

ABSTRACT
Self-cleaving ribozymes are catalytically active RNAs that cleave themselves into a 5′-fragment with a 2′,3′-cyclic phosphate and a 3′-fragment with a 5′-hydroxyl. They are widely applied for the construction of synthetic RNA devices and RNA-based therapeutics. However, the targeted discovery of self-cleaving ribozymes remains a major challenge. We developed a transcriptome-wide method, called cyPhyRNA-seq, to screen for ribozyme cleavage fragments in total RNA extract. This approach employs the specific ligation-based capture of ribozyme 5′-fragments using a variant of the Arabidopsis thaliana tRNA ligase we engineered. To capture ribozyme 3′-fragments, they are enriched from total RNA by enzymatic treatments. We optimized and enhanced the individual steps of cyPhyRNA-seq in vitro and in spike-in experiments. Then, we applied cyPhyRNA-seq to total RNA isolated from the bacterium Desulfovibrio vulgaris and detected self-cleavage of the three predicted type II hammerhead ribozymes, whose activity had not been examined to date. cyPhyRNA-seq can be used for the global analysis of active self-cleaving ribozymes with the advantage to capture both ribozyme cleavage fragments from total RNA. Especially in organisms harbouring many self-cleaving RNAs, cyPhyRNA-seq facilitates the investigation of cleavage activity. Moreover, this method has the potential to be used to discover novel self-cleaving ribozymes in different organisms.
KEYWORDS: RtcB ligase, T4 RNA ligase truncated KQ, RNA pyrophosphohydrolase (RppH), 5ʹ-3ʹ exonuclease Xrn1, next-generation sequencing
Introduction
Non-coding RNAs that catalyse chemical reactions are called ribozymes. They are involved in the catalysis of fundamental biological processes, such as tRNA maturation [1], splicing [2] and translation [3]. Today, 15 natural ribozyme classes are known, each with a unique secondary and tertiary structure [4]. Ten of them are self-cleaving ribozymes [5–12]. These self-cleaving ribozymes carry out a site-specific phosphodiester scission or ligation reaction [4]. The cleavage reaction relies on an SN2-like mechanism wherein the nucleophile 2ʹ-oxygen attacks the adjacent phosphorous centre resulting in a 5′-fragment with a 2′,3′-cyclic phosphate (2′,3′-cP) and a 3′-fragment with a 5′-hydroxyl group (5′-OH). Self-cleaving ribozymes are very abundant and found in all domains of life, but only a few representatives have been linked to a biological function. They are, for example, involved in the rolling circle replication of virus-like RNAs [5], in the metabolite-mediated regulation of gene expression [9] and are parts of mobile genetic elements [13–16]. To date, self-cleaving ribozymes have been mostly discovered serendipitously as a result of studying biological systems. Only a few targeted discovery methods based on enrichment from in vitro transcribed genomic DNA libraries [12,17] or bioinformatic analysis have been described (reviewed in [18]). Most of these methods identified variants of previously known self-cleaving ribozyme structural classes. In 2015, three novel self-cleaving ribozyme classes, namely twister-sister, pistol and hatchet were discovered using comparative genomic analysis [11]. However, the activity of all bioinformatically discovered self-cleaving ribozymes has to be proven individually by biochemical analysis, and many of the computationally predicted candidates did not cleave in these experiments [19]. Here, we present a high-throughput approach utilizing RNA sequencing (RNA-seq) to directly screen for active self-cleaving ribozymes in total RNA.
To apply RNA-seq to extracted RNA molecules, they must be converted into a library [20–22]. Although the preferred enzymes employed to create an RNA-seq library are continuously improving, all library preparation protocols contain several common steps. First, the RNA of interest is ligated to adapter sequences, either at both ends or just at its 3ʹ-end. These adapter sequences are known DNA or RNA oligonucleotides and serve as primer binding sites in subsequent steps. After adapter ligation, a reverse transcription (RT) is performed to create a cDNA. If initially an adapter was only attached to the 3ʹ-end of the RNA, a second adapter sequence needs to be ligated to the cDNA prior to PCR amplification.
The most widely used RNA-seq method for studying short RNAs is based on the ligation of adapters to the 5′-phosphate (5′-P) and 3′-hydroxyl (3′-OH) end of RNAs using T4 RNA ligases [23]. However, 2′,3′-cP and 5′-OH ends, such as those generated by self-cleaving ribozymes, are not recognized by T4 RNA ligases. For this reason, such ligases cannot be used to directly analyse products of self-cleaving ribozymes. However, the Arabidopsis thaliana tRNA ligase (AtRNL) and the Escherichia coli RtcB ligase are able to join RNAs with 2′,3′-cP and 5′-OH ends [24,25]. To initiate ligation, AtRNL first hydrolyzes the 2′,3′-cP into a 2′-P and a 3′-OH and phosphorylates, then adenylates the 5′-OH [24]. The joining of this 5ʹ-adenylated RNA with the 3ʹ-OH leaves a 2′-P at the ligation site, which can be efficiently removed by a specific phosphotransferase, for example Tpt1 from Saccharomyces cerevisiae [26]. In contrast to AtRNL, the RtcB ligase hydrolyzes the 2′,3′-cP end to form a 3′-P and 2′-OH. The resulting 3′-monophosphate (3′-P) is guanylated and then joined to a 5′-OH RNA [27]. Both ligases, AtRNL and RtcB, have previously been cleverly introduced for adapter ligation in NGS [28,29], but have not been applied to self-cleaving ribozyme screenings.
Here, we describe a method, called cyPhyRNA-seq, to capture self-cleaving ribozyme fragments with 2ʹ,3ʹ-cyclic phosphate and 5ʹ-hydroxyl termini. We show the development of cyPhyRNA-seq under controlled conditions in vitro and the enhancement of individual library preparation steps in spike-in experiments, in which we capture in vitro transcribed ribozyme fragments from E. coli total RNA. Finally, we apply cyPhyRNA-seq to capture hammerhead ribozyme (HHR) fragments from Desulfovibrio vulgaris cells. This method can be applied to the global analysis and cleavage pattern investigation of self-cleaving ribozymes in vivo and serve as tool to screen for active self-cleaving ribozymes in different organisms as well as RNA samples derived from a variety of environments.
Results and discussion
Concepts to capture self-cleaving ribozyme fragments by adapter ligation
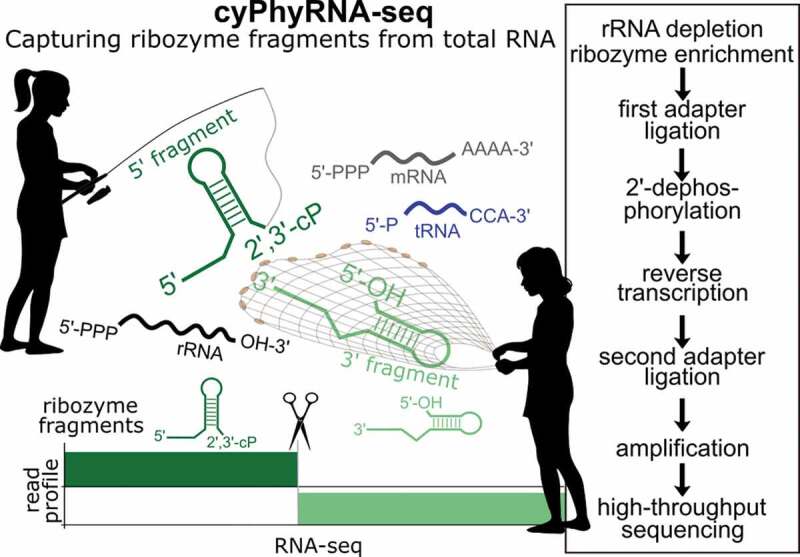
cyPhyRNA-seq includes libraries that are specific for RNAs with 2′,3′-cP (found in 5′-ribozyme fragments) and 5′-OH ends (as found in 3′-ribozyme fragments) and an optional transcriptome analysis to monitor the general expression profile under the investigated growth conditions (Figure 1(a-e)).
Figure 1.
cyPhyRNA-seq method to capture self-cleaving ribozyme fragments. Self-cleavage of a type II hammerhead ribozyme (HHR) results in a 5ʹ-fragment with a 2′,3′-cP and a 3ʹ-fragment with a 5′-OH. For the first ligation reaction, the same 3ʹ-DNA adapter was used in all libraries (a-c). This adapter with a 5ʹ-P was first pre-adenylated (5ʹ-rApp-DNA) with ATP and TS2126 RNL1 (pp indicates a 5ʹ-5ʹ-diphosphate bond). At the 3ʹ-end, the adapter carries an Amino-C7 protection group (indicated by an asterisk*) to avoid self-ligation. (a) Transcriptome analysis using T4 RNL2 TKQ for adapter ligation to all RNAs with 3ʹ-OH. In contrast to the other two methods of capturing ribozyme cleavage fragments, the transcriptome analysis is optional and can possibly provide information about uncleaved ribozymes. (b) Ligation of 5ʹ-rApp-DNA adapter to the 3ʹ-OH end of the enriched 3ʹ-ribozyme cleavage fragments with a 5ʹ-OH using T4 RNL2 TKQ. Since this ligation is not specific for ribozyme fragments, a 5ʹ-OH RNA enrichment is necessary (described in e). (c) Ligation of 5ʹ-rApp-DNA adapter to the 2ʹ,3ʹ-cP end of the 5′-ribozyme cleavage fragment by AtRNL AA followed by the dephosphorylation of the 2ʹ-P at the ligation site by phosphotransferase Tpt1. (d) Reverse transcription of ligation products using SuperScript IV (SSIV) and ligation of a second adapter, carrying an Amino-C6 protection group (indicated by an asterisk*), using T4 RNL1 and PCR amplification with Phusion DNA Polymerase and Illumina compatible primers. (E) Individual steps of cyPhyRNA-seq in total RNA. ‘rRNA depletion, ribozyme enrichment’ step occurs prior to the first adapter ligation. Total RNA for 5ʹ-ribozyme fragment analysis (part c) is rRNA-depleted by precipitation using PEG and salt, followed by a ‘tRNA blocking’ procedure. In case of the 3ʹ-ribozyme fragment analysis (part b), total RNA is enzymatically treated to deplete RNAs with 5ʹ-PPP and 5ʹ-P RNAs, followed by ‘tRNA blocking’. (f) Ligation of the 5ʹ-pre-adenylated DNA adapter (App adapter) to the 2ʹ,3ʹ-cP end of the 5′-hammerhead type I (70% efficiency) or 5ʹ-twister sister (80% efficiency) ribozyme fragment with AtRNL AA. Ligation efficiencies were quantified by dividing the intensity of the ligation product band by the sum of the intensity of the ligation product, adapter and ribozyme fragment bands. (g) Ligation of the 5ʹ-pre-adenylated DNA adapter (App adapter) to the 3′-OH end of the twister, hatchet and HDV ribozyme 3′-fragments by T4 RNL2 TKQ with different efficiencies (twister: 30%, hatchet 65%, HDV: 88%). Ligation efficiencies were calculated as described for part F
For the optional transcriptome library preparation (Figure 1(a)), an approach where a 5′-pre-adenylated DNA adapter is specifically ligated to the 3′-OH end of the RNA by T4 RNA ligase 2 truncated KQ (T4 RNL2 TKQ) can be used [30]. After reverse transcription, the second adapter is ligated to the cDNA by T4 RNL1 followed by PCR amplification [31].
To specifically ligate an adapter to the 5ʹ-OH of the 3ʹ-fragment, we initially investigated E. coli RtcB ligase. As ligation of DNA to RNA by E. coli RtcB is very inefficient [32], only RNA adapters are well-suited for ligation to 5ʹ-OH RNAs. For most ribozyme candidates, the ligation of an RNA adapter by RtcB is feasible (Supplementary Text, Supplementary Figure S1). However, in ribozyme classes, such as hatchet and HDV, the 3ʹ-cleavage fragments still comprise the majority of the ribozyme sequence and, more importantly, retain the active structure [33,34]. For these ribozyme classes, the RNA adapters are cut off by the ribozyme-catalysed nucleophilic attack either immediately after ligation or in subsequent reactions, such as reverse transcription (data not shown). Hence, we abandoned our exploration of RtcB ligase. Instead, we devised an alternative method in which we enriched our total RNA pool for 5′-OH RNAs (explained in the next section) and used T4 RNL2 TKQ for adapter ligation to the 3ʹ-OH (Figure 1(b)).
For cyPhyRNA-seq, we generated an improved variant of AtRNL that combines the two mutations K152A and D726A (AtRNL AA). Previous biochemical studies showed that the introduction of the mutation D726A to the kinase domain results in the loss of phosphorylation activity to 5ʹ-OH ends and that another mutation, K152A to the KxxG motif of the AtRNL, abolishes the enzyme’s ability for adenylation of 5′-P containing nucleic acids [35].
We found that this AtRNL AA variant, in contrast to the wild-type, can neither ligate DNA adapters with 5ʹ-OH nor 5ʹ-P, but efficiently joins a 5ʹ-pre-adenylated DNA adapter to RNAs with 2ʹ,3ʹ-cP (Supplementary Figure S2). Therefore, we used AtRNL AA in our cyPhyRNA-seq approach to increase sensitivity by reducing undesirable ligation reactions to RNAs other than adapter molecules (Figure 1(c)).
Due to the often-complex structures of self-cleaving ribozymes, we first investigated the ligation efficiency of different ribozyme fragments for AtRNL AA (Figure 1(f)) and T4 RNL2 TKQ (Figure 1(g)) in vitro. We observed a ligation efficiency of 70–80% for AtRNL AA and 30–88% for T4 RNL2 TKQ. The lower ligation efficiency of the 3ʹ-twister ribozyme fragment could be due to its three-dimensional structure. The twister ribozyme structure is additionally stabilized by two pseudoknots that can still form in the 3ʹ-cleavage fragment [36] and crystallography studies suggest that the 5ʹ-end of the 3ʹ-ribozyme fragment is buried within its structure and thus could be less available for ligation [37]. Nevertheless, even a ligation efficiency as low as 25% has been shown to be sufficient to capture RNAs with 5′-OH from total RNA [29]. Therefore, we suppose that the ligation efficiencies we observed between 30 and 88% are sufficient to capture ribozyme fragments generated by the majority of self-cleaving ribozymes.
In AtRNL-ligations, a 2ʹ-P is left at the ligation site that can negatively affect the subsequent reverse transcription. Therefore, we removed the 2′-P by Tpt1 [28,38] (Supplementary Figure S3). After ligation and 2ʹ-dephosphorylation, the following steps were identical for all three cyPhyRNA-seq libraries (Figure 1(d)). The ligation products were reverse transcribed, gel-purified to remove excess RT primer and 3ʹ-adapter (first adapter), and then the cDNA was used for second adapter ligation by T4 RNL1 [31] and subsequent DNA amplification.
Capturing self-cleaving ribozyme fragments spiked into E. coli total RNA
After optimization of all individual steps of our screening strategy in vitro, we investigated the efficiency of ribozyme fragment capture from complex RNA populations. Although most cellular RNAs have 5ʹ-P and 3ʹ-OH ends, RNAs with 2ʹ,3ʹ-cP and 5ʹ-OH can be generated independently of self-cleaving ribozymes, e.g. by spontaneous RNA cleavage at any phosphodiester linkage. This inevitable background reaction can be accelerated by millimolar concentrations of divalent metal ions and is also enhanced in less structured regions of an RNA, where the phosphodiester backbone frequently samples an in-line conformation [39]. A number of specific cellular processes, e.g. cleavage by RNA endonucleases [40], and finally, external factors, such as mechanical stress, e.g. during handling of the RNA, can also induce random RNA breaks adding to the pool of 2ʹ,3ʹ-cP and 5ʹ-OH-containing RNAs. Such RNAs would be targeted by cyPhyRNA-seq, and it is important to be able to remove such false positives. Fortunately, self-cleaving ribozymes cleave site-specifically and therefore, sequencing reads with a 2ʹ,3ʹ-cP should always end at exactly the same nucleotide and sequencing reads derived from the ribozyme fragment with 5ʹ-OH should exclusively start at the same nucleotide. This creates distinct profiles of mapped sequencing reads with a coverage often well above that created by mere background degradation. Even if widespread throughout the transcriptome, background degradation occurs at much lower level. Therefore, we expect that 2ʹ,3ʹ-cP and 5ʹ-OH-specific reads generated by these events will be roughly uniformly distributed across the genome.
To investigate the efficiency of capturing ribozyme fragments by cyPhyRNA-seq in the presence of non-ribozyme-derived RNAs with 2ʹ,3ʹ-cP and 5ʹ-OH, we spiked in vitro transcribed 5ʹ-HHR (type I) or 3ʹ-twister (type P1) ribozyme fragments into E. coli total RNA. E. coli total RNA is not only easily accessible, and provides a pool of diverse RNAs with canonical and self-cleaving ribozyme-like ends, but also has no known natural self-cleaving ribozymes. The latter fact allows us to precisely control the amount of ribozyme fragment added to the diverse RNA pool. We compared the cyPhyRNA-seq signal derived from artificially added self-cleaving ribozymes to that of background generated by other RNAs with 2ʹ,3ʹ-cP and 5ʹ-OH ends.
In total RNA samples, rRNAs are the most abundant transcripts (~80%) [41,42] and would dominate the sequencing data in any RNA-seq method ligating an adapter to 3ʹ-OH ends. Due to background degradation, these rRNAs can also be rich sources of 2ʹ,3ʹ-cPs [43]. Therefore, it is necessary to reduce the amount of rRNA in the total RNA pool. A number of rRNA depletion methods and kits are available [44,45]. We chose an approach to selectively remove large RNAs by precipitation with polyethylene glycol (PEG) and salt (Supplementary Figure S4), which can be used for RNA samples from bacteria as well as eukaryotes [46,47]. Removing large RNAs, such as rRNAs and mRNAs, results in an enrichment of shorter RNA species. Although self-cleaving ribozymes are expected to be comparatively short (40–150 nt) based on the assessment of known classes, they could be part of larger mRNA transcripts. This means they would be eliminated by the currently used rRNA depletion strategy. In order to precipitate only rRNAs and thus to prevent the removal of larger mRNAs possibly harbouring ribozymes, alternative rRNA-specific depletion methods could be used if available for the respective organism [48]. However, this is rarely the case when deviating from the few typical model organisms. Therefore, we found the current method most feasible for capturing ribozyme fragments with 2ʹ,3ʹ-cPs. For the capture of ribozyme fragments with 5ʹ-OH, we employed an alternative strategy. As mentioned above, this strategy uses T4 RNL2 TKQ, which ligates all RNAs with 3ʹ-OH ends and thus is not specific for RNAs with 5ʹ-OH ends. Therefore, prior to ligation with T4 RNL2 TKQ, we selectively enriched RNAs with a 5ʹ-OH by removing all RNAs that contain 5ʹ-PPP or 5′-P ends (Supplementary Figure S4). This strategy would leave ribozyme fragments as part of longer RNAs unaffected. Hence, they would be detectable by cyPhyRNA-seq. The conversion of 5′-PPP to 5′-P ends was performed with the RNA pyrophosphohydrolase (RppH) from E. coli [49] and the RNAs with 5′-P ends were removed by the 5′-3′ exonuclease Xrn1 from S. cerevisiae [50]. Hence, this treatment reduces many abundant RNA species from the total RNA pool including rRNAs and mRNAs.
The generated libraries were amplified and subjected to Amplicon-sequencing with read numbers of ~10,000–25,000. In this spike-in experiment, we evaluated another strategy involving the circularization of cDNA (Supplementary Text, Supplementary Figure S5). This method led to a high number of adapter–adapter reads that did not contain any insert [51]. Thus, we used the two-step adapter addition as described in Figure 1(e) for further library preparations.
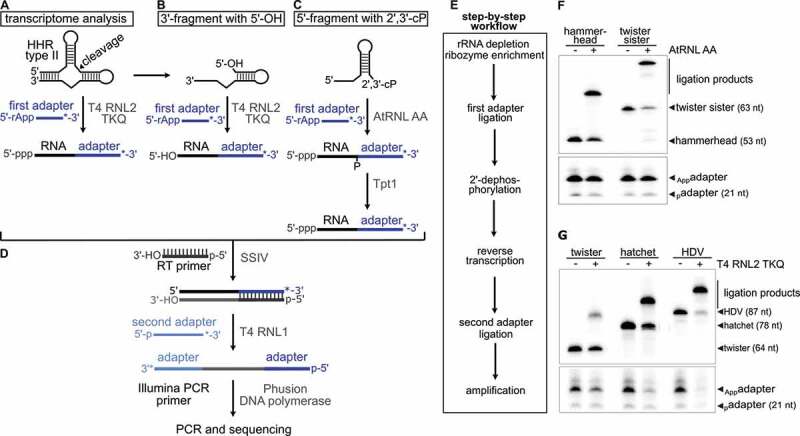
In the Amplicon-sequencing run, we analysed only unique mapping reads filtered by their unique molecular identifier (UMI) tags and assigned them based on annotation to general RNA classes, namely rRNAs, tRNAs, other non-coding RNAs, mRNAs or our added ribozyme cleavage fragments (Figure 2, Supplementary Table S1). In both libraries capturing the 5ʹ- and 3ʹ-ribozyme fragments, the majority of sequencing reads belonged to tRNAs with up to 92% (Figure 2 left). By contrast, only ~4% of the sequencing reads were 5ʹ-HHR fragments (Figure 2 top left) and no reads for 3ʹ-twister ribozyme fragments were detected (Figure 2 bottom left).
Figure 2.
Percent of sequencing reads captured for selected RNA types in E. coli total RNA. E. coli total RNA was spiked with either the 5ʹ-fragment of a HHR (top) or 3ʹ-fragment of a twister ribozyme (bottom) at a ratio of 1:1,000. ‘other ncRNA’ refers to RNAs other than rRNAs, tRNAs and self-cleaving ribozymes added. Strategy with blocking of mature tRNAs (right) reveals a decrease in tRNA and increase of captured 5ʹ-HHR and 3ʹ-twister fragments
To deplete tRNA reads in both library preparations, we included a ‘tRNA blocking’ step (Supplementary Figure S4), in which a hairpin adapter with 3′-NTGG overhang is ligated to mature tRNAs that carry a CCA-end by T4 DNA ligase [52]. This highly specific ligation has yields of 80–90% and is applicable to organisms from all domains of life. The ligation of the hairpin adapter blocks the tRNA ends and eliminates them from the pool of ligatable RNAs. Applying ‘tRNA blocking’ prior to the first adapter ligation reduced tRNA reads from >80% to 41% (Figure 2 top right) or from >90% to 16% (Figure 2 bottom right) and at the same time, reads for 5ʹ-HHR (19%) and 3ʹ-twister ribozyme fragments (6%) increased. We assume the reduction of tRNA reads in the library with AtRNL AA-ligation (Figure 2 top right) is due to the protection of mature tRNAs from degradation at the CCA-end when ‘tRNA blocking’ is performed. ‘tRNA blocking’ effectively decreases the breakdown of tRNA CCA-ends to C- or CC-ends that otherwise appear to be available for ligation by AtRNL AA.
The relative increase in the fraction of rRNA reads (Figure 2 right) can be explained by the extended incubation during ‘tRNA blocking’: It is unlikely that all rRNA molecules are completely degraded by RppH and Xrn1 treatments or eliminated by precipitation. During ‘tRNA blocking’, the comparatively long rRNAs are cleaved and breakdown products are created with sizes that are more similar to the pool of shorter RNAs isolated during cyPhyRNA-seq. Therefore, more short rRNA pieces are isolated and amplified by PCR in ‘tRNA blocking’ than without this treatment. Despite this increase in rRNA reads in libraries with ‘tRNA blocking’, the combined removal of rRNAs either by degradation or precipitation followed by the reduction of mature tRNAs enabled the capturing of ribozyme 3ʹ-fragments and increased the number of reads corresponding to 5ʹ-ribozyme fragments.
While we do capture ribozyme-specific cleavage fragments, we also observe signals that likely correspond to background degradation in mRNA and other non-coding RNAs. However, while in the simple analysis presented here the signals for background degradation might seem high; one has to keep in mind that reads were simply binned into RNA groups based on overlap to annotation without further pre- or post-processing. When these reads are mapped to the genome, they are distributed over many locations. To illustrate this point, in this test case E. coli has an estimated number of 4400 genes. However, we only observe 122 reads in mRNAs and 80 reads in ncRNAs other than ribozymes (Supplementary Table S1) and these reads were distributed over several different mRNAs or ncRNAs. For the HHR fragment, we count 130 reads. Therefore, the number of reads is high for the ribozyme fragments, while for the other RNA types the counted read number is distributed over many different genomic loci, and thus very small per individual locus.
In summary, we were able to capture the spiked ribozyme fragments with cyPhyRNA-seq from E. coli total RNA and significantly improved ribozyme-specific sequencing output by reducing tRNA reads.
Identification of three active type II hammerhead ribozymes in Desulfovibrio vulgaris
Next, we applied our strategy to screen for self-cleaving ribozymes in the bacterium D. vulgaris. We chose this organism because it is cultivatable, fully sequenced and encodes three computationally predicted type II HHRs, whose activity had not been examined. These HHRs are found at distinct genome locations (HHR type II, D. vulgaris loc 1–3, Supplementary Table S2) near different hypothetical and phage-like protein coding genes (Figure 3(a,c)). HHRs [53] and other bacterial self-cleaving ribozyme classes, such as twister, twister sister, pistol and hatchet have also been previously observed to occur in similar genetic contexts in other bacteria [10,11]. Self-cleavage of the three predicted type II-HHRs results in a 5ʹ-fragment of 29–32 nt (depending on the representative) and a 3ʹ-fragment of 39 nt. These fragment lengths correspond to minimal sizes expected for these RNAs and can be detected by cyPhyRNA-seq even if the self-cleaving ribozymes are not part of longer transcripts in vivo (Supplementary Table S2). For the analysis of cyPhyRNA-seq data, only deduplicated and uniquely mapping reads were analysed (Supplementary Table S3). To assess ribozyme cleavage signals and to possibly distinguish them from signals derived from other events such as background cleavage, we 1) evaluated the general read profiles depicting all deduplicated, uniquely mapping reads (Figure 3), 2) plotted only the start and end positions of these reads, and 3) we used a peak calling method to filter these signals (Supplementary Figure S6, S8-11, see below).
Figure 3.
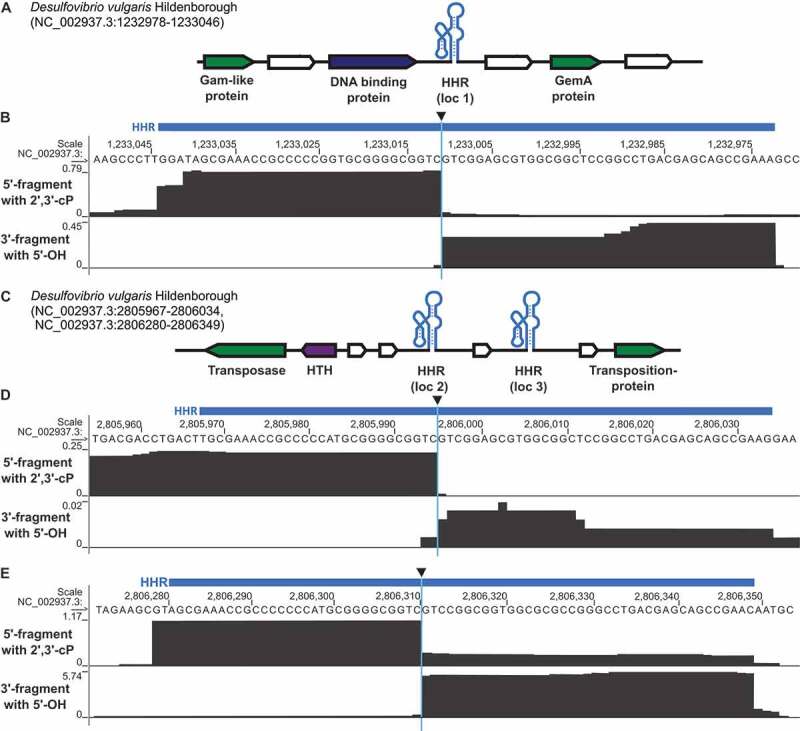
Genomic context of HHR sequences in D. vulgaris and analysis of ribozyme activity using cyPhyRNA-seq. (a) Genetic context of the type II HHR locus loc 1 (NC_002937.3: 1,233,046–1,232,978, antisense strand). For illustration, HHR is shown schematically in its secondary RNA structure (light blue). Nearby protein-coding genes are represented by arrows in white (hypothetical genes with unknown functions), green (protein-coding genes of bacteriophage origin) and dark blue (DNA-binding proteins). The direction of the arrowheads shows on which strand the gene is located. All HHRs and their adjacent genomic context are shown in 5ʹ to 3ʹ direction. (b) Sequencing reads mapped to HHR loc 1, described in a, are represented in grey for the two libraries: 5′- fragment with 2ʹ,3ʹ-cP (top track), 3′-fragment with 5ʹ-OH (bottom track). Reads are deduplicated (single ligation events) and unique (map to only one position in the genome). Blue bar above the sequence: computationally predicted location of the HHR. Black triangle: cleavage site. For each library, number of deduplicated and unique reads covering each position is scaled to the total number of deduplicated and unique reads on the same strand divided by 106 (RPM). Although this is per se only a per library scaling, the fact that all libraries show comparable sequencing depth allows the comparison of normalized values across libraries. (c) Genetic context of the type II-HHR loc 2 (NC_002937.3:2,805,967–2,806,034, sense strand) and the type II HHR loc 3 (NC_002937.3: 2,806,280–2,806,349, sense strand), which are in close proximity to each other. Description is the same as in (a). A gene encoding a helix-turn-helix motif (HTH) is represented in purple. Sequencing reads mapping to the HHR loc 2 shown in (d) and to HHR loc 3 shown in (e), which are described in c. The description is the same as in b. Red box with asterisk highlights area where we find a mixture of reads that is not discernable in this representation: some reads extend through the cleavage site, indicating uncleaved ribozymes, but some reads do start exactly 3ʹ of the cleavage site (see supplementary text, supplementary Figure S6)
As the method to capture 5′-fragments relies on the ligation of the first adapter specifically to RNAs with a 2ʹ,3ʹ-cP, we expect that the reads corresponding to 5′-ribozyme fragments end at the cleavage site. Similarly, reads collected by our method to capture the 3′-fragment with a 5ʹ-OH should start at the cleavage site. In our application of cyPhyRNA-seq to D. vulgaris, the general read profiles at HHR locations (Figure 3(b,d,e) and Supplementary Figure S6, S8) match this assumption. The read profiles show a sharp drop in read number directly at the site of ribozyme cleavage. Thus, an evaluation of read profiles generated by cyPhyRNA-seq can be used to reveal the activity of self-cleaving ribozymes in this organism.
The highest number of uniquely mapping reads were detected for the HHR loc 3 and the lowest number of reads was visible for the nearby HHR loc 2. The fact that most reads only cover the predicted ribozyme motif was surprising to us, but can be explained by the comparatively short read lengths acquired in this sequencing. The average read length mapping to a HHR was <40 nt, with the average length of all deduplicated, uniquely mapping reads being ~55 nt. It is also likely that RNA outside the structured ribozyme motif is more prone to background degradation, which would lead to the detection of shortened reads. Alternatively, it is likewise possible that natural transcription start and termination sites are flanking the ribozyme motif, which would also result in the observed read distribution.
To visualize the start and end of reads within a general read profile more clearly, we created a depiction of where within the genome the first nucleotide of a read (its 5ʹ-end) and last nucleotide of a read (its 3ʹ-end) maps (Supplementary Figure S6, S8-11). This depiction clearly shows that the majority of reads collected at a self-cleaving ribozyme locus end (2ʹ,3ʹ-cP library) or start (5ʹ-OH library) at the cleavage site (Supplementary Figure S6, S8). Only for the HHR loc 2 this analysis of read start and end points is inconclusive, which is likely due to the extremely low number of reads collected at this genomic locus. Other types of RNAs such as tRNAs, rRNAs or mRNAs show a distribution of many start/end positions across their annotated genomic loci without a distinct enrichment at a specific site (Supplementary Figures S9-11).
This representation of start/stop positions enables a clear distinction between reads that span the entire predicted ribozyme and those that only include a ribozyme cleavage fragment. When applying cyPhyRNA-seq to capture the 5ʹ-fragment with a 2ʹ,3ʹ-cP (Figure 1(c)), some reads from the HHR loc 3 become apparent that also start at the cleavage site (Figure 3(e), top track, area highlighted in red box, Supplementary Figure S6C). This can result from unspecific ligation of AtRNL AA. While AtRNL AA does not recognize the more prevalent RNAs with a 5ʹ-P or 5ʹ-PPP as efficiently, our investigations showed that the enzyme is able to ligate the 5ʹ-pre-adenylated DNA adapter to 3ʹ-OH ends of RNAs with a 5ʹ-OH (Supplementary Supplementary Text, Figure S7). This unspecific ligation was only slightly detectable for the HHR loc 3, and not at all for the HHR loc 1 and loc 2. This indicates that this side-reaction does not cause severe problems in assigning RNA ends originating from site-specific ribozyme cleavage in cyPhyRNA-seq. The reads for HHR loc 3 that possibly originate from unspecific AtRNL AA ligation can be explained by the higher expression levels of this ribozyme that is reflected in the higher overall read number for HHR loc 3. A higher number of reads for HHR loc 3 was also observed for the libraries derived from bacteria exposed to heat-shock (see below and Supplementary Figure S8).
When we used the method to capture the 3ʹ-fragment with a 5ʹ-OH (Figure 1(b)), we detected some shorter fragments of ~15-20 nt (Figure 3(b,d,e), bottom tracks) that probably result from the additional treatment with RppH and Xrn1. Alternatively, these shorter reads could indicate that transcripts of HHR loc 1 and loc 2 are less stable and have a shorter half-life in the cell. In case of HHR loc 2, the lower read number detected could reflect an extremely low expression level.
Next, we investigated if the distribution of read start/end positions could be used as an indicator of site-specific RNA cleavage. The enrichment of read-ends over background at a specific site would enable the computational automation of detecting genome regions likely containing self-cleaving ribozymes. More specifically, regions where site-specific RNA cleavage occurs could be distinguished from regions of random RNA background degradation. To automate ribozyme detection, we exploited methods of ‘peak calling’ often used in CLIP-seq data analysis. In CLIP-seq, UV-crosslinking of an RNA-binding protein (RBP) of interest to its target RNA is used to identify RNA-binding sites of RBPs [54]. The crosslinking process leads to noisy datasets due to the plethora of possible interaction sites between RNA and protein. However, not all of these interactions represent true binding sites. Therefore, strict filtering strategies (i.e. peak calling) have to be applied to distinguish signal from noise, especially in genome-wide approaches.
To investigate the applicability of CLIP-seq-peak-calling-methods to cyPhyRNA-seq data from D. vulgaris, we used the well-established peak caller Piranha [55]. This allowed us to exclude ~ 95% of uniquely mapping, deduplicated reads and focus on the remaining 5% for the identification of significant signals that could correspond to ribozyme-specific cleavage (Additional file 1, Supplementary Text). With Piranha, we derived a number of loci that showed distinct peak patterns, which overlap with the annotated ribozymes (Supplementary Figure S6, S8) and other RNA classes (Supplementary Figure S9-11, Additional file 1).
Several self-cleaving ribozyme characteristics allowed us to further filter peaks to distinguish signals typical for these catalytic RNAs from background degradation. Ribozyme-specific peaks are expected to have a narrow average peak width (close to 1) in contrast to tRNAs and rRNAs that have higher average peak widths (Supplementary Text, Additional file 1). An average peak width of 1 indicates that most reads mapping to this genomic area end (or start, depending on the library) at the same position. This is to be expected for site-specific RNA cleavage and can be observed for peaks derived from both sequencing approaches (Supplementary Figure S6, S8).
Because most reads stop or begin at the cleavage site, we expect that the peaks of the 5ʹ- and 3ʹ-fragments of self-cleaving ribozymes map adjacent to each other in the genome. We used this to devise an even more stringent peak filter as peaks derived from site-specific cleavage should not be found more than ± 3 nt apart in the two sequencing libraries (Additional file 1 and Supplementary Text). Indeed, the HHR loc 1 and loc 3 display this expected peak pattern, for HHR loc 2 only a peak in the method for the 5ʹ-fragment was observed (Supplementary Figure S6 and S8). At the same time, this combined filtering strategy immediately eliminated most of the peaks that map to other transcripts such as tRNAs, rRNAs or mRNAs (Supplementary Figures S9-11, Additional file 1). These peaks likely result from background degradation or other unspecific RNA cleavage processes. In these loci, several peaks are found immediately next to each other leading to increased average peak widths and drastically increased peak numbers per annotated RNA type.
cyPhyRNA-seq has the potential to examine self-cleaving ribozyme activity at different growth conditions. This would allow investigating whether ribozymes are regulated in an organism, for example during certain growth phases, nutritional restrictions or other stress conditions. To demonstrate that the method can be used to compare different conditions, we applied heat-shock to one of the samples (Supplementary Figure S8). The results suggest that also under these growth conditions all HHRs are active. However, fewer reads were detected that mapped to HHR loc 2 (Supplementary Figure S8B). Furthermore, more unspecific ligation products were visible for the 5′-fragment method (Supplementary Figure S8C, top track). This could be due to an increase in overall RNA background degradation during the heat-shock. It appears that heat-shock has no influence on HHR activity in D. vulgaris. Applying cyPhyRNA-seq to another condition further illustrates the reproducibility of our method.
Both cultivation conditions showed that only 3–9% of rRNA reads were detectable for all libraries, suggesting that large rRNAs (23S rRNA and 16S rRNA) can be efficiently removed from total RNA with rRNA depletion using PEG8000 and NaCl or RppH and Xrn1 (Supplementary Table S4). The percentage of mRNA reads was 13–22% and 1–6% for other ncRNAs. However, most of the sequencing reads belonged to tRNAs (66–83%). The high number of tRNA reads is not surprising, because tRNAs are the most abundant transcripts (~15% by mass) after rRNAs (~80% by mass) in bacterial cells [41]. Furthermore, they are neither eliminated by precipitation with PEG and salt nor by enzymatic degradation. Nevertheless, ‘tRNA blocking’ only works efficiently for mature tRNAs with intact CCA-end. This leaves all tRNAs with a shortened CCA-end, extended, alternative CCA-ends or tRNAs cleaved elsewhere, e.g. in the anticodon loop, as substrates for first strand ligation during library preparation. This is illustrated by the large number of start/end positions of sequencing reads mapping to tRNAs (Supplementary Figure S9A, middle tracks). In the resulting peak analysis, we therefore observed one wide peak that results from many adjacent start/end signals. These peaks do not represent RNA cleavage events at one specific position, but are distributed over the general area of the anticodon loop and tRNA. Additionally, mature tRNAs with intact CCA-end are effectively reduced by ‘tRNA blocking’ as we mostly observe tRNAs with deficient ends. About 69% of tRNAs are shortened to a CC-end and more than 29% of tRNAs are extended at their CCA-end by at least one additional nucleotide (Supplementary Figure S9C). After observing activity for the type II HHRs by cyPhyRNA-seq, we additionally demonstrated that all of them cleave during in vitro transcription (Supplementary Figure S12).
Conclusion
In the past, self-cleaving ribozymes have been mostly discovered by chance or comparative sequence analysis. Although computational analysis can predict self-cleaving ribozyme candidates, the cleavage activity of individual representatives has to be biochemically confirmed, which limits the scale of candidates that can be investigated. Now, cyPhyRNA-seq offers a high-throughput screening approach for the targeted detection of active self-cleaving ribozymes on a global scale.
cyPhyRNA-seq exploits a biochemical characteristic inherent to every known natural self-cleaving ribozyme: the catalysis of site-specific RNA scission to generate a cleavage fragment containing a 2ʹ,3ʹ-cP and another fragment with a 5ʹ-OH group. We use RNA-seq libraries specific for these RNA-ends and extend established peak calling protocols to filter signal from noise in cyPhyRNA-seq data. This leads to the detection of reads that most likely correspond to RNA fragments derived from site-specific cleavage. Only such reads are characterized by mapping uniquely to a specific genomic location at their 3ʹ-end (for the library that captures RNAs with a 2ʹ,3ʹ-cP) or 5ʹ-end (for the library that enriches RNAs with 5ʹ-OH group) and additionally both ends are expected to be located precisely next to each other or in very close proximity. Thus, using both library preparation methods in parallel enables a more reliable detection of possible self-cleaving ribozyme candidates or site-specific cleavage locations in a transcriptome.
Other biochemistry-based methods have been devised to discover self-cleaving ribozymes. In vitro selection was applied to the human genome and led to the discovery of the CPEB3 ribozyme, an HDV-like ribozyme [17]. Recently, another approach was developed to identify ribozymes in humans, which is based solely on the enrichment of RNAs with 5ʹ-OH using RppH and Xrn1, similar to parts of our method [12]. However, in contrast to our method, this approach does not capture RNAs with 2ʹ,3ʹ-cP, but rather adds a T7 promoter sequence to fragmented gDNA and these DNAs were used for in vitro transcription. While the in vitro transcription of random gDNA fragments has the potential to find self-cleaving RNAs that are only scarcely expressed in vivo, this approach cannot easily detect ribozymes that need protein or metabolite cofactors. cyPhyRNA-seq can be used to screen for active self-cleaving ribozymes in total RNA, even if they need cofactors to be fully active. Moreover, ribozymes that have a very short 3ʹ-ribozyme fragment would hardly be discovered by the approach introduced by Chen et al. [12], which makes the use of capturing methods for both ribozyme fragments in cyPhyRNA-seq more comprehensive. Additionally, cyPhyRNA-seq allows for the investigation of ribozyme activity under different cultivation conditions, for example certain stresses, different life-stages or growth-phases, especially in organisms that contain many self-cleaving ribozymes. Alternatively, reverse transcription quantitative PCR (RT-qPCR) can be used to study differences in expression of known self-cleaving ribozymes [17,56]. Thus, while RT-qPCR seems most useful for isolated ribozyme examples that are known, cyPhyRNA-seq is advantageous when simultaneously investigating several distinct self-cleaving ribozymes in one organism. These analyses by either RT-qPCR or cyPhyRNA-seq could help in deciphering unknown biological roles for some self-cleaving ribozymes.
We used cyPhyRNA-seq to identify ribozyme fragments, but this approach is also applicable to RNAs with identical termini produced in other cellular processes [40] or treatments [57,58]. These cellular processes include cleavage by certain endoribonucleases (e.g. RNase A [59] or MazF [60]) and stress-induction of tRNAs (e.g. oxidative stress) [61]. Several approaches to capture RNAs with 2ʹ,3ʹ-cP have already been described [28,43]. However, their use for the identification of self-cleaving ribozymes was never attempted, but rather these approaches were applied to research questions involving highly abundant RNAs. cyPhyRNA-seq represents a method with the potential to investigate extremely rare RNAs. This is facilitated by an improved ligation of RNAs with 2ʹ,3ʹ-cP ends, for which we generated a variant of the AtRNL (AtRNL AA). In contrast to the wild-type enzyme, this variant has lost its 5ʹ-phosphorylation and adenylation activity. Therefore, the activity of AtRNL AA is limited to the ligation of 5ʹ-pre-adenylated adapter molecules to RNAs with 2ʹ,3ʹ-cP termini resulting in an enhanced ligation. Another improvement of cyPhyRNA-seq is the enrichment for relevant RNAs by enzymatic treatments and ‘tRNA blocking’, which is especially important for capturing 3ʹ-ribozyme fragments. By using the RppH/Xrn1 treatment and ‘tRNA blocking’, we significantly reduced highly abundant RNAs in total RNA, namely rRNAs and tRNAs, allowing the detection of less common RNA species. The use of ‘tRNA blocking’ could be equally beneficial in research questions where, for example, tRNA fragments, RNA-targets of site-specific ribonucleases or snoRNAs are studied, because a targeted decrease in mature tRNAs will lead to an increase in target RNA reads. The potential of our method for the analysis of tRNA fragment expression will be investigated in future work. Furthermore, cyPhyRNA-seq provides the means to capture both ribozyme cleavage fragments, which supplies complementary data for both parts of the catalytic RNA were possible. Several ribozymes classes have one cleavage fragment that could be shorter than 10 nt, if transcription in the natural context starts (in case of HDV and hatchet), or ends (e.g. for hairpin, pistol and twister sister) with the conserved ribozyme RNA. These shorter ribozyme fragments are indistinguishable from other background degradation events creating very short RNAs. Furthermore, the in vivo stability of the 5ʹ- and 3ʹ-ribozyme fragment may vary. The fragment stability could depend on currently understudied factors such as specific recognition by ribonucleases or protection from degradation by their intricate structures. Therefore, it can be beneficial to capture both fragments where possible and thus, increase the chance of detecting as many ribozyme cleavage products as possible.
With its global, transcriptome-wide analysis, cyPhyRNA-seq is well-suited to look at organisms that have a large collection of ribozymes such as Schistosoma mansoni (parasitic worm), Aedes aegypti (mosquito) and Xenopus tropicalis (frog) that contain thousands of self-cleaving ribozyme representatives [53]. Apart from the investigation of cleavage activity of known or predicted ribozymes in an organism, cyPhyRNA-seq also has the potential to be used to discover new self-cleaving ribozyme classes. Therefore, we plan to apply cyPhyRNA-seq to screen for novel self-cleaving ribozymes in diverse organisms and metatranscriptomes.
Methods
Protein expression and purification
The two mutations K152A [35] and D726A [35] were introduced into the wild-type AtRNL sequence of pET28a_AtRNL plasmid (Addgene plasmid #32,242) by site-directed mutagenesis using the PCRBIO HiFi Polymerase (PCR Biosystems) according to manufacturer’s instructions and primers described in Supplementary Table S5. Afterwards, E. coli BL21 (DE3)-RIPL cells were transformed with the resulting pET28a_AtRNL AA plasmid. Protein expression was performed in 400 ml TB [62] containing 30 µg/ml kanamycin and 35 µg/ml chloramphenicol at 30°C until an OD600 of 1.2 was reached. Then, 400 ml of ice-cold TB was added containing antibiotics (see above), ethanol and isopropyl-β-D-1-thiogalactopyranoside (IPTG) to final concentrations of 2% and 0.4 mM, respectively. After cultivation at 16°C and 200 rpm for 20 h, the cells were harvested by centrifugation at 4,000 g and 4°C and stored at −80°C. Cells were resuspended in 30 ml Buffer A [50 mM Tris-HCl (pH 7.5 at 4°C), 500 mM NaCl, 5 mM DTT, 5% glycerol] and 0.2 mg/ml lysozyme was added. Cell suspension was incubated on ice for 30 min and cells were disrupted by sonication for 7 × 10 sec with 60 sec breaks at 70% intensity. After centrifugation at 30,600 g and 4°C for 30 min, supernatant was sterile filtered and loaded onto a HisTrap FastFlow column (1 ml, GE Healthcare) equilibrated with Buffer A. The column was washed with Buffer A containing 5% Buffer B [50 mM Tris-HCl (pH 7.5 at 4°C), 500 mM NaCl, 5 mM DTT, 5% glycerol, 500 mM imidazole]. The elution was performed in three steps using 20%, 50% and 100% of Buffer B. Peak fractions were pooled and purified by size exclusion chromatography (SEC) using a HiLoad 16/60 Superdex 200 pg (GE Healthcare) column equilibrated in Buffer A. Peak fractions were combined and concentrated using a Vivaspin 6 column (30 kDa cut-off).
Tpt1 from Saccharomyces cerevisiae was expressed from pET-24b-TPT1 [38] in E. coli BL21 (DE3) cells. Cells were grown at 37°C in 400 ml LB [62] containing 30 µg/ml kanamycin. At an OD600 of 0.4, overexpression was induced by adding 1 mM IPTG. The cell culture was incubated at 37°C for 2 h, harvested by centrifugation at 4,000 g and 4°C and stored at −80°C. After resuspension in 12 ml Buffer C [20 mM Tris-HCl (pH 7.5 at 4°C), 500 mM NaCl, 4 mM MgCl2, 10% glycerol, 0.5 mM β-mercaptoethanol], cells were lysed by sonication (7x 10 sec with 60 sec breaks at 70% intensity) and centrifuged at 30,600 g and 4°C for 30 min. Sterile filtered supernatant was used for the purification with a HisTrap FastFlow column (1 ml, GE Healthcare) equilibrated in Buffer C. The column was washed with Buffer C containing 5% Buffer D [20 mM Tris-HCl (pH 7.5 at 4°C), 500 mM NaCl, 10% glycerol, 4 mM MgCl2, 0.5 mM β-mercaptoethanol, 500 mM imidazole]. Elution steps contained 20%, 50% and 100% Buffer D. Peak fractions were purified using a HiLoad 16/60 Superdex 75 pg column (GE Healthcare) equilibrated in Buffer E [40 mM Tris-HCl (pH 7.5 at 4°C), 150 mM NaCl, 4 mM EDTA, 8 mM MgCl2, 2 mM DTT]. Peak fractions were combined and concentrated using a Vivaspin 6 column (10 kDa cut-off).
T4 RNL2 truncated KQ and TS2126 RNL1 were expressed and purified as described previously [63,64]. The final protein solutions were diluted with an equal volume of 80% glycerol and stored at −80°C.
RNA synthesis and preparation of oligonucleotides
Ribozyme cleavage fragments (Supplementary Table S2) were generated by T7-based in vitro transcription from PCR products (with T7 promoter) of the respective ribozyme (Supplementary Table S5) [65] in the presence of [α32P]-ATP (Hartmann Analytic). The cleavage fragments were gel-purified as described previously [66]. 3ʹ-DNA adapter
(5ʹ-TGGAATTCTCGGGTGCCAAGG-Amino-C7-3ʹ), RT primer (5′-GCCTTGGCACCCGAGAATTCCA-3′) and circularization-RT primer (5′-P-GATCGTCGGACTGTAGAACTCTGAAC /iSp18/ CACTCA/ iSP18/ GCCTTGGCACCCGAGAATTCCA-3′) (Supplementary Table S5) were radioactively labelled using [γ32P]-ATP (Hartmann Analytic) and T4 Polynucleotide kinase (NEB).
Adenylation of the 3′-adapter using TS2126 RNL1
The 3′-DNA adapter (Supplementary Table S5) was adenylated using the TS2126 RNL1 [67] and purified using phenol/chloroform extraction and ethanol precipitation [68]. For in vitro experiments, the non-radioactive adapter was mixed with 5′-labelled adapter in a ratio of 5:1.
First adapter ligation by AtRNL and 2′-dephosphorylation
For the ligation reaction, 2.5–10 pmol of in vitro transcribed ribozyme RNA (Supplementary Table S2) and twice the amount of adenylated 3′-DNA adapter (Supplementary Table S5) were combined, incubated at 65°C for 5 min and then cooled down on ice. A 10 µl ligation reaction included the RNA-adapter-mix, 1x reaction buffer [20 mM Tris-HCl (pH 7.5 at 23°C), 5 mM MgCl2, 2.5 mM spermidine, 100 µM DTT], 15% PEG and 0.6 µM AtRNL variant AA was incubated at 25°C for 2 h. For in vitro experiments, reactions were stopped by adding 3x RNA loading dye [10 mM Tris/HCl (pH7.6 at 23°C), 80% formamide, 0.25% bromophenol blue and 0.25% xylene cyanol] and separated by PAGE using a 15% denaturing PAA gel containing 8 M urea. To remove the resulting 2′-phosphate at the ligation site, the reactions were incubated with 1x reaction buffer, 10 mM nicotinamide adenine dinucleotide (NAD) and 0.6 µM Tpt1 in a 20 µl reaction at 30°C for 30 min.
First adapter ligation by the T4 RNL2 TKQ
The ligation was carried out with 2.5–10 pmol of in vitro transcribed ribozyme RNA (Supplementary Table S2) and twice the amount of adenylated 3′-DNA adapter (Supplementary Table S5). After incubation of the RNA and adapter at 65°C for 5 min and cool down on ice, 1x T4 RNL buffer (NEB), 20% PEG, 10% DMSO and 1 µM T4 RNL2 TKQ was added to a final volume of 20 µl. The ligation reaction was incubated at 25°C for 16 h. For in vitro experiments, reactions were stopped by adding 3x RNA loading dye and separated by PAGE using a 15% denaturing PAA gel containing 8 M urea.
Reverse transcription and second adapter-ligation by T4 RNL1
To the ligation reactions with AtRNL AA (after 2′-dephosphorylation) and T4 RNL2 TKQ, 0.9 µM non-radioactive and 0.3 µM 5ʹ-labelled RT primer (Supplementary Table S5) was added, incubated at 65°C for 5 min and cooled down on ice. Reverse transcription was performed using the SuperScript IV RT (Thermo Scientific) at 55°C for 30 min. To degrade RNA, 200 mM NaOH was added and the mixture was incubated at 95°C for 3 min. After neutralization with 200 mM HCl, 3x RNA loading dye was added and cDNA was gel-purified using a 15% denaturing PAA gel (with 8 M urea), followed by ethanol precipitation.
Purified cDNA was incubated for 5 min at 65°C and cooled down on ice. Then, 5 µM of second adapter (5ʹ- P-GATCGTCGGACTGTAGAACTCTGAAC-Amino-C6 − 3ʹ, Supplementary Table S5), 1x T4 RNL buffer (NEB), 1 mM ATP, 1 mM hexammine cobalt chloride, 12.5% PEG and 10 U T4 RNL1 (NEB) was added to a final volume of 20 µl. The ligation reaction was incubated 16 h at 16°C followed by heat-inactivation at 65°C for 10 min.
Reverse transcription using the circularization-RT primer and circularization
To the ligation reactions with AtRNL AA (after 2ʹ-dephosphorylation) and T4 RNL2 TKQ, 0.9 µM non-radioactive and 0.3 µM 5′-labelled circularization-RT primer (Supplementary Table S5) [69] was added and incubated at 82°C for 2 min. After cooling down on ice, reverse transcription was performed using the SuperScript IV RT (Thermo Scientific) at 55°C for 30 min. RNA was degraded by incubating the reactions with 200 mM NaOH at 95°C for 3 min. The reaction was neutralized with 200 mM HCl and 3x loading dye was added. Then, cDNA was gel-purified with a 8% PAA gel containing 8 M urea and ethanol precipitated.
After purification, cDNA was incubated for 2 min at 82°C and subsequently circularized using 1x adenylation buffer (50 mM MOPS, 10 mM KCl, 5 mM MgCl2, pH 7.5), 50 µM ATP, 2.5 mM MnCl2, 1 mM DTT and 900 ng TS2126 RNL1 in a 20 µl reaction at 60°C for 2 h, as described before [64,67,69]. TS2126 RNL1 was inactivated at 80°C for 5 min.
DNA amplification with TruSeq Small RNA Illumina adapters
PCR samples (50 µl) included 5 µl circularization or second adapter ligation reaction, 1x GC buffer (Thermo Scientific), 0.5 µM forward and reverse primer (Supplementary Table S5), 200 µM dNTPs and 0.04 U/µl Phusion High-Fidelity DNA Polymerase (Thermo Scientific). The amplification was performed by denaturation at 98°C for 10 sec, annealing at 60°C for 20 sec and elongation at 72°C for 40 sec and 16 cycles. PCR products were analysed on a 3% agarose gel, stained with 250 ng/ml ethidium bromide using the 50 bp ladder (NEB) as size standard.
Preparation of E. coli total RNA and ribozyme fragments for spike-in experiment
E. coli TOP10 cells were grown to an OD600 of 1.2 and centrifuged at 4,000 g and 4°C for 10 min. Total RNA was isolated using TRIzolTM reagent (Thermo Scientific). The HHR 5ʹ-fragment with 2′,3′-cP and twister ribozyme 3ʹ-fragment (Supplementary Table S2) with 5′-OH were produced by in vitro transcription and self-cleavage, PAGE-purified and ethanol precipitated. 1 ng of ribozyme cleavage fragment was added to 1 µg E. coli total RNA and cyPhyRNA-seq library preparations were performed.
Preparation of D. vulgaris total RNA for characterization by cyPhyRNA-seq
180 ml medium 63 (German Collection of Microorganisms and Cell Cultures, DSMZ) was inoculated with 400 µl liquid culture of Desulfovibrio vulgaris (DSM 644) and incubated at 37°C under anaerobic conditions. Daily, pH was measured to monitor cell growth. After 90 h incubation, 50 ml cell culture was transferred to a fresh bottle and incubated at 42°C for 2 h (heat-shock). This cell culture and 50 ml of the main culture (after 92 h incubation, exponential growth phase) were centrifuged for 10 min at 4°C and 4,000 g and cell pellets were resuspended in 2 ml TRIzolTM reagent (Thermo Scientific). After adding TRIzolTM, the samples were frozen immediately in liquid nitrogen and stored at −80°C until needed. The total RNA was isolated according to the manufacturer’s instructions.
Enrichment of short RNAs from total RNA and ‘tRNA blocking’
Short RNA enrichment using polyethylene glycol (PEG8000) and NaCl, was performed for the samples to capture 5′-ribozyme fragments. In a 100 µl reaction, PEG8000 and NaCl were added to the samples to final concentrations of 5% and 0.5 M [46]. Samples were incubated at −20°C for 30 min and subsequently centrifuged at 10,000 g and 4°C for 30 min. The supernatant was transferred to a new tube and three volumes of absolute ethanol were added. After the incubation at −20°C overnight, the samples were centrifuged at 17,000 g for at least 1 h. The supernatant was removed and the pellet was washed with 70% ethanol, air-dried and dissolved in 4–6 µl diethyl pyrocarbonate (DEPC)-water.
Samples to capture the 3ʹ-ribozyme fragments were treated with RppH and Xrn1 to remove RNAs with 5′-PPP and 5′-P. 1 µg of total RNA was incubated with 1x NEBuffer 2, 0.25 U/µl RppH (NEB), 0.05 U/µl Xrn1 (NEB) and 0.02 U/µl RNase inhibitor Murine (NEB) for 15 min at 37°C. The samples were purified using GeneJET RNA Cleanup and Concentration Micro Kit (Thermo Scientific).
Samples were used for ‘tRNA blocking’ with the hairpin adapter (Supplementary Table S5), 30 U T4 DNA Ligase (NEB) and 0.02 U/µl RNase Inhibitor Murine (NEB) as described previously [52]. The hairpin adapter is ligated to the 3ʹ-end of the tRNA. The reactions were incubated at 32°C for 5 h and the enzyme was heat-inactivated at 65°C for 10 min. Samples were purified using phenol/chloroform extraction and ethanol precipitation and dissolved in 5 µl DEPC-H2O [68].
Library preparation for cyPhyRNA-seq
First adapter ligations for 5′- and 3′-ribozyme fragment libraries contained enriched RNA and 40 pmol of pre-adenylated 3′-DNA adapter with unique molecular identifier (UMI, Supplementary Table S5). The UMI is a stretch of random nucleotides (analogous to a barcode) that tags each molecule ligated in the first step of library preparation. In our protocol, we used the UMI to estimate the abundance of an RNA and reduce quantitative bias introduced by PCR [70]. As positive control, in vitro transcribed ribozyme fragments were used for size selection during gel purification after RT (Supplementary Table S2, S5).
For the spike-in experiment, reverse transcription and subsequent circularization were performed with the circularization-RT primer. PCR reactions contained 2.5–5 µl circular cDNA as template and primer with partial Illumina adapter sequences (Supplementary Table S5, Amplicon-EZ sequencing, GENEWIZ). Libraries were purified using the QIAquick PCR purification kit (#28,104, Qiagen), adjusted to a final concentration of 20 ng/µl each and sequenced (~10,000–25,000 reads, GENEWIZ).
Samples of D. vulgaris were used for reverse transcription and second adapter ligation by T4 RNL1. PCR reactions were performed with TruSeq Small RNA Illumina adapters (i7) and 5 µl cDNA directly taken from the second ligation reaction. Libraries were purified using the QIAquick PCR purification kit (#28,104, Qiagen) or solid phase reverse immobilization beads (Vazyme VAHTS DNA Clean Beads) and sequenced with ~30 Mio reads, 150 bp paired-end by HiSeq Illumina sequencing (GENEWIZ).
Annotation of self-cleaving ribozyme candidates in Desulfovibrio vulgaris
A comprehensive annotation for known ribozyme classes in Desulfovibrio vulgaris [71] was generated by applying cmsearch from the infernal package [72] for a set of multiple-sequence alignments. These alignments were derived from Rfam [73,74] or provided by Zasha Weinberg (personal communication) [10,11,73]. After curation and filtering, a final set of predicted ribozyme loci was derived, which was used for downstream analysis of cyPhyRNA-seq data.
Analysis of reads created by amplicon sequencing
Spike-in experiments were analysed based on Amplicon sequencing (Amplicon-EZ, GENEWIZ, Supplementary Table S3). The general workflow applied here consists of following steps: Identification of UMIs, Trimming of barcodes, Mapping of reads and Counting. In detail, reads were preprocessed with UMItools [75] to identify and trim UMI sequences and mark them for downstream counting, followed by trimming of remaining adapters with TrimGalore [76]. An artificial genome was created by adding the spike-in ribozyme fragment sequences (Supplementary Table S2) to the E. coli genome retrieved from https://www.ncbi.nlm.nih.gov/genome/167?genome_assembly_id=753562 together with the corresponding annotation, which was also complemented by the spike-in ribozyme fragment IDs. This genome was indexed and reads were mapped with STAR [77]. Command-line calls for this workflow steps are listed in the Supplementary Text. Unique reads which map only at one position in the genome were extracted and counted with featureCounts [78] followed by manual inspection and deduplication for reads mapping to spiked-in ribozyme fragments.
Analysis of reads created by Illumina HiSeq sequencing
cyPhyRNA-seq with Desulfovibrio vulgaris total RNA on Illumina HiSeq generated ~ 30 M reads per library (Supplementary Table S4). Quality control of raw, trimmed and mapped cyPhyRNA-seq data was conducted with FastQC [79], followed by adapter and quality trimming with Trim Galore [76]. For UMI-based deduplication, trimmed reads were processed with UMI-tools [75] before and after mapping. Reads were mapped against the reference with STAR [77]. After deduplication, unique mapping reads were extracted, counted with featureCounts [78], underwent peak calling [80] and were intersected with our ribozyme annotation for further analysis. The complete workflow is illustrated in Supplementary Figure S13.
UCSC genome browser tracks
Tracks for the UCSC genome browser [81] were generated with in-house scripts following the UCSC manual [82]. Tracks depicting read coverage were normalized for easier comparison as follows: The number of reads covering each position is scaled to the total number of reads on the same strand divided by 106 (reads per million, RPM). Although this is per se only a per sample scaling, the fact that all samples show comparable sequencing depth allows the comparison of normalized values across samples.
Supplementary Material
Funding Statement
This research was supported by a grant of the German Research Foundation(DFG) to C.E.W. (Deutsche ForschungsgemeinschaftLU1889/4) . V.J.O. was supported by a Predoc Award from Leipzig University (jointly given to V.J.O. and C.E.W.) and C.E.W. is also supported by a fellowship of the Peter and Traudl Engelhorn Foundation. Research was also funded in part by the German Federal Ministry for Education and Research (BMBF Bundesministerium für Bildung und Forschung 031A538B, de.NBI/RBC) to P.F.S
Acknowledgments
We thank Elmar Wahle and Zasha Weinberg for scientific discussions and critical reading of this manuscript and Mario Mörl and his lab for scientific discussions, hosting and supporting the growing Weinberg lab. We also thank Sabine Kleinsteuber and Washington Logroño (Helmholtz Center for Environmental Research, Leipzig) for an introduction to anaerobic cultivation of bacteria, Richard Friedrich for cultivating D. vulgaris and Yuliia Lihanova for conducting co-transcriptional cleavage assays of D. vulgaris HHRs.
Disclosure statement
The authors declare that they have no competing interests.
Author’s contributions
V.J.O. designed and conducted experiments, analyzed experimental data, interpreted experimental and bioinformatics data, wrote the paper, C.G. and J.F. were responsible for sequencing data curation and computational analysis, P.F.S. supervised bioinformatics analysis, J.F. conceived and supervised computational analysis and interpreted sequencing data, C.E.W. conceived the project, designed and analyzed experimental data, interpreted experimental and bioinformatics data and wrote the paper with help from all authors.
Data availability
Raw data of cyPhyRNA-seq corresponding to HighSeq Illumina sequencing of D. vulgaris and to Amplicon sequencing from Ribozyme spike-in into E. coli are uploaded to NCBI:http://www.ncbi.nlm.nih.gov/bioproject/728517Link to genome browser for the D. vulgaris data:http://genome-euro.ucsc.edu/cgi-bin/hgTracks?db=hub_62990_DeVu&lastVirtModeType=default&lastVirtModeExtraState=&virtModeType=default&virtMode=0&nonVirtPosition=&position=NC_002937.3%3A1232979%2D1233047&hgsid=269157374_6xbbmG90Mma8tGAAAeBHirYI4aAy
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Guerrier-Takada C, Gardiner K, Marsh T, et al. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell. 1983;35(3):849–857. [DOI] [PubMed] [Google Scholar]
- [2].Peebles CL, Perlman PS, Mecklenburg KL, et al. A self-splicing RNA excises an intron lariat. Cell. 1986;44(2):213–223. [DOI] [PubMed] [Google Scholar]
- [3].Nissen P, Hansen J, Ban N, et al. The structural basis of ribosome activity in peptide bond synthesis. Science. 2000;289(5481):920–930. [DOI] [PubMed] [Google Scholar]
- [4].Jimenez RM, Polanco JA, Lupták A.. Chemistry and biology of self-cleaving ribozymes. Trends Biochem Sci. 2015;40(11):648–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Prody GA, Bakos JT, Buzayan JM, et al. Autolytic processing of dimeric plant virus satellite RNA. Science. 1986;231(4745):1577–1580. [DOI] [PubMed] [Google Scholar]
- [6].Buzayan JM, Gerlach WL, Bruening G.. Satellite tobacco ringspot virus RNA: a subset of the RNA sequence is sufficient for autolytic processing. Proc Natl Acad Sci U S A. 1986;83(23):8859–8862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wu HN, Lin YJ, Lin FP, et al. Human hepatitis delta virus RNA subfragments contain an autocleavage activity. Proc Natl Acad Sci U S A. 1989;86(6):1831–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Saville BJ, Collins RA. A site-specific self-cleavage reaction performed by a novel RNA in neurospora mitochondria. Cell. 1990;61(4):685–696. [DOI] [PubMed] [Google Scholar]
- [9].Winkler WC, Nahvi A, Roth A, et al. Control of gene expression by a natural metabolite-responsive ribozyme. Nature. 2004;428(6980):281–286. [DOI] [PubMed] [Google Scholar]
- [10].Roth A, Weinberg Z, Chen AGY, et al. A widespread self-cleaving ribozyme class is revealed by bioinformatics. Nat Chem Biol. 2014;10(1):56–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Weinberg Z, Kim PB, Chen TH, et al. New classes of self-cleaving ribozymes revealed by comparative genomics analysis. Nat Chem Biol. 2015;11(8):606–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chen Y, Qi F, and Gao F, et al. Hovlinc is a recently evolved class of ribozyme found in human lncRNA. Nat Chem Biol. 2021 [DOI] [PubMed] [Google Scholar]
- [13].Ferbeyre G, Smith JM, Cedergren R. Schistosome satellite DNA encodes active hammerhead ribozymes. Mol Cell Biol. 1998;18(7):3880–3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cervera A, De La Peña M. Eukaryotic penelope-like retroelements encode hammerhead ribozyme motifs. Mol Biol Evol. 2014;31(11):2941–2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Eickbush DG, Eickbush TH. R2 retrotransposons encode a self-cleaving ribozyme for processing from an rRNA cotranscript▿. Mol Cell Biol. 2010;30(13):3142–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bringaud F, Bartholomeu DC, Blandin G, et al. The trypanosoma cruzi L1Tc and NARTc non-LTR retrotransposons show relative site specificity for insertion. Mol Biol Evol. 2006;23(2):411–420. [DOI] [PubMed] [Google Scholar]
- [17].Salehi-Ashtiani K, Lupták A, Litovchick A, et al. A genomewide search for ribozymes reveals an HDV-like sequence in the human CPEB3 gene. Science. 2006;313(5794):1788–1792. [DOI] [PubMed] [Google Scholar]
- [18].Weinberg CE, Weinberg Z, Hammann C. Novel ribozymes: discovery, catalytic mechanisms, and the quest to understand biological function. Nucleic Acids Res. 2019;47(18):9480–9494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Weinberg Z, Lünse CE, Corbino KA, et al. Detection of 224 candidate structured RNAs by comparative analysis of specific subsets of intergenic regions. Nucleic Acids Res. 2017;45(18):10811–10823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wang Z, Gerstein M, Snyder M. RNA-seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kukurba KR, Montgomery SB. RNA sequencing and analysis. Cold Spring Harb Protoc. 2015;2015(11):951–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hrdlickova R, Toloue M, Tian B. RNA-seq methods for transcriptome analysis. Wiley Interdiscip Rev RNA. 2017;8(1):e1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hafner M, Landgraf P, Ludwig J, et al. Identification of microRNAs and other small regulatory RNAs using cDNA library sequencing. Methods. 2008;44(1):3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Englert M, Beier H. Plant tRNA ligases are multifunctional enzymes that have diverged in sequence and substrate specificity from RNA ligases of other phylogenetic origins. Nucleic Acids Res. 2005;33(1):388–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tanaka N, Shuman S. RtcB is the RNA ligase component of an Escherichia coli RNA repair operon. J Biol Chem. 2011;286(10):7727–7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Culver GM, McCraith SM, Consaul SA, et al. A 2ʹ-phosphotransferase implicated in tRNA splicing is essential in Saccharomyces cerevisiae. J Biol Chem. 1997;272(20):13203–13210. [DOI] [PubMed] [Google Scholar]
- [27].Tanaka N, Chakravarty AK, Maughan B, et al. Novel mechanism of RNA repair by RtcB via sequential 2ʹ,3ʹ-cyclic phosphodiesterase and 3ʹ-phosphate/5ʹ-hydroxyl ligation reactions. J Biol Chem. 2011;286(50):43134–43143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Schutz K, Hesselberth JR, Fields S. Capture and sequence analysis of RNAs with terminal 2ʹ,3ʹ-cyclic phosphates. RNA. 2010;16(3):621–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Peach SE, York K, Hesselberth JR. Global analysis of RNA cleavage by 5ʹ-hydroxyl RNA sequencing. Nucleic Acids Res. 2015;43(17):e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Head SR, Komori HK, LaMere SA, et al. Library construction for next-generation sequencing: overviews and challenges. BioTechniques. 2014;56(2):61–64. 66, 68, passim. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Pang YLJ, Abo R, Levine SS, et al. Diverse cell stresses induce unique patterns of tRNA up- and down-regulation: tRNA-seq for quantifying changes in tRNA copy number. Nucleic Acids Res. 2014;42(22):e170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Das U, Chakravarty AK, Remus BS, et al. Rewriting the rules for end joining via enzymatic splicing of DNA 3ʹ-PO4 and 5ʹ-OH ends. Proc Natl Acad Sci U S A. 2013;110(51):20437–20442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zheng L, Falschlunger C, Huang K, et al. Hatchet ribozyme structure and implications for cleavage mechanism. Proc Natl Acad Sci U S A. 2019;116(22):10783–10791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Riccitelli N, Lupták A. HDV family of self-cleaving ribozymes. Prog Mol Biol Transl Sci. 2013;120:123–171. [DOI] [PubMed] [Google Scholar]
- [35].Remus BS, Shuman S. A kinetic framework for tRNA ligase and enforcement of a 2′-phosphate requirement for ligation highlights the design logic of an RNA repair machine. RNA. 2013;19(5):659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Vušurović N, Altman RB, Terry DS, et al. Pseudoknot formation seeds the twister ribozyme cleavage reaction coordinate. J Am Chem Soc. 2017;139(24):8186–8193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gebetsberger J, Micura R. Unwinding the twister ribozyme: from structure to mechanism. Wiley Interdiscip Rev RNA. 2017;8(3):e1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Steiger MA, Kierzek R, Turner DH, et al. Substrate recognition by a yeast 2‘-Phosphotransferase involved in tRNA splicing and by its escherichia coli homolog. Biochemistry. 2001;40(46):14098–14105. [DOI] [PubMed] [Google Scholar]
- [39].Emilsson GM, Nakamura S, Roth A, et al. Ribozyme speed limits. RNA. 2003;9(8):907–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Shigematsu M, Kawamura T, Kirino Y. Generation of 2ʹ,3ʹ-cyclic phosphate-containing RNAs as a hidden layer of the transcriptome. Front Genet. 2018;9:562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Westermann AJ, Gorski SA, Vogel J. Dual RNA-seq of pathogen and host. Nat Rev Microbiol. 2012;10(9):618–630. [DOI] [PubMed] [Google Scholar]
- [42].O’Neil D, Glowatz H, and Schlumpberger M. Ribosomal RNA depletion for efficient use of RNA-seq capacity. Curr Protoc Mol Biol. 2013;Chapter 4:Unit4.19 [DOI] [PubMed] [Google Scholar]
- [43].Shigematsu M, Morichika K, Kawamura T, et al. Genome-wide identification of short 2ʹ,3ʹ-cyclic phosphate-containing RNAs and their regulation in aging. PLoS Genet. 2019;15(11):e1008469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Herbert ZT, Kershner JP, Butty VL, et al. Cross-site comparison of ribosomal depletion kits for Illumina RNAseq library construction. BMC Genomics. 2018;19(1):199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Prezza G, Heckel T, Dietrich S, et al. Improved bacterial RNA-seq by Cas9-based depletion of ribosomal RNA reads. RNA. 2020;26(8):1069–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lu C, Meyers BC, Green PJ. Construction of small RNA cDNA libraries for deep sequencing. Methods. 2007;43(2):110–117. [DOI] [PubMed] [Google Scholar]
- [47].Zhuang F, Fuchs RT, Robb GB. Small RNA expression profiling by high-throughput sequencing: implications of enzymatic manipulation. J Nucleic Acids. 2012;2012:360358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kraus AJ, Brink BG, Siegel TN. Efficient and specific oligo-based depletion of rRNA. Sci Rep. 2019;9(1):12281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Deana A, Celesnik H, Belasco JG. The bacterial enzyme RppH triggers messenger RNA degradation by 5ʹ pyrophosphate removal. Nature. 2008;451(7176):355–358. [DOI] [PubMed] [Google Scholar]
- [50].Stevens A. Purification and characterization of a Saccharomyces cerevisiae exoribonuclease which yields 5ʹ-mononucleotides by a 5ʹ leads to 3ʹ mode of hydrolysis. J Biol Chem. 1980;255(7):3080–3085. [PubMed] [Google Scholar]
- [51].Kawano M, Kawazu C, Lizio M, et al. Reduction of non-insert sequence reads by dimer eliminator LNA oligonucleotide for small RNA deep sequencing. BioTechniques. 2010;49(4):751–755. [DOI] [PubMed] [Google Scholar]
- [52].Erber L, Hoffmann A, Fallmann J, et al. LOTTE-seq (Long hairpin oligonucleotide based tRNA high-throughput sequencing): specific selection of tRNAs with 3ʹ-CCA end for high-throughput sequencing. RNA Biol. 2020;17(1):23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Perreault J, Weinberg Z, Roth A, et al. Identification of hammerhead ribozymes in all domains of life reveals novel structural variations. PLoS Comput Biol. 2011;7(5):e1002031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ule J, Jensen K, Mele A, et al. CLIP: a method for identifying protein-RNA interaction sites in living cells. Methods. 2005;37(4):376–386. [DOI] [PubMed] [Google Scholar]
- [55].Uren PJ, Bahrami-Samani E, Burns SC, et al. Site identification in high-throughput RNA-protein interaction data. Bioinformatics. 2012;28(23):3013–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Webb C-HT, Riccitelli NJ, Ruminski DJ, et al. Widespread occurrence of self-cleaving ribozymes. Science. 2009;326(5955):953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Lindell M, Romby P, Wagner EGH. Lead(II) as a probe for investigating RNA structure in vivo. RNA. 2002;8(4):534–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Twittenhoff C, Brandenburg VB, Righetti F, et al. Lead-seq: transcriptome-wide structure probing in vivo using lead(II) ions. Nucleic Acids Res. 2020;48(12):e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Luhtala N, Parker R. T2 family ribonucleases: ancient enzymes with diverse roles. Trends Biochem Sci. 2010;35(5):253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Schifano JM, Vvedenskaya IO, Knoblauch JG, et al. An RNA-seq method for defining endoribonuclease cleavage specificity identifies dual rRNA substrates for toxin MazF-mt3. Nat Commun. 2014;5(1):3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Thompson DM, Lu C, Green PJ, et al. tRNA cleavage is a conserved response to oxidative stress in eukaryotes. RNA. 2008;14(10):2095–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Sambrook J, Fritsch E, and Maniatis T. Molecular cloning: a laboratory manual: vol. 2. 2nd S.l. Newyork: Cold Spring Harbor; 1989. [Google Scholar]
- [63].Viollet S, Fuchs RT, Munafo DB, et al. T4 RNA ligase 2 truncated active site mutants: improved tools for RNA analysis. BMC Biotechnol. 2011;11(1):72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Seidl CI, Ryan K, Darlix J-L. Circular single-stranded synthetic DNA delivery vectors for microRNA. PLoS ONE. 2011;6(2):e16925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ling ML, Risman SS, Klement JF, et al. Abortive initiation by bacteriophage T3 and T7 RNA polymerases under conditions of limiting substrate. Nucleic Acids Res. 1989;17(4):1605–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Lihanova Y, and Weinberg CE. Biochemical analysis of cleavage and ligation activities of the pistol ribozyme from Paenibacillus polymyxa. RNA Biol. 2021;1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Lama L, Ryan K. Adenylylation of small RNA sequencing adapters using the TS2126 RNA ligase I. RNA. 2016;22(1):155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Sambrook J, and Russell DW. Purification of nucleic acids by extraction with phenol:chloroform. CSH Protoc. 2006. [DOI] [PubMed] [Google Scholar]
- [69].Lorenz C. Analysen zur Temperaturabhängigkeit posttranskriptioneller Modifikationen in bakteriellen tRNAs. Leipzig University; 2020. [Google Scholar]
- [70].Kivioja T, Vähärautio A, Karlsson K, et al. Counting absolute numbers of molecules using unique molecular identifiers. Nat Methods. 2011;9(1):72–74. [DOI] [PubMed] [Google Scholar]
- [71].NCBI . Dsm644 annotation, asm19575v1. [cited 2019 oct 10]. https://www.ncbi.nlm.nih.gov/assembly/GCF_000195755.1/_ASM19575v1
- [72].Nawrocki EP, Eddy SR. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013;29(22):2933–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Kalvari I, Nawrocki EP, Ontiveros-Palacios N, et al. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 2021;49(D1):D192–D200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Rfam . Hammerhead ribozyme type 2, cm: [cited 2019 Oct 10]. http://rfam.xfam.org/family/RF02276/cm.
- [75].Smith T, Heger A, Sudbery I. UMI-tools: modeling sequencing errors in unique molecular identifiers to improve quantification accuracy. Genome Res. 2017;27(3):491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Felix Krueger . TrimGalore. [cited 2020 Sep 9]. https://github.com/FelixKrueger/TrimGalore
- [77].Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–930. [DOI] [PubMed] [Google Scholar]
- [79].Andrews S. Fastqc. [cited 2020 Apr 5]. https://github.com/s-andrews/FastQC.
- [80].The Smith Lab . Piranha — CLIP- and RIP-seq peak caller | The smith lab. [cited 2020 Mar 15]. http://smithlabresearch.org/software/piranha/.
- [81].Navarro Gonzalez J, Zweig AS, Speir ML, et al. The UCSC genome browser database: 2021 update. Nucleic Acids Res. 2021;49(D1):D1046–D1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Zweig AS, Karolchik D, Kuhn RM, et al. UCSC genome browser tutorial. Genomics. 2008;92(2):75–84. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data of cyPhyRNA-seq corresponding to HighSeq Illumina sequencing of D. vulgaris and to Amplicon sequencing from Ribozyme spike-in into E. coli are uploaded to NCBI:http://www.ncbi.nlm.nih.gov/bioproject/728517Link to genome browser for the D. vulgaris data:http://genome-euro.ucsc.edu/cgi-bin/hgTracks?db=hub_62990_DeVu&lastVirtModeType=default&lastVirtModeExtraState=&virtModeType=default&virtMode=0&nonVirtPosition=&position=NC_002937.3%3A1232979%2D1233047&hgsid=269157374_6xbbmG90Mma8tGAAAeBHirYI4aAy