

Abstract
MicroRNA-3662 (miR-3662) is minimally expressed in normal human tissues but is highly expressed in all types of cancers, including breast cancer. As determined with The Cancer Genome Atlas dataset, miR-3662 expression is higher in triple-negative breast cancers (TNBCs) and African American breast cancers than in other breast cancer types. However, the functional role of miR-3662 remains a topic of debate. Here, we found that inhibition or knockout of endogenous, mature miR-3662 in TNBC cells suppresses proliferation and migration in vitro and tumor growth and metastasis in vivo. Functional analysis revealed that, for TNBC cells, knockout of miR-3662 reduces the activation of Wnt/β-catenin signaling. Furthermore, using CRISPR-mediated miR-3662 activation and repression, dual-luciferase assays, and miRNA/mRNA immunoprecipitation assays, we established that HMG-box transcription factor 1 (HBP-1), a Wnt/β-catenin signaling inhibitor, is a target of miR-3662 and is most likely responsible for miR-3662-mediated TNBC cell proliferation. Our results suggest that miR-3662 has an oncogenic function in tumor progression and metastasis via an miR-3662-HBP1 axis, regulating the Wnt /β-catenin signaling pathway in TNBC cells. Since miR-3662 expression occurs a tumor-specific manner, it is a promising biomarker and therapeutic target for patients who have TNBCs with dysregulation of miR-3662, especially African Americans.
Keywords: miR-3662, breast cancer, CRISPR, tumor progression, HBP1
INTRODUCTION
In the United States, breast cancer is the second leading cause of cancer deaths of women, and, in 2021, approximately 281,550 new breast cancer cases will be diagnosed, and 43,600 deaths will occur 1. Invasive breast cancer has a high risk of recurrence to an incurable disease, and clinicians need improved prognostic and therapeutic tools. About 15% to 20% of breast cancers are classified as triple-negative breast cancers (TNBCs) 2, and this subtype is associated with aggressive clinical behavior and poor prognosis 3 due to the lack of hormone receptors as therapeutic targets. Further, more than one-third of patients with TNBCs present with either recurrent or distant metastatic disease 4. However, inherent biological drivers of aggressive and metastatic TNBC remain elusive.
MicroRNAs (miRNAs or miRs), non-coding RNAs with a length of 20–22 nucleotides, are widely distributed in various organisms. MiRNAs regulate gene expression mainly by binding to the 3’-untranslated region (UTR) of their target messenger RNAs (mRNAs) 5. MiRNAs can act as oncogenes or tumor suppressor genes 6, and dysregulation of miRNAs is involved in the initiation and progression of human cancers 7, including breast cancer 8, 9. MiRNAs are also emerging as potential biomarkers of breast cancer for diagnosis, prognosis, and prediction of therapeutic outcomes 6, 8, 10, 11. Early studies identified the high expression levels of hsa-miR-3662 (also miR-3662) in plasma or tumor tissues from patients with lung cancer, suggesting that miR-3662 could serve as a biomarker for diagnosis of lung adenocarcinoma 12, 13. Genome-wide association studies with functional analysis identified miR-3662 as a driver contributing to cell proliferation in acute myeloid leukemia through NF-KB– mediated transcription 14. However, later studies revealed that miR-3662 inhibits hepatocellular carcinoma growth through decreasing the HIF-1α-mediated Warburg effect 15 or upregulating hexokinase 2 (HK2) 16. Also, in aggressive melanoma cells, miR-3662 inhibits the epithelial-mesenchymal transition (EMT) through down-regulation of zinc finger E-box binding homeobox 1 (ZEB1) 17. However, the functional role of miR-3662 in human cancers remains undefined. In the present study, we investigated the role of miR-3662 in TNBC cells in vitro and in vivo. Further, we used multiple approaches to assess the targets of miR-3662 and its transcription regulation in TNBC cells.
RESULTS
Characterization of miR-3662 expression in human primary breast cancer
Mature sequence hsa-miR-3662 arises only from the 5′ arm of the miR-3662 hairpin18. In The Cancer Genome Atlas (TCGA) dataset, miR-3662 expression was found in human carcinomas from bladder, breast, colon, esophagus, head and neck, kidney, liver, lung, pancreas, prostate, rectum, skin, stomach, thyroid, and uterus (Fig. S1A). miR-3662 was minimally expressed in normal epithelial tissues but overexpressed in all carcinomas (Fig. S1A). Notably, the expression of miR-3662 was 3.8-fold higher in breast cancer tissues than in non-cancerous breast tissue (Fig. 1A). Next, we assessed the clinical relevance of miR-3662 in human primary breast cancer. miR-3662 expression was higher in TNBC tissues than in luminal breast cancer tissues (Fig. 1B). Further, miR-3662 expression was higher in African-American breast cancer tissues than in Caucasian and Asian breast cancer tissues (Fig. S1B). High levels of miR-3662 expression were associated with high-stage breast cancers (T2–4) (Fig. S1C). In addition, high expression of miR-3662 was associated with poor survival of patients with breast cancer (Fig. S1D).
Figure 1. miR-3662 KO inhibited cell proliferation and migration of TNBC cells.
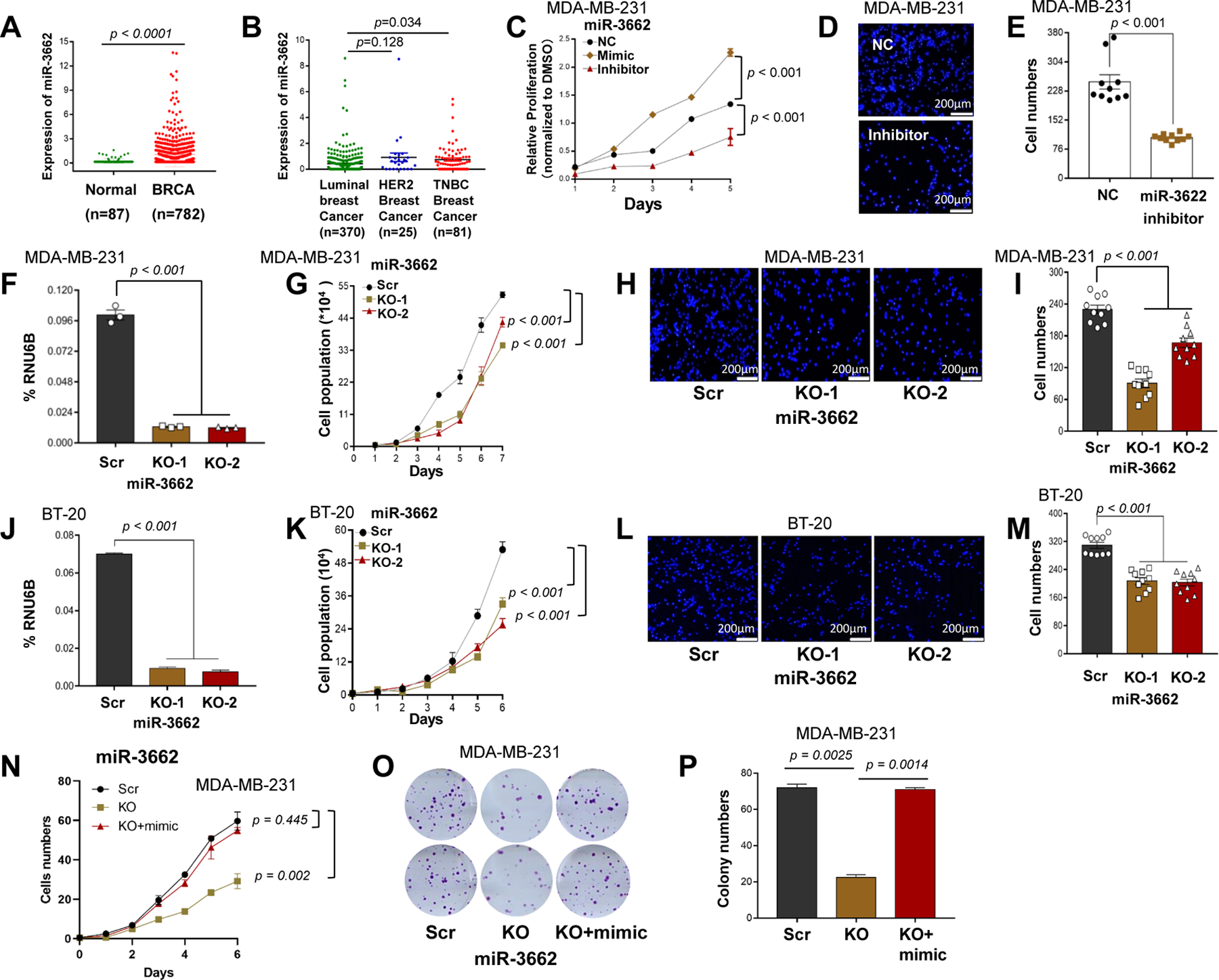
A. The normalized expression levels of miR-3662 in TCGA human primary breast cancers and normal breast tissues. B. Expression of miR-3662 in TCGA human triple-negative breast cancer (TNBC), HER2+ breast cancer, and luminal breast cancer tissues. C-E. Proliferation and migration determined by MTT assays and Transwell assays for MDA-MB-231 cells transfected with an miR-3662 mimic, inhibitor, or NC. F, J. Validation of miR-3662 KO in MDA-MB-231 and BT-20 cells by qPCR. G, K. Proliferation of miR-3662 scrambled control and KO cells for 6 days. H, L. Migration of scrambled control and KO cells for 24 hours as determined by Transwell assays. Images of 10 different 10× fields were captured from each membrane, and the numbers of migratory cells were counted by fluorescence microscopy. I, M. Quantification of rates of cell migration. N, O. Proliferation and colony formation of miR-3662 scrambled control, KO, and miR-3662 mimic-transfected KO cells for 6 days. P. Quantification of colony numbers. Data are presented as means ± standard deviation (SD). P values were determined by a two-tailed t-test or a two-way ANOVA test. NC, negative miRNA control; Scr, scrambled control; KO, knockout. All experiments were repeated three times.
miR-3662 induces proliferation and migration of human TNBC cells
High expression levels of miR-3662 were present in human TNBC cell lines MDA-MD-231, BT-20, and MDA-MD-157, but there were low levels in MDA-MD-468, BT-549, and MDA-MD-453 cells (Figs. S2A and S2B). Also, medium expression levels of miR-3662 were present in HER2+ cell lines BT-474, T47D, and SKBR3 (Figs. S2A and S2B). To explore the role of miR-3662 in human TNBC cell lines, we used specific synthetic antisense inhibitors to sequester the expression of endogenous mature miR-3662 (Fig. S2C) and specific synthetic mimics to mimic endogenous, mature miR-3662 (Fig. S2D). MTT assays revealed that the proliferation rates of MDA-MB-231 and BT-20 cells were reduced when endogenous miR-3662 levels were lowered by an miRNA inhibitor (Fig. 1C and S2E). In contrast, the proliferation rates were increased by addition of the miRNA mimic (Fig. 1C and S2E). Transwell assays showed that the migration rates of MDA-MB-231 and BT-20 cells were low when the endogenous miR-3662 levels were reduced by an miRNA inhibitor (Figs. 1D,1E, and S2F-G).
To validate the role of endogenous miR-3662 in TNBC cells, using CRISPR/Cas9 genome editing, we knocked out the endogenous miR-3662 in MDA-MB-231 and BT-20 cells, an effect confirmed by denaturing gradient gel electrophoresis, Sanger sequencing, and quantitative real-time PCR (qPCR) (Figs. S2H-K, 1F and 1J). For both MDA-MB-231 and BT-20 cells, cell counting assays revealed that the proliferation of miR-3662 knockout (KO) cells was slower than that for scrambled control cells (Figs. 1G and 1K), and Transwell assays revealed that the migration of KO cells was slower than that for scrambled control cells (Figs. 1H-I and 1L-M). Likewise, miR-3662 KO MDA-MB-231 cells showed less colony formation compared to scrambled control cells (Figs. S3A-G). In addition, flow cytometric analysis of cell cycle distribution showed that miR-3662 KO MDA-MB-231 cells had an elevation in the percentages of the G1 population (p=0.0079) and a decline in the percentages of the S population (p=0.0929) (Figs. S3H and S3I). Next, we transiently treated miR-3662 KO cells with an miR-3662 mimic. After treatment, cell growth was rescued by the mimic (Figs. 1N-P). Also, with MDA-MB-231 and BT20 cells, we performed Transwell assays in the presence of the cell cycle blocker, mitomycin C. The migration rates of MDA-MB-231 and BT-20 cells were slower in miR-3662 KO cells than in scrambled control cells (Figs. S4A-D). Likewise, in the presence of mitomycin C, we performed Transwell assays with miR-3662-low expressing MDA-MB-468 and BT-549 cells with an miR-3662 mimic and an antisense inhibitor. The migration rates of these cells were elevated in cells treated with the miR-3662 mimic, but there was no change for cells treated with the miR-3662 antisense inhibitor compared to treatment with a negative miRNA control (Figs. S4E-H). We also assessed the effect of miR-3662 KO on EMT markers in MDA-MB-231 cells. However, in the miR-3662 KO cells, expression levels of the EMT markers E-cadherin, N-cadherin, and vimentin, were not appreciably changed compared to the scrambled control cells (Figs. S4I and S4J). These data indicate that miR-3662 induces cell proliferation and migration of human miR-3662-high expressing TNBC cells.
miR-3662 KO reduces growth and metastasis of human TNBC cells
To address the role of miR-3662 in vivo, we injected miR-3662 scrambled control and KO cells into the fourth mammary fat pads of 8-week-old female NOD SCID gamma (NSG) mice. As shown in Figs. 2A-D and 2I-J, miR-3662 KO reduced tumor growth in the mice bearing MDA-MB-231-luc and BT-20 cells. Further, lung metastases were not evident in mice bearing KO MDA-MB-231-luc cells in contrast to mice bearing scrambled control MDA-MB-231-luc cells (Figs. 2E and 2F). Immunohistochemistry (IHC) staining showed that the expression of Ki67, a biomarker indicating accelerated proliferation, was lower in miR-3662 KO tumors compared with scrambled control tumors (Figs.2G-H and 2K-L).
Figure 2. miR-3662 KO reduced tumor growth and metastasis of TNBC xenografts in NSG mice.
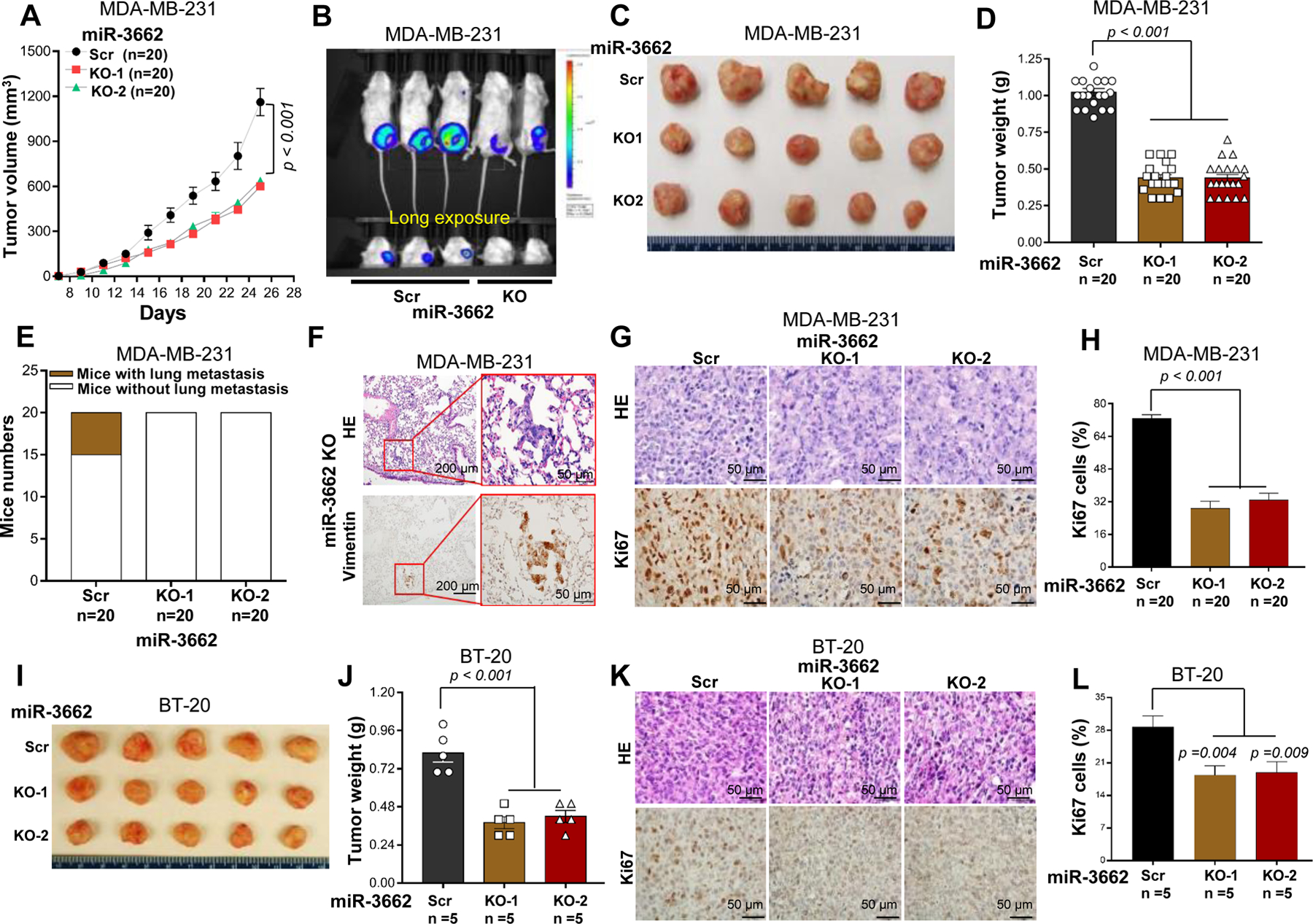
A. Tumor growth in NSG mice subcutaneously injected with miR-3662 scrambled control or KO MDA-MB-231-luc cells. The tumor sizes were measured, and volumes were calculated according to the following formula: volume = (width2 × length)/2. B-D. Representative bioluminescent and optical images and weights of scrambled control and KO MDA-MB-231-luc xenograft tumors and pulmonary metastases at day 45 after transplantation. E. Bar chart of metastatic tumors in the lung. F. Representative H/E and IHC staining with anti-vimentin antibody in metastatic pulmonary lesions. G. Representative H/E and IHC staining with anti-Ki67 antibody in xenograft tumor lesions. H. The percentage of Ki67+ cells in xenograft tumor tissues. For each mouse, at least five 40× fields were counted. I, J. Representative optical images and weights of scrambled control and KO BT-20 xenograft tumors at day 45 after transplantation. K. Representative H/E and IHC staining with anti-Ki67 antibody in xenograft tumors. L. The percentage of Ki67+ cells in xenograft tumors. Data are presented as means ± SD. P values were determined by a two-tailed t-test or a two-way ANOVA test. Scr, scrambled control; KO, knockout. All experiments were repeated two times.
miR-3662 KO attenuates activation of the Wnt/β-catenin signaling pathway
To elucidate the miR-3662-mediated molecular mechanism in the regulation of tumor growth and metastasis, we performed RNA-sequencing (RNA-seq) for miR-3662 scrambled control and KO MDA-MB-231 cells. We used five pathway datasets, including Pathway Interaction, Hallmark Pathway, WikiPathways, Reactome Pathway, and Hallmark and Kyoto Encyclopedia of Genes and Genomes, to identify the candidate pathway list by gene set enrichment analysis (GSEA) in KO cells relative to scrambled control cells. As shown in Figs. S5, S6A-B and 3A, the gene sets were upregulated in the Wnt signal pathway, which was most frequently identified in the top 10 pathways of most datasets. However, we also found other candidate pathways, such as c-Myc and TGFβ signaling, which are also relevant to the Wnt/catenin pathway. Immunofluorescence (IF) analysis showed that nuclear β-catenin (N-β-catenin) expression was lower in KO MDA-MB-231 cells compared with scrambled control cells (Figs. 3B-C). Also, cytoplasmic β-catenin expression was upregulated in KO cells compared with scrambled control cells (Fig. 3C). The ratio of nuclear/cytoplasmic β-catenin was lower in KO cells than in scrambled control cells (Fig. 3C). This observation was validated by Western blots (Figs. 3D and 3E). However, GSK3β. a β-catenin regulator. 19 and its phosphorylation were not appreciably changed, although cyclin D1 and c-Myc were downregulated in KO cells relative to scrambled control cells (Figs. 3D-G). These data suggest that, in TNBC cells, miR-3662 KO leads to less activation of the Wnt/β-catenin signaling pathway.
Figure 3. miR-3662 KO repressed the activation of Wnt/β-catenin signaling in MDA-MB-231 cells.
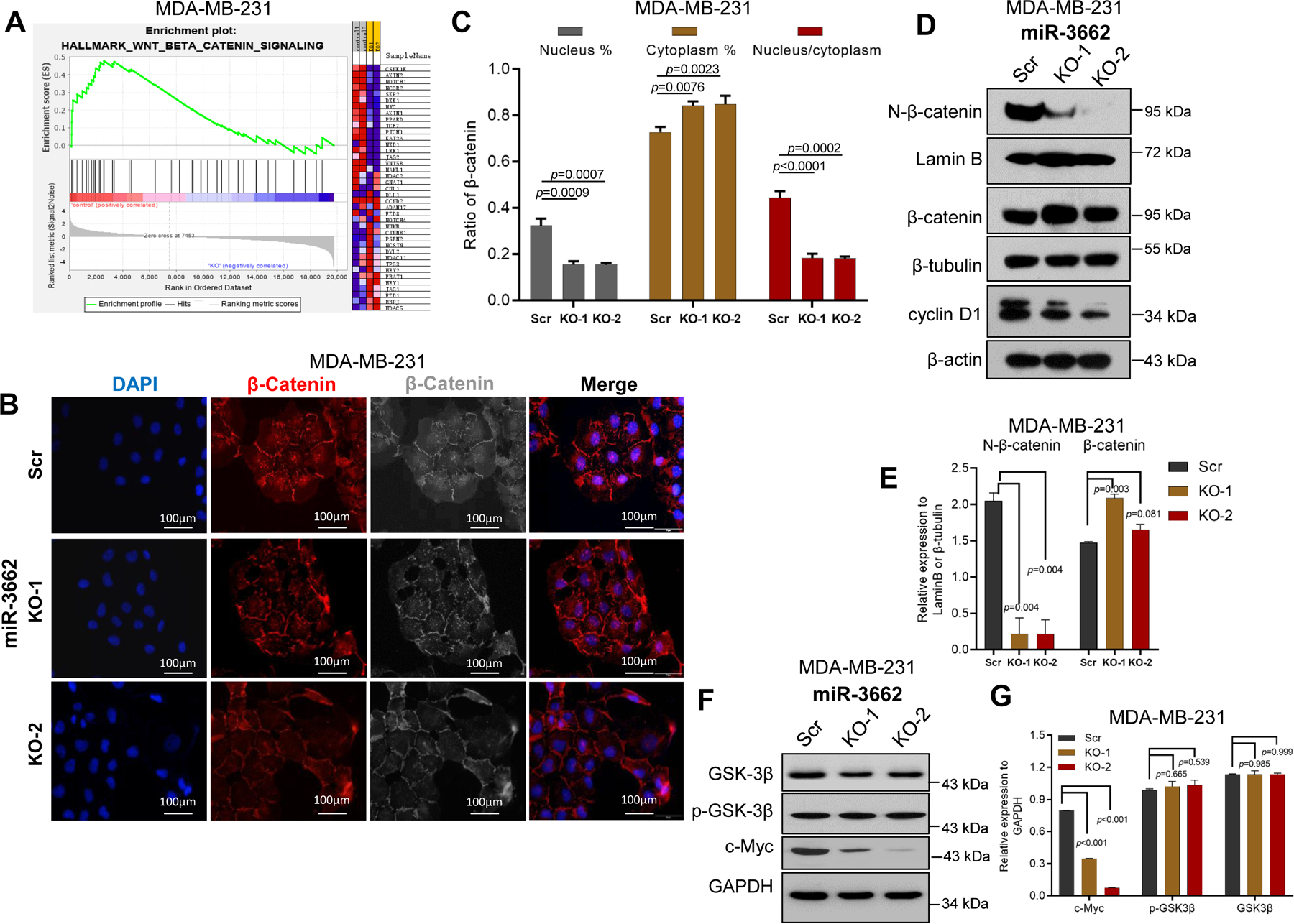
A. Enrichment of the WNT/β-catenin signaling pathway by GSEA analysis in miR-3662 scrambled control MDA-MB-231 cells compared to KO cells. B. Expression levels and nuclear localization of β-catenin by IF analysis in scrambled control and KO cells. C. Quantification of expression levels of nuclear/cytoplasmic β-catenin and their ratio. D, E. Expression levels of N-β-catenin, total β-catenin, and cyclin D1 in scrambled control and KO cells determined by Western blots. There were two bands in cyclin D1 protein expression. Based on cyclin D1 protein size, the lower band is cyclin D1, and the upper band is a non-specific band. F, G. Expression levels of total and phosphorylated GSK3β in scrambled control and KO cells determined by Western blots. Data are presented as means ± SD. P values were determined by a two-tailed t-test. GSEA, gene set enrichment analysis; IF, immunofluorescence; Scr, scrambled control; KO, knockout. All experiments were repeated three times.
HBP1 is a potential target gene of miR-3662 in the Wnt signaling pathway
To screen candidate target genes of miR-3662, we analyzed our RNA-seq data with public datasets. According to the fold change and q-value, 1378 genes were significantly regulated by miR-3662 (Fig. S6A). By scoring algorithm for TargetScan total context score (++) ≤ - 0.15 20 (Supplementary Table S1), 417 genes were identified as potential target genes (Fig. 4A). Next, we predicted 26 candidate targets as determined by combined RNA-seq and Targetscan data (Fig. 4A and Supplementary Table S2). We also analyzed the mRNA expression of these candidate target genes in TCGA dataset. Of the 26 candidate genes, 17 genes were down-regulated, and 4 were upregulated (>1.5-fold change, P< 0.001) in breast cancer tissues compared with normal breast tissues (Fig. 4B). Further, we chose the 17 downregulated genes as potential target genes of miR-3662. To validate the effect of miR-3662 on the downregulated candidate genes, according to the correlation coefficient r value (r ≤ - 0.1) of the candidate genes and miR-3662 in the TCGA dataset, we identified the 5 top candidate genes, HBP1, LEPROT, CSRNP3, ADHFE1, and ZBTB20 (Fig. 4B). However, for MDA-MB-231 and BT20 cells, only HBP1 and LEPROT were upregulated in miR-3662 KO cells compared to scrambled control cells (Figs. 4C, 4G-L, 5C-F, and S6C-D). Also, only HBP1 and LEPROT fragments per kilobase of transcript per million were > 1 in the RNA-seq data. Thus, we selected the two candidate target genes for further analysis. According to the GeneCards dataset (www.genecards.org/), there was a strong gene network among HBP1 and WNT-related proteins (Fig. S6E). In TCGA breast cancer tissues, mRNA expression of HBP1 was inversely related to miR-3662 expression (Fig. S6F). In addition, we determined the expression levels of HBP1 in 10 human breast cancer cell lines. The expression of HBP1 was lower in MDA-MB-231 and BT20 cell lines than in other cell lines (Fig. S6G). Also, in these cells, there was a negative correlation between expression levels of miR-3662 and HBP1 (Fig. S6H).
Figure 4. Identification of HBP1 as a target gene of miR-3662 in MDA-MB-231 cells.
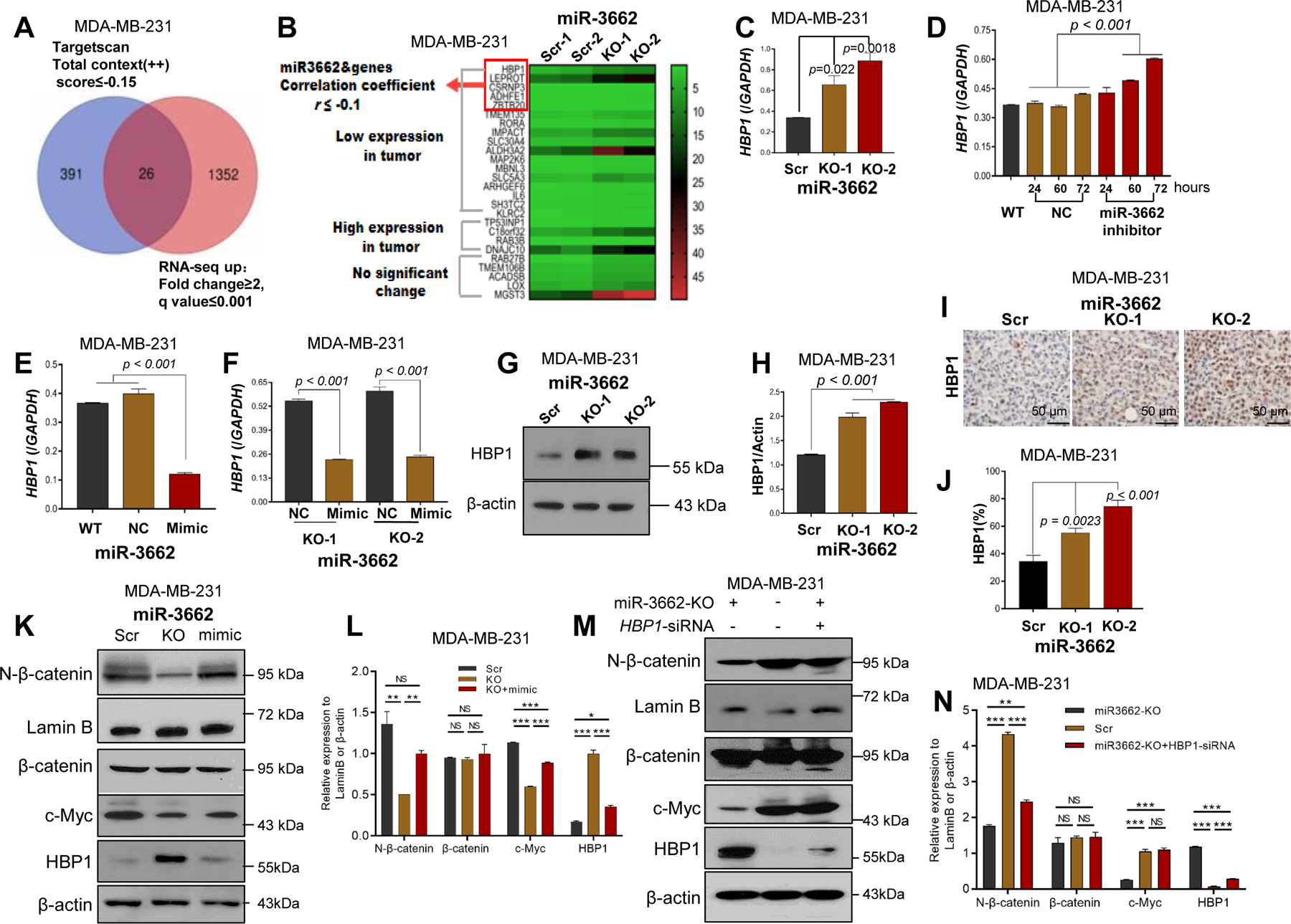
A. Venn diagram showing the intersection of differentially upregulated genes in RNA-seq and target gene predictions from Targetscan. B. Heat-map-based clustering of expression of miR-3662 candidate target genes using our RNA-seq data with TCGA breast cancer dataset. The 26 candidate target genes were divided into three groups according to the expression data from TCGA. The colors of the heat-map represent the expression levels of the 26 candidate genes. The red arrowheads indicate the top candidate 5 genes, for which their expressions were negatively correlated with the expression of miR-3662, and the Spearman’s correlation coefficient was < −0.1 in TCGA dataset. C. qPCR analysis of HBP1 in miR-3662 scrambled control and KO cells. D-F. qPCR analysis of HBP1 in scrambled control or KO cells with miR-3662 mimic or inhibitor. Numbers on the x-axis in D indicate treatment time (hours) with the miR-3662 inhibitor. G, H. Western blot analyses of HBP1 protein expression in scrambled control and KO cells. I, J. Expression of HBP1 protein determined by IHC analysis in scrambled control and KO xenograft tumors from NSG mice. K, L. Western blots and quantitative analyses of expression of HBP1 and Wnt signaling proteins in scrambled control, KO, and miR-3662 mimic-transfected KO cells. M, N. Western blots and quantitative analyses of expression of HBP1 and Wnt signaling proteins in KO, scrambled control, and HBP1 siRNA-transfected KO cells. Data are presented as means ± SD. P values were determined by a two-tailed t-test or a one-way ANOVA test followed by a Dunnett post hoc test. NC, negative miRNA control; Scr, scrambled control; KO, knockout. All experiments were repeated three times.
Figure 5. Validation of miR-3662-HBP1 axis-mediated regulation of the Wnt/β-catenin signaling pathway in BT-20 cells.
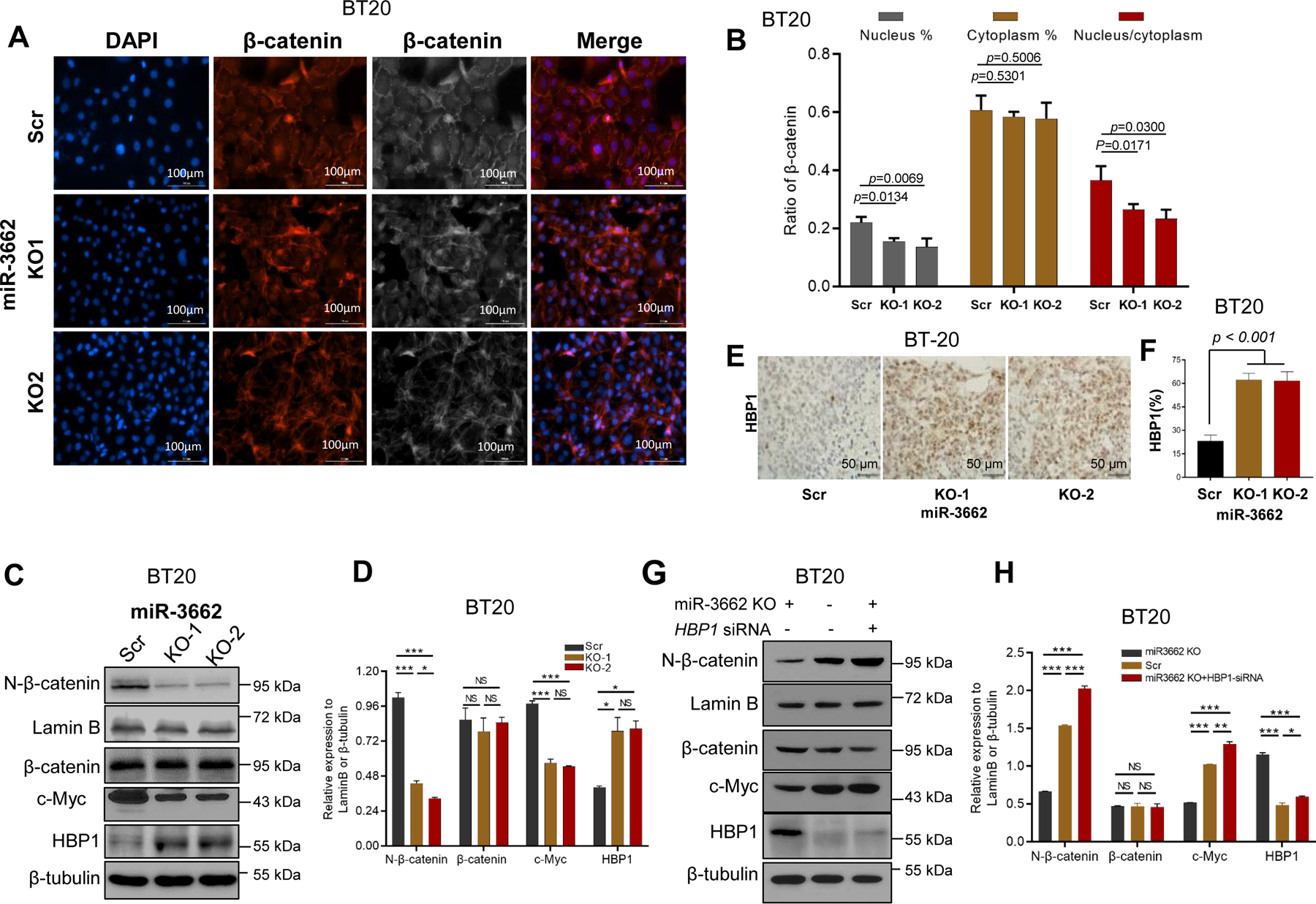
A. Expression levels and nuclear localization of β-catenin by IF analysis in scrambled control and KO cells. B. Quantification of expression levels of nuclear/cytoplasmic β-catenin and their ratio. C, D. Western blot and quantitative analyses of expression of HBP1 and Wnt signaling proteins in miR-3662 scrambled control and KO cells. E, F. Expression of HBP1 protein determined by IHC analysis in scrambled control and KO xenograft tumors from NSG mice. G, H. Western blot and quantitative analyses of expression of HBP1 and Wnt signaling proteins in KO, scrambled control, and HBP1 siRNA-transfected KO cells. Data are presented as means ± SD. P values were determined by a two-tailed t-test. Scr, scrambled control; KO, knockout. All experiments were repeated three times.
To evaluate the effect of miR-3662 on transcription regulation of HBP1, we transiently transfected a negative miRNA control or an miR-3662 inhibitor into scrambled control MDA-MB-231 cells. Upregulation of HBP1 expression was evident after transfection with the inhibitor (Fig. 4D). Likewise, down-regulation of HBP1 expression was evident after transfection with the mimic in either scrambled control cells or KO cells (Figs. 4E and 4F). Also, expression of HBP1 protein was higher in KO cells compared with scrambled control cells (Figs.4G and 4H) and in KO tumors compared with scrambled control tumors from xenograft NSG mice (Figs. 4I-J). To determine whether Wnt/β-catenin signaling is the main pathway contributing to the miR-3662-mediated function, we transiently transfected the miR-3662 mimic into miR-3662 KO MDA-MB-231 cells for rescuing the Wnt/β-catenin signaling. As shown in Figs. 4K-L, the miR-3662 mimic reduced HBP1 expression, leading to rescued N-β-catenin and c-Myc expression. Of note, knockdown of HBP1 by small interfering RNA (siRNA) in miR-3662 KO cells also rescued accumulation of N-β-catenin and expression of c-Myc (Figs. 4M and 4N).
Validation of the miR-3662-HBP1 axis-regulated Wnt/β-catenin signaling pathway
Using an additional TNBC cell line, BT20, we further assessed the miR-3662-HBP1 axis-mediated Wnt/β-catenin regulation. IF analysis validated the lower expression of N-β-catenin in miR-3662 KO BT-20 cells compared with scrambled control cells (Figs. 5A-B). However, cytoplasmic β-catenin expression was not upregulated in KO cells compared with scrambled control cells (Fig. 5B). The nuclear/cytoplasmic β-catenin ratio was lower in KO cells than in scrambled control cells (Fig. 5B). Also, we validated that miR-3662 KO induces HBP1 expression but reduces the accumulation of N-β-catenin and expression of c-Myc in BT-20 cells (Figs. 5C and 5D). In the BT-20 xenograft models, HBP1 expression was higher in KO tumors than in tumors with scrambled control (Figs. 5E and 5F). Due to increased HBP1 by miR-3662 KO, we used HBP1 siRNAs to validate the endogenous HBP1-mediated suppression of Wnt pathway activation and N-β-catenin levels in miR-3662 KO BT20 cells. As shown in Figs. 5G-H, we confirmed the effects of endogenous HBP1 on Wnt pathway activation and N-β-catenin levels. To determine the effect of HBP1 on cell proliferation, we transfected HBP1 siRNAs into miR-3662 KO cells for HBP1 silencing (Figs. 6A and 6B). Cell counting, clone formation, and Transwell assays revealed that reducing HBP1 enhanced proliferation and the migration rate of miR-3662 KO MDA-MB-231 cells (Figs. 6C-G). Thus, silencing of HBP1 in TNBC cells is likely to antagonize the miR-3662 KO-mediated inhibition of cell proliferation.
Figure 6. miR-3662-mediated functional role and post-transcriptional regulation of HBP1.
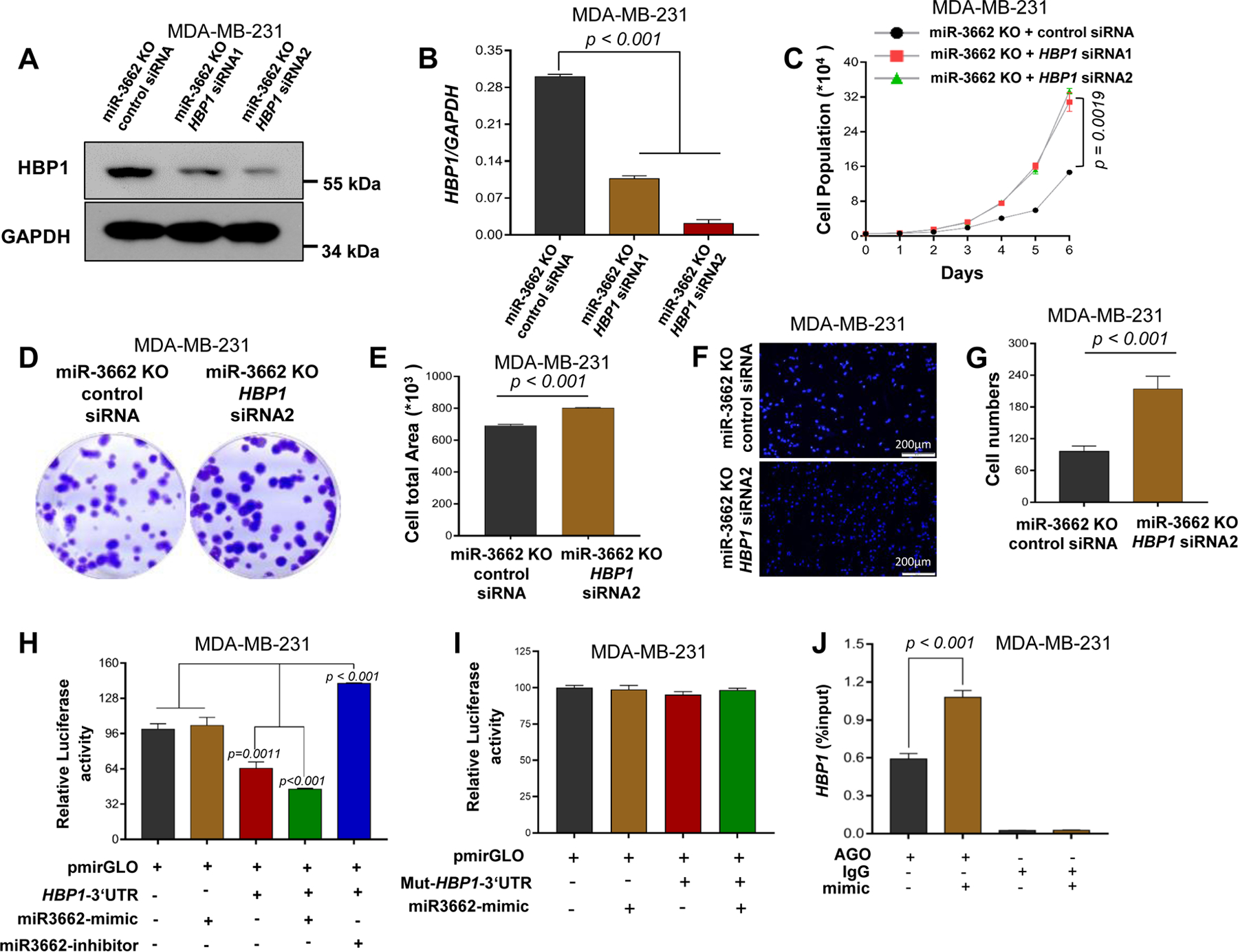
MDA-MB-231 miR3662 KO cells were transfected with either scrambled control siRNA or HBP1-specific siRNA. A. Western blot analysis of HBP1 protein expression. B. qPCR analysis of HBP1 expression. C. Cell proliferation measured by cell counts for 6 days. D, E. Clonogenic cell assay. After 2 weeks, the cloning formation was analyzed with crystal violet staining. F, G. Transwell invasion assays. H. Luciferase activity of HEK293T cells transfected with the miR-3662 mimic or inhibitor. The dual-luciferase reporter assay was performed on aliquots of the samples, and firefly luciferase activity, normalized to Renilla luciferase, was plotted. I. Specificity of the miRNA-HBP1 interaction validated by directed mutagenesis of miRNA-binding sites in HBP1 3′-UTR. J. Interaction analysis of miR-3662 with the 3′-UTRs of HBP1 mRNA by miRNA/mRNA IP assay in MDA-MB-231 cells. Data are presented as means ± SD. P values were determined by a two-tailed t-test or a two-way ANOVA test. siRNA, small interfering RNA; KO, knockout. All experiments were repeated three times.
Post-transcriptional regulation of HBP1 by miR-3662 in human TNBC cells
To determine if transcription of HBP1 is regulated by miR-3662, we predicted the miR-3662-targeting sequences in the 3’-UTR of HBP1. Sequence alignment analysis revealed the potential miR-3662-targeting sites (Fig. S7A). The post-transcriptional regulation of HBP1 by miR-3662 was determined using a luciferase reporting system (Fig. S7A). In HEK293T cells, transfection of the miR-3662 mimic reduced the luciferase activity of the WT HBP1 3’-UTR by 1.41-fold (Fig. 6H). However, deletion of the miR-3662-targeting sequence (CATTTT) in the HBP1 3’-UTR rescued the luciferase activity of HBP1 3’-UTR in HEK293T cells transfected with the miR-3662 mimic (Fig. 6I). In contrast, transfection of the miR-3662 inhibitor induced (2.2-fold) the luciferase activity of the WT HBP1 3’-UTR in HEK293T cells (Fig. 6H).
To determine if endogenous, mature miR-3662 interacts with HBP1 mRNA, we performed an miRNA-mRNA interaction analysis using miRNA target immunoprecipitation (IP) with Ago1/2/3 proteins in MDA-MB-231 cells with or without transfection of the miR-3662 mimic (Fig. S7B). Ago IP analysis showed that, in MDA-MB-231 and BT-20 cells, HBP1 mRNA binds with Ago1/2/3 proteins in the presence of miR-3662 relative to scrambled control miRNA (Fig. 6J and S7C), supporting a direct interaction of miR-3662 with HBP1 mRNA in TNBC cells. However, in MDA-MB-231 and BT-20 cells, no direct interaction of miR-3662 with LEPROT mRNA was evident in the presence or absence of the miR-3662 mimic (Figs. S7D and S7E), suggesting an indirect regulation of LEPROT by miR-3662 in TNBC cells.
Identification of a functional miR-3662-HBP1 axis and its contribution to proliferation and the Wnt pathway of human TNBC cells
To assess the dynamic effect of an miR-3662-HBP1 axis on cell proliferation, we established cell models stably expressing Dox-inducible CRISPR-dCas9-KRAB for CRISPR interference (CRISPRi) and CRISPR-dCas9-VPR for CRISPR activation (CRISPRa) 21 with single-guide RNAs (sgRNAs) for dynamic changes of miR-3662 expression in HEK293T and MDA-MB-231 cells (Figs. 7A and 7B). A U6 promoter-based lentiviral delivery system for sgRNA was designed for the different target regions upstream of the miR-3662 transcription start site 22. In subsequent experiments, the designed sgRNAs were used for CRISPRa and CRISPRi (Fig. 7C). After induction with Dox for 24, 48, or 72 hours, miR-3662 expression was down-regulated in CRISPRi HEK293T cells, whereas miR-3662 expression was steadily upregulated in CRISPRa HEK293T cells (Figs. 7D and 7E). Likewise, after induction with Dox for 1–7 days, miR-3662 expression was down-regulated in CRISPRi MDA-MB-231 cells but upregulated in CRISPRa MDA-MB-231 cells (Figs. 7F-H and 7M). After Dox induction, the mRNA levels of HBP1 were upregulated in CRISPRi MDA-MB-231 cells but down-regulated in CRISPRa MDA-MB-231 cells (Figs. 7I and 7N). To evaluate the efficiency of CRISPRi/a, cell proliferation was analyzed by MTT. Dox induction decreased the proliferation of CRISPRi MDA-MB-231 cells but increased the proliferation of CRISPRa MDA-MB-231 cells (Figs. 7J and 7O), suggesting regulation of cell proliferation through an miR-3662-HBP1 axis in human TNBC cells. Likewise, CRISPRi enhanced protein expression of HBP1, leading to lower expression of N-β-catenin and c-Myc (Figs. 7K-L and S8A). In contrast, CRISPRa reduced protein expression of HBP1, leading to accumulation of N-β-catenin and higher expression of c-Myc (Figs. 7P-Q and S8B).
Figure 7. Regulation of the miR-3662-HBP1 axis and its effect on growth and the Wnt signaling pathway.
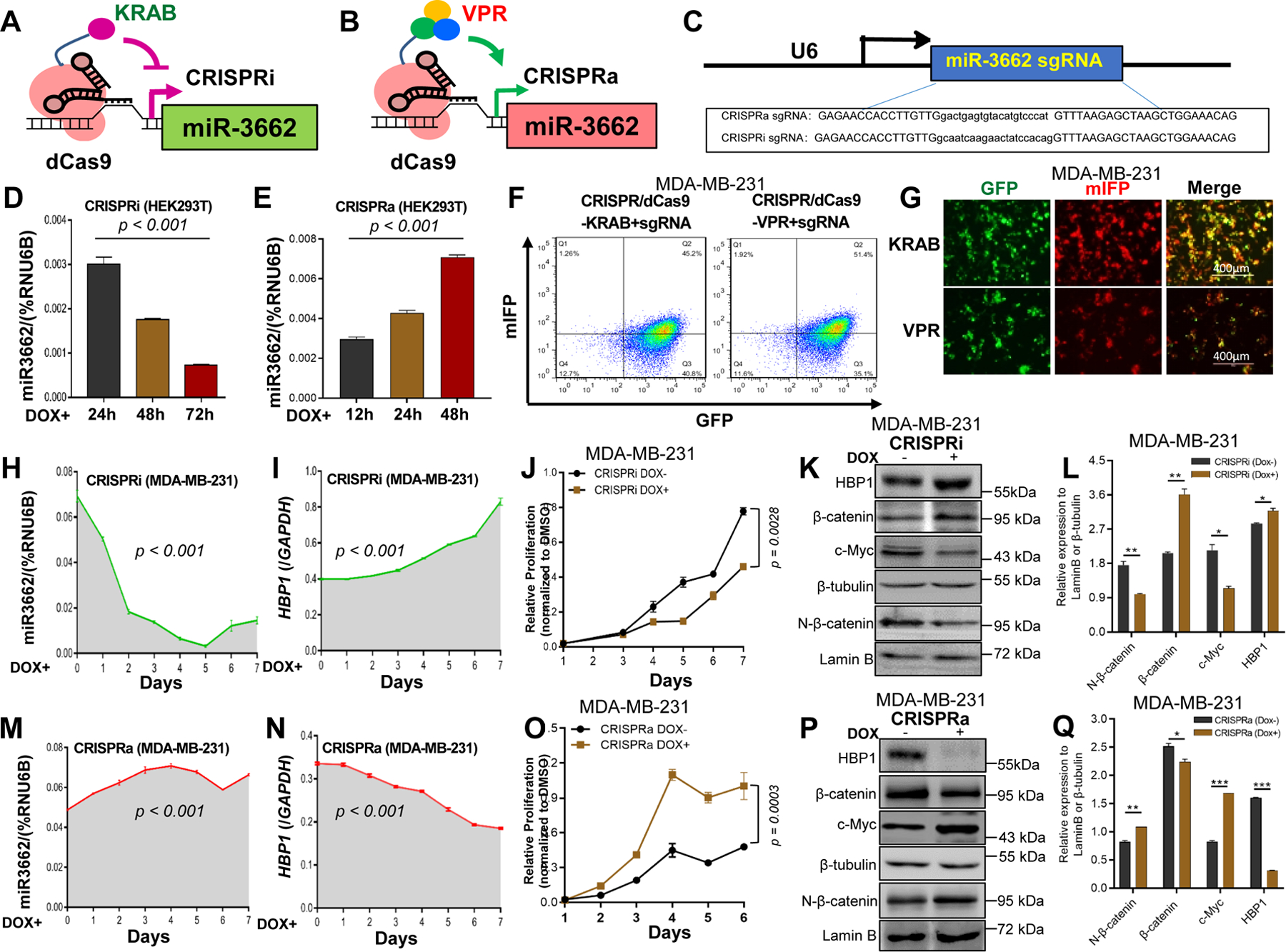
A, B. Schematic illustration of the CRISPRi/a system. C. Design and sequences for CRISPRi/a sgRNAs. D, E. Relative expression levels of mi-R3662 measured by qPCR in CRISPRi/a HEK293T cells after Dox induction (days 0–3). F, G. Expression of dCas9-KRAB, dCas9-VPR, and sgRNA determined by flow cytometric and IF analyses with GFP and mIFP in CRISPRi/a MDA-MB-231 cells. H, M. Expression changes of mi-R3662 in CRISPRi/a MDA-MB-231 cells after Dox induction (days 0–7) measured by qPCR. I, N. Expression changes of HBP1 in CRISPRi/a MDA-MB-231 cells after Dox induction (days 0–7) measured by qPCR. J, O. Changes of proliferation of CRISPRi/a MDA-MB-231 cells after Dox induction (days 1–7) assayed by MTT. K, L. Western blot and quantitative analyses of expression of HBP1 and Wnt signaling proteins in CRISPRi MDA-MB-231 cells before and after Dox induction (days 0 and 4). P, Q. Western blot and quantitative analyses of expression of HBP1 and Wnt signaling proteins in CRISPRa MDA-MB-231 cells before and after Dox induction (days 0 and 4). Data are presented as means ± SD. P values were determined by a two-tailed t-test or a one- or two-way ANOVA test. All experiments were repeated three times.
Although, in TCGA dataset, expression of miR-3662 was higher in TNBCs than in luminal breast cancers, only 50% (3/6) of TNBC cell lines had miR-3662 overexpression. Thus, a miR-3662-mediated oncogenic role may not be specific to the TNBC subtype. To assess whether the miR-3662-regulated Wnt pathway functions in other cancer types, we investigated, with colorectal cancer cell lines, the effect of miR-3662 on cell proliferation, HBP1, and the Wnt pathway using the miR-3662 mimic/antisense inhibitor. As shown in Figs. S2A and S9A, expression levels of miR-3662 in the three colorectal cancer cell lines were lower than in MDA-MB-231 and BT-20 cells, but expression levels of miR-3662 in SW480 and SW837 cells were relatively higher than in HT29 cells. We transiently treated miR-3662-low expressing HT29 cells with the miR-3662 mimic, but, after treatment, there were no changes in cell growth and few changes in HBP1 expression and Wnt signaling activation (Figs. S9B and S9D-F). However, when we transiently treated miR-3662-high expressing SW480 cells with the miR-3662 antisense inhibitor, cell growth was slower than in negative control miRNA-treated cells (Fig. S9C). Also, for SW480 cells, the miR-3662 inhibitor induced expression of HBP1 but reduced accumulation of N-β-catenin and increased expression of c-Myc (Figs. S9D and S9G-H). Thus, miR-3662 may have an oncogenic role in endogenous miR3662 high-expressing cancer cells but may not be specific to TNBC cells.
DISCUSSION
Based on TCGA dataset analysis, miR-3662 is rarely expressed in normal human epithelial tissues but is overexpressed in human cancer tissues. High expression levels of miR-3662 are evident in high-stage or African-American breast cancers. The present study provided evidence that, for TNBC cells, reduction of miR-3662 levels inhibits cell proliferation and migration in vitro and tumor growth and metastasis in vivo, highlighting an oncogenic function of miR-3662. Our functional analysis revealed that, in human TNBC cells, miR-3662 targets the 3’-UTR of HBP1 and post-transcriptionally suppresses HBP1 expression, leading to activation of the Wnt/β-catenin signaling pathway and to subsequent facilitation of tumor progression and metastasis (Fig. S10).
Regarding the role of miR-3662 in cancer cells, previous studies have found conflicting results. In acute myeloid leukemia, miR-3662 is a driver contributing to cell proliferation through NF-KB–mediated transcription 14. However, in hepatocellular carcinoma, miR-3662 is an inhibitor of tumor growth through the ERK and JNK signaling pathways 15 or up-regulation of HK2 16. In addition, for melanoma, miR-3662 is an inhibitor of the aggressive process of epithelial-mesenchymal transition through down-regulation of ZEB1 17. In the present study, our data support an oncogenic role of miR-3662 through activation of Wnt/β-catenin signaling in human TNBC cells. This finding was validated by a combination approach, including miRNA changes by a synthetic mimic and an inhibitor, CRISPR/Cas9 genome editing, and CRISPRi/a epigenome editing. Wnt/β-catenin signaling, a conserved pathway in postnatal evolution, is activated by a Wnt-LRP6-Axin complex, leading to inhibition of β-catenin phosphorylation and thereby to the stabilization of β-catenin, which travels to the nucleus and activates Wnt target gene transcription, such as cyclin D1 and c-Myc 23. Breast cancer was the first cancer to be associated with the Wnt signaling pathway, which is involved in proliferation, metastasis, immune microenvironment regulation, stemness maintenance, therapeutic resistance, and phenotype shaping of cancer 24. However, in our candidate list of miR-3662 target genes, the regulatory components of Wnt/β-catenin signaling pathway have not been identified as direct targets of miR-3662. During screening of potential targets of miR-3662, we identified a binding site of miR-3662 within the 3’-UTR of HBP1, which is likely responsible for miR-3662-mediated activation of the Wnt/β-catenin signaling in human TNBC cells. Although in the present study, the previously reported targets and signaling pathways of miR-3662 were not identified in human TNBC cells, we cannot exclude these findings due to the potential roles and mechanisms of miR-3662 in different organs. Also, HBP1 is not the only miR-3662 target gene in TNBC cells. Other target genes, such as LEPROT (Leptin Receptor Overlapping Transcript), may contribute to the miR-3662-mediated oncogenic function in these cells. LEPROT may be involved in breast cancer development and in aging processes 25. However, our data, derived with TNBC cells, showed no direct interaction of miR-3662 with LEPROT mRNA, suggesting that LEPROT is unlikely to be a direct target of miR-3662.
HBP1, a potential tumor suppressor for various types of cancer, is a transcriptional repressor 26–30. HBP1 is also an inhibitor of Wnt/β-catenin signaling and, in breast cancer cells, negatively regulates proliferation by suppressing DNA binding and transactivation of the TCF-β-catenin complex and transcription of Wnt target genes, such as cyclin D1 and c-Myc 27, 31, 32. The canonical Wnt/β-catenin signaling is activated in more than 50% of patients with breast cancer 33, 34 and is required for TNBC development and progression 35–37. In the present study, we established the relevance of HBP1 with miR-3662-mediated regulation of the Wnt signaling pathway in human TNBC cells. Our data suggest that, in these cells, miR-3662 induces activation of the Wnt/β-catenin signaling pathway through targeting HBP1. To our knowledge, this is the first evidence supporting a functional role for miR-3662-HBP1-Wnt signaling in TNBC progression. However, HBP1 may also inhibit proliferation through direct interaction with other proteins, such as c-Myc and AKT 32. Thus, pathways other than the WNT signaling pathway may also contribute to miR-3662/HBP1-mediated tumor progression.
Conclusion
The present study showed that overexpression of miR-3662 in human breast cancer cells, especially TNBC cells, has an oncogenic function during tumor progression and metastasis. Functional analysis identified an miR-3662-HBP1 axis involving binding of miR-3662 to the 3′-UTR of HBP1, leading to activation of Wnt/β-catenin signaling in TNBC cells. Since, in human cancers, including breast cancers, miR-3662 is present in a tumor-specific manner, it is likely to serve as a biomarker or therapeutic target for breast cancer diagnosis and therapy, particularly for African-American TNBC patients.
MATERIALS AND METHODS
Cell lines, antibodies, and reagents
Human breast cancer cell lines BT-20, BT-474, BT-549, MDA-MB-157, MDA-MB-231, MDA-MB-468, SKBR3, and T-47D; breast epithelial cell line MCF10A; and embryonic kidney epithelial cell line HEK293T were obtained from the American Type Culture Collection (Manassas, VA). The MDA-MB-231-luc cell line was obtained from GenTarget (San Diego, CA). All cell lines were cultured for less than 6 months, authenticated by examining morphology and growth characteristics, and confirmed to be mycoplasma-free. Short tandem-repeat analysis for DNA fingerprinting was also used to verify the cell lines. Cells were maintained in high-glucose Dulbecco’s modified eagle medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin (100 U/L penicillin and 100 U/L streptomycin) (Thermo Fisher Scientific, Waltham, MA), and cultured in a saturated humidity atmosphere containing 5% CO2 at 37°C. Antibodies used were specific for the following targets: HBP1 (Cat No. 11746–1-AP, 1:5000 for Western blots, 1:100 for IHC staining; Proteintech, Rosemont, IL), β-catenin (Cat No. ab32572, 1:5000 for Western blots, 1:500 for IF staining; Abcam, Cambridge, MA), E-cadherin (Cat No. ab76055, 1:3000 for Western blots; Abcam), N-cadherin (Cat No. ab18203, 1:3000 for Western blots; Abcam), vimentin (Cat No. 5741, 1:200 for IHC staining; Cell Signaling Technology, Danvers, MA), GSK-3β (Cat No. 3915, 1:3000 for Western blots; Cell Signaling Technology), p-GSK-3β (Cat No. 9336, 1:3000 for Western blots; Cell Signaling Technology), c-Myc (Cat No. 904401, 1:3000 for Western blots; BioLegend, San Diego, CA), Ki67 (Cat No. ab15580, 1:200 for IHC staining; Abcam), cyclin D1 (Cat No. ab134175, 1:5000 for Western blots, Abcam), lamin B (Cat No. sc-374015, 1:5000 for Western blots; Santa Cruz Biotechnology, Dallas, TX), GAPDH (Cat No. 5174, 1:5000 for Western blots; Cell Signaling Technology), β-actin (Cat No. sc-47778, 1:5000 for Western blots; Santa Cruz Biotechnology), and β-tubulin (Cat No. ab6046, 1:5000 for Western blots; Abcam). Cells (1×105) were transfected with negative scrambled control or miRNA mimic (50 nmol/L) or inhibitor (100 nmol/L) using Lipofectamine 3000 (Thermo Fisher Scientific) for 48 hours. After transfection, cell morphology, viability, and numbers were monitored microscopically daily. Lentiviral vectors were co-transfected with packaging plasmids psPAX2 and pMD2G (Addgene, Cambridge, MA) into HEK293T cells.
Establishment of miR-3662 KO cell lines
The miR-3662 sgRNAs were designed using the online CRISPR design tool (Benchling, San Francisco, CA, https://benchling.com). The pri-miR-3662 region was selected to be targeted by CRISPR/Cas9 genome editing. A ranked list of sgRNAs was generated with specificity and efficiency scores. In two flanking sites of the mature miR-3662 sequence, a pair of sgRNAs with more than 30% specificity and efficiency scores were selected. The pair of oligos for two targeting sites was annealed and ligated to the BbsI-digested pSpCas9(BB)-2A-GFP (pX458) vector (#48138, Addgene). The pX458 plasmid containing each target sgRNA sequence or an pX458 empty vector was transfected into cells with Lipofectamine 3000. After flow cytometry sorting with GFP, 4000 GFP+ cells were seeded into each well of a 10-cm dish. After selection of single colonies, miR-3662 KO colonies were determined by Sanger sequencing with isolated genomic DNA, and miR-3662 expression levels in each clone were validated by qPCR. All sgRNAs were accessed using the online off-target searching tool (Cas-OFFinder, Daejeon, South Korea, http://www.rgenome.net/cas-offinder) 38 (Supplementary Table S3A and S3B). For each KO colony, the potential off-target regions were subjected to Sanger sequencing to exclude an off-target effect, as described previously 39, 40. The scrambled control cells were obtained by sorting Cas9+ (GFP+) cells, which was validated by vertical gels and Sanger sequencing for cells without CRISPR/Cas9 cutting at both on-target and potential off-target loci. The sgRNAs and primers for CRISPR design and DNA construct are shown in Supplementary Table S4.
Generation of inducible CRISPRi and CRISPRa cell lines
Our CRISPRi/a lines were designed by co-expression of piggyBac (PB)-based dead Cas9 (dCas9) effector and sgRNA in the cells. The PB-based dCas9 constructs [(pSLQ1922 for CRISPRi: KRAB-SpdCas9, PiggyBac-Tre3g-KRAB-dCas9 (Bstx1)-2NLS-p2A-sfGFP-SV40polyA/R (EF1a-Zeocin-f2A-rtTA-1); pSLQ1932 for CRISPRa: VPR-SpdCas9, PiggyBac-Tre3g-dcas9-VRP-sfGFP-SV40polyA/R (EF1a-Zeocin-f2A-rtTA-1); both gifts from Dr. Stanley Qi, Stanford)] were used for CRISPR-dCas9-mediated miR-3662 transcription regulation. sgRNAs were cloned into the pLenti U6-spsgTRE3G CMV-mIFP (pSLQ2837, a gift from Dr. Stanley Qi, Stanford), as described previously 22, to generate sgRNA lentiviral constructs. sgRNA target sequences are listed in Supplementary Table S4.
To create stable CRISPRi/a cell lines, reporter cells were seeded at a density of 5×105 per well in 6-well plates and transfected with 3.6 μg PB-based dCas9 construct and 1.4 μg Super PB Transposase construct (Systems Biosciences, Palo Alto, CA) using Lipofectamine 3000. Then, 100 μg/mL Zeocin (Thermo Fisher Scientific) was added for the selection. Next, the CRISPR/dCas9 stably expressing cell lines were transduced with sgRNA lentiviruses (pSLQ2837-gRNAs). After transduction, cells were sorted by fluorescence-activated cell sorting using a BD FACS Aria2 with GFP and mIFP. Finally, the CRISPRi or CRISPRa stably expressing MDA-MD-231 cell lines were established (Supplementary Table S5). Doxycycline (Dox, 1 μg/mL) was supplemented daily to induce expression of dCas9-KRAB and dCas9-VPR in the CRISPRi/a cell lines. The plasmids are summarized in Supplementary Table S4.
Western Blotting
Western blotting was performed as described previously 41, 42. For nuclear proteins, the cells were first incubated in buffer A [10 mmol/L HEPES (pH 7.8), 10 mmol/L KCl, 2 mmol/L MgCl2, 0.1 mmol/L EDTA, 1% NP40, and protease inhibitors], and the pellet was suspended in buffer B [50 mmol/L HEPES (pH 7.8), 300 mmol/L NaCl, 50 mmol/L KCl, 0.1 mmol/L EDTA, 10% (v/v) glycerol, and protease inhibitors] 43.
Quantitative real-time PCR
For miRNA expression, 2 μg of RNA was reverse-transcribed using miScript II RT Kits (QIAGEN, Hilden, Germany) according to the manufacturer’s protocol. Then, cDNA was used as a template for real-time PCR using a LightCycler 480 Real-Time PCR System (Roche Applied Sciences, Indianapolis, IN) with miScript SYBR Green PCR kits (QIAGEN) at 95°C for 2 min, followed by 45 cycles of 95°C for 15 sec and 60°C for 1 min. The relative quantities of miRNA were determined by the comparative method (2−ΔCt) with an RNU6B or miR-16–5p reference, as described previously 44, 45. For mRNA expression of coding genes, relative expression levels were determined by qPCR with SYBR Green Dye (Thermo Fisher Scientific) using the comparative method (2−ΔCt) against endogenous GAPDH in accordance with the manufacturer’s protocol. The qPCR primer sequences are listed in Supplementary Table S4.
Cell proliferation, migration, and cell cycle assays
For the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide) assay (Millipore Sigma, Burlington, MA), cells were plated in 96-well plates (1,000 cells per well, 3 wells per cell line), and MTT assays were conducted each day over a 6-day period. Briefly, 20 μL of MTT solution (2 mg/mL in PBS) was added to the culture medium at a final concentration of 0.5 mg/mL and incubated at 37°C for 4 hours. Then, the supernatants were aspirated, 150 μL of dimethyl sulfoxide was added to each well to dissolve the reaction product of MTT, and the optical density was spectrophotometrically measured at 570 nm (GMI Awareness Stat Fax 4200, Ramsey, MN), with dimethyl sulfoxide as a blank. Viability was expressed as a percentage of the values for vehicle-treated cultures, set to 100%. Cells were plated in 24-well plates (5,000 cells per well, 6 wells per cell line), and cell morphology, viability, and numbers were monitored microscopically daily. The Transwell assays for migration were performed with Transwell inserts with 8.0-μm-pore polycarbonate membranes (Millipore Sigma) as described previously 40. To avoid the effect of cell proliferation on migration analysis, cells were incubated with 10 μg/ml mitomycin C (Sigma-Aldrich) for 2 hours prior to the Transwell migration assay. For evaluation of the cell cycle, after starvation of cells for 48 hours, the cells were collected and subjected to propidium iodide (PI) (50 μg/ml, BD Biosciences) staining and flow cytometry as described previously 41, 42.
Xenogeneic transplantation
NSG mice were purchased from the Jackson Laboratory (Bar Harbor, ME). All animal experiments were performed under the protocol approved by the Institutional Animal Care and Use Committee of University of Alabama at Birmingham (UAB; Birmingham, AL). 1 × 106 cells were subcutaneously injected into the fourth mammary fat pads of 8-week-old female NSG mice. Xenograft tumor growth was assessed for 4 weeks after tumor cell injection. Tumor growth was measured with calipers every other day, and their volume was determined as (width2 × length)/2 43, 46. Xenograft tumor metastasis was monitored by imaging using an IVIS Lumina Series III (PerkinElmer, Akron, OH).
IHC
Xenograft tumors were fixed with 10% neutral buffered formalin for 12–24 hours, then dehydrated with 70% alcohol and embedded in paraffin blocks. ABC kits (Vector Laboratories, Burlingame, CA) were used for immunostaining according to the manufacturer’s protocol.
Dual-luciferase assay
The pmirGLO luciferase reporter vectors (Promega, Madison, WI) were used to construct DNA fragments from the 3’-UTR of HBP1 (Transcript: HBP1–201 ENST00000222574.4) by Pmel and Xbal (Promega) digestion according to the manufacturer’s protocol. The sequences for DNA construction and mutagenesis are listed in Supplementary Table S4. Luciferase activity was measured as described previously 46–48. Briefly, cells were plated at a density of 1× 104 per well into 96-well plates and then transiently co-transfected using Lipofectamine 3000 with luciferase gene reporter vectors (pmirGLO-HBP1–3’-UTR) and miR-3662 mimic (50 nmol/L) or inhibitor (100 nmol/L) according to the manufacturer’s protocol. After transient transfections for 48 hours, cells were washed twice with ice-cold PBS and were lysed in 1 x lysis buffer (Promega) for 15 min on a shaker. The luciferase activity was assessed with a Veritas Microplate Luminometer (TD-20/20 Luminometer, Turner Designs) using a Dual-Luciferase Assay System (Promega).
miRNA/mRNA IP assay
miRNA/mRNA IP was performed by use of miRNA Target IP kits (Active Motif, Carlsbad, CA) according to the manufacturer’s protocol. Briefly, cells (1.5 × 107) were transfected with a negative scrambled control or an miRNA mimic (50 nmol/L) or inhibitor (100 nmol/L) using Lipofectamine 3000 for 24 hours. Transfected cells of each sample were lysed in 150 μL of complete lysis buffer and incubated on ice for 5 min and then at −80°C for 2 hours. Protein G magnetic beads (50 μL) were mixed with 200 μL of BSA solution for 10 min, and then the tubes were placed on a magnet to pellet the beads. After removing the supernatant, 100 μL of wash buffer containing 5 μg of anti-argonaute (Ago)1/2/3 antibody or negative control anti-IgG antibody was incubated for 30 min. The lysate, mixed with 1,000 μL IP buffer, was added to the protein G magnetic beads, incubated overnight at 4°C, and treated with proteinase K to digest protein for 30 min at 55°C. Procedures for purification and detection of mRNA and miRNA are described above. The results of Ago-IP were normalized to that of negative control IgG-IP.
Prediction of miR-3662 target genes
An in silico search for possible miRNA-binding sites in the 3′-UTR of target genes was performed using Targetscan (www.targetscan.org) and Starbase (www.starbase.sysu.edu.cn). The source of potential miR-3662 target genes was selected from TargetScan 20 and our RNA-seq data analysis. Briefly, candidate target genes of miR-3662 were selected from TargetScan due to their total context score (++) ≤ - 0.15. The overlap genes between the TargetScan dataset and our RNA-seq data were regarded as target genes, for which enrichment analysis was conducted using GSEA terms (www.gsea.umt.edu) and KEGG pathways (www.genome.jp/kegg). Based on these results, the miRNA-Gene Ontology (GO)-network and the miRNA-KEGG-network were generated.
RNA-seq
RNA libraries were prepared using TruSeq Stranded mRNA Library Prep Kits (Illumina, San Diego, CA) according to the manufacturer’s protocol. Integrity was assessed with an Agilent 2200 Tapestation instrument (Santa Clara, CA). First-strand cDNA syntheses were performed using random hexamers and ProtoScript II Reverse Transcriptase (New England Biolabs, Ipswich, MA). The libraries were normalized, pooled, and subjected to cluster, and pair-read sequencing was performed for 150 cycles on a HiSeqX10 instrument (Illumina), according to the manufacturer’s instructions. The RNA-seq data were submitted to NCBI GEO (accession No. GSE169128).
RNA interference (RNAi)-mediated gene knockdown
HBP1 and negative control siRNAs (Integrated DNA Technologies, Coralville, IA) were transfected by Lipofectamine 3000 into MDA-MB-231-miR-3662-KO cells. The cells were incubated for 48 hours at 37°C in a CO2 incubator until assayed for gene knockdown.
Bioinformatic analysis
Analysis of the differentially expressed genes (DEGs) was performed by fold change and q-value. miRNA expression data in cancer and normal tissue samples were obtained from Oncomir (www.oncomir.umn.edu) and TCGA (www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga). Coding gene mRNA expression data in cancer and normal tissue samples were obtained from TCGA. The ClueGO (apps.cytoscape.org/apps/cluego), a Cytoscape plug-in, was used to create and visualize a functionally grouped network of terms and pathways for large gene clusters, which were uploaded from a text file or interactively from the Cytoscape network. KEGG (www.genome.jp/kegg/) analysis of the obtained DEGs was performed to retrieve interacting genes and proteins (string-db.org/). The interaction network diagram of DEGs was constructed with Cytoscape software (cytoscape.org/).
Statistical analyses
Differences in outcome between two groups were compared by a two-sided t-test or the Mann-Whitney U test. Analysis of variance (ANOVA), one- and two-way, were used to test for overall differences, followed by a Dunnett post hoc test for differences between groups. All data were entered into an access database using Excel 2016 and analyzed with SPSS (version 25; IBM, Armonk, NY) and GraphPad (Prism 8, San Diego, CA).
Supplementary Material
Acknowledgment
We thank Dr. Donald Hill for editorial assistance in preparing this manuscript. This work was supported by grants from the National Cancer Institute (CA223077, CA238273 and CA242917 for R. Liu) and the Breast Cancer Research Foundation of Alabama (L. Wang). Results are based, in part, upon data generated by TCGA Research Network: http://cancergenome.nih.gov/.
Footnotes
Conflict of interest statement: There are no potential conflicts of interest for disclosure.
References
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin 2021; 71: 7–33. [DOI] [PubMed] [Google Scholar]
- 2.Garrido-Castro AC, Lin NU, Polyak K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov 2019; 9: 176–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res 2007; 13: 4429–4434. [DOI] [PubMed] [Google Scholar]
- 4.Caparica R, Lambertini M, de Azambuja E. How I treat metastatic triple-negative breast cancer. ESMO Open 2019; 4: e000504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Brien J, Hayder H, Zayed Y, Peng C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front Endocrinol (Lausanne) 2018; 9: 402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bertoli G, Cava C, Castiglioni I. MicroRNAs: New Biomarkers for Diagnosis, Prognosis, Therapy Prediction and Therapeutic Tools for Breast Cancer. Theranostics 2015; 5: 1122–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Croce CM, Calin GA. miRNAs, cancer, and stem cell division. Cell 2005; 122: 6–7. [DOI] [PubMed] [Google Scholar]
- 8.van Schooneveld E, Wildiers H, Vergote I, Vermeulen PB, Dirix LY, Van Laere SJ. Dysregulation of microRNAs in breast cancer and their potential role as prognostic and predictive biomarkers in patient management. Breast Cancer Res 2015; 17: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang J, Ahmad A, Sarkar FH. The role of microRNAs in breast cancer migration, invasion and metastasis. Int J Mol Sci 2012; 13: 13414–13437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cittelly DM, Das PM, Spoelstra NS, Edgerton SM, Richer JK, Thor AD et al. Downregulation of miR-342 is associated with tamoxifen resistant breast tumors. Mol Cancer 2010; 9: 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu S, Tao Z, Hai B, Liang H, Shi Y, Wang T et al. miR-424(322) reverses chemoresistance via T-cell immune response activation by blocking the PD-L1 immune checkpoint. Nat Commun 2016; 7: 11406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Powrozek T, Krawczyk P, Kowalski DM, Winiarczyk K, Olszyna-Serementa M, Milanowski J. Plasma circulating microRNA-944 and microRNA-3662 as potential histologic type-specific early lung cancer biomarkers. Transl Res 2015; 166: 315–323. [DOI] [PubMed] [Google Scholar]
- 13.Powrozek T, Mlak R, Dziedzic M, Malecka-Massalska T, Sagan D. Analysis of primary-miRNA-3662 and its mature form may improve detection of the lung adenocarcinoma. J Cancer Res Clin Oncol 2017; 143: 1941–1946. [DOI] [PubMed] [Google Scholar]
- 14.Maharry SE, Walker CJ, Liyanarachchi S, Mehta S, Patel M, Bainazar MA et al. Dissection of the Major Hematopoietic Quantitative Trait Locus in Chromosome 6q23.3 Identifies miR-3662 as a Player in Hematopoiesis and Acute Myeloid Leukemia. Cancer Discov 2016; 6: 1036–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Z, Zuo X, Zhang Y, Han G, Zhang L, Wu J et al. MiR-3662 suppresses hepatocellular carcinoma growth through inhibition of HIF-1alpha-mediated Warburg effect. Cell Death Dis 2018; 9: 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ye J, Xiao X, Han Y, Fan D, Zhu Y, Yang L. MiR-3662 suppresses cell growth, invasion and glucose metabolism by targeting HK2 in hepatocellular carcinoma cells. Neoplasma 2020; 67: 773–781. [DOI] [PubMed] [Google Scholar]
- 17.Zhu L, Liu Z, Dong R, Wang X, Zhang M, Guo X et al. MicroRNA-3662 targets ZEB1 and attenuates the invasion of the highly aggressive melanoma cell line A375. Cancer Manag Res 2019; 11: 5845–5856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hansen TB, Bramsen JB, Kjems J. Re-inspection of small RNA sequence datasets reveals several novel human miRNA genes. PLoS One 2010; 5: e10961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science 1996; 272: 1023–1026. [DOI] [PubMed] [Google Scholar]
- 20.Shi Y, Yang F, Wei S, Xu G. Identification of Key Genes Affecting Results of Hyperthermia in Osteosarcoma Based on Integrative ChIP-Seq/TargetScan Analysis. Med Sci Monit 2017; 23: 2042–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao Y, Xiong X, Wong S, Charles EJ, Lim WA, Qi LS. Complex transcriptional modulation with orthogonal and inducible dCas9 regulators. Nat Methods 2016; 13: 1043–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014; 159: 647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell 2009; 17: 9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene 2017; 36: 1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cicekdal MB, Kazan BT, Tuna BG, Ozorhan U, Ekici ID, Zhu F et al. Effects of two types of energy restriction on methylation levels of adiponectin receptor 1 and leptin receptor overlapping transcript in a mouse mammary tumour virus-transforming growth factor-alpha breast cancer mouse model. Br J Nutr 2021; 125: 1–9. [DOI] [PubMed] [Google Scholar]
- 26.Shih HH, Xiu M, Berasi SP, Sampson EM, Leiter A, Paulson KE et al. HMG box transcriptional repressor HBP1 maintains a proliferation barrier in differentiated liver tissue. Mol Cell Biol 2001; 21: 5723–5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sampson EM, Haque ZK, Ku MC, Tevosian SG, Albanese C, Pestell RG et al. Negative regulation of the Wnt-beta-catenin pathway by the transcriptional repressor HBP1. EMBO J 2001; 20: 4500–4511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berasi SP, Xiu M, Yee AS, Paulson KE. HBP1 repression of the p47phox gene: cell cycle regulation via the NADPH oxidase. Mol Cell Biol 2004; 24: 3011–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yee AS, Paulson EK, McDevitt MA, Rieger-Christ K, Summerhayes I, Berasi SP et al. The HBP1 transcriptional repressor and the p38 MAP kinase: unlikely partners in G1 regulation and tumor suppression. Gene 2004; 336: 1–13. [DOI] [PubMed] [Google Scholar]
- 30.Xiu M, Kim J, Sampson E, Huang CY, Davis RJ, Paulson KE et al. The transcriptional repressor HBP1 is a target of the p38 mitogen-activated protein kinase pathway in cell cycle regulation. Mol Cell Biol 2003; 23: 8890–8901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim J, Zhang X, Rieger-Christ KM, Summerhayes IC, Wazer DE, Paulson KE et al. Suppression of Wnt signaling by the green tea compound (−)-epigallocatechin 3-gallate (EGCG) in invasive breast cancer cells. Requirement of the transcriptional repressor HBP1. J Biol Chem 2006; 281: 10865–10875. [DOI] [PubMed] [Google Scholar]
- 32.Bollaert E, de Rocca Serra A, Demoulin JB. The HMG box transcription factor HBP1: a cell cycle inhibitor at the crossroads of cancer signaling pathways. Cell Mol Life Sci 2019; 76: 1529–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Howe LR, Brown AM. Wnt signaling and breast cancer. Cancer Biol Ther 2004; 3: 36–41. [DOI] [PubMed] [Google Scholar]
- 34.Lin SY, Xia W, Wang JC, Kwong KY, Spohn B, Wen Y et al. Beta-catenin, a novel prognostic marker for breast cancer: its roles in cyclin D1 expression and cancer progression. Proc Natl Acad Sci U S A 2000; 97: 4262–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Geyer FC, Lacroix-Triki M, Savage K, Arnedos M, Lambros MB, MacKay A et al. beta-Catenin pathway activation in breast cancer is associated with triple-negative phenotype but not with CTNNB1 mutation. Mod Pathol 2011; 24: 209–231. [DOI] [PubMed] [Google Scholar]
- 36.Khramtsov AI, Khramtsova GF, Tretiakova M, Huo D, Olopade OI, Goss KH. Wnt/beta-catenin pathway activation is enriched in basal-like breast cancers and predicts poor outcome. Am J Pathol 2010; 176: 2911–2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu J, Prosperi JR, Choudhury N, Olopade OI, Goss KH. beta-Catenin is required for the tumorigenic behavior of triple-negative breast cancer cells. PLoS One 2015; 10: e0117097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bae S, Park J, Kim JS. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 2014; 30: 1473–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, Li X, Liu W, Li B, Chen D, Hu F et al. MicroRNA-1205, encoded on chromosome 8q24, targets EGLN3 to induce cell growth and contributes to risk of castration-resistant prostate cancer. Oncogene 2019; 38: 4820–4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang M, Zhang G, Zhang Y, Cui X, Wang S, Gao S et al. Fibrinogen Alpha Chain Knockout Promotes Tumor Growth and Metastasis through Integrin-AKT Signaling Pathway in Lung Cancer. Mol Cancer Res 2020; 18: 943–954. [DOI] [PubMed] [Google Scholar]
- 41.Wang L, Liu R, Ye P, Wong C, Chen GY, Zhou P et al. Intracellular CD24 disrupts the ARF-NPM interaction and enables mutational and viral oncogene-mediated p53 inactivation. Nat Commun 2015; 6: 5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang W, Yi B, Wang C, Chen D, Bae S, Wei S et al. Silencing of CD24 Enhances the PRIMA-1-Induced Restoration of Mutant p53 in Prostate Cancer Cells. Clin Cancer Res 2016; 22: 2545–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu R, Yi B, Wei S, Yang WH, Hart KM, Chauhan P et al. FOXP3-miR-146-NF-kappaB Axis and Therapy for Precancerous Lesions in Prostate. Cancer Res 2015; 75: 1714–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang G, Zhang W, Li B, Stringer-Reasor E, Chu C, Sun L et al. MicroRNA-200c and microRNA-141 are regulated by a FOXP3-KAT2B axis and associated with tumor metastasis in breast cancer. Breast Cancer Res 2017; 19: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gao S, Wang Y, Wang M, Li Z, Zhao Z, Wang RX et al. MicroRNA-155, induced by FOXP3 through transcriptional repression of BRCA1, is associated with tumor initiation in human breast cancer. Oncotarget 2017; 8: 41451–41464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang L, Liu R, Li W, Chen C, Katoh H, Chen GY et al. Somatic single hits inactivate the X-linked tumor suppressor FOXP3 in the prostate. Cancer Cell 2009; 16: 336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu R, Liu C, Chen D, Yang WH, Liu X, Liu CG et al. FOXP3 Controls an miR-146/NF-kappaB Negative Feedback Loop That Inhibits Apoptosis in Breast Cancer Cells. Cancer Res 2015; 75: 1703–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu R, Wang L, Chen G, Katoh H, Chen C, Liu Y et al. FOXP3 upregulates p21 expression by site-specific inhibition of histone deacetylase 2/histone deacetylase 4 association to the locus. Cancer Res 2009; 69: 2252–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.