

Abstract
Introduction/Aims:
There is considerable heterogenicity in clinical outcomes in Duchenne Muscular Dystrophy (DMD). The aim of current study was to assess whether dystrophin gene (DMD) pathogenic variant location influences upper extremity and lower extremity motor function outcomes in a large prospective cohort.
Methods:
We used longitudinal timed and quantitative motor function measurements obtained from 154 boys with DMD over a 10-year period by the Cooperative International Neuromuscular Research Group Duchenne Natural History Study (CINRG-DNHS) to understand how the trajectories of motor function differ based on proximal versus distal DMD pathogenic variants. Proximal variants were defined as located proximal to 5’ DMD intron 44, and distal variants as those including nucleotides 3’ DMD including intron 44. Distal DMD variants are predicted to alter the expression of short dystrophin isoforms (Dp140, Dp116, and Dp71). We compared various upper extremity and lower extremity motor function in these two groups, after adjusting for total lifetime corticosteroid use.
Results:
The time to loss-of-ambulation and timed motor function measurements of both upper and lower limbs over a ten-year period were comparable between boys with proximal (n = 53) and distal DMD pathogenic variants (n = 101). Age had a significant effect on several motor function outcomes. Boys younger than 7 years of age (n = 49) showed gain in function whereas boys 7 years and older (n = 71) declined, regardless of dystrophin pathogenic variant location.
Discussion:
The longitudinal decline in upper and lower motor functions is independent of proximal versus distal DMD pathogenic variants.
Introduction
Duchenne Muscular Dystrophy (DMD) is a chromosome X-linked genetic disease caused by mutations in the dystrophin gene (DMD) encoding dystrophin protein (1, 2). DMD consists of 79 exons and encodes full-length dystrophin (dp427) and shorter dystrophin isoforms (dp260, dp140, dp116, and dp71) (3). Proximal DMD pathogenic variants (proximal to 5’ DMD intron 44) affect the expression of full-length dystrophin (dp427) and dp260 isoform, whereas distal DMD pathogenic variants (mutations in 3’ DMD including intron 44) are predicted to affect the expression of shorter dystrophin isoforms (dp140, dp116, dp71).
An enduring challenge even in a monogenic disease such as DMD is disease heterogeneity, and variability in clinical outcomes is evident across multiple organs (4–16). We and others have shown that both DMD genotype and genetic modifiers affect motor, respiratory, and cognitive outcomes in DMD. Bello et al. using the Cooperative International Neuromuscular Research Group Duchenne Natural History Study (CINRG-DNHS) cohort showed that boys with a non-sense mutation in DMD lost ambulation at a median age of 11.1 years, compared to those boys with deletions amenable to DMD exon 44 skipping, who lost ambulation at a median age of 14.8 years. Likewise, genetic modifiers such as SPP1, LTBP4 and CD40 affect the age of loss-of-ambulation in DMD; these modifiers have been validated in multiple, independent cohorts of DMD (9–13).
Recently, boys with distal DMD pathogenic variants were shown to have lower pulmonary function measures compared to those with proximal DMD pathogenic variants (17). Similarly, cognitive burden in DMD vary depending between boys with proximal and distal DMD pathogenic variants (14–16, 18–19). Boys with distal pathogenic variants have higher incidence of speech delay, neurodevelopmental disorders like autism, attention-hyperactivity, and poorer performance on neuropsychological assessment.
While skeletal muscle pathology is largely ascribable to lack of full-length Dp427 dystrophin, the role of shorter isoforms in muscle is under-studied and possibly under-recognized; furthermore, the interactions between the central nervous system (where the importance of short dystrophin isoforms is well-established) and muscle are manifold and complex. Whether distal DMD pathogenic variants affect motor outcomes or influence the trajectory of functional decline is not known and forms the scientific rationale for our present work.
The drug pipeline in DMD is rich and newer agents have recently been approved for clinical use. In this context, showing drug efficacy in clinical trials will draw upon knowledge from natural history studies of DMD that is particularly informative of pathophysiological mechanisms. Knowledge gained from natural history studies not only help understand disease heterogeneity, they are also helpful for prognostication during clinical care, optimal stratification of subjects, and in improving clinical trial design in DMD. The latter is important owing to the resource-intense nature of clinical trials in rare diseases such as DMD. With these long-term goals in mind, in the present study, we evaluated the longitudinal trajectory in upper and lower motor strength in proximal and distal DMD pathogenic variants using a large, prospective, well-characterized, international cohort of DMD subjects. We hypothesize that subjects with distal DMD pathogenic variants show decline in motor function earlier compared to those with proximal DMD pathogenic variants.
Methods
Standard protocol approvals, registrations, and patient consents
The study was fully compliant with the Principles of Good Clinical Practice according to the International Conference on Harmonization and the Declaration of Helsinki (2000). Each local Institutional Review Board or Ethics Committee approved the study. Written parental consent and assent from children older than 12 years were obtained prior to study enrollment. The study design and characteristics of the CINRG-DNHS cohort have been previously published (20). The CINRG-DNHS cohort is registered with Clinical Trials.gov, number NCT00468832.
Subject characteristics and subgroups
The inclusion criteria for CINRG-DNHS have been previously published (21). Briefly, study subjects were required to meet at least one of the diagnostic criteria. These criteria included (i) out-of-frame deletion or duplication in DMD using multiplex ligation-dependent probe amplification, (ii) complete DMD sequencing predicting an out-of-frame pathogenic variant in DMD, (iii) absent dystrophin immunofluorescence and/or absent dystrophin on immunoblot, (iv) an X-linked pedigree with an older male sibling with out-of-frame pathogenic variant in DMD or a muscle biopsy demonstrating absent dystrophin. The CINRG-DNHS had two cohorts of subjects, ages 2 to 28 years recruited between 2006 to 2009 (parent CINRG-DNHS) (n = 340), and ages 4 to <8 years) recruited between 2012 to 2016 (the younger cohort) (n = 100).
We used loss-of-ambulation (LoA) data from 212 subjects belonging to the parent CINRG-DNHS (22). Longitudinal upper extremity and lower extremity motor function were analyzed from both cohorts on whom DMD pathogenic variants data was available. Based on DMD pathogenic variant location, we divided the subjects into those with “proximal” pathogenic variants, situated entirely 5’ of DMD intron 44, predicting normal expression of Dp140 (Dp140+), and those with “distal” pathogenic variants as those including nucleotides 3’ DMD including intron 44, predicting potential disruption of Dp140 (Dp140−).
Clinical endpoints assessed during the CINRG-DNHS
All subjects underwent several timed functional tests, quantitative muscle strength assessments, and standardized goniometry techniques by trained clinical evaluators during each study visit as previously described (21). Briefly, the times tests included (i) 6MWT, (ii) time to walk/run 10 meters, and (iii) time to supine-to-stand among others. Isometric strength of elbow flexors and extensors, and knee flexors and extensors using the CINRG Quantitative Measurement System (CQMS) were performed in all participants, regardless of their mobility as previously described (21). Age at LoA was defined as patient-reported and evaluator-verified (where possible) age at full-time wheelchair use, as previously described (22).
Statistical analyses
Statistical tests appropriate for each demographic characteristic were performed comparing those with a proximal versus distal pathogenic variants at the first study visit. These methods included Fisher’s exact tests and one-way analysis-of-variance (ANOVA). The Kaplan-Meier method was used to calculate the median time to LoA. The Cox regression model was used to model DMD pathogenic variant location as the predictor, and corticosteroid use (defined as having at least 1 year of use before LoA versus no treatment or less than 1 year while ambulatory) as a binary co-variate. Analysis of each motor function over time was performed using mixed-effect linear models where the outcome was the dependent variable and age, total lifetime corticosteroid exposure, and DMD pathogenic variant location were the independent variables. Each model included a random effect for participant to account for repeated measurements. The quantitative muscle strength values were square-root transformed for analysis. Grip strength showed a curvilinear relationship, and therefore a quadratic term was included in each model. Lastly, mixed-effect linear models were again used to assess functional outcomes stratified by those <7 years versus those age 7 or greater at the time of assessment. Power calculations for loss-of-ambulation analyses were performed with the “powerSurvEpi” package in R v.3.5.3. Statistical significance was defined at a significance level of 0.05.
Results
Time-to-event analyses of LoA
Of the 212 participants from whom LoA data were analyzed, 66 participants (31%) had proximal DMD pathogenic variants and 146 participants (69%) has distal DMD pathogenic variants. As detailed in Table 1, Cox regression analysis with use of corticosteroids as a co-variate showed no significant statistical difference, although median age at LoA in subjects with distal DMD pathogenic variants was 0.5 years earlier (p = not significant; Figure 1).
Table 1.
Parameters for time-to-event analyses of loss-of-ambulation (LoA) from the parent CINRG-DNHS (n = 212).
| Cox regression factor | Level of factor | N | % | LoA events (n, %) | Median age (years) at LoA (95% CI) | HR (95% CI) | p-value |
|---|---|---|---|---|---|---|---|
| DMD pathogenic variant alocation | Proximal to 5’ DMD intron 44 | 66 | 31% | 34 (52%) | 12.5 (11.2 – 18.1) | 0.85 (0.57 – 1.27) | ns |
| Distal as those including nucleotides 3’ DMD including intron 44 | 146 | 69% | 98 (67%) | 12.0 (11.5 – 13.0) | 1 * | - | |
| Corticosteroid treatment | Untreated or treated for less than one year | 55 | 26% | 48 (87%) | 9.7 (9.0 – 11.0) | 1 * | - |
| Treated at least for 1 year while ambulatory | 157 | 74% | 84 (54%) | 13.5 (12.5 – 14.0) | 0.24 (0.16 – 0.35) | <0.0001 | |
| Total | 212 | 100% | 132 (100%) | 12.0 (11.5 – 13.0) | - | - | |
LoA: loss of ambulation; CI: confidence interval; HR: Hazard Ratio.
A HR of 1 is given for factor levels that are taken as reference in the Cox regression model.
Figure 1.
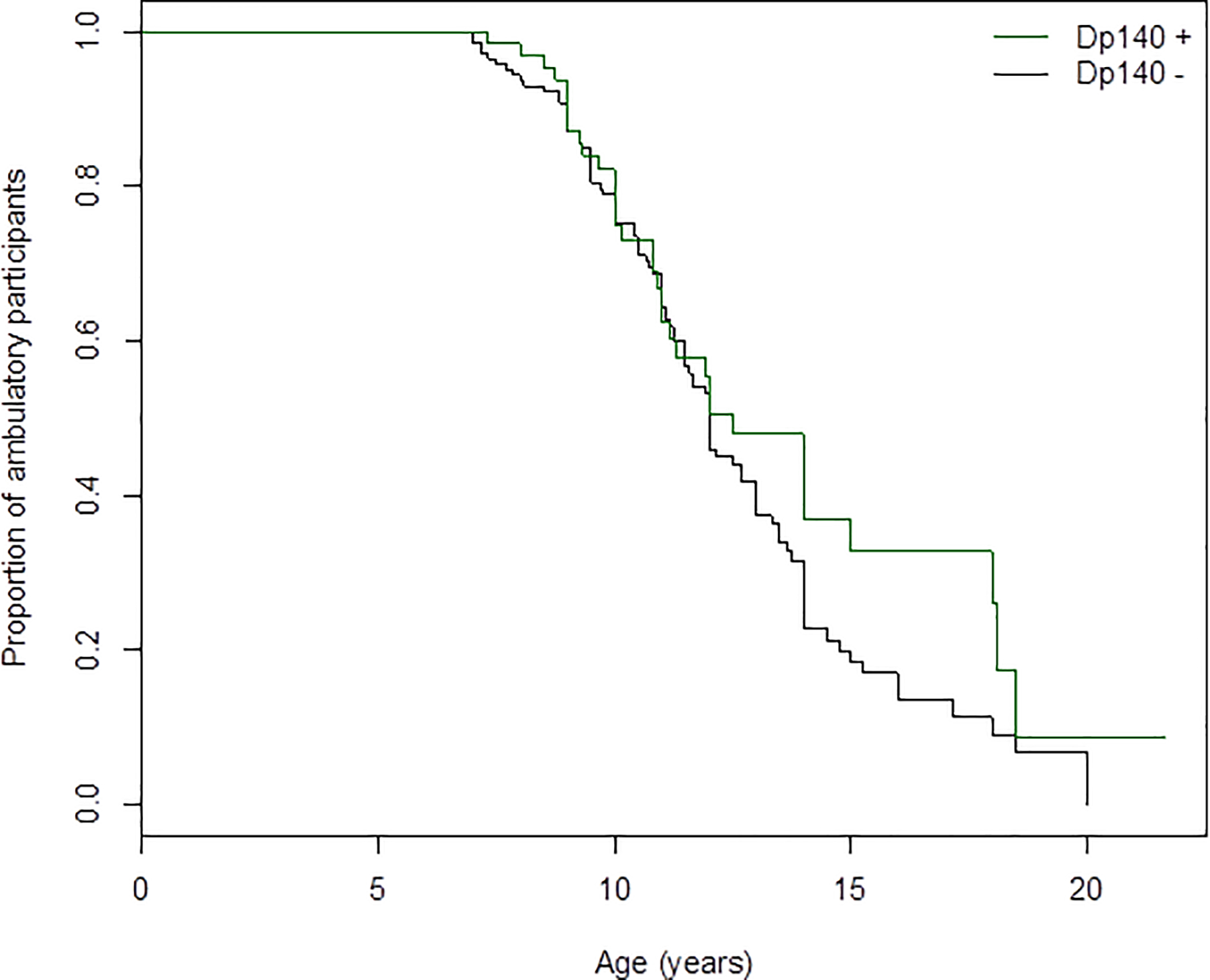
Kaplan-Meier plot of age of loss-of-ambulation based on the proximal DMD pathogenic variants (proximal to 5’ DMD intron 44) versus distal DMD pathogenic variants (as those including nucleotides 3’ DMD including intron 44) from the parent CINRG-DNHS cohort (n = 212).
Characteristics of participants in the longitudinal data
Table 2 summarizes the characteristics of the 154 participants who were included in the longitudinal data analyses. There were 53 boys (34%) with proximal DMD pathogenic variants and 101 boys (66%) with distal DMD pathogenic variants. The mean age and ambulatory status of subjects in the two DMD subgroups were comparable. The mean follow-up period was 4.6 years (SD 2.9 years, range 0 to 8.2 years) in those with proximal DMD pathogenic variants, and 3.6 years (SD 2.8 years, range 0 to 9.9 years) in those with distal DMD pathogenic variants. The lifetime use of corticosteroids was comparable between the two subgroups (data not shown).
Table 2.
Demographic and anthropometric characteristics of the longitudinal cohort at first study visit.
| Characteristic | Proximal DMD pathogenic variants | Distal DMD pathogenic variants | P-value | ||||
|---|---|---|---|---|---|---|---|
| N (%) | Mean ± SD | Median (IQR, Range) | N (%) | Mean ± SD N (%) | Median (IQR, Range) | ||
| Age (years) | 53 | 6.5 ± 1.4 | 6.2 (2.3, 4.8) | 101 | 6.6 ± 1.9 | 6.5 (2.1, 16.3) | 0.72 |
| Calculated height (cm) * | 53 | 116.0 ± 9.6 | 114.9 (14.2, 38.9) | 100 | 117.4 ± 10.8 | 117.8 (13.7, 79.9) | 0.41 |
| Weight (kg) | 53 | 22.3 ± 5.7 | 21.8 (6.8, 27.5) | 101 | 22.3 ± 7.4 | 20.7 (6.6, 52.9) | 0.98 |
| Ambulatory | 0.99 | ||||||
| Yes | 52 (98.1%) | 100 (99.0%) | |||||
| No | 1 (1.9%) | 1 (1.0%) | |||||
| Ethnicity | 0.17 | ||||||
| Hispanic | 1 (1.9%) | 8 (7.9%) | |||||
| Non-Hispanic | 52 (98.1%) | 93 (92.1%) | |||||
| Race | 0.048 | ||||||
| White | 39 (73.6%) | 71 (70.3%) | |||||
| Black | 0 (0%) | 1 (1%) | |||||
| Pacific Islander | 2 (3.8%) | 0 (0%) | |||||
| Asian | 6 (11.3%) | 24 (23.8%) | |||||
| Other | 4 (7.6%) | 2 (2.0%) | |||||
| Unknown | 2 (3.8%) | 3 (3.0%) | |||||
Height calculated from ulna length using Gould equation.
The longitudinal follow-up intervals for subjects with proximal and distal DMD pathogenic variants are listed in Supplemental Table 1. Table 3 summarizes corticosteroid use at first study visit in this cohort.
Table 3:
Descriptive statistics of corticosteroid use at first study visit
| Characteristic | Subjects with proximal DMD pathogenic variants | Subjects with distal DMD pathogenic variants | P-value | ||||
|---|---|---|---|---|---|---|---|
| N | Mean ± SD N (%) | Median (IQR, Range) | N | Mean ± SD N (%) | Median (IQR< Range) | ||
| Total lifetime corticosteroid use (days) | 53 | 355 ± 423 | 208 (563, 1579) | 101 | 266 ± 364 | 82 (468, 1665) | 0.30 |
| Current corticosteroid user | 0.86 | ||||||
| No | 19 (35.9%) | 39 (38.6%) | |||||
| Yes | 34 (64.2%) | 62 (61.4%) | |||||
| Corticosteroid currently used * | 0.96 | ||||||
| None | 19 (35.9%) | 39 (39.0%) | |||||
| Prednisone | 15 (28.3%) | 25 (25.0%) | |||||
| Deflazacort | 11 (20.8%) | 22 (22.0%) | |||||
| Prednisolone | 8 (15.1%) | 14 (14.0%) | |||||
One participant in the distal pathogenic variant group is a current corticosteroid user, but the specific drug is unknown
Timed and quantitative measurements of upper and lower extremities strength based on DMD pathogenic variant location
Functional motor outcomes adjusted for total lifetime corticosteroid use were comparable between proximal and distal DMD pathogenic variants (Table 4). Age had a significant effect on several of the functional motor outcomes.
Table 4:
Assessment of functional outcomes over time with adjustment for total lifetime corticosteroid use
| Outcome | N (Observations) N (Subjects) | DMD pathogenic variant location | DP140 isoform coefficient (P-value) | Age coefficient (p-value) |
|---|---|---|---|---|
| 10 m run/walk velocity (m/sec) | 1061 154 |
Proximal | 0.132 (p=0.31) | −0.103 (p=0.008) |
| Distal | --- | |||
| Standing from supine velocity (rise/sec) | 985 154 |
Proximal | 0.004 (p=0.85) | −0.029 (p<0.001) |
| Distal | --- | |||
| Climbing velocity (task/sec) | 1039 154 |
Proximal | 0.016 (p=0.59) | −0.014 (p=0.13) |
| Distal | --- | |||
| 6MWT distance traveled (m) | 377 93 |
Proximal | −6.8 (p=0.78) | −0.48 (p=0.95) |
| Distal | --- | |||
| Grip strength (lbs.)** | 1009 145 |
Proximal | 0.01 (p=0.90) | 0.217 (p<0.001) |
| Distal | --- | |||
| Elbow extensor strength (lbs.)* | 893 141 |
Proximal | −0.02 (p=0.70) | −0.110 (p<0.001) |
| Distal | --- | |||
| Elbow flexor strength (lbs.)* | 901 141 |
Proximal | −0.04 (p=0.59) | −0.053 (p=0.09) |
| Distal | --- | |||
| Knee extensor strength (lbs.)* | 907 142 |
Proximal | −0.30 (p=0.60) | −0.297 (p<0.001) |
| Distal | --- | |||
| Knee flexor strength (lbs.)* | 895 140 |
Proximal | 0.07 (p=0.49) | 0.0235 (p=0.52) |
| Distal | --- |
Strength outcomes were square-root transformed for analysis, and transformed values are presented here.
This relationship is curvilinear, therefore the coefficient for each DP140 group estimates the instantaneous rate of change in grip strength when all other predictors are zero, rather than the average rate of change over all ages.
We performed additional evaluation of how age affects 6MWT over a 3-year period. Boys 7 years of age and older, over a 3-year period, declined more rapidly, than boys younger than 7 years of age, regardless of DMD pathogenic variant location (Table 5).
Table 5.
Analysis of 3-year change in 6-minute walk test in subjects with proximal and distal DMD pathogenic variants.
| Cohort | Age* | N (obs.) N (indiv.) | Baseline 6MWT (Mean ± SE)** | 6MWT at 3 years (Mean ± SE)** | Calculated 3 year change in 6MWT*** |
|---|---|---|---|---|---|
| All | < 7 years | 189 49 | 353 ± 15 | 439 ± 31 | 85.7 |
| ≥ 7 years | 188 71 | 393 ± 15 | 337 ± 17 | −55.2 | |
| Proximal DMD pathogenic variants | < 7 years | 45 12 | 324 ± 38 | 341 ± 52 | 16.8 |
| ≥ 7 years | 47 22 | 395 ± 29 | 331 ± 34 | −64.4 | |
| Distal DMD pathogenic variants | < 7 years | 144 37 | 363 ± 16 | 454 ± 13 | 91.0 |
| ≥ 7 years | 141 49 | 391 ± 17 | 339 ± 19 | −52.2 |
Note an individual can be in both the <7 and ≥ 7 years sub-cohorts at different times based on age. A participant can contribute assessments while younger than 7 in one model and contribute assessments while 7 or older to the other model.
Mean and SE are predicted values from longitudinal model.
Change over three years is based on the rate of change (coefficient) for the model.
Discussion
Our longitudinal data provides a historical account of the trajectory of motor function decline in DMD. Our hypothesis that DMD subjects with distal DMD pathogenic variants would show decline in motor function earlier that those with proximal DMD pathogenic variants, was not informed by our data. Furthermore, the longitudinal decline in functional motor outcomes did not differ between those with proximal and distal DMD pathogenic variants. The observed trend towards earlier LoA with distal DMD pathogenic variants is similar to those reported previously (19). In a Dutch study focusing on central nervous system (CNS) phenotypes, participants with pathogenic variants in 5’ region of DMD lost ambulation at 11 years (SD 2.6) compared to those with pathogenic variants in the 3’ domain, who lost ambulation at 9.9 years (SD 1.3) (19). We acknowledge that the number of individuals in each category was small, and outliers in either group could skew accurate interpretation of the results.
This potential direction of effect is the same as observed in association with the respiratory phenotype, and may be explained by a worsening of motor or respiratory performance (both mediated by voluntary striated muscles) secondary to some form of CNS involvement, of which there is ample evidence in association with pathogenic variants disrupting Dp140 (5). We propose a viewpoint, supported by literature that individuals with distal DMD pathogenic variants, because of poor executive function, are probably unable to accurately forecast future consequences, or strictly adhere to medical recommendations, or medical treatment. To cite, individuals with DMD who had higher intellectual functioning survived longer (23). Likewise, those with worse intellectual function had poorer long-term outcomes in skeletal, respiratory and cardiac health (24). Altogether, these findings open new perspectives on the tissue-specificity of dystrophin isoforms in mediating end-organ damage. The discordance between genotype-phenotype associations observed in CNS, muscle, and respiratory phenotypes cannot be fully explained in the light of currently available data, and warrants an expansion of “natural history” and observational studies. If we assume that the Hazard Ratio of 0.85 for carrying a proximal DMD pathogenic variant (i.e. 15% per-year reduction of the risk of losing ambulation) is an accurate estimate, and considering that proximal pathogenic variants are observed in 30% of the DMD population, our power calculation estimates a sample size of approximately 1300 non-ambulatory DMD participants to identify this effect with 80% power (1-β = 0.8) and a type-I error (α = 0.05). This large sample size may be reached by future, larger multi-center natural history studies, and/or meta-analyses of existing datasets.
Clinically, it is well known that some skeletal muscles are disproportionately affected compared to others in DMD. Further, timed motor function tests is a read-out of strength of muscle groups, rather than an individual muscle. We therefore reason that a perfect correlation between all timed motor function tests is not biologically plausible. Therefore, it is not incongruous that age was not statistically significant predictor of all timed motor function tests.
Our study has some limitations. The duration of follow-up of individuals with proximal versus distal pathogenic variants were different, and we are therefore taking a nuanced interpretation of our data. Undoubtedly, longitudinal follow-up more equally representing both groups will be highly beneficial. Second, we analyzed DMD pathogenic variant location as a binary variable. This approach makes it more difficult to identify clinically significant pathogenic variant effects in some individuals. Third, for the time to loss of ambulation analysis, we used steroid exposure during ambulation as a covariate defining those who had less than one year of steroid use while ambulatory versus those who had 1 year or greater exposure while ambulatory. This exposure dichotomy is a commonly used method; it does treat participants with many years exposure with those having only 365 days of exposure. Also, LoA data was available in 212 participants compared to longitudinal timed function tests in 154 participants, which may affect data analysis. Last, and a much-needed consideration for future research studies, is to improve the participation of non-white and Hispanic populations to improve research equity.
One of the widely used clinical outcome endpoints in DMD is 6MWT. Earlier studies reported that 6MWT declined annually, and that the trajectory of decline was markedly different in boys younger and older than age 7 years. Boys older than age 7 years showed progressive decline in 6MWT (25–27). These earlier works have shown that age 7 and above appears to be an “inflexion” point and slopes of deterioration in 6MWT, NSAA are evident (25–27). This evidence let us to perform this specific analysis. Further, we wanted to take advantage of the depth of the CINRG data set given that many clinical trials in DMD are focusing on boys of ages 4 to less than 8 years. In concordance with earlier publications, we found that as a cohort, over a three-year period, boys 7 years and older declined more rapidly than boys younger than 7 years. Further, in our cohort, the decline in 6MWT was regardless of DMD pathogenic variant location. The decline in 6MWT also depends on mutation type as reported by Brogna, who showed that individuals with pathogenic variants amendable to exon 44 skipping had better 6MWT at baseline and 36 months, compared to those with pathogenic variants amendable to exon 53 skipping (28). These results, also supported by findings from the CINRG-DNHS (7) and other cohorts (29, 30), suggest that accounting for DMD pathogenic variant type, as well as longer follow-up, may be necessary to fully understand drug efficacy in clinical trials.
In summary, our study along with earlier published literature demonstrates that well-designed natural history studies in rare disease like DMD provides critical insights for clinical trial design.
Supplementary Material
Grant support:
Grants from the U.S. Department of Education/NIDRR (H133B031118 and H133B090001), U.S. Department of Defense (W81XWH-09-1-0592), the National Institutes of Health (UL1RR031988, U54HD053177, UL1RR024992, U54RR026139, G12RR003051, 1R01AR061875, and RO1AR062380), and Parent Project Muscular Dystrophy are gratefully acknowledged.
List of Abbreviations
- DMD
Dystrophin gene
- DMD
Duchenne Muscular Dystrophy
- CINRG
Cooperative International Neuromuscular Research Group
- DNHS
Duchenne Natural History Study
- 6MWT
6-minute Walk Test
- LoA
Loss-of-ambulation
- CQMS
CINRG Quantitative Measurement System
- ANOVA
Analysis-of-variance
- CNS
Central nervous system
Footnotes
Financial disclosure: No financial disclosures related to this work by any of the authors.
Ethical Publication Statement: We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
- 1.Hoffman EP, Brown RH Jr, Kunkel LM. Dystrophin: The Protein Product of the Duchenne Muscular Dystrophy Locus. Cell. 1987;51(6):919–928. [DOI] [PubMed] [Google Scholar]
- 2.Thangarajh M The Dystrophinopathies. Continuum. 2019;25(6):1619–1639. [DOI] [PubMed] [Google Scholar]
- 3.Muntoni F, Torelli S, Ferlini A. Dystrophin and Mutations: One Gene, Several Proteins, Multiple Phenotypes. Lancet Neurol. 2003;2(12):731–740. [DOI] [PubMed] [Google Scholar]
- 4.Bello L, Kesari A, Gordish-Dressman H, et al. Genetic Modifiers of Ambulation in the Cooperative International Neuromuscular Research Group Duchenne Natural History Study. Ann Neurol 2015;77(4):684–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bello L, D’Angelo G, Villa M, et al. Genetic Modifiers of Respiratory Function in Duchenne Muscular Dystrophy. Ann Clin Transl Neurol 2020;7(5):786–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pegoraro E, Hoffman EP, Piva L, et al. SPP1 Genotype Is a Determinant of Disease Severity in Duchenne Muscular Dystrophy. Neurology 2011;76(3):219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bello L, Morgenroth LP, Gordish-Dressman H, et al. DMD Genotypes and Loss of Ambulation in the CINRG Duchenne Natural History Study. Neurology 2016;87(4):401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelley EF, Cross TJ, Snyder EM, et al. Influence of β2 Adrenergic Receptor Genotype on Risk of Nocturnal Ventilation in Patients With Duchenne Muscular Dystrophy. Respir Res 2019;20(1):221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bello L, Flanigan KM, Weiss RB, et al. Association Study of Exon Variants in the NF-κB and TGFβ Pathways Identifies CD40 as a Modifier of Duchenne Muscular Dystrophy. Am J Hum Genet 2016;99(5):1163–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hogarth MW, Houweling PJ, Thomas KC, et al. Evidence for ACTN3 as a Genetic Modifier of Duchenne Muscular Dystrophy. Nat Commun 2017;8:14143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spitali P, Zaharieva I, Bohringer S, et al. TCTEX1D1 Is a Genetic Modifier of Disease Progression in Duchenne Muscular Dystrophy. Eur J Hum Genet 2020;28(6):815–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weiss RB, Vieland VJ, Dunn DM, Kaminoh Y, Flanigan KM, United Dystrophinopathy Project. Long-range Genomic Regulators of THBS1 and LTBP4 Modify Disease Severity in Duchenne Muscular Dystrophy. Ann Neurol. 2018;84(2):234–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flanigan KM, Ceco E, Lamar KM. LTBP4 Genotype Predicts Age of Ambulatory Loss in Duchenne Muscular Dystrophy. Ann Neurol. 2013;73(4):481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thangarajh M, Spurney CF, Gordish-Dressman H, et al. Neurodevelopmental Needs in Young Boys with Duchenne Muscular Dystrophy (DMD): Observations From the Cooperative International Muscular Research Group (CINRG) DMD Natural History Study (DNHS). PLoS Curr 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thangarajh M, Elfring GL, Trifillis P, McIntosh J, Peltz SW, Ataluren Phase 2b Study Group. The Relationship Between Deficit in Digit Span and Genotype in Nonsense Mutation Duchenne Muscular Dystrophy. Neurology. 2018;91(13):e1215–e1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thangarajh M, Hendriksen J, McDermott MP, et al. Relationships Between DMD Mutations and Neurodevelopment in Dystrophinopathy. Neurology 2019;93(17):e1597–e1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bello L, D’Angelo G, Villa M, et al. Genetic Modifiers of Respiratory Function in Duchenne Muscular Dystrophy. Ann Clin Transl Neurol 2020;7(5):786–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ricotti V, Mandy WPL, Scoto M, et al. Neurodevelopmental, Emotional, and Behavioural Problems in Duchenne Muscular Dystrophy in Relation to Underlying Dystrophin Gene Mutations. Dev Med Child Neurol 2016;58(1):77–84. [DOI] [PubMed] [Google Scholar]
- 19.Doorenweerd N, Straathof CS, Dumas EM, et al. Reduced Cerebral Gray Matter and Altered White Matter in Boys With Duchenne Muscular Dystrophy. Ann Neurol 2014;76(3):403–411. [DOI] [PubMed] [Google Scholar]
- 20.McDonald CM, Henricson EK, Abresch RT, et al. The Cooperative International Neuromuscular Research Group Duchenne Natural History Study – A Longitudinal Investigation in the Era of Glucocorticoid Therapy: Design of Protocol and the Methods Used. Muscle Nerve 2013;48(1):32–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McDonald CM, Henricson EK, Abresch RT, et al. Long-term Effects of Glucocorticoids on Function, Quality of Life, and Survival in Patients With Duchenne Muscular Dystrophy: A Prospective Cohort Study. Lancet 2018;391(10119):451–461. [DOI] [PubMed] [Google Scholar]
- 22.Henricson EK, Abresch RT, Cnaan A, et al. The Cooperative International Neuromuscular Research Group Duchenne Natural History Study: Glucocorticoid Treatment Preserves Clinically Meaningful Functional Milestones and Reduces Rate of Disease Progression as Measured by Manual Muscle Testing and Other Commonly Used Clinical Trial Outcome Measures. Muscle Nerve 2013;48(1):55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller G, Tunnecliffe M, Douglas PS. IQ, prognosis and Duchenne muscular dystrophy. Brain Dev. 1985;7(1):7–9. [DOI] [PubMed] [Google Scholar]
- 24.Desguerre I, Christov C, Mayer M, et al. Clinical heterogeneity of duchenne muscular dystrophy (DMD): definition of sub-phenotypes and predictive criteria by long-term follow-up. PLoS One. 2009;4(2):e4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pane M, Mazzone ES, Sormani MP, et al. 6 Minute Walk Test in Duchenne MD Patients With Different Mutations: 12 Month Changes. PLoS One 2014;9(1):e83400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mazzone E, Vasco G, Sormani MP, et al. Functional Changes in Duchenne Muscular Dystrophy: A 12-month Longitudinal Cohort Study. Neurology 2011;77(3):250–256. [DOI] [PubMed] [Google Scholar]
- 27.Mazzone ES, Pane M, Sormani MP, et al. 24 Month Longitudinal Data in Ambulant Boys with Duchenne Muscular Dystrophy. PLoS One 2013;8(1):e52512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brogna C, Coratti G, Pane M, et al. Long-term Natural History Data in Duchenne Muscular Dystrophy Ambulant Patients With Mutations Amenable to Skip Exons 44, 45, 51, and 53. PLoS One 2019;14(6):e0218683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang RT, Barthelemy F, Martin AS, et al. DMD genotype correlations from the Duchenne Registry: Endogenous exon skipping is a factor in prolonged ambulation for individuals with a defined mutation subtype. Hum Mutat 2018;39(9):1193–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van den Bergen JC, Ginjaar HB, Niks EH, Aartsma-Rus A, Verschuuren JJGM. Prolonged Ambulations in Duchenne Patients with a Mutation Amenable to Exon 44 Skipping. J Neuromuscul Dis 2014;1(1):91–94. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.