. Globally, benzyl substituents at position 3 were tolerated (85j–k); the same holds for the insertion of a chlorine atom at position 4 (85l). Unfortunately, none of these derivatives displayed equal or higher activities compared to the three reference drugs. Finally, all derivatives exhibited low cytotoxicity against human colorectal adenocarcinoma (Caco-2), human ileocecal adenocarcinoma (HRT-18), and African green monkey kidney cells (Vero) and were inactive on Human Normal Microflora.
Table 27.
New 7-nitro-thieno[3,2-d]pyrimidine derivatives as anti C. difficile agents.

| |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 |
C. difficile MIC (µg·mL−1) |
|
| ATCC BAA 1870 | ATCC 43255 | ||||
| 85a | H | H |
|
4 | 2 |
| 85b | Me | H |
|
4 | 8 |
| 85c |
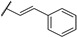
|
H |
|
4 | 16 |
| 85d |
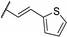
|
H |
|
4 | 4 |
| 85e |
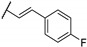
|
H |
|
1 | 2 |
| 85f |
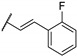
|
H |
|
2 | 4 |
| 85g |
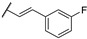
|
H |
|
4 | 4 |
| 85h |
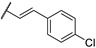
|
H |
|
2 | 2 |
| 85i |
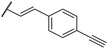
|
H |
|
2 | 4 |
| 85j | Me | Benzyl |
|
2 | 16 |
| 85k | Me | 4-NO2-PhCH2 |
|
2 | 4 |
| 85l | Me | H | Cl | 4 | 8 |
| Vancomycin | - | - | - | 1 | 0.5 |
| Metronidazole | - | - | - | 0.125 | 0.25 |
| Fidaxomicin | - | - | - | 0.0625 | 0.0625 |
Finally, Aly and Saleh synthesized two thieno[3,2-d]pyrimidine derivatives 53a–b and one isomer 52 [39] that were screened on two bacterial strains, namely P. aeruginosa and S. aureus. These compounds inhibited moderately these bacteria compared to tetracyclin, chosen by the authors as the reference drug (Table 28).
Table 28.
Antibacterial activity of thienopyrimidines 52 and 53.
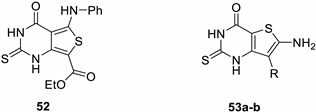
| |||
|---|---|---|---|
| Compounds | R | Inhibition Zone Diameter (mm) | |
| P. aeruginosa | S. aureus | ||
| 52 | - | 13 | 12 |
| 53a | CN | 14 | 14 |
| 53b | CO2Et | 13 | 14 |
| Tetracyclin | - | 28 | 26 |
6. Thienopyrimidines with Antifungal Activity
Nowadays, fungal infections continue to develop in humans and can lead to serious complications in patients with comorbidities causing immunosuppression (acquired immunodeficiency syndrome (AIDS), asthma, cancer, organ transplantation, corticosteroid therapies). The development of medico-surgical practices increases partly the risk of fungal infections. Fungal infections remain a growing scourge, with 150 million severe cases each year and approximately 1.7 million deaths per year worldwide [76]. In the same way as the overuse of antibiotic therapies, the escalating use of antifungals has led to the emergence of multi-drug-resistant fungi. The most threatening fungal pathogens are Aspergillus sp. and Candida sp., especially Candida auris, because of their resistance to most of currently available treatments [76]. To date, three main classes of antimycotic drugs are marketed, namely azoles, echinocandins and polyenes. As a large range of thienopyrimidines could display antibacterial activity (see Section 5), some studies have also evaluated the activity of such compounds on various fungal strains. In this section, only compounds showing antifungal activity at least similar or quite similar to the reference drug included in the study have been reported.
Abu-Hashem et al. evaluated their thienopyrimidin-4-one 68 and 2,4-diones 69 against Candida albicans and Aspergillus niger, by determining an inhibition zone diameter of growth [31]. All derivatives showed quite similar antifungal activity to nystatin. Hydrogen, phenyl, or amine groups in position 3 were well tolerated and led to similar activity on the two fungi, compared to the reference drug (68, 69a, and 69c, Table 29). In addition, replacement of the amino group at position 2 by a sulfur or oxygen atom maintained antifungal activity (68 vs. 69a to 69c). In contrast, replacing the thienopyrimidine moiety with a thienopyridine core caused a loss of potency (71a and 71b), as also observed previously for their antibacterial activity.
Table 29.
Antifungal activity of thienopyrimidines 68–69 and their thienopyridine analogues 71.
| Compound | Inhibition Zone Diameter (mm) a | |
|---|---|---|
| C. albicans | A. niger | |
| 68 | 26 | 30 |
| 69a | 24 | 28 |
| 69b | 22 | 26 |
| 69d | 23 | 25 |
| 71a | 11 | 15 |
| 71b | 13 | 17 |
| Nystatin | 27 | 26 |
a Inhibition diameter was measured after 24 h of incubation.
Aly and Saleh also tested their 2-thioxothienopyrimidin-4-(3H)-ones against two fungal strains, namely Aspergillus flavus and Candida albicans [39]. Compound 55 was inactive against A. flavus and 2-fold less active on A. niger compared to amphotericin B (Table 30). In contrast, compounds 53a and 53b showed similar activity to amphotericin B, although slightly lower.
Table 30.
Antifungal activity of 2-thioxothienopyrimidin-4(3H)-ones 52 and 53a–b.
| Compound | Inhibition Zone Diameter (mm/g of Samples) | |
|---|---|---|
| A. flavus | C. albicans | |
| 52 | 0 | 10 |
| 53a | 13 | 14 |
| 53b | 14 | 13 |
| Amphotericin B | 16 | 19 |
Hafez et al. evaluated their antibacterial thieno[3,2-d]pyrimidines series (see previous section) against Candida albicans (ATCC 15056), Aspergillus flavus (ATCC 24556), and Ganoderma lucidum (ATCC 96918) [73]. Among the tested compounds, seven showed high antifungal activity against the three fungal strains, with MICs from 1 to 7 µmol·mL−1 (Table 31), similar to ketoconazole used as the reference drug. Regarding SAR study, a para-halogenophenyl or a 2-thienyl group (84k to 84m) linked to the pyrazolyl moiety allowed strong antifungal activity (MICs = 1 to 5 µmol·mL−1). Introduction of several heterocyclic substituents at position R was tolerated. Introduction of an isoxazolyl group (84n) slightly decreased the growth inhibition of the three fungal strains (MICs = 4 to 7 µmol·mL−1), whereas the presence of a thioxopyrimidinyl group (84o–q) resulted in the retention of antifungal activity, with a slightly lower activity than the reference drug ketoconazole (MICs = 3 to 5 vs. 2 to 3 µmol·mL−1).
Table 31.
Antifungal activity of thieno[3,2-d]pyrimidin-4-ones 84k–q.
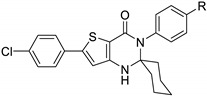
| ||||
|---|---|---|---|---|
| Compound | R | MIC (µmol·mL−1) | ||
| C. albicans | A. flavus | G. lucidum | ||
| 84k |
|
4 | 5 | 4 |
| 84l |
|
2 | 2 | 1 |
| 84m |
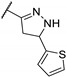
|
3 | 2 | 2 |
| 84n |
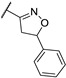
|
7 | 4 | 6 |
| 84o |
|
5 | 4 | 4 |
| 84p |
|
4 | 3 | 3 |
| 84q |
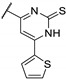
|
4 | 4 | 5 |
| Ketoconazole | - | 3 | 2 | 3 |
In the same way, Shaaban et al. have also studied their thieno[2,3-d]pyrimidines series against three fungal pathogens: Candida albicans, Aspergillus fumigatus, and Rhizopus oryzae [33]. Overall, all derivatives were more active on A. fumigatus and R. oryzae than on C. albicans (Table 32). Among all compounds evaluated, only three compounds (73k–l and 73n) displayed moderate activity against C. albicans, 2-fold lower than the reference drug clotrimazole (MICs = 25 vs. 12.5 µg/mL). In contrast, compounds 73j and 73m–o were twice as active as clotrimazole (MICs = 50 vs. 100 µg/mL).
Table 32.
Antifungal activity of thieno[3,2-d]pyrimidin-4-ones 73j–p.
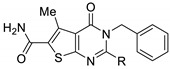
| ||||
|---|---|---|---|---|
| Compound | R | MIC (µg/mL) | ||
| C. albicans | A. fumigatus | R. oryzae | ||
| 73j |
|
100 | 50 | 50 |
| 73k |
|
25 | 100 | 50 |
| 73l |
|
25 | 50 | 100 |
| 73m |
|
100 | 50 | 50 |
| 73n |
|
25 | 50 | 50 |
| 73o | OEt | 50 | 50 | 50 |
| 73p | OH | 100 | 100 | 50 |
| Clotrimazole | - | 12.5 | 100 | 100 |
Temburnikar et al. reported the antifungal activity of two 2,4-dichlorothieno[3,2-d]pyrimidine derivatives against Candida albicans and Cryptococcus neoformans [25] (Figure 9). MIC95 values suggested that the introduction of a bromine atom in β position of the sulfur atom of the thiophene ring improved antifungal activity (86b vs. 86a).
Figure 9.

Structure and antifungal activity of halogenated thieno[3,2-d]pyrimidine derivatives 86.
Thieno[2,3-d]pyrimidine derivatives 76a–d reported by Tolba et al. were also highlighted as potent antifungal agents against Geotrichum candidum, Candida albicans, Trichophyton rubrum, and Aspergillus flavus [70]. Each compound displayed the same antifungal activity as clotrimazole against the four pathogens (MICs = 4.0 to 5.0 µg·mL−1, respectively, Table 33).
Table 33.
Antifungal activity of thieno[2,3-d]pyrimidines 76a–b.
| Compound | Antifungal Activity MIC (µg·mL−1) |
|||
|---|---|---|---|---|
| G. candidum | C. albicans | T. rubrum | A. flavus | |
| 76a | 4.0 | 5.0 | 4.0 | 5.0 |
| 76b | 4.0 | 5.0 | 4.0 | 5.0 |
| 76c | 4.0 | 5.0 | 4.0 | 5.0 |
| 76d | 4.0 | - | 4.0 | 5.0 |
| Clotrimazole | 4.0 | 5.0 | 4.0 | 5.0 |
Thieno[2,3-d]pyrimidine derivatives 80a and 80b, reported having antibacterial activity (see previous section), were also evaluated for their potential antifungal activity against Aspergillus fumigatus (RCMB 002003), Geotrichum candidum (RCMB 052006), Candida albicans (RCMB 005002) and Syncephalastrum racemosum (RCMB 005003) [16]. Their antifungal efficacy was determined by their inhibition zone diameter compared to reference drugs (itraconazole and clotrimazole). Globally, 80a and 80b exhibited moderate activity on the four fungal strains (zone of inhibition = 10.6 to 15.1 mm, Table 34).
Table 34.
Antifungal activity of compounds 80a–b.
| Compound | Inhibition Zone Diameter a (mm) | |||
|---|---|---|---|---|
| A. fumigatus | G. candidum | C. albicans | S. racemosum | |
| 80a | 12.5 ± 0.09 | 12.8 ± 0.1 | 11.0 ± 0.05 | 13.4 ± 0.08 |
| 80b | 15.1 ± 0.01 | 14.4 ± 0.1 | 13.4 ± 0.4 | 10.6 ± 0.2 |
| Itraconazole | 28 ± 0.05 | 27 ± 0.1 | 26 ± 0.02 | 22 ± 0.09 |
| Clotrimazole | 26 ± 0.1 | 23 ± 0.03 | 18 ± 0.1 | 20 ± 0.2 |
a Concentration at 10 mg·mL−1.
7. Thienopyrimidines with Antiviral Activity
7.1. Activity against Influenza A Virus
Influenza or flu is a recurrent respiratory infection in our modern society [77]. As the strains of influenza viruses vary from year to year, their circulation is closely monitored. Although influenza A infection is mostly mild, it can sometimes cause pneumonia and acute respiratory failure. Thus, in addition to the annual influenza vaccine, antiviral drugs remain essential to control influenza epidemics and pandemics. Looking for new anti-influenza molecules, Zhang et al. relied on the structure of pimodivir, an antiviral drug currently evaluated in phase III clinical trials [78]. This antiviral drug inhibits the trimeric RNA-dependent RNA polymerase of the virus. This enzyme is composed of three proteins, named polymerase basic protein 1 (PB1), polymerase basic protein 2 (PB2) and polymerase acidic protein (PA). The PB2 subunit generates 5′-capped RNA fragments from cellular pre-mRNA molecules used as primers for viral transcription [79]. The scaffold developed by Zhang et al. essentially conserved the 7-azaindole core of pimodivir and added a thienopyrimidine ring at position 3 (Table 35). Modifications such as the insertion of a thiophene ring, methylated (compounds 87c and 87d) or not (compounds 87a and 87b), were tolerated and the anti-influenza A activity remained in submicromolar EC50 values (Table 35). The most interesting compounds of this series, 87a and 87b, displayed an in vitro activity comparable to pimodivir (EC50 = 6.5 and 17 nM vs. EC50 = 4 nM for the reference drug). However, almost all compounds except 87c presented quite high cytotoxicity on several cell lines (PBM, CEM, Vero, Huh7, and A549). Cytotoxicity of these compounds could be explained by the oxidation at position 2 of the 7-azaindole ring, engendered by aldehyde-oxidase. Therefore, a nitrogen atom was introduced in position 2 to prevent metabolization. This modification resulted in either maintenance of the antiviral activity (compounds 87e) or a decrease in the antiviral activity (compound 87f).
Table 35.
RSA, anti-influenza A activity, and cytotoxicity of thienopyrimidine derivatives 87a–f.
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | Structure | Anti-Influenza A Activity in A549 Cells EC50 (nM ± SD) | Cytotoxicity CC50 (µM) | |||||
| X | A | PBM | CEM | Vero | Huh7 | A549 | ||
| 87a | C |
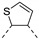
|
6.5 ± 1.1 | 9.3 | 4.2 | 65.9 | 24.0 | 14.5 |
| 87b | C |
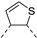
|
17 ± 10 | 64.6 | 31.6 | 74.9 | 68.7 | 14.7 |
| 87c | C |
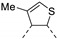
|
29 ± 6 | >100 | >100 | >100 | >100 | >100 |
| 87d | C |
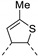
|
27 ± 14 | 78.4 | 12.9 | 90.3 | 31.2 | 41.7 |
| 87e | N |
|
12 ± 1 | >100 | 13.7 | >100 | 62.7 | >100 |
| 87f | N |
|
42 ± 7 | >100 | 67.0 | 52.5 | 70.7 | 32.5 |
| Pimodivir | - | - | 4 ± 2 | >100 | 48.9 | >100 | 95.8 | 20.0 |
7.2. Activity against Hepatitis B Virus
Despite recent therapeutic advances, viral hepatitis still represents a major health issue worldwide [80]. Although a massive action of vaccination against hepatitis B virus (HBV) was carried out in endemic countries [81], the number of new cases remains substantial every year, associated with the development of active chronic forms and cirrhosis. Structurally, HBV is a double-stranded DNA virus and belongs to the hepadnavirus family.
In 2013, Al-Harbi and Abdel-Rahman proposed anti-HBV analogues of acyclovir, notably known for its anti-herpes virus properties [29], by replacing the guanine base by its thienopyrimidinone analogue. Preliminary SAR studies on thienopyrimidinone derivatives revealed that substitution at position 1 by a (2-hydroxyethoxy)methyl chain was essential to increase anti-HBV activity and to reach submicromolar EC50 values (Table 36). Molecule 90a displayed the best activity of this series and similar activity to the antiviral reference drug lamivudine (EC50 = 0.2 and 0.1 µM, respectively). At position 7, several substituents on the cyclohexyl ring were tolerated, such as a methyl or a methoxy group (compounds 90a to 90d).
Table 36.
Thieno[3,2-d]pyrimidine activity against HBV DNA production.
| ||
|---|---|---|
| Compound | R | HBV DNA EC50 (µM) |
| 88 | 4-Me | 1.7 |
| 89 | 4-Me | 1.6 |
| 90a | 4-Me | 0.2 |
| 90b | H | 0.7 |
| 90c | 2-Me | 0.6 |
| 90d | 4-MeO | 0.6 |
| Lamivudine a | - | 0.1 |
a Drug reference.
7.3. Activity against HIV-1
Human immunodeficiency virus (HIV) is a major contributor to the global burden of infectious diseases, with an estimated 38 million people living with this viral infection in 2019 [82]. The same year, the WHO counted 690,000 deaths due to HIV and 1.7 million new cases [83]. It mainly targets activated CD4 T lymphocytes and causes gradual depletion of this cell line, resulting in progressive immune dysfunction [84]. Eventually, progression of the disease reaching the immune deficiency syndrome (AIDS) state makes patients more vulnerable to opportunistic infections such as tuberculosis, pneumonia, cryptococcal meningitis or cytomegalovirus retinitis. Two types of viruses exist, HIV-1 and HIV-2, the former being predominant in humans. Antiretroviral therapy (ART) is the recommended treatment to decrease the viral load to concentrations below the limit of detection of available commercial assays. Unfortunately, resistance mutations to antiretroviral drugs tend to appear under selection pressure and may cause a loss of drug efficacy [85]. Therefore, the need to identify original bioactive compounds and new therapeutic targets remains a major concern.
7.3.1. Thienopyrimidines as Reverse Transcriptase (RT) Inhibitors
A considerable work carried out by Kang et al. allowed the identification of new HIV-1 non-nucleoside reverse transcriptase inhibitors (NNRTIs) with EC50 in the nanomolar range [86]. The starting point of this work was the development of a thieno[3,2-d]pyrimidine scaffold derived from etravirine, a diarylpyrimidine (DAPY) NNRTI especially active against HIV-1. This series of compounds were evaluated for their anti-HIV activity and cytotoxicity in MT-4 cells infected by a wild-type (WT) HIV-1 strain (IIIB) and an HIV-2 strain (ROD). A lead compound 91a was identified with an anti-HIV-1 activity 4-fold better than etravirine (Table 37) and low cytotoxicity (CC50 > 227 µM). Molecule 91a showed good activity against mutant strains, with similar or better activity than etravirine (Table 38) and exhibited favorable in vivo pharmacokinetic parameters in rats. A SAR study demonstrated that the para-cyano group substituting the phenyl ring at position 4 played a key role in increasing antiviral activity (91a vs. 91b). Furthermore, para-substitution of the N-benzylpiperidine group at R2 by several polar groups (unsubstituted sulfamide or amide) increased activity, whereas other groups with lower polarity (substituted sulfamide, fluorine, or ester) caused a drop in potency. Additional SARs based on compound 91a revealed that a para-cyanovinyl substitution of the phenyl ring at position 4 increased non-nucleosidic reverse transcriptase inhibition (91c: EC50 (IIIB) = 1.22 nM) [87]. Compound 91c exhibited better anti-HIV activity than both etravirine and compound 91a against the K103N drug-resistant single mutant, and against F227L + V106A or K103N + Y181C (RES056) double mutants. Replacing the thieno[3,2-d]pyrimidine core by its thieno[2,3-d]pyrimidine isomer led to similar activity against WT HIV-1 strain and a slightly decreased activity on mutant strains compared to compound 91a. Modification of the nitrogen atom position in the aminopiperidine group at position 2 (compound 91d) led to a drop of activity (EC50s in the micromolar range) [87]. Similarly, diaminocyclohexane derivatives 91e and 91f showed a slight decrease in anti-HIV1 activity (EC50 = 7.1 and 10 nM, respectively) [88]. A diaminophenyl group at position 2 caused a loss of potency, with EC50s 10-fold weaker than etravirine (91g vs. ETV) [89]. Finally, various amino-cycloalkyl groups were introduced at position 2. Insertion of 8-azabicyclo[3.2.1]octane at position 2 (compound 91h) was not tolerated whereas azepane (91i), pyrrolidine (91j) or azetidine (91k) derivatives showed moderate to high potency against HIV-1, with EC50 values between 2.20 and 217 nM on NL4-3 cell lines [90]. However, their activity was not evaluated on mutant strains.
Table 37.
Anti-HIV activity of thieno[3,2-d]pyrimidine derivatives 91a–l.
| ||||||
|---|---|---|---|---|---|---|
| Cpd | R1 | R2 | EC50 a (nM) | Ref. | ||
| IIIB | ROD | NL4-3 | ||||
| 91a | CN |
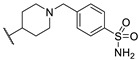
|
1.4 ± 0.4 | >227,890 | 1.16 ± 0.43 | [86,90] |
| 91b | Me |
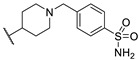
|
3552 ± 848 | >16,460 | - | [86] |
| 91c |
|
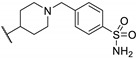
|
1.22 ± 0.26 | >2.30 | - | [87] |
| 91d | CN |
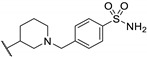
|
>1.17 × 103 | >1.17 | - | [87] |
| 91e | CN |
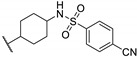
|
7.1 ± 0.5 | >9.287 | - | [88] |
| 91f | CN |
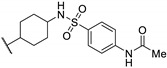
|
10 ± 8 | >3734 | - | [88] |
| 91g | CN |
|
58 ± 29 | >19,390 | - | [89] |
| 91h | CN |
|
- | - | >217 | [90] |
| 91i | CN |
|
- | - | 2.20 ± 0.67 | [90] |
| 91j | CN |
|
- | - | 8.69 ± 2.74 | [90] |
| 91k | CN |
|
- | - | 104 ± 36.8 | [90] |
| 91l | CN |
|
- | - | 4.53 ± 1.30 | [90] |
| ETV | - | - | 5.8 ± 4.0 | - | - | [86] |
a EC50: Concentration of compounds required to achieve 50% protection of TZM-bl cell lines (NL4-3) and MT-4 cell cultures (IIIB and ROD) against HIV-1-induced cytotoxicity, presented as the mean ± standard deviation (SD) and determined by the MTT method.
Table 38.
Anti-HIV-1 activity of thieno[3,2-d]pyrimidine derivatives against HIV-1 mutant strains.
| Cpd | EC50 a (nM) | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|
| L100I | K103N | Y181C | Y188L | E138K | F227L + V106A | RES056 (K103N + Y181C) |
||
| 91a | 3.4 ± 0.6 | 2.9 | 3.2 ± 0.4 | 3.0 ± 0.1 | 2.9 | 4.2 ± 1.2 | 30.6 ± 12 | [86] |
| 91b | 4519 ± 158 | 4937 ± 407 | 4845 ± 118 | 8471 ± 197 | 5505 ± 315 | 4547 ± 13 | >16,462 | [86] |
| 91c | 1.34 ± 0.5 | 0.958 ± 0.07 | 5.00 ± 0.1 | 5.45 ± 0.2 | 4.74 ± 0.2 | 2.70 ± 1.74 | 5.50 ± 0.811 | [87] |
| 91d | >1.17 × 103 | >1.17 × 103 | >1.17 × 103 | >1.17 × 103 | >1.17 × 103 | >1.17 × 103 | >1.17 × 103 | [87] |
| 91e | 424 ± 361 | 70 ± 25 | 428 ± 294 | 675 ± 91 | 45 ± 1 | 3583 ± 241 | >9280 | [88] |
| 91f | 562 ± 487 | 32 ± 2 | 513 ± 415 | 903 ± 248 | 35 ± 1 | 1208 ± 333 | >3727 | [88] |
| 91g | 280 ± 61 | 14 ± 1 | 780 ± 9 | 790 ± 43 | 31 ± 4 | 770 ± 110 | >19,390 | [89] |
| ETV | 5.4 ± 2.1 | 2.4 ± 0.6 | 15.8 ± 2.1 | 20.5 ± 2.9 | 14.4 ± 2.2 | 29.4 ± 7.7 | 17 ± 1.8 | [86] |
a EC50: Concentration of compounds required to achieve 50% protection of MT-4 cell cultures against HIV-1-induced cytotoxicity, as determined by the MTT method.
In addition to these SAR studies, Yang et al. obtained co-crystal structures of 91a and 91c with HIV-1 wild-type (WT) reverse transcriptase (RT) and seven RT variants involved in drug-resistant mutations [91]. Ligand-enzyme interactions between HIV-1 WT reverse transcriptase and compounds 91a or 91c are presented in Figure 10. Several ligand–receptor interactions were common to both compounds: two hydrogen bonds between the primary sulfonamide group and Lys104 and Val106, one hydrogen bond of the NH-linker of the thienopyrimidine core and the piperidine ring with Lys101, and hydrophobic interactions with the phenoxy group and several aromatic amino-acids (e.g., Tyr188 and Trp219). In contrast, differences were observed, notably the hydrogen bond between the cyano group of 91b and Tyr188, and the interaction of the nitrogen atom of the piperidine ring of 91b with Lys103 and Pro236 via a hydrogen bond with a water molecule.
Figure 10.

Co-crystallized structure of compounds 91a (A) and 91b (B) in the HIV-1 wild-type (WT) reverse transcriptase (RT) active site (PDB: 6C0J (91a) and 6C0N (91b)). The enzyme is represented as pale cyan cartoon mode and the compound is shown as stick representation. Key bonding interactions are indicated as yellow dotted lines.
Further works carried out in the thieno[3,2-d]pyrimidine series allowed the identification of new NNRTI candidates. Sang et al. developed new thienopyrimidine derivatives based on biphenyl diarylpyrimidines (DAPYs) with nanomolar potency (EC50 = 7.8 to 526.2 nM) [28]. Ortho-substitution of the phenyl ring at position 4 was very well tolerated compared to meta-substitution that led to a drop of activity (EC50 = 7.8 nM for compound 92a and 87.7 nM for compound 92b, Table 39). Ortho-substitution was also in favor of high selectivity indexes compared to the reference drugs nevirapine and etravirine (SI = 28,346 vs. > 50 and > 833). Di-substitution was tolerated too. Compound 92a displayed activity against HIV-1 similar to etravirine and 40-fold higher than nevirapine. Compounds 92c and 92f were almost 2-fold less active than di-fluorinated derivatives (compounds 92d and 92e), suggesting that only small substituents promote potency. All compounds presented lower cytotoxicity than the reference drugs nevirapine and etravirine (CC50 > 18.5 µM). Interestingly, all compounds demonstrated a better potency on HIV-1 mutant strains than nevirapine. However, none of these molecules presented a better activity against HIV-1 mutant strains than etravirine, except 92a. Indeed, this latter compound showed similar activities on K103N and E138K, in the same range as etravirine (Table 40). Another SAR study on this series was reported thereafter [92]. The results demonstrated that only R5 and R6 substitutions were tolerated. An ortho-fluorinated substituent showed strong activity against HIV-1 IIIB strains in MT-4 cells (7: EC50 = 11 nM). Introduction of ortho-substituents such as a chlorine atom or a methyl group or introduction of meta-substituents such as a fluorine, a chlorine atom, or a methyl group led to moderate activity, better than or similar to nevirapine, but lower than etravirine. Then, combined substitutions at R5 and R6 were studied (Table 39). Four compounds, 92h to 92k, revealed a strong anti-HIV-1 activity (EC50 = 14 to 29 nM), similar or slightly weaker than etravirine. Concerning their activity on HIV mutant strains, 92h to 92k demonstrated lower potency than etravirine but a better activity than nevirapine.
Table 39.
Activity of thieno[3,2-d]pyrimidine derivatives 92a–k against HIV-1 (IIIB) and HIV-2 (ROD) strains in MT-4 cells.
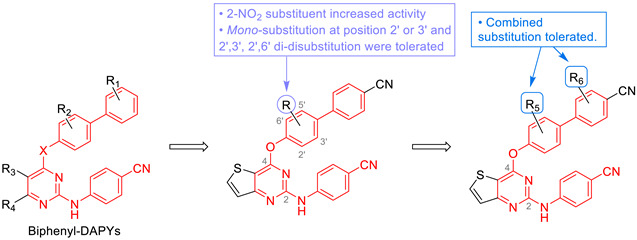
| ||||||
|---|---|---|---|---|---|---|
| Compound | R | R5 | R6 | EC50 a | Ref. | |
| HIV-1 (IIIB) (nM) | ROD (µM) | |||||
| 92a | 2′-NO2 | - | - | 7.8 ± 3.7 | >107.2 | [28] |
| 92b | 3′-NO2 | - | - | 87.7 ± 30.6 | >34.4 | [28] |
| 92c | 2′,3′-(Me)2 | - | - | 38 ± 12.7 | 1.4 ± 0.3 | [28] |
| 92d | 2′,3′-F2 | - | - | 13.5 ± 5.6 | >104.3 | [28] |
| 92e | 2′,6′-F2 | - | - | 12.3 ± 2.1 | >37.8 | [28] |
| 92f | 2′-Me-6′-Cl | - | - | 34.5 ± 16.2 | >70.3 | [28] |
| 92g | - | H | 2-F | 11 ± 8 | - | [92] |
| 92h | - | 3-Me | 2-F | 29 ± 5 | - | [92] |
| 92i | - | 3-Cl | 3-Cl | 21 ± 10 | - | [92] |
| 92j | - | 2-Me | 2-Me | 14 ± 5 | - | [92] |
| 92k | - | 2-Me | 3-Me | 17 ± 12 | - | [92] |
| NVP | - | - | - | 309.4 ± 57.7 | >4.0 | [28] |
| ETV | - | - | - | 5.5 ± 4.1 | >2.0 | [28] |
a EC50: The effective concentration required to protect the cell against viral cytopathic by 50% in MT-4 cells.
Table 40.
Activity of thieno[3,2-d]pyrimidine derivatives 92 against clinical HIV-1 mutant strains in MT-4 cells.
| Cpd | EC50 a (nM) | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|
| L100I | K103N | Y181C | Y188L | E138K | F227L + V106A | K103N + Y181C | ||
| 92a | 18.2 ± 3.9 | 5.5 ± 0.6 | 55.1 ± 0.6 | >15,140 | 6.5 ± 0.5 | ≥40,000 | >20,000 | [28] |
| 92c | 23.2 ± 4.2 | 33.8 ± 4.2 | 57.1 ± 10.6 | 50.7 ± 17.0 | 42.3 ± 4.2 | 253.6 ± 8.5 | 152.2 ± 42.3 | [28] |
| 92d | 52.0 ± 8.3 | 9.4 ± 2.1 | 58.2 ± 33.3 | 228 ± 41.6 | 17.0 ± 0.2 | 603 ± 21 | >2037.1 | [28] |
| 92e | 47.8 ± 8.3 | 8.7 ± 2.7 | 120.6 ± 18.7 | >20,308 | 16.6 ± 2.1 | ≥7233.70 | >78,552.4 | [28] |
| 92f | 28.4 ± 16.2 | 30.4 ± 6.1 | 48.7 ± 14.2 | 107.5 ± 42 | 52.7 ± 6.1 | 405.6 ± 6.1 | 148.0 ± 20.3 | [28] |
| 92g | 610 ± 270 | 90 ± 10 | 630 ± 490 | - | 130 ± 110 | - | - | [92] |
| 92h | 440 ± 450 | 210 ± 130 | 710 ± 10 | - | 400 ± 70 | - | - | [92] |
| 92i | 940 ± 50 | >20,000 | 660 ± 90 | - | 40 ± 30 | - | - | [92] |
| 92j | 130 ± 80 | 20 ± 10 | 30 ± 10 | - | 40 ± 10 | - | - | [92] |
| 92k | 1970 ± 660 | 130 ± 10 | >19000 | - | 160 ± 120 | - | - | [92] |
| NVP | 2102 ± 751 | >10,075.0 | >15,866.7 | >15,866.7 | 210 ± 26 | >15,866.7 | >15,866.7 | [28] |
| ETV | 7.1 ± 2.8 | 3.2 ± 0.5 | 12.0 ± 1.4 | 20.0 ± 7.6 | 6.5 ± 5.8 | 15.2 ± 16.1 | 55.3 ± 9.2 | [28] |
a EC50: The effective concentration required to protect the cell against viral cytopathic by 50% in MT-4 cells.
Kankanala et al. focused their research on the catalytic domain of the HIV reverse transcriptase (RT) enzyme, named HIV RT-associated ribonuclease H (RNase H) [30]. Indeed, RT presents two distinctive active sites, an N-terminal DNA polymerase site and a C-terminal RNase H site. Four carboxylic residues interacting with two Mg2+ metal ions activate RNase. Therefore, a chelating group with the ability to complex two Mg2+ metal ions is essential to inhibit RNase. Inhibitors of RT RNase H described in the literature presented a metal chelate site and a peripheral hydrophobic group. Based on this model, Kankanala et al. synthesized a series of 3-hydroxythienopyrimidine-2,4-diones that were evaluated for their anti-RNase potential (Table 41). The first thiophene derivatives 93a exhibited a potent activity with submicromolar IC50s and no polymerase (pol) inhibition. In contrast, a moderate integrase strand-transfer (INST) inhibition (IC50 = 4.5 μM), a low antiviral activity (EC50 = 11 μM) and an acceptable cytotoxicity on MAGI cells (CC50 = 28 μM) were observed. Regarding other derivatives, the para-halogen substitution of the phenyl ring at position 6 could cause a drop of either antiviral potency or cytotoxicity (compounds 93b and 93c). Regarding compound 93c, para-chloro substitution led to a selectivity improvement, with an increase in RNase H inhibition (IC50 = 0.07 μM) and a decrease in INST inhibition. The main drawback of molecule 93c was its low antiviral activity. To address this, bioisosteric analogues were synthesized and evaluated. Compound 94a displayed similar activities to compound 93a on RNase and INST. Replacing the phenyl group with a benzyl group at position 6 increased RNase inhibition (IC50 = 0.043 μM for compound 94b). Substitution at position 5 by a phenyl ring, a hydrogen atom, or a methyl group led to submicromolar anti-RNase IC50 values (compounds 94c, 94e–f). In contrast, compound 94d displayed the best activity in this series (IC50 = 0.040 μM). Substitution of both positions 5 and 6 maintained RNase H inhibition (compounds 94g–h). To summarize, all compounds showed better RNase H inhibition and lower integrase strand-transfer activity than raltegravir, the reference drug chosen by the authors.
Table 41.
Hydroxythienopyrimidine activities against RT, INST, and anti-HIV-1 cells.
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Compounds | R1 | R2 | R3 | RT IC50 a (μM) | INST IC50 a (μM) | MAGI Antiviral | ||
| RNase H | Pol | EC50 b (μM) | CC50 c (μM) | |||||
| 93a | Ph | - | - | 0.10 ± 0.06 | >10 | 4.5 ± 0.8 | 11 ± 2 | 28 ± 0.2 |
| 93b | 4-F-Ph | - | - | 0.20 ± 0.1 | >10 | 17 ± 4 | >20 | >100 |
| 93c | 4-Cl-Ph | - | - | 0.070 ± 0.05 | >10 | 23 ± 6 | >20 | >100 |
| 94a | - | Ph | H | 0.084 ± 0.006 | >10 | 2.2 ± 0.4 | >20 | >100 |
| 94b | - | Benzyl | H | 0.043 ± 0.008 | >10 | 5.0 ± 1 | >20 | >100 |
| 94c | - | H | Ph | 0.10 ± 0.03 | >10 | 1.3 ± 0.1 | 14 ± 1 | >100 |
| 94d | - | H | 4-Cl-Ph | 0.040 ± 0.02 | >10 | 2.1 ± 0.3 | 7.4 ± 0.3 | >100 |
| 94e | - | H | H | 0.20 ± 0.03 | >10 | 35 ± 6 | 18 ± 1 | 54 ± 6 |
| 94f | - | H | Me | 0.10 ± 0.02 | >10 | 12 ± 2 | >20 | 81 ± 6 |
| 94g | - | Me | Me | 0.10 ± 0.03 | >10 | 8.4 ± 1 | 8.9 ± 1 | 62 ± 1 |
| 94h | - | CO2H | Me | 0.10 ± 0.02 | >10 | 7.9 ± 1 | >20 | >100 |
| Raltegravir | - | - | - | >10 | ND d | 0.65 | 0.030 ± 0.005 | ND d |
a Concentration of compounds inhibiting the target enzyme by 50%. b Concentration of compounds inhibiting virus replication by 50%. c Concentration of compounds resulting in 50% cell death. d ND = not determined. All assay results expressed as a mean ± standard deviation from at least two independent experiments.
7.3.2. Anti-HIV Thienopyrimidines with Other Mechanisms
Using a cell-based full-replication assay, Kim et al. identified 2-(phenylsulfonylmethyl)-thieno[3,2-d]pyrimidine derivatives able to inhibit the HIV-1 replication [20]. These compounds presented EC50 values in the micromolar or submicromolar range. The best compounds 95a and 95i, bearing a para-methoxypyridin-3-yl group at R1 (Table 42), presented a strong activity against HIV-1 virus replication (EC50 = 25 and 14 nM, respectively). Other para-substituents, such as halogen atoms (95b) or carbonyl group (95c) led to a loss of potency, highlighting that an electron-donating group at this position could be in favour of a good activity profile. Several derivatives bearing a heterocyclic group (pyridine (95d), morpholine (95e), thiophene (95f), or furan (95g)) or a substituted phenyl ring at R1 (95h) also showed lower activity on anti-HIV-1 replication. Other modifications at R2 were carried out by replacing the linker between the thienopyrimidine core and the 3-chlorophenyl group. Replacing the sulfone with a sulfide doubled the activity (95i vs. 95a). Other modifications of the sulfone group (replacement by a carbon (95j), a carbonyl group (95k), etc.) led to a slightly to strongly decreased anti-HIV activity. Afterward, substituents on the phenyl ring at R2 were modulated. Meta-substitution was tolerated (95i), whereas di-substitution caused a loss of potency (compound 95l). None of these compounds presented noticeable cytotoxicity on CEMx174-LTR-GFP CG8 cells and HeLa-LTR-GFP cells. The target involved in the HIV-1 replication inhibition remains to be identified.
Table 42.
Antiviral activity of thienopyrimidine derivatives 95a–l against HIV-1.
| ||||
|---|---|---|---|---|
| Compound | R1 | X | R2 | EC50 a (µM) |
| 95a |
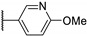
|
SO2 | H | 0.025 |
| 95b |
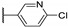
|
SO2 | H | 3.2 |
| 95c |
|
SO2 | H | 0.915 |
| 95d |
|
SO2 | H | 0.485 |
| 95e |
|
SO2 | H | 0.59 |
| 95f |
|
SO2 | H | 0.140 |
| 95g |
|
SO2 | H | 0.775 |
| 95h |
|
SO2 | H | 0.103 |
| 95i |
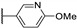
|
S | H | 0.014 |
| 95j |
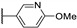
|
CH2 | H | 1.6 |
| 95k |
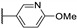
|
CO | H | 0.20 |
| 95l |
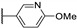
|
S | Cl | 0.728 |
| Nevirapine b | - | - | 0.150 | |
a EC50 is the concentration of compound that inhibits HIV-1 replication by 50%. The values are the geometric mean of two determinations; all individual values are within 25% of the mean. b Nevirapine was used as a positive control.
During antiretroviral therapy, residual viraemia is present in the body. A quiescent form of the HIV-1 genome still replicates and persists in some CD4+ T cells [93]. Thus, research efforts focused on the eradication of these latently infected cells. In 2019, Vargas et al. demonstrated the inhibitory activity of various compounds on signaling pathways that blocked the reversal of HIV-1 latency [94]. Screening of compounds was carried out in the absence or presence of three mechanically distinct latency reversal agents (LRAs) called prostratin, panobinostat, and JQ-1. Compound PF-3758309 (Figure 11) which targets p-21-activated kinase 4 (PAK4), was identified as an inhibitor of LRAs. This means that it blocked the latency-reversing activity of prostratin, panobinostat, and JQ-1 (IC50 values equal to 0.07, 0.4, and 0.1 nM, respectively) in the HIV-1-latent 24ST1NLESG cell line. Furthermore, PF-3758309 revealed good selectivity. In addition, this molecule blocked cellular transcription of HIV-1 and virus reactivation in CD4+ T cells. Due to its mode of action, PF-3758309 could be associated to antiretroviral therapy to reduce the immune activation of CD4+ T caused by the presence of a lower rate of viraemia.
Figure 11.
Structure of PF-3758309, an inhibitor of HIV-1 reversal latency.
Overall, due to their structural relationship with purine bases, thienopyrimidines have been particularly studied as potential antiviral agents. The main works in this area have focused on the HIV virus and several compounds showed antiviral activity on HIV-1 or HIV-2 with micromolar or submicromolar values. Among the studied targets, several works focused on the reverse transcriptase of the virus, leading to the discovery of potent inhibitors, notably against various resistant strains.
8. Conclusions
After an in-depth study of the literature, thienopyrimidine emerges as an attractive scaffold in medicinal chemistry with a wide array of pharmacological properties. In this review, we have reported the distinct strategies currently used to access thienopyrimidine derivatives, as well essential information to design novel anti-infective agents and optimize their structures.
Among the different routes of synthesis studied, the construction of the pyrimidine ring from aminothiophene derivatives is the most used synthetic pathway. Introduction of various substituents on the pyrimidine and on the thiophene ring is quite easy, which allows access to a wide range of modulations. Moreover, SAR analysis reveals that thieno[3,4-d]pyrimidine derivatives are little studied as, to our knowledge, only one compound has been reported to have antibacterial and antifungal activities (compound 52). In addition, antibacterial, antifungal and antitubercular agents are mostly thieno[2,3-d]thienopyrimidine derivatives, whereas compounds with antiviral activity are mainly represented by thieno[3,2-d]pyrimidines. Concerning antiparasitic agents, both thieno[2,3-d]pyrimidines or thieno[3,2-d]pyrimidines were reported. Finally, most of the compounds with anti-infective properties were identified after phenotypic screening and only few targets involved in their biological activity have been reported to date. When confirmed, enzymes have been identified as the main targets of these derivatives (protease, transferase, transcriptase, etc.).
Funding
This research was funded by grants from the Agence Nationale de la Recherche (ANR Plasmodrug 18-CE18-0009-01).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Litvinov V.P., Dotsenko V.V., Krivokolysko S.G. Thienopyridines: Synthesis, Properties, and Biological Activity. Russ. Chem. Bull. 2005;54:864–904. doi: 10.1007/s11172-005-0333-1. [DOI] [Google Scholar]
- 2.Ali E.M.H., Abdel-Maksoud M.S., Oh C.-H. Thieno[2,3-d]Pyrimidine as a Promising Scaffold in Medicinal Chemistry: Recent Advances. Bioorganic Med. Chem. 2019;27:1159–1194. doi: 10.1016/j.bmc.2019.02.044. [DOI] [PubMed] [Google Scholar]
- 3.Gramec D., Peterlin Mašič L., Sollner Dolenc M. Bioactivation Potential of Thiophene-Containing Drugs. Chem. Res. Toxicol. 2014;27:1344–1358. doi: 10.1021/tx500134g. [DOI] [PubMed] [Google Scholar]
- 4.Maillard L.T., Bertout S., Quinonéro O., Akalin G., Turan-Zitouni G., Fulcrand P., Demirci F., Martinez J., Masurier N. Synthesis and Anti-Candida Activity of Novel 2-Hydrazino-1,3-Thiazole Derivatives. Bioorganic Med. Chem. Lett. 2013;23:1803–1807. doi: 10.1016/j.bmcl.2013.01.039. [DOI] [PubMed] [Google Scholar]
- 5.Fersing C., Boudot C., Pedron J., Hutter S., Primas N., Caroline D., Bourgeade-Delmas S., Sournia-Saquet A., Moreau A., Cohen A., et al. 8-Aryl-6-Chloro-3-Nitro-2-(Phenylsulfonylmethyl)Imidazo[1,2-a]Pyridines as Potent Antitrypanosomatid Molecules Bioactivated by Type 1 Nitroreductases. Eur. J. Med. Chem. 2018;157:115–126. doi: 10.1016/j.ejmech.2018.07.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fersing C., Basmaciyan L., Boudot C., Pedron J., Hutter S., Cohen A., Caroline D., Primas N., Laget M., Casanova M., et al. Nongenotoxic 3-Nitroimidazo[1,2-a]Pyridines Are NTR1 Substrates That Display Potent in Vitro Antileishmanial Activity. ACS Med. Chem. Lett. 2018;10:34–39. doi: 10.1021/acsmedchemlett.8b00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fersing C., Boudot C., Castera-Ducros C., Pinault E., Hutter S., Paoli-Lombardo R., Primas N., Pedron J., Seguy L., Bourgeade-Delmas S., et al. 8-Alkynyl-3-Nitroimidazopyridines Display Potent Antitrypanosomal Activity against Both T. b. Brucei and Cruzi. Eur. J. Med. Chem. 2020;202:112558. doi: 10.1016/j.ejmech.2020.112558. [DOI] [PubMed] [Google Scholar]
- 8.Fersing C., Boudot C., Paoli-Lombardo R., Primas N., Pinault E., Hutter S., Castera-Ducros C., Kabri Y., Pedron J., Bourgeade-Delmas S., et al. Antikinetoplastid SAR Study in 3-Nitroimidazopyridine Series: Identification of a Novel Non-Genotoxic and Potent Anti-T. b. Brucei Hit-Compound with Improved Pharmacokinetic Properties. Eur. J. Med. Chem. 2020;206:112668. doi: 10.1016/j.ejmech.2020.112668. [DOI] [PubMed] [Google Scholar]
- 9.Bosson-Vanga H., Primas N., Franetich J.-F., Lavazec C., Gomez L., Ashraf K., Tefit M., Soulard V., Dereuddre-Bosquet N., Le Grand R., et al. A New Thienopyrimidinone Chemotype Shows Multistage Activity against Plasmodium falciparum, Including Artemisinin-Resistant Parasites. Microbiol. Spectr. 2021;9:e00274-21. doi: 10.1128/Spectrum.00274-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pullarao B., Sharif S.D.K., Kumar D.R., Ramachandran D. Design, Synthesis and Biological Evaluation of Thiophene Based Pyrimidin-4-One Derivatives as New Type of Antimicrobial Agents. Asian J. Chem. 2016;28:1997–2000. doi: 10.14233/ajchem.2016.19861. [DOI] [Google Scholar]
- 11.Ortikov I.S., Turdibaev Z.É., Islamova Z.I., Élmuradov B.Z., Abdurazakov A.S., Bektemirov A.M., Osipova S.O., Khushbaktova Z.A., Syrov V.N., Shakhidoyatov K.M. Search for Bactericides Among Derivatives of Deoxyvasicinone, Mackinazolinone, and Thienopyrimidinones. Pharm. Chem. J. 2017;51:456–464. doi: 10.1007/s11094-017-1633-0. [DOI] [Google Scholar]
- 12.Habib N.S., Soliman R., El-Tombary A.A., El-Hawash S.A., Shaaban O.G. Synthesis and Biological Evaluation of Novel Series of Thieno[2,3-d]Pyrimidine Derivatives as Anticancer and Antimicrobial Agents. Med. Chem. Res. 2013;22:3289–3308. doi: 10.1007/s00044-012-0324-3. [DOI] [PubMed] [Google Scholar]
- 13.Kanawade S.B., Toche R.B., Rajani D.P. Synthetic Tactics of New Class of 4-Aminothieno[2,3-d]Pyrimidine-6-Carbonitrile Derivatives Acting as Antimicrobial Agents. Eur. J. Med. Chem. 2013;64:314–320. doi: 10.1016/j.ejmech.2013.03.039. [DOI] [PubMed] [Google Scholar]
- 14.Woodring J.L., Patel G., Erath J., Behera R., Lee P.J., Leed S.E., Rodriguez A., Sciotti R.J., Mensa-Wilmot K., Pollastri M.P. Evaluation of Aromatic 6-Substituted Thienopyrimidines as Scaffolds against Parasites That Cause Trypanosomiasis, Leishmaniasis, and Malaria. Med. Chem. Commun. 2015;6:339–346. doi: 10.1039/C4MD00441H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shao X., AbdelKhalek A., Abutaleb N.S., Velagapudi U.K., Yoganathan S., Seleem M.N., Talele T.T. Chemical Space Exploration around Thieno[3,2-d]Pyrimidin-4(3H)-One Scaffold Led to a Novel Class of Highly Active Clostridium Difficile Inhibitors. J. Med. Chem. 2019;62:9772–9791. doi: 10.1021/acs.jmedchem.9b01198. [DOI] [PubMed] [Google Scholar]
- 16.Aly H.M., Saleh N.M., Elhady H.A. Design and Synthesis of Some New Thiophene, Thienopyrimidine and Thienothiadiazine Derivatives of Antipyrine as Potential Antimicrobial Agents. Eur. J. Med. Chem. 2011;46:4566–4572. doi: 10.1016/j.ejmech.2011.07.035. [DOI] [PubMed] [Google Scholar]
- 17.Desroches J., Kieffer C., Primas N., Hutter S., Gellis A., El-Kashef H., Rathelot P., Verhaeghe P., Azas N., Vanelle P. Discovery of New Hit-Molecules Targeting Plasmodium Falciparum through a Global SAR Study of the 4-Substituted-2-Trichloromethylquinazoline Antiplasmodial Scaffold. Eur. J. Med. Chem. 2017;125:68–86. doi: 10.1016/j.ejmech.2016.09.029. [DOI] [PubMed] [Google Scholar]
- 18.De Schutter J.W., Morrison J.P., Morrison M.J., Ciulli A., Imperiali B. Targeting Bacillosamine Biosynthesis in Bacterial Pathogens: Development of Inhibitors to a Bacterial Amino-Sugar Acetyltransferase from Campylobacter Jejuni. J. Med. Chem. 2017;60:2099–2118. doi: 10.1021/acs.jmedchem.6b01869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mavrova A.T., Vuchev D., Anichina K., Vassilev N. Synthesis, Antitrichinnellosis and Antiprotozoal Activity of Some Novel Thieno[2,3-d]Pyrimidin-4(3H)-Ones Containing Benzimidazole Ring. Eur. J. Med. Chem. 2010;45:5856–5861. doi: 10.1016/j.ejmech.2010.09.050. [DOI] [PubMed] [Google Scholar]
- 20.Kim J., Kwon J., Lee D., Jo S., Park D.-S., Choi J., Park E., Hwang J.Y., Ko Y., Choi I., et al. Serendipitous Discovery of 2-((Phenylsulfonyl)Methyl)-Thieno[3,2-d]Pyrimidine Derivatives as Novel HIV-1 Replication Inhibitors. Bioorganic Med. Chem. Lett. 2014;24:5473–5477. doi: 10.1016/j.bmcl.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 21.Sławiński J., Żołnowska B., Pirska D., Kędzia A., Kwapisz E. Synthesis and Antibacterial Activity of Novel 4-Chloro-2-Mercaptobenzenesulfonamide Derivatives. J. Enzym. Inhib. Med. Chem. 2013;28:41–51. doi: 10.3109/14756366.2011.625024. [DOI] [PubMed] [Google Scholar]
- 22.Patel A. Modi Synthesis and Biological Evaluation of Schiff Base Involving Thieno[2,3-d] Pyrimidine Moiety as Antimicrobial Agents. RJLBPCS. 2019;5:31–41. doi: 10.26479/2019.0505.03. [DOI] [Google Scholar]
- 23.Giri T., Sailaja G., Laxminarayana E., Thirumala Chary M., Ramesh M. Synthesis and Antibacterial Activity of Novel 4-{4-(Methylamino)Thieno[3,2-d]Pyrimidin-2-Yl}-Benzohydrazide Derivatives. Russ. J. Gen. Chem. 2017;87:1275–1280. doi: 10.1134/S1070363217060238. [DOI] [Google Scholar]
- 24.Tharikoppula G., Eppakayala L., Maringanti T.C., Kamalapuram C., Kudle K.R. Synthesis and Antibacterial Activity of Thienopyrimidine Amide Derivatives. Asian J. Chem. 2017;29:1515–1521. doi: 10.14233/ajchem.2017.20545. [DOI] [Google Scholar]
- 25.Temburnikar K.W., Zimmermann S.C., Kim N.T., Ross C.R., Gelbmann C., Salomon C.E., Wilson G.M., Balzarini J., Seley-Radtke K.L. Antiproliferative Activities of Halogenated Thieno[3,2-d]pyrimidines. Bioorganic Med. Chem. 2014;22:2113–2122. doi: 10.1016/j.bmc.2014.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prabhakar V., Babu Kondra S., Maddula S.R. Synthesis, Structural Elucidation of Novel Thieno[2,3-d]Pyrimidine Core Unit Containing 1,2,4-Triazoles and Thiophenes as Potent Antimicrobial Activity. Org. Chem. Curr. Res. 2016;5:1000169. doi: 10.4172/2161-0401.1000169. [DOI] [Google Scholar]
- 27.Prabhakar V., Babu S.J., Siva Jyothi S.V.L. Synthesis, Structural Elucidation and Anti-Bacterial Evaluation of Some Novel Heterocyclic Molecules Derived from Thieno[2,3-d]pyrimidine as a Core Unit. Org. Chem. Curr. Res. 2016;5:1000172. doi: 10.4172/2161-0401.1000172. [DOI] [Google Scholar]
- 28.Sang Y., Han S., Han S., Pannecouque C., De Clercq E., Zhuang C., Chen F. Follow On-Based Optimization of the Biphenyl-DAPYs as HIV-1 Nonnucleoside Reverse Transcriptase Inhibitors against the Wild-Type and Mutant Strains. Bioorganic Chem. 2019;89:102974. doi: 10.1016/j.bioorg.2019.102974. [DOI] [PubMed] [Google Scholar]
- 29.Al-Harbi R.A.K., Abdel-Rahman A.A.H. Synthesis and Anti-HBV Activity of 2-(Methylthio)Thieno[3,2-d]Pyrimidin- 4(1H)-One Analogues of ACV. Der Pharma Chem. 2013;5:1–7. [Google Scholar]
- 30.Kankanala J., Kirby K.A., Huber A.D., Casey M.C., Wilson D.J., Sarafianos S.G., Wang Z. Design, Synthesis and Biological Evaluations of N-Hydroxy Thienopyrimidine-2,4-Diones as Inhibitors of HIV Reverse Transcriptase-Associated RNase H. Eur. J. Med. Chem. 2017;141:149–161. doi: 10.1016/j.ejmech.2017.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abu-Hashem A.A., Abu-Zied K.M., El-Shehry M.F. Synthetic Utility of Bifunctional Thiophene Derivatives and Antimicrobial Evaluation of the Newly Synthesized Agents. Mon. Chem. 2011;142:539–545. doi: 10.1007/s00706-011-0456-z. [DOI] [Google Scholar]
- 32.Dewal M.B., Wani A.S., Vidaillac C., Oupický D., Rybak M.J., Firestine S.M. Thieno[2,3-d]Pyrimidinedione Derivatives as Antibacterial Agents. Eur. J. Med. Chem. 2012;51:145–153. doi: 10.1016/j.ejmech.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shaaban O.G., Issa D.A.E., El-Tombary A.A., Abd El Wahab S.M., Abdel Wahab A.E., Abdelwahab I.A. Synthesis and Molecular Docking Study of Some 3,4-Dihydrothieno[2,3-d]Pyrimidine Derivatives as Potential Antimicrobial Agents. Bioorganic Chem. 2019;88:102934. doi: 10.1016/j.bioorg.2019.102934. [DOI] [PubMed] [Google Scholar]
- 34.Endo Y., Kawai K., Asano T., Amano S., Asanuma Y., Sawada K., Onodera Y., Ueo N., Takahashi N., Sonoda Y., et al. 2-(Isopropylamino)Thieno[3,2-d]Pyrimidin-4(3H)-One Derivatives as Selective Phosphodiesterase 7 Inhibitors with Potent in Vivo Efficacy. Bioorganic Med. Chem. Lett. 2015;25:1910–1914. doi: 10.1016/j.bmcl.2015.03.031. [DOI] [PubMed] [Google Scholar]
- 35.González Cabrera D., Le Manach C., Douelle F., Younis Y., Feng T.-S., Paquet T., Nchinda A.T., Street L.J., Taylor D., de Kock C., et al. 2,4-Diaminothienopyrimidines as Orally Active Antimalarial Agents. J. Med. Chem. 2014;57:1014–1022. doi: 10.1021/jm401760c. [DOI] [PubMed] [Google Scholar]
- 36.Cohen A., Suzanne P., Lancelot J.-C., Verhaeghe P., Lesnard A., Basmaciyan L., Hutter S., Laget M., Dumètre A., Paloque L., et al. Discovery of New Thienopyrimidinone Derivatives Displaying Antimalarial Properties toward Both Erythrocytic and Hepatic Stages of Plasmodium. Eur. J. Med. Chem. 2015;95:16–28. doi: 10.1016/j.ejmech.2015.03.011. [DOI] [PubMed] [Google Scholar]
- 37.Chambhare R.V., Khadse B.G., Bobde A.S., Bahekar R.H. Synthesis and Preliminary Evaluation of Some N-[5-(2-Furanyl)-2-Methyl-4-Oxo-4H-Thieno[2,3-d]Pyrimidin-3-Yl]-Carboxamide and 3-Substituted-5-(2-Furanyl)-2-Methyl-3H-Thieno[2,3-d]Pyrimidin-4-Ones as Antimicrobial Agents. Eur. J. Med. Chem. 2003;38:89–100. doi: 10.1016/S0223-5234(02)01442-3. [DOI] [PubMed] [Google Scholar]
- 38.Abdel Hamid A.M., Shehta W. Synthesis of Some Novel Furan-Tagged Thienopyrimidine Derivatives as Antibacterial Agents: Synthesis of Some Novel Furan-Tagged Thienopyrimidine Derivatives as Antibacterial Agents. J. Heterocycl. Chem. 2019;56:485–492. doi: 10.1002/jhet.3423. [DOI] [Google Scholar]
- 39.Aly H.M., Saleh N.M. Utility of a Pyrimidine Thione Derivative in the Synthesis of New Fused Pyrimido[4,5-d]Pyrimidine, Pyrido[2,3-d]Pyrimidine and Different Types of Thienopyrimidine Derivatives. Int. J. Adv. Res. 2014;2:694–702. [Google Scholar]
- 40.Malaria. [(accessed on 28 August 2020)]. Available online: https://www.who.int/news-room/fact-sheets/detail/malaria.
- 41.World Health Organization . World Malaria Report 2020: 20 Years of Global Progress and Challenges. World Health Organization; Geneva, Switzerland: 2020. [Google Scholar]
- 42.Ariey F., Witkowski B., Amaratunga C., Beghain J., Langlois A.-C., Khim N., Kim S., Duru V., Bouchier C., Ma L., et al. A Molecular Marker of Artemisinin-Resistant Plasmodium Falciparum Malaria. Nature. 2014;505:50–55. doi: 10.1038/nature12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okombo J., Chibale K. Recent Updates in the Discovery and Development of Novel Antimalarial Drug Candidates. Med. Chem. Comm. 2018;9:437–453. doi: 10.1039/C7MD00637C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu J., Chen T., Liu J., Ma R., Lu W., Huang J., Li H., Li J., Jiang H. 2-(3,4-Dihydro-4-Oxothieno[2,3-d]Pyrimidin-2-Ylthio) Acetamides as a New Class of Falcipain-2 Inhibitors. 3. Design, Synthesis and Biological Evaluation. Molecules. 2009;14:785–797. doi: 10.3390/molecules14020785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rosenthal P.J. Falcipain Cysteine Proteases of Malaria Parasites: An Update. Biochim. Et Biophys. Acta (BBA)—Proteins Proteom. 2020;1868:140362. doi: 10.1016/j.bbapap.2020.140362. [DOI] [PubMed] [Google Scholar]
- 46.Bekono B.D., Ntie-Kang F., Owono Owono L.C., Megnassan E. Targeting Cysteine Proteases from Plasmodium Falciparum: A General Overview, Rational Drug Design and Computational Approaches for Drug Discovery. Curr. Drug Targets. 2018;19:501–526. doi: 10.2174/1389450117666161221122432. [DOI] [PubMed] [Google Scholar]
- 47.Lee B.J., Singh A., Chiang P., Kemp S.J., Goldman E.A., Weinhouse M.I., Vlasuk G.P., Rosenthal P.J. Antimalarial Activities of Novel Synthetic Cysteine Protease Inhibitors. Antimicrob. Agents Chemother. 2003;47:3810–3814. doi: 10.1128/AAC.47.12.3810-3814.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Edlin C.D., Morgans G., Winks S., Duffy S., Avery V.M., Wittlin S., Waterson D., Burrows J., Bryans J. Identification and In-Vitro ADME Assessment of a Series of Novel Anti-Malarial Agents Suitable for Hit-to-Lead Chemistry. ACS Med. Chem. Lett. 2012;3:570–573. doi: 10.1021/ml300091c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barrows R.D., Hammill J.T., Tran M.C., Falade M.O., Rice A.L., Davis C.W., Emge T.J., Rablen P.R., Kiplin Guy R., Knapp S. Evaluation of 1,1-Cyclopropylidene as a Thioether Isostere in the 4-Thio-Thienopyrimidine (TTP) Series of Antimalarials. Bioorganic Med. Chem. 2020;28:115758. doi: 10.1016/j.bmc.2020.115758. [DOI] [PubMed] [Google Scholar]
- 50.Fletcher S., Avery V.M. A Novel Approach for the Discovery of Chemically Diverse Anti-Malarial Compounds Targeting the Plasmodium Falciparum Coenzyme A Synthesis Pathway. Malar. J. 2014;13:343. doi: 10.1186/1475-2875-13-343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fletcher S., Lucantoni L., Sykes M.L., Jones A.J., Holleran J.P., Saliba K.J., Avery V.M. Biological Characterization of Chemically Diverse Compounds Targeting the Plasmodium Falciparum Coenzyme A Synthesis Pathway. Parasites Vectors. 2016;9:589. doi: 10.1186/s13071-016-1860-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weidner T., Lucantoni L., Nasereddin A., Preu L., Jones P.G., Dzikowski R., Avery V.M., Kunick C. Antiplasmodial Dihetarylthioethers Target the Coenzyme A Synthesis Pathway in Plasmodium Falciparum Erythrocytic Stages. Malar. J. 2017;16:192. doi: 10.1186/s12936-017-1839-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kikuchi H., Yamamoto K., Horoiwa S., Hirai S., Kasahara R., Hariguchi N., Matsumoto M., Oshima Y. Exploration of a New Type of Antimalarial Compounds Based on Febrifugine. J. Med. Chem. 2006;49:4698–4706. doi: 10.1021/jm0601809. [DOI] [PubMed] [Google Scholar]
- 54.Gonzàlez Cabrera D., Douelle F., Le Manach C., Han Z., Paquet T., Taylor D., Njoroge M., Lawrence N., Wiesner L., Waterson D., et al. Structure–Activity Relationship Studies of Orally Active Antimalarial 2,4-Diamino-Thienopyrimidines. J. Med. Chem. 2015;58:7572–7579. doi: 10.1021/acs.jmedchem.5b01156. [DOI] [PubMed] [Google Scholar]
- 55.Garrido A., Lepailleur A., Mignani S.M., Dallemagne P., Rochais C. HERG Toxicity Assessment: Useful Guidelines for Drug Design. Eur. J. Med. Chem. 2020;195:112290. doi: 10.1016/j.ejmech.2020.112290. [DOI] [PubMed] [Google Scholar]
- 56.van der Watt M.E., Reader J., Churchyard A., Nondaba S.H., Lauterbach S.B., Niemand J., Abayomi S., van Biljon R.A., Connacher J.I., van Wyk R.D.J., et al. Potent Plasmodium Falciparum Gametocytocidal Compounds Identified by Exploring the Kinase Inhibitor Chemical Space for Dual Active Antimalarials. J. Antimicrob. Chemother. 2018;73:1279–1290. doi: 10.1093/jac/dky008. [DOI] [PubMed] [Google Scholar]
- 57.Woodring J.L., Behera R., Sharma A., Wiedeman J., Patel G., Singh B., Guyett P., Amata E., Erath J., Roncal N., et al. Series of Alkynyl-Substituted Thienopyrimidines as Inhibitors of Protozoan Parasite Proliferation. ACS Med. Chem. Lett. 2018;9:996–1001. doi: 10.1021/acsmedchemlett.8b00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trypanosomiasis, Human African (Sleeping Sickness) [(accessed on 8 November 2021)]. Available online: https://www.who.int/news-room/fact-sheets/detail/trypanosomiasis-human-african-(sleeping-sickness)
- 59.Johnston S.R.D., Leary A. Lapatinib: A Novel EGFR/HER2 Tyrosine Kinase Inhibitor for Cancer. Drugs Today. 2006;42:441–453. doi: 10.1358/dot.2006.42.7.985637. [DOI] [PubMed] [Google Scholar]
- 60.Bell A.S., Yu Z., Hutton J.A., Wright M.H., Brannigan J.A., Paape D., Roberts S.M., Sutherell C.L., Ritzefeld M., Wilkinson A.J., et al. Novel Thienopyrimidine Inhibitors of Leishmania N -Myristoyltransferase with On-Target Activity in Intracellular Amastigotes. J. Med. Chem. 2020;63:7740–7765. doi: 10.1021/acs.jmedchem.0c00570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tuberculosis (TB) [(accessed on 6 August 2021)]. Available online: https://www.who.int/news-room/fact-sheets/detail/tuberculosis.
- 62.Rashmi P., Nargund L.V.G., Hazra K., Chandra J.N.N.S. Thienopyrimidines as Novel Inhibitors of Mycobacterium Tuberclosis: Synthesis and In-Vitro Studies. Arch. Pharm. Pharm. Med. Chem. 2011;344:459–465. doi: 10.1002/ardp.201000394. [DOI] [PubMed] [Google Scholar]
- 63.Ananthan S., Faaleolea E.R., Goldman R.C., Hobrath J.V., Kwong C.D., Laughon B.E., Maddry J.A., Mehta A., Rasmussen L., Reynolds R.C., et al. High-Throughput Screening for Inhibitors of Mycobacterium Tuberculosis H37Rv. Tuberculosis. 2009;89:334–353. doi: 10.1016/j.tube.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Harrison G.A., Mayer Bridwell A.E., Singh M., Jayaraman K., Weiss L.A., Kinsella R.L., Aneke J.S., Flentie K., Schene M.E., Gaggioli M., et al. Identification of 4-Amino-Thieno[2,3-d]Pyrimidines as QcrB Inhibitors in Mycobacterium Tuberculosis. mSphere. 2019;4:e00606-19. doi: 10.1128/mSphere.00606-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lu X., Williams Z., Hards K., Tang J., Cheung C.-Y., Aung H.L., Wang B., Liu Z., Hu X., Lenaerts A., et al. Pyrazolo[1,5-a]Pyridine Inhibitor of the Respiratory Cytochrome Bcc Complex for the Treatment of Drug-Resistant Tuberculosis. ACS Infect. Dis. 2019;5:239–249. doi: 10.1021/acsinfecdis.8b00225. [DOI] [PubMed] [Google Scholar]
- 66.Antibiotic Resistance. [(accessed on 23 July 2021)]. Available online: https://www.who.int/news-room/fact-sheets/detail/antibiotic-resistance.
- 67.Campylobacter. [(accessed on 30 August 2021)]. Available online: https://www.who.int/news-room/fact-sheets/detail/campylobacter.
- 68.Luangtongkum T., Jeon B., Han J., Plummer P., Logue C.M., Zhang Q. Antibiotic Resistance in Campylobacter: Emergence, Transmission and Persistence. Future Microbiol. 2009;4:189–200. doi: 10.2217/17460913.4.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.de Candia M., Altamura C., Denora N., Cellamare S., Nuzzolese M., De Vito D., Voskressensky L.G., Varlamov A.V., Altomare C.D. Physicochemical Properties and Antimicrobial Activity of New Spirocyclic Thieno[2,3-d]Pyrimidin-4(3H)-One Derivatives. Chem. Heterocycl. Comp. 2017;53:357–363. doi: 10.1007/s10593-017-2057-1. [DOI] [Google Scholar]
- 70.Tolba M.S., El-Dean A.M.K., Ahmed M., Hassanien R., Farouk M. Synthesis and Antimicrobial Activity of Some New Thienopyrimidine Derivatives. Arkivoc. 2017;2017:229–243. doi: 10.24820/ark.5550190.p010.226. [DOI] [Google Scholar]
- 71.Saddik A.A., Kamal El-Dean A.M., El-Said W.A., Hassan K.M., Abbady M.S. Synthesis, Antimicrobial, and Anticancer Activities of a New Series of Thieno[2,3-d] Pyrimidine Derivatives: Synthesis of New Series from Thieno[2,3-d]Pyrimidine and Study the Antimicrobial and Anticancer Activities for Some Compounds. J. Heterocycl. Chem. 2018;55:2111–2122. doi: 10.1002/jhet.3256. [DOI] [Google Scholar]
- 72.Bhagchand J., Santra S. Prasanta Kumar Santra Synthesis and Evaluation of Antimicrobial Activity of Pyrimidine Derivatives. Asian J. Pharm. Clin. Res. 2019;12:156–163. doi: 10.22159/ajpcr.2019.v12i5.30919. [DOI] [Google Scholar]
- 73.Hafez H.N., El-Gazzar A.-R.B.A., Zaki M.E.A. Simple Approach to Thieno[3,2-d]Pyrimidines as New Scaffolds of Antimicrobial Activities. Acta Pharm. 2016;66:331–351. doi: 10.1515/acph-2016-0029. [DOI] [PubMed] [Google Scholar]
- 74.Mathias F., Curti C., Montana M., Bornet C., Vanelle P. Management of Adult Clostridium Difficile Digestive Contaminations: A Literature Review. Eur. J. Clin. Microbiol. Infect. Dis. 2019;38:209–231. doi: 10.1007/s10096-018-3419-z. [DOI] [PubMed] [Google Scholar]
- 75.CDC Most Cases of C. Difficile Occur While Taking Antibiotics or Soon after. [(accessed on 31 August 2021)]; Available online: https://www.cdc.gov/cdiff/index.html.
- 76.Antifungal Resistance|Fungal Diseases|CDC. [(accessed on 22 July 2021)]; Available online: https://www.cdc.gov/fungal/antifungal-resistance.html.
- 77.Krammer F., Smith G.J.D., Fouchier R.A.M., Peiris M., Kedzierska K., Doherty P.C., Palese P., Shaw M.L., Treanor J., Webster R.G., et al. Influenza. Nat. Rev. Dis. Primers. 2018;4:1–21. doi: 10.1038/s41572-018-0002-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang H., Zhou L., Amichai S., Zandi K., Cox B., Schinazi R., Amblard F. Novel Influenza Polymerase PB2 Inhibitors for the Treatment of Influenza A Infection. Bioorganic Med. Chem. Lett. 2019;29:126639. doi: 10.1016/j.bmcl.2019.126639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Engelhardt O.G., Fodor E. Functional Association between Viral and Cellular Transcription during Influenza Virus Infection. Rev. Med. Virol. 2006;16:329–345. doi: 10.1002/rmv.512. [DOI] [PubMed] [Google Scholar]
- 80.The Lancet Towards Elimination of Viral Hepatitis by 2030. Lancet. 2016;388:308. doi: 10.1016/S0140-6736(16)31144-8. [DOI] [PubMed] [Google Scholar]
- 81.Nelson N.P., Easterbrook P.J., McMahon B.J. Epidemiology of Hepatitis B Virus Infection and Impact of Vaccination on Disease. Clin. Liver Dis. 2016;20:607–628. doi: 10.1016/j.cld.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.UNAIDS Data 2020. [(accessed on 19 October 2020)]. Available online: https://www.unaids.org/en/resources/documents/2020/unaids-data.
- 83.HIV/AIDS. [(accessed on 1 October 2020)]. Available online: https://www.who.int/news-room/fact-sheets/detail/hiv-aids.
- 84.Maartens G., Celum C., Lewin S.R. HIV Infection: Epidemiology, Pathogenesis, Treatment, and Prevention. Lancet. 2014;384:258–271. doi: 10.1016/S0140-6736(14)60164-1. [DOI] [PubMed] [Google Scholar]
- 85.WHO|HIV Drug Resistance Report 2019. [(accessed on 19 October 2020)]. Available online: http://www.who.int/hiv/pub/drugresistance/hivdr-report-2019/en/
- 86.Kang D., Fang Z., Li Z., Huang B., Zhang H., Lu X., Xu H., Zhou Z., Ding X., Daelemans D., et al. Design, Synthesis, and Evaluation of Thiophene[3,2-d]Pyrimidine Derivatives as HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors with Significantly Improved Drug Resistance Profiles. J. Med. Chem. 2016;59:7991–8007. doi: 10.1021/acs.jmedchem.6b00738. [DOI] [PubMed] [Google Scholar]
- 87.Kang D., Fang Z., Huang B., Lu X., Zhang H., Xu H., Huo Z., Zhou Z., Yu Z., Meng Q., et al. Structure-Based Optimization of Thiophene[3,2-d]Pyrimidine Derivatives as Potent HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors with Improved Potency against Resistance-Associated Variants. J. Med. Chem. 2017;60:4424–4443. doi: 10.1021/acs.jmedchem.7b00332. [DOI] [PubMed] [Google Scholar]
- 88.Kang D., Ding X., Wu G., Huo Z., Zhou Z., Zhao T., Feng D., Wang Z., Tian Y., Daelemans D., et al. Discovery of Thiophene[3,2-d]Pyrimidine Derivatives as Potent HIV-1 NNRTIs Targeting the Tolerant Region I of NNIBP. ACS Med. Chem. Lett. 2017;8:1188–1193. doi: 10.1021/acsmedchemlett.7b00361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kang D., Wang Z., Chen M., Feng D., Wu G., Zhou Z., Jing L., Zuo X., Jiang X., Daelemans D., et al. Discovery of Potent HIV -1 Non-nucleoside Reverse Transcriptase Inhibitors by Exploring the Structure–Activity Relationship of Solvent-exposed Regions I. Chem. Biol. Drug Des. 2019;93:430–437. doi: 10.1111/cbdd.13429. [DOI] [PubMed] [Google Scholar]
- 90.Kang D., Zhang H., Wang Z., Zhao T., Ginex T., Luque F.J., Yang Y., Wu G., Feng D., Wei F., et al. Identification of Dihydrofuro[3,4-d]Pyrimidine Derivatives as Novel HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors with Promising Antiviral Activities and Desirable Physicochemical Properties. J. Med. Chem. 2019;62:1484–1501. doi: 10.1021/acs.jmedchem.8b01656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yang Y., Kang D., Nguyen L.A., Smithline Z.B., Pannecouque C., Zhan P., Liu X., Steitz T.A. Structural Basis for Potent and Broad Inhibition of HIV-1 RT by Thiophene[3,2-d]Pyrimidine Non-Nucleoside Inhibitors. eLife. 2018;7:e36340. doi: 10.7554/eLife.36340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sang Y., Han S., Pannecouque C., De Clercq E., Zhuang C., Chen F. Conformational Restriction Design of Thiophene-Biphenyl-DAPY HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors. Eur. J. Med. Chem. 2019;182:111603. doi: 10.1016/j.ejmech.2019.111603. [DOI] [PubMed] [Google Scholar]
- 93.Richman D.D., Margolis D.M., Delaney M., Greene W.C., Hazuda D., Pomerantz R.J. The Challenge of Finding a Cure for HIV Infection. Science. 2009;323:1304–1307. doi: 10.1126/science.1165706. [DOI] [PubMed] [Google Scholar]
- 94.Vargas B., Giacobbi N.S., Sanyal A., Venkatachari N.J., Han F., Gupta P., Sluis-Cremer N. Inhibitors of Signaling Pathways That Block Reversal of HIV-1 Latency. Antimicrob. Agents Chemother. 2018;63:e01744-18. doi: 10.1128/AAC.01744-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.