

Abstract
Hendra virus (HeV) and Nipah virus (NiV) are bat-borne zoonotic paramyxoviruses identified in the mid- to late 1990s in outbreaks of severe disease in livestock and people in Australia and Malaysia, respectively. HeV repeatedly re-emerges in Australia while NiV continues to cause outbreaks in South Asia (Bangladesh and India), and these viruses have remained transboundary threats. In people and several mammalian species, HeV and NiV infections present as a severe systemic and often fatal neurologic and/or respiratory disease. NiV stands out as a potential pandemic threat because of its associated high case-fatality rates and capacity for human-to-human transmission. The development of effective vaccines, suitable for people and livestock, against HeV and NiV has been a research focus. Here, we review the progress made in NiV and HeV vaccine development, with an emphasis on those approaches that have been tested in established animal challenge models of NiV and HeV infection and disease.
Keywords: henipavirus, Hendra virus, Nipah virus, vaccine, subunit vaccine, henipavirus countermeasures
INTRODUCTION
Nipah virus (NiV) and Hendra virus (HeV) are bat-borne viral zoonoses that were discovered in the mid- to late 1990s in outbreaks of severe disease in livestock and people in Australia (HeV) and Malaysia [NiV-Malaysia (NiV-M)] (1). They are the prototype members of the genus Henipavirus in the family Paramyxoviridae (2). NiV outbreaks have also been recorded in Bangladesh and India by a closely related strain, NiV-Bangladesh (NiV-B) (3). Three other henipaviruses are also recognized: Cedar virus (CedV) as an isolate and Ghana virus (GhV) and Mojiang virus (MojV) known only from sequence data (4–7). Both NiV and HeV are highly pathogenic in a broad range of mammalian hosts that are capable of infecting and causing severe disease in humans, monkeys, pigs, horses, cats, dogs, ferrets, hamsters, and guinea pigs and that span six mammalian orders including bats, although bats do not exhibit disease when infected (8–21). In contrast, CedV is nonpathogenic in well-characterized models of HeV and NiV disease including ferrets and hamsters (4, 22). The pathogenic potential of GhV and MojV is unknown.
Several species of Pteropus fruit bats are the natural reservoir hosts of NiV, HeV, and CedV (4, 23–27). NiV- or HeV-mediated disease has not been reported in wild or experimentally infected bats (13, 28–30). NiV and HeV infections in people and many animals manifest as severe systemic and often fatal neurologic and/or respiratory diseases (31–33). Both NiV and HeV are regarded as transboundary biological threats to both human and animal health and are classified as biosafety level 4 (BSL-4) select agents (34, 35). NiV and henipaviral diseases are included in the World Health Organization (WHO) R&D Blueprint list of priority pathogens with epidemic potential that need research attention (36). This review summarizes the important characteristics of the NiV and HeV pathogens, the modes of virus transmission, and the immunization strategies being developed against them.
Emergence and Outbreaks of Hendra and Nipah Viruses
In 1994 in the Brisbane suburb of Hendra, Australia, an outbreak of severe respiratory disease resulted in the deaths of 14 horses and their trainer, along with the nonfatal infection of 7 other horses and 1 other person. This led to the discovery of a novel paramyxovirus initially termed equine morbillivirus, now known as HeV (37–39). The first known cases of HeV in horses and a human actually occurred a few months prior, where one person became ill after assisting in the necropsies of two horses later shown to have died from HeV (40, 41). This individual experienced a relapsed fatal encephalitis caused by HeV 13 months later (42). HeV has since re-emerged in Australia 62 times with a total of 104 horse deaths (fatal or euthanized), along with 4 human fatalities of 7 cases (43). Every recorded occurrence of HeV in Australia has involved horses, all resulting in a severe or fatal disease, and all cases of human infection were acquired from virus-shedding horses (31, 44).
In 1998, an outbreak of encephalitis among pig farmers in Peninsular Malaysia occurred and a virus was isolated from samples of cerebrospinal fluid (CSF) of two patients who had died; cells infected with this virus cross-reacted with antibodies against HeV (45). Genetic studies revealed a new paramyxovirus that was closely related to HeV, and it was named Nipah after the village in Malaysia where one of the patients had lived (45). There were 265 cases of human infection with 105 fatalities in Malaysia and 11 cases and 1 fatality among abattoir workers in Singapore (46, 47). This outbreak was controlled through the culling of more than 1 million pigs, resulting in significant economic impacts to the region (48, 49).
A genetically similar but distinct strain of NiV was identified as the causative agent of fatal encephalitis in people in Bangladesh (NiV-B) (3, 50). Since 2001, nearly annual occurrences of human NiV-B infections have occurred in Bangladesh, and there have been three outbreaks In India (51–54). The recent 2018 NiV outbreak in Kerala, India, was significant, having occurred in a new geographic region far from locations in Bangladesh and India where all prior outbreaks had occurred and with a case fatality rate of 91% (51). In 2014, an outbreak of NiV-M encephalitis occurred in the Philippines with 9 fatalities of 11 human cases of acute encephalitis and influenza-like illness or meningitis in another 6 individuals (55). Altogether, there have been over 650 cases of human NiV infection (combined ~60% fatality rate) in South Asia and Southeast Asia in five countries (54, 56).
Transmission of Hendra and Nipah Viruses
The routes of transmission of virus infection to humans from animals are different for HeV and NiV, with horses the only spillover host of HeV in Australia, while for NiV it was pigs in Malaysia and horses in the Philippines (Figure 1). However, human NiV infections in Bangladesh, India, and the Philippines also include bat-to-human and human-to-human transmission (57–60). Transmission routes of HeV and NiV to animals are likely urine from infected bats contaminating pastures or pigsties and/or virus-contaminated fruit spat from bats that is ingested (61, 62) (Figure 1). Recoverable virus is shed in the urine of experimentally infected bats and can also be detected in throat and rectal swabs (13, 28–30). Pooled urine samples from flying foxes are also routinely used to detect and isolate henipaviruses (4, 13, 23, 27, 63–65).
Figure 1.
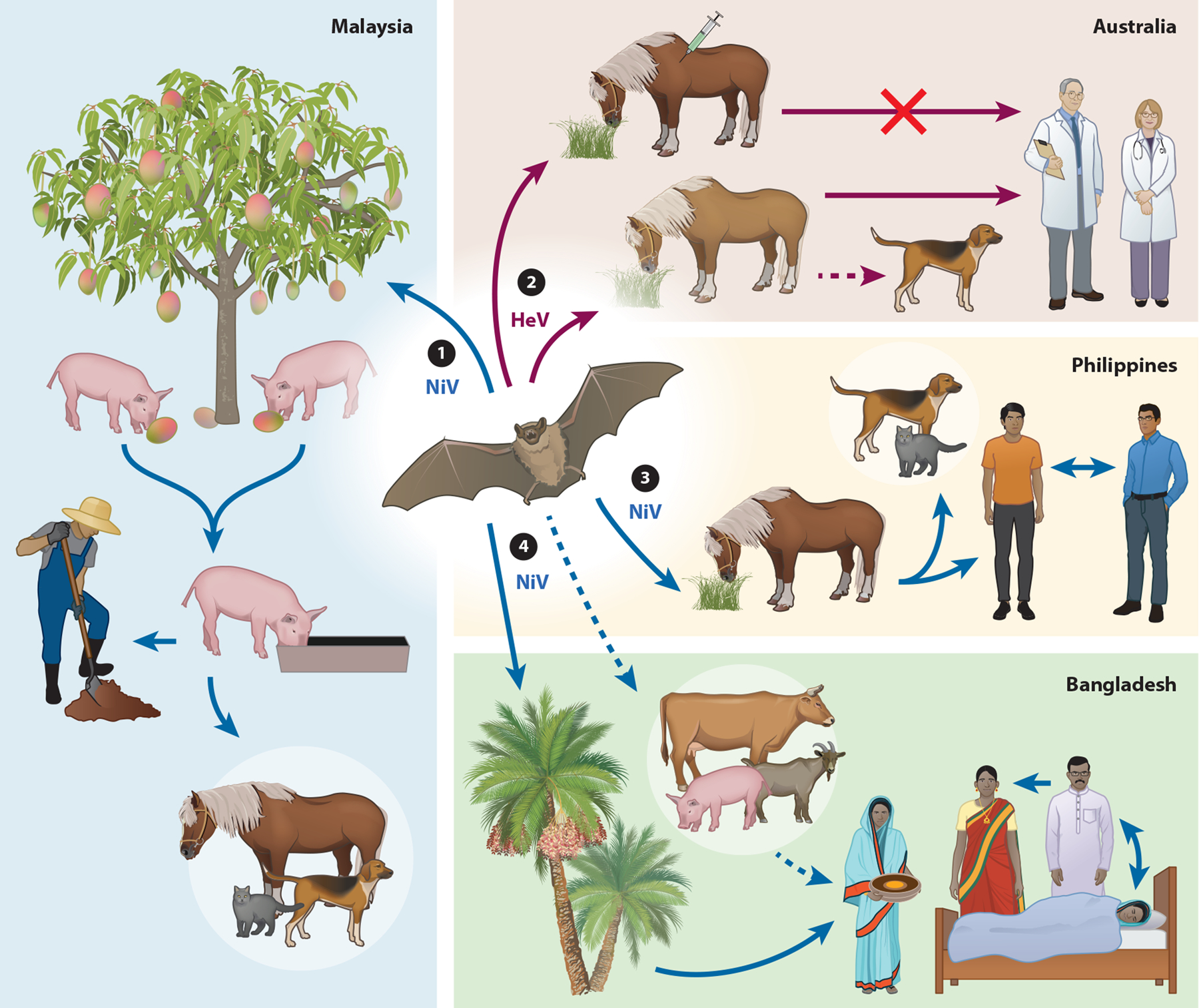
Nipah virus (NiV) and Hendra virus (HeV) modes of transmission in different countries. The transmission routes of NiV in Malaysia (left), Philippines (middle right), Bangladesh (bottom right), and HeV (top right) are depicted. Solid lines represent transmission that has been observed and documented, and dashed lines represent suspected transmission in natural conditions. Fruit bats are the natural reservoirs of NiV and HeV. ① Pigs are infected by consuming partially eaten or contaminated fruit from infected bats (urine, saliva) and transmit NiV to other pigs, pig farmers, or other animals (dogs, cats, and horses) through close or direct contact. ② Horses can be infected from grazing in contaminated pastures and transmit HeV to humans and on occasion domestic dogs through close contact. A One Health vaccine approach was developed for vaccination of horses in Australia with the dual purpose of saving horses from lethal HeV infection and preventing HeV transmission from horses to humans. ③ NiV is transmitted to humans through close contact with infected horses. NiV transmission to humans, cats, and dogs appears to have occurred following close contact with or consumption of infected horse meat. Human-to-human NiV transmission can occur through close contact. ④ Bat-to-human NiV transmission occurs through consumption of contaminated date palm sap. Human-to-human transmission can occur through close contact with infected patients. Humans may also become infected through contact with infected animals. Figure adapted with permission from Reference 171.
It was previously suggested that infected horses could transmit HeV to people during the feeding of ill animals (38). Also, the majority of all HeV-infected horse cases have involved a single animal, suggesting that HeV is not readily transmitted between horses, and multiple horse outbreaks are likely via contamination of fomites (43, 66). The transmission risk of HeV from infected horses to humans appears to be virus-contaminated fluids or tissues during examination procedures and/or the necropsy of horses (31, 67) (Figure 1). Indeed, all cases of human HeV infection have been associated with postmortem examination of horses or close contact with ill horses (31, 38, 42, 68). In Malaysia, it was contact with infected pigs or fresh infected pig products that was required for transmission of virus to humans (45, 69, 70) (Figure 1). NiV shedding in respiratory fluids of infected pigs suggested that it probably spread among farmed animals by aerosol droplets or direct contact (16, 71, 72). In Bangladesh, the transmission of NiV from bats to people has been linked to the consumption of virus-contaminated fresh date palm sap, and bats will consume sap during its collection (57, 73, 74). Domestic animals have also been linked to NiV infection in people in Bangladesh from unwell animals (cows and goats) and pigs (50, 59). Human- to-human transmission of NiV has been well documented in Bangladesh and India (52, 58–60, 75) (Figure 1). A study of human NiV-B cases in Bangladesh spanning 14 years reported that of 248 cases studied, one-third were caused by human-to-human transmission (56). Human-to-human transmission of NiV-M was not apparent in Malaysia (76, 77), whereas in the Philippines’ NiV-M outbreak, human cases were linked to horse slaughtering and horse meat consumption or exposure to other human patients, indicating both horse-to-human and human-to-human transmission (55) (Figure 1). The NiV-B outbreak in Kerala had a very high rate of human-to-human transmission (22 of 23 cases) at three different hospital locations (51).
Naturally acquired NiV infections were also recorded in cats, dogs, and horses in the initial Malaysian outbreak (Figure 1), and serological evidence of natural NiV infection in dogs was linked to outbreak farms (11, 61, 78). In the Philippines, both dogs and cats were linked to NiV-M infection, with cats dying after eating horse meat and dogs having NiV-neutralizing antibodies (55) (Figure 1). In Australia, a dog was found to be seropositive for HeV and later euthanized but showed no signs of disease, and a second HeV-positive dog was identified in 2013 following exposure to blood from an infected horse (79) (Figure 1). Dogs are susceptible to experimental HeV infection and shed virus but show little evidence of clinical illness (80).
Entry and Tropism of Nipah and Hendra Viruses
NiV and HeV are enveloped viruses containing an unsegmented, single-stranded, negative-sense RNA genome (2). Figure 2a is an illustration of the viral particle and the associated viral proteins. The genomes of HeV and NiV, and also CedV, GhV, and MojV, are considerably longer than the genomes of other paramyxoviruses, at greater than 18 kb. Henipavirus genomes encode 6 structural proteins: nucleoprotein (N), phosphoprotein (P), matrix protein (M), fusion glycoprotein (F), attachment glycoprotein (G), and the polymerase protein (L) (Figure 2a). The N, P, and L proteins comprise the replication complex. The P gene undergoes RNA editing to produce 2 additional nonstructural proteins, V and W, that are interferon (IFN) antagonists (81–84). The C protein is transcribed from a second open reading frame in the P gene (Figure 2a). NiV has been central to understanding the V, W, P, and C protein roles in antagonizing the innate immune responses via a diverse set of mechanisms (85, 86). Recent in vivo studies with recombinant NiV variants have further defined the varying importance of these nonstructural proteins in pathogenesis, but only a lack of the V protein results in a nonlethal infection (87–89). The henipavirus virion bears surface projections composed of the F and G glycoproteins that are anchored in the viral membrane and together mediate infection of host cells, and they are the major antigens of vaccine strategies (1) (Figure 2a). The F glycoprotein facilitates membrane fusion between the virus and host cell. The G glycoprotein consists of a characteristic stalk with a globular head that engages entry receptors on host cells, leading to the fusion activation of F and virus infection. The native structure of G is a tetramer while F is a trimer, and together they are the key determinants of infection and tropism (90–92). Models of the soluble ectodomain of the HeV G (HeV-sG) as a dimer and tetramer and the soluble ectodomain of the NiV F (NiV-sF) as a trimer are shown in Figure 2b. NiV and HeV utilize the host cell proteins ephrin-B2 and ephrin-B3 for entry (93–96). Ephrin-B2 and ephrin-B3 are members of a large family of ligands that bind to Eph receptors and are highly sequence conserved among mammals (97, 98). Ephrin-B2 expression is prominent in the vasculature of multiple organs, whereas ephrin-B3 is found predominantly in the nervous system (99–101). The ability of HeV and NiV to use these ephrins as receptors provided explanations of their broad host and tissue tropism (32, 33, 102). The NiV and HeV G head domain structures alone and in complex with ephrin-B2 and ephrin-B3 receptors have been determined (103–106). The structures of both the NiV and HeV F in their prefusion conformation have also been determined (107, 108). These studies have provided insights into understanding the virus entry receptors and host tropism features of the viruses on the molecular level and also facilitated further structural studies of henipavirus G and F glycoproteins in complexes with specific virus-neutralizing antibodies, providing valuable information that has aided vaccine design and choice (109, 110).
Figure 2.

Henipavirus structure and genome organization and models of the G and F glycoprotein soluble ectodomains, Hendra virus (HeV-sG) and Nipah virus (NiV-sF), respectively, and their complexes with respective NiV and HeV cross-reactive neutralizing monoclonal antibodies m102.3 (anti-G) and 5B3 (anti-F). (a) Schematic representation of a henipavirus particle with the structural proteins depicted in different colors (left) and the henipavirus genome (right). HeV and NiV P genes encode 3 nonstructural proteins: The C protein is expressed from an alternative start site, and the V and W proteins are expressed following the addition of one or two G residues at the messenger RNA editing site, respectively (right). (b, left) HeV-sG shown as a dimer solvent-accessible surface view with one monomer (cyan) overlaid with the monoclonal antibody m102.3 CDR-H3 loop (red) at the receptor binding site, and the other monomer (magenta) in complex with m102.3 Fab, which has an identical heavy chain and a similar light chain, that was used in place of the m102.4 monoclonal antibody (mAb) in the structural solution of the complex (109). The HeV-sG consists of amino acids 76–604, and the structures of the two globular head domains of HeV-sG are derived from the crystal structure (103, 172). The stalk regions of each G monomer (residues 77–136) are modeled (173). The light chain of m102.3 Fab is colored in yellow, and the heavy chain is colored in red. (b, middle) The HeV-sG tetramer surface view is modeled with one dimer (cyan and magenta) in front and the other dimer (blue and green) in back. N-linked glycans are gray spheres. (b, right) Structural model of the NiV-sF trimer in complex with the 5B3 Fab derived from the cryo–electron microscopy structure (110). The NiV-sF consists of amino acid residues 1–494 with a FLAG tag (DYKDDDK) introduced between residues L104-V105 and a C-terminal GCN4 motif. Each monomer of NiV-sF is in a different shade of blue, 5B3 heavy chain is in red, and light chain is in gold. N-linked glycans are illustrated in gray.
Nipah Virus and Hendra Virus Infection in Humans and Animals
Human NiV and HeV infections are generally accepted to occur via the oronasal route, and the incubation periods for both have been estimated to be 1 to 2 weeks (31, 51, 111). Acute infection in people is a systemic infection likely via hematogenous spread of the virus from the respiratory system (112). In general, HeV and NiV disease onset is characterized by fever, myalgia, shortness of breath, and cough (38, 111). Human HeV infections have resulted in both fatal respiratory or encephalitic disease and also recovery from infection (31, 38, 42, 68). The predominant clinical feature in the NiV-M outbreak in Malaysia was encephalitis, but respiratory symptoms were also common with fever, cough, and headache (47, 111, 112). The clinical presentation of NiV-B infections in Bangladesh also includes severe respiratory disease. In the 2018 NiV-B outbreak in Kerala, 83% of cases presented with acute respiratory distress syndrome (ARDS) (51, 113). Central findings of human NiV and HeV infection are a widespread endothelial cell tropism and systemic vasculitis, with prominent parenchymal cell infection in most major organs with the brain and lung significantly affected (45, 112, 114). Human NiV and HeV infections can also take a protracted course following apparent recovery, and some patients can experience late-onset encephalitis or relapsed encephalitis can occur in patients who previously recovered (42, 115). Relapsed encephalitis caused by NiV appears to result from a recrudescence of virus replication in the central nervous system (CNS), with cases presenting from a few months to as long as 11 years later (116–118). Recrudescence of virus has important implications for vaccine development.
The development of animal models of NiV and HeV infection and pathogenesis has been a major focus since the late 1990s and an essential component of vaccine development and testing. Also, the approval process of countermeasures for NiV and HeV would fall under the animal rule requirement set forth by the US Food and Drug Administration (FDA) in 2002 as an alternative licensing pathway for countermeasures against highly pathogenic agents when human efficacy studies are not feasible or ethical (119, 120). Several animal models of NiV and HeV infection have emerged that well reflect the pathogenesis seen in infected people, which includes a systemic vasculitis with both respiratory and neurological diseases. Detailed reviews of NiV and HeV infections of a variety of mammalian species have recently been published (33, 121–123). It is generally accepted that the pathogenic processes of NiV and HeV infection in the hamster, ferret, and African green monkey (AGM) best reflect the pathogenesis observed in humans, whereas the most appropriate models for livestock are the horse and pig themselves.
VACCINATION
The attachment and fusion glycoproteins of paramyxoviruses such as measles, mumps, and parainfluenza viruses are the viral antigens to which virtually all neutralizing antibodies are directed (124–126). Likewise, immunization strategies for NiV and HeV have largely targeted their G and F glycoproteins.
Passive Immunization Strategies
Early passive immunization studies in the hamster model demonstrated that polyclonal antiserums or mouse monoclonal antibodies (mAbs) to NiV F or G could provide complete protection against NiV-M or HeV when administered before and immediately after virus infection (10, 127, 128). These studies demonstrated a major role of a viral glycoprotein-specific antibody in protection.
Recombinant human antibody technology was used to generate a potent cross-neutralizing mAb against NiV and HeV (m102.4) (129, 130). The m102.4 mAb epitope maps to the ephrin receptor binding site of G and blocks virus infection (see the left side of Figure 2b), and it can neutralize NiV-M, NiV-B, and HeV (8, 109). The m102.4 mAb provided complete protection from NiV-M-mediated disease in ferrets as a single 50 mg dose administered 10 h post-challenge (8). In the AGM model, m102.4 administered as two 20 mg/kg doses, intravenously, at 10 h and again on day 3, on days 1 and 3 (days 1/3), or on days 3/5, after HeV challenge [4 × 105 50% tissue culture infectious dose (TCID50)] by intratracheal (i.t.) administration, protected 100% of treated subjects (131). All treated subjects seroconverted against HeV F glycoprotein with a rise in antibody titer over time, indicating all animals had become infected with HeV and recovered, whereas untreated control subjects succumbed to HeV disease and failed to mount a protective immune response. No clinical signs were evident at any time in the early treatment groups; although neurological symptoms were observed in subjects in the late treatment group (days 3/5), all later recovered from infection. There was no HeV antigen or virus-specific histopathology detected in the lung or brain at the conclusion of the study in any treated subject, and infectious virus could not be recovered from any tissue. A similar study evaluated m102.4 against NiV-M disease in the AGM model at several time points following virus challenge (5 × 105 PFU), including a late cohort where treatment was initiated at the onset of clinical illness (day 5) (132). All subjects became infected after challenge, and all subjects that received m102.4 survived infection and all controls succumbed to disease. Subjects in the late day 5/7 treatment group exhibited disease, but all recovered. A comparative study in AGMs using NiV-M and NiV-B [5 × 105 PFU divided by i.t. and intranasal (i.n.) administration] revealed that NiV-B caused a more aggressive disease, with a shortened time to death and higher virus loads in tissues and fluids (133). When m102.4 was tested in this model, all subjects in the days 1/3 and days 3/5 post-infection treatment groups survived NiV-B challenge, but subjects in the days 5/7 treatment group succumbed, indicating a shorter therapeutic window in treating NiV-B infection (133). Another well-characterized, humanized mouse mAb, 5B3 (h5B3.1), that is cross-reactive to the F glycoprotein of NiV and HeV and binds a prefusion conformation epitope on F, preventing membrane fusion, was recently tested (110, 134) (Figure 2b). The h5B3.1 mAb was given to ferrets in 20 mg/kg doses by intraperitoneal (i.p.) injection, at 1 to several days post-challenge, with either NiV or HeV (~5 × 103 PFU) delivered i.n. (135). All subjects that received h5B3.1 after infection were protected from disease and had increasing neutralizing antibody titers, whereas all controls died. No pathology was observed, and no infectious virus could be isolated at the study endpoint. Altogether, these studies demonstrate that passive immunization with mAbs can provide therapeutic benefit and allow the infected host an extended period to mount a protective immune response. The findings from these experiments were also important because they suggest that vaccine approaches designed to induce adequate neutralizing antibody responses to NiV and HeV should be effective.
The m102.4 mAb producing cell line was provided to the Queensland Government, Australia, to produce the mAb for compassionate use in future cases of high-risk human HeV infection. To date, 14 individuals exposed to either HeV in Australia (n = 13) or NiV in the United States (n = 1) have been given high-dose m102.4 therapy (15–20 mg/kg) by emergency use protocols, and all have remained well. In Australia, m102.4 was used in a randomized, controlled phase I study in healthy adults (136). The study included four single and one repeat dosing groups, and the m102.4 mAb was found to be safe and well tolerated, with a half-life ranging between ~16.5 and 27 days, and no observed immunogenicity was reported. Two doses of 20 mg/kg (days 1/3) were as well tolerated as a single dose. This study’s findings will aid in the design of future dosing regimens of mAbs for evaluating their ability to prevent and/or treat HeV and NiV human infections.
Active Immunization Strategies
A variety of immunization strategies have been developed to prevent NiV and HeV infection including several live-recombinant virus vectors, protein subunit, and virus-like particle (VLP) approaches, and all target the virus attachment and entry steps of infection by employing the G and/or F glycoprotein antigens. Here we summarize these various vaccination countermeasure approaches to NiV and HeV infection (Tables 1 and 2).
Table 1.
Virus vectored vaccine strategies for NiV and HeV
| Approach | Name(s) | Animal model | Vaccination dose, route, schedule | Adjuvant | Challenge | Challenge dose, route, schedule | Survival | Correlate(s) of immunity/protection | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Poxvirus | NYVAC-NiV-F and/or -G | Hamster | 2 doses at 1 × 107 PFU, s.c., 1 month apart | None | NiV-M | 1 × 103 PFU, i.p., 3 months later | 100% | NAb response, viral RNA | 127 |
| ALVAC-NiV-F and/or -G | Pigs | 2 doses at 1 × 108 PFU, i.m., 2 weeks apart | None | NiV-M | 2.5 × 105 PFU, i.n., 28 days later | 100% | NAb response, viral RNA, infectious virus, viral shedding, cytokine production | 140 | |
| ALVAC-HeV-F and/or -G | Hamster | 2 doses at 7.4 or 5.4 log10 CCID50, s.c., 3 weeks apart | None | HeV | 1 × 103 LD50, i.p., 21 days later | 89% and 63% | NAb response, viral RNA, viral antigen, viral shedding | 141 | |
| Ponies | 2 doses at 6 log10 CCID50, i.m., 3 weeks apart | None | NT | NA | NA | High NAb titers | |||
| MVA-NiV-sG and/or MVA-NiV-G | IFNAR−/− mice | 1 or 2 doses at 1 × 108 PFU, i.m., 3 weeks apart | None | NT | NA | NA | High serum IgG titers, NiV-G-specific CD8 and CD4 T cells | 142 | |
| VSV | VSV-NiV-F and/or -G | Mice | 5 × 103 PFU, i.n. or i.m. | None | NT | NA | NA | High NAb titers | 144 |
| Hamster | 1 × 106 infectious particles, i.m. | NiV-M | 1 × 105 TCID50, i.p., 32 days later | 100% | NAb response, viral RNA, viral antigen | 145 | |||
| VSV-NiV-B F and/or G | Ferret | 1 × 107 PFU, i.m. | None | NiV-M | 5 × 103 PFU, i.n., 28 days later | 100% | Serum IgG response, viral RNA, viral antigen | 146 | |
| AGM | NiV-B | 5 × 105 PFU, i.t. and i.n., 28 days later | NAb response, viral RNA, viral antigen | 147 | |||||
| VSV-ZEBOV-GP-NiV F, G, or N | Hamster | 1 × 105 PFU, i.p. | None | NiV-M | 1 × 103 LD50, i.p., 28 days later | 100% | NAb response, viral RNA, infectious virus | 148 | |
| AGM | 1 × 107 PFU, i.m. | 1 × 105 TCID50, i.t., 29 days later | NAb response, viral RNA, infectious virus, viral shedding | 149 | |||||
| VSV-HeV-G | Mice | 1 × 105 PFU, i.m. | None | NT | NA | NA | Serum IgG, Nab response | 150 | |
| AAV | AAV8 NiV.G | Mice | 2 × 1010 genome particles, i.m. or 1 × 1010 genome particles, i.d. | None | NT | NA | NA | Serum IgG, Nab response | 151 |
| Hamster | 6 × 1011 genome particles, i.m. | NiV-M | 1 × 104 PFU, i.p., 5 weeks later | 100% | Serum IgG, NAb response, viral RNA, viral antigen | ||||
| HeV | 50% | ||||||||
| Adenovirus | ChAdOx1 NiV-B G | Hamster | 2 doses at 1 × 108 IU, i.m., 28 days apart | None | NiV-B | 5.3 × 105 TCID50, i.p., 28 days later | 100% | NAb response, viral RNA, infectious virus, virus shedding | 153 |
| 1 × 108 IU, i.m. | NiV-M | 6.8 × 104 TCID50, i.p., 28 days later | 100% | ||||||
| HeV | 6 × 103 TCID50, i.p., 28 days later | 33% | |||||||
| Measles virus | rMV-Ed-G or rMV-HL-G | Hamster | 2 doses at 2 × 104 TCID50, i.p., 3 weeks apart | None | NiV-M | 1 × 103 TCID50, i.p., 1 week later | 100% | Serum IgG response | NA |
| rMV-Ed-G | AGM | 2 doses at 1 × 105 TCID50, s.c., 4 weeks apart | 1 × 105 TCID50, i.p., 1 week later | Serum IgG response, viral RNA | 154 | ||||
| Inactivated RABV | RABV-HeV-G | Mice | 3 doses at 10 μg, i.m., 2 weeks apart | None | NT | NA | NA | High NAb titers, serum IgG response | 150 |
| RABV-NiV-B G | 2 doses at 10 μg, i.m., 4 weeks apart | 155 | |||||||
| RABV | RABV-NiV-F and/or -G | Mice | 1 × 106.5 FFU, oral | None | NT | NA | NA | Serum IgG, Nab response | 156 |
All NiV glycoprotein vaccines employ the NiV-M strain unless otherwise indicated.
Abbreviations: AAV, adeno-associated virus; AGM, African green monkey; CCID50, 50% cell culture infectious dose; ChAdOx1, chimpanzee adenovirus Oxford 1; F, fusion glycoprotein; FFU, focus forming units; G, attachment glycoprotein; HeV, Hendra virus; i.d., intradermal; i.m., intramuscular; i.n., intranasal; i.p., intraperitoneal; i.t., intratracheal; IFNAR, interferon receptor; IgG, immunoglobulin G; IU, infectious unit; LD50, 50% lethal dose; MVA, modified vaccinia virus Ankara; NA, not applicable; NAb, neutralizing antibody; NiV, Nipah virus; NiV-B, Nipah virus Bangladesh; NiV-M, Nipah virus Malaysia; NT, not tested; PFU, plaque forming unit; RABV, rabies virus; rMV-Ed, recombinant measles virus Edmonston; rMV-HL, recombinant measles virus HL; s.c., subcutaneous; sF, F glycoprotein soluble ectodomain; sG, G glycoprotein soluble ectodomain; TCID50, 50% tissue culture infectious dose; VSV, vesicular stomatitis virus; ZEBOV-GP, Zaire ebolavirus glycoprotein.
Table 2.
VLP, subunit, and nucleic acid–based vaccine strategies for NiV and HeV
| Approach | Name | Animal model | Immunization dose, route, schedule | Adjuvant | Challenge | Challenge dose, route, schedule | Survival | Immune correlate(s) of survival | Reference |
|---|---|---|---|---|---|---|---|---|---|
| VLPs | VLPs-NiV M/F/G | Mice | 2 doses at 1.75, 3.5, 7, or 14 μg and 6 μg, s.c., 2 weeks apart | None | NT | NA | NA | High NAb titers | 159 |
| Hamster | 1 dose or 3 doses at 30 μg, i.m., 3 weeks apart | Alhydrogel/MPLA or Alhydrogel/CpG | NiV-M | 1.6 × 104 PFU (3-dose trial) or 3.3 × 104 PFU (1-dose trial), i.p., 28 days later | 100% | NAb response, viral RNA | 160 | ||
| Subunit vaccines | NiV-sG | Cat | 3 doses at 100 μg, s.c., 2 weeks apart | CSIRO triple adjuvant | NiV-M | 5 × 102 TCID50, s.c., 2 months later | 100% | NAb response, viral antigen, viral genome | 14 |
| HeV-sG | Cat | 3 doses at 100 μg, s.c., 2 weeks apart | CSIRO triple adjuvant | NiV-M | 5 × 102 TCID50, s.c., 2 months later | 100% | NAb response, viral antigen, viral genome | 14 | |
| 2 doses at 50, 25, or 5 μg, i.m., 3 weeks apart | CpG/Alhydrogel | 5 × 104 TCID50, o.n., 2 weeks later | 100% | Serum IgG, NAb response, viral RNA, viral shedding, infectious virus | 162 | ||||
| Ferret | 2 doses at 100, 20, or 4 μg, i.m., 20 days apart | CpG | HeV | 5 × 103 TCID50, o.n., 3 weeks later | 100% | NAb response, viral RNA, infectious virus | 163 | ||
| CpG/Alhydrogel | NiV-B | 5 × 104 TCID50, 20 days later or 12 months later | 100% | viral RNA, viral antigen, infectious virus | 164 | ||||
| AGM | 2 doses at 100, 50, or 10 μg, i.m., 3 weeks apart | CpG/Alhydrogel | NiV-M | 1 × 105 TCID50, i.t., 3 weeks later | 100% | Serum IgG, NAb response, viral RNA, viral antigen, infectious virus | 165 | ||
| 2 doses at 100 μg, i.m., 3 weeks apart | Alhydrogel or CpG/Alhydrogel | HeV | 5 × 105 PFU, i.t., 21 days later | 100% | NAb response, viral RNA | 166 | |||
| Horse | 2 doses at 100 or 50 μg, i.m., 3 weeks apart | Zoetis | HeV | 2 × 106 TCID50, o.n., 28 or 194 days later | 100% | NAb response, viral RNA, viral antigen, infectious virus | 167 | ||
| Pig | 2 doses of 2 mL preformulation, i.m., 3 weeks apart | Zoetis | HeV | 5 × 105 PFU, i.n., 35 days later | Partial | NAb response, viral RNA, infectious virus, viral shedding | 168 | ||
| NiV-M | 0% | ||||||||
| Nucleic acid–based vaccine | HeV-sG mRNA LNP | Hamster | 10 or 30 μg, i.m. | None | NiV-M | 1 × 105 TCID50, i.p., 30 days later | 30% or 70% | Serum IgG, NAb response | 169 |
All NiV glycoprotein vaccines employ the NiV-M strain.
Abbreviations: AGM, African green monkey; CSIRO, Commonwealth Scientific and Industrial Research Organisation; F, fusion glycoprotein; G, attachment glycoprotein; HeV, Hendra virus; i.m., intramuscular; i.n., intranasal; i.p., intraperitoneal; i.t., intratracheal; IgG, immunoglobulin G; LNP, lipid nanoparticle; M, matrix protein; MPLA, monophosphoryl lipid A; mRNA, messenger RNA; NA, not applicable; NAb, neutralizing antibody; NiV, Nipah virus; NiV-B, Nipah virus Bangladesh; NiV-M, Nipah virus Malaysia; NT, not tested; o.n., oronasal; PFU, plaque forming unit; s.c., subcutaneous; sG, G glycoprotein soluble ectodomain; TCID50, 50% tissue culture infectious dose; VLP, virus-like particle.
Poxvirus vectored.
Poxviruses have a long history as a platform for the expression of heterologous genes to study protein function and serve as vaccine candidates as a live-attenuated viral vaccine platform capable of inducing both cell-mediated and humoral immune responses (137). The F and G glycoproteins of NiV and HeV were functionally characterized using recombinant vaccinia viruses in the early 2000s (138, 139). The first NiV vaccine tested used a highly attenuated vaccinia virus strain (NYVAC) encoding either the F or G glycoproteins from NiV-M (127). Hamsters were vaccinated by subcutaneous (s.c.) injection in a prime-boost strategy with NYVAC-NiV-F or NYVAC-NiV-G, individually and in combination, and then 3 months later challenged i.p. with NiV-M. Vaccination yielded complete protection from NiV-M with no detection of viral RNA, and control subjects succumbed 7–10 days after challenge (127) (Table 1). Another poxvirus-based approach was examined as a vaccine for pigs using canarypox (ALVAC) vaccine vectors encoding either NiV-M F or G glycoprotein (140). A prime-boost strategy with ALVAC-NiV-F or ALVAC-NiV-G vectors was tested alone or in combination in pigs. The animals were then challenged 28 days later with NiV-M via i.n. administration. All vaccinated animals survived NiV-M challenge as determined by the lack of NiV RNA and infectious virus from nasal washes, pharyngeal swabs, and a variety of sampled organs (140).
ALVAC-vectored vaccines encoding HeV F or G glycoprotein for potential use in horses were also examined (141). ALVAC-HeV-F or ALVAC-HeV-G vectors were combined and first used to vaccinate hamsters at a high or low dose of each vector, by s.c. injection, and then challenged with HeV by i.p. administration. Vaccination did not result in complete protection, with 8 out of 9 subjects in the high-dose group and 5 out of 8 subjects in the low-dose group surviving challenge. No signs of disease were noted, and viral antigen or viral RNA could not be detected in survivors. Nine ponies vaccinated using the same prime-boost regimen were able to develop high cross-neutralizing antibody titers to HeV and NiV-M at day 28 after vaccination. Although ponies were not challenged, most animals yielded titers of at least 1:32 and were considered likely protective (141).
More recently, a modified vaccinia virus Ankara (MVA) vector encoding NiV-M G glycoprotein and a soluble version of G (NiV-sG) were examined in interferon receptor α and β (IFNAR−/−) knockout mice (142) (Table 1). IFNAR−/− mice were immunized once with MVA-NiV-G or MVA-NiV-sG or prime-boosted. IFNAR−/− mice developed high serum immunoglobulin G (IgG) titers to NiV-G and also generated NiV-G-specific CD8 and CD4 T cells following vaccination. MVA-NiV-sG vaccination induced rapid and significantly higher amounts of NiV-G epitope-specific CD8 T cells compared with the MVA-NiV-G candidate vaccine, suggesting superior immunogenicity. Together, these immunization studies with poxvirus vectors highlight that both T cell and B cell responses play a role in an adaptive immune response to NiV and HeV. However, detailed studies on the adaptive immune responses in animal experiments with henipaviruses have been limited. Future studies evaluating the role of NiV-specific T cells will be important because two human survivors of NiV-B infection in the 2018 outbreak in Kerala showed marked elevation of activated CD8+ T cells, which coincided with virus clearance (143).
Vesicular stomatitis virus vectored.
Recombinant vesicular stomatitis virus (rVSV) vectors as a vaccine platform suitable for single immunization strategies to potentially meet emergency use requirements have been tested by several groups (Table 1). A method of using two defective VSVΔG vectors each expressing only the NiV G or F glycoprotein was devised using VSV G glycoprotein complementation that can generate replication-defective VSV vectors that could elicit NiV-neutralizing antibodies (144). Using this technique, researchers tested rVSV vaccines expressing either NiV-M F or G glycoproteins (VSV-ΔG-NiVG, VSV-ΔG-NiVF) in hamsters by intramuscular (i.m.) vaccination (145). Hamsters were then challenged 32 days later with NiV-M by i.p. administration. All vaccinated animals survived lethal infection with no clinical signs of disease. No viral RNA or viral antigen could be detected in the sampled tissues when compared with controls, and there was a lack of an anamnestic immune response in vaccinated subjects following challenge, suggesting the induction of sterilizing immunity.
Another study used rVSV-ΔG vectors expressing NiV-B F or G glycoprotein and also tested them as single-injection vaccinations in NiV-M-challenged ferrets (146). Ferrets were vaccinated i.m. with rVSV-NiV-B F or rVSV-NiV-B G complemented with VSV G or a mix of both vectors, rVSV-NiV-B F/G, that was generated as a complementing pair in the absence of VSV G and then challenged at 28 days with NiV-M by i.n. administration. All vaccinated ferrets were completely protected against NiV-M challenge. Although viral RNA was detected in blood at day 6 post-challenge in 2 of 5 animals in each group, those levels were 100 times lower than in the unvaccinated controls, and by day 21 no viral RNA was detected (146). In a second study, rVSV-NiV-B F and rVSV-NiV-B G were assessed separately and in combination in AGMs (147). Cohorts were vaccinated with the rVSVs by i.m. injection and challenged 28 days later with NiV-B divided between the i.t. and i.n. routes (147). Complete protection was recorded from NiV-B disease with no gross pathology and no detectable NiV antigen in lung or spleen tissues. Viral RNA was detected in nasal and oral swabs of the vaccinated groups, but no viral RNA could be detected in blood samples.
Replication-competent rVSV-NiV-M F or G vectors, generated by the retention of the envelope glycoprotein from Zaire ebolavirus (ZEBOV-GP), which allowed virus stocks to be propagated (rVSV-ZEBOV-GP-NiVF, rVSV-ZEBOV-GP-NiVG, and rVSV-ZEBOV-GP-NiVN), have also been tested (148). These rVSVs were used to immunize hamsters by i.p. administration and were challenged 28 days later with NiV-M. All subjects vaccinated with either the NiV F or G glycoprotein encoding rVSV vectors were completely protected with no clinical disease or pathology, whereas those vaccinated with the NiV N protein were only partially protected (2 of 6 animals) with no clinical signs of disease and the other subjects succumbed to infection. The protective efficacy of the rVSV-ZEBOV-GP-NiVG was also tested in AGMs, where vaccinated subjects were challenged with NiV-M by i.t. administration 29 days later (149). All vaccinated subjects were protected from lethal challenge and showed no signs of clinical disease, no viral RNA was detected in the blood or oral and nasal swabs, and no infectious virus could be recovered. Another study using a rVSV vector expressing a codon-optimized HeV G gene together with an inactivated counterpart was evaluated in mice for humoral immune responses only as a comparator to a recombinant rabies virus vaccine encoding HeV G as a HeV vaccine candidate (150). Here, the live rVSV vectors induced greater levels of HeV G-specific antibodies and higher levels of HeVneutralizing antibodies than did the recombinant rabies virus vectors (see the section titled Rabies Virus Vectored).
Adeno-associated virus and adenovirus vectored.
Adeno-associated virus (AAV) vectors as a vaccine platform against infectious diseases, particularly viral pathogens, have been explored. AAV is a small, single-stranded DNA virus in the family Parvoviridae. Immunization of hamsters with an AAV vector expressing NiV-M G glycoprotein (AAV8 NiV.G) by i.m. injection demonstrated complete protection from a challenge of NiV-M by i.p. administration, and no signs of clinical disease were recorded (151) (Table 1). Neutralizing antibodies to NiV were induced, no viral RNA or viral antigen was detected in any of the sampled tissues, and there was only a moderate anamnestic response observed in a single subject, suggestive of potential sterilizing immunity. However, in a cross-protection study, AAV8 NiV.G protected only 50% of hamsters challenged with HeV.
Chimpanzee adenoviral (ChAd) vectors circumvent issues of the preexisting immunity observed with human adenovirus vectors (152). Adenoviruses are double-stranded DNA viruses in the family Adenoviridae. An engineered replication-deficient ChAd vector, Oxford 1 (ChAdOx1), was tested as a NiV/HeV vaccine (153). Here, ChAdOx1 encoding NiV-B (ChAdOx1 NiV-B) G glycoprotein was used to vaccinate hamsters by i.m. injection, either as a single dose or as a prime-boost protocol. Hamsters were challenged by i.p. administration with NiV-B 42 days following the booster or the single vaccination. Neutralizing antibodies were detectable, and all vaccinated hamsters were protected against lethal disease with no lung pathology, suggesting that a single dose of ChAdOx1 NiV-B was sufficient to completely protect against NiV-B. No viral RNA in the lung tissue and no viral shedding in oropharyngeal swabs could be detected, and no infectious virus could be isolated. A second cohort using a single dose of ChAdOx1 NiV-B to vaccinate hamsters was trialed, and these animals were challenged 28 days later with NiV-M or HeV. All vaccinated animals were protected from lethal NiV-M challenge, but 4 out of 6 hamsters succumbed to HeV disease between days 5 and 7 post-challenge. Neither virus shedding in oropharyngeal swabs nor infectious virus was detected in the lung or brain tissues of NiV-M-challenged vaccinated hamsters. In contrast, infectious virus was detected in the lung tissues of 75% of the HeV-challenged vaccinated animals. The lower cross-protection observation using NiV G vaccination followed by HeV challenge was not unexpected, as it was previously shown that when the G glycoprotein (as a recombinant soluble subunit immunogen) of either HeV or NiV was used to vaccinate cats, both could completely protect against lethal NiV-M challenge, and that the HeV-sG elicited greater heterologous neutralizing antibody responses in comparison to NiV-sG (14).
Measles virus vectored.
Recombinant measles virus vectors based on the HL (rMV-HL) and Edmonston (rMV-Ed) measles virus strains have also been explored in which they encoded the NiV-M G glycoprotein (rMV-HL-G and rMV-Ed-G) (154) (Table 1). Hamsters were immunized twice by i.p. administration of rMV-HL-G or rMV-Ed-G. All vaccinated animals produced NiV G-specific antibody titers after the booster immunization. Animals were challenged 1 week after the second immunization with NiV-M by i.p. administration. All immunized hamsters exhibited no clinical symptoms and survived challenge. The study was extended to non-human primates (NHPs), where 2 AGMs were immunized twice by s.c. injection with rMV-Ed-G. Subjects were challenged 2 weeks after the second immunization with NiV-M by i.p. administration. Here, immunization completely protected the AGMs with no observed clinical disease and no detectable pathological changes, and no viral RNA could be detected in sampled tissues. Although this was a small study, the safety profile and success of the live-attenuated measles virus vaccine suggests that a recombinant platform encoding the NiV G glycoprotein as a NiV vaccine candidate is promising and should induce a balanced and long-lasting immune response against NiV.
Rabies virus vectored.
A rabies virus (RABV) SAD B19 vaccine strain, BNSP333, expressing HeV or NiV G glycoproteins has been evaluated (150, 155). Recombinant BNSP333 encoding either the wild-type or a codon-optimized HeV G gene, together with their inactivated counterparts, was used in mice (150) (Table 1). Mice were immunized by i.m. injection with a single dose of the RABV-based vectors or with 3 doses of their inactivated versions. The inactivated RABV-based vectors induced higher and more rapid HeV G-specific antibody responses and higher neutralizing antibody titers than their live counterparts. The inactivated RABV-coHeV-G induced cross-neutralizing antibodies against NiV. A similar study used the BNSP333 vector expressing NiV-B G glycoprotein (RABV-NiV-BG) (Table 1) and elicited NiV G-specific neutralizing antibodies (155).
Recently, the recombinant RABV Evelyn-Rokitnicki-Abelseth (ERA) strain (rERAG333E) expressing either NiV-M F or G glycoproteins was evaluated in mice and pigs (156) (Table 1). This vector, rERAG333E, serves as an oral vaccine in dogs. Here, mice were orally immunized with RABV-NiV-F or RABV-NiV-G either individually or in combination. Pigs were also immunized in a similar manner but with 2 doses of each vector either alone or in combination. RABV-NiV-F and/or RABV-NiV-G immunization induced NiV F- and G-specific IgG antibody responses and neutralizing antibodies in both mice and pigs with the combination vaccine inducing higher titers. Although not suitable for human use, the live-attenuated rERAG333E vector is of interest as a potential veterinary vaccine for NiV because it is already approved for use in some animals and could be adapted for emergency use to protect against NiV infection in livestock, particularly swine.
Many of these virus-vectored vaccines for NiV are promising candidates because of their established safety profiles and ease of genetic modification. Several of these virus-vectored vaccines also require no adjuvants, and some are clearly efficacious as a single immunization strategy, suitable features for emergency use circumstances. In addition, several of these platforms are able to induce both cell-mediated and humoral immune responses, which may also be desirable but as yet are not fully explored in the development of vaccines for NiV and HeV. Although animals immunized with viral vectors encoding the NiV G glycoprotein and challenged with the homologous virus were completely protected, cross-protection studies with some of these vaccines against a HeV challenge were less effective. For example, only 50% of AAV8 NiV.G-immunized hamsters or 33% of ChAdOx1 NiV-B G glycoprotein–immunized hamsters were protected from a lethal HeV challenge (151, 153). In addition, the ALVAC-HeV-F and ALVAC-HeV-G vaccination studies showed that these vectors did not provide 100% protection in hamsters challenged with HeV, perhaps due to either a suboptimal immunization dose or the immunization route (141).
Virus-like particles.
VLPs have been explored as a vaccine platform because of the resemblance of their surface structure, dimensions, and compositions to authentic virus yet are of high safety because of the lack of viral genetic material. Earlier studies revealed that the M protein of NiV was capable of orchestrating the formation and budding of NiV VLPs when expressed in cells that appeared structurally similar to authentic NiV virions, and these VLPs could also incorporate other viral proteins such as the F and G glycoproteins (157, 158). VLPs composed of NiV M, F, and G were used to vaccinate mice s.c. at weeks 0, 2, and 4 and demonstrated they could induce high neutralizing antibody titers by day 35 (159) (Table 2). NiV VLPs were later used in NiV-M challenge studies either alone or in combination with adjuvant, monophosphoryl lipid A (MPLA) and Alhydrogel™ (15 μg/50 μg) or CpG and Alhydrogel (40 μg/50 μg) (160). Hamsters were vaccinated i.m. either as a single dose or as a 3-dose regimen and then challenged via i.p. administration of NiV-M at 28 days or 58 days, respectively. In all cohorts, 100% of the vaccinated animals survived with no clinical signs of disease and no detection of viral RNA in any of the sampled tissues, regardless of the presence of adjuvant. VLPs are thus an alternative means, with inherent safety, of producing an inactivated whole virus vaccine from an otherwise highly pathogenic virus.
Subunit vaccine.
The HeV-sG subunit vaccine has been extensively evaluated in several studies. Here, a brief summary of earlier reports is made, but the focus is on studies in NHPs and livestock. Recombinant HeV-sG and NiV-sG can elicit a potent neutralizing antibody response and were first tested as vaccine immunogens in the feline model (14, 161) (Table 2). Both HeV-sG and NiV-sG vaccination of cats completely protected against lethal NiV-M challenge, and HeV-sG elicited greater heterologous neutralizing titers than did NiV-sG, demonstrating that a single subunit vaccine may be effective against both NiV and HeV (14). Other studies using lower doses of HeV-sG (Table 2) demonstrated that a pre-challenge neutralizing titer of 1:32 could protect against NiV-M (162). Additional studies in ferrets showed that low doses of HeV-sG could protect against HeV and NiV-B (163, 164) (Table 2). Also, a longevity study showed that vaccinated ferrets challenged with NiV-B at 14 months post-immunization, with pre-challenge neutralizing titers of 1:16 to 1:128, were also protected (164).
The HeV-sG vaccine has been extensively evaluated in AGMs (Table 2). In a cross-protection study, 100 μg, 50 μg, or 10 μg doses of HeV-sG in combination with Alhydrogel and CpG were administered i.m. as a prime-boost, on days 0 and 21. Pre-challenge 50% neutralization titers ranged from 1:28 to 1:379. All subjects were challenged with NiV-M by i.t. administration on day 42. All vaccinated subjects were completely protected, displaying no clinical signs of disease, and no viral RNA could be detected in blood and tissues and no infectious virus was isolated (165). Similarly, HeV-sG vaccination HeV challenge in AGMs has also been performed. Using a prime-boost regimen, AGMs were vaccinated twice, 3 weeks apart, by i.m. injection with 100 μg HeV-sG with Alhydrogel or HeV-sG with Alhydrogel and CpG, and then challenged 3 weeks later with HeV by i.t. administration (166). All vaccinated animals were completely protected from clinical disease, and no HeV RNA or viral antigen could be detected in swabs, blood, or tissues, and notably HeV-sG formulated in only Alhydrogel protected (166).
The efficacy and inherent safety of the HeV-sG subunit led to its development as an equine vaccine to prevent HeV infection of horses and also reduce the risk of HeV transmission to people, as a One Health concept (Figure 1). HeV-sG, formulated in an approved equine adjuvant (Zoetis, Inc.), was evaluated in two efficacy studies; the first tested 50 μg and 100 μg doses of the same HeV-sG used in prior animal studies to vaccinate horses, and the second used 100 μg doses of HeV-sG produced in Chinese hamster ovary cells (Zoetis, Inc.). Two vaccinations were given by i.m. administration 3 weeks apart. All horses in these efficacy studies were challenged by oronasal inoculation with HeV (Table 2). Seven horses were challenged at 28 days and 3 horses were challenged at 194 days after the second immunization. All vaccinated horses remained clinically healthy following challenge; pre-challenge neutralization titers ranged from 1:128 to more than 4,096 in horses challenged 21 days after vaccination and only from 1:16 to 1:32 in horses challenged at 6 months. There was no gross or histologic evidence of infection in any of the vaccinated horses at study completion, and all tissues examined were negative for viral antigen, with no viral genome detected in any tissue. In 9 of 10 vaccinated horses, HeV nucleic acid was not detected in daily nasal, oral, or rectal swab samples or from blood, urine, or fecal samples collected before euthanasia, no recoverable virus was present, and no rise in antibody titer was detected in any vaccinated horse following challenge (167). The HeV-sG horse vaccine (Equivac® HeV) was launched by Zoetis, Inc., in November 2012 on a minor use permit by the regulatory authority, the Australian Pesticides and Veterinary Medicines Authority (APVMA), and is the first commercially developed and deployed vaccine against a BSL-4 agent. All vaccinated horses are microchipped, and a database is maintained. Equivac HeV received full registration by the APVMA in 2015. To date, more than 765,000 doses of Equivac HeV vaccine have been administered to more than 179,000 unique horses, and laboratory-confirmed HeV infections in horses have since occurred only in unvaccinated animals.
Studies showed HeV-sG as a NiV vaccine in the pig model (which is a non-lethal challenge model) was much less effective in comparison to results observed in the cat, ferret, NHP, and horse, and HeV-sG was only partially protective against HeV challenge and unprotective against NiV-M in the pig (168). These experiments also indicated that both humoral and cellular immune responses were required for protection of swine against NiV and HeV. Here, pigs were immunized with HeV-sG in a proprietary adjuvant (Zoetis, Inc.), and subjects were challenged with HeV or NiV via i.n. administration (Table 2). HeV-sG-vaccinated pigs developed neutralizing titers ranging from 1:160 to 1:320 to HeV, but only partial protection was achieved with reduced viral RNA in tissues and no recoverable virus, and there was no reduction of viral shedding in nasal washes. These HeV-sG-vaccinated pigs did not develop neutralizing antibodies to NiV-M that were considered protective (low), nor did they have measurable activation of cellular immune memory. Only a comparative group of pigs that were first orally infected (vaccinated) with NiV (and recovered) were subsequently protected against an i.n. rechallenge with NiV. This group of pigs developed protective antibody levels and cell-mediated immune memory responses (168).
Single-dose lipid nanoparticle mRNA, HeV-sG vaccine.
More recently, messenger RNA (mRNA)-based vaccines have emerged as an attractive vaccine strategy because of safety, efficacy, and rapid implementation features. In a recent study, the efficacy of an mRNA vaccine approach was assessed in a NiV-M animal challenge model (169). mRNA transcripts encoding HeV-sG were complexed with lipid nanoparticles (LNPs) to generate HeV-sG mRNA LNP. Two groups of 10 hamsters were vaccinated with a single dose of HeV-sG mRNA LNP at either 10 μg or 30 μg by i.m. injection. Subjects were challenged with NiV-M by i.p. administration 30 days post-vaccination (Table 2). The HeV-sG mRNA LNP was only partially protective, with 3 hamsters from the low-dose group and 7 hamsters from the high-dose group surviving challenge. Of the surviving animals, signs of clinical disease were observed in 2 low-dose group and 6 high-dose group hamsters; however, disease symptoms were gone by study termination. NiV N gene RNA levels in the blood and a variety of tissues in surviving hamsters were lower compared with nonsurvivors, but NiV RNA copy levels were not different compared with controls. No anti-NiV IgG or virus-neutralizing activity was detected in vaccinated animals prior to challenge; however, all post-challenge survivors were positive for anti-NiV IgG antibodies, and all survivors (in both groups) had similar neutralizing titers ranging from 1:160 to 1:640. Euthanized animals had little to undetectable neutralizing activity, highlighting the correlation of this immune response to protection. Although promising, the partial efficacy of HeV-sG mRNA LNP observed in this study suggests that further optimization of vaccination route, addition of an adjuvant, and/or a prime-boost regimen is needed.
SUMMARY AND FUTURE PERSPECTIVES
The frequency of henipavirus outbreaks and human infections is a significant global health concern. A promising passive immunization strategy has been developed using a human mAb, m102.4, shown effective in the NHP challenge model, which has also been administered numerous times to people by compassionate use protocol and has successfully completed a phase I safety trial in Australia. In addition, the Equivac HeV vaccine is available, targeting the protection of horses and also people by breaking the chain of HeV transmission to people, and is an example of a One Health approach to counter an infectious disease threat. Over the past 15 years, nearly a dozen NiV and HeV vaccine approaches have been trialed in animal challenge models, and many show promise as effective human-use vaccines. Recently, the formation of the Coalition for Epidemic Preparedness Innovations (CEPI), a global partnership between public and private organizations, was undertaken with the goals of developing vaccines against emerging infectious diseases and offering equitable access to those vaccines (170). Indeed, without the support of CEPI, the prospects of having a NiV or HeV vaccine suitable for use in people, at a deployable stage in the event of a significant outbreak, would have remained academic. Research teams can now capitalize on the large body of basic and preclinical vaccine development data on a half-dozen important emerging viral threats including NiV and, with the support of CEPI, can develop vaccine candidates for clinical use and future licensure. Several of the NiV human vaccine candidates described in this review are now supported by CEPI.
ACKNOWLEDGMENTS
C.C.B. is supported by National Institutes of Health (NIH) grant AI142764 and Coalition for Epidemic Preparedness Innovations grant to Aurobindo Pharma USA Inc. The authors thank Dr. Kai Xu, NIH, Bethesda, Maryland, and Ha V. Dang and Dr. David Veesler, University of Washington, Seattle, Washington, for providing the glycoprotein models in Figure 2.
Footnotes
DISCLOSURE STATEMENT
C.C.B. is a US federal employee and co-inventor on US and foreign patents pertaining to soluble forms of HeV and NiV G and F glycoproteins and monoclonal antibodies against HeV and NiV whose assignee is the United States as represented by the Henry M. Jackson Foundation for the Advancement of Military Medicine (Bethesda, Maryland). The soluble forms of the HeV and NiV G glycoproteins are licensed to Zoetis, Inc., and Aurobindo Pharma USA Inc. M.A. declares no competing interests.
LITERATURE CITED
- 1.Eaton BT, Broder CC, Middleton D, Wang LF. 2006. Hendra and Nipah viruses: different and dangerous. Nat. Rev. Microbiol 4:23–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang L-F, Mackenzie JS, Broder CC. 2013. Henipaviruses. In Fields Virology, ed. Knipe DM, Howley PM, pp. 1070–85. Philadelphia: Lippincott Williams & Wilkins [Google Scholar]
- 3.Harcourt BH, Lowe L, Tamin A, Liu X, Bankamp B, et al. 2005. Genetic characterization of Nipah virus, Bangladesh, 2004. Emerg. Infect. Dis 11:1594–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marsh GA, de Jong C, Barr JA, Tachedjian M, Smith C, et al. 2012. Cedar virus: a novel Henipavirus isolated from Australian bats. PLOS Pathog. 8:e1002836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drexler JF, Corman VM, Gloza-Rausch F, Seebens A, Annan A, et al. 2009. Henipavirus RNA in African bats. PLOS ONE 4:e6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu Z, Yang L, Yang F, Ren X, Jiang J, et al. 2014. Novel Henipa-like virus, Mojiang Paramyxovirus, in rats, China, 2012. Emerg. Infect. Dis 20:1064–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maes P, Amarasinghe GK, Ayllon MA, Basler CF, Bavari S, et al. 2019. Taxonomy of the order Mononegavirales: second update 2018. Arch. Virol 164:1233–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bossart KN, Zhu Z, Middleton D, Klippel J, Crameri G, et al. 2009. A neutralizing human monoclonal antibody protects against lethal disease in a new ferret model of acute Nipah virus infection. PLOS Pathog. 5:e1000642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geisbert TW, Daddario-DiCaprio KM, Hickey AC, Smith MA, Chan YP, et al. 2010. Development of an acute and highly pathogenic nonhuman primate model of Nipah virus infection. PLOS ONE 5:e10690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guillaume V, Wong KT, Looi RY, Georges-Courbot MC, Barrot L, et al. 2009. Acute Hendra virus infection: analysis of the pathogenesis and passive antibody protection in the hamster model. Virology 387:459–65 [DOI] [PubMed] [Google Scholar]
- 11.Hooper P, Zaki S, Daniels P, Middleton D. 2001. Comparative pathology of the diseases caused by Hendra and Nipah viruses. Microbes Infect. 3:315–22 [DOI] [PubMed] [Google Scholar]
- 12.Hooper PT, Westbury HA, Russell GM. 1997. The lesions of experimental equine morbillivirus disease in cats and guinea pigs. Vet. Pathol 34:323–29 [DOI] [PubMed] [Google Scholar]
- 13.Middleton DJ, Morrissy CJ, van der Heide BM, Russell GM, Braun MA, et al. 2007. Experimental Nipah virus infection in pteropid bats (Pteropus poliocephalus). J. Comp. Pathol 136:266–72 [DOI] [PubMed] [Google Scholar]
- 14.Mungall BA, Middleton D, Crameri G, Bingham J, Halpin K, et al. 2006. Feline model of acute Nipah virus infection and protection with a soluble glycoprotein-based subunit vaccine. J. Virol 80:12293–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rockx B, Bossart KN, Feldmann F, Geisbert JB, Hickey AC, et al. 2010. A novel model of lethal Hendra virus infection in African green monkeys and the effectiveness of ribavirin treatment. J. Virol 84:9831–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weingartl H, Czub S, Copps J, Berhane Y, Middleton D, et al. 2005. Invasion of the central nervous system in a porcine host by Nipah virus. J. Virol 79:7528–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Westbury HA, Hooper PT, Brouwer SL, Selleck PW. 1996. Susceptibility of cats to equine morbillivirus. Aust. Vet. J 74:132–34 [DOI] [PubMed] [Google Scholar]
- 18.Westbury HA, Hooper PT, Selleck PW, Murray PK. 1995. Equine morbillivirus pneumonia: susceptibility of laboratory animals to the virus. Aust. Vet. J 72:278–79 [DOI] [PubMed] [Google Scholar]
- 19.Wong KT, Grosjean I, Brisson C, Blanquier B, Fevre-Montange M, et al. 2003. A golden hamster model for human acute Nipah virus infection. Am. J. Pathol 163:2127–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li M, Embury-Hyatt C, Weingartl HM. 2010. Experimental inoculation study indicates swine as a potential host for Hendra virus. Vet. Res 41:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marianneau P, Guillaume V, Wong T, Badmanathan M, Looi RY, et al. 2010. Experimental infection of squirrel monkeys with Nipah virus. Emerg. Infect. Dis 16:507–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schountz T, Campbell C, Wagner K, Rovnak J, Martellaro C, et al. 2019. Differential innate immune responses elicited by Nipah virus and Cedar virus correlate with disparate in vivo pathogenesis in hamsters. Viruses 11:291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chua KB, Koh CL, Hooi PS, Wee KF, Khong JH, et al. 2002. Isolation of Nipah virus from Malaysian Island flying-foxes. Microbes Infect. 4:145–51 [DOI] [PubMed] [Google Scholar]
- 24.Halpin K, Young PL, Field HE, Mackenzie JS. 2000. Isolation of Hendra virus from pteropid bats: a natural reservoir of Hendra virus. J. Gen. Virol 81:1927–32 [DOI] [PubMed] [Google Scholar]
- 25.Rahman SA, Hassan SS, Olival KJ, Mohamed M, Chang LY, et al. 2010. Characterization of Nipah virus from naturally infected Pteropus vampyrus bats, Malaysia. Emerg. Infect. Dis 16:1990–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reynes JM, Counor D, Ong S, Faure C, Seng V, et al. 2005. Nipah virus in Lyle’s flying foxes, Cambodia. Emerg. Infect. Dis 11:1042–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anderson DE, Islam A, Crameri G, Todd S, Islam A, et al. 2019. Isolation and full-genome characterization of Nipah viruses from bats, Bangladesh. Emerg. Infect. Dis 25:166–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Halpin K, Hyatt AD, Fogarty R, Middleton D, Bingham J, et al. 2011. Pteropid bats are confirmed as the reservoir hosts of henipaviruses: a comprehensive experimental study of virus transmission. Am. J. Trop. Med. Hyg 85:946–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williamson MM, Hooper PT, Selleck PW, Gleeson LJ, Daniels PW, et al. 1998. Transmission studies of Hendra virus (equine morbillivirus) in fruit bats, horses and cats. Aust. Vet. J 76:813–18 [DOI] [PubMed] [Google Scholar]
- 30.Williamson MM, Hooper PT, Selleck PW, Westbury HA, Slocombe RF. 2000. Experimental Hendra virus infection in pregnant guinea-pigs and fruit bats (Pteropus poliocephalus). J. Comp. Pathol 122:201–7 [DOI] [PubMed] [Google Scholar]
- 31.Playford EG, McCall B, Smith G, Slinko V, Allen G, et al. 2010. Human Hendra virus encephalitis associated with equine outbreak, Australia, 2008. Emerg. Infect. Dis 16:219–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wong KT, Ong KC. 2011. Pathology of acute henipavirus infection in humans and animals. Pathol. Res. Int 2011:567248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Geisbert TW, Feldmann H, Broder CC. 2012. Animal challenge models of henipavirus infection and pathogenesis. Curr. Top. Microbiol. Immunol 359:153–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.CDC (Cent. Dis. Control Prev.). 2009. Biosafety in Microbiological and Biomedical Laboratories, ed. Chosewood LC, Wilson DE. Washington, DC: Natl. Inst. Health. 5th ed. [Google Scholar]
- 35.ARS (Agricult. Res. Serv.). 2018. Henipavirus Gap Analysis. Washington, DC: US Dep. Agricult. [Google Scholar]
- 36.Sweileh WM. 2017. Global research trends of World Health Organization’s top eight emerging pathogens. Global Health 13:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murray K, Rogers R, Selvey L, Selleck P, Hyatt A, et al. 1995. A novel morbillivirus pneumonia of horses and its transmission to humans. Emerg. Infect. Dis 1:31–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Selvey LA, Wells RM, McCormack JG, Ansford AJ, Murray K, et al. 1995. Infection of humans and horses by a newly described morbillivirus. Med. J. Aust 162:642–45 [DOI] [PubMed] [Google Scholar]
- 39.Murray K, Selleck P, Hooper P, Hyatt A, Gould A, et al. 1995. A morbillivirus that caused fatal disease in horses and humans. Science 268:94–97 [DOI] [PubMed] [Google Scholar]
- 40.Hooper PT, Gould AR, Russell GM, Kattenbelt JA, Mitchell G. 1996. The retrospective diagnosis of a second outbreak of equine morbillivirus infection. Aust. Vet. J 74:244–45 [DOI] [PubMed] [Google Scholar]
- 41.Rogers RJ, Douglas IC, Baldock FC, Glanville RJ, Seppanen KT, et al. 1996. Investigation of a second focus of equine morbillivirus infection in coastal Queensland. Aust. Vet. J 74:243–44 [DOI] [PubMed] [Google Scholar]
- 42.O’Sullivan JD, Allworth AM, Paterson DL, Snow TM, Boots R, et al. 1997. Fatal encephalitis due to novel paramyxovirus transmitted from horses. Lancet 349:93–95 [DOI] [PubMed] [Google Scholar]
- 43.Queensland Gov. 2019. Summary of Hendra virus incidents in horses. Queensland Government. https://www.business.qld.gov.au/industries/service-industries-professionals/service-industries/veterinary-surgeons/guidelines-hendra/incident-summary
- 44.Barclay AJ, Paton DJ. 2000. Hendra (equine morbillivirus). Vet. J 160:169–76 [DOI] [PubMed] [Google Scholar]
- 45.Chua KB, Goh KJ, Wong KT, Kamarulzaman A, Tan PS, et al. 1999. Fatal encephalitis due to Nipah virus among pig-farmers in Malaysia. Lancet 354:1257–59 [DOI] [PubMed] [Google Scholar]
- 46.Chua KB, Bellini WJ, Rota PA, Harcourt BH, Tamin A, et al. 2000. Nipah virus: a recently emergent deadly paramyxovirus. Science 288:1432–35 [DOI] [PubMed] [Google Scholar]
- 47.Paton NI, Leo YS, Zaki SR, Auchus AP, Lee KE, et al. 1999. Outbreak of Nipah-virus infection among abattoir workers in Singapore. Lancet 354:1253–56 [DOI] [PubMed] [Google Scholar]
- 48.Ahmad K 2000. Malaysia culls pigs as Nipah virus strikes again. Lancet 356:230. [DOI] [PubMed] [Google Scholar]
- 49.Lam SK, Chua KB. 2002. Nipah virus encephalitis outbreak in Malaysia. Clin. Infect. Dis 34:S48–51 [DOI] [PubMed] [Google Scholar]
- 50.Hsu VP, Hossain MJ, Parashar UD, Ali MM, Ksiazek TG, et al. 2004. Nipah virus encephalitis reemergence, Bangladesh. Emerg. Infect. Dis 10:2082–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arunkumar G, Chandni R, Mourya DT, Singh SK, Sadanandan R, et al. 2019. Outbreak investigation of Nipah virus disease in Kerala, India, 2018. J. Infect. Dis 219:1867–78 [DOI] [PubMed] [Google Scholar]
- 52.Chadha MS, Comer JA, Lowe L, Rota PA, Rollin PE, et al. 2006. Nipah virus-associated encephalitis outbreak, Siliguri, India. Emerg. Infect. Dis 12:235–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harit AK, Ichhpujani RL, Gupta S, Gill KS, Lal S, et al. 2006. Nipah/Hendra virus outbreak in Siliguri, West Bengal, India in 2001. Indian J. Med. Res 123:553–60 [PubMed] [Google Scholar]
- 54.Luby SP, Hossain MJ, Gurley ES, Ahmed BN, Banu S, et al. 2009. Recurrent zoonotic transmission of Nipah virus into humans, Bangladesh, 2001–2007. Emerg. Infect. Dis 15:1229–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ching PK, de los Reyes VC, Sucaldito MN, Tayag E, Columna-Vingno AB, et al. 2015. Outbreak of henipavirus infection, Philippines, 2014. Emerg. Infect. Dis 21:328–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nikolay B, Salje H, Hossain MJ, Khan A, Sazzad HMS, et al. 2019. Transmission of Nipah virus—14 years of investigations in Bangladesh. N. Engl. J. Med 380:1804–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Luby SP, Rahman M, Hossain MJ, Blum LS, Husain MM, et al. 2006. Foodborne transmission of Nipah virus, Bangladesh. Emerg. Infect. Dis 12:1888–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gurley ES, Montgomery JM, Hossain MJ, Bell M, Azad AK, et al. 2007. Person-to-person transmission of Nipah virus in a Bangladeshi community. Emerg. Infect. Dis 13:1031–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Luby SP, Gurley ES, Hossain MJ. 2009. Transmission of human infection with Nipah virus. Clin. Infect. Dis 49:1743–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Homaira N, Rahman M, Hossain MJ, Epstein JH, Sultana R, et al. 2010. Nipah virus outbreak with person-to-person transmission in a district of Bangladesh, 2007. Epidemiol. Infect 138:1630–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Field H, Young P, Yob JM, Mills J, Hall L, Mackenzie J. 2001. The natural history of Hendra and Nipah viruses. Microbes Infect. 3:307–14 [DOI] [PubMed] [Google Scholar]
- 62.Field HE. 2016. Hendra virus ecology and transmission. Curr. Opin. Virol 16:120–25 [DOI] [PubMed] [Google Scholar]
- 63.Edson D, Field H, McMichael L, Vidgen M, Goldspink L, et al. 2015. Routes of Hendra virus excretion in naturally-infected flying-foxes: implications for viral transmission and spillover risk. PLOS ONE 10:e0140670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Peel AJ, Wells K, Giles J, Boyd V, Burroughs A, et al. 2019. Synchronous shedding of multiple bat paramyxoviruses coincides with peak periods of Hendra virus spillover. Emerg. Microbes Infect 8:1314–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barr J, Smith C, Smith I, de Jong C, Todd S, et al. 2015. Isolation of multiple novel paramyxoviruses from pteropid bat urine. J. Gen. Virol 96:24–29 [DOI] [PubMed] [Google Scholar]
- 66.Middleton D 2014. Hendra virus. Vet. Clin. North Am. Equine Pract 30:579–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mahalingam S, Herrero LJ, Playford EG, Spann K, Herring B, et al. 2012. Hendra virus: an emerging paramyxovirus in Australia. Lancet Infect. Dis 12:799–807 [DOI] [PubMed] [Google Scholar]
- 68.Hanna JN, McBride WJ, Brookes DL, Shield J, Taylor CT, et al. 2006. Hendra virus infection in a veterinarian. Med. J. Aust 185:562–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Parashar UD, Sunn LM, Ong F, Mounts AW, Arif MT, et al. 2000. Case-control study of risk factors for human infection with a new zoonotic paramyxovirus, Nipah virus, during a 1998–1999 outbreak of severe encephalitis in Malaysia. J. Infect. Dis 181:1755–59 [DOI] [PubMed] [Google Scholar]
- 70.Amal NM, Lye MS, Ksiazek TG, Kitsutani PD, Hanjeet KS, et al. 2000. Risk factors for Nipah virus transmission, Port Dickson, Negeri Sembilan, Malaysia: results from a hospital-based case-control study. Southeast Asian J. Trop. Med. Public Health 31:301–6 [PubMed] [Google Scholar]
- 71.Mohd Nor MN, Gan CH, Ong BL. 2000. Nipah virus infection of pigs in peninsular Malaysia. Rev. Sci. Tech 19:160–65 [DOI] [PubMed] [Google Scholar]
- 72.Middleton DJ, Westbury HA, Morrissy CJ, van der Heide BM, Russell GM, et al. 2002. Experimental Nipah virus infection in pigs and cats. J. Comp. Pathol 126:124–36 [DOI] [PubMed] [Google Scholar]
- 73.Khan MS, Hossain J, Gurley ES, Nahar N, Sultana R, Luby SP. 2010. Use of infrared camera to understand bats’ access to date palm sap: implications for preventing Nipah virus transmission. Ecohealth 7:517–25 [DOI] [PubMed] [Google Scholar]
- 74.Rahman MA, Hossain MJ, Sultana S, Homaira N, Khan SU, et al. 2012. Date palm sap linked to Nipah virus outbreak in Bangladesh, 2008. Vector-Borne Zoonotic Dis. 12:65–72 [DOI] [PubMed] [Google Scholar]
- 75.Sazzad HM, Hossain MJ, Gurley ES, Ameen KM, Parveen S, et al. 2013. Nipah virus infection outbreak with nosocomial and corpse-to-human transmission, Bangladesh. Emerg. Infect. Dis 19:210–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tan CT, Tan KS. 2001. Nosocomial transmissibility of Nipah virus. J. Infect. Dis 184:1367. [DOI] [PubMed] [Google Scholar]
- 77.Mounts AW, Kaur H, Parashar UD, Ksiazek TG, Cannon D, et al. 2001. A cohort study of health care workers to assess nosocomial transmissibility of Nipah virus, Malaysia, 1999. J. Infect. Dis 183:810–13 [DOI] [PubMed] [Google Scholar]
- 78.Mills JN, Alim AN, Bunning ML, Lee OB, Wagoner KD, et al. 2009. Nipah virus infection in dogs, Malaysia, 1999. Emerg. Infect. Dis 15:950–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kirkland PD, Gabor M, Poe I, Neale K, Chaffey K, et al. 2015. Hendra virus infection in dog, Australia, 2013. Emerg. Infect. Dis 21:2182–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Middleton DJ, Riddell S, Klein R, Arkinstall R, Haining J, et al. 2017. Experimental Hendra virus infection of dogs: virus replication, shedding and potential for transmission. Aust. Vet. J 95:10–18 [DOI] [PubMed] [Google Scholar]
- 81.Ciancanelli MJ, Volchkova VA, Shaw ML, Volchkov VE, Basler CF. 2009. Nipah virus sequesters inactive STAT1 in the nucleus via a P gene-encoded mechanism. J. Virol 83:7828–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rodriguez JJ, Horvath CM. 2004. Host evasion by emerging paramyxoviruses: Hendra virus and Nipah virus V proteins inhibit interferon signaling. Viral. Immunol 17:210–19 [DOI] [PubMed] [Google Scholar]
- 83.Shaw ML, Cardenas WB, Zamarin D, Palese P, Basler CF. 2005. Nuclear localization of the Nipah virus W protein allows for inhibition of both virus- and toll-like receptor 3-triggered signaling pathways. J. Virol 79:6078–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shaw ML, Garcia-Sastre A, Palese P, Basler CF. 2004. Nipah virus V and W proteins have a common STAT1-binding domain yet inhibit STAT1 activation from the cytoplasmic and nuclear compartments, respectively. J. Virol 78:5633–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Basler CF. 2012. Nipah and Hendra virus interactions with the innate immune system. Curr. Top. Microbiol. Immunol 359:123–52 [DOI] [PubMed] [Google Scholar]
- 86.Pelissier R, Iampietro M, Horvat B. 2019. Recent advances in the understanding of Nipah virus immunopathogenesis and anti-viral approaches. F1000Research 8:1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Satterfield BA, Borisevich V, Foster SL, Rodriguez SE, Cross RW, et al. 2019. Antagonism of STAT1 by Nipah virus P gene products modulates disease course but not lethal outcome in the ferret model. Sci. Rep 9:16710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Satterfield BA, Cross RW, Fenton KA, Agans KN, Basler CF, et al. 2015. The immunomodulating V and W proteins of Nipah virus determine disease course. Nat. Commun 6:7483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Satterfield BA, Cross RW, Fenton KA, Borisevich V, Agans KN, et al. 2016. Nipah virus C and W proteins contribute to respiratory disease in ferrets. J. Virol 90:6326–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chang A, Dutch RE. 2012. Paramyxovirus fusion and entry: multiple paths to a common end. Viruses 4:613–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Aguilar HC, Iorio RM. 2012. Henipavirus membrane fusion and viral entry. Curr. Top. Microbiol. Immunol 359:79–94 [DOI] [PubMed] [Google Scholar]
- 92.Navaratnarajah CK, Generous AR, Yousaf I, Cattaneo R. 2020. Receptor-mediated cell entry of paramyxoviruses: mechanisms, and consequences for tropism and pathogenesis. J. Biol. Chem 295:2771–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bishop KA, Stantchev TS, Hickey AC, Khetawat D, Bossart KN, et al. 2007. Identification of Hendra virus G glycoprotein residues that are critical for receptor binding. J. Virol 81:5893–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bonaparte MI, Dimitrov AS, Bossart KN, Crameri G, Mungall BA, et al. 2005. Ephrin-B2 ligand is a functional receptor for Hendra virus and Nipah virus. PNAS 102:10652–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Negrete OA, Levroney EL, Aguilar HC, Bertolotti-Ciarlet A, Nazarian R, et al. 2005. EphrinB2 is the entry receptor for Nipah virus, an emergent deadly paramyxovirus. Nature 436:401–5 [DOI] [PubMed] [Google Scholar]
- 96.Negrete OA, Wolf MC, Aguilar HC, Enterlein S, Wang W, et al. 2006. Two key residues in ephrinB3 are critical for its use as an alternative receptor for Nipah virus. PLOS Pathog. 2:e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Drescher U 2002. Eph family functions from an evolutionary perspective. Curr. Opin. Genet. Dev 12:397–402 [DOI] [PubMed] [Google Scholar]
- 98.Bossart KN, Tachedjian M, McEachern JA, Crameri G, Zhu Z, et al. 2008. Functional studies of host-specific ephrin-B ligands as Henipavirus receptors. Virology 372:357–71 [DOI] [PubMed] [Google Scholar]
- 99.Gale NW, Baluk P, Pan L, Kwan M, Holash J, et al. 2001. Ephrin-B2 selectively marks arterial vessels and neovascularization sites in the adult, with expression in both endothelial and smooth-muscle cells. Dev. Biol 230:151–60 [DOI] [PubMed] [Google Scholar]
- 100.Poliakov A, Cotrina M, Wilkinson DG. 2004. Diverse roles of eph receptors and ephrins in the regulation of cell migration and tissue assembly. Dev. Cell 7:465–80 [DOI] [PubMed] [Google Scholar]
- 101.Pasquale EB. 2008. Eph-ephrin bidirectional signaling in physiology and disease. Cell 133:38–52 [DOI] [PubMed] [Google Scholar]
- 102.Wong KT, Tan CT. 2012. Clinical and pathological manifestations of human henipavirus infection. Curr. Top. Microbiol. Immunol 359:95–104 [DOI] [PubMed] [Google Scholar]
- 103.Xu K, Chan YP, Rajashankar KR, Khetawat D, Yan L, et al. 2012. New insights into the Hendra virus attachment and entry process from structures of the virus G glycoprotein and its complex with ephrin-B2. PLOS ONE 7:e48742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xu K, Rajashankar KR, Chan YP, Himanen JP, Broder CC, Nikolov DB. 2008. Host cell recognition by the henipaviruses: crystal structures of the Nipah G attachment glycoprotein and its complex with ephrin-B3. PNAS 105:9953–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bowden TA, Aricescu AR, Gilbert RJ, Grimes JM, Jones EY, Stuart DI. 2008. Structural basis of Nipah and Hendra virus attachment to their cell-surface receptor ephrin-B2. Nat. Struct. Mol. Biol 15:567–72 [DOI] [PubMed] [Google Scholar]
- 106.Bowden TA, Crispin M, Harvey DJ, Jones EY, Stuart DI. 2010. Dimeric architecture of the Hendra virus attachment glycoprotein: evidence for a conserved mode of assembly. J. Virol 84:6208–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wong JJ, Paterson RG, Lamb RA, Jardetzky TS. 2016. Structure and stabilization of the Hendra virus F glycoprotein in its prefusion form. PNAS 113:1056–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xu K, Chan YP, Bradel-Tretheway B, Akyol-Ataman Z, Zhu Y, et al. 2015. Crystal structure of the pre-fusion Nipah virus fusion glycoprotein reveals a novel hexamer-of-trimers assembly. PLOS Pathog. 11:e1005322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xu K, Rockx B, Xie Y, Debuysscher BL, Fusco DL, et al. 2013. Crystal structure of the Hendra virus attachment G glycoprotein bound to a potent cross-reactive neutralizing human monoclonal antibody. PLOS Pathog. 9:e1003684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dang HV, Chan YP, Park YJ, Snijder J, Da Silva SC, et al. 2019. An antibody against the F glycoprotein inhibits Nipah and Hendra virus infections. Nat. Struct. Mol. Biol 26:980–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Goh KJ, Tan CT, Chew NK, Tan PS, Kamarulzaman A, et al. 2000. Clinical features of Nipah virus encephalitis among pig farmers in Malaysia. N. Engl. J. Med 342:1229–35 [DOI] [PubMed] [Google Scholar]
- 112.Wong KT, Shieh WJ, Kumar S, Norain K, Abdullah W, et al. 2002. Nipah virus infection: pathology and pathogenesis of an emerging paramyxoviral zoonosis. Am. J. Pathol 161:2153–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hossain MJ, Gurley ES, Montgomery JM, Bell M, Carroll DS, et al. 2008. Clinical presentation of Nipah virus infection in Bangladesh. Clin. Infect. Dis 46:977–84 [DOI] [PubMed] [Google Scholar]
- 114.Wong KT, Robertson T, Ong BB, Chong JW, Yaiw KC, et al. 2009. Human Hendra virus infection causes acute and relapsing encephalitis. Neuropathol. Appl. Neurobiol 35:296–305 [DOI] [PubMed] [Google Scholar]
- 115.Tan CT, Goh KJ, Wong KT, Sarji SA, Chua KB, et al. 2002. Relapsed and late-onset Nipah encephalitis. Ann. Neurol 51:703–8 [DOI] [PubMed] [Google Scholar]
- 116.Wong SC, Ooi MH, Wong MN, Tio PH, Solomon T, Cardosa MJ. 2001. Late presentation of Nipah virus encephalitis and kinetics of the humoral immune response. J. Neurol. Neurosurg. Psychiatry 71:552–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chong HT, Tan CT. 2003. Relapsed and late-onset Nipah encephalitis, a report of three cases. Neurol. J. Southeast Asia 8:109–12 [Google Scholar]
- 118.Abdullah S, Chang LY, Rahmat K, Goh KT, Tan CT. 2012. Late-onset Nipah virus encephalitis 11 years after the initial outbreak: a case report. Neurol. Asia 17:71–74 [Google Scholar]
- 119.Aebersold P 2012. FDA experience with medical countermeasures under the animal rule. Adv. Prev. Med 2012:507571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Crawford LM. 2002. New drug and biological drug products; evidence needed to demonstrate effectiveness of new drugs when human efficacy studies are not ethical or feasible. Fed. Reg 67:37988–98 [PubMed] [Google Scholar]
- 121.de Wit E, Munster VJ. 2015. Animal models of disease shed light on Nipah virus pathogenesis and transmission. J. Pathol 235:196–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Dhondt KP, Horvat B. 2013. Henipavirus infections: lessons from animal models. Pathogens 2:264–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rockx B 2014. Recent developments in experimental animal models of Henipavirus infection. Pathog. Dis 71:199–206 [DOI] [PubMed] [Google Scholar]
- 124.Rubin SA, Sauder CJ, Carbone KM. 2013. Mumps virus. In Fields Virology, ed. Knipe DM, Howley PM, pp. 1024–41. Philadelphia: Lippincott Williams & Wilkins [Google Scholar]
- 125.Griffin DE. 2013. Measles virus. In Fields Virology, ed. Knipe DM, Howley PM, pp. 1042–69. Philadelphia: Lippincott Williams & Wilkins [Google Scholar]
- 126.Karron RAC, Collins PL. 2013. Parainfluenza viruses. In Fields Virology, ed. Knipe DM, Howley PM, pp. 996–1023. Philadelphia: Lippincott Williams & Wilkins [Google Scholar]
- 127.Guillaume V, Contamin H, Loth P, Georges-Courbot MC, Lefeuvre A, et al. 2004. Nipah virus: vaccination and passive protection studies in a hamster model. J. Virol 78:834–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Guillaume V, Contamin H, Loth P, Grosjean I, Courbot MC, et al. 2006. Antibody prophylaxis and therapy against Nipah virus infection in hamsters. J. Virol 80:1972–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhu Z, Dimitrov AS, Bossart KN, Crameri G, Bishop KA, et al. 2006. Potent neutralization of Hendra and Nipah viruses by human monoclonal antibodies. J. Virol 80:891–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhu Z, Bossart KN, Bishop KA, Crameri G, Dimitrov AS, et al. 2008. Exceptionally potent cross-reactive neutralization of Nipah and Hendra viruses by a human monoclonal antibody. J. Infect. Dis 197:846–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bossart KN, Geisbert TW, Feldmann H, Zhu Z, Feldmann F, et al. 2011. A neutralizing human monoclonal antibody protects African green monkeys from Hendra virus challenge. Sci. Transl. Med 3:105ra03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Geisbert TW, Mire CE, Geisbert JB, Chan YP, Agans KN, et al. 2014. Therapeutic treatment of Nipah virus infection in nonhuman primates with a neutralizing human monoclonal antibody. Sci. Transl. Med 6:242ra82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mire CE, Satterfield BA, Geisbert JB, Agans KN, Borisevich V, et al. 2016. Pathogenic differences between Nipah virus Bangladesh and Malaysia strains in primates: implications for antibody therapy. Sci. Rep 6:30916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chan YP, Lu M, Dutta S, Yan L, Barr J, et al. 2012. Biochemical, conformational, and immunogenic analysis of soluble trimeric forms of henipavirus fusion glycoproteins. J. Virol 86:11457–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mire CE, Chan YP, Borisevich V, Cross RW, Yan L, et al. 2019. A cross-reactive humanized monoclonal antibody targeting fusion glycoprotein function protects ferrets against lethal Nipah virus and Hendra virus infection. J. Infect. Dis 2019:jiz515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Playford EG, Munro T, Mahler SM, Elliott S, Gerometta M, et al. 2020. Safety, tolerability, pharmacokinetics, and immunogenicity of a human monoclonal antibody targeting the G glycoprotein of henipaviruses in healthy adults: a first-in-human, randomised, controlled, phase 1 study. Lancet Infect. Dis 20:445–54 [DOI] [PubMed] [Google Scholar]
- 137.Prow NA, Jimenez Martinez R, Hayball JD, Howley PM, Suhrbier A. 2018. Poxvirus-based vector systems and the potential for multi-valent and multi-pathogen vaccines. Expert Rev. Vaccines 17:925–34 [DOI] [PubMed] [Google Scholar]
- 138.Bossart KN, Wang LF, Eaton BT, Broder CC. 2001. Functional expression and membrane fusion tropism of the envelope glycoproteins of Hendra virus. Virology 290:121–35 [DOI] [PubMed] [Google Scholar]
- 139.Bossart KN, Wang LF, Flora MN, Chua KB, Lam SK, et al. 2002. Membrane fusion tropism and heterotypic functional activities of the Nipah virus and Hendra virus envelope glycoproteins. J. Virol 76:11186–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Weingartl HM, Berhane Y, Caswell JL, Loosmore S, Audonnet JC, et al. 2006. Recombinant Nipah virus vaccines protect pigs against challenge. J. Virol 80:7929–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Guillaume-Vasselin V, Lemaitre L, Dhondt KP, Tedeschi L, Poulard A, et al. 2016. Protection from Hendra virus infection with Canarypox recombinant vaccine. NPJ Vaccines 1:16003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kalodimou G, Veit S, Jany S, Kalinke U, Broder CC, et al. 2019. A soluble version of Nipah virus glycoprotein G delivered by vaccinia virus MVA activates specific CD8 and CD4 T cells in mice. Viruses 12:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Arunkumar G, Devadiga S, McElroy AK, Prabhu S, Sheik S, et al. 2019. Adaptive immune responses in humans during Nipah virus acute and convalescent phases of infection. Clin. Infect. Dis 69:1752–56 [DOI] [PubMed] [Google Scholar]
- 144.Chattopadhyay A, Rose JK. 2011. Complementing defective viruses that express separate paramyxovirus glycoproteins provide a new vaccine vector approach. J. Virol 85:2004–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lo MK, Bird BH, Chattopadhyay A, Drew CP, Martin BE, et al. 2014. Single-dose replication-defective VSV-based Nipah virus vaccines provide protection from lethal challenge in Syrian hamsters. Antivir. Res 101:26–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mire CE, Versteeg KM, Cross RW, Agans KN, Fenton KA, et al. 2013. Single injection recombinant vesicular stomatitis virus vaccines protect ferrets against lethal Nipah virus disease. Virol. J 10:353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mire CE, Geisbert JB, Agans KN, Versteeg KM, Deer DJ, et al. 2019. Use of single-injection recombinant vesicular stomatitis virus vaccine to protect nonhuman primates against lethal Nipah virus disease. Emerg. Infect. Dis 25:1144–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.DeBuysscher BL, Scott D, Marzi A, Prescott J, Feldmann H. 2014. Single-dose live-attenuated Nipah virus vaccines confer complete protection by eliciting antibodies directed against surface glycoproteins. Vaccine 32:2637–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Prescott J, DeBuysscher BL, Feldmann F, Gardner DJ, Haddock E, et al. 2015. Single-dose live-attenuated vesicular stomatitis virus-based vaccine protects African green monkeys from Nipah virus disease. Vaccine 33:2823–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kurup D, Wirblich C, Feldmann H, Marzi A, Schnell MJ. 2015. Rhabdovirus-based vaccine platforms against henipaviruses. J. Virol 89:144–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ploquin A, Szecsi J, Mathieu C, Guillaume V, Barateau V, et al. 2013. Protection against henipavirus infection by use of recombinant adeno-associated virus–vector vaccines. J. Infect. Dis 207:469–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ewer K, Sebastian S, Spencer AJ, Gilbert S, Hill AVS, Lambe T. 2017. Chimpanzee adenoviral vectors as vaccines for outbreak pathogens. Hum. Vaccines Immunother 13:3020–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.van Doremalen N, Lambe T, Sebastian S, Bushmaker T, Fischer R, et al. 2019. A single-dose ChAdOx1-vectored vaccine provides complete protection against Nipah Bangladesh and Malaysia in Syrian golden hamsters. PLOS Negl. Trop. Dis 13:e0007462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Yoneda M, Georges-Courbot MC, Ikeda F, Ishii M, Nagata N, et al. 2013. Recombinant measles virus vaccine expressing the Nipah virus glycoprotein protects against lethal Nipah virus challenge. PLOS ONE 8:e58414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Keshwara R, Shiels T, Postnikova E, Kurup D, Wirblich C, et al. 2019. Rabies-based vaccine induces potent immune responses against Nipah virus. NPJ Vaccines 4:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Shuai L, Ge J, Wen Z, Wang J, Wang X, Bu Z. 2020. Immune responses in mice and pigs after oral vaccination with rabies virus vectored Nipah disease vaccines. Vet. Microbiol 241:108549. [DOI] [PubMed] [Google Scholar]
- 157.Ciancanelli MJ, Basler CF. 2006. Mutation of YMYL in the Nipah virus matrix protein abrogates budding and alters subcellular localization. J. Virol 80:12070–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Patch JR, Crameri G, Wang LF, Eaton BT, Broder CC. 2007. Quantitative analysis of Nipah virus proteins released as virus-like particles reveals central role for the matrix protein. Virol. J 4:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Walpita P, Barr J, Sherman M, Basler CF, Wang L. 2011. Vaccine potential of Nipah virus-like particles. PLOS ONE 6:e18437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Walpita P, Cong Y, Jahrling PB, Rojas O, Postnikova E, et al. 2017. A VLP-based vaccine provides complete protection against Nipah virus challenge following multiple-dose or single-dose vaccination schedules in a hamster model. NPJ Vaccines 2:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Bossart KN, Crameri G, Dimitrov AS, Mungall BA, Feng YR, et al. 2005. Receptor binding, fusion inhibition, and induction of cross-reactive neutralizing antibodies by a soluble G glycoprotein of Hendra virus. J. Virol 79:6690–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.McEachern JA, Bingham J, Crameri G, Green DJ, Hancock TJ, et al. 2008. A recombinant subunit vaccine formulation protects against lethal Nipah virus challenge in cats. Vaccine 26:3842–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Pallister J, Middleton D, Wang LF, Klein R, Haining J, et al. 2011. A recombinant Hendra virus G glycoprotein-based subunit vaccine protects ferrets from lethal Hendra virus challenge. Vaccine 29:5623–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Pallister JA, Klein R, Arkinstall R, Haining J, Long F, et al. 2013. Vaccination of ferrets with a recombinant G glycoprotein subunit vaccine provides protection against Nipah virus disease for over 12 months. Virol. J 10:237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Bossart KN, Rockx B, Feldmann F, Brining D, Scott D, et al. 2012. A Hendra virus G glycoprotein subunit vaccine protects African green monkeys from Nipah virus challenge. Sci. Transl. Med 4:146ra07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Mire CE, Geisbert JB, Agans KN, Feng YR, Fenton KA, et al. 2014. A recombinant Hendra virus G glycoprotein subunit vaccine protects nonhuman primates against Hendra virus challenge. J. Virol 88:4624–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Middleton D, Pallister J, Klein R, Feng YR, Haining J, et al. 2014. Hendra virus vaccine, a One Health approach to protecting horse, human, and environmental health. Emerg. Infect. Dis 20:372–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Pickering BS, Hardham JM, Smith G, Weingartl ET, Dominowski PJ, et al. 2016. Protection against henipaviruses in swine requires both, cell-mediated and humoral immune response. Vaccine 34:4777–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lo MK, Spengler JR, Welch SR, Harmon JR, Coleman-McCray JD, et al. 2019. Evaluation of a single-dose nucleoside-modified messenger RNA vaccine encoding Hendra virus-soluble glycoprotein against lethal Nipah virus challenge in Syrian hamsters. J. Infect. Dis 2019:jiz553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Gouglas D, Christodoulou M, Plotkin SA, Hatchett R. 2019. CEPI: driving progress towards epidemic preparedness and response. Epidemiol. Rev 41:28–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Clayton BA. 2017. Nipah virus: transmission of a zoonotic paramyxovirus. Curr. Opin. Virol 22:97–104 [DOI] [PubMed] [Google Scholar]
- 172.Colgrave ML, Snelling HJ, Shiell BJ, Feng YR, Chan YP, et al. 2011. Site occupancy and glycan compositional analysis of two soluble recombinant forms of the attachment glycoprotein of Hendra virus. Glycobiology 22:572–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc 4:363–71 [DOI] [PubMed] [Google Scholar]