

ABSTRACT
Cyclin-dependent kinase 9 (CDK9) phosphorylates RNA polymerase II to promote productive transcription elongation. Here we show that short-term CDK9 inhibition affects the splicing of thousands of mRNAs. CDK9 inhibition impairs global splicing and there is no evidence for a coordinated response between the alternative splicing and the overall transcriptome. Alternative splicing is a feature of aggressive prostate cancer (CRPC) and enables the generation of the anti-androgen resistant version of the ligand-independent androgen receptor, AR-v7. We show that CDK9 inhibition results in the loss of AR and AR-v7 expression due to the defects in splicing, which sensitizes CRPC cells to androgen deprivation. Finally, we demonstrate that CDK9 expression increases as PC cells develop CRPC-phenotype both in vitro and also in patient samples. To conclude, here we show that CDK9 inhibition compromises splicing in PC cells, which can be capitalized on by targeting the PC-specific addiction androgen receptor.
KEYWORDS: Cyclin-dependent kinase 9, splicing, bioinformatics, prostate cancer, o-glcnac transferase
Introduction
Compounds targeting cyclin-dependent kinase 9 (CDK9) have shown success in clinical trials, and it is important to understand why cancer cells are so addicted to high CDK9 activity [1–3]. There are several Phase I–III clinical trials assessing CDK9 inhibitors as cancer therapy [4–6]. CDK9 phosphorylates carboxy-terminal domain (CTD) of RNA polymerase II (RNA Pol II) to promote productive transcription elongation in all cells, and the kinase is particularly important for the expression of genes driven by super-enhancers [7]. In general, mRNAs encoding for pro-proliferative and anti-apoptotic proteins have short half-lives, and high levels of transcription is required to maintain their robust expression [8]. A decrease in the global transcription has been postulated as the major mechanism of action for the anti-tumour effects of CDK9 inhibitors. However, all cells depend on active transcription, which proposes that cancer cells have additional features that explain their dependency on high CDK9 activity.
Utilization of the combinatorial lethal screens is an excellent strategy to discover synergistic dependencies. It is known that co-targeting of CDK9 and BET bromodomain proteins induces strong combinatorial effects on cancer cells due to the global suppression of transcription [9]. We recently described a cancer cell-selective synergistic lethal interaction between combined inhibition of O-GlcNAc transferase (OGT) and CDK9 [10]. Compromised CDK9 activity renders cancer cells dependent on OGT, and by identifying the processes OGT is important for, we may be able to design more rational combinatorial treatment strategies.
OGT glycosylates thousands of nucleocytoplasmic proteins to regulate their functions [11–14]. We have shown that OGT is overexpressed in aggressive prostate cancer and that the enzyme coordinates with c-MYC to promote proliferation of cancer cells [15,16]. OGT directly modifies many transcription factors and also RNA Pol II [17]. Overall, OGT is positioned to regulate transcriptional adaptations to CDK9 inhibition in prostate cancer cells.
In this study, we show that CDK9 inhibition induces transcriptional activation of the spliceosome mRNAs in an OGT-dependent manner. As a single agent treatment, CDK9 inhibitors compromise spliceosome activity with particularly strong effects on intron retention and exon skipping. In short-term, the effects elicited by CDK9 inhibitors are not enhanced by targeting OGT, and overall the CDK9 inhibitor-induced alternative splicing is likely a defect as there is no coordination between the treatment-induced alternative splicing and the transcriptional program. Furthermore, we show that inhibition of CDK9 compromises expression of androgen-receptor (AR) due to defective splicing, and development of castration-resistant prostate cancer (CRPC) is associated with increased CDK9 expression. Our data proposes that CRPC patients might respond well to CDK9 targeting therapies.
Results
Targeting CDK9 induces OGT-dependent increase in the spliceosome mRNAs
It is well established that targeting CDK9 leads to a decrease in transcription, but it is not known if cells also mount an adaptive transcriptional program when CDK9 activity is compromised. Combined inhibition of OGT (OSMI-2) and CDK9 (AT7519) induces strong anti-proliferative effects on prostate cancer cells [10], so we hypothesized that OGT is important for the transcriptional response to CDK9 inhibition. To identify the mRNAs and pathways that are induced in response to CDK9 inhibition (AT7519) in an OGT-dependent manner, we used both OGT small molecule inhibitor (OSMI-2) and OGT knockdown (siRNA), and performed RNA-seq. We selected all mRNAs whose expression was increased significantly in comparison to the untreated sample (adjusted p-value < 0.01). There was 332 mRNAs whose abundance was increased in response to CDK9 inhibitor treatment but not increased when CDK9 inhibitor was combined either with OSMI-2 or OGT knockdown (Fig. 1A).
Figure 1.
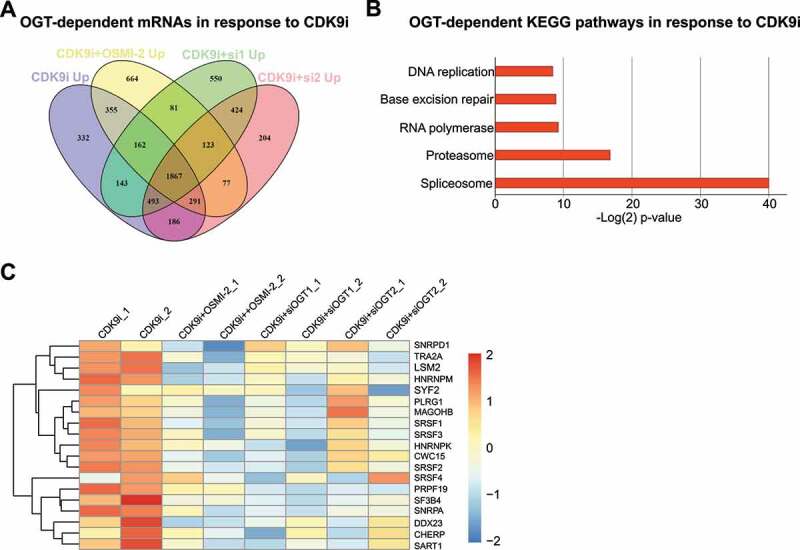
Inhibition of CDK9 activity increases the levels of spliceosome mRNAs in an OGT-dependent manner. A) Venn diagram showing CDK9/OGT inhibition effects on mRNA upregulation. Cells were treated for 4 hours with 0.5 µM AT7519, 40 µM OSMI-2 or OGT knockdown (si1 and si2 for 48 hours) before RNA-seq. Transcripts whose levels were statistically significantly increased are shown (p < 0.01). B) KEGG pathway enrichment analysis of mRNAs, which were selectively increased in response to CDK9 inhibition (332 mRNAs). C) OGT inhibition using OSMI-2 or OGT knockdown (siOGT) block AT7519 induced increase in the spliceosomal mRNAs discovered in B.
To determine if the CDK9 inhibitor-induced, OGT-dependent mRNAs are enriched for any particular process, we performed a KEGG pathway enrichment analysis. Strikingly, we identified a single biological process ‘spliceosome’ that is highly significantly enriched after the acute CDK9 inhibitor treatment in an OGT-dependent manner (Figs. 1B and 1 C). In searching for the potential mechanism how OGT could regulate these genes, we noted that over 80% of the promoters of the genes constituting the ‘spliceosome’ gene signature harbour an O-GlcNAc peak that is rapidly erased in response to OGT inhibition (OSMI-2), as determined by the biological triplicate ChIP-seq experiments (Suppl. Fig. 1). These data propose that when transcription is compromised due to decrease in CDK9 activity, spliceosome becomes more important. Reciprocally, we hypothesize that if the spliceosome-activity is compromised, CDK9 becomes more important, which we moved on to assess next.
We took the CDK9 inhibitor-induced, OGT-dependent ‘spliceosome’ gene signature of 19 mRNAs, and asked if these genes harbour any alterations in prostate cancer patient samples. Half of the assessed prostate cancer patients in the TCGA dataset [18] harbour alterations (mutation, amplification or altered mRNA expression) in at least one of the genes. As hypothesized above, patients who have altered spliceosome-signature also show significantly higher expression of CDK9 (p = 1e-11, Suppl. Fig. 2A). Altered activity of the spliceosome machinery through amplifications, deletions and mutations may lead to increased stress to the transcription machinery. This implies that the compromised spliceosome activity selects for cancer cells with higher CDK9 expression. In addition, we noted that the patients who harbour alterations in the ‘spliceosome’ gene signature have poor prognosis, as measured by Gleason Score, biochemical recurrence and other metrics of the aggressive prostate cancer (Suppl. Figs. 2B and 2 C). These data propose that spliceosome aberrations, which are frequent in aggressive prostate cancer [19] or increased CDK9 expression, may be used as a biomarker to identify patients benefiting from CDK9 targeting therapies.
Figure 2.
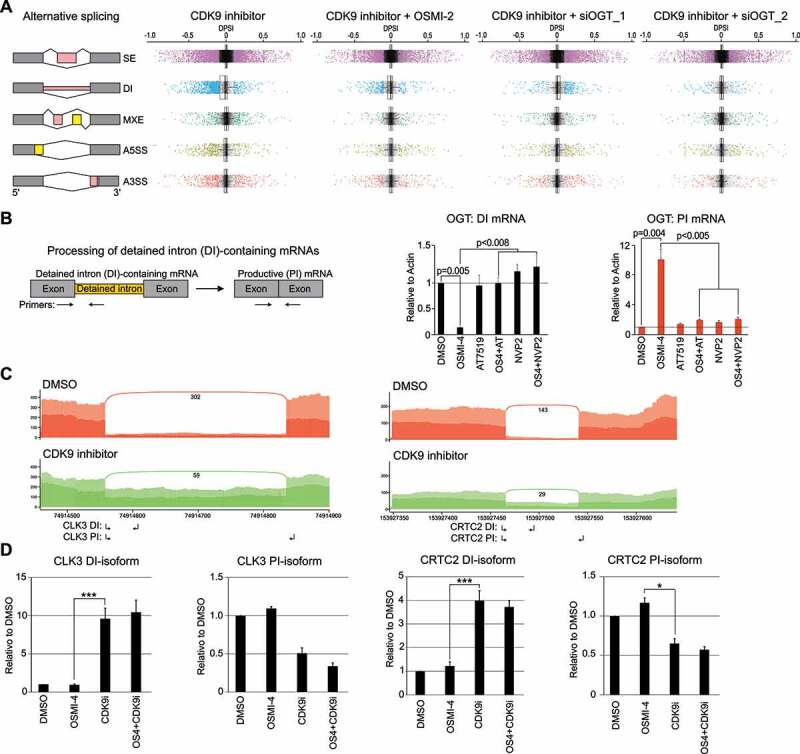
CDK9 inhibition results in gross alternative splicing and increases intron retention. A) Left: Main types of alternative splicing events: SE: exon skipping, DI: Detained intron, MXE: mutually exclusive exons, A5SS: alternative 5′ splice sites and A3SS: alternative 3′ splice sites. Right: Changes in alternative splicing events after 4 hour treatment with CDK9 inhibitor alone (0.5 µM AT7519) or in combination with OGT targeting approach (40 µM OSMI-2 or OGT knockdown (siOGT 48 hours prior) represented as boxplots. Colour coded dots represent splicing events that reach significance. B) Left: RT-qPCR assay for the detection of OGT mRNA that contains detained intron (left) and productive isoform (right); small arrows indicate the primers used. Cells were treated as indicated for 4 hours and analysed using RT-qPCR (average of 3 biological replicates with SEM; Student’s t-test was used to assess the significance). C) The histograms show poly(A) reads for both DMSO and AT7519 treatments. The Y-axis is the number of reads normalized to read density, and the number of junction spanning reads for detained intron are also shown. D) Validation of intron retention using RT-qPCR. Cells were treated as indicated for 4 hours and analysed using RT-qPCR (primers used are indicated in C with small arrows). DI: Detained intron; PI: productive isoform. Data shown are average of 2–3 biological replicates with SEM and Student’s t-test was used to assess the significance, *, P < 0.05; ***, P < 0.001)
Overall, our data shows that CDK9 inhibition induces upregulation of the spliceosome mRNAs in an OGT-dependent manner. However, we did not yet know if the spliceosome activity is altered when CDK9 activity is compromised.
CDK9 inhibition leads to global dysregulation of the spliceosome activity
To determine the global alternative splicing landscape in response to CDK9 and OGT inhibition, we employed rMATS algorithm [20]. Five main alternative splicing patterns, including alternative 3′ and 5′ splice sites (A3’SS and A5’SS, respectively), mutually exclusive exons (MXE), detained introns (DI) and skipped exons (SE) were evaluated (Fig. 2A). Splicing events with inclusion-level difference ≥ 10%, and FDR < 0.1 were determined as differential alternative splicing events.
Acute treatment (4 hours) with CDK9 inhibitor resulted in 5000 alternative splicing events (Fig. 2A, Suppl. Fig. 3). We did not detect clear coordination between any of the alternative splicing events and total mRNA levels (Suppl. Fig. 4). CDK9 inhibition has a pronounced effect on global DI levels, which is partially reversed upon OGT inhibition by OSMI-2 or OGT knockdown (Fig. 2A). OGT inhibitor OSMI-2 was recently reported to affect intron detention in HEK cells [21]. We used the same compound and also OGT knockdown, but only observed modest effects on alternative splicing (Suppl. Fig. 3). The discrepancy between the previous data and our study may be explained due to different treatment time (longer in the previous study) and different model systems used. Most notably, prostate cancer cells express high levels of OGT and enzymes producing OGT’s co-substrate UDP-GlcNAc [22,23]. However, there was one highly significant mRNA that was differentially spliced also in our study when OGT was inhibited, and that was OGT itself. We used RT-qPCR to confirm this effect: OGT intron 4 is rapidly spliced away to promote generation of the productive isoform when OGT activity is depleted and this response was completely blocked by CDK9 inhibition (Fig. 2B). CDK9 activity is therefore essential for the intron removal in the OGT mRNA.
Figure 3.
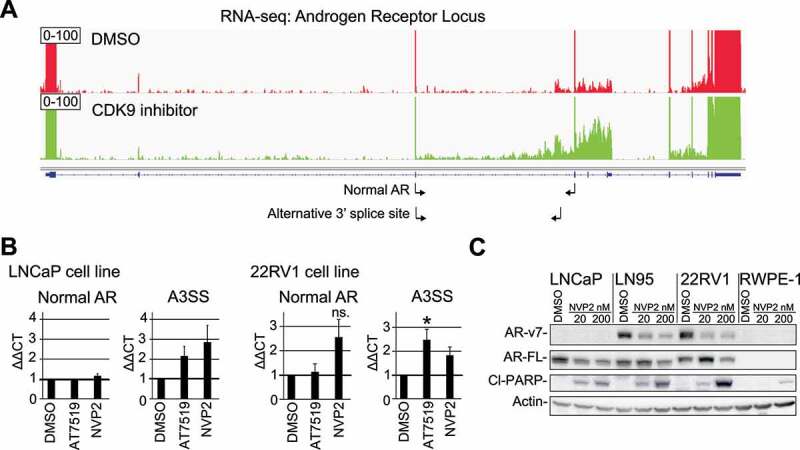
CDK9 inhibition leads to loss of AR-v7 protein expression in CRPC cells. A) Treatment with CDK9 inhibitor AT7519 leads to increased intronic reads in the AR mRNA. Integrative genomics viewer was used to visualize RNA-seq data for the AR locus. Note the scaling to highlight changes in the intronic reads. B) CDK9 inhibition leads to alternative splicing of the AR mRNA. Cells were treated as indicated for 4 hours and analysed using RT-qPCR. The primers used are indicated in 3A below the AR gene using small arrows. Data shown are average of 4 biological replicates with SEM; Student’s t-test was used to assess the statistical significance; *, P < 0.05. C) Cells were treated as indicated for 24 hours and analysed using western blot
Figure 4.
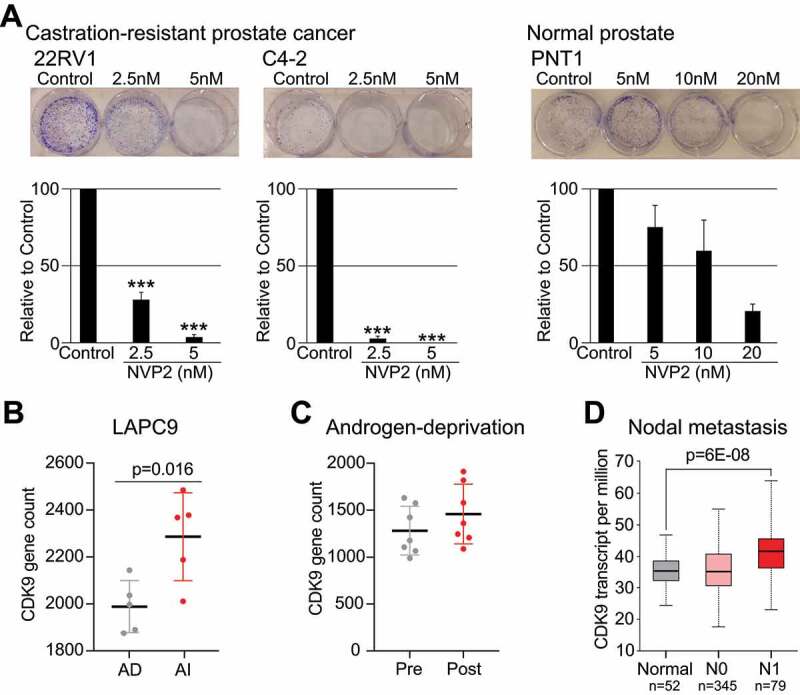
High CDK9 activity is required for proliferation of castration-resistant prostate cancer (CRPC) cells. A) Colony formation assay of CRPC (22RV1 and C4-2) and normal prostate (PNT1) cells treated with CDK9 inhibitor. Data shown are average of four biological replicates with SEM; Student’s t-test was used to assess the statistical significance. B) CDK9 expression in LAPC9 xenografts that are androgen-dependent (AD) and androgen-independent (generated using data from GSE88752). C) CDK9 expression in before (pre) and after (post) androgen ablation therapy for 5–6 months (generated using data from GSE48403). E) CDK9 expression in normal, non-metastatic (N0) and metastasis to 1–3 axillary lymph nodes. The figure was generated using UALCAN [34]
The acute CDK9 inhibition particularly affected exon skipping and intron retention, and the latter should lead to loss of the productive isoforms (Fig. 2A). For validation, we selected two genes, CLK3 and CRTC2, which showed prominent increased intron retention in response to CDK9 inhibition (Fig. 2C). We confirmed the increased intron retention using RT-qPCR and observed 4–10 fold increase (Fig. 2D). As expected, this led to a prominent (2 fold) depletion of the productive isoform. OGT inhibition did not reverse the CDK9 inhibitor effects (Fig. 2D and Suppl. Figs. 5 and 6). We noted that OGT knockdown increased both detained intron and productive isoforms of these mRNAs, potentially representing cellular compensation mechanism to decreased OGT expression.
To summarize our data so far, targeting CDK9 results in a gross dysregulation of the spliceosome activity in prostate cancer cells. In these cells, CDK9 is an important regulator of global splicing and overrides the effects elicited by OGT inhibition. Our data also shows that CDK9 inhibitor-induced alternative splicing is not coordinated with the changes in the transcriptional program acutely, which implies that the effects on splicing are not adaptive mechanism in the short term. Based on these data, depleting OGT activity does not override the effects of CDK9 inhibitor on alternative splicing. We propose that identification of prostate cancer cell-specific features may enable design of rational drug combinations with CDK9 inhibitors.
CDK9 inhibition compromises splicing of the androgen receptor mRNA
We hypothesized that targeting CDK9 might affect the expression of androgen receptor (AR), the major drug target in prostate cancer. Based on our alternative splicing-data, CDK9 inhibition results in utilization of an alternative 3ʹ splice site in the AR mRNA (p = 0.0003 based on rMATS analysis). More careful inspection of the entire AR gene revealed robust increase in the intronic reads, which may also indicate utilization of an alternative poly-adenylation site (Fig. 3A). We also noted accumulation of reads in the region of the so called cryptic exons after the third major exon. AR cryptic exons are known to generate various AR splice variants that are expressed in patient samples and have been associated with aggressive prostate cancer [24,25].
We designed primer pairs to rigorously assess if CDK9 inhibition alters alternative 3ʹ splice site utilization, and in addition we obtained primers reported previously by the land-mark paper on the clinically relevant AR variants by Hu et al. [2009, 24]. Interestingly, acute depletion of CDK9 activity using two structurally unrelated inhibitors, AT7519 [26] and NVP2 [27] prominently increased the generation of the AR splice variants, while the short term treatments did not have effects on the levels of the normal AR mRNA in LNCaP cells (Fig. 3B, left and Suppl. Fig. 7). We note that the relative abundance of any of these splice variants is low in comparison to the normal AR mRNA, which is expected because LNCaP cells are not known to express AR variants in the basal condition. Therefore, we performed the same experiment in a model of CRPC (22RV1), a cell line that is known to express AR variants due to inclusion of the cryptic exons [24]. Importantly, both CDK9 inhibitors significantly increased the AR splice variants that are generated through the alternative 3ʹ splice site or inclusion of the cryptic exons (Fig. 3B, right and Suppl. Fig. 7). Based on the RNA-seq and RT-qPCR data, we conclude that the short-term CDK9 inhibition compromises intron-exon definition in the AR gene.
We hypothesized that the difficulties in the splice site recognition might in long term result in the loss of AR protein expression because many of these splice variants result in a premature stop codon [24]. It is also possible that by targeting CDK9 we promote generation of the ligand-independent, clinically relevant AR-v7 variant (P3 in the Suppl. Fig. 7). We, therefore, evaluated CDK9 inhibitor effects on both AR full length and AR-v7 variant using western blotting. Even a low dose of the CDK9 inhibitor NVP2 led to a loss of AR full-length protein in LNCaP cells (Fig. 3C). In the CRPC cells (22RV1 and LN95), we noted that the low dose CDK9 inhibitor results in a prominent decrease of the AR-v7 protein but increases AR full length. We confirmed the effects on AR-v7 using AT7519 (Suppl. Fig. 8). Importantly, we observed the effects on AR and AR-v7 using CDK9 inhibitor doses that do not induce cell death activation as measured by PARP cleavage in normal prostate cells (RWPE-1, Fig. 3C). AR-v7 is an alternatively spliced variant that generates androgen-independent form of the key drug target in prostate cancer [28]. Accordingly, CDK9 inhibition sensitized prostate cancer cells to androgen deprivation (Suppl. Fig. 9). To conclude, CDK9 inhibition induces global changes in splicing with striking effects on the CRPC-specific AR-v7.
CRPC cells are addicted on high CDK9 activity
Due to the gross effects on splicing and AR, a major driver of CRPC, we hypothesized that CRPC cells are addicted on high CDK9 activity when compared to their normal counterparts. Depletion of CDK9 activity completely blocked the ability of CRPC cells to form colonies and over 8-times higher dose of the inhibitor was required to see similar effects on normal prostate cells (Fig. 4A). We detected the effects on proliferation using very low doses of the CDK9 inhibitor, and therefore propose that these effects are due to the transcriptional stress as observed above using higher CDK9 inhibitor doses and shorter treatment times. These data show that high CDK9 activity is important for the survival of the CRPC cells in vitro, and we moved on to evaluate CDK9 expression in patient samples.
We asked if CDK9 expression is altered in prostate cancer cells after development of anti-androgen resistance. First, we noted that CDK9 expression is not altered in primary prostate cancer when compared to adjacent tissue (Suppl. Fig. 10). Second, we evaluated CDK9 expression in LAPC9 mouse model of androgen-independent prostate cancer and also in prostate cancer patient samples before and after androgen ablation-therapy. CDK9 expression was significantly increased as LAPC9 model developed androgen-independence and the expression was also increased in prostate cancer patient samples after anti-androgen therapy (Figs. 4B and 4 C). Third, CDK9 expression is also increased in prostate cancers that metastasize to lymph nodes (Fig. 4D). Overall, CDK9 expression is increased after androgen-ablation and is associated with aggressive prostate cancer.
Discussion
Here we show for the first time that targeting CDK9 results in a gross dysregulation of the spliceosome activity. We aimed to explain why combined inhibition of OGT and CDK9 is toxic to prostate cancer cells by focusing on the transcriptional networks that are activated in an OGT-dependent manner when CDK9 activity is compromised. This approach revealed that OGT is required for the transcriptional activation of the mRNAs that constitute the spliceosome (Fig. 1B). However, simultaneous targeting of both OGT and CDK9 did not further increase the number of alternative splicing events when compared to CDK9 inhibition alone, and it is possible that the combinatorial effects become more prominent after a longer treatment time. Our study is the first to detect gross changes in the spliceosome activity in response to CDK9 inhibition. CDK9 phosphorylates Ser-2 on the carboxy-terminal domain of RNA polymerase II, which is known to be important for the coordination of transcription and splicing [3,29]. However, this is the first study to report that targeting CDK9 actually results in the robust alternative splicing affecting thousands of mRNAs (Fig. 2A). Our data propose that spliceosome becomes a point of vulnerability when CDK9 activity is compromised.
CDK9 inhibitors selectively kill cancer cells, despite CDK9 being important for maintaining transcription in all cells, and hence it is important to re-visit genomics data to explain why cancer cells would be particularly addicted on high CDK9 activity. Prostate cancer patients whose tumours have genetic alterations in the genes encoding for the spliceosome components show significantly increased expression of CDK9 (Suppl. Fig. 2). Interestingly, myeloid cancers have a high frequency of mutations in the spliceosome genes [30] and the same tumours are also highly susceptible to CDK9 inhibition [1]. Spliceosome mRNAs also have high alteration frequency in aggressive prostate cancer [19]. Based on our data, CDK9 inhibition-induced changes in splicing are rather a defective than adaptive response (Suppl. Fig. 4), and further targeting of splicing would likely be toxic to normal cells as well. Therefore, combining CDK9 inhibitors with compounds aimed at a specific feature cancer cells are addicted to, including AR, should be prioritized. Overall, underlying genetic alterations may enable selecting the right patients to be treated with the CDK9 inhibitors.
In the future, it is important to simultaneously profile the active transcription using nucleotide labels, splicing and the overall transcriptome to form a comprehensive picture of how the major RNA Pol II kinases affect the transcriptional programs in both normal and cancer cells. Finally, utilization of proteomic approaches to identify the factors OGT acts on to remodel the CDK9inhibitor-induced proteome will enable direct targeting of these responses.
Materials and methods
Cell culture
LNCaP, C4-2, 22RV1, PC3 and RWPE-1 cells were obtained from ATCC, PNT1 cells were from the European Collection of Authenticated Cell Cultures, while LN95 were a kind gift from Professor Stephen R. Plymate (University of Washington). Cell lines were maintained as recommended by the provider, and LN95 cells were maintained in phenol red-free RPMI supplemented with charcoal-stripped serum.
Compounds and assays
OSMI-4, NVP2 and AT7519 were obtained from MedChem Express. ATP levels in cells were assessed using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega). OSMI-2 was a gift from Professor Suzanne Walker (Harvard Medical School). Knockdown experiments were performed using RNAiMax reagent (Sigma). OGT targeting siRNAs were from ThermoFisher Scientific: siOGT_1 s16094 and siOGT_2 s16095. For all statistical analyses presented in the manuscript, p-values are from two-sided analysis unless otherwise specified. Colony formation assays were performed in a 12-well plate, and cells were plated 1000 cells per well. After 1–2 days, cells were treated with compounds of interest for one week. Crystal violet staining was performed as described previously [31].
Protein and mRNA profiling
Samples for western blotting were prepared as previously described [15]. Antibodies used are as follows: PARP (9532) from Cell Signaling Technology, AR (ab108341) and actin (ab49900) from Abcam, and AR-v7 was from Revmab Biosciences (31-1109-00). RNA isolation was performed using the illustraMiniSpin-kit (GE Healthcare) according to the manufacturer’s instructions, and cDNA was synthesized using the qScript cDNA Synthesis Kit (Quantabio).
RNA-Seq data analysis
Libraries were prepared by the Norwegian High Throughput Sequencing Centre using a Strand-specific TruSeq RNA-seq library prep kit. For each condition, two biological replicates were used. Samples were multiplexed and paired-end sequenced in four lanes with 150 bp read length. Sequencing reads were aligned to Hg19 (GRCh37) using STAR [32]. Raw counts of reads were mapped to genes using HTSeq counts (http://htseq.readthedocs.io/en/master/count.html) and differential expression analysis performed on duplicate samples using DESEQ2 [33]. Genes with a p-value of 0.01 were considered significant. RNA-seq BAM files were normalized and converted to BigWig for visualization on Integrated Genome Viewer.
Analysis of alternative splicing
Reads were aligned to indexed human reference genome hg19 with GENCODE V25 annotation by STAR 2.7.3a [32] using a 2-pass model to improve the detection of splicing events. The rMATS v4.1.0 [20] was used to determine the differential alternative splicing events between different conditions. The bioconductor package Rcwl was used to perform reproducible analysis through the Common Workflow Language (CWL)[35]. Five types of alternative splicing events based on the GENCODE gene annotation were evaluated, including skipped exon (SE), alternative 5′ splice site (A5SS), alternative 3′ splice site (A3SS), mutually exclusive exons (MXE) and detained intron (DI). The events with an inclusion-level difference ≥ 10%, and FDR < 0.1 were determined as differential alternative splicing events.
Supplementary Material
Acknowledgments
HMI is grateful for the funding from the Academy of Finland (Decision nos. 331324 and 335902). In addition, the work was supported by the John Black Charitable Foundation and the Norwegian Research Council (230559). The authors wish to acknowledge CSC – IT Center for Science, Finland, for the computational resources of some of the work reported here.
Funding Statement
This work was supported by the Academy of Finland grants 331324 and 335902 granted to HMI. In addition, the work was supported by the John Black Charitable Foundation and the Norwegian Research Council (230559).
Disclosure statement
No competing interests to declare.
Data availability
RNA-seq data has been deposited to the Gene Expression Omnibus (GSE169090).
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Boffo S, Damato A, Alfano L, et al. CDK9 inhibitors in acute myeloid leukemia. J Exp Clin Cancer Res. 2018;37:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chou J, Quigley DA, Robinson TM, et al. Transcription-Associated Cyclin-Dependent Kinases as Targets and Biomarkers for Cancer Therapy. Cancer Discov. 2020;10:351–370. [DOI] [PubMed] [Google Scholar]
- [3].Parua PK, Fisher RP.. Dissecting the Pol II transcription cycle and derailing cancer with CDK inhibitors. Nat Chem Biol. 2020;16:716–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ghia P, Scarfo L, Perez S, et al. Efficacy and safety of dinaciclib vs ofatumumab in patients with relapsed/refractory chronic lymphocytic leukemia. Blood. 2017;129:1876–1878. [DOI] [PubMed] [Google Scholar]
- [5].Cidado J, Boiko S, Proia T, et al. AZD4573 Is a Highly Selective CDK9 Inhibitor That Suppresses MCL-1 and Induces Apoptosis in Hematologic Cancer Cells. Clin Cancer Res. 2020;26:922–934. [DOI] [PubMed] [Google Scholar]
- [6].Wu T, Qin Z, Tian Y, et al. Recent Developments in the Biology and Medicinal Chemistry of CDK9 Inhibitors: an Update. J Med Chem. 2020;63:13228–13257. [DOI] [PubMed] [Google Scholar]
- [7].Minzel W, Venkatachalam A, Fink A, et al. Small Molecules Co-targeting CKIalpha and the Transcriptional Kinases CDK7/9 Control AML in Preclinical Models. Cell. 2018;175:171–185 e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lam LT, Pickeral OK, Peng AC, et al. Genomic-scale measurement of mRNA turnover and the mechanisms of action of the anti-cancer drug flavopiridol. Genome Biol. 2001;2:RESEARCH0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lu H, Xue Y, Yu GK, et al. Compensatory induction of MYC expression by sustained CDK9 inhibition via a BRD4-dependent mechanism. Elife. 2015;4:e06535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Itkonen HM, Poulose N, Steele RE, et al. Inhibition of O-GlcNAc Transferase Renders Prostate Cancer Cells Dependent on CDK9. Mol Cancer Res. 2020;18:1512–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hart GW. Nutrient regulation of signaling and transcription. J Biol Chem. 2019;294:2211–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yang X, Qian K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol. 2017;18:452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hanover JA, Krause MW, Love DC. Bittersweet memories: linking metabolism to epigenetics through O-GlcNAcylation. Nat Rev Mol Cell Biol. 2012;13:312–321. [DOI] [PubMed] [Google Scholar]
- [14].Itkonen HM, Loda M, Mills IG. O-GlcNAc Transferase - An Auxiliary Factor or a Full-blown Oncogene? Mol Cancer Res. 2021;19:555–564. [DOI] [PubMed] [Google Scholar]
- [15].Itkonen HM, Minner S, Guldvik IJ, et al. O-GlcNAc transferase integrates metabolic pathways to regulate the stability of c-MYC in human prostate cancer cells. Cancer Res. 2013;73:5277–5287. [DOI] [PubMed] [Google Scholar]
- [16].Itkonen HM, Urbanucci A, Martin SE, et al. High OGT activity is essential for MYC-driven proliferation of prostate cancer cells. Theranostics. 2019;9:2183–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Brian A, Lewis DL. O-GlcNAc Transferase Activity is Essential for RNA Pol II Pausing in a Human Cell-Free Transcription System. bioRxiv. 2020. [Google Scholar]
- [18].N; Cancer Genome Atlas Research . The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015;163:1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhang D, Hu Q, Liu X, et al. Intron retention is a hallmark and spliceosome represents a therapeutic vulnerability in aggressive prostate cancer. Nat Commun. 2020;11:2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Shen S, Park JW, Lu ZX, et al. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc Natl Acad Sci U S A. 2014;111:E5593–5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tan ZW, Fei G, Paulo JA, et al. O-GlcNAc regulates gene expression by controlling detained intron splicing. Nucleic Acids Res. 2020;48:5656–5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Itkonen HM, Mills IG. N-linked glycosylation supports cross-talk between receptor tyrosine kinases and androgen receptor. PLoS One. 2013;8:e65016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Itkonen HM, Engedal N, Babaie E, et al. UAP1 is overexpressed in prostate cancer and is protective against inhibitors of N-linked glycosylation. Oncogene. 2015;34:3744–3750. [DOI] [PubMed] [Google Scholar]
- [24].Hu R, Dunn TA, Wei S, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Paschalis A, Sharp A, Welti JC, et al. Alternative splicing in prostate cancer. Nat Rev Clin Oncol. 2018;15:663–675. [DOI] [PubMed] [Google Scholar]
- [26].Squires MS, Feltell RE, Wallis NG, et al. Biological characterization of AT7519, a small-molecule inhibitor of cyclin-dependent kinases, in human tumor cell lines. Mol Cancer Ther. 2009;8:324–332. [DOI] [PubMed] [Google Scholar]
- [27].Olson CM, Jiang B, Erb MA, et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat Chem Biol. 2018;14:163–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sharp A, Coleman I, Yuan W, et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J Clin Invest. 2019;129:192–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Decker TM, Forne I, Straub T, et al. Analog-sensitive cell line identifies cellular substrates of CDK9. Oncotarget. 2019;10:6934–6943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. [DOI] [PubMed] [Google Scholar]
- [31].Barkovskaya A, Seip K, Prasmickaite L, et al. Inhibition of O-GlcNAc transferase activates tumor-suppressor gene expression in tamoxifen-resistant breast cancer cells. Sci Rep. 2020;10:16992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chandrashekar DS, Bashel B, Balasubramanya SAH, et al. UALCAN: a Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia. 2017;19:649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hu Q, Hutson A, Liu S, Morgan M, Liu Q. Bioconductor toolchain for reproducible bioinformatics pipelines using Rcwl and Rcwl Pipelines. Bioinformatics. 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data has been deposited to the Gene Expression Omnibus (GSE169090).