

Abstract
Statins are HMG-CoA reductase inhibitors prescribed for lowering cholesterol. They can also inhibit inflammatory responses by suppressing isoprenylation of small G proteins. Consistent with this, we previously found that fluvastatin suppresses IgE-mediated mast cell function. However, some studies have found that statins induced pro-inflammatory cytokines in macrophages and NK cells. In contrast to IgE signaling, we show that fluvastatin augments IL-33-induced TNF and IL-6 production by mast cells. This effect required the key mast cell growth factor, stem cell factor (SCF). Treatment of IL-33-activated mast cells with mevalonic acid or isoprenoids reduced fluvastatin effects, suggesting fluvastatin acts at least partly by reducing isoprenoid production. Fluvastatin also enhanced IL-33-induced NF-κB transcriptional activity and promoted neutrophilic peritonitis in vivo, a response requiring mast cell activation. Other statins tested did not enhance IL-33 responsiveness. Therefore, this work supports observations of unexpected pro-inflammatory effects of some statins and suggests mechanisms by which this may occur. Because statins are candidates for repurposing in inflammatory disorders, our work emphasizes the importance of understanding the pleiotropic and possible unexpected effects of these drugs.
Keywords: Mast cell, statin, IL-33, inflammation, allergy
1. Introduction
Mast cells play a sentinel role as early-acting innate immune cells. Although they have a variety of functions in defense against pathogens, parasites, and toxins, they are best known as effector cells in Type 1 hypersensitivities (1, 2). It is well established that mast cells are activated during allergic responses by IgE cross-linking. More recently, a role for IL-33 stimulation has also been emphasized (3–5). Due to the importance of mast cells in allergies and allergic asthma, many therapeutics target them. Most drugs fall into one of three categories: those inhibiting mast cell mediators, such as antihistamines and leukotriene antagonists; those that target IgE-mediated activation (e.g., omalizumab); or anti-inflammatory corticosteroids (6, 7). Although these treatments are effective, some are expensive, have adverse side effects, or fail to target other types of mast cell activation such as the IL-33 pathway. Therefore, a goal of our work has been to determine if some FDA-approved drugs might be repurposed for mast cell-associated diseases.
Statins are a widely prescribed class of cholesterol-lowering drugs. Within the U.S., 1 in 4 adults over the age of 40 are prescribed statins for hypercholesterolemia or cardiovascular disease, making them one of the most commonly used drugs (8). Statins act by targeting HMG-CoA reductase (HMGCR) through competitive inhibition, which decreases mevalonate synthesis and subsequently cholesterol production (9). Mevalonate is also metabolized to isoprenoids used to modify proteins, including members of the Ras family that play a vital role in cell signaling (10). Due to their relative safety and ability to decrease isoprenoid synthesis, statins are considered for use in inflammatory disease.
Previously, statins have been shown to suppress inflammation in mouse models of airway disease (11, 12) and have been debated as a potential asthma therapeutic (13–21). In mast cells, fluvastatin has been shown to reduce IgE-mediated degranulation (22, 23). Previously, we found that fluvastatin and other statins inhibit IgE-induced inflammatory cytokine production in mast cells, decrease the severity of systemic anaphylaxis in a mouse model, and induce mast cell apoptosis and autophagy (23, 24). These effects were found to be driven by blockade of geranylgeranyl transferase. In contrast, some studies have shown that statins can enhance inflammatory functions of immune cells. Statin-treated macrophages stimulated with LPS had augmented inflammasome signaling, resulting in increased IL-1β release (25). Similarly, NK cells co-stimulated with statins and IL-2 had enhanced pro-inflammatory cytokine production and cytotoxic effects on tumor cells (26). Related to this, defective isoprenoid production due to mevalonate kinase deficiency (MKD), commonly known as hyper IgD syndrome, causes periodic fevers characterized by elevated plasma IL-1β (27). While it has been hypothesized that the lack of cholesterol or IgG was contributing to elevated IL-1β, loss of HMG-CoA reductase pathway intermediates have been suggested to contribute (28, 29). We now show that fluvastatin unexpectedly increased IL-33-mediated mast cell inflammatory responses in vitro and in vivo. These data emphasize the importance of understanding both the pleiotropic effects of statins and the fundamental contributions of isoprenoid lipids to inflammation.
2. Methods
2.1. Animals
Mouse strain C57BL/6J or BALB/cJ breeding pairs were purchased from The Jackson Laboratory (Bar Harbor, ME) and colonies were maintained in a specific pathogen-free facility. Bone marrow was extracted from mice at a minimum of 10 weeks of age. IL-33-induced peritonitis studies were also conducted mice of 10–16 weeks of age, under protocols approved by Virginia Commonwealth University's Institutional Animal Care and Use Committee. Both sexes of mice were used for in vivo experiments, with comparisons matched to sex. In vitro data shown are from female BMMC; male BMMC yielded similar outcomes.
2.2. Mouse mast cell culture
Mouse bone marrow-derived mast cells (BMMC) were derived from female mouse femur and tibia bone marrow cultured in complete RPMI 1640 medium (cRPMI; Invitrogen Life Technologies, Carlsbad, CA) containing 10% FBS, 2mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 1mM sodium pyruvate, and 1mM HEPES (Corning, Corning, NY). Media was supplemented with supernatant from WEHI-3B cells and BHK-MKL cells that contain IL-3 (1 ng/ml) and SCF (15 ng/ml) respectively, as assessed by ELISA. BMMC were used after 21–28 days of culture. These cultures are >95% mast cells, based on flow cytometry analysis for c-Kit and FcεRI expression. Peritoneal mast cells were collected from C57BL/6J mice by peritoneal lavage with PBS supplemented with 1mM EDTA. Peritoneal cells were cultured in cRPMI supplemented with 10 ng/ml of recombinant mouse IL-3 and SCF. Cells were used after 7–10 days of culture.
2.3. Human mast cell culture
Human mast cell studies were conducted with approval of the Internal Review Board at the University of South Carolina. Skin samples were obtained from the Cooperative Human Tissue Network of the National Cancer Institute or the National Disease Research Interchange. Skin mast cells were prepared and cultured as described previously (30, 31). Cells were used after purity was greater than 95% mast cells, as determined by toluidine blue staining and surface FcεRI expression assessed by flow cytometry.
2.4. Cytokines and reagents
Recombinant mouse IL-3, SCF, and IL-33 were purchased from Shenandoah Biotechnology (Warwick, PA) for in vitro studies. For in vivo studies and human mast cells, carrier free recombinant mouse IL-33 and human IL-33 were purchased from Biolegend (San Diego, CA). Mevalonic acid, pravastatin, lovastatin, simvastatin, atorvastatin, and zaragozic acid were purchased from Sigma-Aldrich (St. Louis, MO). Fluvastatin sodium salt was purchased from Tocris Bioscience, part of Bio-techne (Minneapolis, MN). Farnesyl pyrophosphate and geranylgeranyl pyrophosphate were purchased from Echelon (Salt Lake City, UT). Purified anti-mouse TNFα (hence referred to as TNF) (clone MP6-XT22), anti-mouse TNF receptor1 and 2 (clones 55R-593 and TR75–32.4), APC-coupled anti-mouse IL-33Ra, PE-coupled anti-mouse Ly6G, APC-coupled anti-mouse CD45, PE-coupled anti-mouse CD117, FITC-coupled anti-mouse Ly6C, FITC-coupled anti-mouse CD11b FITC (clone M1/70), APC-Cy7-coupled anti-mouse F4/80 (clone BM8), Alexa Fluor 647-coupled anti-mouse Siglec-F (clone S17007L), APC-coupled anti-mouse FcεRIα (clone MAR-1), PE-coupled anti-mouse B220 (clone RA3–6B2), BV421-coupled anti-mouse CCR3 (clone J073E5), PE-Cy5-coupled anti-mouse CD4 (clone: GK1.5), FITC-coupled anti-mouse CD8a (clone 53–6.7), and corresponding isotype controls were purchased from Biolegend (San Diego, CA). PE-coupled anti-mouse Ly6G (clone 1A8), PE-coupled anti-mouse c-KIT (clone ACK45), APC-coupled anti-human phospho-ERK, BV421-coupled anti-human phospho-Akt, PE-coupled anti-human phospho-Btk, relevant isotype controls, and purified rat anti-mouse CD16/CD32 (clone 2.4G2) for blocking were all purchased from BD Biosciences (San Jose, CA).
2.5. Cytokine and chemokine measurements
BMMC were washed and resuspended in cRPMI at a concentration of 1×106 cells/ml with IL-3 (10 ng/ml) and SCF (50 ng/ml) unless otherwise stated. Cultures were stimulated with IL-33 (100 ng/mL) for 16 hours and supernatant was assessed for IL-6 and TNF levels using ELISA kits from Biolegend (San Diego, CA).
2.6. RT-qPCR
RNA was extracted from cell cultures treated with fluvastatin (20 μM) or vehicle using TRIzol reagent (Life Technologies, Grand Island, NY). Following RNA extraction, cDNA was produced using the qScript RNA cDNA Synthesis Kit (Quanta Biosciences, Gaithersburg, MD) using manufacturers' protocol. Amplification and qPCR analysis were conducted using Bio-Rad CFX96 Touch™ Real-Time PCR Detection System (Hercules, CA) and SYBR® Green detection. The reactions performed contained cDNA, PerfeCTa SYBR Green SuperMix (Quanta Biosciences, Gaithersburg, MD) and primers for the gene of interest and housekeeping genes. For IL-6 quantification, primers for IL-6 (5'-TCCAGTTGCCTTCTTGGGAC-3' and 5'-GTGTAATTAAGCTCCGACTTG-3') and the housekeeping gene Gapdh (5'-GATGACATCAAGAAGGTGGTG-3'and 5'-GCTGTAGCCAAATTCGTTGTC-3') obtained from Eurofins MWG Operon (Huntsville, AL). Amplification conditions were as follows: 95°C for 2 mins, followed by 40 cycles at 95°C for 15 s, 55°C for 30 s, and 60°C for 1 min. mRNA levels in each sample were normalized to the level of housekeeping gene mRNA using the Relative Livak Method (ΔΔCt).
2.7. Luciferase Assay
BMMC were transfected with vectors GL4.74[hRluc/TK](1.2 μg) and pGL4.32[luc2p/NFkB RE/Hygro](6 μg) encoding Renilla luciferase under the HSV-TK promoter and firefly luciferase under NFκB response elements. Transfections were performed with Amaxa Nucleofector (Lonza; Allendale, NJ, USA) on program T-005 in DMEM containing 20% FBS and 50 mM HEPES (pH 7.5). Cells were used 48 hours after transfection. Cultures contained IL-3 (10 ng/ml) and SCF (50 ng/ml) +/− vehicle or fluvastatin (20 μM) for 24 hours and were then activated with IL-33 (100 ng/ml) for 2 hours. Cells were lysed and luciferase activity was measured with the Dual-Luciferase Reporter Assay System and a Glomax 20/20 Luminometer (Promega, Madison, WI). Ratios of Firefly luciferase to Renilla luciferase were normalized to DMSO-treated, unstimulated samples.
2.8. Flow cytometric analysis
Cells were cultured at 1×106 cells/ml in cRPMI with the indicated conditions before staining. Afterward, cells were washed, pelleted, and resuspended in PBS containing 3% fetal calf serum and 0.1% sodium azide (FACS buffer) with the indicated antibodies and anti-CD16/32 clone 2.4G2. Samples were incubated at 4° C for 45 minutes, washed, and resuspended in FACS buffer. Samples were then analyzed for expression of surface molecules. To identify cell lineages from peritoneal lavage, forward scatter height and area were used to exclude doublets and the following markers were used: mast cells (FcεRIα/c-Kit-positive), T cells (CD4+ or CD8+), B cells (B220+), neutrophils (Ly6Ghigh, Ly6Clow/−), macrophages (CD11b+, F4/80+), eosinophils (Siglec F+, CCR3+). For viability staining, cells were cultured at 1×106 cells/ml in cRPMI with the indicated treatments before staining. Cells (2×105) were stained with propidium iodide (10 ng/test) immediately before analysis. For intracellular staining of phosphorylated proteins, cells were cultured at 1×106 cells/ml in cRPMI with the indicated treatments prior to activation. Cells were stimulated with IL-33 (100 ng/ml) for the stated time before fixation with 1.6% paraformaldehyde for 20 minutes. Cells were then washed with PBS and fixed with 100% methanol for 10 minutes at 4°C. Following fixation; cells were washed and stained with the indicated antibody at room temperature for 45 minutes. Cells were rewashed and resuspended in FACS buffer prior to analysis. All samples were acquired on a BD FACSCelesta and analyzed using BD FACSDIVA™ (BD Biosciences, Franklin Lakes, NJ).
2.9. Western Blotting
Cells were cultured at 1×106 cells/ml in cRPMI with IL-3 (10 ng/ml) and SCF (50 ng/ml) with 20μM fluvastatin or vehicle control for 24 hours. Cultures were then resuspended at 3×106 cells/ml and activated with IL-33 (100 ng/ml). Lysates were collected using Lysis Buffer (Cell Signaling Technology, Danvers, MA) containing 1.5x ProteaseArrest (G-Biosciences, Maryland Heights, MO). The Pierce BCA protein assay kit (Thermo Scientific, Waltham, MA) was used to determine protein concentrations. Lysates were separated on 4–20% Mini-Protean TGX Gels (Bio-Rad, Hercules, CA) followed by transfer onto a nitrocellulose membrane. Following transfer, membranes were blocked using Blocker Casein Buffer (Thermo Scientific, Rockford, IL) diluted 1:2 in tris-buffered saline (TBS) for 1 hour at room temperature, and then incubated overnight at 4°C in primary antibody resuspended in Blocker Casein Buffer diluted 1:2 with TBS/0.1% TWEEN-20 (TBS-T). Membranes were washed with TBS-T several times prior to incubation with dye-labeled secondary goat antibodies against mouse and rabbit IgG (LI-Cor, Lincoln, NE), also diluted in Blocker Casein Buffer/TBS-T. Membranes were imaged on an Odyssey CLx infrared scanner (Li-Cor, Lincoln, NE).
2.10. Measurement of neutrophil recruitment and cytokines in vivo
C57BL/6J mice were injected intraperitoneally (i.p.) with vehicle or fluvastatin (0.5 mg/mouse) 24 hours and 1 hour prior to i.p. injection with 1 μg of IL-33. Four hours later, peritoneal lavage and cardiac puncture were performed. Peritoneal cells were stained as described above and analyzed by flow cytometry. Precision Count Beads™ (San Diego, CA) from Biolegend were used to count the absolute cell number. Plasma was collected by cardiac puncture, and cytokine levels were analyzed by ELISA.
2.11. Statistical analyses
p values were calculated with GraphPad Prism software by paired or unpaired two-tailed Studenťs t-test when comparing 2 samples. For a comparison of 3 or more samples, ANOVA was performed followed by Tukey's post hoc test. P values of <0.05 were considered statistically significant. Data are expressed as mean ± standard error of mean (SEM) with statistical significance: *p< .05, **p< .01, and ***p< .001.
3. Results
3.1. Fluvastatin augments IL-33-induced cytokine production by mast cells.
Recent publications have demonstrated statins can unexpectedly augment IL-1β and TNF production from LPS-stimulated macrophages (25, 32) and enhance NK cell cytotoxic effects (26). Because IL-33 signaling is an important mast cell stimulus in allergic disease that also shares MyD88-mediated signaling with TLR receptors (33), we tested the effect of fluvastatin on IL-33-mediated mast cell function. BMMC were cultured with fluvastatin prior to activation with IgE/antigen crosslinking (IgE XL) or IL-33 stimulation. In agreement with our previous work, fluvastatin suppressed IgE-mediated IL-6 and TNF secretion. In contrast, fluvastatin significantly enhanced IL-6 and TNF production by IL-33-stimulated BMMC (Figure 1A).
Figure 1. Fluvastatin has opposing effects on IgE and IL-33-mediated cytokine.
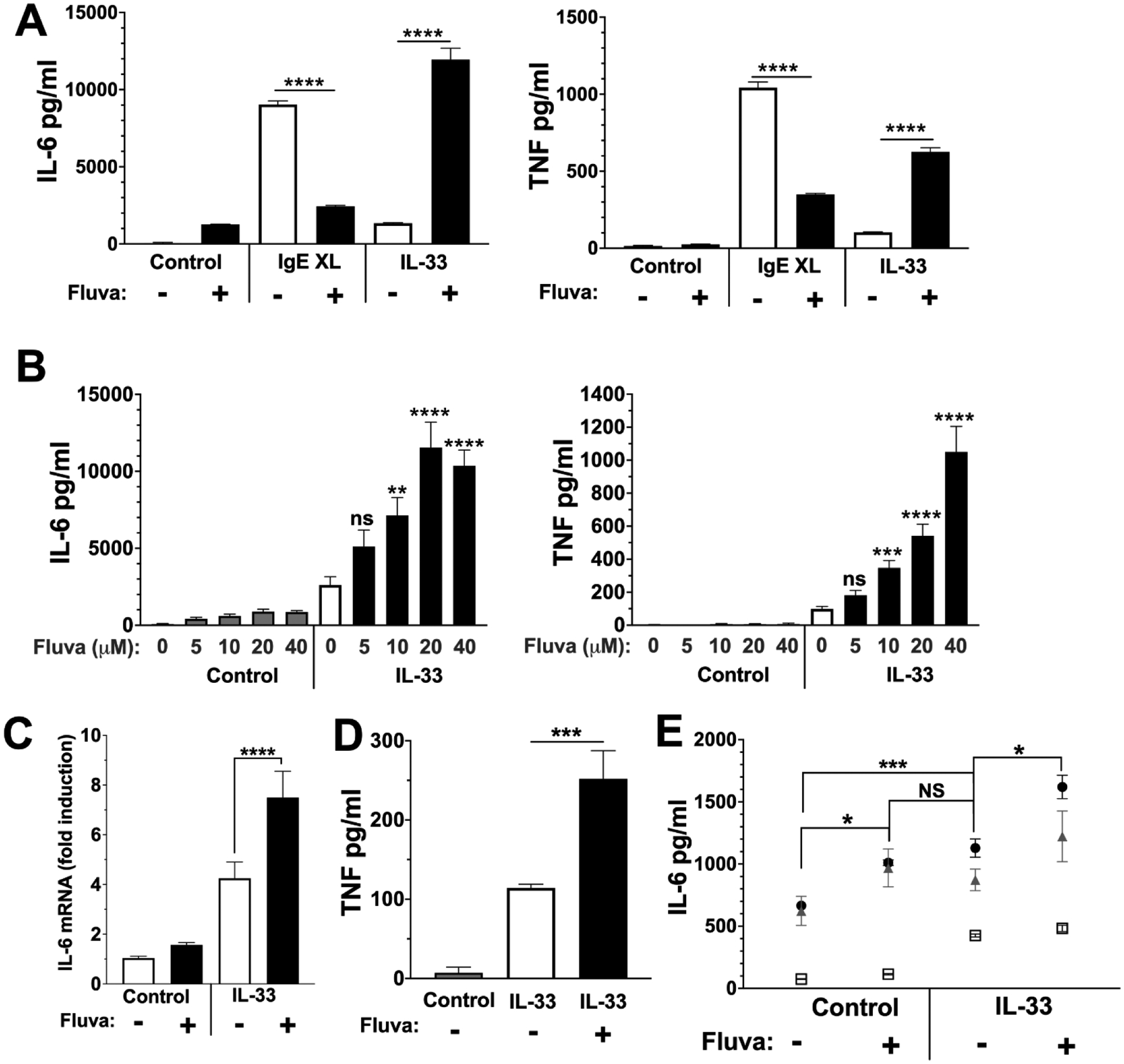
(A) BMMC were treated with anti-DNP IgE ± 20 μM Fluvastatin for 24 hours prior to IgE XL or IL-33 stimulation. Supernatants were collected 16 hours after activation and cytokines were measured via ELISA. Data are means ± SEM of 3 populations, representative of 2 independent experiments. (B) BMMC were treated with the indicated dose of fluvastatin for 24 hours prior to activation. Cells were then stimulated with IL-33 (100 ng/ml) for 16 hours, and cytokines were measured via ELISA. Data are means ± SEM of 9 populations from at least 2 independent experiments. (C) BMMC were treated as described in (A), with RNA collected 4 hours after IL-33 stimulation. RT-qPCR was used to measure IL-6 mRNA expression. Data are means ± SEM of 3 populations. (D) Peritoneal mast cells or (E) Skin-derived human mast cells from 3 donors were treated with 20 μM fluvastatin or DMSO for 24 hours prior to stimulation with 100 ng/ml of IL-33 stimulation. Supernatants were collected 16 hours after activation and cytokines were measured by ELISA. Data are means ± SEM, with each icon representing a donor.
To clarify this effect, BMMC were cultured with varying doses of fluvastatin (0–40 μM) for 24 hours (Figure 1B) before activation with IL-33. Fluvastatin effects on TNF and IL-6 were dose-dependent, with concentrations above 10 μM eliciting significant enhancement that generally increased with concentration. IL-33-induced IL-6 mRNA levels were also elevated in fluvastatin-treated mast cells in comparison to the control (Figure 1C).
Since mast cell phenotype is influenced by the microenvironment and BMMC mature in vitro, it was important to determine if fluvastatin effects were due to culture conditions (34). Comparable to BMMC cultures, fluvastatin augmented IL-33-induced TNF production by mouse peritoneal mast cells, which differentiate in vivo (Figure 1D). Human skin mast cells (HuMC) are similar to mouse peritoneal mast cells in their mature phenotype and expansion from a mature population (35). We found that HuMC stimulated with IL-33 produced little IL-6 in the absence of fluvastatin, but levels increased with fluvastatin treatment (Figure 1E). These data show that fluvastatin consistently increases cytokine production in IL-33-stimulated mouse and human mast cells.
3.2. Fluvastatin effects are SCF-dependent but not reliant on TNF production or changes in surface receptor expression.
Pro-inflammatory statin effects on NK cells are dependent on the mitogen IL-2 (26). Stem cell factor (SCF) is a mast cell mitogen that also augments activation by IgE or IL-33 (36, 37). We therefore determined whether SCF has a role in fluvastatin effects on IL-33-induced function. BMMC were cultured in IL-3 with or without SCF for three days prior to the addition of fluvastatin for 24 hours and subsequent IL-33 stimulation. As shown in Figure 2, fluvastatin had no effect on IL-33-induced IL-6 and little effect on TNF secretion in the absence of SCF. Further, SCF effects were concentration-dependent, with an optimal effect at 50 ng/ml (Figure 2).
Figure 2. Fluvastatin effects are SCF-dependent.
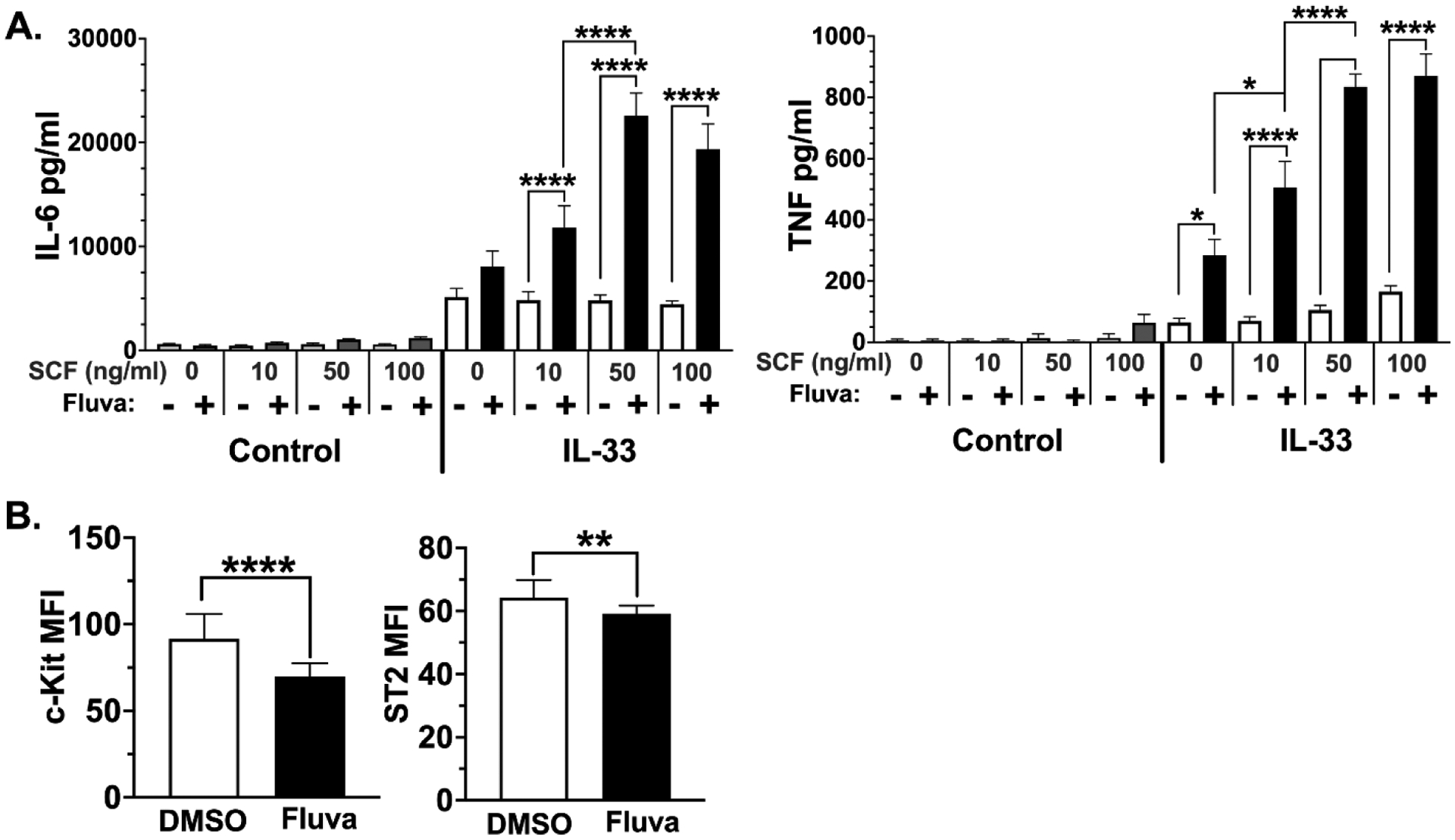
(A) BMMC were cultured in IL-3 ± SCF (50 ng/ml) for three days. Fluvastatin (20 μM) was added for 24 hours prior to IL-33 activation for 16 hours. Cytokines were measured via ELISA. Data are means ± SEM of 6 populations from 2 independent experiments. (B) BMMC were cultured for 24 hours in media containing DMSO (vehicle control) or fluvastatin (20 μM), and surface expression of c-Kit and ST2 was measured by flow cytometry. Data are means ± SD from 17 samples of BMMC populations.
Since SCF and IL-33 signaling are controlled by their surface receptors, we next determined if fluvastatin altered expression of CD117 and ST2, the receptors for SCF and IL-33, respectively. Flow cytometry analysis after 24 hours of culture in fluvastatin showed that both receptors were modestly reduced, an effect that would not explain increased signaling (Figure 2B).
After establishing the effects of fluvastatin on IL-33-stimulated mast cells, we next sought what factors augment IL-6 and TNF secretion. One possibility is that fluvastatin-enhanced TNF secretion may create a feedback loop that enhances IL-6 production. Unlike most cytokines, TNF can be pre-formed and stored in mast cell granules, allowing rapid release during degranulation (38). Also, TNF is known to increase IL-6 production from skeletal muscle cells (39). To determine if increased IL-6 production is secondary to TNF signaling, anti-TNF or anti-TNF receptor 1 and 2 neutralizing antibodies were added to cultures prior to IL-33 stimulation (Figure 3). TNF blockade did not alter fluvastatin effects on IL-6. These data support the conclusion that fluvastatin effects on IL-6 are not secondary to TNF-mediated feedback.
Figure 3. TNF blockade does not prevent fluvastatin effects.
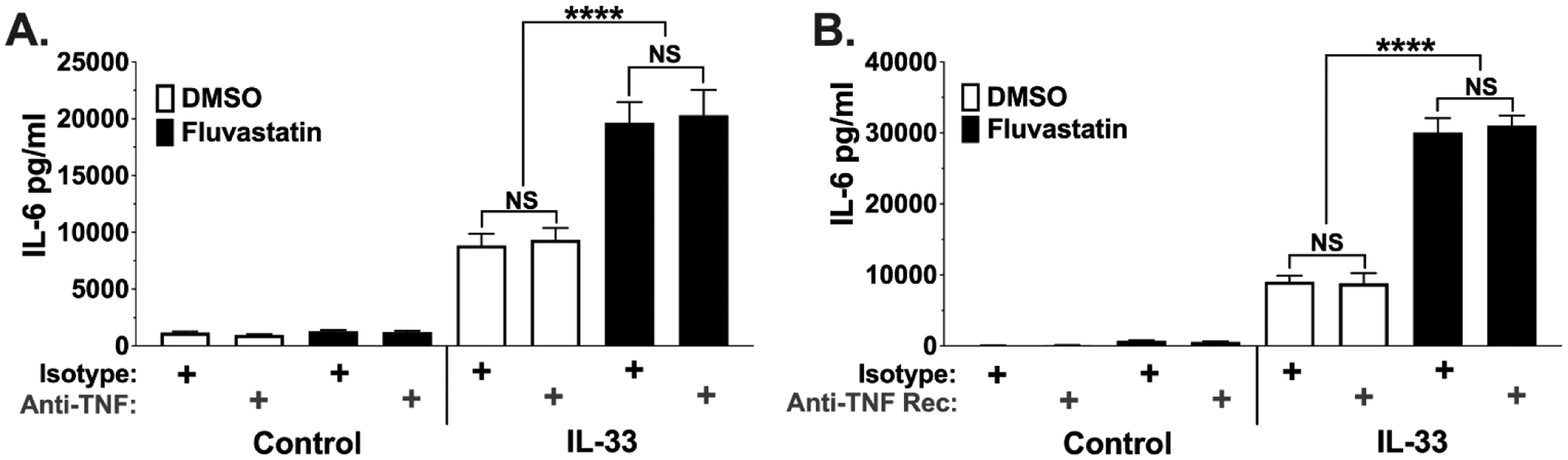
BMMC were cultured +/−fluvastatin for 24 hours as described in Figure 1. Cells were then activated +/− IL-33 (100ng/ml) for 16 hours in the presence of (A) anti-TNF (5ng/ml), (B) anti-TNF receptor 1 and 2 (100 ng/ml), or isotype control antibodies. Culture supernatants were analyzed by ELISA. Data shown are means ± SEM from each of 3 BMMC populations analyzed in triplicate.
3.3. Fluvastatin-mediated increase in cytokine production is linked to the isoprenoid pathway.
Fluvastatin suppresses cholesterol production by competitively inhibiting the enzyme HMGCR, an apical rate-limiting step in cholesterol synthesis (Figure 4A). Blockade of this enzyme not only decreases cholesterol synthesis, but also reduces production of the isoprenoids farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP) used to modify Ras family G-proteins and other factors (10) (Figure 4A). Because this post-translational modification is needed for cellular localization, isoprenoid inhibition can disrupt many signaling cascades. We therefore determined which aspects of the pathway are involved in enhancing cytokine production.
Figure 4. Fluvastatin-mediated increase in cytokine production depends on inhibiting the isoprenoid arm of the HMG-CoA pathway.
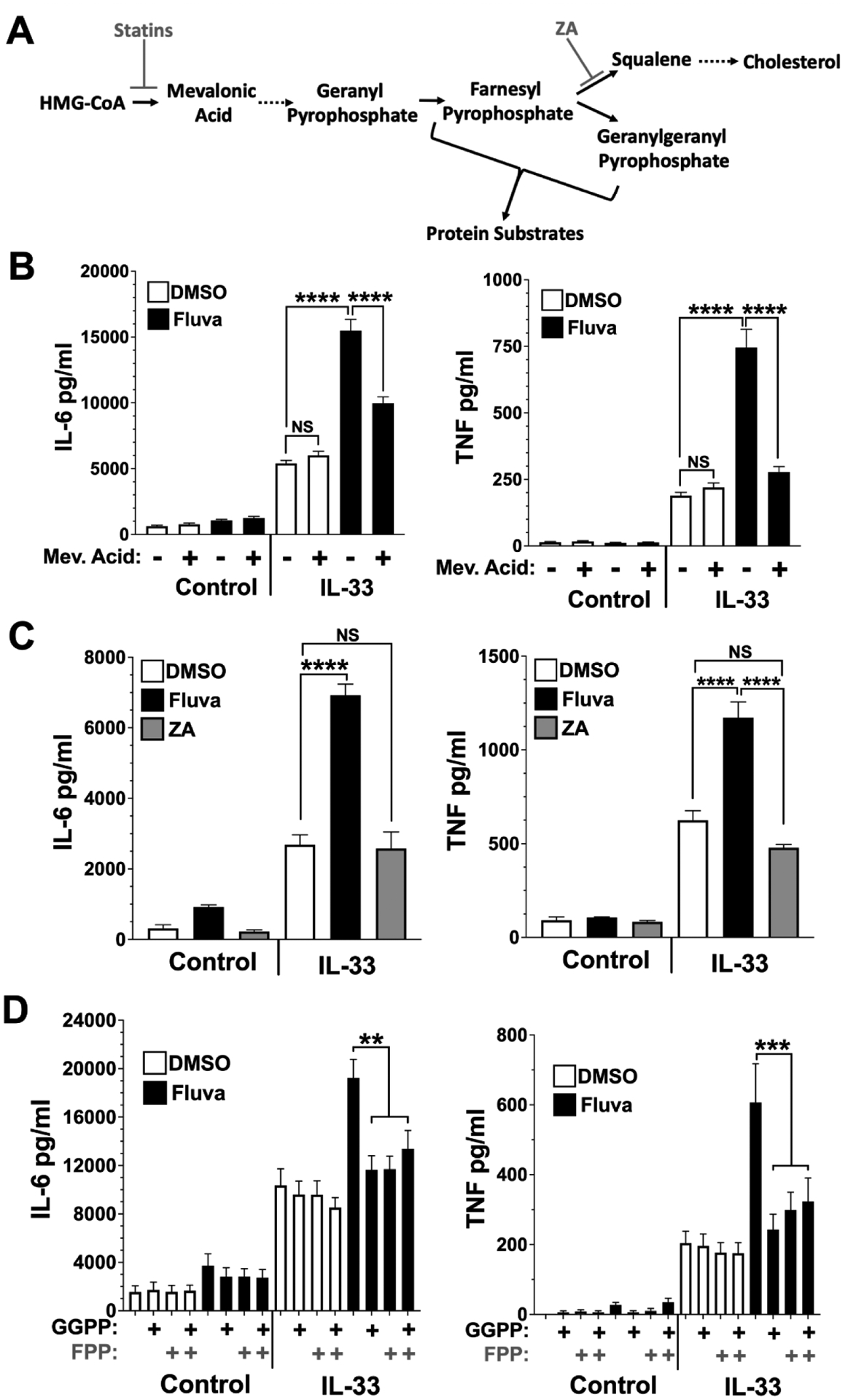
(A) A simplified depiction of cholesterol synthesis and the inhibitors used. Dashed arrows indicate summarized pathway steps in which intermediates are not shown. Inhibitors are indicated by gray font. (B) BMMC were cultured in vehicle or fluvastatin (20 μM) ± mevalonic acid (1 mM). (C) BMMC were cultured for 24 hrs in the presence of ZA or fluvastatin at 20 μM. (D) BMMC were cultured for 24 hrs in ± fluvastatin (20 μM) in the presence or absence of GGPP, ± FPP at 20 μM. Cultures were then activated with IL-33 (100 ng/ml) for 16 hours, and cytokines were measured by ELISA. Data shown are means ± SEM from 6 (B, D) or 14 (C) samples from at least 2 separate experiments.
We first determined if fluvastatin effects are linked to the intended target, HMGCR. BMMC were co-cultured with fluvastatin with or without mevalonic acid, the product of the HMGCR reaction, prior to stimulation with IL-33 (Figure 4B). The ability of fluvastatin to enhance IL-6 and TNF secretion was greatly diminished by mevalonic acid, suggesting that the drug effects are due to HMGCR suppression.
We next explored whether a decrease in cholesterol synthesis is responsible for fluvastatin-mediated IL-33 augmentation. BMMC were cultured for 24 hours with zaragozic acid (ZA), an inhibitor of squalene synthase, which is distal to HMGCR and isoprenoid synthesis. Unlike fluvastatin, zaragozic acid did not alter IL-33-induced TNF and IL-6 production (Figure 4C). Thus, loss of cholesterol per se does not appear to enhance cytokine production.
Isoprenoid synthesis lies between HMGCR and squalene synthase and is important for many signaling pathways. To determine the importance of isoprenoid depletion by fluvastatin, BMMC were co-cultured in fluvastatin with or without FPP and/or GGPP prior to IL-33 stimulation. The addition of either FPP or GGPP reversed fluvastatin effects, reducing IL-6 and TNF secretion (Figure 4D). These data support the hypothesis that fluvastatin effects are due to reduced availability of GGPP and FPP, which may contribute to loss of isoprenylation.
3.4. Fluvastatin alters signaling proteins downstream of IL-33 stimulation.
We next examined fluvastatin effects on IL-33 signaling, by observing changes in downstream phosphorylation events. Mast cells cultured with fluvastatin 24 hours prior to activation with IL-33 were analyzed by flow cytometry (Figure 5A). Like our IgE signaling study (23), IL-33-induced ERK and Akt phosphorylation was reduced by fluvastatin in comparison to vehicle control. Because we and others have found that ERK inhibition reduces IL-33-mediated cytokine production (40, 41) reduced ERK activation is an unlikely explanation for fluvastatin effects. While IL-33-induced Akt phosphorylation has generally been associated with promoting IL-33 function (3, 42–45), the role of IL-33-Akt signaling in mast cells is not clear. Therefore, we tested Akt inhibitors for their ability to mimic fluvastatin. As shown in Figure 5B, two different Akt inhibitors enhanced IL-33-mediated IL-6 production, much like fluvastatin. In contrast, Akt inhibition had no effect on TNF production. These data support the hypothesis that fluvastatin-mediated Akt inhibition could contribute to increasing IL-6 production. The role of Akt function in IL-33 signaling that selectively controls IL-6 production warrants further study.
Figure 5. Fluvastatin alters IL-33 induced signaling.
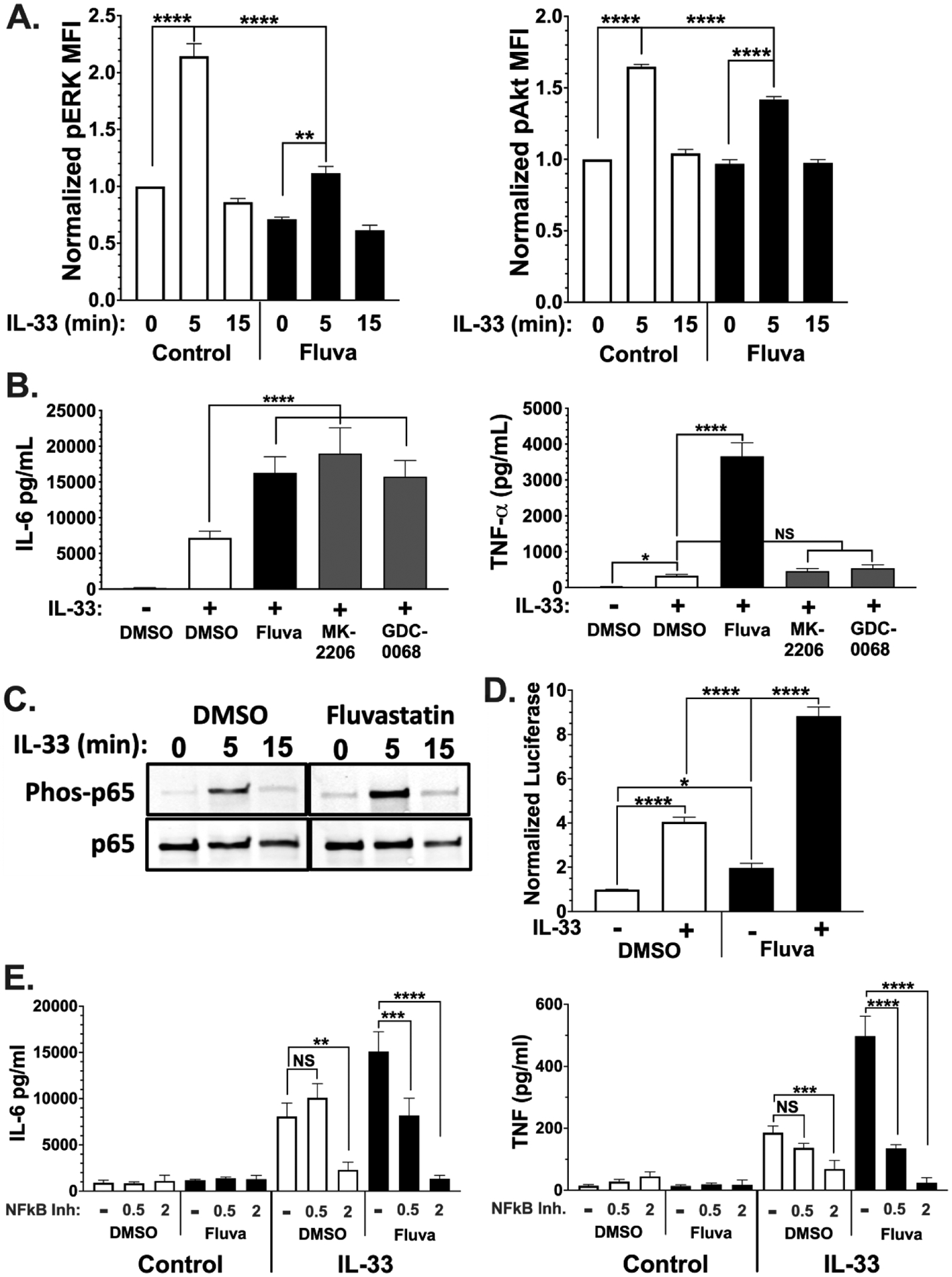
(A) BMMC were cultured for 24 hrs in the presence of vehicle or fluvastatin (20 μM) followed by activation with IL-33 (100 ng/ml) for 0–15 minutes. Samples were fixed and stained with the indicated antibodies and analyzed via flow cytometry. MFI of phosphoprotein was normalized to vehicle 0' time point. Data shown are means ± SEM from 3 populations, representative of 2 experiments. (B) BMMC were cultured for 24 hours with the indicated chemicals prior to IL-33 stimulation for 16 hours. Culture supernatants were analyzed by ELISA. The Akt inhibitors MK-2206 and GDC-0068 were used at 3 μM and 5 μM, respectively. Data shown are means ± SEM from 3 populations. (C) Samples were lysed and analyzed by western blot. Data shown are from 1 of 3 experiments that yielded similar outcomes. (D) BMMC were transfected with vectors encoding luciferase genes from Renilla reniformis under HSV-TK promoter and Firefly under NF-κB response elements. Cultures were then treated vehicle or fluvastatin for 24 hours prior to activation IL-33 for 2 hours. Ratios of Firefly to Renilla luciferase were normalized to DMSO unstimulated. (E) BMMC were cultured in the presence of vehicle or fluvastatin ± bay11–7082. Cells were activated with IL-33 for 16 hours, and cytokines were measured by ELISA. Data are means ± SEM of 9 (D) or 8 (E) populations from 3 independent experiments.
Downstream of these kinases, NF-κB is a critical component in IL-33-induced mast cell function (46). In striking contrast to our study of IgE signaling, fluvastatin increased IL-33-mediated NF-κB p65 phosphorylation (Figure 5C). This effect appeared to be functionally important, as a reporter assay showed greater NF-κB-mediated transcription. BMMC were transfected with a plasmid vector encoding Firefly luciferase under control of NF-κB response elements or a control plasmid prior to fluvastatin treatment and IL-33 stimulation. IL-33 induced luciferase production, an effect that was more than doubled by fluvastatin (Figure 5D). This indicates that fluvastatin increases IL-33-mediated NF-κB transcriptional activity.
Although the data show p65 activation and NF-κB transcriptional activity are elevated, the link between fluvastatin enhancement of IL-33-induced NF-κB function and increased cytokine production is correlative. To test the hypothesis that enhanced NF-κB function is required for fluvastatin effects, we used varying concentrations of an NF-κB inhibitor (Bay11–7082). At concentrations too low to inhibit IL-33-mediated cytokine secretion (0.5μM), the NF-κB inhibitor reversed fluvastatin effects (Figure 5D). These data suggest that fluvastatin increases IL-33-mediated NF-κB activity, resulting in greater IL-6 and TNF production.
3.5. Fluvastatin enhances IL-33-mediated inflammation in vivo.
Gunnar Nilsson's lab has shown that intraperitoneal IL-33 injection elicits neutrophil recruitment requiring mast cell-dependent TNF secretion (47). To test if fluvastatin enhances mast cell function in vivo, we injected mice with fluvastatin prior to IL-33 treatment (Figure 6A). Peritoneal cellular infiltration was assessed by flow cytometry of lavage fluid. We found that fluvastatin did not alter the number of CD45+ immune cells in the peritoneum (Figure 6B). As previously reported, IL-33 induced neutrophil infiltration. This was significantly enhanced by fluvastatin. Moreover, fluvastatin alone induced neutrophil recruitment commensurate with IL-33 (Figure 6C). This stimulatory effect was selective for neutrophils; fluvastatin decreased the percentage of several lineages, including mast cells, macrophages, CD4, CD8, and B cells (Figures 6C and 6D). Since only macrophages decreased in absolute number (DMSO = 6.7×105 cells; fluvastatin = undetected), the decreased percentage of other lineages was apparently due to neutrophil influx that shifted lineage percentages. IL-33 alone also decreased the percentage and number of macrophages but had no effect on any other lineage (Figures 6C and 6D). While we could not detect TNF in plasma, IL-33 increased plasma IL-6 levels. Fluvastatin induced plasma IL-6 and enhanced the effect of IL-33. Peritoneal lavage fluid showed similar changes in IL-6 without detectable TNF (Figure 6E). These data show that fluvastatin can enhance IL-33-mediated inflammation in vivo. In addition to promoting neutrophil infiltration, both fluvastatin and IL-33 decreased macrophages in the peritoneum, perhaps due to emigration.
Figure 6. Fluvastatin enhances IL-33-mediated cytokine production and neutrophil recruitment in vivo.
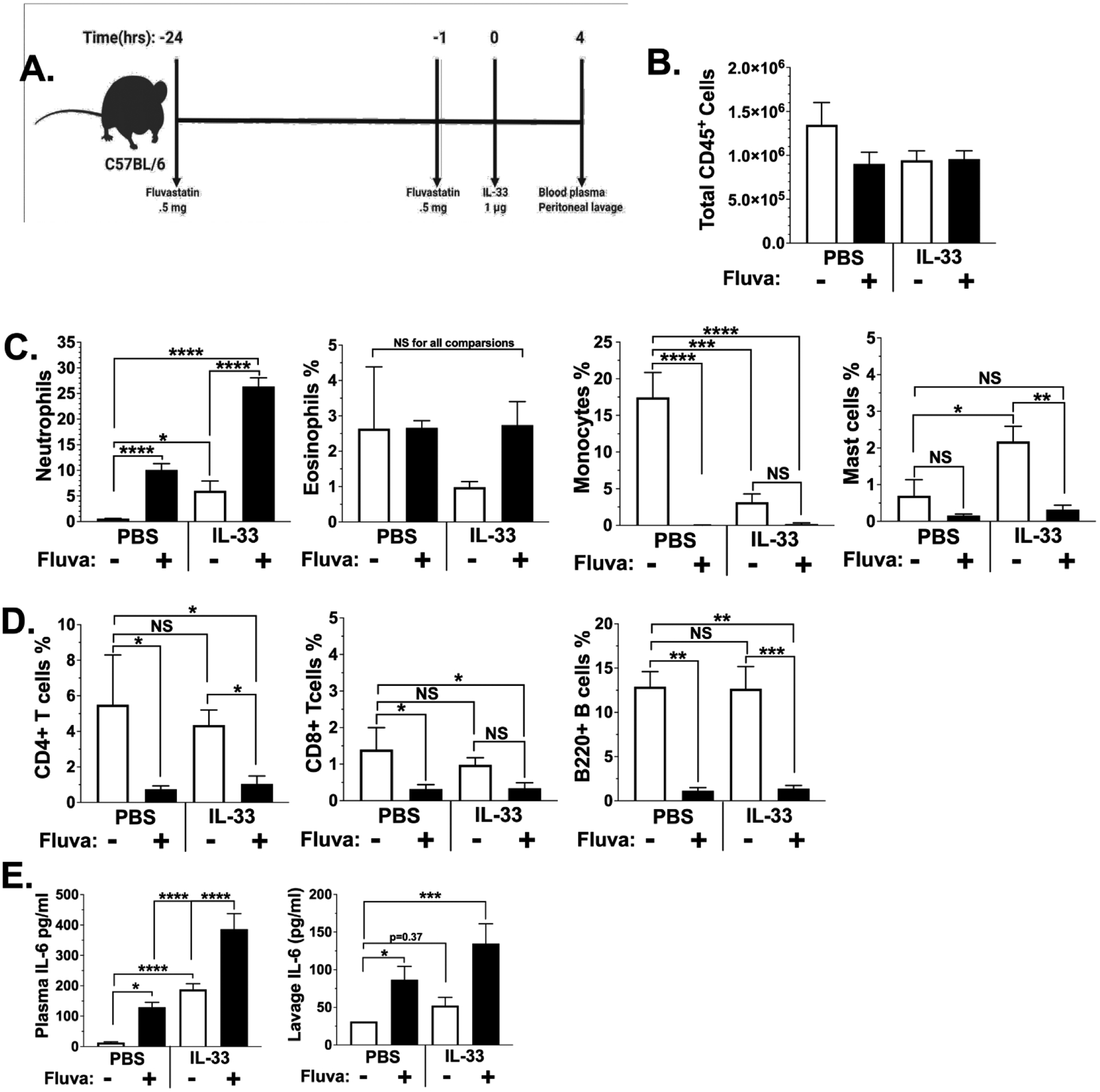
(A) C57BL/6J mice were injected i.p. with vehicle or fluvastatin (0.5 mg/mouse) 24 hours and again 1 hour prior to IL-33 (1 μg/mouse) or PBS. Schematic created with BioRender. Peritoneal lavage and cardiac puncture were performed 4 hours after IL-33 injection. (B) Cells from the peritoneal lavage were stained with anti-CD45 and the percentage positive cells was used to calculate total cell numbers in peritoneal lavage fluid. (C) The indicated innate immune lineages were assessed by flow cytometry. (D) T cell and B cell populations were assessed by flow cytometry. CD11b, Ly6C, and Ly6G and analyzed using flow cytometry. (E) Plasma and peritoneal lavage fluid were collected and assessed for IL-6 via ELISA. Data are means ± SEM of n=5 mice, analyzed by ANOVA, and representative of 3 independent experiments.
4. Discussion
Although statins are currently approved as a cholesterol-lowering drug, several studies have noted their immunomodulatory effects. Statins have been indicated as a treatment for inflammatory diseases such as autoimmune disorders, asthma, and cancer (13, 14, 20, 48–60). Statins have also been specifically implicated as a possible treatment for mast cell-driven disease. Cerivastatin and atorvastatin have been shown to inhibit human mast cell proliferation and IgE-induced degranulation (61). Fluvastatin has also been shown to inhibit IgE-induced degranulation in RBL-2H3 cells, and we previously demonstrated that fluvastatin inhibits IgE-mediated mast cell activation in vitro and in vivo (22, 23) and induces apoptosis after 3–4 days of exposure (24). Since IL-33 is associated with allergic diseases and is a known mast cell activator, we determined the effect of fluvastatin on IL-33-stimulated mast cells.
Fluvastatin appears to be acting on HMGCR as intended, based on the ability of mevalonic acid to reverse the augmentation. Further, suppressing cholesterol production with zaragozic acid did not enhance cytokine secretion, suggesting cholesterol loss is not required for fluvastatin effects. Downstream of the statin target HMGCR, farnesyl transferase and geranylgeranyl transferase couple isoprenoid lipids to many proteins, including small G proteins in the Ras Family (10). We previously found that statin effects on IgE-mediated mast cell function and survival were reversed by GGPP but not by FPP (23, 24). In the present study, both GGPP and FPP reversed fluvastatin-mediated cytokine enhancement. These data suggest the ability of fluvastatin to augment cytokine production is due to loss of geranylgeranyl transferase and farnesyl transferase actions on a pathway that restricts IL-33-mediated cytokine secretion. Despite fluvastatin actions likely being due to HMGCR inhibition, we were unable to replicate its effects with pitavastatin, pravastatin, or lovastatin, which had no consistent effect on IL-33-mediated TNF and IL-6 secretion (data not shown). This incongruence is like our earlier study of statin effects on FcεRI signaling, which also showed considerable drug variability (23, 24). Currently the reason for this variability is unknown. It could relate to drug uptake or degradation. These differences emphasize the importance of understanding the selective effects of drugs with the same mechanism of action.
When searching for changes in IL-33 signaling, we found reductions in ERK and Akt phosphorylation that mirrored our previous findings with IgE-mediated signals. In contrast, fluvastatin greatly increased NF-κB function, an effect that appeared to be necessary for augmented TNF and IL-6 production (Figure 5). Akt1/2 suppression by fluvastatin might partly explain how the drug can have opposite effects on IL-33- and IgE-induced function. While Akt has stimulatory effects downstream of the IgE receptor (62), it inhibits NF-κB function in LPS-stimulated monocytes (70, 95, 96). Because ST2 signaling mirrors LPS-induced signals, decreased Akt activity may allow greater NFκB function. In support of this, Akula et al. found that GGT deletion reduced the suppressive function of the KRAS-PI3K/Akt pathway, allowing greater NF-κB activation (25). Thus, one hypothesis is that statin-mediated HMGCR inhibition diminishes isoprenylated KRAS availability, resulting in reduced Akt-mediated suppression of NF-κB. In agreement with this possibility, Akt suppresses GSK3 α/β function by phosphorylating serine residues (63). Because GSK-3β promotes NF-κB transcriptional activity (64), reduced Akt function may equate to greater GSK-3β and NF-κB activation. Delineating these pathways in IL-33 signaling could reveal novel aspects of mast cell regulation. For example, Drube and coworkers demonstrated that PI3K inhibition reduces IL-33-induced cytokine production in BMMC (65). Because PI3K is often linked to Akt activation, one might logically conclude that Akt promotes cytokine production. However, many pathways lie downstream of PI3K and the inhibitory role for Akt in TLR signaling supports the possibility that Akt restricts rather than enhances PI3K-induced mast cell function.
Stem cell factor plays an essential role in mast cell function. It drives mast cell maturation, proliferation, migration, and enhances mast cell responses to IL-33 (36, 66). We found that fluvastatin had little effect in the absence of SCF (Figure 2). However, the mechanism explaining SCF function is unknown. SCF and IL-33 co-stimulation augments STAT3, ERK1/1, JNK2, and NF-κB activation in mast cells (36). While fluvastatin inhibited ERK phosphorylation, NF-κB transcriptional activity was augmented. We propose that statin-mediated inhibition of the Akt pathway alleviates suppression on NF-κB signaling, which is then promoted by SCF. More experiments are needed to test this hypothesis.
Fluvastatin effects were consistent on mouse peritoneal mast cells and human skin mast cells in vitro and in a mast cell-dependent model of IL-33-induced peritonitis. We found that fluvastatin enhanced both IL-6 production and neutrophil recruitment. Although the previous models found that IL-33-mediated neutrophil recruitment is TNF-dependent (47), we were unable to detect significant TNF in plasma or peritoneal lavage fluid. TNF detection may be hampered in our assays by the short half-life of the cytokine. It was interesting to note that fluvastatin alone elicited neutrophil influx, macrophage loss, and increased plasma IL-6, suggesting the drug has potent immunomodulatory effects in naïve animals. These effects were more notable when combined with IL-33, supporting the in vitro findings that fluvastatin increases IL-33-induced signaling pathways.
Our work corroborates what some in vitro studies have found: that statins are pro-inflammatory in the proper context. These conform to findings on the rare genetic disease Mevalonate Kinase Deficiency (MKD). Those who suffer from MKD cannot metabolize mevalonate in the cholesterol synthesis pathway, terminating the cholesterol synthesis pathway similarly to statin blockade of HMGCR. These patients display hypocholesterolemia as expected, but also increased IgD and low IgG levels, and have periodic fever syndrome driven by high cytokines levels (27). It is thought that either the build-up of mevalonate or the lack of small G-protein isoprenylation causes innate immune cells to produce high levels of cytokines (28, 29). Our study suggests that mast cells might also produce pro-inflammatory cytokines in these patients, but more evidence is needed from human samples.
In summary, our data demonstrate an enhancing effect of fluvastatin on IL-33-mediated mast cell function. These data indicate important differences between IL-33 and IgE receptor signaling that warrant further investigation, since these pathways are rational targets for allergic disease. This unexpected outcome appears to be specific to fluvastatin, despite the drug acting on HMGCR as intended. Other statins tested showed no enhancing effects. While the mechanism can be at least partly ascribed to increased NF-κB function, why other statin family drugs do not have the same activity is unclear. This disparity emphasizes the importance of understanding drug-specific inhibitory and stimulatory effects statins have on immune function. Our data are consistent with work showing statins can enhance LPS-mediated IL-1β and TNF production by macrophages (25, 32). Because LPS/TLR4 signaling shares features of IL-33/ST2, this offers some direction in understanding pro-inflammatory statin effects, particularly the possibility that inhibiting Akt may have opposite effects in some signaling pathways. Understanding drug-specific effects in the statin class has direct relevance to the current COVID-19 pandemic, since statins are being tested in multiple studies and appear to reduce the risk of severe disease or death from COVID-19, especially among in-patient subjects (reviewed in (67)). Our work suggests that the choice of statin may matter significantly and needs to be included when interpreting these studies.
5. Conclusions
We find that fluvastatin has unexpected stimulatory effects on IL-33-mediated mast cell activation. Fluvastatin augmented IL-33-induced TNF and IL-6 production consistently in mouse and human mast cells through a process that was dependent on inhibiting isoprenylation and enhancing NF-κB function. While IL-6 and TNF were both enhanced, some mechanisms differed, since Akt inhibition reproduced fluvastatin effects on IL-6 but not TNF. The stimulatory effects of fluvastatin were also consistent in vivo, where the drug increased IL-33-mediated neutrophil recruitment and systemic IL-6 production. These results suggest that despite its inhibitory effects on FcεRI, fluvastatin has an opposite impact on IL-33. Other statins do not appear to have this enhancing effect. While statins are logical candidates for treating inflammatory disorders, these findings emphasize the need to understand drug-specific effects and how these drugs affect differing signaling cascades.
Highlights:
Fluvastatin surprisingly enhances IL-33-induced mast cell IL-6 and TNF secretion.
These effects are consistent on mouse and human cell and in vivo.
Fluvastatin effects are mediated by blocking isoprenylation.
Fluvastatin inhibits IL-33-induced Akt activation and is mimicked by Akt inhibitors.
Fluvastatin enhances NFκB activation by IL-33.
6. Acknowledgements
This work was supported by NIH grants R01AI138495 and R21AI138494 awarded to JJR.
Abbreviations:
- BMMC
bone marrow-derived mast cell
- FPP
farnesyl pyrophosphate
- GGPP
geranylgeranyl pyrophosphate
- HMG-CoA
3-hydroxy-3-methylglutaryl-coenzyme A
- HuMC
human mast cells
- MKD
mevalonate kinase deficiency
- NF-κB
nuclear factor kappa light chain enhancer of activated B cells
- SCF
stem cell factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors have no competing interests to disclose.
All authors agree with submission of this paper to Cellular Immunology
8. References
- 1.SJ. G 2016. The Mast Cell-IgE Paradox: From Homeostasis to Anaphylaxis. Am J Pathol. Feb;186(2):212–24.: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galli SJ, T. M 2012. IgE and mast cells in allergic disease. Nat Med. May 4;18(5):693–704. : [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parveen S, Saravanan DB, Saluja R, and Elden BT. 2019. IL-33 mediated amplification of allergic response in human mast cells. J Recept Signal Transduct Res 39: 359–367 [DOI] [PubMed] [Google Scholar]
- 4.Ho LH, Ohno T, Oboki K, Kajiwara N, Suto H, Iikura M, Okayama Y, Akira S, Saito H, Galli SJ, and Nakae S. 2007. IL-33 induces IL-13 production by mouse mast cells independently of IgE-FcepsilonRI signals. J Leukoc Biol 82: 1481–1490 [DOI] [PubMed] [Google Scholar]
- 5.Ali S, Huber M, Kollewe C, Bischoff SC, Falk W, and Martin MU. 2007. IL-1 receptor accessory protein is essential for IL-33-induced activation of T lymphocytes and mast cells. Proc Natl Acad Sci U S A 104: 18660–18665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siebenhaar F, R. F, Bischoff SC, Gibbs BF, Maurer M. 2018. Mast Cells as Drivers of Disease and Therapeutic Targets. Trends Immunol. Feb;39(2):151–162.: [DOI] [PubMed] [Google Scholar]
- 7.Theoharides TC, T. I, Ren H 2019. Recent advances in our understanding of mast cell activation - or should it be mast cell mediator disorders? Expert Rev Clin Immunol Jun;15(6):639–656.: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salami JA, W. H, Valero-Elizondo J, Spatz ES, Desai NR, Rana JS, Virani SS, Blankstein R, Khera A, Blaha MJ, Blumenthal RS, Lloyd-Jones D, Nasir K. 2017. National Trends in Statin Use and Expenditures in the US Adult Population From 2002 to 2013: Insights From the Medical Expenditure Panel Survey. JAMA Cardiol Jan 1;2(1):56–65: [DOI] [PubMed] [Google Scholar]
- 9.Stancu C, S. A 2001. Statins: mechanism of action and effects. J Cell Mol Med. Oct-Dec;5(4):378–87.: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Montecucco F, M. F 2009. Update on statin-mediated anti-inflammatory activities in atherosclerosis. Semin Immunopathol. Jun;31(1):127–42.: [DOI] [PubMed] [Google Scholar]
- 11.Zeki AA, Bratt JM, Chang KY, Franzi LM, Ott S, Silveria M, Fiehn O, Last JA, and Kenyon NJ. 2015. Intratracheal instillation of pravastatin for the treatment of murine allergic asthma: a lung-targeted approach to deliver statins. Physiol Rep 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee CS, Yi EH, Lee JK, Won C, Lee YJ, Shin MK, Yang YM, Chung MH, Lee JW, Sung SH, and Ye SK. 2013. Simvastatin suppresses RANTES-mediated neutrophilia in polyinosinic-polycytidylic acid-induced pneumonia. Eur Respir J 41: 1147–1156 [DOI] [PubMed] [Google Scholar]
- 13.Thomson NC 2017. Clinical Studies of Statins in Asthma and COPD. Curr Mol Pharmacol 10: 60–71 [DOI] [PubMed] [Google Scholar]
- 14.Bhattacharjee D, Chogtu B, and Magazine R. 2015. Statins in Asthma: Potential Beneficial Effects and Limitations. Pulm Med 2015: 835204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu JN, Suh DH, Yang EM, Lee SI, Park HS, and Shin YS. 2014. Attenuation of airway inflammation by simvastatin and the implications for asthma treatment: is the jury still out? Exp Mol Med 46: e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeki AA, Oldham J, Wilson M, Fortenko O, Goyal V, Last M, Last A, Patel A, Last JA, and Kenyon NJ. 2013. Statin use and asthma control in patients with severe asthma. BMJ Open 3.3752054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walker DY, and Edwards KL. 2013. Statins in the treatment of asthma. Am J Health Syst Pharm 70: 1661–1669 [DOI] [PubMed] [Google Scholar]
- 18.Silva D, Couto M, Delgado L, and Moreira A. 2012. A systematic review of statin efficacy in asthma. J Asthma 49: 885–894 [DOI] [PubMed] [Google Scholar]
- 19.Huang CC, Chan WL, Chen YC, Chen TJ, Chou KT, Lin SJ, Chen JW, and Leu HB. 2011. Statin use in patients with asthma: a nationwide population-based study. Eur J Clin Invest 41: 507–512 [DOI] [PubMed] [Google Scholar]
- 20.Cowan DC, Cowan JO, Palmay R, Williamson A, and Taylor DR. 2010. Simvastatin in the treatment of asthma: lack of steroid-sparing effect. Thorax 65: 891–896 [DOI] [PubMed] [Google Scholar]
- 21.Menzies D, Nair A, Meldrum KT, Fleming D, Barnes M, and Lipworth BJ. 2007. Simvastatin does not exhibit therapeutic anti-inflammatory effects in asthma. J Allergy Clin Immunol 119: 328–335 [DOI] [PubMed] [Google Scholar]
- 22.Fujimoto M, Oka T, Murata T, Hori M, and Ozaki H. 2009. Fluvastatin inhibits mast cell degranulation without changing the cytoplasmic Ca2+ level. Eur J Pharmacol 602: 432–438 [DOI] [PubMed] [Google Scholar]
- 23.Kolawole EM, McLeod JJ, Ndaw V, Abebayehu D, Barnstein BO, Faber T, Spence AJ, Taruselli M, Paranjape A, Haque TT, Qayum AA, Kazmi QA, Wijesinghe DS, Sturgill JL, Chalfant CE, Straus DB, Oskeritzian CA, and Ryan JJ. 2016. Fluvastatin Suppresses Mast Cell and Basophil IgE Responses: Genotype-Dependent Effects. J Immunol 196: 1461–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paez PA, K. M, Taruselli MT, Ajith S, Dailey JM, Kee SA, Haque TT, Barnstein BO, McLeod JJA, Caslin HL, Kiwanuka KN, Fukuoka Y, Le QT, Schwartz LB, Straus DB, Gewirtz DA, Martin RK, Ryan JJ. 2020. Fluvastatin Induces Apoptosis in Primary and Transformed Mast Cells. J Pharmacol Exp Ther. Jul;374(1):104–112.: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akula MK, Shi M, Jiang Z, Foster CE, Miao D, Li AS, Zhang X, Gavin RM, Forde SD, Germain G, Carpenter S, Rosadini CV, Gritsman K, Chae JJ, Hampton R, Silverman N, Gravallese EM, Kagan JC, Fitzgerald KA, Kastner DL, Golenbock DT, Bergo MO, and Wang D. 2016. Control of the innate immune response by the mevalonate pathway. Nat Immunol 17: 922–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gruenbacher G, G. H, Nussbaumer O, Nussbaumer W, Rahm A, Thurnher M. 2010. IL-2 costimulation enables statin-mediated activation of human NK cells, preferentially through a mechanism involving CD56+ dendritic cells. Cancer Res. Dec 1;70(23):9611–20.: [DOI] [PubMed] [Google Scholar]
- 27.Jeyaratnam J, F. J 2020. Management of Mevalonate Kinase Deficiency: A Pediatric Perspective. Front Immunol. Jun 5;11:1150: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuijk LM, B. J, Koster J, Waterham HR, Frenkel J, Coffer PJ. 2008. HMG-CoA reductase inhibition induces IL-1beta release through Rac1/PI3K/PKB-dependent caspase-1 activation. Blood Nov 1;112(9):3563–73.: [DOI] [PubMed] [Google Scholar]
- 29.Kuijk LM, M. S, Schellens I, Waterham HR, Rijkers GT, Coffer PJ, Frenkel J. 2008. Statin synergizes with LPS to induce IL-1beta release by THP-1 cells through activation of caspase-1. Mol Immunol. Apr;45(8):2158–65.: [DOI] [PubMed] [Google Scholar]
- 30.McHale C, M. Z, Deppen J, Gomez G. 2018. Interleukin-6 potentiates FcepsilonRI-induced PGD(2) biosynthesis and induces VEGF from human in situ-matured skin mast cells. Biochim Biophys Acta Gen Subj. May;1862(5):1069–1078.: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McHale C, M. Z, Gomez G 2019. Human Skin-Derived Mast Cells Spontaneously Secrete Several Angiogenesis-Related Factors. Front Immunol. Jun 25;10:1445.: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bekkering S, A. R, Novakovic B, Kourtzelis I, van der Heijden CDCC, Li Y, Popa CD, Ter Horst R, van Tuijl J, Netea-Maier RT, van de Veerdonk FL, Chavakis T, Joosten LAB, van der Meer JWM, Stunnenberg H, Riksen NP, Netea MG. 2018. Metabolic Induction of Trained Immunity through the Mevalonate Pathway. Cell Jan 11;172(1–2):135–146.e9. : [DOI] [PubMed] [Google Scholar]
- 33.Jang TY, K. Y 2015. Interleukin-33 and Mast Cells Bridge Innate and Adaptive Immunity: From the Allergologisťs Perspective. Int Neurourol J. Sep;19(3):142–50.: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frossi B, M. F, Sibilano R, Danelli L, Pucillo CEM. 2018. Is it time for a new classification of mast cells? What do we know about mast cell heterogeneity? Immunol Rev. Mar;282(1):35–46.: [DOI] [PubMed] [Google Scholar]
- 35.Kambe N, Kambe M, Kochan JP, and Schwartz LB. 2001. Human skin-derived mast cells can proliferate while retaining their characteristic functional and protease phenotypes. Blood 97: 2045–2052 [DOI] [PubMed] [Google Scholar]
- 36.Drube S, Heink S, Walter S, Lohn T, Grusser M, Gerbaulet A, Berod L, Schons J, Dudeck A, Freitag J, Grotha S, Reich D, Rudeschko O, Norgauer J, Hartmann K, Roers A, and Kamradt T. 2010. The receptor tyrosine kinase c-Kit controls IL-33 receptor signaling in mast cells. Blood 115: 3899–3906 [DOI] [PubMed] [Google Scholar]
- 37.Iwaki S, Tkaczyk C, Satterthwaite AB, Halcomb K, Beaven MA, Metcalfe DD, and Gilfillan AM. 2005. Btk plays a crucial role in the amplification of Fc epsilonRI-mediated mast cell activation by kit. J Biol Chem 280: 40261–40270 [DOI] [PubMed] [Google Scholar]
- 38.Gordon JR, G. S 1991. Release of both preformed and newly synthesized tumor necrosis factor alpha (TNF-alpha)/cachectin by mouse mast cells stimulated via the Fc epsilon RI. A mechanism for the sustained action of mast cell-derived TNF-alpha during IgE-dependent biological responses. J Exp Med. Jul 1;174(1):103–7.: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Podbregar M, L. M, Prelovsek O, Mars T. 2013. Cytokine response of cultured skeletal muscle cells stimulated with proinflammatory factors depends on differentiation stage. ScientificWorldJournal. 2013:617170.: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abebayehu D, Spence AJ, Qayum AA, Taruselli MT, McLeod JJ, Caslin HL, Chumanevich AP, Kolawole EM, Paranjape A, Baker B, Ndaw VS, Barnstein BO, Oskeritzian CA, Sell SA, and Ryan JJ. 2016. Lactic Acid Suppresses IL-33-Mediated Mast Cell Inflammatory Responses via Hypoxia-Inducible Factor-1alpha-Dependent miR-155 Suppression. J Immunol 197: 2909–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tung HY, P. B, Huang SK, Zhou Y. 2014. Murine mast cells secrete and respond to interleukin-33. J Interferon Cytokine Res. Mar;34(3):141–7.: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakajima S, Ishimaru K, Kobayashi A, Yu G, Nakamura Y, Oh-Oka K, Suzuki-Inoue K, Kono K, and Nakao A. 2019. Resveratrol inhibits IL-33-mediated mast cell activation by targeting the MK2/3-PI3K/Akt axis. Sci Rep 9: 18423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ndaw VS, Abebayehu D, Spence AJ, Paez PA, Kolawole EM, Taruselli MT, Caslin HL, Chumanevich AP, Paranjape A, Baker B, Barnstein BO, Haque TT, Kiwanuka KN, Oskeritzian CA, and Ryan JJ. 2017. TGF-beta1 Suppresses IL-33-Induced Mast Cell Function. J Immunol 199: 866–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luo X, L. C, Wang Y, Yang G, Xu Y, Li G, Liao F, Tan S. 2020. Interleukin-33 Promotes Th2/Th17 Response in Eosinophilic and Non-Eosinophilic Nasal Polyps. ORL J Otorhinolaryngol Relat Spec. 82(1):34–39. : [DOI] [PubMed] [Google Scholar]
- 45.Yang Z, G. X, Wang J, Xu L, Zheng Y, Xu Y. 2018. Interleukin-33 enhanced the migration and invasiveness of human lung cancer cells. Onco Targets Ther. Feb 16;11:843–849.: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Milovanovic M, V. V, Radosavljevic G, Jovanovic I, Pejnovic N, Arsenijevic N, Lukic ML. 2012. IL-33/ST2 axis in inflammation and immunopathology. Immunol Res. Apr;52(1–2):89–99.: [DOI] [PubMed] [Google Scholar]
- 47.Enoksson M, Moller-Westerberg C, Wicher G, Fallon PG, Forsberg-Nilsson K, Lunderius-Andersson C, and Nilsson G. 2013. Intraperitoneal influx of neutrophils in response to IL-33 is mast cell-dependent. Blood 121: 530–536 [DOI] [PubMed] [Google Scholar]
- 48.Braganza G, Chaudhuri R, McSharry C, Weir CJ, Donnelly I, Jolly L, Lafferty J, Lloyd SM, Spears M, Mair F, and Thomson NC. 2011. Effects of short-term treatment with atorvastatin in smokers with asthma--a randomized controlled trial. BMC Pulm Med 11: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cardwell CR, Mc Menamin U, Hughes CM, and Murray LJ. 2015. Statin use and survival from lung cancer: a population-based cohort study. Cancer Epidemiol Biomarkers Prev 24: 833–841 [DOI] [PubMed] [Google Scholar]
- 50.Dai Y, Khanna P, Chen S, Pei XY, Dent P, and Grant S. 2007. Statins synergistically potentiate 7-hydroxystaurosporine (UCN-01) lethality in human leukemia and myeloma cells by disrupting Ras farnesylation and activation. Blood 109: 4415–4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fehr T, Kahlert C, Fierz W, Joller-Jemelka HI, Riesen WF, Rickli H, Wuthrich RP, and Ammann P. 2004. Statin-induced immunomodulatory effects on human T cells in vivo. Atherosclerosis 175: 83–90 [DOI] [PubMed] [Google Scholar]
- 52.Henslee AB, and Steele TA. 2018. Combination statin and chemotherapy inhibits proliferation and cytotoxicity of an aggressive natural killer cell leukemia. Biomark Res 6: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hothersall EJ, Chaudhuri R, McSharry C, Donnelly I, Lafferty J, McMahon AD, Weir CJ, Meiklejohn J, Sattar N, McInnes I, Wood S, and Thomson NC. 2008. Effects of atorvastatin added to inhaled corticosteroids on lung function and sputum cell counts in atopic asthma. Thorax 63: 1070–1075 [DOI] [PubMed] [Google Scholar]
- 54.Kanda H, Yokota K, Kohno C, Sawada T, Sato K, Yamaguchi M, Komagata Y, Shimada K, Yamamoto K, and Mimura T. 2007. Effects of low-dosage simvastatin on rheumatoid arthritis through reduction of Th1/Th2 and CD4/CD8 ratios. Mod Rheumatol 17: 364–368 [DOI] [PubMed] [Google Scholar]
- 55.Khush KK, and Waters DD. 2006. Effects of statin therapy on the development and progression of heart failure: mechanisms and clinical trials. J Card Fail 12: 664–674 [DOI] [PubMed] [Google Scholar]
- 56.Maneechotesuwan K, Ekjiratrakul W, Kasetsinsombat K, Wongkajornsilp A, and Barnes PJ. 2010. Statins enhance the anti-inflammatory effects of inhaled corticosteroids in asthmatic patients through increased induction of indoleamine 2, 3-dioxygenase. J Allergy Clin Immunol 126: 754–762 e751 [DOI] [PubMed] [Google Scholar]
- 57.Pezeshkpoor F, Farid Hosseini R, Rafatpanah H, Shakerian B, Jabbari F, Zandkarimi MR, Yousefzadeh H, Sadri H, Bahrami A, and Zamani MA. 2012. Efficacy of atorvastatin and antihistamines in comparison with antihistamines plus placebo in the treatment of chronic idiopathic urticaria: a controlled clinical trial. Iran J Allergy Asthma Immunol 11: 236–240 [PubMed] [Google Scholar]
- 58.Sassano A, Katsoulidis E, Antico G, Altman JK, Redig AJ, Minucci S, Tallman MS, and Platanias LC. 2007. Suppressive effects of statins on acute promyelocytic leukemia cells. Cancer Res 67: 4524–4532 [DOI] [PubMed] [Google Scholar]
- 59.Thomson NC, Charron CE, Chaudhuri R, Spears M, Ito K, and McSharry C. 2015. Atorvastatin in combination with inhaled beclometasone modulates inflammatory sputum mediators in smokers with asthma. Pulm Pharmacol Ther 31: 1–8 [DOI] [PubMed] [Google Scholar]
- 60.Zhang J, Yang Z, Xie L, Xu L, Xu D, and Liu X. 2013. Statins, autophagy and cancer metastasis. Int J Biochem Cell Biol 45: 745–752 [DOI] [PubMed] [Google Scholar]
- 61.Krauth MT, Majlesi Y, Sonneck K, Samorapoompichit P, Ghannadan M, Hauswirth AW, Baghestanian M, Schernthaner GH, Worda C, Muller MR, Sperr WR, and Valent P. 2006. Effects of various statins on cytokine-dependent growth and IgE-dependent release of histamine in human mast cells. Allergy 61: 281–288 [DOI] [PubMed] [Google Scholar]
- 62.Kitaura J, Asai K, Maeda-Yamamoto M, Kawakami Y, Kikkawa U, and Kawakami T. 2000. Akt-dependent cytokine production in mast cells. J Exp Med 192: 729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Duda P, A. S, Abrams SL, Steelman LS, Martelli AM, Cocco L, Ratti S, Candido S, Libra M, Montalto G, Cervello M, Gizak A, Rakus D, McCubrey JA. 2020. Targeting GSK3 and Associated Signaling Pathways Involved in Cancer. Cells Apr 30;9(5):1110: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hoesel B, S. J 2013. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer. Aug 2;12:86: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Drube S, Kraft F, Dudeck J, Muller AL, Weber F, Gopfert C, Meininger I, Beyer M, Irmler I, Hafner N, Schutz D, Stumm R, Yakovleva T, Gaestel M, Dudeck A, and Kamradt T. 2016. MK2/3 Are Pivotal for IL-33-Induced and Mast Cell-Dependent Leukocyte Recruitment and the Resulting Skin Inflammation. J Immunol 197: 3662–3668 [DOI] [PubMed] [Google Scholar]
- 66.Galli SJ, Tsai M, Wershil BK, Tam SY, and Costa JJ. 1995. Regulation of mouse and human mast cell development, survival and function by stem cell factor, the ligand for the c-kit receptor. Int Arch Allergy Immunol 107: 51–53 [DOI] [PubMed] [Google Scholar]
- 67.Fedson DS 2020. Statin Treatment of COVID-19. Am J Cardiol. 136: 171–173 [DOI] [PMC free article] [PubMed] [Google Scholar]