

Abstract
The androgen receptor (AR) is important in the development of both experimental and human bladder cancer. However, the role of AR in bladder cancer growth and progression is less clear, with literature indicating that more advanced stage and grade disease are associated with reduced AR expression. To determine the mechanisms underlying these relationships, we profiled AR-expressing human bladder cancer cells by AR ChIP-seq and complementary transcriptomic approaches in response to in vitro stimulation by the synthetic androgen R1881. In vivo functional genomics consisting of pooled shRNA or pooled ORF libraries was employed to evaluate 97 genes that recapitulate the direction of expression associated with androgen stimulation. Interestingly, we identified CD44, the receptor for hyaluronic acid, a potent biomarker and driver of progressive disease in multiple tumor types, as significantly associated with androgen stimulation. CRISPR-based mutagenesis of androgen response elements (ARE) associated with CD44 identified a novel silencer element leading to the direct transcriptional repression of CD44 expression. In human bladder cancer patients, tumor AR and CD44 mRNA and protein expression were inversely correlated, suggesting a clinically relevant AR-CD44 axis. Collectively, our work describes a novel mechanism partly explaining the inverse relationship between AR and bladder cancer tumor progression and suggests that AR and CD44 expression may be useful for prognostication and therapeutic selection in primary bladder cancer.
Keywords: Androgen, CD44 Molecule, Bladder Cancer, Transcription, ChIP-seq, Functional Genomics
INTRODUCTION
Bladder cancer is the sixth most common cancer type, with over 81,000 new cases expected to be diagnosed in 2020 (1). Notably, bladder cancer has a very strong male predominance, and this has been associated in part with greater environmental exposures such as smoking (2). However, adjusting for these confounding factors does not fully explain the greater risk in males (2). One explanation may be differences in the expression, or action, of sex steroid hormones such as androgens and estrogens (3). In support of this hypothesis is a significant body of evidence from experimental (4-7) and human studies (3) implicating the androgen receptor (AR) as a key molecule contributing to urothelial carcinogenesis. It has previously been shown that both male and female bladder cancer express AR, although females with bladder cancer have tumors which express AR to a lesser degree (2).
In contrast to the data implicating AR in carcinogenesis, there is uncertainty on the association of AR with clinicopathological features and patient outcome in established bladder cancer (8). Several studies have examined AR expression at the RNA and protein levels and most, but not all, have found highly heterogenous and overall lower AR expression in bladder cancer than normal urothelium (3-5,8-10). Furthermore, increased AR expression was typically associated with lower tumor grade and stage (3-5,8-10) both of which have significant prognostic ability in bladder cancer (11). These observations in bladder cancer run contrary to those of prostate cancer, where increased AR expression is relatively homogenous in cells throughout a tumor and a poor prognostic indicator in patients (12).
Given the putative protective role of AR in bladder cancer progression, we sought to further understand the molecular basis underlying these observations. We reasoned that an investigation of the underlying AR genomic regulation using transcriptomic analysis, further shaped by functional genomic assessments, would allow for identification of genes directly regulated by AR that have mechanistic importance in controlling bladder cancer biology. In this study, we show that activated AR directly inhibits expression of Cluster of differentiation 44 (CD44), a transmembrane receptor for hyaluronic acid, that has been strongly implicated in tumor progression in a number of tumor types, including bladder cancer (13). Our observations provide a novel association between AR and CD44, potentially explaining the protective role of AR in bladder cancer.
MATERIALS AND METHODS
Cell Lines
293FT cells (Invitrogen, Carlsbad, CA, USA) were utilized for viral packaging and maintained according to manufacturer instructions (e.g. DMEM + 10% FBS + sodium pyruvate). The UMUC3 cell line was maintained as previously described and as suggested by ATCC (14). Briefly, UMUC3 cells were maintained in MEM media with 10% FBS and Sodium Pyruvate. Single cell cloning accomplished via dilution cloning of the UMUC3 cell line led to the derivation of the UMUC3-c31 (clone 31) clonal derivative cell line which was maintained similarly to the parental cell line. For all experiments, medium was aspirated and exchanged for phenol-red free media supplemented with 10% charcoal stripped serum (CSS; Sigma-Aldrich; St. Louis, MO, USA) for a minimum of 24 hours prior to experiment initiation to wash out endogenous androgens. All cells were serially passaged by trypsinization and maintained at 37° C and 5% CO2 in a humidified atmosphere.
Proliferation
UMUC3-parental and UMUC3-c31 cells were plated in 96-well plates using CSS to androgen deprive the cells. 24-hours after plating, cells were treated with a titration of R1881 or ethanol (vehicle control) diluted in CSS. Cells were incubated for 72-hours under standard conditions. Alamar blue was added to each well, and cells incubated for 2-hours before being analyzed using a BioTek Synergy H1 (Winooski, VT, USA) for analysis.
Animals
All animal studies were performed in an AALAC-approved facility with approval of the University of Colorado – Anschutz Medical Campus Institutional Animal Care and Use Committee (IACUC). Male nude mice 8-10 weeks of age were used for all experiments. The number of mice used per group is noted in all specific experiments.
Castration
Mice were anesthetized using inhaled isoflurane (3% isoflurane with supplemental oxygen). Mice were placed in dorsal recumbency. The scrotum of the mice was cleansed with alternating iodine and ethanol swabs. A midline incision on the scrotum was made and the testicles visualized. Testicles were isolated, expressed, and cautery used to bisect the testicular neurovascular bundle. Incisions were closed with 4-0 VICRYL (VWR, Radnor, PA, USA) interrupted sutures and subcutaneous tissues closed with an autoclip (Braintree Scientific, Braintree, MA, USA). Mice were administered Buprenorphine SR (Zoopharm-Wildlife Pharmaceuticals; Laramie, WY, USA) and recovered on room air. Control mice received a sham procedure where the testicles were expressed, replaced, and the incision immediately closed as described above. Tumor volume was determined as previously described using the V = (S2 ∙ L) / 2 method, where L is the longest dimension measured and S is the perpendicular measurement.
ChIP-Seq
UMUC3-c31 cells were grown to 70-80% confluency and treated with vehicle control (ethanol; n=4) or 10nM R1881 (n=5; Perkin Elmer; Waltham, MA, USA) for 24 hours (Supplemental Methods). Cells were fixed with a 1% formaldehyde solution (Sigma; St. Louis, MO, USA) prior to lysis with a modified RIPA buffer. Chromatin was sheared using a Diagenode Bioruptor (Denville, NJ, USA). Input control samples were not treated with antibody to allow for background peak subtraction. Cleared suspensions were incubated with α-AR antibody (Santa Cruz Biotechnology; Dallas, TX, USA; SC-816X) and protein A/G PLUS-Agarose beads. Cross links were reversed using a basic elution buffer followed by Proteinase K and RNase A treatment (Qiagen; Germantown, MD, USA). DNA was purified using the QIAquick PCR purification kit (Qiagen) according to manufacturer instructions.
DNA fragmentation was confirmed by running eluted DNA on a 1.5% agarose gel with ethidium bromide and visualized using a Bio-Rad Chemidoc MP Imaging System (Hercules, CA, USA). Western blots to confirm immunoprecipitation, as described below, were performed to confirm enrichment.
Purified ChIP-DNA was submitted to the Genome Technology Access Center at Washington University of St. Louis for 1x50 sequencing. Reads were aligned to the reference human genome (hg38) using Novoalign and duplicate reads were removed. Peaks were identified using the narrow peak detection MACS2 algorithm using a negative input sample to accurately estimate background peaks. Candidate peaks were those considered uniquely identified in the AR treated samples. Motifs were identified using RSAT oligo-analysis and candidate motifs were those with a binomial significance score > 10. All relevant files have been uploaded to the NCBI Gene Expression Omnibus (GEO) repository (Series GSE147940; GSE147939).
Microarray
UMUC3-c31 cells were treated with vehicle control (ethanol; n=5) or 10nM R1881 (n=5). RNA was extracted and purified using the Qiagen RNeasy kit according to manufacturer instructions (Supplemental Methods). Purified RNA was submitted to the Genomics and Microarray Shared Resource at the University of Colorado – Anschutz Medical Campus for transcriptome measurement using PrimeView Human Gene Expression Arrays (HG-219; ThermoFisher Scientific; Waltham, MA, USA) in triplicate. Expression values were normalized and summarized into probesets using robust multichip analysis (RMA). For each probeset, a linear model was used to test for differential expression between samples treated with AR and controls using the lmFit function from the limma package (v3.34.9) in R (v3.4.3). A false discovery rate (FDR) was implemented to account for multiple testing and a probeset was considered differentially expressed if the FDR value <0.05. The Affymetrix HG-U219 (v35) annotation was used to link probeset ID to gene symbol. All relevant files have been uploaded to the NCBI Gene Expression Omnibus (GEO) repository (Series GSE147940; GSE147938).
ChIP-Seq & RNA Expression Integration
To determine which AR binding sites have a functional effect and regulate gene transcription we identified genes which were a candidate in both the ChIP-Seq and the differential expression analyses. We performed this integration at the gene symbol level. Enrichment analysis on GO-terms was performed on these AR regulated candidates using EnrichR (15).
Enrichment Analyses
For each of the three different candidate lists: 1) AR targeted genes (ChIP-Seq candidates), 2) differentially expressed genes (array candidates) and 3) AR regulated genes (intersection of ChIP-seq and array candidates), enrichment analysis on GO-terms was performed using EnrichR (15). For the intersection of genes identified by ChIP-seq and microarray, all GO pathways found in the Venn intersection with an FDR < 0.1 were compared against all Venn segments.
Western Blotting
Whole cell lysates were derived using a modified RIPA buffer with protease inhibitors (Supplemental Methods). Protein concentration was measured using a BCA kit (ThermoScientific) and BioTek Synergy H1 plate reader. Protein concentrations were normalized prior to running on 4-20% Tris-Glycine protein gels (Invitrogen; Carlsbad, CA, USA) and transferred to PVDF. Membranes were blocked with 5% nonfat dry milk solution prior to antibody incubation. The following antibodies were used for analysis: 1) α-AR antibody (D6F11); 2) β-actin (13E5); 3) CD44 (156-3C11); 4) α-rabbit IgG, HRP linked; 5) α-mouse IgG, HRP linked. All antibodies were purchased from Cell Signaling Technology (Dancers, MA, USA). Membranes were developed using SuperSignal West Pico Plus ECL (Thermo Scientific) and imaged on a Bio-Rad Chemidoc MP system. Relative protein concentrations were determined by image analysis using ImageJ.
Real-Time qPCR
RNA was isolated, purified, and quantified as described above. RNA was reverse transcribed using the Bio-Rad iScript cDNA synthesis kit. Quantitative real-time PCR (qPCR) was performed in triplicate using iTaq SYBR green mastermix (Bio-Rad) in 10ul reaction volumes on an Applied Biosystems (Foster City, CA, USA) QuantStudio6 with QuantStudio Realtime PCR software. Primers (Supplemental Table 1) were purchased from Integrated DNA Technologies (IDT; Coralville, IA, USA). Measurements from triplicate Ct values were normalized to β2-microglobulin, averaged, and reported using the ΔΔCt method.
siRNA
siRNA non-targeting (NTC) and AR were purchased from Dharmacon (Lafayette, CO, USA) and re-suspended according to manufacturer instructions. Cells were plated for 24-hours in standard media before transfection with 10nM siRNA complexes using Lipofectamine RNAiMax according to manufacturer instructions. After 72-hours of incubation with siRNA complexes, RNA was isolated as described above in preparation for qPCR analysis of AR.
ORF Functional Screen
To refine the list of genes overexpressed with R1881 stimulation, a library of pooled ORF clones (AR ORF sub-library) was utilized (Supplemental Methods). The AR ORF sub-library was generated using clones from the CCSB-Broad Lentiviral Expression Library (Dharmacon) by the Functional Genomics Facility at the University of Colorado – Anschutz Medical Campus. This library consisted of 63 clones (Supplemental Table 2 ORF-library) that were individually grown, pooled, and plasmid isolated using Qiagen Plasmid Plus Maxi kit. Pooled plasmids were packed into lentiviral particles and transduced into UMUC3-c31 cells. Due to the variable size of ORFs, a novel PCR based method of deconvoluting the library was developed (Supplemental Methods). Statistical analysis was performed similarly to the shRNA screen described below.
shRNA Functional Genomic Screen
A functional library of shRNAs (AR shRNA sub-library) targeting genes with decreased expression as observed post-R1881 treatment was developed. The AR shRNA sub-library (Supplemental Table 3) was generated from The RNAi Consortium shRNA library (Sigma-Aldrich) by the Functional Genomics Facility of University of Colorado – Anschutz Medical Campus (functionalgenomicsfacility.org; Supplemental Methods). The pooled shRNA library was packaged into lentiviral particles, transduced into UMUC3-c31, and incubated for 96 hours prior to injection into castrated mice. 1x106 transduced cells were injected subcutaneously into the dorsal flank of each mouse. Tumors were measured biweekly and mice euthanized when the tumor reached a largest diameter of 1cm. Genomic DNA (gDNA) was isolated from snap frozen tumors using the Gentra Puregene kit (Qiagen). Next generation sequencing of the shRNA cassette (primers detailed in Supplemental Table 4 was performed by 1x151 sequencing on a HiSeq4000 at the Genomics and Microarray Shared Resource at the University of Colorado – Anschutz Medical Campus. Reads were trimmed, aligned to known shRNA sequences, and quantified as counts. A negative binomial likelihood ratio test with shrinkage estimators for dispersion was implemented in DESeq2 to identify differentially expressed genes between groups while adjusting for time. The Benjamini Hochberg false discovery rate (FDR) was used to correct for multiple comparisons and significance was assessed based on an FDR adjusted p-value <0.05.
CRISPR targeted screen of proposed AREs
ChIP peaks associated with potential CD44 AREs were identified and gRNAs designed to saturate these peaks (Supplemental Table 5) were designed and created by the Functional Genomics Facility at the University of Colorado – Anschutz Medical Campus (Supplemental Methods). 131 oligonucleotides, corresponding to 121 ChIP peaks, 10 non-targeting control gRNA (NTC-gRNA), and 10 CDS targeting gRNA (CD44-CDS-gRNA) were developed. Lentiviral particles were prepared as described above.
UMUC3-c31 cells were transduced with the lentiviral pool. 96 hours after transduction, media was converted to CSS with puromycin for selection of cells containing the gRNA. 24 hours later, cells were treated with 10nM R1881 (n = 5) or vehicle control (n = 5). Puromycin selection was halted after 72 hours of treatment. After 10 days of R1881 treatment, cells were prepared for cell sorting. Briefly, cells were trypsinized, washed in 1x PBS, and re-suspended in FACS buffer, and stained with CD44-PE (Biolegend, clone IM7; San Diego, CA, USA). CD44+ cells were sorted into CD44hi and CD44lo expressing populations (Figure 5 and Supplemental Figure 3) using MoFlo XDP100 (Beckman Coulter; Indianapolis, IN 46268). gDNA was isolated from the pooled populations as described above. The gRNAs were PCR amplified using unique custom primers (Supplemental Table 6). Samples were analyzed by Novogene (Sacramento, CA, USA) using PE150 sequencing on a HiSeq4000. gRNAs were mapped using CRISPR-AnalyzeR v1.5 (16). CD44hi and CD44lo populations were compared using two-way ANOVA and multiple t-tests with a two-stage step-up Benjamini, Krieger, and Yekutieli FDR comparisons with an FDR cutoff of 0.05 for differential gRNA presence.
Figure 5: CRISPR based functional evaluation of putative CD44 regulatory AREs.

A pooled CRISPR library targeting AREs associated with CD44 was developed to saturate all potential AREs. A) A schematic of the experimental methodology of targeting potential CD44-AREs with CRISPR. UMUC3-c31 cells were transduced with the library before being treated with vehicle control (ethanol) or 10nM of R1881 in CSS media. B) UMUC3-c31 cells were sorted by FACS 1, 3, and 5 days post treatment into CD44low, CD44int, and CD44high groups and gDNA prepared for NGS of the gRNA. Volcano plots of CD44 population comparisons on Day 5 sorted samples shows a significant number of differential gRNAs present between the CD44low (C) and CD44high (D) populations. E) Three gRNAs found to have the most significant read count changes at Day 5 between populations with different CD44 expression.
CRISPR based validation of AR-CD44 AREs
CRISPR gRNAs found to have a significant impact on CD44 expression were chosen for further evaluation. Single sgRNAs were cloned into a lentiCRISPRv2-GFP backbone (as described in Supplemental Methods), lentiviral particles produced, and UMUC3-c31 transduced. Cells were expanded for 7 days prior to FACS analysis for CD44+GFP+ cells. Individual clones were expanded in FBS containing media to best ensure clonogenic survival. Once sufficient outgrowth was achieved, cell media was changed to CSS media and treated with R1881 as described above. Clones were screened by qPCR, ChIP-PCR, and western blotting to assess changes in CD44 expression (Supplemental Table 7).
Analysis of publicly available datasets
cBioPortal (www.cbioportal.org) was used to access the publicly available Bladder Urothelial Carcinoma (Firehose Legacy) dataset and queried for AR and CD44 mRNA expression data. Graphs presented were prepared by cBioPortal. Neoadjuvant chemotherapy data (17,18) for AR and CD44 was analyzed by linear regression using GraphPad Prism 8.
Immunohistochemistry
Contiguous sections of previously defined bladder cancer TMA’s were utilized (19) as described in Supplemental Methods. Antigen retrieval using pH6 citrate buffer followed by incubation with CD44 (1:100; clone: E7K2Y; Cell Signaling Technology, catalog #37259; Danvers, MA, USA) or AR (1:400; clone D6F11; Cell Signaling Technology, catalog #5153) antibody. AR and CD44 immunostaining was scored semi-quantitatively using a range of 0 (no staining) to 3 (intense staining). AR and CD44 expression data were analyzed for the primary tumor and any relevant metastases present on the TMA.
Statistics
Relevant statistical processes for ChIP-seq, microarray analysis, and analysis of next generation sequencing data are referenced above in association with the procedures being analyzed. Further statistical analysis was performed using Prism 8 (GraphPad Software; San Diego, CA, USA). Unpaired t-test, one-sample t-test, simple linear regression, and ANOVA were performed with Prism. For all statistical analyses where a specific FDR or p-value is not denoted a cutoff of p<0.05 was used to assess statistical significance.
RESULTS
Development of AR sensitive human bladder cancer cells
AR expression is heterogenous in human bladder tumors and established cell lines (9,20). To isolate AR positive cells, we utilized the well characterized bladder cancer cell line UMUC3 and single cell cloned it by limiting dilution. 42 clones were isolated, expanded, and whole cell lysates evaluated for AR expression by western blotting. 11 clones were found to express some AR while 32 were essentially negative. We observed a strong increase in AR protein expression when UMUC3 parental (UMUC3-P) cells were grown in phenol-red free charcoal stripped serum (CSS) cell culture media compared to complete serum containing media (FBS; fetal bovine serum). UMUC3-clone 31 (UMUC3-c31) was found to have a significant increase in AR expression, as measured by western (Supplemental Figure 1A), and RNA expression (Supplemental Figure 1B) when grown in CSS. To examine if increased AR expression of UMUC3-c31 has an impact on androgen driven proliferation, UMUC3-P and UMUC3-c31 cells were supplemented with synthetic androgen (R1881) and found to have similar growth upon stimulation (Supplemental Figure 1C). Treatment of UMUC3-c31 with R1881 increased expression of the androgen responsive gene FKBP prolyl isomerase 5 (FKBP5; Supplemental Figure 1D) while siRNA targeting AR decreased FKBP5 expression (Supplemental Figure 1E) (21). Intact mice (n = 5 per group) were challenged with UMUC3-P and UMUC3-c31 and observed to have similar (p > 0.05) growth characteristics in vivo (Supplemental Figure 1F). However, we observed that UMUC3-c31 tumor growth in castrated male mice (n = 5 mice per group; experiments repeated 3 times; n = 15 mice total per group) was significantly (p < 0.05) increased compared to intact mice (Supplemental Figure 1G). Therefore, we concluded UMUC3-c31 is an appropriate tool/reagent to study AR biology in bladder cancer due to its high AR expression and clonal derivation which minimizes the molecular noise generated by heterogeneity.
Defining the AR ChIP-seq and transcriptional landscape in human bladder cancer cells
To understand the regulatory network of AR in bladder cancer, we performed AR ChIP-seq using the UMUC3-c31 cell line. We identified 3599 unique genes with at least a single AR associated peak following R1881 stimulation (Figure 1A). Peak enrichment was primarily associated with non-promoter elements, such as distal intergenic elements (Figure 1B and Figure 1C). In general, each ChIP-seq peak was associated with 1-2 genes (Figure 1D). We observed a majority of AR associated peaks in intronic and distal intergenic sequences suggesting AR is playing a strong role controlling enhancer and silencer elements in bladder cancer (Figure 1C) similar to previous observations of AR in prostate cancer (22). We identified several potential AR motifs (Figure 1E) including the AGAACA motif previously identified in prostate cancer (22,23), further supporting the validity of our model and ChIP-seq data. We primarily observed GO pathways (FDR < 0.1) associated signal transduction and cytoskeletal organization (Figure 1F), similar to processes observed in other tumor types upon AR activation (22,23). AR ChIP peaks were associated with the canonical AR target gene FKBP5 (Figure 1G). Importantly, these data are the first AR ChIP-seq exploration in bladder cancer and provide evidence for the significance of AR regulation in bladder cancer.
Figure 1: Characterization of the AR regulon in bladder cancer.

UMUC3-c31 cells were stimulated with 10nM R1881 (n = 5) or vehicle control (n = 4) for 24 hours prior to analysis by ChIP-seq. Control peaks were background subtracted from five independent biological replicates performed. A) ChIP peaks from a representative biological replicate, minus background peaks, depicted across the human genome. B) Representation of ChIP peaks in association to the transcription start site of all genes. C) Graphical summary of ChIP-peak association with common promoter and genomic elements. D) Distribution of ChIP peaks in association with genes identified to be regulated by AR upon R1881 stimulation. E) Motif analysis showing bladder cancer specific AR binding motifs with the total number of sites shown. F) Gene Ontology (GO) pathway analysis of genes regulated by AR when stimulated by R1881 (FDR < 0.1). G) Representative description of ChIP peaks associated with the canonical AR target gene FKBP5 (NM_004117.4) accompanied by H3K4Me1, H3K4Me3, H3K27Ac, and DNaseI Signal.
To understand the transcriptomic alterations driven by AR stimulation, microarray analysis using UMUC3-c31 cells stimulated with 10nM R1881 for 24 hours was performed. We identified 250 unique genes (311 significant probesets) with an FDR < 0.05 (Figure 2A; Supplemental Table 8; including the canonical AR target gene FKBP5 (21). There was a preference for target gene expression to be increased with R1881 rather than inhibited (Figure 2B), again matching previous observations (12,24). Many GO-biological processes were found to be significantly (FDR < 0.05) regulated by AR signaling (Figure 2C). Pathways regulating proliferation, migration, and apoptosis are similar to those previously described to be associated with AR in prostate cancer (12,25). These data provide evidence that transcriptomic changes due to AR stimulation in bladder cancer are broad, with the potential existence of many biological processes regulated by AR.
Figure 2: Characterization of the AR transcriptome in response to AR stimulation by R1881 in UMUC3-c31 cells.

Cells were stimulated with 10nM R1881 (n = 5), or ethanol vehicle control (n = 5), for 24 hours followed by RNA isolation and microarray analysis. A) Volcano plot depicting fold change (log2) and statistical significance with red dots representing a significant difference (FDR < 0.05) between vehicle control and R1881 stimulated samples (n = 3/group). B) Unsupervised hierarchical clustering of the 250 genes found to be significantly (FDR < 0.05) differentially expressed. C) Gene Ontology (GO) analysis (FDR < 0.01) of the 250 genes identified.
To define an AR regulatory network in bladder cancer, we integrated the genes identified in the ChIP-seq and microarray assays. Together, we identified 97 genes with significant differential expression and at least a single associated ChIP-peak upon R1881 stimulation (Figure 3A; Supplemental Table 9). We observed 85 up-regulated genes and 12 down-regulated genes (Supplemental Table 9). 24 GO-biological processes were identified from the interaction of ChIP-seq and microarray (Figure 3B). To identify the individual components of the combined GO-processes (ChIP + Array; Intersection), ChIP and microarray datasets were reanalyzed restricted to the 24 processes identified in the combined data set. We did not observe a large correlation between processes identified individually in ChIP or microarray when compared to the intersection. These analyses suggest that some of the genes present in the intersection are not directly, or primarily, regulated by AR on a transcriptional level even though they are significantly influenced by stimulation with androgen. Therefore, it is necessary to define the genes for which AR directly regulates transcription and the biological processes those targets govern.
Figure 3: Description of 97 genes putatively regulated directly by AR in bladder cancer.

A) Genes identified in ChIP-seq and microarray were compared to define 97 genes putatively regulated directly by AR. B) Gene Ontology (GO) pathways were determined for the 97 gene overlap from the Venn diagram (FDR < 0.1). The GO pathways, in identical order, were analyzed across all the 3599 ChIP-seq specific genes and 250 microarray specific genes for comparison across modalities.
Functional evaluation of candidate AR effectors
We developed two functional genomics approaches to recapitulate AR activity in mice. In vivo experimentation allows for long term androgen deprivation and a more translational context for our initial findings. We first identified 85 genes up-regulated by R1881 stimulation and matched them with available ORF (over expressing; open reading frame) constructs from the CCSB-Broad Lentiviral Expression library. We identified 63 genes with available ORF constructs (Supplemental Table 2). However, 22 genes that were upregulated by R1881 did not have available ORF constructs; these genes were not included in the ORF pooled library. The ORF pool was transduced into UMUC3-c31 cells prior to subcutaneous challenge in intact (n = 6) and castrated (n = 4) male mice.
A novel probe-based qPCR methodology (Supplemental Methods, Supplemental Figure 2A, and Supplemental Table 10) was developed to deconvolute the construct counts in harvested tumors generated from cells harboring this pooled ORF library. We identified 52 of 63 constructs in the resulting tumors with P4HA3 (prolyl 4-hydroxylase subunit alpha-3) as the predominant construct. However, P4HA3 was present in both the intact (90.91%) and castrated (94.97%) tumors (Supplemental Figure 2B). None of the gene counts were found to be significantly differentially expressed between the intact and castrated groups (Supplemental Figure 2C).
Next, we sought to recapitulate the effects of the 12 genes downregulated with R1881 stimulation using an shRNA pool. In addition, the 22 genes up-regulated by R1881 which did not have available ORFs, were included in the shRNA pool. An shRNA pooled lentiviral library of the 34 genes was constructed (n = 5 shRNA/gene; Supplemental Table 3). β-actin and 4 non-targeting control shRNA constructs were included as internal controls. UMUC3-c31 cells were transduced with this shRNA library and injected subcutaneously into intact (n = 9) and castrated (n = 7) male mice (Figure 4A). Castrated mice were observed to have significantly smaller tumors than intact mice (p < 0.05; Figure 4B) which is consequential since UMUC3-c31 cells (non-transduced) show significantly (p < 0.05) enhanced growth when grown in castrated mice (Supplemental Figure 1G). Next generation sequencing of the shRNA cassette of the resulting tumors was performed (Supplemental Table 4). We identified 28 shRNA constructs, covering 20 unique genes, which had significantly (FDR < 0.1) different counts between the intact and castrated tumors (Figure 4C and Supplemental Table 11). Genes with multiple significant shRNA constructs whose directionality of count accumulation did not match (e.g. one shRNA enriched and one depleted) were excluded (Figure 4D). Constructs whose accumulation did not recapitulate the directionality of the array data were excluded (Figure 4D). Filtering resulted in 7 potential AR regulated genes (Figure 4E).
Figure 4: Functional screening of 12 AR down-regulated gene candidates.
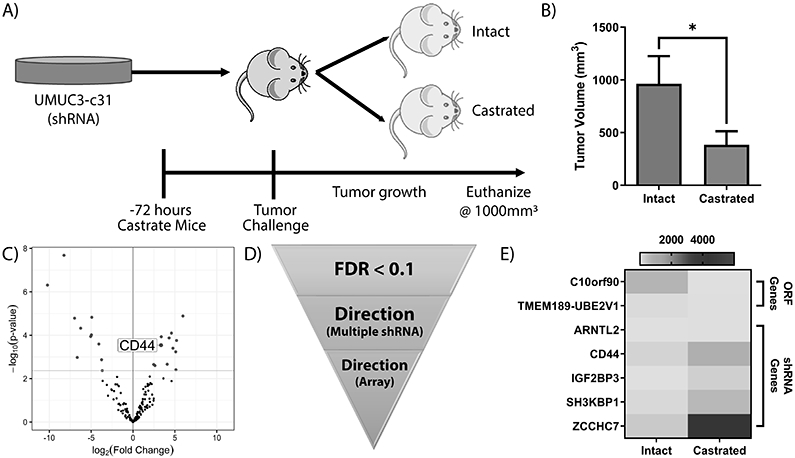
The 97 genes identified via ChIP-seq and transcriptomic approaches were screened using pooled ORF and shRNA libraries. A) Schematic showing experimental design of the ORF and shRNA screens. UMUC3-c31 cells were transduced with pooled libraries, selected, and injected into castrated or intact mice. B) Final tumor volume of mice (intact n = 6; castrated n = 4) challenged with the pooled AR-shRNA library show a significant inhibition in tumor volume associated with castration (p<0.05; two-tailed unpaired t-test). Tumors were resected and prepared for NGS using library specific methods. C) Volcano plot depicting shRNA constructs significantly varying between sham and castrated tumors. D) Filtering schema of shRNA constructs. Genes with multiple significant shRNA constructs with contradictory accumulation were removed from analysis. Genes not recapitulating the R1881 induced expression as measured by microarray were removed from analysis. E) Heatmap of filtered significant (FDR < 0.1) genes identified from the pooled screen. Genes with more than one shRNA construct were averaged for graph.
We were particularly intrigued by CD44 for several reasons. First, CD44 is an important mediator of increased bladder cancer aggressiveness (13,19,26) and an established cancer stem cell driver in multiple tumor types (13). Second, it is a potential therapeutic target via classical antibody targeting and novel approaches such as nanoparticles (27-30). Thirdly, prostate and breast cancer cell lines preferentially express AR or CD44 with an inverse association present in both tumor types (31,32). Taken together this compelled us to investigate the potentially novel relationship of AR and CD44 further. However, to our knowledge, it has yet to be shown that AR can directly regulate CD44 transcription, even though AR overexpression experiments have shown decreased CD44 expression (32).
Validation of CD44 as a direct target of AR
CD44 expression decreased upon R1881 stimulation (Supplemental Table 8). To determine if CD44 was directly regulated by AR, a lentiviral CRISPR library to mutate potential AREs was developed (Figure 5A). 123 gRNAs were designed (Supplemental Table 5) to saturate and disrupt AR binding to the 12 potential AREs associated with CD44 as determined using our own and publicly available data (33-35). Non-targeting control (gRNA-NTC) and positive controls targeting the CD44 coding sequence (gRNA-CD44-CS) were included. The pooled CRISPR library was transduced into UMUC3-c31 cells and subsequently stimulated with R1881 or vehicle control. Cells were sorted by FACS based on CD44 expression (Figure 5B). As expected, UMUC3-c31 cells showed a reduction in CD44 expression as early as one day post-R1881 treatment (Figure 5B and Supplemental Figure 3A). In general, R1881 stimulation led to an increase in CD44low cells compared to CSS (Supplemental Figure 3B). Genomic DNA (gDNA) was isolated from CD44low, CD44int and CD44high cells. PCR amplification of the gRNA was performed for NGS analysis (Supplemental Table 6). Read counts of the resulting gRNA data were normalized and gRNA counts were compared statistically between CSS and R1881 for CD44low (Figure 5C) and CD44high (Figure 5D) populations. Based on these analyses, 9 gRNAs were significantly (p < 0.05) different between CSS and R1881 in the CD44high or CD44low cells (Figure 5D). As expected, cells with gRNA targeting the CD44 coding sequence were enriched in CD44low cells. Similar to shRNA filtering, AREs with conflicting gRNA accumulation between CD44low and CD44high cells were excluded. We identified 3 AREs with potential CD44 silencing function (named CD44.006, CD44.008, and CD44.009; Figure 5E). The sgRNA with the strongest effect for each ARE was chosen for further validation.
Validation of specific CD44 ARE disruption in regulating CD44 transcription
The ChIP sites denoted as CD44.006, CD44.008, and CD44.009 are depicted in association with H3K4Me1, H3K4Me3, H3K27Ac, and DNase I Signal (accessible chromatin) in relation to their position of the genomic CD44 sequence (Figure 6A; Supplemental Figure 4A-C). sgRNAs were cloned into a LentiCRISPR-v2 vector expressing GFP as a reporter. Two non-targeting control (NTC1 and NTC2) gRNAs were similarly transduced into UMUC3-c31 cells creating 5 independent cell lines. Flow cytometry was used to single cell sort GFP+ (sgRNA containing) cells for clonogenic expansion and validation. Sanger sequencing (Supplemental Table 11) of selected clones showed CRISPR-mediated insertions in UMUC3-c31.006 (CD44.006; Figure 6B) and UMUC3-c31.008 (CD44.008; Figure 6C). Sanger sequencing of CD44.009 clones showed disruption of the CD44 coding sequence, and thus these cells were excluded from further analysis. The insertion identified in UMUC3-c31.006 was found to interrupt the conserved partial AR motif (AGAAC) identified in Figure 1E and previously described in prostate cancer (22,23). However, UMUC3-c31.008 was not associated with a known AR motif. Nevertheless, the CRISPR based mutation observed in UMUC3-c31.008 is associated with the canonical signal transducer and activator of transcription 3 (STAT3) motif (ATTTC) as determined by MotifMap (36). STAT3 and AR have previously been described as acting in an androgen dependent manner to regulate/modulate AR target genes (37-39). To evaluate the impact of ARE mutation on CD44 expression, we exposed the 4 remaining cell lines to R1881 and evaluated Endothelin 2 (EDN2), a known positive control for AR activity (40). We found EDN2 increased with R1881 as expected in all cell lines (Figure 6D). Similarly, we observed a significant decrease (p < 0.05) in CD44 expression in both UMUC-c31.NTC1 and UMUC-c31.NTC2 control cell lines; mirroring the original observations in UMUC3-c31 cells. In contrast, CD44 expression did not change in UMUC3-c31.006 and increased significantly (p < 0.05) in UMUC3-c31.008, in the presence of R1881 (Figure 6D). This indicates both AREs are suppressors of CD44 expression and defines a mechanism of AR mediated CD44 inhibition. Next, we performed ChIP-PCR for both AR-CD44 AREs and FKBP5 (Figure 6E-G; Supplemental Figure 4D). CRISPR based mutagenesis at CD44-006 and CD44-008 did not alter AR binding to the FKBP5 ARE investigated but did alter target AR binding at the novel CD44 sites. Finally, CD44 protein expression was found to decrease significantly in UMUC-c31.NTC1 and UMUC-c31.NTC2 but not in UMUC3-c31.006 and UMUC3-c31.008 (Figure 6H). In conclusion, we have identified CD44 as a direct target for AR, and defined a novel ARE-silencer responsible for the direct regulation of CD44 transcription.
Figure 6: Validation of CD44 based AREs using single gRNA.

To verify that mutation of the putative ARE was effective, single gRNAs identified from the pooled screened were further validated. A) Schematic of the CD44 gene, ChIP-peaks, H3K4Me1, H3K4Me3, H3K27Ac, and DNAseI Signal (DNAse I Hypersensitivity) (e.g. active/open chromatin), and associated gRNAs used for further validation highlighted (red = CD44-006; blue = CD44-008; and purple = CD44-009). B) Alignment of CD44-006 and (C) alignment of CD44-008 UMUC3-clones showing mutated ARE located within introns. Black boxes show the CRISPR mediated insertion. The magenta box (B) shows the ‘AGAAC’ AR partial-motif broken by the CRISPR mediated insertion. The light blue box (C) shows the ‘ATTTC’ STAT3 motif broken by the CRISPR mediated insertion. D) qPCR validation of ARE disruption was validated by measuring EDN2 (positive control) and CD44 RNA expression (n = 4 per group). ChIP-PCR of the CD44-006 (E), CD44-008 (F), or a control ARE (G; FKBP5, control) are shown (n = 3–4 per group; 3 replicate experiments). ARE targeting significantly altered AR binding, but CRISPR based ARE modification did not alter a distant control site (G; FKBP5). H) Western blot of stable gRNA expressing UMUC3-c31 cells with densitometry of the resulting blot depicted as relative fold change in respect to vehicle control. Two-tailed unpaired t-test with p< 0.05 were used to assess D, E, F, and G. A one-sample t-test comparing each group to ‘1’ (no change in expression) with a p < 0.05 was used for (H).
Association of AR with CD44 expression in patients with bladder cancer
We next sought to determine if any evidence exists for the relationship between AR signaling and CD44 in human bladder cancer tumors. Analyzing TCGA data via cBioPortal, we found that CD44 expression significantly (p < 0.05) inversely correlated with AR expression in muscle invasive bladder cancer (Figure 7A) (41). Additionally, using data from a study of neoadjuvant chemotherapy (17,18), similar trends were observed pre and post-chemotherapy (Figure 7B). To assess protein associations, contiguous sections of bladder cancer TMAs were stained for AR and CD44. The TMA consists of patient’s primary bladder cancer and matched metastatic disease when available (19). Representative images of AR and CD44 expression in primary tumor and metastases are presented (Figure 7C; Supplemental Figure 5). In non-metastatic primary bladder cancer, no statistically significant (p > 0.05) difference in CD44 expression was observed (Figure 7D). Interestingly, the primary tumors of patients with AR-positive disease have a significantly (p < 0.05) lower CD44 expression compared to patients with AR-negative disease (Figure 7E). In contrast, the metastases from these same patients show no difference (p > 0.05) in CD44 expression as a function of AR expression (Figure 7E). When matched primary-metastases were compared for CD44 expression, an overall increase in CD44 expression, independent of AR status, was observed (Figure 7F). These data strongly support the clinical relevance in human bladder cancer of the mechanistic link we identified between AR and CD44 in our models.
Figure 7: RNA and protein assessment of AR-CD44 associations in patients with bladder cancer.

A) RNA expression correlation between AR and CD44 from bladder cancer TCGA dataset via www.cBioPortal.org (n = 408 samples). B) Regression of pre and post-neoadjuvant chemotherapy of patients with invasive bladder cancer for AR and CD44 RNA expression (n = 115). C) Representative sections of AR and CD44 IHC stained from contiguous sections are presented. D) CD44 stain intensity was not statistically different (p > 0.05) between patients based on AR expression. E) Patient primary and metastatic tumors were evaluated for AR and CD44 staining. A significant (bar; p < 0.05) decrease in CD44 staining was observed in those patients with AR positive primary disease compared to those with AR negative disease. In contrast, no difference in CD44 intensity was observed in metastatic sections. F) CD44 staining intensity in patients with matched primary-metastatic tumors was evaluated based on AR status. Overall, CD44 increased (p > 0.05) in metastatic tumors independent of AR status in the primary or metastatic tumor.
DISCUSSION
Bladder cancer is an androgen responsive tumor (4-7). Herein, we established the UMUC3-c31 cell line and analyzed by ChIP-seq and microarray to identify 97 genes transcriptionally altered by AR. Microarray technology led us to discover CD44, a biomarker of progressive disease, as a direct target of AR transcriptional regulation. In the future, it is possible that by using more comprehensive transcriptional technologies, such as RNA-seq, additional genes directly regulated by AR may be discovered. Utilizing multiple functional genomic approaches, we validate 2 potential AREs associated with CD44. AR expression in human bladder cancer tumors decreases with advanced stage and grade (8,11), indicating a possible role for AR as an inhibitor of tumor progression (Supplemental Figure 6A-D).
Despite overall AR positivity of the primary tumor, clonal CD44 expression, and emergence of AR+CD44+ cells, allows for metastatic progression (42). Since CD44 is an important contributor to EMT and metastasis (13,26,43), increased CD44 expression in metastases, independent of AR expression, may be explained by selective pressures in the primary tumors that drive expression and counteract the suppressive influences of AR. Herein, we have identified two putative silencer AREs associated with CD44. However, the enhancer/silencer function of these AREs is likely complex. Huang et al. recently surveyed epigenetic profiles to identify putative suppressors (44). They found that many genomic elements may act as shared enhancers or silencers depending on the transcription factor repertoire bound, leading to a mechanism which fine tunes gene expression through common enhancer/silencer elements (44). In addition to the AREs we defined herein, it is likely that other transcription factors, epigenomic modifiers, and co-factors regulate CD44 transcription. In breast cancer, it has been shown that ER and AR compete for the same DNA binding sequences (45). Additionally, the pioneer factor FOXA1 can act in cooperation with GATA3 and PPARγ to drive luminal phenotypes in bladder cancer (46). The ability of AR and FOXA1 to interact through the NFI family of transcription factors suggests plausible mechanisms behind these clinical observations (47). A recent report found that muscle invasive bladder cancer in males was associated with higher androgen response pathways scores were typically associated with a luminal phenotype suggesting that AR signaling may be partially responsible for luminal differentiation (48). Interestingly, CD44 expression was found to be inhibited in males, further suggesting an association between AR signaling and CD44 expression in muscle invasive bladder cancer. The authors also suggested the effect was independent of age, and thus the effect may be due to AR expression rather than endogenous androgens, as androgen levels are known to drop in older men (48).
CD44 is associated with epithelial-mesenchymal transition (EMT), a key biomarker of cancer stem cells (26). The EMT phenotype is a biomarker of chemoresistance in patients undergoing neoadjuvant chemotherapy for muscle invasive bladder cancer, and thus CD44 may be a driver of chemoresistance (43). Increased CD44 expression is also associated with IL-6 driven STAT3 activation which promotes an invasive phenotype associated with EMT (26). While UMUC3-c31.008 (CD44-008) does not include a canonical AR motif it does encode the conserved STAT3 motif ATTTC. This finding suggest a model supported by publications which have shown through immunofluorescence (38) and reciprocal co-immunoprecipitation (37,39) that AR and STAT3 interact to regulate AR target genes on conserved AREs for genes such as prostate-specific antigen (PSA; KLK3). Furthermore, STAT3 has been shown to directly interact with CD44 (reviewed in (42)) and has even been suggested as a therapeutic for CRPC when combined with enzalutamide (38). Hence, it is plausible that AR and STAT3 interact, or compete, to integrate CD44 expression. These processes may become exacerbated during tumorigenesis, progression and chemoresistance leading to poor outcomes. In its antagonistic form, this interaction may also be responsible for the clonal selection of AR+CD44+ cells discussed above.
We previously investigated the role of CD44 in bladder cancer progression (13,19), but never in the context of androgen-sensitive disease. CD44 is the receptor for hyaluronic acid and osteopontin (19,49). Hyaluronan synthase 2 (HAS2) is an isozyme responsible for the synthesis of hyaluronic acid (HA) and has previously been shown to be a poor prognostic factor in bladder cancer (49-52). Even though we did not identify HAS2 as an AR regulated gene in this study, it is plausible that other regulatory mechanisms may lead to increased HA production, thus facilitating autocrine or paracrine CD44 activation, thus leading to CD44 activation as described in previous studies (53-55). The development of an autocrine or paracrine feedback loop, allowing survival of AR+CD44low cells, may partially explain the lack of difference in CD44 expression observed in bladder cancer metastases.
Here, we describe how AR directly regulates CD44 transcription through interaction with a novel CD44-associated silencer. It has been widely described that the AR expressing and sensitive LNCaP prostate cancer cell line has very low expression of CD44 (56). Conversely, the PC3 and DU145 cell lines, which lack AR expression, have higher CD44 expression (32,57). Srinivasan et al. have recently reported that overexpression of AR in PC3 leads to decreased CD44 expression, and characteristics associated with stemness, suggesting a causal AR-CD44 link exists in prostate cancer (32). These associations support our conclusions that AR inhibits CD44 expression on the transcriptional level and that CD44 suppression may be applicable to other tumors, such as prostate cancer. Granted, AR is typically associated with increased aggressiveness in prostate cancer, but it is plausible that decreased CD44 may still be part of the AR regulon (12,25,58). As we have suggested, it is plausible that AR activation overall is a poor prognostic factor in prostate, but decreased CD44 expression may be an important component of AR transcriptional regulation.
Even though androgen deprivation therapy (ADT) may inhibit bladder cancer carcinogenesis (3,9), increased AR appears to be protective once bladder cancer is established (11). Consequentially, these data suggest that activation of AR in bladder cancer may inhibit disease progression post-diagnosis. Our data further suggests that AR activation may lead to continual suppression of CD44 leading to a mechanistic explanation why males are more likely to develop bladder cancer, but develop less aggressive tumors than females (48). To this end, it is plausible that AR stimulation, via supraphysiologic androgen (SPA) (59) or bipolar androgen therapy (BAT) (60) may provide a benefit to AR positive bladder cancer patients. Although paradoxical, there is limited evidence suggesting some patients may benefit from these therapies (61-63) and bladder cancer provides a unique context for investigation due to the observations that AR may be protective of bladder cancer progression (11). Due to the lack of clinically viable therapeutics targeting CD44 (13), other therapeutic avenues must be explored.
It has been previously documented, across multiple tumor types, that AR signaling is complex, sometimes contradictory, and typically context and tissue/tumor type dependent (8,64,65). Our data enriches this discussion by showing that AR activation leads to the binding of silencer elements associated with CD44 in bladder cancer. At this time, these observations are restricted to bladder cancer, but it is plausible that they extend to other AR sensitive tumor types such as prostate. Our data suggests that further investigation is necessary to determine the consequences, and role of, AR activity in bladder cancer progression, so selective use of AR-targeted therapy can be determined.
Supplementary Material
SIGNIFICANCE STATEMENT.
This study describes novel androgen response elements that suppress CD44 and an expected inverse correlation of AR-CD44 expression observed in human bladder tumors.
ACKNOWLEDGEMENTS
The authors would like to thank the National Institute of Health and the National Cancer Institute (grant number CA143971) for funding this study. We thank the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine for help with genomic analysis. The Center is partially supported by NCI Cancer Center Support Grant #P30 CA91842 to the Siteman Cancer Center and by ICTS/CTSA Grant# UL1 TR000448 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research. This publication is solely the responsibility of the authors and does not necessarily represent the official view of NCRR or NIH. We would also like to thank the Genomics and Microarray Shared Resource at the University of Colorado – Anschutz Medical Campus for their expertise in sequencing. We would like to thank Scott Cramer for additional ChIP-seq data. We would like to thank Dr. Andreii Rozhok for his aid in mapping and interpreting the shRNA functional genomic NGS screening data. We would also like to thank Dr. Divya Sahu (Department of Pathology – University of California, San Diego) for her aid in preparation of the immunohistochemistry data. We would like to thank Michael R. Freeman (Division of Cancer Biology and Therapeutics, Departments of Surgery & Biomedical Sciences, Cedars-Sinai Medical Center) for his critical review of the manuscript.
Financial Support: NIH grant CA143971 to D.T
ABBREVIATIONS
- ADT
Androgen Deprivation Therapy
- AR
Androgen Receptor
- ARE
Androgen Response Element
- BAT
Bipolar Androgen Therapy
- CD44
Cluster of Differentiation 44
- ChIP
Chromatin Immunoprecipitation
- CSS
Charcoal Stripped Serum
- EDN2
Endothelin 2
- EMT
Epithelial-Mesenchymal Transition
- FDR
False Discovery Rate
- FKBP5
FK506 Binding Protein 5
- gDNA
Genomic DNA
- gRNA
guide-RNA
- HA
Hyaluronic Acid
- HAS2
Hyaluronan Synthase 2
- NGS
Next Generation Sequencing
- NTC
Non-Targeting Control
- ORF
Open Reading Frame
- P4HA3
Prolyl 3-Hydroxylase subunit alpha-3
- PSA
Prostate Specific Antigen
- SPA
Supraphysiologic Androgen
- STAT3
Signal transducer and activator of transcription 3
- TMA
Tissue Microarray
Footnotes
Conflict of Interest: The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70:7–30 [DOI] [PubMed] [Google Scholar]
- 2.Janisch F, Shariat SF, Schernhammer E, Rink M, Fajkovic H. The interaction of gender and smoking on bladder cancer risks. Curr Opin Urol 2019;29:249–55 [DOI] [PubMed] [Google Scholar]
- 3.Izumi K, Taguri M, Miyamoto H, Hara Y, Kishida T, Chiba K, et al. Androgen deprivation therapy prevents bladder cancer recurrence. Oncotarget 2014;5:12665–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li P, Chen J, Miyamoto H. Androgen Receptor Signaling in Bladder Cancer. Cancers (Basel) 2017;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kashiwagi E, Ide H, Inoue S, Kawahara T, Zheng Y, Reis LO, et al. Androgen receptor activity modulates responses to cisplatin treatment in bladder cancer. Oncotarget 2016;7:49169–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Agarwal N, Dancik GM, Goodspeed A, Costello JC, Owens C, Duex JE, et al. GON4L Drives Cancer Growth through a YY1-Androgen Receptor-CD24 Axis. Cancer Res 2016;76:5175–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanguedolce F, Cormio L, Carrieri G, Calo B, Russo D, Menin A, et al. Role of androgen receptor expression in non-muscle-invasive bladder cancer: a systematic review and meta-analysis. Histol Histopathol 2019:18189 [DOI] [PubMed] [Google Scholar]
- 8.Inoue S, Mizushima T, Miyamoto H. Role of the androgen receptor in urothelial cancer. Mol Cell Endocrinol 2018;465:73–81 [DOI] [PubMed] [Google Scholar]
- 9.Izumi K, Ito Y, Miyamoto H, Miyoshi Y, Ota J, Moriyama M, et al. Expression of androgen receptor in non-muscle-invasive bladder cancer predicts the preventive effect of androgen deprivation therapy on tumor recurrence. Oncotarget 2016;7:14153–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee ES, Son DS, Kim SH, Lee J, Jo J, Han J, et al. Prediction of recurrence-free survival in postoperative non-small cell lung cancer patients by using an integrated model of clinical information and gene expression. Clin Cancer Res 2008;14:7397–404 [DOI] [PubMed] [Google Scholar]
- 11.Ide H, Inoue S, Miyamoto H. Histopathological and prognostic significance of the expression of sex hormone receptors in bladder cancer: A meta-analysis of immunohistochemical studies. PLoS One 2017;12:e0174746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lonergan PE, Tindall DJ. Androgen receptor signaling in prostate cancer development and progression. J Carcinog 2011;10:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sottnik JL, Theodorescu D. CD44: A metastasis driver and therapeutic target. Oncoscience 2016;3:320–1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Said N, Smith S, Sanchez-Carbayo M, Theodorescu D. Tumor endothelin-1 enhances metastatic colonization of the lung in mouse xenograft models of bladder cancer. J Clin Invest 2011;121:132–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 2013;14:128- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Winter J, Schwering M, Pelz O, Rauscher B, Zhan T, Heigwer F, et al. CRISPRAnalyzeR: Interactive analysis, annotation and documentation of pooled CRISPR screens. bioRxiv 2017 [Google Scholar]
- 17.Seiler R, Ashab HAD, Erho N, van Rhijn BWG, Winters B, Douglas J, et al. Impact of Molecular Subtypes in Muscle-invasive Bladder Cancer on Predicting Response and Survival after Neoadjuvant Chemotherapy. Eur Urol 2017;72:544–54 [DOI] [PubMed] [Google Scholar]
- 18.Seiler R, Gibb EA, Wang NQ, Oo HZ, Lam HM, van Kessel KE, et al. Divergent Biological Response to Neoadjuvant Chemotherapy in Muscle-invasive Bladder Cancer. Clin Cancer Res 2019;25:5082–93 [DOI] [PubMed] [Google Scholar]
- 19.Ahmed M, Sottnik JL, Dancik GM, Sahu D, Hansel DE, Theodorescu D, et al. An Osteopontin/CD44 Axis in RhoGDI2-Mediated Metastasis Suppression. Cancer Cell 2016;30:432–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Overdevest JB, Knubel KH, Duex JE, Thomas S, Nitz MD, Harding MA, et al. CD24 expression is important in male urothelial tumorigenesis and metastasis in mice and is androgen regulated. Proc Natl Acad Sci U S A 2012;109:E3588–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Magee JA, Chang L-w, Stormo GD, Milbrandt J. Direct, Androgen Receptor-Mediated Regulation of the FKBP5 Gene via a Distal Enhancer Element. Endocrinology 2006;147:590–8 [DOI] [PubMed] [Google Scholar]
- 22.Wilson S, Qi J, Filipp FV. Refinement of the androgen response element based on ChIP-Seq in androgen-insensitive and androgen-responsive prostate cancer cell lines. Sci Rep 2016;6:32611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shaffer PL, Jivan A, Dollins DE, Claessens F, Gewirth DT. Structural basis of androgen receptor binding to selective androgen response elements. Proc Natl Acad Sci U S A 2004;101:4758–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shiota M, Yokomizo A, Naito S. Increased androgen receptor transcription: a cause of castration-resistant prostate cancer and a possible therapeutic target. J Mol Endocrinol 2011;47:R25–41 [DOI] [PubMed] [Google Scholar]
- 25.Ferraldeschi R, Welti J, Luo J, Attard G, de Bono JS. Targeting the androgen receptor pathway in castration-resistant prostate cancer: progresses and prospects. Oncogene 2015;34:1745–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu CT, Lin WY, Chen WC, Chen MF. Predictive Value of CD44 in Muscle-Invasive Bladder Cancer and Its Relationship with IL-6 Signaling. Ann Surg Oncol 2018;25:3518–26 [DOI] [PubMed] [Google Scholar]
- 27.Huang WY, Lin JN, Hsieh JT, Chou SC, Lai CH, Yun EJ, et al. Nanoparticle Targeting CD44-Positive Cancer Cells for Site-Specific Drug Delivery in Prostate Cancer Therapy. ACS Appl Mater Interfaces 2016;8:30722–34 [DOI] [PubMed] [Google Scholar]
- 28.Gadhoum SZ, Madhoun NY, Abuelela AF, Merzaban JS. Anti-CD44 antibodies inhibit both mTORC1 and mTORC2: a new rationale supporting CD44-induced AML differentiation therapy. Leukemia 2016;30:2397–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeLisser HM. CD44: target for antiangiogenesis therapy. Blood 2009;114:5114–5 [DOI] [PubMed] [Google Scholar]
- 30.Crow AR, Song S, Suppa SJ, Ma S, Reilly MP, Andre P, et al. Amelioration of murine immune thrombocytopenia by CD44 antibodies: a potential therapy for ITP? Blood 2011;117:971–4 [DOI] [PubMed] [Google Scholar]
- 31.Riaz N, Idress R, Habib S, Azam I, Lalani E-NM. Expression of Androgen Receptor and Cancer Stem Cell Markers (CD44(+)/CD24(−) and ALDH1(+)): Prognostic Implications in Invasive Breast Cancer. Transl Oncol 2018;11:920–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Srinivasan D, Senbanjo L, Majumdar S, Franklin RB, Chellaiah MA. Androgen receptor expression reduces stemness characteristics of prostate cancer cells (PC3) by repression of CD44 and SOX2. J Cell Biochem 2018;120:2413–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Decker KF, Zheng D, He Y, Bowman T, Edwards JR, Jia L. Persistent androgen receptor-mediated transcription in castration-resistant prostate cancer under androgen-deprived conditions. Nucleic Acids Res 2012;40:10765–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robinson JL, Macarthur S, Ross-Innes CS, Tilley WD, Neal DE, Mills IG, et al. Androgen receptor driven transcription in molecular apocrine breast cancer is mediated by FoxA1. EMBO J 2011;30:3019–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Castro MA, de Santiago I, Campbell TM, Vaughn C, Hickey TE, Ross E, et al. Regulators of genetic risk of breast cancer identified by integrative network analysis. Nat Genet 2016;48:12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Daily K, Patel VR, Rigor P, Xie X, Baldi P. MotifMap: integrative genome-wide maps of regulatory motif sites for model species. BMC Bioinformatics 2011;12:495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsuda T, Junicho A, Yamamoto T, Kishi H, Korkmaz K, Saatcioglu F, et al. Cross-talk between signal transducer and activator of transcription 3 and androgen receptor signaling in prostate carcinoma cells. Biochem Biophys Res Commun 2001;283:179–87 [DOI] [PubMed] [Google Scholar]
- 38.Thaper D, Vahid S, Kaur R, Kumar S, Nouruzi S, Bishop JL, et al. Galiellalactone inhibits the STAT3/AR signaling axis and suppresses Enzalutamide-resistant Prostate Cancer. Sci Rep 2018;8:17307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ueda T, Bruchovsky N, Sadar MD. Activation of the androgen receptor N-terminal domain by interleukin-6 via MAPK and STAT3 signal transduction pathways. J Biol Chem 2002;277:7076–85 [DOI] [PubMed] [Google Scholar]
- 40.Moses MA, Kim YS, Rivera-Marquez GM, Oshima N, Watson MJ, Beebe KE, et al. Targeting the Hsp40/Hsp70 Chaperone Axis as a Novel Strategy to Treat Castration-Resistant Prostate Cancer. Cancer Res 2018;78:4022–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robertson AG, Kim J, Al-Ahmadie H, Bellmunt J, Guo G, Cherniack AD, et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017;171:540–56 e25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen C, Zhao S, Karnad A, Freeman JW. The biology and role of CD44 in cancer progression: therapeutic implications. J Hematol Oncol 2018;11:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hensley PJ, Kyprianou N, Purdom MS, He D, DiCarlo V, Wang C, et al. Predictive value of phenotypic signatures of bladder cancer response to cisplatin-based neoadjuvant chemotherapy. Urol Oncol 2019;37:572 e1– e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang D, Petrykowska HM, Miller BF, Elnitski L, Ovcharenko I. Identification of human silencers by correlating cross-tissue epigenetic profiles and gene expression. Genome Res 2019;29:657–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.D'Amato NC, Gordon MA, Babbs B, Spoelstra NS, Carson Butterfield KT, Torkko KC, et al. Cooperative Dynamics of AR and ER Activity in Breast Cancer. Mol Cancer Res 2016;14:1054–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Warrick JI, Walter V, Yamashita H, Chung E, Shuman L, Amponsa VO, et al. FOXA1, GATA3 and PPAR Cooperate to Drive Luminal Subtype in Bladder Cancer: A Molecular Analysis of Established Human Cell Lines. Sci Rep 2016;6:38531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grabowska MM, Elliott AD, DeGraff DJ, Anderson PD, Anumanthan G, Yamashita H, et al. NFI transcription factors interact with FOXA1 to regulate prostate-specific gene expression. Mol Endocrinol 2014;28:949–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Jong JJ, Boormans JL, van Rhijn BWG, Seiler R, Boorjian SA, Konety B, et al. Distribution of Molecular Subtypes in Muscle-invasive Bladder Cancer Is Driven by Sex-specific Differences. Eur Urol Oncol 2020;3:420–3 [DOI] [PubMed] [Google Scholar]
- 49.Oldenburg D, Ru Y, Weinhaus B, Cash S, Theodorescu D, Guin S. CD44 and RHAMM are essential for rapid growth of bladder cancer driven by loss of Glycogen Debranching Enzyme (AGL). BMC Cancer 2016;16:713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guin S, Pollard C, Ru Y, Ritterson Lew C, Duex JE, Dancik G, et al. Role in tumor growth of a glycogen debranching enzyme lost in glycogen storage disease. J Natl Cancer Inst 2014;106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guin S, Ru Y, Agarwal N, Lew CR, Owens C, Comi GP, et al. Loss of Glycogen Debranching Enzyme AGL Drives Bladder Tumor Growth via Induction of Hyaluronic Acid Synthesis. Clin Cancer Res 2016;22:1274–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Richmond CS, Oldenburg D, Dancik G, Meier DR, Weinhaus B, Theodorescu D, et al. Glycogen debranching enzyme (AGL) is a novel regulator of non-small cell lung cancer growth. Oncotarget 2018;9:16718–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ahrens T, Sleeman JP, Schempp CM, Howells N, Hofmann M, Ponta H, et al. Soluble CD44 inhibits melanoma tumor growth by blocking cell surface CD44 binding to hyaluronic acid. Oncogene 2001;20:3399–408 [DOI] [PubMed] [Google Scholar]
- 54.Kamikura DM, Khoury H, Maroun C, Naujokas MA, Park M. Enhanced transformation by a plasma membrane-associated met oncoprotein: activation of a phosphoinositide 3'-kinase-dependent autocrine loop involving hyaluronic acid and CD44. Mol Cell Biol 2000;20:3482–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ghatak S, Hascall VC, Markwald RR, Misra S. Stromal hyaluronan interaction with epithelial CD44 variants promotes prostate cancer invasiveness by augmenting expression and function of hepatocyte growth factor and androgen receptor. J Biol Chem 2010;285:19821–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stevens JW, Palechek PL, Griebling TL, Midura RJ, Rokhlin OW, Cohen MB. Expression of CD44 isoforms in human prostate tumor cell lines. Prostate 1996;28:153–61 [DOI] [PubMed] [Google Scholar]
- 57.Draffin JE, McFarlane S, Hill A, Johnston PG, Waugh DJ. CD44 potentiates the adherence of metastatic prostate and breast cancer cells to bone marrow endothelial cells. Cancer Res 2004;64:5702–11 [DOI] [PubMed] [Google Scholar]
- 58.Heidegger I, Brandt MP, Heck MM. Treatment of non-mestastatic castration resistant prostate cancer in 2020: What is the best? Urol Oncol 2020 [DOI] [PubMed] [Google Scholar]
- 59.Chatterjee P, Schweizer MT, Lucas JM, Coleman I, Nyquist MD, Frank SB, et al. Supraphysiological androgens suppress prostate cancer growth through androgen receptor-mediated DNA damage. J Clin Invest 2019;130:4245–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Teply BA, Wang H, Luber B, Sullivan R, Rifkind I, Bruns A, et al. Bipolar androgen therapy in men with metastatic castration-resistant prostate cancer after progression on enzalutamide: an open-label, phase 2, multicohort study. Lancet Oncol 2018;19:76–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schweizer MT, Antonarakis ES, Denmeade SR. Bipolar Androgen Therapy: A Paradoxical Approach for the Treatment of Castration-resistant Prostate Cancer. Eur Urol 2017;72:323–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lam HM, Corey E. Supraphysiological Testosterone Therapy as Treatment for Castration-Resistant Prostate Cancer. Front Oncol 2018;8:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mohammad OS, Nyquist MD, Schweizer MT, Balk SP, Corey E, Plymate S, et al. Supraphysiologic Testosterone Therapy in the Treatment of Prostate Cancer: Models, Mechanisms and Questions. Cancers (Basel) 2017;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carceles-Cordon M, Kelly WK, Gomella L, Knudsen KE, Rodriguez-Bravo V, Domingo-Domenech J. Cellular rewiring in lethal prostate cancer: the architect of drug resistance. Nat Rev Urol 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Narayanan R, Coss CC, Dalton JT. Development of selective androgen receptor modulators (SARMs). Mol Cell Endocrinol 2018;465:134–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.