

Abstract
Background & Aims:
Human non-alcoholic fatty liver disease (NAFLD) is characterized at early stages by hepatic steatosis, which may progress to nonalcoholic steatohepatitis (NASH) when the liver displays microvesicular steatosis, lobular inflammation, and pericellular fibrosis. The secretin (SCT)/secretin receptor (SCTR) axis promotes biliary senescence and liver fibrosis in cholestatic models through downregulation of miR-125b signaling. We aim to evaluate the effect of disrupting biliary SCT/SCTR/miR-125b signaling on hepatic steatosis, biliary senescence and liver fibrosis in NAFLD/NASH.
Approach & Results:
In vivo, 4 wk male WT, Sct-/- and Sctr-/- mice were fed a control diet (CD) or high-fat diet (HFD) for 16 wks. The expression of SCT/SCTR/miR-125b axis was measured in human NAFLD/NASH liver samples and HFD mouse livers by immunohistochemistry (IHC) and qPCR. Biliary/hepatocyte senescence, ductular reaction and liver angiogenesis were evaluated in mouse liver and human NAFLD/NASH liver samples. miR-125b target lipogenesis genes in hepatocytes were screened and validated by custom RT2 Profiler PCR array and luciferase assay. Biliary SCT/SCTR expression was increased in human NAFLD/NASH samples and in livers of HFD mice, whereas the expression of miR-125b was decreased. Biliary/hepatocyte senescence, ductular reaction, and liver angiogenesis were observed in human NAFLD/NASH samples as well as HFD mice, which were decreased in Sct-/- and Sctr-/- HFD mice. Elovl1 is a lipogenesis gene targeted by miR-125b, and its expression was also decreased in HFD mouse hepatocytes following Sct or Sctr knockout. Bile acid profile in fecal samples have the greatest changes between WT mice and Sct-/-/Sctr-/- mice.
Conclusion:
The biliary SCT/SCTR/miR-125b axis promotes liver steatosis by upregulating lipid biosynthesis gene Elovl1. Targeting the biliary SCT/SCTR/miR-125b axis may be key for ameliorating phenotypes of human NAFLD/NASH.
Keywords: hepatic steatosis, miR-125b, ELOVL1, ductular reaction, angiogenesis
Introduction
Nonalcoholic fatty liver disease (NAFLD), characterized by hepatic steatosis, is an alarming public health concern and is considered the most common liver disease worldwide (1). Simple steatosis can progress to nonalcoholic steatohepatitis (NASH) when the liver displays microvesicular steatosis, hepatocellular ballooning, lobular inflammation and pericellular fibrosis (2). NASH is a growing cause of liver cirrhosis and hepatocellular carcinoma ultimately leading to liver failure (3).
During cholestasis, cholangiocytes are the key link between biliary damage and fibrosis that characterizes chronic hepatobiliary injury, through the products of their activation like secretin (SCT) (4–6). Recent evidence indicates that cholangiocytes play a role in the pathogenesis of NAFLD through activation of biliary proliferation (ductular reaction, DR) and subsequent liver fibrosis (7, 8). One study found that secretin receptor (SCTR) knockout mice are resistant to high-fat diet (HFD)-induced obesity and exhibit impaired intestinal lipid absorption (9); however, the role of the SCT/SCTR axis in NAFLD/NASH progression remains unknown.
Liver steatosis is determined by de novo lipogenesis, lipid uptake, fatty acid β-oxidation, and very low-density lipoprotein (VLDL) secretion (10). Lipogenesis is regulated by acetyl-CoA being converted to malonyl-CoA by acetyl-CoA carboxylase (ACC), and malonyl-CoA is then converted to palmitate by fatty acid synthase (FASN) (11). Elevated lipogenesis, in combination with excess adipose fatty acid release, is a significant contributor to NAFLD (12). Elongation of very-long-chain fatty acids 1 (ELOVL1) is key for the synthesis of Cer d24:0 and Cer d24:1(13); however, the relevance of ELOVL1 in NAFLD and NASH remains unknown.
miRNAs, which are small non-coding RNAs that regulate gene expression via RNA silencing and post-transcriptional modification, may regulate lipid metabolism in the liver (14, 15). The SCT/SCTR axis stimulates biliary senescence and liver fibrosis during cholestasis by downregulation of miR-125b (6). miR-125b inhibits fat accumulation in the liver during HFD (16) and regulates vascular endothelial growth factor-A (VEGF-A) expression during cholestasis (17). Liver angiogenesis increases in NAFLD and contributes to inflammation, fibrosis, and hepatocellular carcinoma development (18). The present study was conducted using a HFD fed mouse model, as well as human NAFLD and NASH samples to evaluate the role of the SCT/SCTR/miR-125b axis in NAFLD, and to explore the potential target lipogenesis genes of miR-125b.
Materials and methods
Materials
Reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated. The antibodies for SCT and SCTR were obtained from Bioss (Woburn, MA) and Abcam (Cambridge, MA). The antibodies for cytokeratin-19 (CK-19), desmin, cyclin-dependent kinase inhibitor 2A (p16), VEGF-A, cluster of differentiation 68 (CD68) and von Willebrand factor (vWF) were obtained from Abcam (Cambridge, MA). Cluster of differentiation 31 (CD31) antibody was purchased from R&D (Minneapolis, MN). The monoclonal antibody for F4/80 was purchased from Cell Signaling Technology (Danvers, MA). The two different antibodies for ELOVL1 were purchased from both Thermo Fisher Scientific (Mountain View, CA) and Abcam (Cambridge, MA). Commercially available enzyme-linked immunosorbent assay (ELISA) kits for measuring SCT levels were purchased from Phoenix Pharmaceuticals (Burlingame, CA). ELISA kits to measure transforming growth factor-β1 (TGF-β1) levels in serum and cholangiocyte supernatant were purchased from Abcam. The mirVana™ miRNA Isolation Kit for RNA isolation was purchased from Ambion (Mountain View, CA). Selected mouse and human PCR primers were purchased from Qiagen (Valencia, CA). The iScript cDNA Synthesis Kit (170–8891) and iTaq Universal SYBR Green Supermix were purchased from Bio-Rad (Hercules, CA). The information on real-time PCR (qPCR) primers used is listed in the Supplemental Table 1.
Animal models
Animal procedures were performed according to protocols approved by the Baylor Scott & White Health and Indiana University School of Medicine Institutional Animal Care and Use Committees. C57/BL6 wild-type (WT) mice were purchased from Charles River (Wilmington, MA). Sct-/- and Sctr-/- mouse colonies are established in our facility (19); mice were originally obtained from Billy K. C. Chow, School of Biological Sciences, The University of Hong Kong, Hong Kong. Sctr-/- mice did not differ in locomotor activity or energy expenditure compared with WT mice (9). We did not observe any specific hepatic phenotypes in Sct-/- and Sctr-/- compared to WT mice. Mice were maintained in microisolator cages in a temperature-controlled environment with 12:12-hour light-dark cycles. Studies were performed in 4-week old male WT, Sct-/- and Sctr-/- mice fed either a control diet (CD, Envigo, Indianapolis, IN) consisting of standard chow and reverse osmosis water or a HFD (0.2% cholesterol, high-fat trans-fat with 45% calories from fat; Envigo, Indianapolis, IN) coupled with a high fructose corn syrup equivalent (55% fructose, 45% glucose, w/w) dissolved in reverse osmosis water for 16 wk (20). In all groups, liver and body weight and liver to body weight ratio were measured; serum, total liver, feces, cholangiocytes, hepatocytes and cholangiocyte/hepatocyte supernatants were collected.
Human samples
Liver samples from healthy human (n=4), NAFLD (n=9) and NASH (n=9) were purchased from Sekisui Xeno Tech (Kansas City, KS). Another 5 NASH liver samples were obtained from Dr. Burcin Ekser under the Institutional Review Board (IRB) approved protocol at Indiana University School of Medicine. Patient demographics are listed in Supplemental Table 2. Human serum samples (Control n=24, NAFLD n=10, NASH [stage 1 n=10, stage 2 n=10, stage 3 n=10, stage 4 n=59]) (Supplemental Table 3) were obtained from either Dr. Burcin Ekser under the IRB approved protocol at Indiana University School of Medicine; Dr. Wasim Dar under the protocol HSC-MS-12–0652 approved by UTHealth, McGovern Medical School IRB and Committee for the Protection of Human Subjects in Houston, Texas; or Dr. Niharika Samala under the IRB approved observational study of patients with NAFLD at Indiana University School of Medicine.
Detailed descriptions for other experimental procedures are described in the Supplemental Information.
Statistical analysis
All data are expressed as the mean ± SD. Differences between groups were analyzed by unpaired Student’s t test when two groups were analyzed and one-way ANOVA when more than two groups were analyzed, followed by an appropriate post hoc test. The level of significance was set at P < 0.05.
Results
SCT levels and biliary expression of the SCT/SCTR axis increases in patients with NAFLD/NASH and HFD-fed mice
To address the relevance of SCT and SCTR to human NAFLD and NASH, we measured the expression of SCT and SCTR in liver sections from human subjects. NAFLD and NASH samples showed increased biliary SCT and SCTR immunoreactivity compared to control (Figure 1A, Supplementary Figure 1A). Additionally, the mRNA expression of SCT and SCTR (in total liver samples) and serum SCT levels were higher in NAFLD and NASH compared to control (Figure 1B-C). The biliary immunoreactivity, as well as the mRNA expression of Sct and Sctr was higher in WT HFD compared to WT CD mice (Figure 1D-E, Supplementary Figure 1B). A very small number of periportal hepatocytes expressing SCTR were observed in liver sections from human NASH (but not NAFLD) and in WT mice fed HFD (Supplementary Figure 1A-B). SCT serum and cholangiocyte supernatant levels were increased in WT HFD compared to WT CD mice (Figure 1F). SCT levels were increased in the supernatant of a human primary cholangiocyte cell line (HIBEpiCs) treated with FFAs (oleate acid (OA), palmitate acid (PA) or stearate acid (SA)) (Supplementary Figure 1C) along with increased SCT and SCTR expression (Supplementary Figure 1D) when compared to control.
Figure 1. Biliary SCT/SCTR expression is increased in human NAFLD and HFD mice.
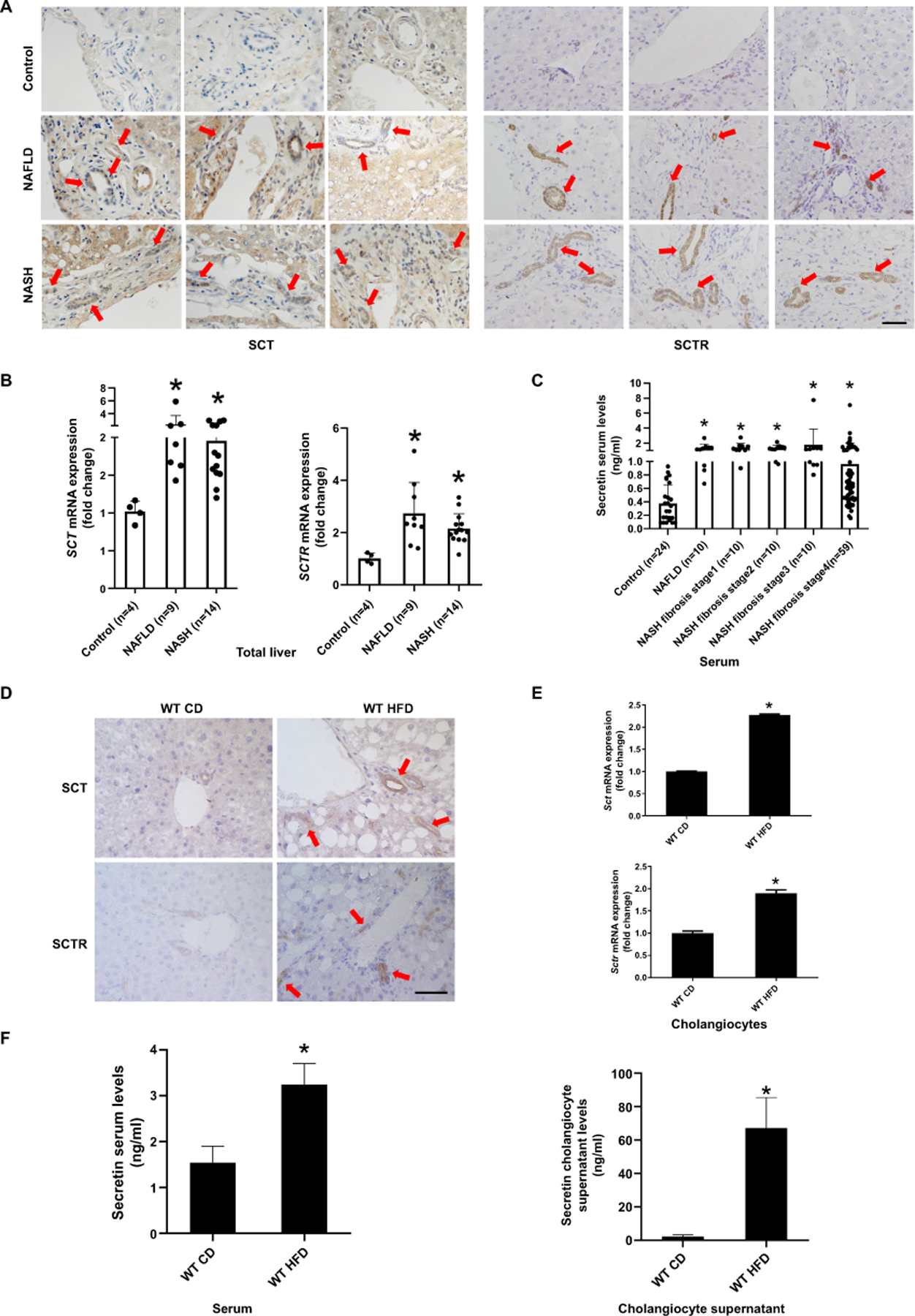
(A) Representative IHC images for SCT and SCTR in liver from patients with NAFLD (n=9) and NASH (n=14) and control (n=4) (40×; scale bar, 25 μm). (B) mRNA expression of SCT and SCTR in human NAFLD (n=9) and NASH (n=15) liver. (C) SCT levels in NAFLD (n=10) and NASH (stage 1–4; n=89) patients’ serum. (D) Representative images of SCT and SCTR expression in liver sections from WT CD and WT HFD mice (n=3; 40×; scale bar, 25 μm), E) mRNA expression of Sct and Sctr in isolated mouse cholangiocytes (n=3 reactions from samples obtained from a cumulative cell preparation from 8 mice). (F) SCT levels in mouse serum and cholangiocyte supernatant (n=3 reactions from samples obtained from a cumulative cell preparation from 8 mice). Data are mean ± SD. *P <0.05 versus Control or WT CD.
Knockout of the SCT/SCTR axis protects against HFD-induced liver injury and steatosis
H&E staining demonstrated that, after 16 wk of HFD feeding, WT mice displayed overt NAFLD phenotypes: mixed micro- and macrovesicular steatosis involving 80–90% of hepatocytes; increased portal lymphocytes with interface activity, pericellular fibrosis, increased inflammation with lipogranulomas, and prominent Kupffer cell numbers; these phenotypes were ameliorated in Sct-/- and Sctr-/- HFD mice (Figure 2A). WT HFD mice had increased serum levels of aspartate aminotransferase (AST), alanine aminotransferase (ALT), and alkaline phosphatase (ALK PHOS) compared to control mice, which were decreased (while AST not significant) in Sct-/- and Sctr-/- HFD mice compared to WT HFD mice (Supplemental Table 4). Additionally, Sct-/- and Sctr-/- HFD mice displayed a smaller gain in body weight and lower liver to body weight ratio compared to WT HFD mice (Supplemental Table 4). Furthermore, Sct-/- and Sctr-/- HFD mice displayed a milder degree of hepatic steatosis compared to WT HFD mice (Figure 2B). The mRNA expression of the fatty acid oxidation genes, Pparα, Acsl1, and Cpt1α, was significantly lower in hepatocytes from WT HFD mice compared to WT CD mice (Figure 2C); however, the expression of these genes was significantly increased in hepatocytes from both Sct-/- and Sctr-/- HFD mice compared to WT HFD mice (Figure 2C). The mRNA expression of adipogenesis genes Pparγ, Fasn, and Acacα was increased in hepatocytes from WT HFD mice compared to WT CD mice, which was reversed in Sct-/- and Sctr-/- HFD mice (Figure 2D). Interestingly, the expression of fatty acid oxidation genes was decreased in cholangiocytes from WT HFD mice compared with WT CD mice, but increased in Sct-/- and Sctr-/- HFD mice (Figure 2E).
Figure 2. SCT or SCTR knockout protects against HFD-induced steatosis.
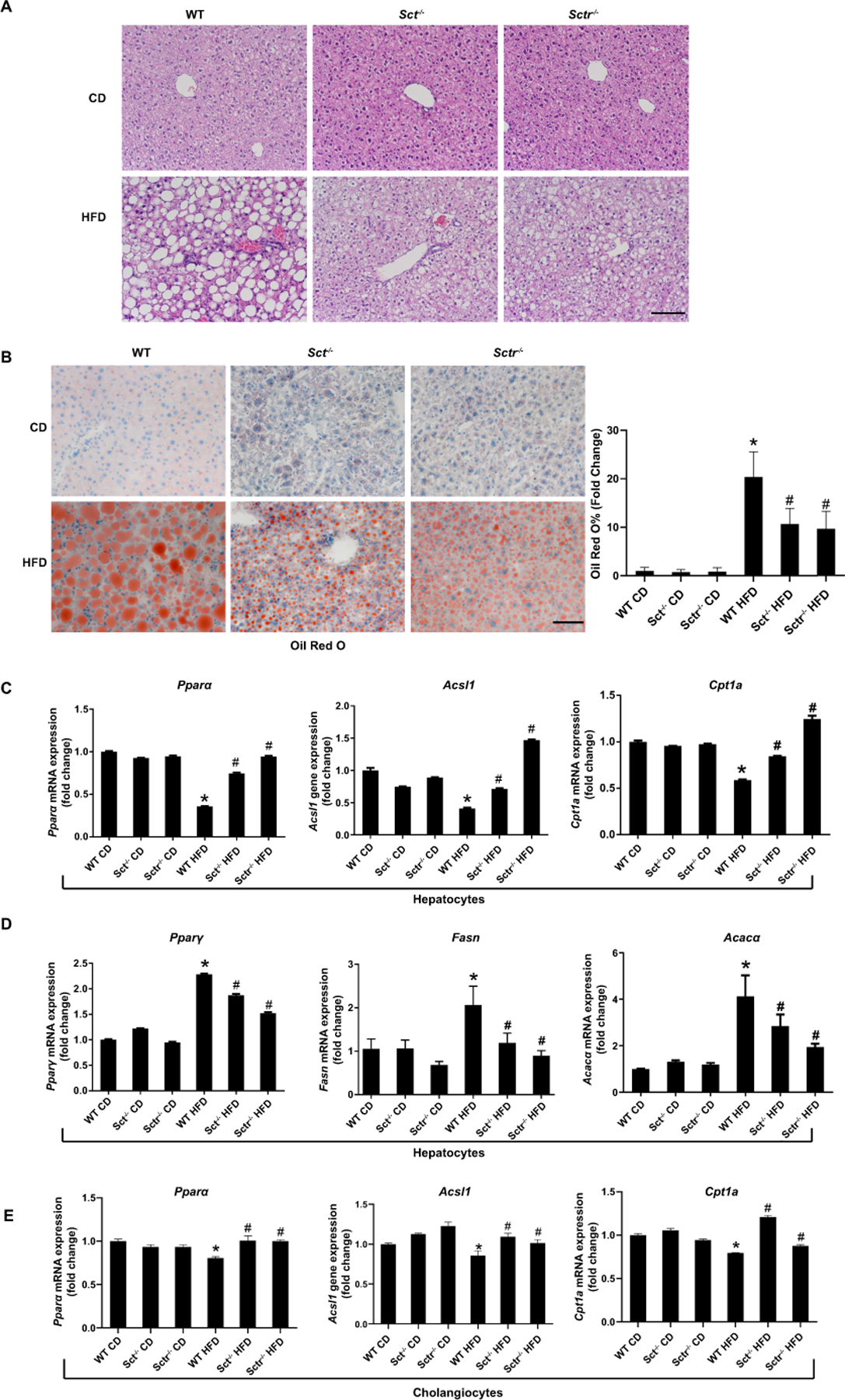
(A) Representative H&E images in liver from Sct-/- and Sctr-/- HFD mice (n=8; 20×; scale bar, 50 μm). (B) Representative images of Oil Red O staining in Sct-/- and Sctr-/- HFD mice. (n=24 from 8 different mice; 20×; scale bar, 50 μm). (C) Fatty acid oxidation genes Pparα, Acsl1 and Cpt1α expression in hepatocytes from Sct-/- and Sctr-/- HFD mice (n=3 reactions from samples obtained from a cumulative cell preparation from 8 mice). (D) The expression of adipogenesis gene Pparγ, Fasn and Acacα in hepatocytes from Sct-/- and Sctr-/- HFD mice (n=3 reactions from samples obtained from a cumulative cell preparation from 8 mice). (E) The expression fatty acid oxidation gene (Pparα, Acsl1 and Cpt1α) expression in cholangiocytes from Sct-/- and Sctr-/- HFD mice (n=3 reactions from samples obtained from a cumulative cell preparation from 8 mice). Data are mean ± SD. *P <0.05 versus WT CD, #P <0.05 versus WT HFD.
SCT/SCTR axis downregulates biliary miR-125b in NAFLD and NASH
Cholangiocyte SCT/SCTR signaling stimulates biliary senescence and liver fibrosis in cholestasis by downregulation of miR-125b (6), and miR-125b may inhibit fat accumulation during HFD (16). We demonstrated that miR-125b expression is much higher in cholangiocytes compared to hepatocytes (Supplementary Figure 2A), and miR-125b expression was decreased in total liver samples from NAFLD and NASH patients compared to control (Figure 3A). In mouse models, the expression of miR-125b was decreased in cholangiocytes from WT HFD mice, which returned to values similar to those of WT CD mice, in Sct-/- and Sctr-/- HFD mice (Figure 3B). Thus, we aimed to elucidate the mechanisms by which the biliary SCT/SCTR/miR-125b axis (21) regulates hepatocyte lipogenesis. miR-125b mimic and inhibitor were transfected into human hepatocytes (HHs) (Supplementary Figure 2B). miR-125b mimic decreased lipogenesis in transfected HHs treated with FFAs, whereas the miR-125b inhibitor increased lipogenesis (Supplementary Figure 2C). To further explore the paracrine effect of cholangiocyte-derived miR-125b on hepatocyte lipogenesis, we performed TargetScan and KEGG bioinformatic analyses and found 29 lipid biosynthesis genes potentially targeted by miR-125b (Figure 3C). By generating a custom RT2 Profiler PCR array, we found that 14 of the 29 genes increased in WT HFD mouse hepatocytes compared to WT CD mice, and decreased in Sct-/- and Sctr-/- HFD mouse hepatocytes compared to WT HFD mice (Figure 3D-E). These 14 genes were further assessed by IPA to identify new and novel targets that have not previously been published. Our results identified 9 novel genes (Ifng, St8sia4, Adh1a3, Acer2, Pgap3, Lpcat4, Elovl1, St6galnac6, and Lclat1) that are most likely targeted by miR-125b (Supplementary Figure 3A). To verify these observations, qPCR was performed to measure the levels of these 9 genes. Sct-/- and Sctr-/- HFD mice had reduced expression of these genes in hepatocytes compared to WT HFD mice (Figure 3F). These findings identify a novel pathway whereby SCT/SCTR mediates hepatocyte lipogenesis via paracrine miR-125b-dependent signaling.
Figure 3. SCT/SCTR axis downregulates biliary miR-125b that targets lipogenesis genes in NAFLD.
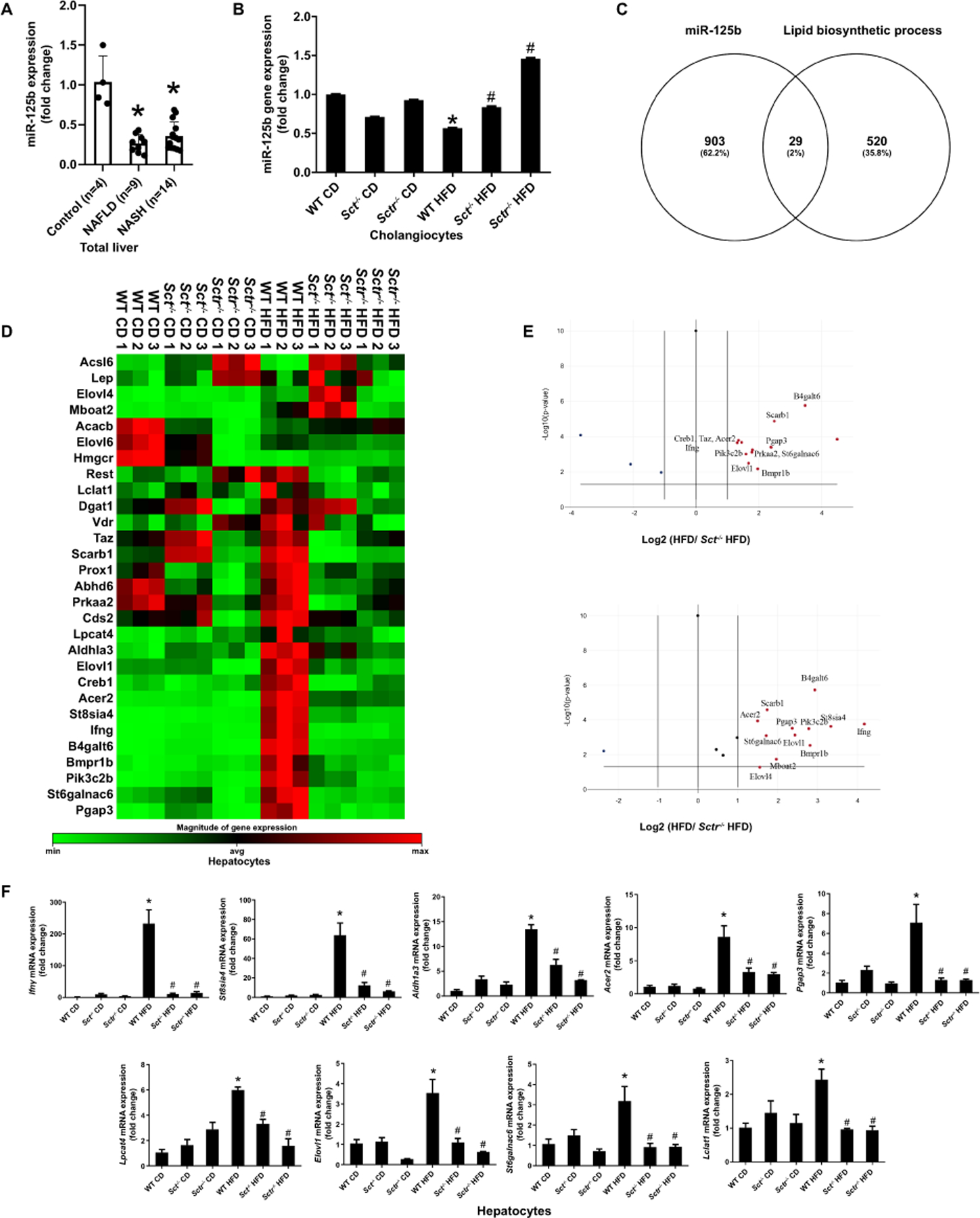
(A) miR-125b expression in liver of NAFLD (n=9) and NASH (n=14). (B) miR-125b expression in cholangiocytes from selected mouse groups (n=3 reactions from samples obtained from a cumulative cell preparation from 8 mice). (C) Venn map of predicted miR-125b target lipid biosynthetic process genes by TargetScan and KEGG. (D) Custom RT2 Profiler PCR array was used to analyze miR-125b potential target lipid biosynthetic process genes expression between WT HFD and Sct-/-/Sctr-/- HFD mouse in hepatocytes (n=3 reactions from samples obtained from a cumulative cell preparation from 8 mice). (E) The Volcano Plot identifies significant gene expression changes. (F) The expression of Ifng, St8sia4, Adh1a3, Acer2, Pgap3, Lpcat4, Elovl1, St6galnac6 and Lclat1 in mouse hepatocytes (n=3 reactions from samples obtained from a cumulative cell preparation from 8 mice). Data are mean ± SD. *P <0.05 versus Control or WT CD; #P <0.05 versus WT HFD.
Downregulation of miR-125b promotes steatosis by upregulating hepatocyte ELOVL1 in NAFLD
We further validated the genes targeted by miR-125b in human NAFLD and NASH, as well as HFD fed mice using TargetScan for both mouse and human sequences, to identify targets with species consistency, and found 5 genes (Acer2, St8sia4, St6galnac6, Elovl1 and Lclat1) from the 9 previously identified for further analysis (Supplementary Figure 3B). Dual luciferase reporter gene assay verified the miR-125b target genes, and the results demonstrated that luciferase signaling was decreased in hepatocytes treated with miR-125b mimic/pmirGLO-Acer2, miR-125b mimic/pmirGLO-Elovl1, miR-125b mimic/pmirGLO-Lclat and miR-125b mimic/pmirGLO-St8sia4 compared with negative control (NC) treatment (Figure 4A). The luciferase activity of the Elovl1 MUT 3’UTR was not significantly different in miR-125b mimic and NC transfected Huh7, indicating that miR-125b specifically binds to ELOVL1 (Figure 4B). We observed increased mRNA expression of ELOVL1 in HHs treated with cholangiocyte supernatant from WT HFD mice when compared with supernatant from WT CD mice, which was reduced in HHs treated with cholangiocyte supernatant from Sct-/- and Sctr-/- HFD mice (Supplementary Figure 3C). Furthermore, ELOVL1 protein expression increased in NAFLD and NASH patients’ liver compared with control (Figure 4C). In WT HFD mice, the ELOVL1 expression was increased in hepatocytes compared with WT CD mice; however, knockout of Sct or Sctr decreased ELOVL1 expression in hepatocytes following HFD feeding (Figure 4E). The immunoreactivity of ELOVL1 was increased in NAFLD and NASH patients compared with control (Figure 4D). There was also enhanced immunoreactivity for ELOVL1 in hepatocytes from WT HFD mice compared with WT CD mice, which was reversed by knockout of Sct and Sctr (Figure 4F).
Figure 4. miR-125b promotes steatosis by upregulating Elovl1.
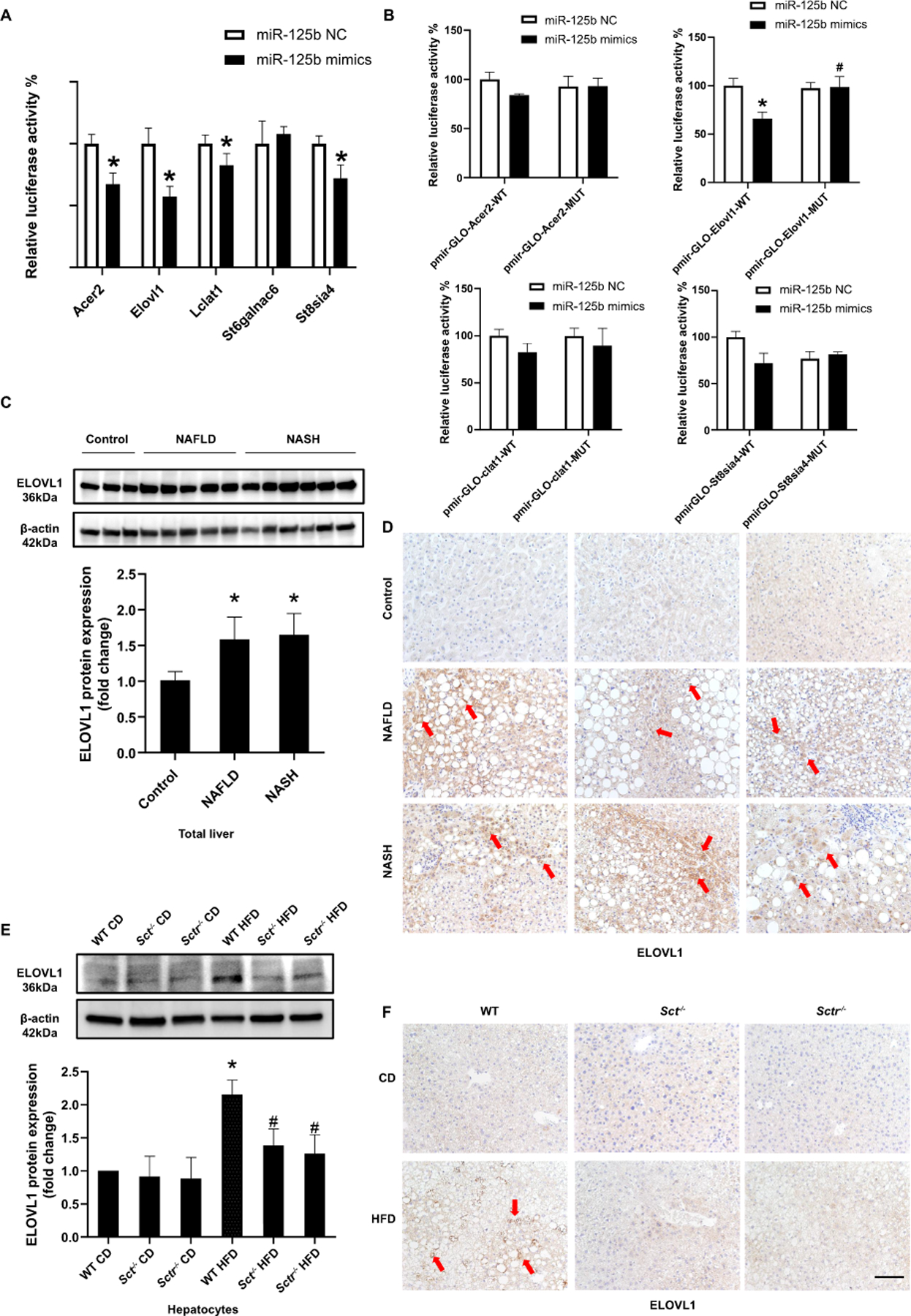
(A) Luciferase activities were detected after post-transfection of pmirGLO vectors (Acer2, St8sia4, St6galnac6, Elovl1 and Lclat1) and miR-125b mimic transfection in Huh7 cells (n=3). (B) Luciferase activities were detected after post-transfection of pmirGLO vectors (Acer2, St8sia4, Elovl1 and Lclat1 MUT 3’UTR) and miR-125b mimic transfection in Huh7 cells (n=3). (C) ELOVL1 expression in NAFLD (n=9) and NASH (n=14) patients (n=4) was detected by western blot. (D) Representative IHC images for ELOVL1 in liver from patients with NAFLD (n=9) and NASH (n=14) and control (n=4) (20×; scale bar, 50 μm). (E) ELOVL1 expression was detected by western blot in mouse isolated hepatocytes (n=3 reactions from samples obtained from a cumulative cell preparation from 8 mice). (F) Representative IHC images for ELOVL1 in mouse livers (n=8; 20×; scale bar, 50 μm). Data are mean ± SD. *P <0.05 versus Control, WT CD or mimic negative control; #P <0.05 versus WT HFD.
Knockout of the SCT/SCTR axis reduces HFD-induced intrahepatic bile duct mass (IBDM) and liver inflammation
By immunohistochemistry (IHC) for CK-19 (co-stained with CD68), IBDM increased in NAFLD samples that did not show a significant increase in CD68 immunoreactivity (Figure 5A), which suggests that IBDM precedes liver inflammation in NAFLD to NASH progression. There was increased IBDM in WT HFD mice compared to WT CD mice, which was reduced in Sct-/- and Sctr-/- HFD mice (Figure 5B).
Figure 5. HFD-induced intrahepatic bile duct mass (IBDM) precedes inflammation and is reduced by SCT and SCTR knockout.
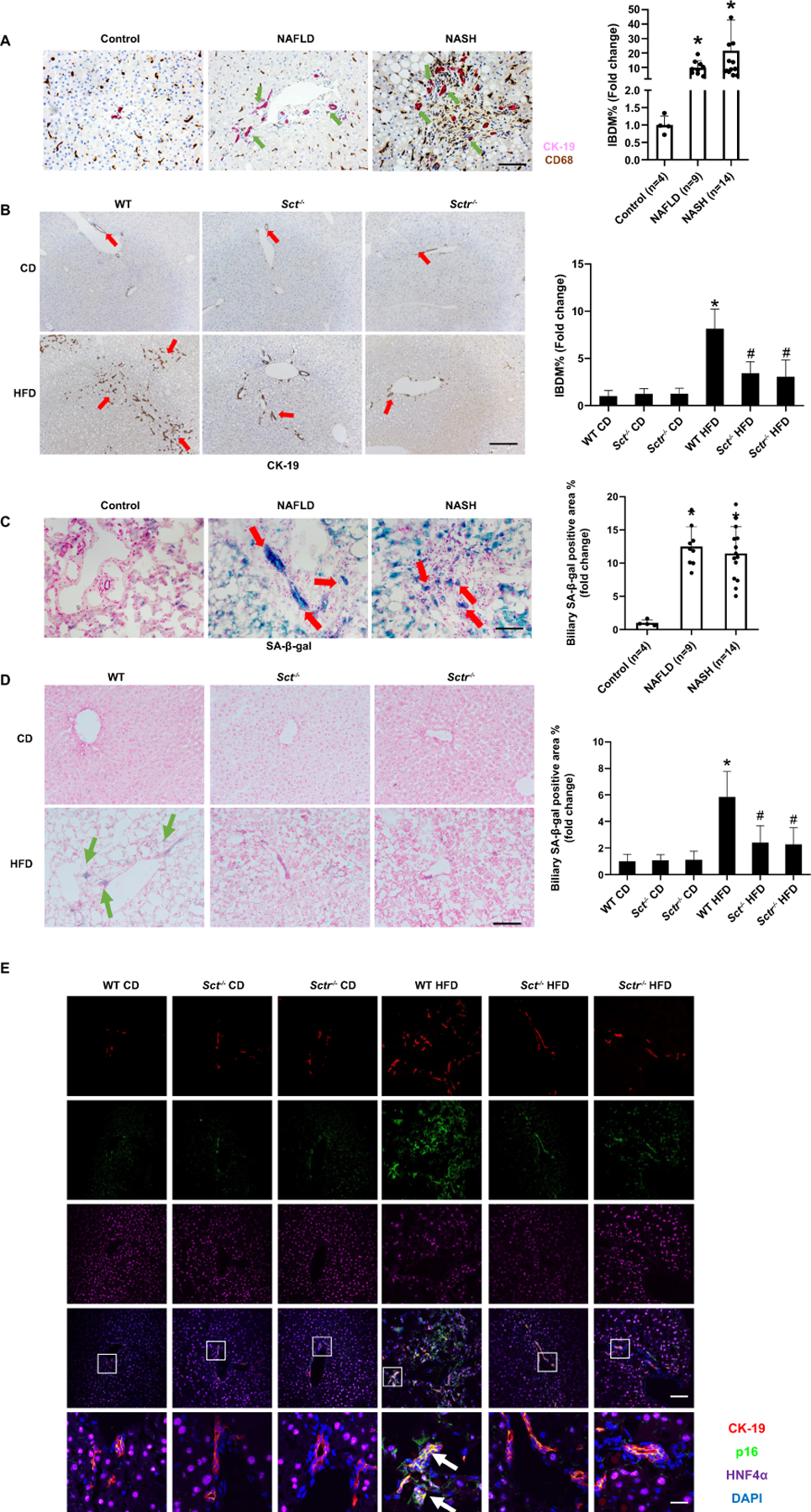
(A) Representative IHC images for CK-19 in liver from patients with NAFLD (n=9) and NASH (n=14) and control (n=4) (10×; scale bar, 100 μm)). (B) Representative IHC images for CK-19 in liver from Sct-/- and Sctr-/- HFD mice (n=24 from 8 different mice; 10×; scale bar, 100 μm). (C) Biliary senescence in liver from NAFLD (n=9) and NASH (n=14) patients was measured by SA-β-gal staining (20×; scale bar, 50 μm). (D) Measurement of biliary senescence in Sct-/- and Sctr-/- HFD mouse liver by SA-β-gal staining (n=24 from 8 different mice; 20×; scale bar, 50 μm). (E) Immunofluorescence for p16 /CK-19 in Sct-/- and Sctr-/- HFD mouse livers (n=8; upper four panels: Orig. magn. ×20, scale bar 50 μm; lower panel: Orig. magn. ×100, scale bar 10 μm; red CK-19, green p16, purple HNF4α, blue DAPI; white arrow: p16 positive cholangiocytes). Data are mean ± SD. *P <0.05 versus Control or WT CD; #P <0.05 versus WT HFD.
Knockout of the SCT/SCTR axis reduces HFD-induced biliary and hepatocyte senescence
SA-β-gal staining demonstrated enhanced biliary and hepatocyte senescence in NAFLD and NASH patients compared to control human samples (Figure 5C). Biliary and hepatocyte senescence increased in WT HFD mice compared to WT CD mice, which was significantly decreased in Sct-/- and Sctr-/- HFD mice (Figure 5D, Supplementary Figure 4A). By immunofluorescence, there was enhanced immunoreactivity for p16 in cholangiocytes (co-stained with CK-19) and in hepatocytes (co-stained with HNF4α) from WT HFD mice compared to WT CD (Figure 5E, Supplementary Figure 4B), which decreased in Sct-/- and Sctr-/- HFD mice. The mRNA expression of the senescence markers, Cdkn2a, Cdkn1a and Ccl2, increased in cholangiocytes from WT HFD mice compared to WT CD, but significantly decreased in Sct-/- and Sctr-/- HFD mice (Supplementary Figure 4C). In vitro, the expression of Cdkn2c and Cdkn1a increased in immortalized murine cholangiocyte lines (IMCLs) treated with OA, PA or SA compared to control, which was significantly reduced in Sctr-shRNA transfected IMCLs (Supplementary Figure 4D).
Knockout of the SCT/SCTR axis decreases HFD-induced liver angiogenesis and inflammation
We next evaluated the expression of (i) VEGF-A (a trophic factor for biliary mitosis and liver fibrosis (17, 21)); and (ii) angiogenic factors that stimulate biliary proliferation and liver fibrosis (22). We demonstrated: (i) enhanced immunoreactivity and mRNA expression for VEGF-A in human NAFLD and NASH compared to control (Figure 6A); (ii) increased expression of vWF in NAFLD and NASH patients compared to control (Figure 6B); and (iii) enhanced VEGF-A immunoreactivity and mRNA expression in WT HFD mice compared to WT CD, which was decreased in Sct-/- and Sctr-/- HFD mice (Figure 6C). Furthermore, CD31 immunoreactivity and mRNA expression in total liver were significantly higher in WT HFD mice compared with WT CD, but significantly decreased in Sct-/- and Sctr-/- HFD mice (Figure 6D). Similarly, the mRNA expression of Vwf was increased in WT HFD mice compared with WT CD mice but decreased in Sct-/- and Sctr-/- HFD mice (Supplementary Figure 5A). In addition, there was an increased number of F4/80-positive cells and mRNA expression of Il1β, Il6, Il33 and Tnfα from WT HFD mice compared with WT CD (Figure 6E, Supplementary Figure 5B), which was decreased in Sct-/- and Sctr-/- HFD mice.
Figure 6. Knockout of the SCT/SCTR decreases HFD-induced liver angiogenesis and inflammation.
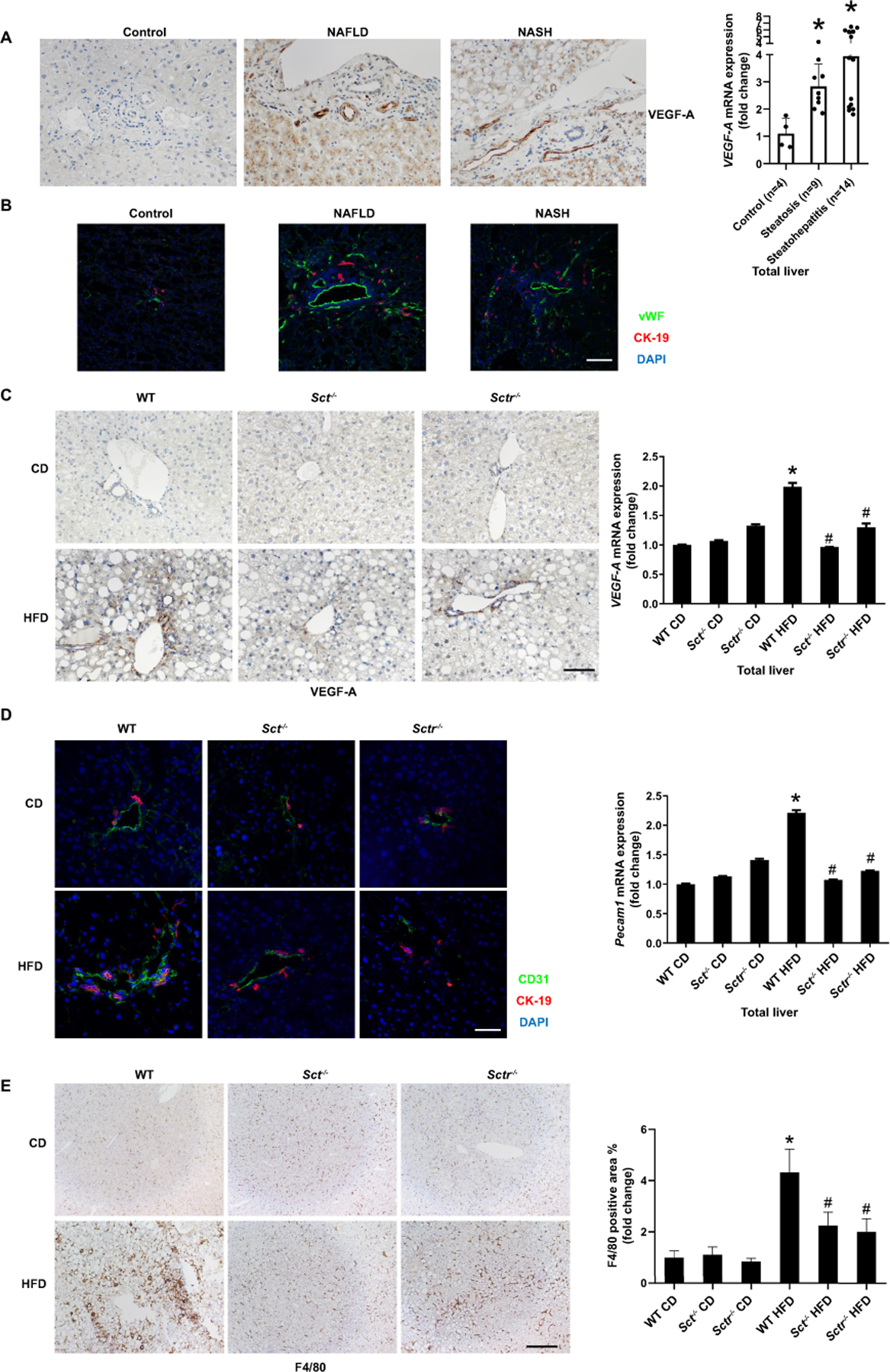
(A) VEGF-A expression in livers from NAFLD (n=9) and NASH (n=14) was measured by immunohistochemistry (20×; scale bar, 50 μm) and by qPCR. (B) Liver angiogenesis in liver samples from NAFLD (n=9) and NASH (n=14) was observed by immunofluorescence of vWF (20×; scale bar, 50 μm). (C) VEGF-A expression in Sct-/- and Sctr-/- HFD mouse liver was measured by immunohistochemistry (n=3; 20×; scale bar, 50 μm) and qPCR. (D) Angiogenesis in mouse liver samples was observed by immunofluorescence of CD31 (n=3; 40×; scale bar, 25 μm) and qPCR of Pecam1 in total liver (n=3). (E) Liver inflammation was detected by immunohistochemistry for F4/80 in Sct-/- and Sctr-/- HFD mice (n=24 from 8 different mice; 10×; scale bar, 100 μm). Data are mean ± SD. *P <0.05 versus WT CD or Control, #P <0.05 versus WT HFD.
Knockout of the SCT/SCSR axis reduces HFD-induced liver fibrosis and biliary TGF-β1 levels
There was a significant increase in collagen deposition in WT HFD mice compared to WT CD mice, which was significantly reduced in Sct-/- and Sctr-/- HFD mice (Figure 7A). The mRNA expression of fibrosis markers increased in total liver samples (Figure 7B) from WT HFD mice compared to WT CD mice, but decreased in Sct-/- and Sctr-/- HFD mice. Desmin and α-SMA immunoreactivity increased in WT HFD mice compared with WT CD mice (Figure 7C), but this was decreased in Sct-/- and Sctr-/- HFD mice (Figure 7C). The expression of collagen I was increased in WT HFD mice compared with WT CD mice, but decreased in Sct-/- HFD mice (Supplementary Figure 6A). TGF-β1 levels were significantly increased in serum and cholangiocyte supernatants from WT HFD mice compared to WT CD mice but decreased in Sct-/- and Sctr-/- HFD mice (Figure 7D). Human hepatic stellate cells (HHSteCs) were treated with in vivo isolated cholangiocyte supernatants to evaluate the paracrine effect of cholangiocyte-secreted factors (e.g. TGF-β1) on HSC activation. Interestingly, there was increased expression of fibrosis markers (ACTA2, TGF-β1, COL1A1 and FN1) in HHSteCs treated with cholangiocyte supernatant from WT HFD mice when compared with supernatant from WT CD mice (Supplementary Figure 6B). However, the fibrosis gene expression was reduced in HHSteCs treated with cholangiocyte supernatant from Sct-/- and Sctr-/- HFD mice (Figure 7D). Biliary mRNA expression of Col1a1 Tgf-β1 and Fn1 was significantly increased in WT HFD mice compared to WT CD mice, but decreased in Sct-/- and Sctr-/- HFD mice (Supplementary Figure 6C). In vitro, the expression of Col1a1, Tgf-β1 and Fn1 was increased in IMCLs treated with OA, PA or SA compared to control, which was reduced in Sctr-shRNA transfected IMCLs (Supplementary Figure 6D).
Figure 7. Knockout of the SCT/SCTR axis reduces HFD induced liver fibrosis.
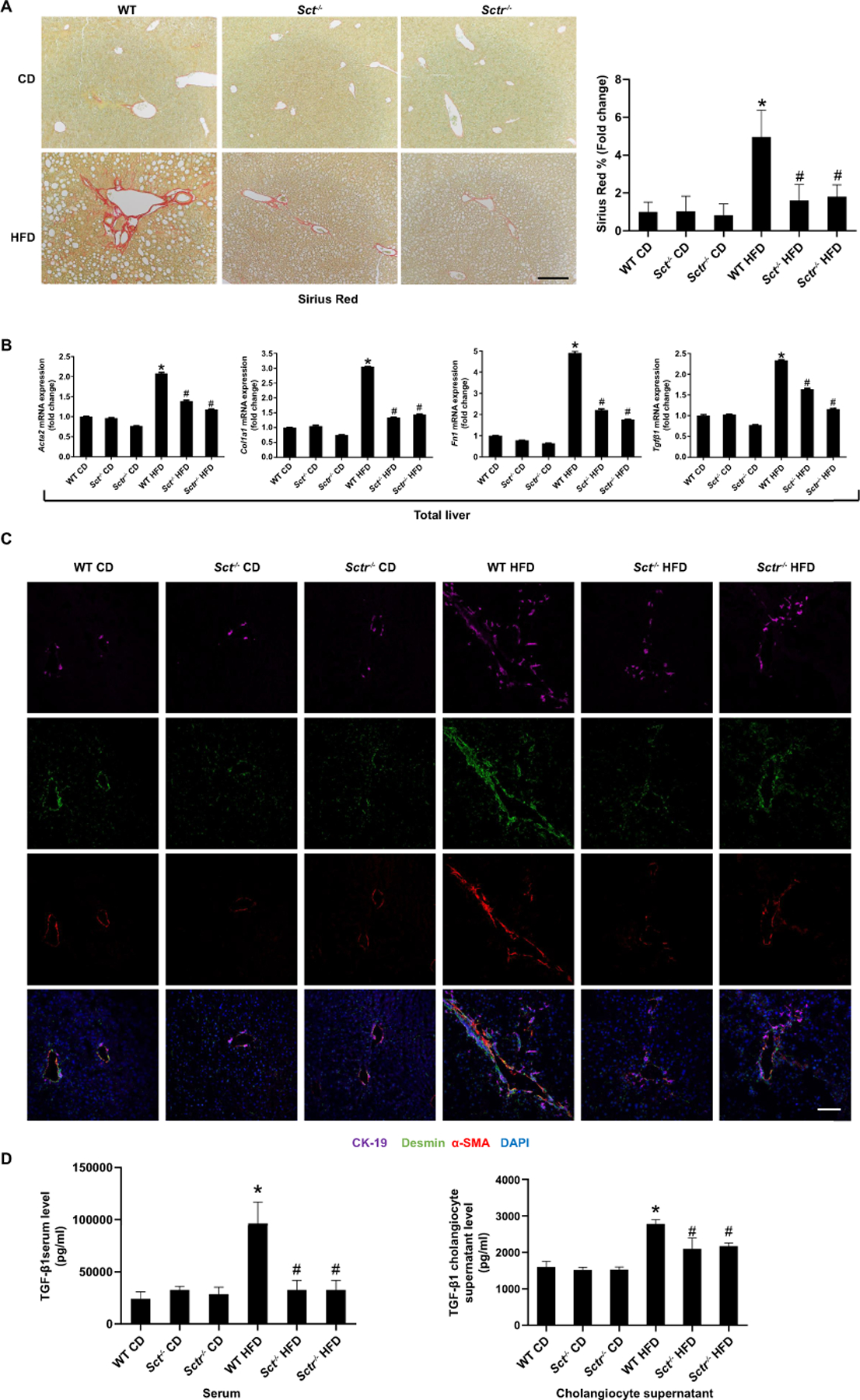
(A) Measurement of collagen deposition by Sirius Red staining in Sct-/- and Sctr-/- HFD mice liver (n=24 from 8 different mice; 10×; scale bar, 100 μm). (B) mRNA expression of fibrotic markers in Sct-/- and Sctr-/- HFD mouse total liver; n=3. (C) Immunofluorescence for α-SMA/desmin in mouse livers (n=8; 20×; scale bar, 50 μm). (D) TGF-β1 levels in mouse serum and cholangiocyte supernatant (n=3 reactions from samples obtained from a cumulative cell preparation from 8 mice). Data are mean ± SD. *P <0.05 versus WT CD, #P <0.05 versus WT HFD.
Knockout of the SCT/SCTR axis alters fecal total bile acid levels, bile acid composition and subsequent signaling pathways
Bile acid data showed that fecal bile acid levels and composition are more prominently altered than the serum or liver bile acid levels and composition in Sct/Sctr knockout mice compared to WT mice (Supplementary Table 5-10). Of note, Sct or Sctr knockout increased total bile acid fecal excretion (Supplementary Table 9), but also significantly increased the level of secondary bile acids that are metabolized by the gut microbiota (e.g. ωMCA and DCA), thus demonstrating that Sct/Sctr knockout may modify the gut microbiota (Supplementary Table 6). While not significant, total bile acid levels were reduced in the liver but enhanced in the serum of Sct and Sctr knockout mice fed HFD compared to WT HFD mice (Supplementary Table 8, Supplementary Table 10). The expression of ASBT (apical Na+‐dependent bile acid transporter, upregulated by secretin) gene, Slc10a2, was increased in cholangiocytes from WT HFD mice compared to WT CD mice, which was reversed in Sct-/- or Sctr-/- HFD mice (Supplementary Figure 7A), and loss of biliary ASBT may decrease cholehepatic shunting thus leading to the aforementioned changes in hepatic and serum total bile acid levels. Furthermore, the expression of Cyp7b1 was decreased in hepatocytes from WT HFD mice compared to WT CD mice, but was increased in hepatocytes from Sct-/- and Sctr-/- HFD mice (Supplementary Figure 7B), which may also account for the enhanced serum and fecal bile acid levels.
Discussion
The SCT/SCTR axis exerts pleiotropic effects, including regulation of fluid homeostasis (23) and development of obesity (24). The SCT/SCTR axis, only expressed by cholangiocytes in the liver (25), stimulates ductular reaction/senescence (by an autocrine loop) (21, 26) and liver fibrosis (by a paracrine loop) (5) in cholestasis (4). SCT stimulates lipolysis and lipid absorption in adipose cells (24) and Sctr knockout mice are resistant to HFD-induced obesity (9). The liver constitutes an essential organ in lipid metabolism and is a central regulator of lipid homeostasis (27). In this study, we provide data indicating that biliary SCT/SCTR signaling is associated with changes in biliary senescence (by an autocrine loop) and liver steatosis, fibrosis and angiogenesis (by paracrine mechanisms) in NAFLD/NASH. Sct or Sctr knockout decreases HFD-induced liver steatosis by regulating miR-125b/ELOVL1 expression in hepatocytes. HFD-induced biliary/hepatocyte senescence, DR, liver fibrosis and angiogenesis were reduced by Sct or Sctr knockout, and Sct or Sctr knockout increased total bile acid fecal excretion in HFD mice.
In human NAFLD/NASH and HFD-fed mice, we observed increased biliary expression of the SCT/SCTR and enhanced SCT serum levels, providing the first evidence that the SCT/SCTR axis is linked to NAFLD/NASH. Furthermore, in vitro cholangiocytes treated with FFAs displayed increased SCT secretion supporting the specific, direct interaction of FFAs with cholangiocytes. Taken together with our previous studies showing that enhanced SCT/SCTR expression induces DR and liver fibrosis in cholestasis (4, 28), our work suggests that enhanced SCT/SCTR signaling regulates the phenotypes (e.g., liver inflammation, steatosis and fibrosis) of NAFLD/NASH.
Although we have previously demonstrated that SCT and SCTR are only expressed by cholangiocytes in control human liver and are upregulated during cholestatic liver injury, we wanted to evaluate if there is a change in hepatocyte SCT and SCTR expression in models of steatosis and steatohepatitis. We observed that a small number of periportal hepatocytes express SCTR in both human NASH and WT mice fed HFD; however, whether these SCTR positive hepatocytes are hepatocytes acquiring biliary phenotypes, or they are cholangiocytes that are transdifferentiating to hepatocytes needs to be further studied.
Hepatic steatosis, defined by triglyceride accumulation in hepatocytes, is the earliest manifestation of NAFLD (29). Sct and Sctr knockout ameliorated HFD-induced hepatic steatosis. During hepatic lipid homeostasis, de novo lipogenesis is instrumental in the development of hepatic steatosis (30). Studies using stable isotope tracers suggest that one important characteristic of NAFLD is abnormally elevated hepatocyte lipogenesis (31). Our data shows that the expression of lipogenesis genes was reversed in Sct-/- or Sctr-/- HFD hepatocytes compared to WT HFD. Studies have shown that miR-125b is also implicated in different liver diseases (32). miR-125b promotes HSC activation by activating RhoA signaling in CCl4 or BDL mouse models (32). miR-125b promotes the NF-κB-mediated inflammatory response in hepatocytes from NAFLD (33). We have previously demonstrated that SCT/SCTR mediated downregulation of miR-125b promotes biliary proliferation and liver fibrosis during cholestasis (6, 21), whereas overexpression of miR-125b protects HFD-induced hepatic steatosis (16). In our study, miR-125b expression was decreased in human NAFLD/NASH liver and WT HFD mouse cholangiocytes, which was reversed by Sct or Sctr knockout. In addition, we found the expression of miR-125b is increased in cholangiocytes compared to hepatocytes. Therefore, we propose that SCT-dependent miR-125b plays a role in the modulation of NAFLD phenotypes by paracrine mechanisms. It is known that cell–cell communication is mediated via extracellular vesicle (EV)-delivered miRNAs (34), suggesting a possible paracrine cross-talk between cholangiocytes and hepatocytes likely mediated by SCT/SCTR/miR-125b signaling.
Our data verified that the lipogenesis gene Elovl1 is a target of miR-125b. ELOVL1 is increased in NASH Pten null livers, which increases liver lipogenesis (35). This study identifies that changes in a signaling pathway specific to cholangiocytes (i.e., SCT/SCTR axis) modulate, in a paracrine fashion, hepatic steatosis during NAFLD/NASH. Nevertheless, our studies do not exclude the possibility that SCT effects on hepatic steatosis may be due to impaired intestinal lipid absorption (9), but this issue will be elucidated by feeding cholangiocyte-specific Sctr-/- mice with HFD. Our data herein demonstrates that miR-125b targets ELOVL1 to regulate hepatocytes lipogenesis, indicating that the biliary SCT/SCTR axis has a direct effect on hepatic steatosis.
SCT-regulated miR-125b targets VEGF-A in models of cholestasis (17), and we noted increased VEGF-A expression and angiogenesis in human NAFLD/NASH. Angiogenesis, defined as new vessel formation from pre-existing vessels, is a key event in NAFLD progression (36) and pathologic angiogenesis increases with NASH (37). VEGF-A expression and angiogenesis were increased in HFD fed mice but reduced with knockout of Sct or Sctr. These findings suggest that biliary SCT/SCTR/miR-125b axis regulates angiogenesis in NAFLD/NASH. The hepatic inflammatory response is an important driving force of NAFLD progression to NASH (38). We found that depletion of Sct or Sctr reduced proinflammatory responses in HFD fed mice, demonstrating the paracrine role of the SCT/SCTR axis in regulating inflammatory events during NASH development. Portal inflammatory infiltrate is significantly associated with periportal DR in NAFLD (39), and DR in NAFLD accompanies more advanced histological impairments (8). HFD-induced DR was reduced in Sct-/- or Sctr-/- mice, and other studies have demonstrated that: (i) SCT stimulates biliary proliferation in cholestasis (21); and (ii) knockout of Sct or Sctr, or treatment of cholestatic rodents with a SCTR antagonist decreases DR (5, 6, 21). We observed DR in human NAFLD samples without significant inflammatory infiltrate, suggesting that DR precedes liver inflammation in NAFLD/NASH. These findings further highlight the paracrine role of the biliary SCT/SCTR axis in NAFLD/NASH development. Aside from DR, senescent cholangiocytes play various roles in aggravated liver inflammation and fibrosis by secreting senescence-associated secretory phenotypes (SASPs) (4, 40, 41). Evidence suggests that senescence is involved cholangiopathy pathogenesis (42), and our lab has shown that Sctr-/- mice have reduced biliary senescence in cholestatic models (5, 6). In this study, cholangiocyte senescence was observed in human NAFLD/NASH (43) and HFD fed mice, but was significantly decreased in Sct-/- or Sctr-/- HFD mice. FFAs can induce cholangiocyte damage in NAFLD (7). Interestingly, our data found that in vitro knockdown of cholangiocyte Sctr decreased senescence following FFAs treatment, providing further evidence that loss of SCT/SCTR signaling reduces biliary damage during NAFLD progression. One study has demonstrated that hepatocyte senescence correlates closely with fibrosis stage and predicts progression in NAFLD (44). Herein, we found that hepatocyte senescence increased in human NAFLD/NASH and HFD-fed mice, and Sct or Sctr knockout decreases HFD-induced hepatocyte senescence.
Fibrosis is associated with long-term outcomes in NASH (45), and the presence of advanced fibrosis, including bridging fibrosis and cirrhosis, has the highest risk of mortality related to liver disease (46). In NAFLD/NASH, fibrosis can be followed by steatosis (47), angiogenesis (18), portal inflammation (38) and biliary senescence (48). After HFD feeding, WT mice showed enhanced hepatic fibrosis, which was decreased in Sct-/- or Sctr-/- HFD mice. HSC activation is crucial for the deposition of the extracellular matrix during liver fibrosis, which was observed in HHSteCs treated with cholangiocyte supernatant from WT HFD mice (containing higher levels of TGF-β1), but this was not noted in HHSteCs treated with cholangiocyte supernatant from Sct-/- or Sctr-/- HFD mice (containing little to no TGF-β1). These findings support our hypothesis that biliary SCT/SCTR signaling promotes hepatic fibrosis.
NAFLD and NASH might be linked to dysregulation of cholesterol metabolism through the alternative pathway of bile acid synthesis that is mediated by Cyp7b1 (49). We analyzed the bile acid profile in liver, serum and feces samples in our mice fed CD or HFD. We found that knockout of Sct or Sctr increases total bile acid fecal excretion and modulates secondary bile acid metabolized by the gut microbiota. Interestingly, the ASBT gene, Slc10a2, expression was increased in WT HFD cholangiocytes but decreased in Sct-/- or Sctr-/- HFD mice, showing reduced cholehepatic shunting. We hypothesize that a similar mechanism maybe occurring in the intestines, whereby the increased bile acid fecal excretion may be due to reduced enterohepatic circulation regulated by SCT/ASBT in the intestines, but this warrants further studies. Parallel with previous findings (50), we demonstrated that Cyp7b1 expression was decreased in WT HFD hepatocytes, whereas Sct or Sctr knockout reverses the expression of Cyp7b1, which indicates Sct or Sctr knockout alleviate HFD-induced liver injury by altering bile acid pathway.
In conclusion, we have demonstrated that cholangiocytes play a key role in regulating hepatic steatosis during NAFLD. We demonstrate that the biliary SCT/SCTR/miR-125b axis plays an important role in regulating biliary senescence, steatosis, angiogenesis, inflammation, and hepatic fibrosis in NAFLD/NASH (Figure 8). Targeting the biliary SCT/SCTR/miR-125b axis may be key for ameliorating phenotypes of human NAFLD/NASH.
Figure 8. Working model of biliary SCT/SCTR/miR-125b axis regulation of biliary damage, steatosis, and hepatic fibrosis in NAFLD/NASH.
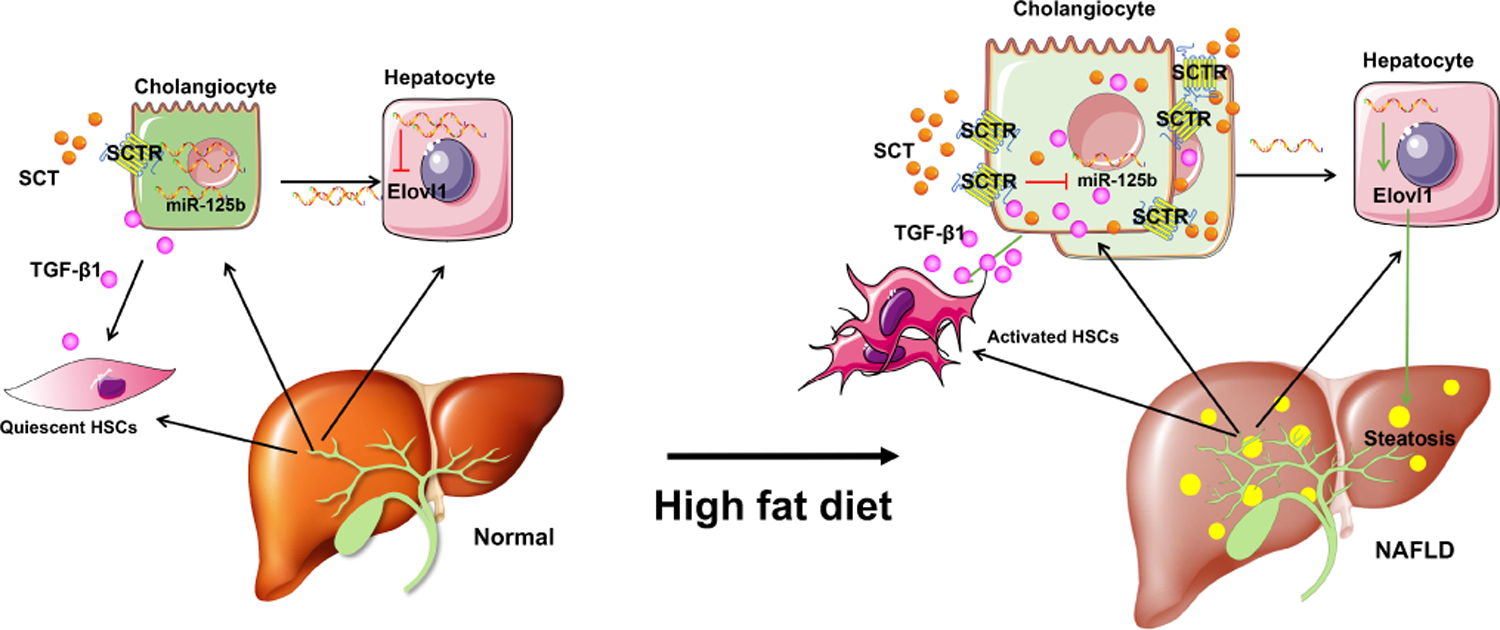
Biliary SCT and SCTR expression is increased in NAFLD. SCT/SCTR downregulation biliary miR-125b promotes steatosis by targeting lipogenesis gene ELOVL1 in NAFLD. Increased cholangiocyte-derived TGF-β1 induces HSC activation by paracrine in NAFLD.
Supplementary Material
Acknowledgment:
We would like to thank Dr. Naga Chalasani for providing human NAFLD/NASH serum samples under the IRB approved observational study of patients with NAFLD/NASH at Indiana University School of Medicine.
Financial support statement: This work was supported by the Hickam Endowed Chair, Gastroenterology, Medicine (GA), Indiana University, the Indiana University Health – Indiana University School of Medicine Strategic Research Initiative (GA and HF); the VA Merits 5I01BX000574 to GA and 1I01BX003031 to HF from the United States Department of Veteran’s Affairs, Biomedical Laboratory Research and Development Service and NIH grants DK108959 and DK119421 (HF), DK076898, DK107310 and DK110035 to GA, and SG. R01 DK121330–01 and R01 DK122796–01 to WD. This material is the result of work supported by resources at the Central Texas Veterans Health Care System, Temple, TX, Richard L. Roudebush VA Medical Center, Indianapolis, IN, and Medical Physiology, Medical Research Building, Temple, TX. The views expressed in this article are those of the authors and do not necessarily represent the views of the Department of Veterans Affairs.
Abbreviations:
- ACTA2 (α-SMA)
Actin Alpha 2, Smooth Muscle (α-smooth muscle actin)
- Acacα
acetyl-CoA carboxylase alpha
- Acer2
Alkaline ceramidase 2
- Acsl1
long chain Acyl CoA synthetase
- Aldh1a3
Aldehyde dehydrogenase 1 family member A3
- CCL2
C-C motif chemokine ligand 2
- CD68
cluster of differentiation 68
- CK-19
cytokeratin-19
- Col1a1
collagen type I alpha 1
- Cdkn1a (p21)
cyclin dependent kinase inhibitor 1a
- Cdkn2a (p16)
cyclin dependent kinase inhibitor 2a
- Cdkn2c (p18)
cyclin dependent kinase inhibitor 2c
- Cpt1α
carnitine palmitoyl-transferase 1α
- Cyp7b1
oxysterol 7α-hydroxylase
- ELOVL1
Elongation of very-long-chain fatty acids 1
- ELISA
enzyme linked immunosorbent assay
- Fasn
fatty acid synthase
- FFAs
free fatty acids
- Fn1
fibronectin1
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HHs
Human hepatocytes
- HIBEpiCs
human primary cholangiocyte cell line
- HHSteCs
human hepatic stellate cells
- HSCs
hepatic stellate cells
- IBDM
intrahepatic bile duct mass
- Ifnγ
Interferon Gamma
- IMCLs
immortalized murine cholangiocyte lines
- Lclat1
Lysocardiolipin Acyltransferase 1
- LCM
laser capture microdissection
- Lpcat4
Lysophosphatidylcholine Acyltransferase 4
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- OA
Oleic acid
- PA
Palmitic acid
- Pecam1 (CD31)
platelet endothelial cell adhesion molecule 1
- Pgap3
Post-GPI Attachment To Proteins Phospholipase 3
- Pparα
peroxisome proliferator activated receptor α
- Pparγ
peroxisome proliferator activated receptor γ
- PSC
primary sclerosing cholangitis
- SA
Stearate acid
- Sct
secretin
- Sctr
secretin receptor
- St6galnac6
ST6 N-Acetylgalactosaminide Alpha-2,6-Sialyltransferase 6
- St8sia4
ST8 Alpha-N-Acetyl-Neuraminide Alpha-2,8-Sialyltransferase 4
- Slc10a2 (ASBT)
solute carrier family 10 member 2
- TGF-β1
transforming growth factor-β1
- VEGF-A
vascular endothelial growth factor-A
- vWF
von Willebrand factor
- WT
wild-type
Footnotes
Conflict of interest statement: The authors have declared that no conflict of interest exists.
REFERENCES
- 1.Sanyal AJ. Past, present and future perspectives in nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol 2019;16:377–386. [DOI] [PubMed] [Google Scholar]
- 2.Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc 1980;55:434–438. [PubMed] [Google Scholar]
- 3.Estes C, Razavi H, Loomba R, Younossi Z, Sanyal AJ. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018;67:123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu N, Meng F, Invernizzi P, Bernuzzi F, Venter J, Standeford H, Onori P, et al. The secretin/secretin receptor axis modulates liver fibrosis through changes in transforming growth factor-beta1 biliary secretion in mice. Hepatology 2016;64:865–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu N, Meng F, Zhou T, Venter J, Giang TK, Kyritsi K, Wu C, et al. The Secretin/Secretin Receptor Axis Modulates Ductular Reaction and Liver Fibrosis through Changes in Transforming Growth Factor-beta1-Mediated Biliary Senescence. Am J Pathol 2018;188:2264–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou T, Wu N, Meng F, Venter J, Giang TK, Francis H, Kyritsi K, et al. Knockout of secretin receptor reduces biliary damage and liver fibrosis in Mdr2(-/-) mice by diminishing senescence of cholangiocytes. Lab Invest 2018;98:1449–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Natarajan SK, Ingham SA, Mohr AM, Wehrkamp CJ, Ray A, Roy S, Cazanave SC, et al. Saturated free fatty acids induce cholangiocyte lipoapoptosis. Hepatology 2014;60:1942–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sorrentino P, Tarantino G, Perrella A, Micheli P, Perrella O, Conca P. A clinical-morphological study on cholestatic presentation of nonalcoholic fatty liver disease. Dig Dis Sci 2005;50:1130–1135. [DOI] [PubMed] [Google Scholar]
- 9.Sekar R, Chow BK. Secretin receptor-knockout mice are resistant to high-fat diet-induced obesity and exhibit impaired intestinal lipid absorption. FASEB J 2014;28:3494–3505. [DOI] [PubMed] [Google Scholar]
- 10.Liu Y, Jiang L, Sun C, Ireland N, Shah YM, Liu Y, Rui L. Insulin/Snail1 axis ameliorates fatty liver disease by epigenetically suppressing lipogenesis. Nat Commun 2018;9:2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith GI, Shankaran M, Yoshino M, Schweitzer GG, Chondronikola M, Beals JW, Okunade AL, et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J Clin Invest 2020;130:1453–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014;146:726–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohno Y, Suto S, Yamanaka M, Mizutani Y, Mitsutake S, Igarashi Y, Sassa T, et al. ELOVL1 production of C24 acyl-CoAs is linked to C24 sphingolipid synthesis. Proc Natl Acad Sci U S A 2010;107:18439–18444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ambros V. The functions of animal microRNAs. Nature 2004;431:350–355. [DOI] [PubMed] [Google Scholar]
- 15.Rottiers V, Naar AM. MicroRNAs in metabolism and metabolic disorders. Nat Rev Mol Cell Biol 2012;13:239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang ZC, Liu Y, Xiao LL, Li SF, Jiang JH, Zhao Y, Qian SW, et al. Upregulation of miR-125b by estrogen protects against non-alcoholic fatty liver in female mice. J Hepatol 2015;63:1466–1475. [DOI] [PubMed] [Google Scholar]
- 17.Kennedy LL, Hargrove LA, Graf AB, Francis TC, Hodges KM, Nguyen QP, Ueno Y, et al. Inhibition of mast cell-derived histamine secretion by cromolyn sodium treatment decreases biliary hyperplasia in cholestatic rodents. Lab Invest 2014;94:1406–1418. [DOI] [PubMed] [Google Scholar]
- 18.Hammoutene A, Rautou PE. Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J Hepatol 2019;70:1278–1291. [DOI] [PubMed] [Google Scholar]
- 19.Chu JY, Chung SC, Lam AK, Tam S, Chung SK, Chow BK. Phenotypes developed in secretin receptor-null mice indicated a role for secretin in regulating renal water reabsorption. Mol Cell Biol 2007;27:2499–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mells JE, Fu PP, Kumar P, Smith T, Karpen SJ, Anania FA. Saturated fat and cholesterol are critical to inducing murine metabolic syndrome with robust nonalcoholic steatohepatitis. J Nutr Biochem 2015;26:285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glaser S, Meng F, Han Y, Onori P, Chow BK, Francis H, Venter J, et al. Secretin stimulates biliary cell proliferation by regulating expression of microRNA 125b and microRNA let7a in mice. Gastroenterology 2014;146:1795–1808 e1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gaudio E, Barbaro B, Alvaro D, Glaser S, Francis H, Ueno Y, Meininger CJ, et al. Vascular endothelial growth factor stimulates rat cholangiocyte proliferation via an autocrine mechanism. Gastroenterology 2006;130:1270–1282. [DOI] [PubMed] [Google Scholar]
- 23.Chu JY, Lee LT, Lai CH, Vaudry H, Chan YS, Yung WH, Chow BK. Secretin as a neurohypophysial factor regulating body water homeostasis. Proc Natl Acad Sci U S A 2009;106:15961–15966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miegueu P, Cianflone K, Richard D, St-Pierre DH. Effect of secretin on preadipocyte, differentiating and mature adipocyte functions. Int J Obes (Lond) 2013;37:366–374. [DOI] [PubMed] [Google Scholar]
- 25.Alpini G, Ulrich CD 2nd, Phillips JO, Pham LD, Miller LJ, LaRusso NF. Upregulation of secretin receptor gene expression in rat cholangiocytes after bile duct ligation. Am J Physiol Gastrointest Liver Physiol 1994;266:G922–928. [DOI] [PubMed] [Google Scholar]
- 26.Glaser S, Lam IP, Franchitto A, Gaudio E, Onori P, Chow BK, Wise C, et al. Knockout of secretin receptor reduces large cholangiocyte hyperplasia in mice with extrahepatic cholestasis induced by bile duct ligation. Hepatology 2010;52:204–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nguyen P, Leray V, Diez M, Serisier S, Le Bloc’h J, Siliart B, Dumon H. Liver lipid metabolism. J Anim Physiol Anim Nutr (Berl) 2008;92:272–283. [DOI] [PubMed] [Google Scholar]
- 28.Kennedy L, Francis H, Invernizzi P, Venter J, Wu N, Carbone M, Gershwin ME, et al. Secretin/secretin receptor signaling mediates biliary damage and liver fibrosis in early-stage primary biliary cholangitis. FASEB J 2019;33:10269–10279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ligabue G, Besutti G, Scaglioni R, Stentarelli C, Guaraldi G. MR quantitative biomarkers of non-alcoholic fatty liver disease: technical evolutions and future trends. Quant Imaging Med Surg 2013;3:192–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ipsen DH, Lykkesfeldt J, Tveden-Nyborg P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol Life Sci 2018;75:3313–3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Diraison F, Moulin P, Beylot M. Contribution of hepatic de novo lipogenesis and reesterification of plasma non esterified fatty acids to plasma triglyceride synthesis during non-alcoholic fatty liver disease. Diabetes Metab 2003;29:478–485. [DOI] [PubMed] [Google Scholar]
- 32.You K, Li SY, Gong J, Fang JH, Zhang C, Zhang M, Yuan Y, et al. MicroRNA-125b Promotes Hepatic Stellate Cell Activation and Liver Fibrosis by Activating RhoA Signaling. Mol Ther Nucleic Acids 2018;12:57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Q, Yu K, Cao Y, Luo Y, Liu Y, Zhao C. miR-125b promotes the NF-kappaB-mediated inflammatory response in NAFLD via directly targeting TNFAIP3. Life Sci 2021;270:119071. [DOI] [PubMed] [Google Scholar]
- 34.Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 2007;9:654–659. [DOI] [PubMed] [Google Scholar]
- 35.Muir K, Hazim A, He Y, Peyressatre M, Kim DY, Song X, Beretta L. Proteomic and lipidomic signatures of lipid metabolism in NASH-associated hepatocellular carcinoma. Cancer Res 2013;73:4722–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iwakiri Y, Shah V, Rockey DC. Vascular pathobiology in chronic liver disease and cirrhosis - current status and future directions. J Hepatol 2014;61:912–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tamaki Y, Nakade Y, Yamauchi T, Makino Y, Yokohama S, Okada M, Aso K, et al. Angiotensin II type 1 receptor antagonist prevents hepatic carcinoma in rats with nonalcoholic steatohepatitis. J Gastroenterol 2013;48:491–503. [DOI] [PubMed] [Google Scholar]
- 38.Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol 2018;15:349–364. [DOI] [PubMed] [Google Scholar]
- 39.Gadd VL, Skoien R, Powell EE, Fagan KJ, Winterford C, Horsfall L, Irvine K, et al. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 2014;59:1393–1405. [DOI] [PubMed] [Google Scholar]
- 40.Sasaki M, Miyakoshi M, Sato Y, Nakanuma Y. Chemokine-chemokine receptor CCL2-CCR2 and CX3CL1-CX3CR1 axis may play a role in the aggravated inflammation in primary biliary cirrhosis. Dig Dis Sci 2014;59:358–364. [DOI] [PubMed] [Google Scholar]
- 41.Wan Y, Meng F, Wu N, Zhou T, Venter J, Francis H, Kennedy L, et al. Substance P increases liver fibrosis by differential changes in senescence of cholangiocytes and hepatic stellate cells. Hepatology 2017;66:528–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakanuma Y, Sasaki M, Harada K. Autophagy and senescence in fibrosing cholangiopathies. J Hepatol 2015;62:934–945. [DOI] [PubMed] [Google Scholar]
- 43.Kennedy L, Hargrove L, Demieville J, Bailey JM, Dar W, Polireddy K, Chen Q, et al. Knockout of l-Histidine Decarboxylase Prevents Cholangiocyte Damage and Hepatic Fibrosis in Mice Subjected to High-Fat Diet Feeding via Disrupted Histamine/Leptin Signaling. Am J Pathol 2018;188:600–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aravinthan A, Scarpini C, Tachtatzis P, Verma S, Penrhyn-Lowe S, Harvey R, Davies SE, et al. Hepatocyte senescence predicts progression in non-alcohol-related fatty liver disease. J Hepatol 2013;58:549–556. [DOI] [PubMed] [Google Scholar]
- 45.Loomba R, Chalasani N. The Hierarchical Model of NAFLD: Prognostic Significance of Histologic Features in NASH. Gastroenterology 2015;149:278–281. [DOI] [PubMed] [Google Scholar]
- 46.Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, Harrison SA, et al. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018;67:328–357. [DOI] [PubMed] [Google Scholar]
- 47.McPherson S, Hardy T, Henderson E, Burt AD, Day CP, Anstee QM. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: implications for prognosis and clinical management. J Hepatol 2015;62:1148–1155. [DOI] [PubMed] [Google Scholar]
- 48.Chiba M, Sasaki M, Kitamura S, Ikeda H, Sato Y, Nakanuma Y. Participation of bile ductular cells in the pathological progression of non-alcoholic fatty liver disease. J Clin Pathol 2011;64:564–570. [DOI] [PubMed] [Google Scholar]
- 49.Pandak WM, Kakiyama G. The acidic pathway of bile acid synthesis: Not just an alternative pathway. Liver Res 2019;3:88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kakiyama G, Marques D, Martin R, Takei H, Rodriguez-Agudo D, LaSalle SA, Hashiguchi T, et al. Insulin resistance dysregulates CYP7B1 leading to oxysterol accumulation: a pathway for NAFL to NASH transition. J Lipid Res 2020;61:1629–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.