

Abstract
The aim of this study was to investigate the effect of nucleotide‐binding oligomerization domain (NOD)‐like receptor family CARD domain containing 5 (NLRC5) in cardiac hypertrophy, and to explore the mechanism implicated in this effect Cardiac hypertrophy was induced in neonatal rat cardiac myocytes using 1 μM of angiotensin II (Ang II) for 12, 24 and 48 h. Overexpression of NLRC5 was induced in H9C2 cells, and the NLRC5 + Ang II–treated cells were exposed to SC9 and 3‑methyladenine (3MA). An immunofluorescence assay was used for α‐actinin staining, and quantitative real‐time reverse transcriptase‐polymerase chain reaction (qRT‐PCR) was performed for NLRC5, atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) determination. Western blot analysis was applied to measure the levels of NLRC5, microtubule‐associated protein 1A/1B‐light chain 3 type I (LC3I), LC3II, sequestosome 1 (p62), protein kinase B (AKT), phosphorylated Akt (pAKT), mammalian target of rapamycin (mTOR) and phosphorylated mTOR (pmTOR). The level of NLRC5 was significantly decreased after Ang II treatment in cardiomyocytes, but the levels of ANP and BNP were increased. Overexpression of NLRC5 reduced the cell size, downregulated the levels of ANP and BNP, increased LC3II / LC3I, but decreased p62 in Ang II–induced cardiomyocyte hypertrophy. In addition, the results from Western blot showed that overexpression of NLRC5 distinctly decreased the ratios of pAKT/AKT and pmTOR/mTOR in cardiomyocyte hypertrophy. SC79 and 3MA significantly downregulated the ratio of LC3I/LC3II but increased the level of p62 in NLRC5 + Ang II–treated cells. These results provide a possible novel therapeutic strategy for cardiac hypertrophy that might be useful in a clinical setting.
Keywords: autophagy, cardiac hypertrophy, nod‐like receptor family CARD domain containing 5, protein kinase B/mammalian target of rapamycin pathway
1. INTRODUCTION
Heart failure (HF) is the final result in the pathogenesis of many cardiovascular diseases, such as heart defects, coronary vascular disease, valvular heart disease, heart infections or cardiomyopathy. 1 Although there has been some progress in the management of HF, 2 mortality is still high. 3
Pathological cardiac hypertrophy is a compensatory response of the heart when the haemodynamic overload is prolonged or abnormal. 4 It has been demonstrated that cardiac hypertrophy is an independent risk factor for cardiovascular events. 5 In addition, compelling evidence has been adduced that pathological hypertrophy is usually related to fibrosis, increased production of pro‐inflammatory cytokines, impaired normal pathways and suppressed autophagy. 4 Therefore, exploring this pathogenesis is of great importance for cardiac hypertrophy prevention and treatment.
Autophagy plays a critical role in maintaining cellular homeostasis, but the prolonged repression of autophagy could result in the impairment of homeostasis associated with excessive accumulation of damaged, non‐recycled cellular constituents. 6 It has been reported that induction of autophagy attenuates immune response impairment and alleviates respiratory exacerbations of various lung diseases. 7 Moreover, augmentation of autophagy decreased the infarct size and alleviated adverse left ventricular remodelling in patients with myocardial infarction. 8 Unfortunately, it is a challenge to maintain moderate autophagy in clinical practice because of the apparent all or none extent of autophagy induction and the inherent side effects in each intervention. 9 Therefore, novel remedies most suitable for maintaining autophagy are of great importance. Therefore, a new therapeutic strategy needs to be found for maintaining autophagy in a most suitable way.
Nod‐like receptors (NLRs), which are a form of intracellular receptors, play a pivotal role in innate immune response. 10 A member of the NLR family, NLRC5, has been shown to regulate both inflammatory responses and cell death. 11 NLRC5 was reported to be of great importance in homeostatic control of innate immunity by negatively regulating nuclear factor‐κB and type I interferon signalling. 12 Besides, NLRC5 deficiency induced myocardial injury in high‐fat diet mice via activation of the TLR4/NF‐κB pathway and suggested NLRC5 was implicated in alleviating myocardial damage. 13 However, the role of NLRC5 in cardiac hypertrophy remains poorly understood.
Therefore, angiotensin II was used to induce cardiac hypertrophy in neonatal rat cardiac myocytes in the current study, and then the effect of NLRC5 on cardiac hypertrophy, as well as its underlying mechanism action, was investigated further.
2. METHODS
2.1. Cell culture and cardiac hypertrophy induction
The rat cardiac myocytes (H9C2) were obtained from the Cell Bank of Chinese Academy of Sciences, and maintained in Dulbecco's modified Eagle's medium (Gibco), supplemented with 10% foetal bovine serum (Gibco), 100 U/ml of penicillin and 100 µg/ml of streptomycin (Invitrogen) at 37°C and 5% CO2. H9C2 cells were treated with angiotensin II (Ang II, 1 μM; Sigma‐Aldrich) for 12, 24 and 48 h.
2.2. Overexpression of NLRC5
The polymerase chain reaction (PCR) was used to amplify cDNA encoding NLRC5, which was subcloned into the vector (Invitrogen). Next, Lip2000 (Thermo Fisher) was used to transfect the NLRC5 overexpression plasmid into H9C2 cells following the manufacturer's instructions. H9C2 cells were then treated with 1 μM of Ang II (Sigma‐Aldrich) for 24 h. Next NLRC5 + Ang II–treated cells were treated with SC9 (1 μg/ml and 4 μg/ml) and SMA.
2.3. Immunofluorescence assay
H9C2 cells were fixed with 4% formaldehyde, permeated with 0.1% Triton X‐100 in PBS and stained with α‐actinin antibody (1:100, 3134S; Cell Signaling Technology) overnight at 4°C. The cells were cultured with secondary Cy3‐conjugated goat anti‐mouse antibody (1:100; Jackson ImmunoResearch Laboratories, Inc) for 60 min at 37°C, and then 4, 6‐diamidino‐2‐phenylindole was used to observe the nuclei. The surface areas were assessed by Image‐Pro Plus 6.0. Next, an automated optical inspection was applied for automatic cell boundary tracing, and the cell area was converted by the tool COUNT/SIZE according to the ruler taken under 400X magnification.
2.4. Quantitative real‐time reverse transcriptase‐polymerase chain reaction (qRT‐PCR)
The total mRNA was isolated by TRIzol reagent (Invitrogen) from H9C2 cells, and then cDNAs were reverse‐transcribed using cDNA Synthesis Kit (Invitrogen). Quantitative RT‐PCR was performed with a thermocycler profile at 95°C for 10 min, 40 cycles of denaturation at 95°C for 15 s, and 63°C for 40 s. The expression levels of indicated genes were normalized to GAPDH expression. The relative expressions of indicated genes were quantified by the use of the 2−ΔΔCT method. 14 Primers used for qRT‐PCR are shown in Table 1.
TABLE 1.
Primer pairs for qRT‐PCR
| Genes | Primers | |
|---|---|---|
| NLRC5 | 5'‐CGCTCTGTGGCCACTTTCAG‐3' (forward) | 5'‐TGCCCGCTGTGAGACTTCAT −3' (reverse) |
| ANP | 5'‐GCCGGTAGAAGATGAGGTCA‐3' (forward) | 5'‐GGGCTCCAATCCTGTCAATC‐3' (reverse) |
| BNP | 5'‐TCTGCTCCTGCTTTTCCTTA‐3' (forward) | 5'‐GAACTATGTGCCATCTTGGA‐3' (reverse) |
| GAPDH | 5'‐CAAGCTCATTTCCTGGTATGAC‐3' (forward) | 5'‐CAGTGAGGGTCTCTCTCTTCCT‐3' (reverse) |
2.5. Western blot analysis
Total proteins from H9C2 cells were isolated using RIPA lysis buffer (Beyotime). Then the proteins were electrophoresed by 10% SDS‐PAGE, transferred to polyvinylidene difluoride membranes and blocked with 5% non‐fat milk, followed by incubation with primary monoclonal antibodies including NLRC5 (1:1000; Abcam), LC3I (1:100; Abcam), LC3II (1:100; Abcam), p62 (1:1000; Abcam), pAKT (1:1000; Abcam), AKT (1:800; Abcam), pmTOR (1:100; Abcam) and mTOR (1:1000; Abcam) overnight. Subsequently, the blots were incubated for 1 h with horseradish peroxidase–conjugated secondary goat anti‐rabbit IgG antibodies (1:1000; Invitrogen). GAPDH was used as an internal loading control. Chemoluminescence was used for quantification of protein expressions.
2.6. Statistical analysis
SPSS (version 17.0) (SPSS, Inc,) was used for all statistical analyses. The data were expressed as mean ± standard deviation. One‐way analysis of variance was utilized for comparison between groups, followed by Tukey's multiple comparison tests. The statistical significance was p < 0.05.
3. RESULTS
3.1. Ang II treatment downregulates NLRC5 expression in rat cardiomyocytes
To investigate the effect of Ang II on rat cardiomyocytes, the levels of NLRC5, ANP and BNP were measured after 12‐, 24‐ and 48 h treatment. As shown in Figure 1A,B, Ang II application substantially reduced the level of NLRC5 when compared to the control group (p < 0.001). In addition, the relative level of NLRC5 was significantly reduced after 24 h and 48 h Ang II treatment compared with 12 h treatment (p < 0.05 or p < 0.01). Compared with the control group, the relative levels of ANP and BNP were markedly increased after Ang II treatment (p < 0.001), and the inhibitory effect of Ang II on ANP and BNP was time‐dependent, respectively (Figure 1C,D). These results indicated that Ang II inhibited NLRC5 expression in rat cardiomyocytes.
FIGURE 1.

Ang II application represses NLRC5 expression in rat cardiomyocytes. A, The relative expression levels of NLRC5 after 12‐, 24‐ and 48 h Ang II treatment were measured using qRT‐PCR. B, The protein levels of NLRC5 after 12‐, 24‐ and 48 h Ang II treatment were determined by Western blot analysis. C‐D, The relative expression levels of ANP and BNP after 12‐, 24‐ and 48 h Ang II treatment were measured using qRT‐PCR. NLRC5, NOD‐like receptor family CARD domain containing 5; ANP, atrial natriuretic peptide; BNP, brain natriuretic peptide; Con, control group; Ang II, angiotensin II–treated group. ***p < 0.001, compared with the control group; and # p < 0.05 and ## p < 0.01, compared with the 12‐h Ang II–treated group
3.2. NLRC5 overexpression attenuates Ang II–induced cardiomyocyte hypertrophy
To identify the role of NLRC5 in Ang II–induced cardiomyocyte hypertrophy, overexpression of NLRC5 was induced. Figure 2A,B shows that overexpression of NLRC5 in H9C2 cells substantially elevated the relative expression and protein level of NLRC5 (p < 0.01). Next, cell size was assessed with α‐actinin immunostaining. The results from the immunofluorescence assay indicated that the size of cardiomyocytes in the NLRC5 overexpression group did not change significantly compared with the vector group. Ang II induced increase in cardiomyocyte size, but NLRC5 overexpression inhibited the increase in cell size (Figure 2C). Moreover, NLRC5 overexpression significantly inhibited the transcription level of ANP (p < 0.01), but Ang II markedly increased the levels of ANP and BNP compared with the vector group (p < 0.01). Overexpression of NLRC5 clearly decreased the transcription levels of ANP and BNP in Ang II–treated cells (p < 0.01) (Figure 2D,E). The results showed that NLRC5 attenuated Ang II–induced cardiomyocyte hypertrophy.
FIGURE 2.

Overexpression of NLRC5 inhibits Ang II–induced cardiomyocyte hypertrophy. A, The relative expression level of NLRC5 after NLRC5 treatment was determined by qRT‐PCR. B, The protein expression level of NLRC5 after NLRC5 treatment was determined by Western blot analysis. C, The α‐actinin immunostaining was performed by the immunofluorescence assay after Ang II and NLRC5 treatment. D‐E, The transcription levels of ANP and BNP were determined by qRT‐PCR assay after Ang II and NLRC5 treatment. NLRC5, NOD‐like receptor family CARD domain containing 5; ANP, atrial natriuretic peptide; BNP, brain natriuretic peptide; Ang II, angiotensin II–treated group. ## p < 0.01, compared with the vector group; and **p < 0.01, compared with the Ang II–treated group
3.3. Overexpression of NLRC5 promotes autophagy
It has been reported that autophagy plays a critical role in cardiomyocyte hypertrophy. Therefore, to investigate whether overexpression of NLRC5 affects autophagy in hypertrophic cardiomyocytes, autophagy‐related proteins were assessed (Figure 3). Western blot analysis showed that no significant difference was observed in the ratio of LC3II/LC3I and p62 between NLRC5 overexpression and vector groups. In addition, Ang II clearly suppressed the ratio of LC3I/LC3II but increased the level of p62 (p < 0.01). However, overexpression of NLRC5 increased LC3II / LC3I (p < 0.01), but diminished the increase in p62 in Ang II–induced cardiomyocyte hypertrophy (p < 0.01). These results suggested that overexpression of NLRC5 promoted autophagy in Ang II–induced cardiac hypertrophy.
FIGURE 3.

Overexpression of NLRC5 promotes autophagy in hypertrophic cardiomyocytes. Autophagy‐related proteins including LC3I, LC3II and p62 were assessed by Western blot analysis after Ang II and NLRC5 treatment. NLRC5, NOD‐like receptor family CARD domain containing 5; LC3, microtubule‐associated protein 1A/1B‐light chain 3; p62, sequestosome 1; Ang II, angiotensin II–treated group. ## p < 0.01, compared with the vector group; and **p < 0.01, compared with the Ang II–treated group
3.4. Overexpression of NLRC5 inhibits Akt/mTOR signalling pathway
Akt and mTOR were considered as examples of key proteins in the regulation of autophagy. Thus, a further experiment was performed to investigate the mechanism of NLRC5 overexpression in cardiomyocyte hypertrophy. Figure 4 shows that NLRC5 overexpression had no significant effect on pAKT/AKT and pmTOR/mTOR in normal cardiomyocytes. The ratios of pAKT/AKT and pmTOR/mTOR were significantly elevated after Ang II stimulation compared with the vector group (p < 0.01), while overexpression of NLRC5 distinctly decreased the ratios of pAKT/AKT and pmTOR/mTOR in Ang II–induced cardiomyocyte hypertrophy (p < 0.01). These findings showed that overexpression of NLRC5 attenuated Ang II–induced cardiomyocyte hypertrophy through inhibiting Akt/mTOR signalling pathway.
FIGURE 4.

Overexpression of NLRC5 attenuated Ang II–induced cardiomyocyte hypertrophy through inhibiting Akt/mTOR signalling pathway. The protein expression levels of pAKT, AKT, pmTOR and mTOR were determined by Western blot analysis. NLRC5, NOD‐like receptor family CARD domain containing 5; AKT, protein kinase B; pAKT, phosphorylated Akt; mTOR, mammalian target of rapamycin; pmTOR, phosphorylated mTOR; Ang II, angiotensin II–treated group. ## p <0.01, compared with the vector group; and **p < 0.01, compared with the Ang II–treated group
3.5. Akt agonist reverses the effect of NLRC5 alleviating cardiac hypertrophy
The Akt agonist SC79 15 and an AKT inhibitor 3MA 16 were used in NLRC5 + Ang II–treated cells. The results from the immunofluorescence assay showed that the cardiomyocyte size was visibly increased after SC79 and 3MA treatment compared with NLRC5 + Ang II–treated cells, and 4 μg/ml of SC79 had the strongest effect on increasing cell size (Figure 5A). Figure 5B,C shows that the transcription level of ANP and BNP was patently increased after SC79 and 3MA treatment compared with the NLRC5 + Ang II–treated group (p < 0.05 or p < 0.01). In Figure 5D, SC79 and 3MA significantly downregulated the protein level of LC3II/LC3I but increased the level of p62 when compared to the NLRC5 + Ang II–treated group (p < 0.01). These results suggested that Akt agonist reversed the effect of NLRC5 alleviating cardiac hypertrophy. Although 3MA was an inhibitor of Akt, it should alleviate cardiac hypertrophy. However, 3MA dramatically caused cardiac hypertrophy because 3MA is an inhibitor of autophagy.
FIGURE 5.
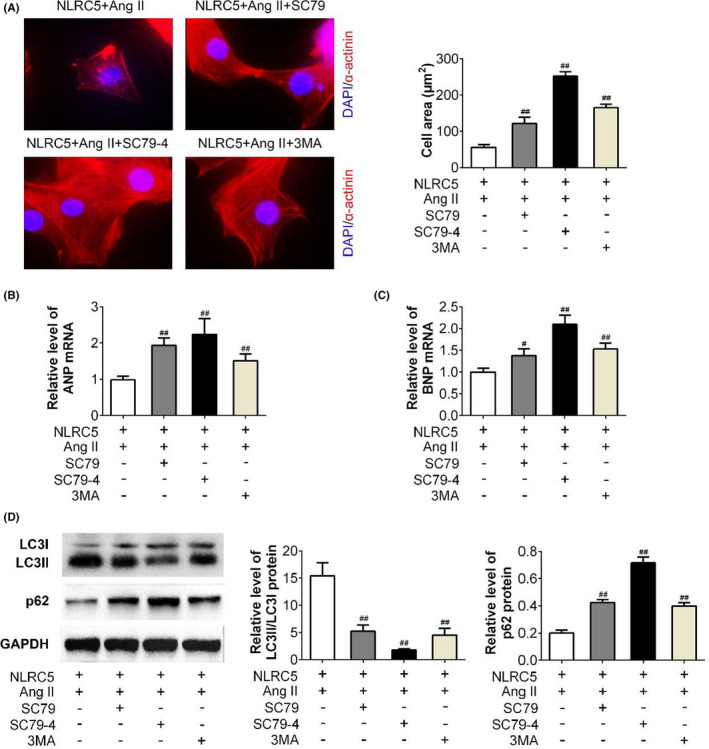
Akt agonist reverses the effect of NLRC5 on cardiac hypertrophy. A, The α‐actinin immunostaining was performed by the immunofluorescence assay after SC79 and 3MA in NLRC5 + Ang II–treated cells. B‐C, The transcription levels of ANP and BNP were determined by qRT‐PCR assay after SC79 and 3MA in NLRC5 + Ang II–treated cells. D, the ratio of LC3II/LC3I and level of p62 were determined by Western blot analysis after SC79 and 3MA in NLRC5 + Ang II–treated cells. NLRC5, NOD‐like receptor family CARD domain containing 5; LC3, microtubule‐associated protein 1A/1B‐light chain 3; ANP, atrial natriuretic peptide; BNP, brain natriuretic peptide; p62, sequestosome 1; Ang II, angiotensin II–treated group. ## p < 0.01, compared with the NLRC5 + Ang II–treated group
4. DISCUSSION
Cardiac hypertrophy is an important pathological process in the development of HF. 4 Although a large body of research has revealed that molecules may protect or damage the myocytes, the prognosis of patients with cardiac hypertrophy is still unclear. 17 Thus, the in‐depth underlying mechanism warrants further study. In the current study, the level of NLRC5 was significantly decreased after Ang II treatment in cardiomyocytes. Overexpression of NLRC5 attenuated Ang II–induced cardiomyocyte hypertrophy by reducing cell size, decreasing the transcription levels of ANP and BNP and promoting autophagy through inactivation of Akt/mTOR signalling pathway. These data suggested that NLRC5 could alleviate cardiac hypertrophy by promoting autophagy via inhibition of AKT/mTOR pathway.
Increasing evidence has suggested that the loss of autophagy may lead to disruptiom of physiological protein homeostasis, and promote intracellular oxidative stress, thus contributing to progression of cardiac hypertrophy and HF. 18 Deficiency of REDD1 in cardiomyocytes led to deterioration in cardiac hypertrophy by inhibiting autophagic markers including LC3BII and p62, but the autophagy enhancer rapamycin dramatically inhibited cardiac hypertrophy, suggesting that REDD1 attenuated cardiac hypertrophy by promoting autophagy. 19 It was reported that oridonin increased the expression of LC3II but decreased p62 expression in Ang II–caused cardiac hypertrophy, and the results showed that the effect of oridonin was to attenuate cardiac hypertrophy by enhancement of autophagy. 20 In the present study, Ang II clearly suppressed the ratio of LC3II/LC3I but promoted the accumulation of the p62. However, NLRC5 treatment increased the ratio of LC3II/LC3I but repressed the increase in p62. These findings indicated that NLRC5 alleviated cardiac hypertrophy by enhancing autophagy in cardiomyocytes.
Akt and mTOR were important autophagy‐related proteins. In addition, pressure overload partially inhibits the Akt/mTOR pathway and downstream substrates after 8 weeks, which could distinguish physiological from pathological hypertrophy. 21 A recent study demonstrated that the levels of phosphorylated AKT and mTOR were increased after pressure overload, but oridonin application blocked the effect of pressure overload. 20 Rapamycin attenuated pressure overload‐caused cardiac hypertrophy induced in mice by inactivation of mTOR. 22 Wang and his colleagues 23 demonstrated that spontaneous hypertension in rats induced an increase in cardiac Akt and pmTOR, while atorvastatin application reversely decreased the levels of Akt and pmTOR, suggesting that atorvastatin promoted cardiac autophagy through inactivation of the Akt/mTOR pathway. Consistent with these previous results, the data from this study showed a significant increase in the protein levels of pAKT/AKT and pmTOR/mTOR after Ang II stimuli, but NLRC5 distinctly decreased the levels of pAKT/AKT and pmTOR/mTOR, indicating that NLRC5 attenuated cardiac hypertrophy by augmentation of autophagy via inhibiting Akt/mTOR pathway.
In view of all of the above findings, the current study indicates that NLRC5 ameliorates Ang II–induced cardiac hypertrophy in vitro; further in vivo studies are warranted to confirm this. In addition the mechanism whereby NLRC5 affects the AKT/mTOR pathway remains to be investigated in depth.
In conclusion, NLRC5 might ameliorate cardiac hypertrophy by promoting autophagy via inhibition of AKT/mTOR pathway, and this finding provides a possible novel clinical therapeutic strategy that could be useful for patients with cardiac hypertrophy.
CONFLICT OF INTERESTS
The authors state that there are no conflicts of interest to disclose.
AUTHORS' CONTRIBUTIONS
Ba Bayinsilema and Abudoukelimu Mayila designed the study and supervised the data collection. Yankai Guo analysed the data and interpreted the data. Jie Xu, Shifeng Xing and GuiQiu Cao prepared the manuscript for publication and reviewed the draft of the manuscript. All authors have read and approved the manuscript.
ETHICAL APPROVAL
Not applicable.
ACKNOWLEDGEMENT
Not applicable.
Bayinsilema B, Mayila A, Guo Y, Xu J, Xing S, Cao G. NLRC5 enhances autophagy via inactivation of AKT/mTOR pathway and ameliorates cardiac hypertrophy. Int J Exp Path.2022;103:23–30. doi: 10.1111/iep.12427
Funding information
This work was supported by the Science and Technology Program of Xinjiang Uighur Autonomous Region (Grant No. 2020E0274).
DATA AVAILABILITY STATEMENT
All data generated or analysed during this study are included in this published article.
REFERENCES
- 1. Coronel R, De Groot JR, Van Lieshout JJ. Defining heart failure. Cardiovasc Res. 2001;50:419‐422. [DOI] [PubMed] [Google Scholar]
- 2. Hunt SA, Abraham WT, Chin MH, et al. ACC/AHA 2005 Guideline update for the diagnosis and management of chronic heart failure in the adult. Circulation. 2005;2005:112. [Google Scholar]
- 3. Levy D, Kenchaiah S, Larson MG, et al. Long‐term trends in the incidence of and survival with heart failure. N Engl J Med. 2002;347:1397‐1402. [DOI] [PubMed] [Google Scholar]
- 4. Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol. 2016;97:245‐262. [DOI] [PubMed] [Google Scholar]
- 5. Katholi RE, Couri DM. Left Ventricular hypertrophy: major risk factor in patients with hypertension: update and practical clinical applications. Int J Hypertens. 2011;2011:495349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stroikin Y, Dalen H, Brunk UT, Terman A. Testing the “garbage” accumulation theory of ageing : mitotic activity protects cells from death induced by inhibition of autophagy. Biogerontology. 2005;6:39‐47. [DOI] [PubMed] [Google Scholar]
- 7. Pehote G, Vij N. Autophagy augmentation to alleviate immune response dysfunction, and resolve respiratory and COVID‐19 exacerbations. Cells. 2020;9:1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Buss SJ, Riffel JH, Katus HA, Hardt SE. Augmentation of autophagy by mTOR‐inhibition in myocardial infarction: when size matters. Autophagy. 2010;6:304‐306. [DOI] [PubMed] [Google Scholar]
- 9. Sciarretta S, Zhai P, Volpe M, Sadoshima J. Pharmacological modulation of autophagy during cardiac stress. J Cardiovasc Pharmacol. 2012;60:235‐241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen GY, Shaw MH, Kim YG, Nunez G. NOD‐like receptors: role in innate immunity and inflammatory disease. Annu Rev Pathol. 2009;4:365‐398. [DOI] [PubMed] [Google Scholar]
- 11. Benko S, Magalhaes JG, Philpott DJ, Girardin SE. NLRC5 limits the activation of inflammatory pathways. J Immunol. 2010;185:1681‐1691. [DOI] [PubMed] [Google Scholar]
- 12. Cui J, Zhu L, Xia X, et al. NLRC5 negatively regulates the NF‐κB and type I interferon signaling pathways. Cell. 2010;141:483‐496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ma S, Xie X. NLRC5 deficiency promotes myocardial damage induced by high fat diet in mice through activating TLR4/NF‐κB. Biomed Pharmacother. 2017;91:755‐766. [DOI] [PubMed] [Google Scholar]
- 14. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods. 2001;25:402‐408. [DOI] [PubMed] [Google Scholar]
- 15. Feng H, Cheng X, Kuang J, et al. Apatinib‐induced protective autophagy and apoptosis through the AKT‐mTOR pathway in anaplastic thyroid cancer. Cell Death Dis. 2018;9:1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guan G, Yang L, Huang W, et al. Mechanism of interactions between endoplasmic reticulum stress and autophagy in hypoxia/reoxygenation‐induced injury of H9c2 cardiomyocytes. Mol Med Rep. 2019;20:350‐358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Olivetti G, Cigola E, Maestri R, Lagrasta C, Corradi D, Quaini F. Recent advances in cardiac hypertrophy. Cardiovasc Res. 2000;45:68‐75. [DOI] [PubMed] [Google Scholar]
- 18. Sciarretta S, Maejima Y, Zablocki D, Sadoshima J. The role of autophagy in the heart. Annu Rev Physiol. 2018;80:1‐26. [DOI] [PubMed] [Google Scholar]
- 19. Liu C, Xue R, Wu D, et al. REDD1 attenuates cardiac hypertrophy via enhancing autophagy. Biochem Biophys Res Comm. 2014;454:215‐220. [DOI] [PubMed] [Google Scholar]
- 20. Xu M, Wan C, Huang S, et al. Oridonin protects against cardiac hypertrophy by promoting P21‐related autophagy. Cell Death Dis. 2019;10:403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kemi OJ, Ceci M, Wisloff U, et al. Activation or inactivation of cardiac Akt/mTOR signaling diverges physiological from pathological hypertrophy. J Cell Physiol. 2008;214:316‐321. [DOI] [PubMed] [Google Scholar]
- 22. Mcmullen JR, Sherwood MC, Tarnavski O, et al. Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation. 2004;109:3050‐3055. [DOI] [PubMed] [Google Scholar]
- 23. Wang W, Wang H, Geng Q, et al. Augmentation of autophagy by atorvastatin via Akt/mTOR pathway in spontaneously hypertensive rats. Hypertens Res. 2015;38:813‐820. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analysed during this study are included in this published article.