

ABSTRACT
In plants, recent studies have revealed that N6-methyladenosine (m6A) methylation of mRNA has potential regulatory functions of this mRNA modification in many biological processes. m6A methyltransferase, m6A demethylase and m6A-binding proteins can cause differential phenotypes, indicating that m6A may have critical roles in the plant. In this study, we depicted the m6A map of sea buckthorn (Hippophae rhamnoides Linn.) transcriptome. Similar to A. thaliana, m6A sites of sea buckthorn transcriptome is significantly enriched around the stop codon and within 3ʹ-untranslated regions (3ʹUTR). Gene ontology analysis shows that the m6A modification genes are associated with metabolic biosynthesis. In addition, we identified 13,287 different m6A peaks (DMPs) between leaf under drought (TR) and control (CK) treatment. It reveals that m6A has a high level of conservation and has a positive correlation with mRNA abundance in plants. GO and KEGG enrichment results showed that DMP modification DEGs in TR were particularly associated with ABA biosynthesis. Interestingly, our results showed three m6A demethylase (HrALKBH10B, HrALKBH10C and HrALKBH10D) genes were significantly increased following drought stress, which indicated that it may contributed the decreased m6A levels. This exhaustive m6A map provides a basis and resource for the further functional study of mRNA m6A modification in abiotic stress.
KEYWORDS: Sea buckthorn, N6-methyladenosine, drought stress, m6A modification, m6A demethylase
Introduction
As a rising novel epigenetic modification, N6-methyladenosine (m6A) of mRNA has been widely studied in mammals and plants [1–3]. As one component of a methyltransferase complex, N6-adenosine methyltransferase like 3 (METTL3) were considered as a methyltransferase of m6A, which identified in Saccharomyces cerevisiae, Drosophila melanogaster, Homo sapiens and Arabidopsis thaliana [4–7]. Expect, methyltransferase complex also contained methyltransferase-like 14 (METTL14) and Wilms tumour 1-associated protein (WTAP), which combined with METTL3 to catalyse m6A RNA methylation in mammals [8,9]. All these N6-adenosine methyltransferases can affect organism growth and development, including embryo, shoot, root, leaf and floral [2]. On the contrary, past researches revealed that FTO and ALKBH5 were confirmed as two m6A RNA demethylases in mammalian development [10]. Then, thirteen m6A demethylases (ALKBH family) are also studied in Arabidopsis [11,12]. For example, AtALKBH9B can remove m6A modification of single-stranded RNA in vitro. In addition, several RNA-binding proteins (RBPs), which play a role in recognizing m6A marks, have been identified in animals and Arabidopsis. All these findings reveal the reversible RNA modification can regulate functions of its target mRNA.
In recent researches, transcriptome-wide m6A methylation have been identified in Arabidopsis, rice and maize [3,13–15]. For instance, the m6A content of mRNA varies across different tissues in Arabidopsis. As an N6-adenosine methyltransferase, MTA is important for shoot, root, leaf and floral development of Arabidopsis [2]. An m6A eraser protein (ALKBH10B) can influence floral transition by regulating transcript levels of SPL3, SPL9 and FT [11]. Then, m6A reader proteins (ECT2, ECT3 and ECT4) can also regulate plant organogenesis [16]. With the development of m6A-seq, m6A modification was found in many species in response to environmental stress [17–21]. For example, the m6A can enhance the salt tolerance of Arabidopsis by regulating the stability of salt-tolerant transcripts [22]. Similarly, m6A-seq reveal that N6-methyladenosine can also regulate the salt tolerance of sweet sorghum [23]. In response to heat stress, the reader ECT2 can relocate to stress granules in the cell of Arabidopsis [24]. As a grave global ecological problem, drought stress has a great impact on plant development. In Arabidopsis, levels of m6A methyltransferase, m6A demethylase and m6A-binding proteins were not significantly modulated by drought stresses. However, drought stresses can up-regulate the expression level of m6A demethylase in maize [19]. It implied the different roles of mA in different species in the response to drought stress.
As a deciduous shrub, sea buckthorn (Hippophae rhamnoides Linn.) are widely grown and cultivated in Asia and Europe [25–27]. Now, sea buckthorn has been widely used to protect the environment because of its light-tolerant and drought-tolerant properties. The physiological responses of sea buckthorn seedlings under drought stresses have been studies on previous researches, which showed that epigenetic modification and post-translational modification may play important roles in sea buckthorn to drought stress [28]. In this study, to further explore the roles of mA in plants response to drought stress, we show transcriptome wide mA profiling in sea buckthorn seedlings and leaf under CK and TR condition. We found that mA is significantly enriched around the stop codon region and within 3ʹUTRs in sea buckthorn, which is similar to those in A. thaliana. m6A deposition and mRNA levels have a positive correlation, which indicates a regulatory role of m6A in plant gene expression. In general, as compared with the normal sample, we detected 13,287 DMPs within mRNAs. Intriguingly, the DEGs harbouring DMPs were involved in many important biological pathways associated with photosynthesis.
Results
m6A methylation is abundant in sea buckthorn mRNA
We used the m6A-targeted antibody and performed the MeRIP-seq to obtain the transcriptome-wide m6A map of the whole H. rhamnoides plants. We obtained almost 30 million reads per library (input and IP libraries). After quality control of raw data, 90% clean reads were uniquely mapped into the sea buckthorn reference genome. exomePeak2 software package was used to identify the m6A peaks between the IP and control samples. We totally detected 18,102 m6A enriched peaks in sea buckthorn leaf samples, which have an average length of 331 bp. The minimum length of peaks was 50 bp. Based on these results, we found that the H. rhamnoides transcriptome contains 0.7 m6A peaks per 1,000 nucleotides, which are similar to those obtained in mammals. And, most genes contain one or two m6A peaks (Figure 1(a)).
Figure 1.
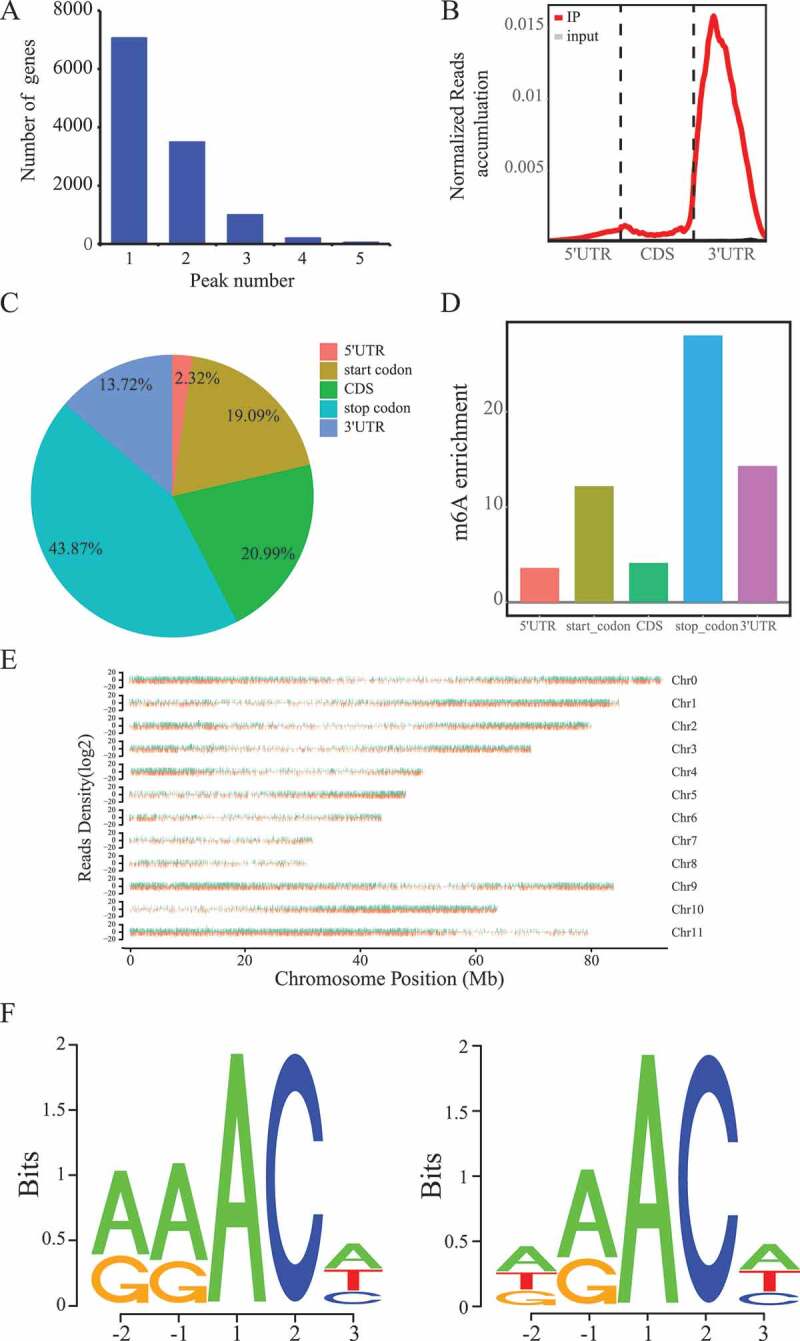
Overview of m6A methylome in sea buckthorn. (a) Number of genes with different m6A peak numbers. (b) Accumulation of m6A-IP and input reads of transcripts. Each transcript contained three parts: 5ʹ UTRs, CDS and 3ʹ UTRs. (c) The m6A peak distribution in different genic regions. (d) The m6A peak distribution along a metagene. Enrichment scores are calculated as (n/N)/p; n, number of peaks belonging to each category; N, number of total peaks; p, proportion of each category within the genome by length. (e) Distribution of m6A peaks across sea buckthorn chromosomes. (f) The RRACH and DRACH conserved sequence motifs of m6A peak regions
We investigated the m6A peaks distribution according to gene annotations. Among them, most reads of m6A peaks were localized around 3ʹUTRs (Figure 1(b)). This distribution tread was similar to those in Arabidopsis. The result of the locations of m6A on transcripts showed that m6A peaks are abundant around the stop codon (43.87%), followed by the coding regions (20.99%), start codon (19.09%) and then 3ʹ UTRs (13.72%), which consistent with the distribution of reads (Figure 1(c)). Then, we also observed that m6A peaks is significantly enriched around the stop codon (enrichment score > 25) by segment normalization of each gene portion (Figure 1(d)). Whole-genome density of m6A modification peaks revealed that m6A peaks have different distribution modes across chromosomes (Figure 1(e)). Similarly, m6A peaks have a higher density at the telomeric ends compared with other regions. Further analysis showed that the average density of chromosome 9 was the highest whereas chromosome 8 had the lowest average density (Figure 1€). These distribution modes among chromosomes indicated that m6A modification may have a relationship with chromatin state. Based on the analysis of m6A consensus sequence, we found that 15,387 (94.39%) m6A peaks have at least one RRACH motif in the CK group and 10,873 (94.15%) m6A peaks have at least one RRACH motif in the TR group (Fig. 1F). The same sequence motif, which is observed in eukaryotic mRNA, may be essential for the formation of m6A modification.
Characterization of mRNAs containing m6A modification
Past researches revealed that m6A is critical for plant development. To further analyse the functional roles of m6A modification in H. rhamnoides, we performed gene ontology (GO) enrichment analysis of genes containing m6A modification using the enrichment tool at HRIA. We found that most of these genes are involved in metabolic biosynthesis (Figure 2(a)). To determine whether the m6A distribution patterns of genes have a relationship with its GO categories, we divided genes into three subgroups (PeakStart: m6A peaks around start codon, PeakStop: m6A peaks around stop codon, PeakBoth: m6A peaks around both start and stop codons) based on the distribution of mA peaks. GO enrichment analysis of each subgroup showed that mA-containing genes in PeakStart and PeakBoth didn’t have enrichment terms. But, mA-containing genes of PeakStop exhibit high enrichment of metabolic process, signal transduction and RNA binding (Figure 2(b)). Certainly, we identified a series of metabolic-related genes carrying mA peaks. For instance, Chalcone Synthase (HrCHS), Chalcone isomerase (HrCHI) and flavanone 3-hydroxylase (HrF3H) contained mA peaks around stop codon, which plays important roles in flavonoid biosynthesis (Figure 2(c)). Phytoene synthase (HrPSY), which also contained mA peaks at stop codon, play an important role in phytoene biosynthesis. The results suggest that mA mRNA methylation may regulate metabolites biosynthesis in plants.
Figure 2.
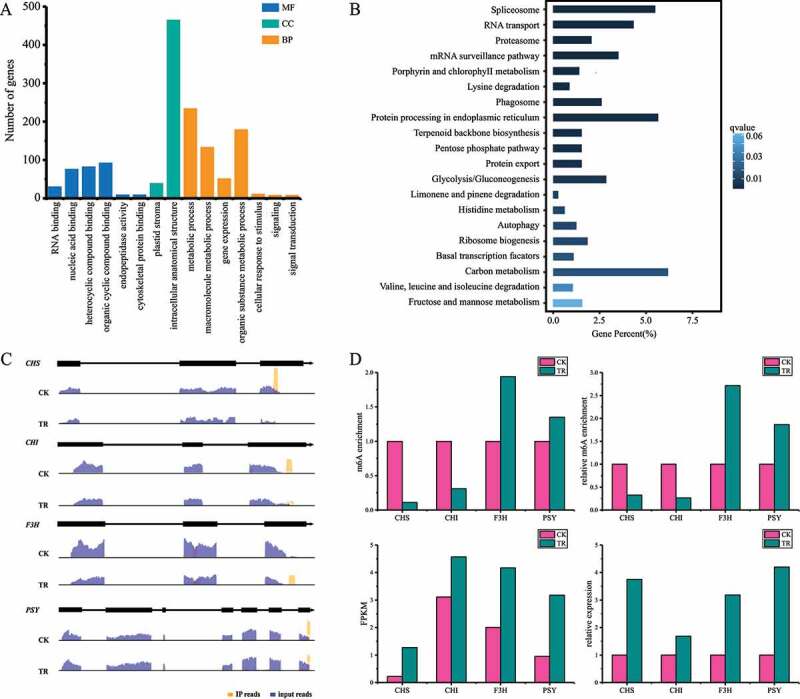
Enrichment analysis of genes with m6A peaks. (a) GO enrichment of genes with m6A peaks. MF: Molecular Function, CC: Cellular Component, BP: Biological Process. (b) KEGG enrichment of genes with m6A peaks. (c) Examples of genes with m6A peaks at stop codon
m6A methylation level is decreased in sea buckthorn under drought stress
To determine the change of m6A methylation in sea buckthorn response to drought stress, we profiled the m6A methylomes of the sea buckthorn leaf under CK and TR treatments. We performed the comparison of m6A profiles of CK and TR and identified 16,302 and 11,549 m6A differential methylated peaks (DMPs) in CK and TR, revealed a significant decrease in TR compared to CK. The Venn diagram identifies 7,496 common m6A peaks both in CK and TR (Figure 3(a)). Different analysis showed that 13,212 m6A methylation peaks were considered as DMP, including 6609 up-regulated DMS and 6603 down-regulated sites (Figure 3(b)). Then, we detected 9,596 transcripts that have differentially m6A methylation peaks (DMPs) in TR compared to CK. In detail, 6609 up-regulated DMPs contained 2174 up-regulated genes, 2932 down-regulated genes and 96 unaltered genes. 6603 up-regulated DMP contained 2106 up-regulated genes, 3077 down-regulated genes and 100 unaltered genes. More than 40% DMPs are located in stop codon regions (Figure 3(c)). Further analysis of the whole transcriptome of CK and TR, we found that m6A modification can certainly affect genes expression levels (Figure 3(d)). Therefore, we performed gene ontology (GO) enrichment analysis of genes containing DMPs to explore functional characteristics of DMPs in the context of genic location. The GO enrichment analysis of up-regulated DMP contained genes showed that the biological processes of blue light signalling pathway (GO:0009785), electron transport chain (GO:0022900) and photosynthesis, light reaction (GO:0019684) were significantly enriched (Figure 3(c)). The GO enrichment analysis of up-regulated DMS contained genes showed that the cellular component of the cell wall (GO:0005618) was significantly enriched. But, the GO enrichment analysis of down-regulated DMPs contained genes showed that the biological processes of protein ubiquitination (GO:0016567), nuclear-transcribed mRNA catabolic process (GO:0000956), cell wall modification (GO:0042545) were significantly enriched (Figure 3(e)). Otherwise, KEGG analysis reveal that up- and down-regulated DMPs contained genes were enriched metabolic pathway (Figure 3(f)).
Figure 3.
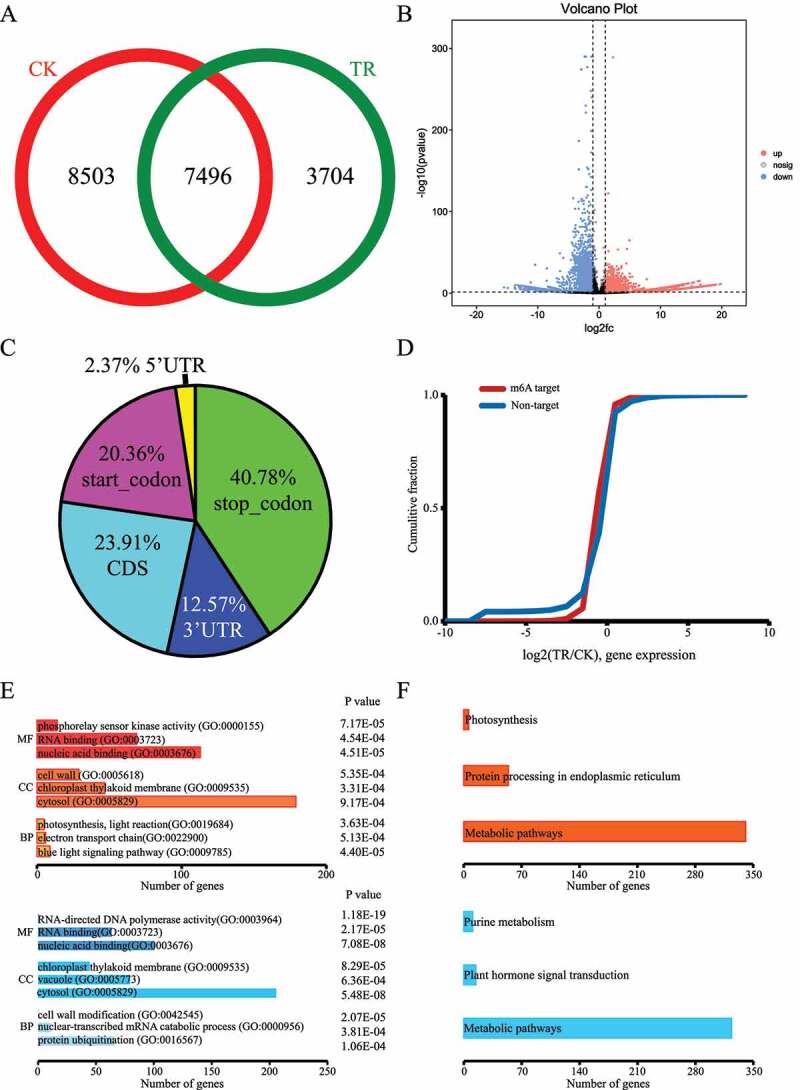
Overview of m6A methylation of sea buckthorn leaf under CK (control) and TR (drought) treatment. (a) Numbers of group-common and group-specific m6A peaks between CK and TR. (b) volcano analysis of differentially m6A methylation peaks. (c) Distribution of differentially m6A methylation peaks across the mRNAs. (d) Cumulative distribution of mRNA expression changes between CK and TR for m6A-modified genes and non-target genes. (e) Gene ontology enrichment terms for the genes with DMPs between CK and TR. (f) KEGG enrichment pathways of the genes with DMPs between CK and TR
The m6A pathway modulates genes responses to drought stress
To explore the roles of m6A methylation in sea buckthorn response to drought stress, we profiled the modification between normal and drought stress treatment leaf samples. We performed co-differential analysis to further associate m6A modification and gene expression. Finally, 541 DMGs were identified as significant co-differential genes with DMPs, including 228 up-regulated genes and 313 down-regulated genes (Figure 4), Supplementary Tables 1 and 2). Previous studies also showed that m6A modification has a great change during plant development and under abiotic stress. Furthermore, GO and KEGG analysis research revealed that up-regulated genes were significantly enriched in abscisic acid metabolic process, response to ethylene, fatty acid metabolic process, negative regulation of abscisic acid-activated signalling pathway (Supplementary Table 3). For example, CRTISO (Sph_LG3G002144) plays an important role in response to drought stress by affecting ABA biosynthesis in Arabidopsis. In addition, CYP707A2 (Sph_LG2G000371) encode ABA 8′-hydroxylases have hyper- and hypo- m6A peaks in TR samples, which decrease the ABA levels in Arabidopsis (Figure 4). The methylation levels of m6A peaks of CRTISO mRNA have a two-fold increment in TR samples compared to CK samples. However, the transcripts level of CRTISO in TR samples was higher than in CK samples. RMA1H1 (Sph_LG9G000969) was hypermethylated in TR, which can be induced by dehydration and conferred strongly enhanced tolerance to drought stress in Arabidopsis plants. Together, all these results indicate that m6A methylation can regulate the expression level of ABA-related genes to candidate the response to drought stress in sea buckthorn.
Figure 4.
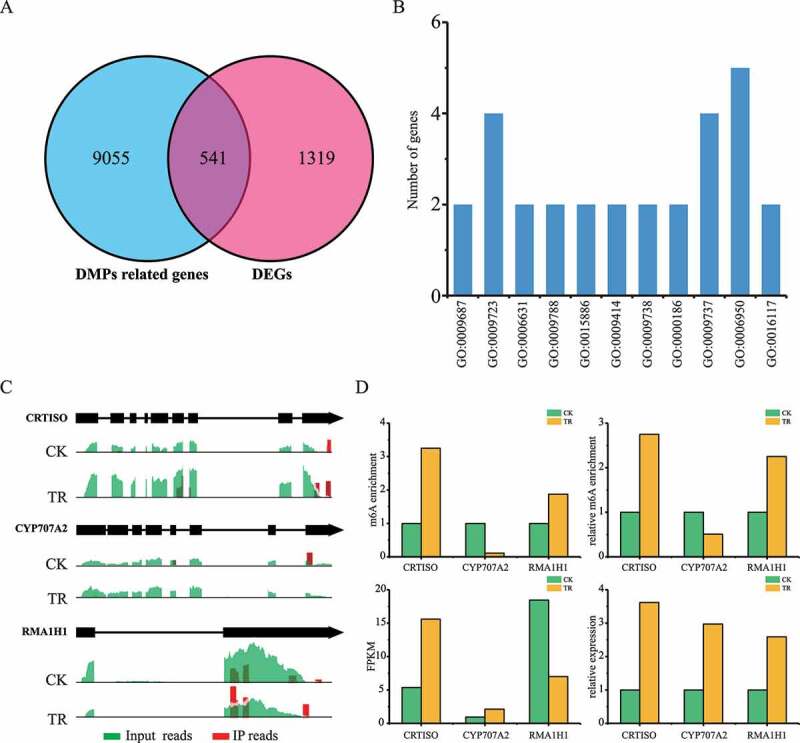
Analysis of DMP-related gene and DEGs between sea buckthorn leaf under CK (control) and TR (drought) treatment. DEGs: differentially expression genes, DMP: differentially m6A methylation peaks. (a) Venn map of DMP related gene and DEGs. (b) Gene ontology enrichment terms of the DEGs with DMPs. (c) Examples of DEGs with DMPs at stop codon. (d) Expression level of DEGs with DMPs at stop codon
The m6A pathway modulates proteins responses to drought stress
In addition, we determined whether m6A affects the protein level in the drought response process by characterizing the m6A methylome and proteome in CK and TR groups. First, we selected 9,596 genes with DMPs, which didn’t have change at transcripts level during drought stress. Next, we focused on whether the m6A level of selected 9,596 genes is associated with its protein levels (Supplementary Table 4). Using the proteomic data that MALDI‐TOF‐TOF to deeply profile the leaf proteome under control and drought stress, we found that the m6A level and protein expression level of some genes have a correlation between TR and CK (Supplementary Table 5). For example, we observed a decrease in m6A methylation in the 3′ UTR and 5′ UTR of the Photosystem II Subunit (PsbS), Oxygen-evolving enhancer protein (PSBO), ribulose bisphosphate carboxylase/oxygenase activase (RCA1) are which correlated with a decreased protein level in TR, but no significant change at mRNA levels. Contrarily, aminomethyltransferase (AMT) has increased m6A methylation level in its mRNA and increased protein level in TR compared with CK. All these results suggested m6A modification may regulated protein levels of key genes to response to drought stress in plants.
ALKBH10B may contribute to the decreased m6A methylation level in TR samples
Genome-wide m6A methyltransferases, demethylases and binding proteins in sea buckthorn were identified and constructed a phylogenetic tree using ML methods (Figure 5(a)). And, chromosome distribution analysis showed these genes were widely distributed in 12 chromosomes (Figure 5(b)). Based on the RNA-seq data, we quantify the expression abundance of m6A related genes in the leaves of sea buckthorn under control, drought stress and rehydration treatment. Interestingly, three ALKBH10B homologs (HrALKBH10B, HrALKBH10C and HrALKBH10D) were significantly upregulated by drought stress and downregulated by rehydration (Figure 5(c)). Meanwhile, the m6A abundance in samples under drought stress was significantly lower than that in samples under control treatment, suggesting global m6A hypomethylation induced by drought stress. According to the expression level of m6A demethylase genes (Figure 5(d)), we considered that the m6A demethylase genes most likely induced decreased m6A methylation during drought stress in sea buckthorn.
Figure 5.
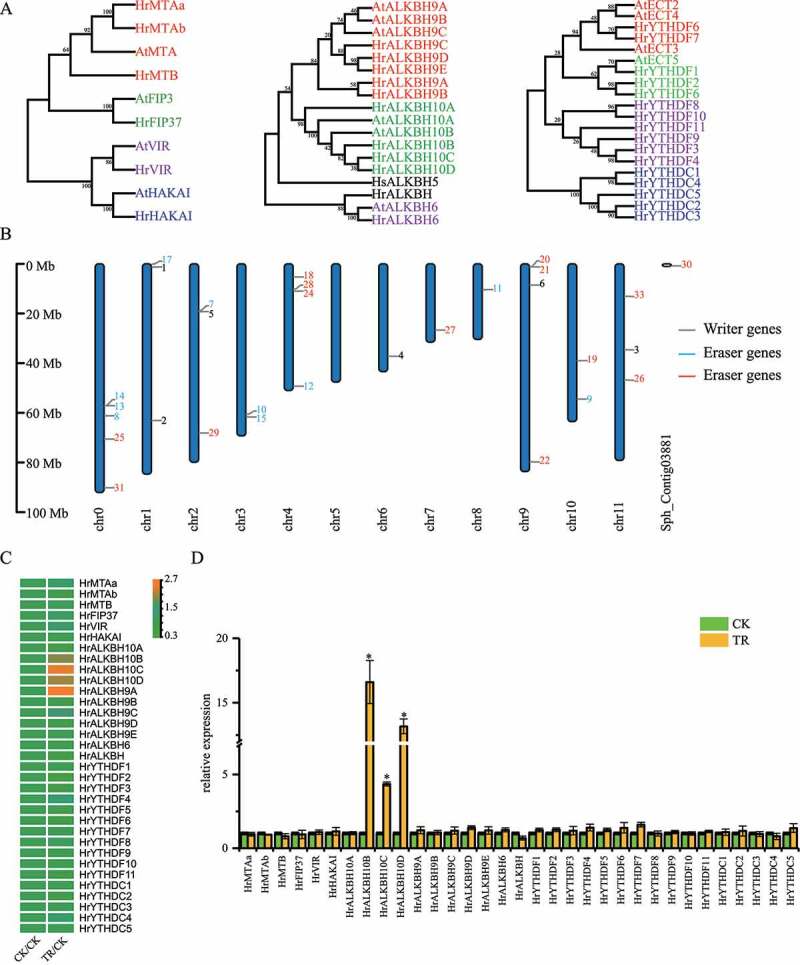
Expression analyses of sea buckthorn m6A methyltransferases, demethylases and binding proteins. (a) Phylogenetic analysis of sea buckthorn m6A RNA methyltransferases (left), demethylases (middle) and binding proteins (right). (b) Chromosome distribution of m6A methyltransferases, demethylases and binding proteins genes. (c) Expression level of sea buckthorn m6A methyltransferases, demethylases and binding proteins genes in leaf under CK, TR and RE. Arabidopsis genes (AtMTA, AtFIP3, AtVIR, AtHAKAI, AtALKBH9A, AtALKBH9B, AtALKBH9C, AtALKBH10A, AtALKBH10B, AtALKBH6, AtECT2, AtECT3, AtECT4, AtECT5) were used as a comparison. (d) Relative mRNA levels of sea buckthorn m6A methyltransferases, demethylases and binding proteins genes in leaf under CK and TR
Discussion
Recently, several research studies have illustrated the changes and roles of m6A methylation in the eukaryotic, including mice, human, Arabidopsis, rice, maize and tomato [3,14,19,29–33]. Since the finding of methyltransferase and demethylase proteins, m6A was considered as a dynamic and reversible RNA modification [34,35]. For example, m6A methyltransferases play an important role in plant development and regulate shoot stem cell fate of Arabidopsis [16]. It is been shown that m6A modification can dynamically regulate cellular responses to heat shock, ultraviolet light and oxidative stress in mammal [36–38]. However, the study about m6A modification in plant under drought stress was lacking. In this research, we used the sea buckthorn to present the dynamic m6A modification in response to drought stress. We found that m6A is of high abundance in leaf tissue of sea buckthorn. This resource indicated that mA modification may play a functional role in sea buckthorn leaf.
In these sea buckthorn leaf mA-seq data, we discovered that the features of the mA distribution in sea buckthorn are similar to those in Arabidopsis 3. m6A in sea buckthorn transcriptome is significantly enriched at stop codon. These results indicate that distribution features of the m6A were conserved in plants. In mammals, the RRACH motif is the most common motif of m6A methylation peaks, which has also been found in Arabidopsis and maize [3,15,19]. A recent study about motif analysis of all m6A modification peaks of sea buckthorn showed that most peaks have high enrichment in canonical RRACH motif and DRACH motif. These are common between sea buckthorn and Arabidopsis likely indicated that the conservative of these two m6A methylation motifs between two species, which can be clearly clarified by further functional analysis of m6A-related proteins. m6A modification normally is enriched stop codons and 3′ UTR to impact mRNA expression level. According to the distribution of m6A modification peaks, we found some genes with m6A peaks around their stop codon were involved in the metabolic process, including flavonoid and porphyrin and chloroghyII.
A recent study revealed that m6A modification may candidate plant response to abiotic stress. Here, we found a great change in m6A modification of sea buckthorn leaf under drought stress, indicating that m6A may be considered as an important role in drought responses. Importantly, transcripts with m6A modification have a great change compared with transcripts with non-mA modification, which showed that m6A modification can regulate transcripts level during drought stress. This result has also been found in two maize strains (B73 and Han21) [19]. Furthermore, enrichment analysis revealed that DEGs with m6A methylation was associated with some important biological processes, implying the role of m6A modification in regulating expression of gene-related drought response. As an important gene involved in ABA biosynthesis pathway, a homolog of CRTISO (Sph_LG3G002144) were hypermethylated in leaf under drought stress, which plays an important role in Arabidopsis under drought stress [39]. Compared with leaf under control, CYP707A2 (Sph_LG2G000371) have hyper- and hypo-m6A peaks in TR samples, which can decrease the ABA levels in Arabidopsis [40]. All these results indicated that m6A may contribute to drought response by ABA biosynthesis in sea buckthorn leaf. In addition, RMA1H1 (Sph_LG9G000969) were increased and gained m6A peaks in TR. Overexpression RMA1H1 conferred strongly enhanced tolerance to drought stress in transgenic Arabidopsis plants [41]. So, m6A modification may regulate the expression of key genes to enhance the drought stress tolerance in sea buckthorn.
Drought response is governed by genetic and epigenetic regulation in the plant. In this study, we firstly contrasted a transcriptome-wide m6A modification map of sea buckthorn and showed the role of m6A modification in response to drought stress. These results provide resources for further uncovering m6A modification functions in abiotic stress response and plant metabolism.
Materials and methods
Plant material
Plant growth and analyses generally followed the procedures described in past research [42]. Two-year-old sea buckthorn plants were cultured in a greenhouse with controlled light, temperature and humidity conditions (16 h dark, 8 h light; 25°C under day, 20°C under dark, 60–70% relative humidity) for three months. Then, sea buckthorns plants were treated with water deficit conditions for 10d (25–30% field capacity). The control group was treated with 75–85% field capacity by watering (CK). The leaf, stem and root tissue were collected and mixed as seedlings sample. And, leaf under control (CK) and water deficit condition (TR) were collected and immediately frozen in liquid nitrogen.
RNA extraction and library construction
We extracted total RNA from sea buckthorn seedlings sample, leaf under control condition (CK), leaf under water deficit condition (TR) using Trizol (Invitrogen) following a published protocol. Then, we tested the quality of extracted total RNA by Nanodrop and gel electrophoresis. Polyadenylated RNA was extracted and fragmented to 200nt using RNA Fragmentation Reagents (Ambion). Fragmented RNA and m6A antibody were incubated in IP buffer for 3 h at 4°C. We eluted the mixture with elution buffer and treated the eluted RNA with RNasin following the manufacturer’s instructions. According to published protocol, we constructed immunoprecipitated RNA and input RNA library using mRNA Sample Prep Kit (Illumina) for each sample. Thus, we performed m6A-seq and input RNA-seq with NovaSeq 6000 by paired-end strategy.
Analysis of m6A-seq
According to the past research [20,33,43], we analysed sequence data using bioinformatic software. Firstly, we used FastQC to remove the adapter and low-quality data (Q < 25) of raw data from IP RNA-seq and input RNA-seq. Using HISAT2 software [44], we mapped the clean data from the input and IP-sequenced samples to the sea buckthorn reference genome (http://hipp.shengxin.ren/). Based on the background information (input RNA-seq data), we used R package (exomePeak2) to identify m6A peaks (P-value < 1e-5). Following past researches [15,20], we estimated the distribution of m6A in the different parts of the gene and transcripts. The conserved sequences of identified m6A peaks were found by MEME Suite (MEME-chip) and HOMER [45]. The de novo motifs were uncovered by MEME-chip software with setting the motif length (3–8bp and 8–15bp). Then we used Homer software to establish the enrichment significance of RRACH and DRACH in sequences of m6A peaks. We used methods in past research to study the distribution pattern of the m6A peaks [20,23]. PeakAnnotator (version 2.0) was performed to annotate m6A peaks to the sea buckthorn annotation file [46]. The m6A site differential algorithm was used for the identification of differentially methylated peaks (DMPs) between the CK and TR sample with a criterion of P-value < 0.05 and fold change ≥ 2.
Analysis of RNA-seq data
Previous studies reveal that the input sequencing reads in the m6A-seq can be used for RNA-seq analysis. Briefly, tophat2 and cufflinks were used for the reads mapping and assembly of each sample. We calculated the fragments per kilobase of exon per million mapped fragments (FPKM) as Gene expression level using Cuffdiff [47]. The FPKM were used to identify differentially expressed genes (DEGs) with a cut-off criterion (log2FC ≥ 1 and P-value < 0.05). The P values of each DEG were also adjusted to control the false discovery rate (FDR) by Benjamini and Hochberg’s approach. The Gene Ontology (GO) and KEGG pathway enrichment analysis of m6A modified genes and DEGs with DMPs was performed based on the tool in HRIA. The significance values of GO and KEGG pathways with P-value < 0.05 were considered significantly enrichment terms.
Identification, phylogenetic and expression analysis of the m6A methyltransferases, demethylases and binding proteins genes in sea buckthorn
Based on the sequence of the mA methyltransferases, demethylases and binding proteins, we identified their homologs in sea buckthorn by BLAST homology searches. The reference genome sequence and annotations of sea buckthorn were downloaded from the Hippophae rhamnoides Information Archive (http://hipp.shengxin.ren). MEGA 7.0 were used for constructing a maximum likelihood phylogenetic tree of m6A methyltransferases, demethylases and binding proteins with 1000 bootstrap replicates. The expression level of all these genes in CK, TR and RE groups was calculated.
m6A-IP-qPCR
According to past research [33], we performed m6A-IP-qPCR for selected genes. Briefly, RNA fragmentation buffer (10 mM Tris-HCl, pH 7.0 and 10 mM ZnCl2) was used to fragment intact mRNAs into ~ 300 nucleotide-long fragments by incubation at 95°C for 25 s. We purified fragmented mRNAs using standard ethanol precipitation and resuspended with 250 μL of DEPC-treated water. Then, 50 μL of fragmented mRNAs and 2.5 μg of anti-m6A polyclonal antibody (Synaptic Systems) were incubated at 4°C for 2 h in IP buffer. 5 μL of fragmented mRNAs was taken as the input control. The mixture and 10 μL of Dynabeads protein-A (Invitrogen) were subsequently incubated at 4°C for 2.5 h. After washing with high-salt buffer, the m6A-containing fragments and 6.5 mM N6-methyladenosine were incubated in 100 μL of IP buffer at 4°C for 2 h. Then, both the m6A-containing mRNAs and the input mRNAs were submitted to quantitative RT-PCR. The primers were listed in Supplementary Table 6. The value for the immunoprecipitated sample was normalized against that for the input. Relative m6A enrichment in a specific region of a transcript was calculated using the 2−ΔΔCt method. The experiment was performed with three biological replicates.
Measurement of gene expression levels using real-time qPCR
We used the plant RNA extraction kit (Tiangen, DP441) to extract total RNAs from the sea buckthorn leaves. The extracted RNAs were used for reverse transcription reaction by the PrimeScript™ RT reagent Kit (Takara, RR036A). The synthesized cDNAs were then employed as templates for PCR amplification using the TB Green Fast qPCR Mix (RR430S) and StepOne Plus Real-Time PCR System (Applied Biosystems) in the following program: 94°C for 10 min, followed by 40 cycles of 94°C for 15 s and 60°C for 30 s. Transcription levels of selected genes were quantified by the cycle threshold 2−ΔΔCt method. Sea buckthorn 18S rRNA gene was applied to normalize the expression values. The primers for PCR amplification are listed in Supplementary Table 6. The experiment was conducted with three biological replicates.
Supplementary Material
Acknowledgments
This work was supported by the Fundamental Research Funds of CAF (CAFYBB2020SY006) and the Joint Funds of the National Natural Science Foundation of China (U2003116).
Funding Statement
This work was supported by the Fundamental Research Funds of CAF [CAFYBB2020SY006]; the Joint Funds of the National Natural Science Foundation of China [U2003116].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–206. [DOI] [PubMed] [Google Scholar]
- [2].Yue H, Nie X, Yan Z, et al. N6-methyladenosine regulatory machinery in plants: composition, function and evolution. Plant Biotechnol J. 2019;17(7):1194–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Luo GZ, MacQueen A, Zheng G, et al. Unique features of the m6A methylome in Arabidopsis thaliana. Nat Commun. 2014;5(1):5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Barbieri I, Tzelepis K, Pandolfini L, et al. Promoter-bound METTL3 maintains myeloid leukaemia by m(6) A-dependent translation control. Nature. 2017;552(7683):126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Xia H, Zhong C, Wu X, et al. Mettl3 mutation disrupts gamete maturation and reduces fertility in Zebrafish. Genetics. 2018;208(2):729–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Liu S, Zhuo L, Pan T, et al. METTL3 plays multiple functions in biological processes. Am J Cancer Res. 2020;10:1631–1646. [PMC free article] [PubMed] [Google Scholar]
- [7].Shen L, Liang Z, Gu X, et al. N(6)-methyladenosine RNA modification regulates shoot stem cell fate in arabidopsis. Dev Cell. 2016;38(2):186–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Huang J, Dong X, Gong Z, et al. Solution structure of the RNA recognition domain of METTL3-METTL14 N(6)-methyladenosine methyltransferase. Protein Cell. 2019;10(4):272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Liu J, Yue Y, Han D, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10(2):93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Aik W, Scotti JS, Choi H, et al. Structure of human RNA N(6)-methyladenine N6-methyladenine demethylase ALKBH5 provides insights into its mechanisms of nucleic acid recognition and demethylation. Nucleic Acids Res. 2014;42(7):4741–4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Duan HC, Wei LH, Zhang C, et al. ALKBH10B is an RNA N(6)-methyladenosine N6-methyladenosine demethylase affecting Arabidopsis floral transition. Plant Cell. 2017;29(12):2995–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Martinez-Perez M, Aparicio F, Lopez-Gresa MP, et al. Arabidopsis m(6)A m6 A demethylase activity modulates viral infection of a plant virus and the m(6)A m6 A abundance in its genomic RNAs. Proc Natl Acad Sci U S A. 2017;114(40):10755–10760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Parker MT, Knop K, Sherwood AV, et al. Nanopore direct RNA sequencing maps the complexity of Arabidopsis mRNA processing and m(6)A modification. Elife. 2020;9. DOI: 10.7554/eLife.49658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhang F, Zhang YC, Liao JY, et al. The subunit of RNA N6-methyladenosine methyltransferase OsFIP regulates early degeneration of microspores in rice. PLoS Genet. 2019;15(5):e1008120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Du X, Fang T, Liu Y, et al. Global profiling of N(6) N6-methyladenosine methylation in maize callus induction. Plant Genome. 2020;13(2):e20018. [DOI] [PubMed] [Google Scholar]
- [16].Arribas-Hernandez L, Simonini S, Hansen MH, et al. Recurrent requirement for the m(6) A-ECT2/ECT3/ECT4axis in the control of cell proliferation during plant organogenesis. Development. 2020;147:dev189134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hu J, Cai J, Park SJ, et al. N(6) N6-methyladenosine mRNA methylation is important for salt stress tolerance in Arabidopsis. Plant J. 2021;106(6):1759–1775. [DOI] [PubMed] [Google Scholar]
- [18].Huong TT, Ngoc LNT, Kang H.. Functional characterization of a putative RNA demethylase ALKBH6 in Arabidopsis growth and abiotic stress responses. Int J Mol Sci. 2020;21(18):6707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Miao Z, Zhang T, Qi Y, et al. Evolution of the RNA N(6)-methyladenosine 6-methyladenosine methylome mediated by genomic duplication. Plant Physiol. 2020;182(1):345–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Liu G, Wang J, Hou X.. Transcriptome-wide N(6)-methyladenosine (m(6)A) methylome profiling of heat stress in Pak-choi (Brassica rapa ssp. chinensis). Plants (Basel). 2020;9:1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lu L, Zhang Y, He Q, et al. MTA, an RNA m(6)A methyltransferase, enhances drought tolerance by regulating the development of trichomes and roots in poplar. Int J Mol Sci. 2020;21:2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Anderson SJ, Kramer MC, Gosai SJ, et al. N(6)-methyladenosine inhibits local ribonucleolytic cleavage to stabilize mRNAs in Arabidopsis. Cell Rep. 2018;25(5):1146–57 e3. [DOI] [PubMed] [Google Scholar]
- [23].Zheng H, Sun X, Li J, et al. Analysis of N6-methyladenosine reveals a new important mechanism regulating the salt tolerance of sweet sorghum. Plant Sci. 2021;304:110801. [DOI] [PubMed] [Google Scholar]
- [24].Wei LH, Song P, Wang Y, et al. The m(6)A m6 A reader ECT2 controls trichome morphology by affecting mRNA stability in Arabidopsis. Plant Cell. 2018;30(5):968–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhang G, Diao S, Zhang T, et al. Identification and characterization of circular RNAs during the sea buckthorn fruit development. RNA Biol. 2019;16(3):354–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhang G, Chen D, Zhang T, et al. Transcriptomic and functional analyses unveil the role of long non-coding RNAs in anthocyanin biosynthesis during sea buckthorn fruit ripening. DNA Res. 2018;25(5):465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].He C, Zhang G, Zhang J, et al. Integrated analysis of multiomic data reveals the role of the antioxidant network in the quality of sea buckthorn berry. FASEB J. 2017;31(5):1929–1938. [DOI] [PubMed] [Google Scholar]
- [28].He CY, Zhang GY, Zhang JG, et al. Physiological, biochemical, and proteome profiling reveals key pathways underlying the drought stress responses of Hippophae rhamnoides. Proteomics. 2016;16(20):2688–2697. [DOI] [PubMed] [Google Scholar]
- [29].Luo Z, Zhang Z, Tai L, et al. Comprehensive analysis of differences of N6-methyladenosine N6-methyladenosine RNA methylomes between high-fat-fed and normal mouse livers. Epigenomics. 2019;11(11):1267–1282. [DOI] [PubMed] [Google Scholar]
- [30].Liu J, Li K, Cai J, et al. Landscape and regulation of m(6)A and m(6)Am methylome across human and mouse tissues. Mol Cell. 2020;77(2):426–40 e6. [DOI] [PubMed] [Google Scholar]
- [31].Tao X, Chen J, Jiang Y, et al. Transcriptome-wide N(6) 6-methyladenosine methylome profiling of porcine muscle and adipose tissues reveals a potential mechanism for transcriptional regulation and differential methylation pattern. BMC Genomics. 2017;18(1):336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Batista PJ. The RNA modification N(6)-methyladenosine and its implications in human disease. Genomics Proteomics Bioinformatics. 2017;15(3):154–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhou L, Tian S, Qin G. RNA methylomes reveal the m(6) A-mediated regulation of DNA demethylase gene SlDML2 in tomato fruit ripening. Genome Biol. 2019;20(1):156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ping XL, Sun BF, Wang L, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24(2):177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Reichel M, Koster T, Staiger D. Marking RNA: m6A writers, readers, and functions in Arabidopsis. J Mol Cell Biol. 2019;11(10):899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zhou J, Wan J, Gao X, et al. Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature. 2015;526(7574):591–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Robinson M, Shah P, Cui YH, et al. The role of dynamic m(6) m6 A RNA methylation in photobiology. Photochem Photobiol. 2019;95(1):95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Fry NJ, Law BA, Ilkayeva OR, et al. Holley, mansfield KD. N6-methyladenosine N6-methyladenosine is required for the hypoxic stabilization of specific mRNAs. RNA. 2017;23(9):1444–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hussain A, Mun BG, Imran QM, et al. Nitric oxide mediated transcriptome profiling reveals activation of multiple regulatory pathways in Arabidopsis thaliana. Front Plant Sci. 2016;7:975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kushiro T, Okamoto M, Nakabayashi K, et al. The Arabidopsis cytochrome P450 CYP707A encodes ABA 8ʹ-hydroxylases: key enzymes in ABA catabolism. EMBO J. 2004;23(7):1647–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lee HK, Cho SK, Son O, et al. Drought stress-induced Rma1H1, a RING membrane-anchor E3 ubiquitin ligase homolog, regulates aquaporin levels via ubiquitination in transgenic arabidopsis plants. Plant Cell. 2009;21(2):622–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Gao G, Lv Z, Zhang G, et al. An ABA-flavonoid relationship contributes to the differences in drought resistance between different sea buckthorn subspecies. Tree Physiol. 2020;41(5):744–755. [DOI] [PubMed] [Google Scholar]
- [43].Wang YN, Jin HZ. Transcriptome-wide m(6)A methylation in skin lesions from patients with psoriasis vulgaris. Front Cell Dev Biol. 2020;8:591629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kim D, Paggi JM, Park C, et al. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37(8):907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bailey TL, Johnson J, Grant CE, et al. The MEME suite. Nucleic Acids Res. 2015;43(W1):W39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Salmon-Divon M, Dvinge H, Kairi Tammoja BP. PeakAnalyzer: genome-wide annotation of chromatin binding and modification loci. BMC Bioinformatics. 2010;11(1):415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.