

Abstract
Remarkable progress has been made in the development of new therapies for cancer, dramatically changing the landscape of treatment approaches for several malignancies and continuing to increase patient survival. Accordingly, adverse effects of cancer therapies that interfere with the continuation of best-possible care, induce life-threatening risks or lead to long-term morbidity are gaining increasing importance. Cardiovascular toxic effects of cancer therapeutics and radiation therapy are the epitome of such concerns, and proper knowledge, interpretation and management are needed and have to be placed within the context of the overall care of individual patients with cancer. Furthermore, the cardiotoxicity spectrum has broadened to include myocarditis with immune checkpoint inhibitors and cardiac dysfunction in the setting of cytokine release syndrome with chimeric antigen receptor T cell therapy. An increase in the incidence of arrhythmias related to inflammation such as atrial fibrillation can also be expected, in addition to the broadening set of cancer therapeutics that can induce prolongation of the corrected QT interval. Therefore, cardiologists of today have to be familiar not only with the cardiotoxicity associated with traditional cancer therapies, such as anthracycline, trastuzumab or radiation therapy, but even more so with an ever-increasing repertoire of therapeutics. This Review provides this information, summarizing the latest developments at the juncture of cardiology, oncology and haematology.
Cancer-related diseases have been on the rise and cancer-related mortality has been on the decline, leading to a profound increase in the number of survivors of cancer over the past three decades1. With this change has come greater recognition of the importance of the adverse effects of cancer therapies, some of the most important being cardiovascular in nature. Pre-existing cardiovascular disease can likewise complicate and even lead to the termination of cancer therapy (especially if it is not managed appropriately). Therefore, an important interaction exists between these two disease entities and their management. Considering the ageing of the general population, these dynamics are expected to increase in the years to come2. Preparing individuals and society for this future is an important goal, and its pursuit has started in the form of the emerging field of cardio-oncology.
Ewer and Ewer provided a classic overview of the field of cardio-oncology in 2010, with an update in 2015 (REFS3,4). Since then, the focus of cardio-oncology might not have shifted much, but the field of view has certainly become much broader, including not only cardiotoxicity but also many other cardiovascular diseases, especially vascular toxicity and arrhythmias (FIG. 1; Supplementary Fig. 1). This change is in large part related to the progress in cancer therapeutics from chemical compounds in the twentieth century to targeted agents around the turn of the millennium and to immunotherapies in the past decade (FIG. 2). This Review provides an updated overview of cardiotoxicity and arrhythmias associated with cancer therapies; vascular toxic effects are covered in a separate Review in this Issue5. As applicable, references will be made to available guidelines and consensus documents from various societies (Supplementary Tables 1–7) to reflect and discuss currently published consensus recommendations.
Fig. 1 |. Outline of cardiovascular toxic effects associated with cancer therapies.
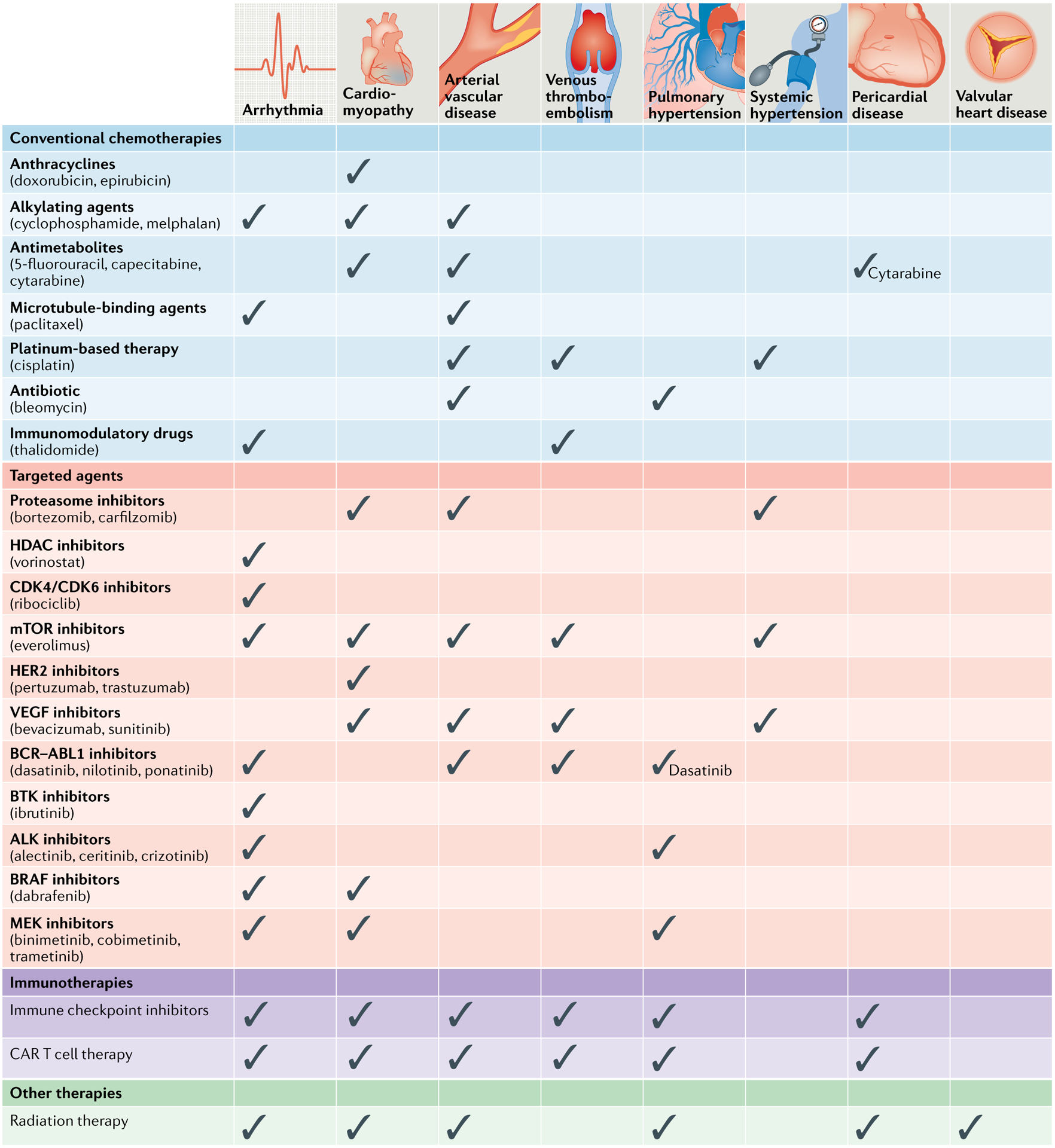
Numerous cancer therapies have been associated with adverse effects and complications across the entirety of the cardiovascular system. As illustrated, some therapies have a very confined and others have a very broad cardiovascular toxicity profile. Classic chemical compounds are shown in blue, targeted therapies are shown in pink, immunotherapies are shown in purple and radiation therapy is shown in green. CAR, chimeric antigen receptor; HDAC, histone deacetylase; MEK, MAPK/ERK kinase; mTOR, mechanistic target of rapamycin; VEGF, vascular endothelial growth factor.
Fig. 2 |. Timeline of cancer therapy development.

The timeline presents landmarks in the development of cancer therapeutics. Three main eras can be distinguished on the basis of the type of agent: classic chemical compounds (shown in blue), targeted therapies (shown in pink) and immunotherapies (shown in purple). ACT, adoptive T cell therapy; ALL, acute lymphoblastic leukaemia; BCG, bacillus Calmette–Guérin; CAR, chimeric antigen receptor; CDK, cyclin-dependent kinase; CTLA4, cytotoxic T lymphocyte antigen 4; HDAC, histone deacetylase; HL, Hodgkin lymphoma; HPV, human papillomavirus; ICI, immune checkpoint inhibitor; MART1, melanoma antigen recognized by T cells 1; mTOR, mechanistic target of rapamycin; NHL, non-Hodgkin lymphoma; PD1, programmed cell death 1; TCR, T cell receptor.
Cardiotoxicity of cancer therapies
Over the years, the term ‘cardiotoxicity’ has encompassed many specific disease entities and is very much in need of a universal definition. This need holds true for cancer therapy-related cardiomyopathies. The unifying element for these conditions is a decline in cardiac function, with differences in the defining criteria. Mechanistically, a decline in cardiac function can be due to direct (endogenous) cardiomyocyte damage (termed in this Review as ‘cancer therapy-related type I or primary (toxic) cardiomyopathy’), to alterations in perfusion, innervation or hormonal milieu (termed in this Review as ‘cancer therapy-related type II or secondary (indirect) cardiomyopathy’) or to inflammatory cell infiltration in the myocardium (termed ‘cancer therapy-related type III cardiomyopathy or myocarditis’) (BOX 1). Although cardiomyopathies associated with cancer therapies are rarely mediated by one single mechanism, this classification might serve the ultimate goals of fostering the proper selection of care and achieving the best possible outcomes.
Box 1 |. Proposal for a pathophysiology-based classification of cancer therapy-related cardiomyopathies.
A decline in cardiac function in patients with cancer can occur as a consequence of direct toxic effects of cancer therapies on the myocardium (primary or type I cardiomyopathy) or secondary to other alterations that translate into a reduction in cardiac function (secondary or type II cardiomyopathy). Non-toxic or non-reactive primary inflammatory myocarditis is a unique subtype of cancer therapy-related cardiomyopathy (type III), and requires immunosuppressive treatment. In type II scenarios, treatment of the underlying or contributing abnormality (such as coronary or valvular heart disease) is crucial to the restoration of cardiac function, whereas for type I scenarios, heart failure therapy is essential. The table shows the cancer therapies that have been associated with each type of cardiomyopathy, as well as the diagnosis and management strategies.
| Characteristic | Cancer therapy-related cardiomyopathy | ||
|---|---|---|---|
| Type I | Type II | Type III | |
| Definition | Direct impairing effect on the myocardium | Indirect impairing effect on the myocardium | Impairing effect owing to myocarditis |
| Risk with cancer therapy | |||
| Doxorubicin | Yes | Yes | Yes (toxic or reactive) |
| Cyclophosphamide | Yes | Yes | Yes (toxic or reactive) |
| 5-Fluorouracil | Yes | Yes | NR |
| HER2 (ERBB2) inhibitors | Yes | Unclear | NR |
| VEGF inhibitors | Yes (TKIs) | Yes | Unclear |
| ICIs | Possible | Possible | Yes (immunomediated) |
| Radiation therapy | Yes (at high dose) | Yes | Yes (toxic or reactive) |
| Diagnosis | |||
| Imaging | Echocardiography, cardiac MRI, MUGA scan | (Stress) echocardiography, (stress) cardiac MRI, nuclear stress test, CT coronary angiography, vasoreactivity studies | Cardiac MRI, PET, echocardiography |
| Biomarkers | Cardiac troponins, natriuretic peptides (especially long term) | Thyroid function studies, cytokines, catecholamines ECG abnormalities (e.g. ST-segment shifts, T-wave inversions) | Cardiac troponins, natriuretic peptides, ECG abnormalities (e.g. heart block, ectopy) |
| Management | |||
| Treatment | Stop cancer therapy, β-blocker(carvedilol), ACE inhibitor, ARB, spironolactone | Stop cancer therapy, therapy directed at the underlying cause (e.g. correction of myocardial ischaemia or valve disease) | Stop cancer therapy; for ICI therapy, anti-inflammatory and immunosuppressive therapy, supportive care as needed (e.g. ECMO) |
| Prevention | Screening for and optimal treatment of comorbidities, exercise; for anthracyclines, cardiovascular medications (carvedilol or nebivolol, ACE inhibitor, ARB or spironolactone, statins, dexrazoxane) | Screening for and optimal treatment of predisposing conditions, dose and type of administration; for radiation therapy, dose reduction (e.g. shielding, positioning or proton beam) | Screening for and optimal treatment of comorbidities (efficacy not proven), early detection with biomarkers |
ACE, angiotensin-converting enzyme; ARB, angiotensin-receptor blocker; ECG, electrocardiogram; ECMO, extracorporeal membrane oxygenation; ICI, immune checkpoint inhibitor; MUGA, multigated acquisition; NR, not reported; PET, positron emission tomography; TKI, tyrosine kinase inhibitor; VEGF, vascular endothelial growth factor.
Type I cardiomyopathy
Cancer therapy-related type I cardiomyopathies can occur with various cancer therapeutics. They are a consequence of direct toxic effects of cancer therapies on the myocardium and represent the prototypical toxic cardiomyopathy.
Conventional chemotherapies.
Conventional chemotherapeutics are chemical compounds intended to kill tumour cells by interfering with their high metabolic demand and mitotic activity. One of the most effective and prominent examples is anthracyclines, which intercalate between base pairs of DNA or RNA strands and thereby inhibit DNA or RNA synthesis6. Furthermore, anthracyclines inhibit topoisomerase IIα, an important enzyme for DNA transcription and replication. Other effects include induction of iron-mediated oxidative stress that damages DNA, proteins and lipids, as well as histone modification that deregulates epigenomic and transcriptomic responses.
Cardiotoxicity is a dose-limiting adverse effect of anthracycline therapy (TABLE 1). Common terminology has been to label any evidence of cardiac injury occurring during and within 1 week of active cancer therapy as acute cardiotoxicity and thereafter as chronic cardiotoxicity, with either early or late onset (that is, within or after 1 year of completion of cancer treatment)7. Acute anthracycline-related cardiotoxicity is a rare event, seen in less than 5% of patients. This cardiomyopathy pres ents with electrocardiogram (ECG) changes (in 20–30% of patients) and arrhythmias (up to 3% of patients), mainly sinus tachycardia but supraventricular tachycardia, heart block and ventricular arrhythmias can occur as well, leading to palpitations, presyncope and syncope, and even cardiac arrest. Acute declines in cardiac function can be seen as well, presenting with dyspnoea to the point of heart failure (HF)8. Finally, some patients develop pericarditis and have chest pain in addition to shortness of breath9. Pathologically, acute anthracycline-related cardiotoxicity resembles an acute toxic myocarditis with cardiomyocyte damage, inflammatory infiltrates and interstitial oedema10.
Table 1 |.
Leading cardiovascular toxic effects of conventional chemotherapies and radiation therapy
| Therapy | Cancer therapy indications (label and off-label) | Toxicity | |||
|---|---|---|---|---|---|
| Cardiac | Arrhythmia | Vascular | Other | ||
| Anthracyclines | |||||
| Doxorubicin | ALL, bladder cancer, breast cancer, endometrial carcinoma, Ewing sarcoma, hepatocellular cancer, HL, leukaemia or lymphoma, metastatic solid tumours, multiple myeloma, NHL, osteosarcoma, SCLC, soft-tissue sarcoma, thymoma, uterine sarcoma, Waldenstrom macroglobulinaemia | +++ | +++ | – | US black box warning: cardiomyopathy, secondary malignancy, extravasation and tissue necrosis, myelosuppression |
| Epirubicin | Breast cancer, gastric cancer, oesophageal cancer, osteosarcoma, soft-tissue sarcoma | +++ | +++ | – | |
| Idarubicin | AML | +++ | +++ | – | |
| Mitoxantrone | AML, HL, NHL, prostate cancer, stem cell transplantation | ++ | +++ | ++ | |
| Alkylating agents | |||||
| Busulfan | Essential thrombocythaemia, haematopoietic stem cell conditioning regimen, polycythaemia vera | – | +++ | +++ | Oedema, pericardial effusion, even tamponade |
| US black box warning: bone marrow suppression | |||||
| Cyclophosphamide | ALL, breast cancer, CLL, Ewing sarcoma, HL, multiple myeloma, NHL, SCLC, stem cell transplant conditioning | ++ | + | + | Bone marrow suppression |
| Ifosfamide | Bladder cancer, cervical cancer, Ewing sarcoma, HL, NHL, osteosarcoma, ovarian cancer, soft-tissue sarcoma, testicular cancer, thymoma | + | – | – | US black box warning: myelotoxicity, CNS toxicity, nephrotoxicity, haemorrhagic cystitis |
| Melphalan | Amyloidosis, multiple myeloma, ovarian cancer, HL, stem cell transplant conditioning (lymphomas) | – | ++ | – | US black box warning: bone marrow suppression, hypersensitivity, secondary malignancy |
| Antimetabolites | |||||
| 5-Fluorouracil | Anal carcinoma, bladder cancer, breast cancer, cervical cancer, colorectal cancer, oesophageal cancer, gastric cancer, hepatobiliary cancer, pancreatic cancer, squamous cell carcinomas | ++ | ++ | +++ | Bone marrow suppression |
| Capecitabine | Anal carcinoma, breast cancer, colorectal cancer, gastric cancer, hepatobiliary cancer, oesophageal cancer, ovarian, fallopian peritoneal cancer, pancreatic cancer, cancer of unknown primary | + | ++ | ++ | Bone marrow suppression, oedema |
| US black box warning: warfarin interaction | |||||
| Clofarabine | ALL, AML | + (high dose or in combination with cyclophosphamide) | +++ | – | Bone marrow suppression, pericardial effusion, capillary leak syndrome, hypotension, hypertension |
| Cytarabine | ALL, AML, acute promyelocytic leukaemia, CLL, CNS lymphoma, HL, meningeal leukaemia, NHL | Not defined | – | – | Bone marrow suppression, pericarditis |
| Gemcitabine | Adenocarcinoma of unknown primary, breast cancer, bladder cancer, cervical cancer, head and neck cancer, hepatobiliary cancer, HL, malignant pleural mesothelioma, NHL, NSCLC, ovarian cancer, pancreatic cancer, sarcomas, SCLC, testicular cancer, uterine cancer | – | – | + | Bone marrow suppression, oedema |
| Microtubule-binding agents | |||||
| Paclitaxel | Adenocarcinoma of unknown primary, bladder cancer, breast cancer, cervical cancer, head and neck cancers, Kaposi sarcoma, NSCLC, oesophageal and gastric cancer, ovarian cancer, penile cancer, SCLC, soft-tissue sarcoma, testicular germ cell tumours, thymoma | + | ++ | ++ | Oedema, hypotension, flushing |
| US black box warning: hypersensitivity reaction and bone marrow suppression | |||||
| Docetaxel | Adenocarcinoma of unknown primary, bladder cancer, breast cancer, Ewing sarcoma, head and neck cancers, NSCLC, oesophageal and gastric cancer, ovarian cancer, prostate cancer, SCLC, soft-tissue sarcoma | + | + | + | Neurotoxicity, bone marrow suppression, oedema, pericardial effusion, hypotension |
| US black box warning: fluid retention, neutropenia, hypersensitivity, hepatic function impairment, increased mortality | |||||
| Vinblastine | Bladder cancer, HL, melanoma, NSCLC, soft-tissue sarcoma, testicular cancer | – | – | + | Bone marrow suppression, pulmonary toxicity |
| Vincristine | ALL, CNS tumours, HL, NHL, Ewing sarcoma, gestational trophoblastic tumours, multiple myeloma, ovarian cancer, primary CNS lymphoma, SCLC, thymoma | – | – | + | Oedema, hypotension |
| Platinum-based drugs | |||||
| Cisplatin | Bladder cancer, breast cancer, cervical cancer, endometrial carcinoma, oesophageal and gastric cancer, head and neck cancer, HL, malignant pleural mesothelioma, multiple myeloma, NHL, osteosarcoma, ovarian cancer, penile cancer, SCLC, testicular cancer | + | + | ++ | US black box warning: myelosuppression, nephrotoxicity, peripheral neuropathy |
| Oxaliplatin | Biliary adenocarcinoma, CLL, cancer of unknown primary, colorectal cancer, neuroendocrine tumours (carcinoid), NHL, ovarian cancer, oesophageal/gastric cancers, pancreatic cancer, testicular cancer | – | + | ++ | Bone marrow suppression, oedema, peripheral neuropathy and neurotoxicity |
| US black box warning: hypersensitivity and anaphylactic reactions | |||||
| Antitumour antibiotics | |||||
| Bleomycin | HL, testicular cancer, ovarian germ cell cancer | – | – | + | Phlebitis |
| US black box warning: pulmonary toxicity, idiosyncratic reaction | |||||
| Immunomodulatory drugs | |||||
| Lenalidomide | CLL, diffuse large B cell lymphoma, mantle cell lymphoma, multiple myeloma, myelodysplastic syndrome | + | ++ | ++ | Oedema |
| US black box warning: fetal risk, haemotoxicity, arterial and venous thromboembolic events | |||||
| Thalidomide | Multiple myeloma, systemic light chain amyloidosis, Waldenstrom macroglobulinaemia | – | + | +++ | Oedema |
| US black box warning: risk in pregnancy and risk of fetal malformation, thromboembolic events | |||||
| Radiation therapy | |||||
| Mainly external beam | Breast cancer, gastric cancer, head and neck cancer, lung cancer, lymphoma, oesophageal cancer, prostate cancer, testicular cancer | ++ | + | ++ | Valvular heart disease, pericarditis with/without constriction, restrictive cardiomyopathy |
Based on data from Micromedex (IBM, NY, USA) and Lexicomp (Wolters Kluwer, Netherlands). Frequency of cardiovascular toxic effects: –, not reported; +, uncommon (<1%); ++, common (1–10%); +++, very common (>10%). ALL, acute lymphoblastic leukaemia; AML, acute myeloid leukaemia; CLL, chronic lymphocytic leukaemia; CNS, central nervous system; HL, Hodgkin lymphoma; NHL, non-Hodgkin lymphoma; NSCLC, non-small-cell lung cancer; SCLC, small-cell lung cancer.
Chronic anthracycline-related cardiotoxicity is histopathologically characterized by vacuole formation, myofibril dropout, necrosis and fibrosis11. Importantly, these changes predate the declines in ejection fraction and can be seen in myocardial biopsy samples, ranging from mild to severe, while nuclear or echocardio-graphic imaging still indicates normal cardiac function parameters12. Moreover, even changes (increases) in myocardial injury early after anthracycline exposure do not necessarily correlate with changes (decreases) in left ventricular ejection fraction (LVEF) as assessed by either imaging modality12. Nevertheless, a multigated acquisition scan-based study indicated that declines in cardiac function can be noted in certain individuals after a cumulative doxorubicin dose of 200 mg/m2 and indicates the risk of progression through the HF stages13. The risk of HF progression gains particular meaning when one considers studies that present anthracycline-related cardiomyopathy as one of the worst types of cardiomyopathy14. However, data published in 2017 indicate a prognostic profile of anthracycline-related cardiomyopathy on a par with that of dilated cardiomyopathy, and an unrelenting decline in cardiac function does not have to be the norm with current regimens of cardiomyopathy and HF treatment15. The epidemiological scope of anthracycline-related cardiomyopathy is also in flux, with the reported incidence ranging from 0% to 57%, attributable to differences in study populations, definitions and tests used over time16.
The mechanisms of anthracycline-related cardiotoxicity have some overlap with its anticancer effects but also have unique and specific differences. For instance, anthracyclines inhibit topoisomerase IIβ and show a particular predilection for mitochondria in cardiomyocytes17. Mitochondrial injury is seemingly one of the cardinal elements of anthracycline-related cardiotoxicity, and damage to mitochondrial DNA has been proposed to be responsible for the long-term risk of cardiomyopathy associated with anthracycline exposure18–21. Other studies have indicated that anthracyclines preferentially affect progenitor cells and thereby reduce the regenerative potential of the (injured) myocardium, the consequences of which then emerge over time, especially with any additional stressors22–24.
Targeted cancer therapies.
The pharmacological action of classic chemotherapeutics is not very specific and, therefore, the potential to harm normal cells is fairly high. Consequently, therapies that specifically target the malignant molecular fingerprint were designed with the aim of yielding higher success rates with lower complication rates. A classic example is trastuzumab, a humanized antibody directed against HER2 (also known as ERBB2), which is overexpressed in 15–20% of breast cancers25 (BOX 2). Like other oncogenes, HER2 signalling increases cancer cell proliferation, tumour growth and metastatic spread; HER2 inhibition, therefore, translated into revolutionary clinical success26,27.
Box 2 |. Targeted cancer therapies.
ALK inhibitors
ALK is an oncogene encoding a protein involved in cell growth. Mutated forms of the ALK gene and protein have been found, for instance, in non-small-cell lung cancer and anaplastic large-cell lymphoma, in which ALK inhibitors are used.
BRAF inhibitors
The serine/threonine-protein kinase BRAF is a member of the RAF family and a downstream target of RAS in the mitogen-activated protein kinase (MAPK) signalling pathway. Activating mutations in BRAF have been described in a few cancers, such as V600E in melanoma and non-small-cell lung cancer. BRAF inhibitors can be combined with MAPK/ERK kinase (MEK) inhibitors to extend the time to resistance and the tumour and survival responses.
CDK inhibitors
Cyclin-dependent kinases (CDKs) phosphorylate and thereby regulate the activity of proteins that are important for progression through the cell cycle and cell division. CDK4 and CDK6 inhibitors are used to interrupt this action and thereby the proliferation of cancer cells, which are more likely to have disturbances in CDK4 and CDK6, such as hormone receptor-positive breast cancer cells.
EGFR and HER2 inhibitors
The four main members of the human epidermal growth factor receptor (EGFR) family, EGFR (also known as HER1), HER2 (also known as ERBB2), ERBB3 (also known as HER3) and ERBB4 (also known as HER4), regulate the growth, survival and differentiation of various cells via multiple intracellular signal transduction pathways after ligand-mediated association of two receptors (dimerization). HER2-directed therapy is extremely important in breast oncology and includes drugs that inhibit the extracellular domains (pertuzumab and trastuzumab) or the intracellular tyrosine kinase domain (lapatinib) of the receptor. Inhibitors targeting the tyrosine kinase domain of EGFR (such as erlotinib) are used in lung oncology.
HDAC inhibitors
Histone deacetylase (HDAC) inhibitors interfere with the actions of HDACs, which are enzymes involved in the remodelling of chromatin and have an important role in the epigenetic regulation of gene expression and the regulation of the activity of non-histone proteins through hypoacetylation. These drugs are approved by the FDA for use in T cell lymphoma and myeloma.
MEK inhibitors
The classic MAPK signalling pathway is important for cell growth and division. This pathway is activated, for example, by growth factors such as EGF, and entails the sequential activation of RAS, RAF, MAPK/ERK1 (MEK1), MEK2, ERK1 and ERK2. Aberrant activation occurs through gain-of-function mutations in RAS and RAF gene family members, which are among the most frequently mutated genes in human cancer. MEK inhibitors lock non-phosphorylated MEK1 and MEK2 into a catalytically inactive state that is not related to the ATP-binding pocket of the proteins, which reduces the risk of off-target effects. These drugs are used primarily in patients with melanoma.
MET
MET is a receptor tyrosine kinase that, after binding with its ligand, hepatocyte growth factor, activates the MAPK and other intracellular signalling pathways involved in cell proliferation, motility, migration and invasion. Overactivation of MET via mutation, amplification or protein overexpression has been documented in various human malignancies. MET inhibitors interfere with the tyrosine kinase activity of MET and are approved for use in patients with thyroid, renal cell or hepatocellular cancer.
mTOR inhibitors
Mechanistic target of rapamycin (mTOR) is a protein kinase that forms two types of mTOR complexes (mTORC). mTORC1 suppresses catabolic processes (such as autophagy) and activates anabolic pathways (such as protein synthesis), thereby supporting cell growth. mTOR inhibitors inhibit mTORC1, thereby shifting cancer cell metabolism to a status unfavourable for cell growth. These drugs are prescribed for patients with renal cell cancer or breast cancer.
Topoisomerase inhibitors
As polymerases separate DNA strands for transcription of gene information or duplication, the remaining portions of the DNA strands become more densely coiled. Topoisomerases cleave and relax hypercoiled DNA segments and subsequently reattach the cleaved ends. On the basis of their cleaving either one or both strands of DNA, topoisomerases are designated as type I or type II, respectively, and so are their inhibitors. Topoisomerase inhibition leads to the formation of irreversible covalent crosslinks between topoisomerases and DNA, thereby stalling DNA expression, duplication and integrity.
VEGF inhibitors
Vascular endothelial growth factors (VEGFs) have an important role in the formation of new vessels (angiogenesis), thereby supporting tumour growth and metastasis. VEGF inhibitors interfere with this aspect of tumour growth and bind to VEGFA (bevacizumab), trap VEGF subtypes (aflibercept), bind to VEGF receptor 2 (VEGFR2) (ramucirumab) or inhibit VEGFR2 tyrosine kinase activity (pazopanib, sorafenib and sunitinib). These agents are indicated for patients with renal cell cancer or thyroid cancer.
Oncogenes.
Oncogenes encode proteins that can transform cells into tumour cells. All but a few are derived from normal cellular genes (proto-oncogenes), and activation of a proto-oncogene into an oncogene generally involves a gain-of-function mutation.
However, much at odds with the promise of ‘smarter and safer designer drugs’, trastuzumab caused declines in cardiac function and even HF in nearly 30% of patients in early seminal clinical trials28 (TABLE 2). Studies thereafter revealed an incidence of trastuzumab-related cardiotoxicity of 15–20% and of HF of <5%. Nonetheless, declines in cardiac function ≥10% can be seen in 40–45% of patients receiving trastuzumab in consecutive patient datasets29,30. However, Ewer and Lippman pointed out unique differences between trastuzumab-related and anthracycline-related cardiotoxicity, leading to the terminology of cancer therapy-related type II (alternative) cardiotoxicity and cancer therapy-related type I (classic) cardiotoxicity, respectively31. A key differentiating element in these definitions is the recovery of cardiac function after cessation of trastuzumab therapy. However, the average LVEF of the original cohort of patients remained approximately 5% below baseline levels. Approximately 10% of patients had a LVEF that was more than 10% lower than the baseline level, and 20% of patients with a normal LVEF at baseline had a LVEF of less than 50% after treatment32. Other studies have indicated that as many as 75% of patients exposed to trastuzumab therapy might have an irreversible decline in cardiac function33. Overall, 20% of patients experience an interruption of their trastuzumab therapy, and only half of these patients are able to resume the therapy, with a 15–40% likelihood of a recurrent drop in LVEF32,34,35. These data outline the remarkable burden that trastuzumab-related cardiomyopathy can have in patients with breast cancer.
Table 2 |.
Leading cardiovascular toxic effects of targeted cancer therapies
| Therapy | Cancer therapy indications (label and off-label) | Toxicity | |||
|---|---|---|---|---|---|
| Cardiac | Arrhythmia | Vascular | Other | ||
| Proteasome inhibitors | |||||
| Bortezomib | Follicular lymphoma, mantle cell lymphoma, multiple myeloma, systemic light chain amyloidosis, T cell lymphoma, Waldenstrom macroglobulinaemia | ++ | + | + | Bone marrow suppression, hypotension |
| Carfilzomib | Multiple myeloma, Waldenstrom macroglobulinaemia | ++ | – | +++ | Bone marrow suppression, oedema, pulmonary hypertension |
| HDAC inhibitors | |||||
| Panobinostat | Multiple myeloma | – | +++ | ++ | Bone marrow suppression, peripheral oedema, orthostatic hypotension |
| US black box warning: severe and fatal cardiac ischaemic events, severe arrhythmias and ECG changes, severe diarrhoea | |||||
| Romidepsin | T cell lymphoma | – | ++ | ++ | Bone marrow suppression, oedema, hypotension |
| Vorinostat | T cell lymphoma | – | ++ | ++ | Bone marrow suppression, peripheral oedema |
| CDK4/CDK6 inhibitors | |||||
| Abemaciclib | Breast cancer | – | – | + | – |
| Ribociclib | Breast cancer | – | ++ | – | Peripheral oedema, syncope |
| mTOR inhibitors | |||||
| Everolimus | Breast cancer, neuroendocrine tumours, RCC | ++ | ++ | ++ | Bone marrow suppression, peripheral oedema, hypertension |
| Temsirolimus | RCC | – | – | +++ | Bone marrow suppression, peripheral oedema, hypertension |
| Monoclonal antibodies (target) | |||||
| Alemtuzumab (anti-CD52) | B cell CLL, aplastic anaemia, T cell lymphocytic and prolymphocytic leukaemia | + | ++ | ++ | Peripheral oedema, hypotension, hypertension |
| US black box warning: autoimmunity, infusion reactions, and malignancies, myelosuppression, infection, stroke | |||||
| Rituximab (anti-CD20) | Burkitt lymphoma, CLL, CNS lymphoma, HL, NHL, Waldenstrom macroglobulinaemia | + | + | ++ | Peripheral oedema, hypertension, hypotension, flushing |
| US black box warning: infusion reasons, mucocutaneous reactions, progressive multifocal leukoencephalopathy | |||||
| Cetuximab (anti-EGFR/HERl) | Colorectal cancer, head and neck cancer, penile cancer, squamous cell skin cancer (KRAS wild type) | – | + | ++ | US black box warning: cardiopulmonary arrest, infusion reactions |
| Necitumumab (anti-EGFR/HERl) | NSCLC | – | – | +++ | US black box warning: cardiopulmonary arrest, hypomagnesaemia |
| Panitumumab (anti-EGFR/HERl) | Colorectal cancer, KRAS wild type | – | – | + | US black box warning: dermatological toxicity |
| Pertuzumab (anti-HER2/ERBB2) | Breast cancer | ++ | – | – | Peripheral oedema |
| US black box warning: cardiotoxicity, birth defects | |||||
| Trastuzumab (anti-HER2/ERBB2) | Breast cancer, gastric cancer | +++ | ++ | – | Peripheral oedema |
| US black box warning: cardiotoxicity, pulmonary toxicity, infusion reactions, birth defects | |||||
| Aflibercept (anti-VEGF–VEGFR2) | Metastatic colorectal cancer | + | – | ++ | Hypertension, reversible posterior leukoencephalopathy syndrome, thrombotic microangiopathy |
| US black box warning: haemorrhage, GI tract perforation, compromised wound healing | |||||
| Bevacizumab (anti-VEGF-VEGFR2) | Glioblastoma, persistent/recurrent/metastatic cervical cancer, metastatic colorectal cancer, (non-squamous) NSCLC, ovarian (epithelial), fallopian tube or primary peritoneal cancer, metastatic RCC | ++ | – | +++ | Oedema, hypotension, hypertension, reversible posterior leukoencephalopathy syndrome, thrombotic microangiopathy |
| US black box warning: haemorrhage, GI tract perforation, compromised wound healing | |||||
| Ramucirumab (anti-VEGF-VEGFR2) | Metastatic NSCLC, metastatic gastric cancer, metastatic colorectal cancer | – | – | ++ | Hypertension, reversible posterior leukoencephalopathy syndrome, thrombotic microangiopathy |
| Multi-target kinase inhibitors (primary target) | |||||
| Erlotinib (EGFR/HER1) | NSCLC, pancreatic cancer | – | ++ | +++ | Oedema, pulmonary toxicity |
| Osimertinib (EGFR/HER1) | NSCLC | ++ | ++ | – | Bone marrow suppression, pulmonary toxicity |
| Dacomitinib (EGFR/HER1) | NSCLC | – | – | ++ | Pulmonary toxicity |
| Lapatinib (HER2/ERBB2) | Breast cancer | ++ | + | – | Pulmonary toxicity |
| US black box warning: hepatotoxicity | |||||
| Axitinib (VEGFR1–VEGFR3) | RCC, thyroid cancer | + | – | ++ | Hypertension, haemorrhage |
| Lenvatinib (VEGFR1– VEGFR3 | Hepatocellular cancer, RCC, thyroid cancer | ++ | ++ | ++ | Peripheral oedema, hypertension |
| Pazopanib (VEGFR1–VEGFR3) | RCC, soft-tissue carcinoma, thyroid cancer | +++ | +++ | ++ | Peripheral oedema, hypertension, thrombotic microangiopathy, bleeding, pulmonary toxicity |
| US black box warning: hepatotoxicity | |||||
| Sorafenib (VEGFR1–VEGFR3) | Angiosarcoma, hepatocellular cancer, RCC, thyroid cancer, GIST | ++ | + | ++ | Hypertension, bleeding |
| Sunitinib (VEGFR1–VEGFR3) | GIST, pancreatic neuroendocrine tumours, RCC, soft-tissue sarcoma, thyroid cancer | +++ | + | +++ | Hypertension, thrombotic microangiopathy, increased creatine kinase level |
| US black box warning: hepatotoxicity | |||||
| Vandetanib (VEGFR) | Thyroid cancer | ++ | +++ | ++ | Hypertensive crisis, bleeding, pulmonary toxicity |
| US black box warning: QT interval prolongation, torsades de pointes, sudden cardiac death | |||||
| Regorafenib (VEGFR2) | Colorectal cancer, GIST, hepatocellular carcinoma | – | – | ++ | Hypertension |
| US black box warning: hepatotoxicity | |||||
| Bosutinib (BCR–ABL) | Philadelphia chromosome-positive CML | – | ++ | ++ | Oedema, chest pain, pericardial effusion, hypertension |
| Dasatinib (BCR–ABL1) | Philadelphia chromosome-positive ALL and CML, GIST | – | ++ | ++ | Oedema, pulmonary hypertension |
| Imatinib (BCR–ABL1) | Philadelphia chromosome-positive ALL and CML, GIST, myelodysplastic syndrome, melanoma, stem cell transplant for CML | + | ++ | ++ | Palpitations, oedema, chest pain, subdural haematoma |
| Nilotinib (BCR–ABL1) | Philadelphia chromosome-positive ALL and CML, GIST | − | ++ | +++ | Oedema, hyperglycaemia, hypercholesterolaemia |
| US black box warning: QT interval prolongation, sudden cardiac death | |||||
| Ponatinib (BCR–ABL1) | Philadelphia chromosome-positive ALL and CML | ++ | +++ | +++ | Bone marrow suppression, bleeding, hypertensive crisis, thrombotic microangiopathy |
| US black box warning: hepatotoxicity, arterial occlusion, venous thromboembolism | |||||
| Ibrutinib (BTK) | CLL, mantle cell lymphoma, marginal zone lymphoma, Waldenstrom macroglobulinaemia | – | +++ | – | Oedema, hypertension, subdural haematoma |
| Alectinib (ALK) | NSCLC | – | +++ | ++ | Oedema, pulmonary toxicity |
| Brigatinib (ALK) | NSCLC | – | ++ | – | Hypertension, pulmonary toxicity |
| Ceritinib (ALK) | NSCLC | – | +++ | – | Pericarditis, pericardial effusion, pulmonary toxicity |
| Crizotinib (ALK) | NSCLC | – | +++ | ++ | Oedema, pulmonary toxicity |
| Lorlatinib (ALK) | NSCLC | – | ++ | – | Oedema, pulmonary toxicity |
| Dabrafenib (BRAF) | Melanoma, NSCLC, thyroid cancer | ++ | – | – | Bleeding, oedema, hypertension |
| Encorafenib (BRAF) | Melanoma | – | + | – | Facial paresis |
| Vemurafenib (BRAF) | Melanoma, NSCLC | – | +++ | – | Oedema, nephrotoxicity, hypertension |
| Gilteritinib (FLT3) | Relapsed or refractory FLT3+AML | ++ | ++ | – | Oedema, hypotension, hypertension, pericardial effusion, pericarditis |
| Ruxolitinib (JAK) | Myelofibrosis, polycythaemia vera | – | + | – | Oedema |
| Cabozantinib (MET) | Hepatocellular carcinoma, RCC, thyroid cancer | – | ++ | ++ | Bone marrow suppression, hypertension |
| Binimetinib (MEK) | Melanoma | ++ | – | ++ | Oedema, bleeding, pulmonary toxicity, hypertension |
| Cobimetinib (MEK) | Melanoma | +++ | – | – | Bleeding, hypertension |
| Trametinib (MEK) | Melanoma, NSCLC, thyroid cancer | +++ | ++ | – | Oedema, bleeding, pulmonary toxicity, hypertension |
Based on data from Micromedex (IBM, NY, USA) and Lexicomp (Wolters Kluwer, Netherlands). Frequency of cardiovascular toxic effects: –, not reported; +, uncommon (<1%); ++, common (1–10%); +++, very common (>10%). ALL, acute lymphoblastic leukaemia; AML, acute myeloid leukaemia; CDK, cyclin-dependent kinase; CLL, chronic lymphocytic leukaemia; CML, chronic myeloid leukaemia; CNS, central nervous system; ECG, electrocardiogram; EGFR, epidermal growth factor receptor; GI, gastrointestinal; GIST, gastrointestinal stromal tumour; HDAC, histone deacetylase; HL, Hodgkin lymphoma; JAK, Janus kinase; MEK, MAPK/ERK kinase; NHL, non-Hodgkin lymphoma; NSCLC, non-small-cell lung cancer; mTOR, mechanistic target of rapamycin; RCC, renal cell carcinoma; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor.
Elevations in the circulating levels of cardiac troponin (cTn) seem to identify those patients receiving trastuzumab who are at risk of an irreversible decline in cardiac function36. Although plasma cTn level elevations have been noted in experimental studies with trastuzumab37, these elevations are usually seen at the transition from anthracycline therapy to trastuzumab therapy. This observation highlights the previously described anthracycline–trastuzumab interaction, whereby trastuzumab impairs the repair response to anthracycline in cardiomyocytes4, which can then translate into cardiac injury and dysfunction. Inhibition of HER2 in the presence of other potent stressors to the myocardium, such as ischaemia and/or high afterload, can be similarly detrimental. Therefore, trastuzumab therapy can unmask any injury or stress on the myocardium that leads to the upregulation and activation of the HER2 stress response pathway. The interplay between various risk factors and the state of dependence of the myocardium on the HER2 signalling pathway might also provide an explanation for the seemingly contradictory findings between clinical trials and real-world registries. For instance, the risk of cardiotoxicity was low and confined to the active treatment period in the HERA trial38, whereas an increasing risk of HF after trastuzumab therapy has been seen in the SEER–Medicare database of patients39. Of further note, in the SEER–Medicare database, the 3-year risk of HF was lower with anthracycline therapy than with trastuzumab therapy, but was highest when both were combined39. Similar observations were made in the Cancer Research Network as well as other registry-based studies40. In summary, the long-term cardiovascular implications of trastuzumab therapy remain to be defined.
Other HER2-directed therapies, such as lapatinib, pertuzumab and trastuzumab–emtansine, are associated with a lower risk of cardiotoxicity than trastuzumab. Furthermore, dual trastuzumab–pertuzumab HER2-directed therapy is not associated with a higher risk of cardiotoxicity than trastuzumab therapy alone41–43. Of interest, the reported LVEF decline with the tyrosine kinase inhibitors (TKIs) afatinib and osimertinib, which target epidermal growth factor receptor (EGFR; also known as HER1), has been attributed to inhibition of HER2 in addition to EGFR inhibition44.
TKIs are the second major group of targeted cancer therapies. These drugs interfere with the transfer of a phosphate group to a tyrosine residue of a protein, a critical regulatory cue in signalling pathways that control cell function, proliferation and survival45. A prominent example of a TKI is imatinib, which neutralizes the BCR–ABL1 fusion protein, the molecular fingerprint of Philadelphia chromosome-positive haematological cancers, such as chronic myeloid leukaemia46. Unexpectedly, cases of HF were reported in patients receiving imatinib, and in vivo and in vitro experiments indicated direct cardiotoxicity potential47. Activation of the endoplasmic reticulum stress response, collapse of the mitochondrial membrane potential, release of cytochrome c into the cytosol and reduction in cellular ATP content were the originally implicated mechanisms leading to cardiomyocyte death47. Over the years, experimental studies have both supported and challenged these initial observations19,48,49. In clinical practice, cardiomyopathy and HF are very rarely seen with imatinib therapy (incidence of ≤1%)50,51.
Philadelphia chromosome.
Named after the city in which it was discovered in 1960 as the first tumour-specific chromosomal change in the form of a shortened chromosome 22 as a result of a reciprocal translocation that leads to the oncogenic BCR–ABL1 gene fusion, which has a causal role in the malignant transformation of white blood cell precursors; the Philadelphia chromosome is found in 90% of patients with chronic myeloid leukaemia.
Endoplasmic reticulum stress response.
Disruption of endoplasmic reticulum function leads to impairment of protein folding, accumulation of unfolded and misfolded proteins and risk of cell toxicity. The cell reacts to this endoplasmic reticulum stress by initiating the unfolded protein response to increase the capacity of the cell to handle and/or eliminate the accumulating unfolded or misfolded proteins or to initiate apoptosis.
As outlined in Supplementary Fig. 2, the incidence of cardiovascular toxicity differs considerably between different TKIs, and various mechanisms for the cardiotoxicity have been proposed over the years47,52–58. Intuitively, the thought has been that cardiotoxicity is the consequence of drug promiscuity (that is, a function of the number of kinases inhibited)47,52,55,56. However, experimental studies support the sentinel kinase theory and, as shown in Supplementary Fig. 2, the TKIs affecting the vascular endothelial growth factor (VEGF) and MAPK/ERK kinase (MEK) signalling pathways might be the TKIs associated with the highest risk of cardiotoxicity clinically52,59. Furthermore, elegant studies have shown a remarkable spectrum of changes in the heart and cardiomyocytes even with TKIs targeting a single kinase60. These changes encompass not only downregulation but also upregulation of kinase gene expression and activity. For instance, erlotinib might be associated with a low risk of cardiotoxicity not necessarily because of the sole inhibition of EGFR but rather because of the upregulation of signal transducer and activator of transcription 3 (STAT3) signalling, allowing adaptive fatty acid metabolism to maintain cardiac function60. Likewise, studies in human inducible pluripotent stem cell-derived cardiomyocytes point towards insulin receptor signalling as a compensatory pathway in therapies inhibiting VEGF signalling61. Therefore, TKI-associated cardiotoxicity is complex and might be best assessed in an integrative (systems biology) manner62–64.
Sentinel kinase theory.
The theory that inhibition of one specific enzyme among all the enzymes that catalyse the transfer of a phosphate group from ATP onto a tyrosine, serine or threonine residue of a protein (kinome) is responsible for a specific action.
Management and prevention.
Consensus documents and guidelines on cardiotoxicity with cancer therapy (Supplementary Tables 1–3) generally agree that before starting any (potentially) cardiotoxic therapy, all patients should undergo a baseline assessment of cardiac function, with echocardiography as the preferred imaging modality (the American Society Echocardiography and European Association of Cardiovascular Imaging (ASE/EACI) recommend 3D echocardiography or 2D contrast echocardiography, plus global longitudinal strain (GLS), plus cTn measurement)65, an assessment of any potential cardiovascular diseases and risk factors and optimal control of any of the cardiovascular abnormalities identified (ASE/EACI recommend cardiology referral in the case of any abnormal baseline parameter, for discussion of the pros and cons of cancer therapy and the initiation of cardioprotective strategies)65. In this way, patients with cancer are approached in a manner similar to a preanaesthesia medical evaluation.
Recommendations for on-therapy and after-therapy evaluations have varied for anthracycline and non-anthracycline regimens. This difference is on the basis of the expected differences in cardiac function dynamics with these medications: a drop after therapy with anthracyclines versus a drop during therapy with non-anthracyclines. For therapies involving anthracyclines, the ASE/EACI consensus recommendation is to reassess all outlined parameters (LVEF, GLS and cTn) at completion and at 6 months after therapy, and if the cumulative doxorubicin-equivalent dose is >240 mg/m2, repeated measurements of LVEF, GLS and cTn should be performed before each additional dose of 50 mg/m2 (REF.65). For cardiotoxicity related to non-anthracycline therapies, the ASE/EACI consensus is follow-up every 3 months with the outlined parameters while the patient is receiving cancer therapy, with two exceptions: patients receiving TKIs or VEGF inhibitors, who should have an additional early follow-up at 1 month, and patients with previous anthracycline exposure, who should have an additional evaluation at 6 months65. For a surveillance strategy that is based on cTn levels, measurements are to be taken before and/or 24 h after each cycle of cancer therapy. Of note, the cumulative incidence of cTn level elevation increases with each cycle and can be seen with any form of high-dose chemotherapy. Patients who have a persistent elevation of cTn levels at 1 month of follow-up seem to be at the highest risk of cardiovascular events (mainly cardiomyopathy, HF and arrhythmias)66. For GLS, a 15% relative change is considered to represent subclinical left ventricular dysfunction, but imaging should be repeated within 2–3 weeks65. For LVEF, a drop of 10% from baseline to <53% is considered to represent cardiac dysfunction. Importantly, the load dependency of these measures needs to be taken into consideration65.
The course of action for patients with abnormal GLS at either the relative threshold or the absolute threshold is not defined at present but clinical trials are ongoing to address this question in patients receiving anthracycline or non-anthracycline therapy, such as the SUCCOUR67 and TACTIC68 trials. In patients with reduced cardiac function and/or HF, treatment according to AHA/ACC HF stages is recommended69,70 (Supplementary Table 4). A critical question is whether to continue cancer therapy and by which parameters and cut-off values this should be decided. At present, there is no consensus that the LVEF should be ≥40% for cancer therapy in general71, whether the LVEF cut-off level can be as low as 30% even with anthracycline therapy72 or whether the LVEF should be >45% for patients receiving anthracyclines73, and whether cancer therapy should be stopped if a LVEF decline of a certain degree to a certain level is recognized (for example, ≥10% decline to a LVEF of <50%)72 and other causes cannot be identified74. Tests and prediction models of risk, reversibility and prognosis of cardiotoxicity would be extremely helpful. An illustrating example is the utility of cTn levels in defining which patients are at risk of irreversible LVEF decline with trastuzumab therapy, as outlined earlier.
The modes of prevention of cancer therapy-related cardiotoxicity have varied drastically. For anthracyclines, the considerations have been the use of epirubicin instead of doxorubicin, although studies have suggested that when adjusted for equivalent dose, no significant difference is seen between these two drugs; prolonged infusion rates to reduce peak circulating concentrations of the drug; use of liposomal formulations to reduce myocardial accumulation; concomitant use of dexrazoxane, which was introduced as an iron chelator but also has cardioprotective effects through interaction with topoisomerase IIβ17; and use of an alternative, non-anthracycline-based therapy, which might or might not have equivalent anticancer efficacy. Various cardiovascular disease medications, especially the second-generation and third-generation β-blockers carvedilol and nebivolol, angiotensin-converting enzyme inhibitors, angiotensin-receptor blockers, spironolactone and statins, have been shown to have a preventive effect against anthracycline-related cardiomyopathy, although not unequivocally7,75. Additional novel approaches to test in future studies include the use of erythropoietin, which might act through the progenitor cell pool76–78. Another potential approach is the use of patient-specific, inducible pluripotent stem cell-derived cardiomyocytes to identify patients at high risk of cardiotoxicity with anthracyclines79,80. However, the long turnaround time for this test currently does not allow the expeditious decision-making that is often needed for cancer therapy. Finally, although some gene variants predisposing to cancer therapy-related cardiotoxicity have been defined and their use for patient screening and for selecting therapy is conceptually attractive, testing for these variants has not yet been adopted in clinical practice63.
Effective cardioprotective therapies have not been defined for trastuzumab-induced cardiomyopathy, because the two trials conducted so far (one on bisoprolol and perindopril and one on candesartan) did not meet their primary end points81,82. Although initial reports suggested that cessation of trastuzumab therapy suffices for the recovery of cardiac function, other studies indicated that institution of guideline-directed cardiovascular therapy helps to decrease the burden of irreversible cardiac decline30,83. Re-exposure to trastuzumab after recovery of cardiac function has been deemed possible, although a redecline might occur and LVEF needs to be followed up serially. Severe increases in blood pressure (systolic blood pressure >180 mmHg) should be avoided because experimental studies and clinical reports show that the risk of decompensating HF is increased when (very) high afterload conditions are combined with HER2 inhibition84,85. This recommendation is also important for patients receiving VEGF-inhibitor therapies (see the next section). These considerations are important for the concept of the (functional) cardiovascular reserve capacity, which is a very simple but important and practical framework for the general approach to patients with cancer at risk of cardiomyopathy and HF. Related conceptual models that predict risk on the basis of this concept remain to be validated. The role of improving the cardiovascular reserve before, during and after cancer therapy has been addressed in previous reviews and a 2019 AHA statement86,87.
Type II cardiomyopathy
In contrast to cancer therapy-related type I cardiomyopathies, in cancer therapy-related type II cardiomyopathies, factors other than a direct toxic effect on cardiomyocytes are the main reason for or contribute substantially to a decline in cardiac function. Recognizing these factors is important for patient management and outcomes.
Conventional chemotherapies.
Therapy with 5-fluorouracil (5-FU) and capecitabine has been associated with cardiotoxicity in up to 20–30% of patients (depending on the patient population studied and criteria used)88 (TABLE 1). Induction of profound and diffuse vasoconstriction that involves the coronary microcirculation is one possible mechanism of cardiotoxicity, especially in patients who show a rather quick recovery and have a type of cardiomyopathy referred to as Takotsubo syndrome89–96. In other patients, permanent damage can evolve as a consequence of vasospasm-related myocardial infarction (MI) or direct toxic injury to the myocardium and the vasculature97. Indeed, direct injury to cardiomyocytes, even similar to anthracycline-related damage, has been reported with 5-FU98–102. This direct cardio toxic effect has been attributed to several mechanisms, among them induction of oxidative stress and metabolic derangements in cardiomyocytes103,104. 5-FU is catabolized to fluoroacetate, which interferes with Krebs cycle activity, leading to depletion of high-energy phosphates that are critical for normal cardiac function105,106. Importantly, the metabolism of 5-FU is controlled by dihydropyrimidine dehydrogenase (DPD), and deficiencies in this enzyme have been associated with an increased risk of 5-FU-related toxic effects, although the link with cardiotoxicity remains debated107,108. Arguing against a link with DPD deficiency is the occurrence of 5-FU-related cardiotoxicity in patients with normal DPD activity104. Nonetheless, treatment with the 5-FU prodrug tegafur (5-fluoropyrimidine) in combination with the DPD inhibitor uracil, which allows the delivery of 5-fluoropyrimidine while blocking the generation of toxic metabolites, has been shown to reduce 5-FU-related cardiotoxicity104,108.
Targeted therapies.
HF presentations and declines of cardiac function, even presentations of Takotsubo cardiomyopathy, have been reported with VEGF inhibitors, such as bevacizumab. Given that bevacizumab does not have the confounding aspects of multitarget effects of TKIs and has not been shown to be directly toxic to cardiomyocytes, one might conclude that inhibition of the VEGF signalling pathway alone suffices to induce ‘cardiotoxicity’ and via effects different from conventional chemotherapy-induced cell toxicity109–111. As reviewed in detail previously111, inhibition of the VEGF pathway impairs vascular reactivity and the angiogenic response to ischaemia and increased afterload conditions in the heart. This effect might explain the relevance of coronary artery disease (CAD) and hypertension as risk factors for VEGF inhibitor-related cardiomyopathy. However, any pre-existing or evolving, absolute or relative, structural or functional coronary (micro) vascular deficit can result in a risk of cardiomyopathy with VEGF-inhibitor therapy111. Of note, whereas capillary regression is seen in endocrine organs rapidly after initiation of VEGF-inhibitor therapy, this regression is not observed in the heart112–115. Therefore, a decline in cardiac function with VEGF-inhibitor therapy might not be seen unless additional stressors increase the activity of (and/or the demand for a compensatory response via) the VEGF pathway.
Additional pathways of interest include the insulin receptor pathway, which can serve a compensatory role when VEGF signalling is inhibited61, and the platelet-derived growth factor subunit-β pathway, which has a critical role in pericyte viability and is a critical off-target pathway contributing to sunitinib-related cardiotoxicity. Sunitinib, which inhibits multiple receptor tyrosine kinases, including VEGF receptors and platelet-derived growth factor receptors, destabilizes the coronary microvascular endothelial network and reduces the coronary flow reserve and cardiac contractile reserve.
Immunotherapies.
Immunomodulatory strategies have been developed with the aim to train the host immune cells to target and destroy cancer cells. One type of cancer immunotherapy, known as chimeric antigen receptor (CAR) T cell therapy, is based on the recognition by engineered T cells of signature surface antigens on cancer cells116,117 (BOX 3). The first CAR T cell strategy that was developed targeted melanoma-associated antigen 3 (MAGEA3)118. Despite no signal for toxicity in preclinical testing, two patients who received this therapy died of HF within a few days119 (TABLE 3). Severe acute myocarditis with T cell-mediated cardiac injury was the underlying histopathology. Intriguingly, this effect was not related to cardiac expression of MAGEA3 but instead was caused by cross-reactive recognition of titin, a striated muscle-specific protein120.
Box 3 |. CAR T cell therapy.
T cells are normally activated when the T cell receptor (TCR) recognizes an antigen that is presented in conjunction with MHC class I or class II complexes. By contrast, engineered chimeric antigen receptors (CARs) recognize the antigen in a manner similar to that of an antibody, in an MHC-unrestricted manner. CARs consist of an extracellular antigen-recognition domain (most commonly a monoclonal antibody single-chain variable region that targets a tumour-associated antigen) linked to a T cell signalling transmembrane domain (such as an intracellular fragment of the TCR CD3ζ chain in ‘first-generation’ CARs) that anchors the chimeric receptor in the cell membrane and activates the T cell when the extracellular portion recognizes the target cell expressing the specific antigen, thereby linking recognition to activation. Activation is mediated by the intracellular fraction, which stimulates T cell proliferation, cytokine secretion and cytolysis to eliminate the target cell. Therefore, expression of engineered CARs on T cells allows for the control of T cell targeting of tumour cells with high degree of specificity. To generate CAR T cells, T cells are harvested from the patient (or from an allogeneic donor) and are transfected with a gene-therapy vector (such as a lentivirus) encoding the CAR construct.
The first CAR T cell strategy that was developed targeted melanoma-associated antigen 3 (MAGEA3), the first human tumour-associated antigen to be specifically recognized by CD8+ T cells. Another asset in favour of this approach was the expression of MAGEA3 in a wide variety of neoplasms, particularly melanoma and non-small-cell lung cancer, but not in normal tissues except the testes and placenta (where this antigen is not presented to CD4+ T cells and CD8+ T cells).
HER2 (also known as ERBB2 or neu) was chosen early on as another promising tumour-associated antigen target for CAR T cells. The hope was for the broader application of this approach as a therapeutic strategy for all cancers expressing HER2, including breast, gastric, colon, renal and ovarian cancer. By contrast, CD19 was chosen as a target for CAR T cell therapy because its expression is confined to B cells and the levels are much higher than those of any other markers in B cell leukaemias and lymphomas. Furthermore, any potential depletion of B cells that might arise as a consequence of the anti-CD19 CAR T cell therapy was thought to be beneficial to avoid any potential antibody response to the engineered CAR T cells.
Table 3 |.
Leading cardiovascular toxic effects of cancer immunotherapies
| Therapy (target) | Cancer therapy indications (label and off-label) | Toxicity | |||
|---|---|---|---|---|---|
| Cardiac | Arrhythmia | Vascular toxicity | Other | ||
| Immune checkpoint inhibitors | |||||
| Ipilimumab (anti-CTLA4) | Colorectal cancer, melanoma, RCC, SCLC | + | + | + | US black box warning: immune-mediated adverse reactions |
| Nivolumab (anti-PD1) | Colorectal cancer, HNSCC, hepatocellular carcinoma, HL, melanoma, NSCLC, RCC, SCLC, urothelial carcinoma | + | + | ++ | Immune-mediated adverse events, peripheral oedema, hypertension |
| Pembrolizumab (anti-PD1) | Cervical cancer, gastric cancer, HNSCC, hepatocellular carcinoma, HL, melanoma, Merkel cell carcinoma, NSCLC, primary mediastinal large B cell lymphoma, urothelial carcinoma | + | + | + | Immune-mediated adverse events, oedema, pericarditis, pericardial effusion |
| Atezolizumab (anti-PDL1) | Breast cancer (triple negative), NSCLC, SCLC, urothelial carcinoma | + | – | + | Immune-mediated adverse events, peripheral oedema |
| Avelumab (anti-PDL1) | Merkel cell carcinoma, urothelial carcinoma | + | – | – | Immune-mediated adverse events, peripheral oedema, hypertension |
| Durvalumab (anti-PDL1) | Non-small-cell carcinoma, urothelial carcinoma | + | – | – | Immune-mediated adverse events, peripheral oedema |
| CAR T cell therapy | |||||
| Tisagenlecleucel (anti-CD19) | ALL, diffuse large B cell lymphoma | ++ | +++ | ++ | Hypotension, hypertension |
| US black box warning: cytokine release syndrome, neurotoxicity | |||||
Based on data from Micromedex (IBM, NY, USA) and Lexicomp (Wolters Kluwer, Netherlands). Frequency of cardiovascular toxic effects: –, not reported; +, uncommon (<1%); ++, common (1–10%); +++, very common (>10%). ALL, acute lymphoblastic leukaemia; CAR, chimeric antigen receptor; CTLA4, cytotoxic T lymphocyte antigen 4; HL, Hodgkin lymphoma; HNSCC, head and neck squamous cell carcinoma; NSCLC, non-small-cell lung cancer; PD1, programmed cell death 1; PDL1, programmed cell death 1 ligand 1; RCC, renal cell carcinoma; SCLC, small-cell lung cancer.
The first CAR T cell therapy directed against HER2 was also associated with toxicity, inducing the development of acute respiratory failure, shock and cardiac arrest within 12 h (REF.121). Diffuse alveolar damage was seen on autopsy and was thought to be the initial insult that was then followed by multiorgan ischaemia and systemic haemorrhagic microangiopathy. On-target and off-tumour reactivity against HER2 in normal pulmonary tissue on first-pass clearance in the lungs with subsequent pneumonitis and cytokine storm was postulated as the underlying mechanism of anti-HER2 CAR T cell toxicity. However, the associated adverse effects might also have been a consequence of the dose because this patient received the highest permitted number of cells. In agreement with this idea, subsequent studies with a different HER2-specific CAR T cell therapy at much lower doses (and without conditioning chemotherapy) have proven it to be safe122.
The currently best-studied type of CAR T cell therapy is directed against CD19 and is approved by the FDA as tisagenlecleucel (Kymriah) for children and young adults with relapsed or resistant B cell acute lymphoblastic leukaemia and adults with relapsed or refractory diffuse large B cell lymphoma116. The adverse effect profile associated with anti-CD19 CAR T cell therapy is extensive, but the best-known adverse effect of this therapy, and of any CAR T cell therapy, is cytokine release syndrome (CRS)116,123–125.
Cytokine release syndrome.
(CRS). A systemic inflammatory response that can be triggered by a variety of factors such as infections, antibody-based immunotherapies and chimeric antigen receptor T cell therapy. CRS is caused by the rapid release of a large amount of cytokines into the circulation, leading to fever, nausea, headache, rash, tachycardia, hypotension and respiratory distress.
The cardiovascular sequelae with CRS in the setting of CAR T cell therapy include tachycardia (with mild CRS) and hypotension, arrhythmias and decreased cardiac ejection fraction (with severe CRS)125. Cardiac arrest is rare, but can occur even 1 week after therapy initiation, as reported in one patient in conjunction with a precipitous drop in LVEF116,123–125. The cardiac function dynamics in CRS are thought to be similar to those seen in patients with systemic inflammatory response syndrome or sepsis but can differ, with differences in the cytokine profile and a general lack of endotoxin exposure126. Tumour necrosis factor (TNF) and IL-1β are considered the two leading cytokines accounting for the drop in LVEF in sepsis, via nitric oxide-dependent and nitric oxide-independent alteration of myocardial contractility127. Counterintuitively, a reduction in LVEF is not a sign of poor prognosis in patients with sepsis, and the presence of new-onset left ventricular dysfunction does not increase the risk of long-term HF outcomes in severe sepsis and septic shock128. Indeed, patients who survived septic shock showed a dynamic LVEF profile with decline during the acute phase and recovery after 7–10 days, whereas LVEF remained static in patients who subsequently died129. This observation seems to be the consequence of cardiac remodelling, that is, an increase in ventricular compliance that leads to ventricular dilatation, which subsequently accounts for a lower calculated LVEF while stroke volume remains the same and cardiac output is not depressed. These dynamics reduce the likelihood of a myocardial hypercontractile response in the hyperdynamic circulatory state characteristic of septic shock, which translates into poorer outcomes in these patients. In agreement with this concept, β-blocker therapy in patients with septic shock leads to improved outcomes130. Importantly, although protected from hyperdynamic circulatory exhaust, the hearts of patients who survive sepsis remain responsive to catecholamine stimulation during septic shock and show increased contractility and cardiac performance with dobutamine therapy131. These details of cardiac function dynamics and their relationship with outcomes have not been fully described in patients with CRS.
A second type of T cell-directed immunotherapy, known as bispecific T cell engager therapy (BiTE therapy), can lead to a similar spectrum of complications as CAR T cell therapy, but not as commonly or severely132,133.
Bispecific T cell engager therapy.
(BiTE therapy). BiTE antibody constructs are designed to create an immunologic synapse between an effector T cell and a tumour cell by simultaneously binding to the T cell activation molecule CD3 and a tumour-associated antigen, which is CD19 on B cells in the case of blinatumomab (approved for the treatment of B cell acute lymphoblastic leukaemia).
A third and the leading type of cancer immunotherapy at present is immune checkpoint inhibition (BOX 4). Immune checkpoint inhibitors (ICIs) are a unique type of antibody-based targeted therapies. This approach leverages the principles of immunosurveillance, its under-pinning by cancer cells and its reactivation by targeting the ‘breaks’ or ‘checkpoints’ of effector T cells134–136. Although the main cardiotoxicity associated with ICIs is myocarditis, Takotsubo syndrome and global cardiomyopathies have also been reported in patients receiving ICIs137,138. The mechanisms of these types of ICI-related cardiotoxicity are not entirely clear. As in all patients with myocarditis, various stressors could have a contributing role139. As in other patients with Takotsubo syndrome or global cardiomyopathy, the main clinical presentations are acute coronary syndrome and acutely decompensated HF139.
Box 4 |. Immune checkpoint inhibitors.
T cell activation is modulated not only by co-stimulation but also co-inhibition pathways to prevent an excessive immune response. These pathways can be exploited by tumour cells to escape immune-mediated destruction. Immune checkpoint inhibitors (ICIs) are molecules that target T cell inhibition pathways, such as the cytotoxic T lymphocyte antigen 4 (CTLA4) and programmed cell death 1 (PD1) pathways, thereby reversing the immune tolerance of the T cells towards tumour cells and promoting T cell antitumour activity.
CTLA4, the first target used for clinical ICI therapy, interacts with the same surface molecules on antigen-presenting cells that interact with the co-stimulatory signal CD28: CD80 and CD86. CTLA4 directly competes with CD28 for the binding sites, but with much higher affinity. In contrast to the signalling of CD28 promoting T cell activation, CTLA4 signalling blocks the T cell response. CTLA4 is stored in intracellular vesicles in the T cell, which are transported to the cell surface on antigen-driven engagement of the T cell receptor. This relocation usually occurs within 2 days of T cell activation and affects both CD4+ T cells and CD8+ T cells in the lymphatic tissues. Therefore, antibodies interfering with CTLA4 are directed to T cells that interact with antigen-presenting cells in lymphoid organs in the early stage of the adaptive immune response and, most of all, to T helper cells. In addition, CTLA4 is constitutively expressed and has a vital role in regulatory T cells. Therefore, CTLA4 inhibition has a profoundly negative effect on this anti-inflammatory T cell population.
The immune checkpoint that became the second and more prevalent target for clinical therapy is the PD1–PD1 ligand 1 (PDL1) system. This pathway is a negative regulator of T cell activity in peripheral tissues, including tumours. PD1 is expressed in all inflammatory cells, including monocytes, dendritic cells, natural killer cells, B cells and T cells. In the tumour, PD1 is expressed in activated tumour-infiltrating (mainly CD4+) T cells. In addition, PD1 is highly expressed on regulatory T cells, which also tend to infiltrate tumours densely. PDL1, which triggers the inhibitory signal in these immune cells on binding to PD1, is expressed by various tissue and cancer cells, as well as by tumour-infiltrating macrophages. Antigen-presenting cells also express PDL2, another ligand for PD1 with functions overlapping those of PDL1.
Radiation therapy.
Radiation therapy has always been an integral part of cancer treatment. The effectiveness of radiation therapy against cancer cells is mediated primarily by induction of DNA damage that then leads to cell senescence and cell death140. Generation of oxidative and nitrosative stress with modification of various cell molecules and structures has an additive effect on these outcomes.
Cell senescence.
A process defined as irreversible cell cycle arrest, driven by a variety of mechanisms, including telomere shortening, other forms of genotoxic stress, mitogens or inflammatory cytokines, that culminate in the activation of the tumour suppressor p53 and/or the cyclin-dependent kinase inhibitor p16.
The cardiomyopathy seen with radiation therapy is of the restrictive subtype (TABLE 1). This cardiomyopathy typically presents as HF with preserved ejection fraction141. Importantly, cardiomyocytes are fairly resistant to radiation injury142. However, cardiomyocytes are not immune to damage to their DNA and organelles; oxidative stress and metabolic abnormalities can also evolve with radiation therapy143. Experimental studies have demonstrated degeneration of cardiomyocytes in irradiated hearts144,145, but this cardiomyocyte damage seemingly follows changes in the coronary microcirculation146. The first response in radiation-related cardiotoxicity is activation of the coronary microvascular endothelium (to a large part related to the activation of the nuclear factor-κB (NF-κB) signalling pathway), with an increase in the expression of chemoattractant and adhesion molecules, which favours leukocyte infiltration. In addition, an increase in vascular permeability leads to the extravasation of blood content such as fibrin and its deposition in the interstitium as amyloid-like structures147. Increased endothelial dysfunction together with a reduction in thrombomodulin levels contributes to thrombus formation. Depending on the extent of radiation-induced injury, enfacement and swelling of endothelial cells can also be seen, further contributing to microvascular obstruction. Capillary density might not change or might even slightly increase during the acute phase. However, over time, the proliferative (angiogenic) response of coronary microvascular endothelial cells is exhausted, and the area of the functionally competent microvasculature is reduced. This loss of microvasculature can result in ischaemia and cardiomyocyte loss with replacement fibrosis. Myocardial fibrosis is further provoked by the inflammatory response and premature senescent changes in tissue fibroblasts. These changes along with activation of the transforming growth factor-β (TGFβ)–SMAD signalling pathway in these fibroblasts induce the production of excessive amounts of collagen. A similar type of inflammatory and fibrotic injury response to radiation therapy can be observed on the valves and the pericardium148,149. Pericardial and valvular disease have long been known to contribute to HF, the final common pathway of radiation-induced heart disease. Defining the relative contributions of ischaemia, restriction, constriction, volume overload and pressure overload is important for the treatment of patients with radiation-induced heart disease but might not always be possible. Some of these factors can be differentiated by dose exposure, because several studies have revealed that the risk of pericarditis (and therefore its long-term complications) is low with dose exposures below 35–40 Gy (REF.150).
Management and prevention.
The identification and management of factors that contribute to or drive cancer therapy-related type II cardiomyopathies are essential for the management of these conditions. These factors differ by the type of cancer therapy, as outlined in the following paragraphs. As a common principle, cancer therapies contributing to the cardiotoxicity should be discontinued at least until cardiac function recovers and the precipitating or contributing factors are controlled. Resumption of these therapies is subject to risk–benefit assessment and discussion (with close follow-up of patients).
A main element in the management of 5-FU-induced or capecitabine-induced cardiotoxicity is potent vasodilatory therapy. Nitrates might suffice on the epicardial level but can be insufficient on the coronary microcirculatory level151. Calcium-channel blockers (for example, diltiazem or long-acting nifedipine) are more efficacious in this regard. A history of cardiac disease (in particular ischaemic heart disease) significantly increases the risk of 5-FU-induced cardiotoxicity152. Some data suggest that renal insufficiency rather than age (>55 years) is also a risk factor for 5-FU-induced cardiotoxicity104,151. The mode of administration of the cancer therapeutic is an important factor: the risk of cardiotoxicity is higher with continuous administration (over 2 days) than with bolus infusion (over 3 h); the latter is, therefore, a preventive strategy153. In addition, patients at risk of cardiomyopathy, especially those with previous events, should be given vasodilatory therapy. However, this approach might not provide full protection, and continuous ECG monitoring is advised. In some patients in whom left ventricular function decline is highly suspected in the absence of traditional clinical signs and symptoms, on-therapy follow-up with measurement of plasma B-type natriuretic peptide (BNP) levels and/or by echocardiography might prove useful. Other approaches include the use of alternative preparations of 5-FU, such as tegafur–uracil and tegafur–gimeracil–oteracil (known as S-1)104. Uridine triacetate (Vistogard) was approved in 2015 for the treatment of life-threatening 5-FU-related and capecitabine-related toxicity154. This compound delivers high concentrations of uridine, which competes with 5-FU metabolites154.
For VEGF-inhibitor therapy, the evaluation and treatment of any possible contributing factor is the best approach, as discussed earlier and in greater detail else-where111,155 (Supplementary Fig. 3). Proper management of hypertension is a general principle for the prevention of HF, but especially in patients receiving VEGF-inhibitor therapy, and an argument can be made for aiming towards the SPRINT156 blood pressure target of <130/80 mmHg in these patients. Other conditions of increased cardiac afterload, such as aortic stenosis, might not be as easily amenable to therapy, especially if they do not yet meet the criteria for intervention but are still severe enough to trigger a hypertrophic response in the myocardium and a reduced cardiovascular flow reserve. The same reduction in the cardiovascular flow reserve might be present in patients with diabetes before any cancer therapy, and correction might also not be possible in these patients, especially not in a short time. Although a clinical history of CAD suffices as a risk factor for VEGF inhibitor-related cardiomyopathy, whether a history of MI suffices as the sole critical element leading to VEGF inhibitor-related cardiomyopathy or whether, for instance, the extent of baseline and inducible ischaemia should be defined is unknown157–159.
Cardiovascular flow reserve.
The capacity of the coronary vascular bed to increase blood flow maximally to the myocardium, often expressed as a ratio with regard to baseline blood flow.
The treatment of choice for CRS grade 3 or greater in patients receiving CAR T cell therapy is an IL-6 antagonist (such as tocilizumab or siltuximab)125. The prophylactic use of these agents might prevent the development of CRS and is currently under investigation. A concern is that this strategy could negate the main anticancer effect of the CAR T cell therapy. The same concerns apply to prednisone, an anti-inflammatory glucocorticoid therapy that is recommended for severe CRS. In patients with evidence of circulatory compromise (shock), haemodynamic support with vasopressors is also recommended160.
For patients receiving radiation therapy, reduction of dose exposure is the best intervention. Some experimental studies have indicated a benefit of statin and angiotensin-converting enzyme-inhibitor therapy, and anti-inflammatory and antioxidant therapies are theoretically attractive, but none of these approaches has been proven in clinical practice. These strategies should be tested, but challenges include defining the optimal treatment window and covering the diverse spectrum of cardiac disease associated with radiation therapy. These concerns apply to any strategies newly identified in preclinical studies, including TGFβ receptor type 1 inhibitors, sestrin 2 inducers, recombinant neuregulin 1 and miR-21 inhibitors161. The ASE/EAVI and the Society for Cardiovascular Angiography and Interventions provide consensus algorithms for follow-up after radiation therapy162,163 (Supplementary Fig. 4).
Type III cardiomyopathy
Conventional chemotherapies.
The classic example of conventional chemotherapy that can induce myocarditis is cyclophosphamide164. Particularly at high doses, cyclophosphamide can cause haemorrhagic myocarditis165. The threshold dose for cyclophosphamide-induced myocarditis is not defined. A dosage of >270 mg/kg for 1–4 days or doses of ≥1.55 g/m2 are considered to be associated with a substantial risk of cardiotoxicity166. However, doses as low as 100 mg/kg can generate cardiotoxicity166. At an in-between dose of >150 mg/kg, the incidence of acute HF is 7–33%166. Interindividual variation in metabolism might be a factor contributing to the differences in incidence. Metabolites of cyclophosphamide can induce endothelial capillary injury with oedema, haemorrhage and thrombosis166. Tachyarrhythmias can be induced as a result of myocardial injury, and more advanced stages present as HF. Cyclophosphamide therapy can also induce pericardial effusion, even with acutely life-threatening tamponade. Progressive myocardial mechanical failure can also evolve. Mortality in patients with cyclophosphamide-induced myocarditis is 2–17%166.
Targeted cancer therapies.
Immune mechanisms have been suggested to contribute to the anticancer effects of trastuzumab167, but whether and to what degree immune mechanisms contribute to trastuzumab-induced cardiomyopathy is not known; at present, only little experimental evidence is available168. Only one case has been reported of fulminant acute myocarditis with the TKI sorafenib, which led to cardiogenic shock with a fatal outcome169. This patient also had myositis, a constellation of conditions more commonly seen with ICI therapy.
Immunotherapies.
ICIs can induce a broad spectrum of immune-related adverse events that differ on the basis of similarities and differences between therapies targeted at cytotoxic T lymphocyte antigen 4 (CTLA4) or at the programmed cell death 1 (PD1)–PD1 ligand 1 (PDL1) axis170–172. The incidence of immune-related adverse events is generally higher with CTLA4 inhibition and highest (>50%) with combined CTLA4 and PDL1 inhibition173. Usually, colitis, dermatitis and pneumonitis are the earliest and most common organ presentations (in descending order, all with incidence of >10%). However, myocarditis, which has been reported with all types of ICIs, is associated with the highest mortality (40% in a 2018 systematic review and meta-analysis)174,175. Precise estimates of the incidence of ICI-induced myocarditis are evolving and expected to rise beyond the currently reported rates of up to 1% with increasing awareness176. The severest forms of ICI-induced myocarditis are prone to attract clinical attention and encompass decompensated HF, cardiogenic shock and sudden cardiac death. Myocardial biopsy, when performed, is often but not always positive for myocarditis. Sampling bias and sampling error are inherent limitations that confound the conclusions. However, one cannot exclude the possibility that profound, global declines in cardiac function can develop even in the absence of florid myocarditis177. Furthermore, in one of the first pooled analyses of patients with ICI-induced cardiotoxicity (n = 30), late gadolinium enhancement (an indicator of myocardial fibrosis) was seen on cardiac MRI in only 23% of patients and myocardial oedema was seen in only 33% of patients177. Apical ballooning was diagnosed in 14% of the patients, and among patients with available data on LVEF changes, complete reversibility of LVEF decline was seen in only 50% of them138,177.
By contrast, cardiac function (assessed by echocardiography) remained fairly normal despite evolving fulminant myocarditis with ICI use in the first reported cases of this entity178. ECG changes, including various forms of conduction block, ventricular ectopy and ventricular tachycardia (VT), and elevation of circulating cTn levels seemed to be more sensitive indicators of myocarditis. Circulating BNP and amino-terminal pro-BNP levels are also recognized as sensitive markers of myocarditis and might even be superior to cTn levels for detecting all forms of ICI-related cardiomyopathy, including those associated with global or regional (apical) cardiac function decline that does not fulfil imaging or tissue criteria for myocarditis138,177. Therefore, at least three different forms of cardiac function abnormalities with putatively different pathological mechanisms can evolve in patients undergoing ICI therapy.
Mechanistically, ICI-induced, immune-related adverse events such as myocarditis can be caused by one or a combination of the following factors: direct binding of ICIs to target molecules on non-lymphocytic cells, with downstream immune activation; formation of new T cells or reactivation of exhausted T cells against tumour antigens that cross-react with off-target tissues; generation of autoantibodies and production of pro-inflammatory cytokines136. Interestingly, PD1-deficient mice have a dramatically reduced lifespan that is compensated by cross-breeding with Rag1−/− mice, which lack mature B cells and T cells, indicating that an immune mechanism has an important role in the effects of genetic PD1 deficiency179. Dilated cardiomyopathy was a striking feature and accounted for the premature death of PD1-deficient mice. Although fibrotic reactions were seen sporadically, the ventricular walls of these mice appeared otherwise relatively normal, and scattered degeneration of cardiomyocytes was seen only on electron microscopy, with disarrayed and disrupted myofilaments and irregularly shaped mitochondria throughout the ventricular walls179. Subsequent studies revealed that the dilated cardiomyopathy was caused by the generation of autoantibodies against cTnI expressed on cardiomyocytes180. Of interest, tumours in humans can express cTn and other muscle-specific proteins, such as desmin and titin. However, whether the expression of these proteins (and autoantibodies against them) is causally involved in ICI-related myocarditis (and/or cardiomyopathy) has not been confirmed. PDL1 expression on non-haematopoietic cells, mainly endothelial cells, has an important role in providing protection against cytotoxic T cells181–184. This protection is particularly relevant in the setting of non-self-antigen expression in the heart secondary to a viral infection. The PD1–PDL1 system is also upregulated in the setting of other intrinsic modes of myocardial injury that induce inflammation, such as myocardial ischaemia and MI, probably to prevent inflammatory over-reactivity against cardiac tissue185. Identifying patients vulnerable to ICI-induced myocarditis (and ICI-related cardiomyopathy) secondary to the upregulation of the PD1–PDL1 system or to other mechanisms is an important current and future need186.
Management and prevention.
One of the main prerequisites for the appropriate management of ICI-related myocarditis is the knowledge and anticipation of this possible complication. Clinical presentation differs, and subtle signs and symptoms need to be adequately interpreted. Waiting until HF and cardiogenic shock develop to initiate management is suboptimal, especially because any culprit cancer therapy should preferably be discontinued as soon as possible. Management is mainly supportive, which can entail inotropic therapy and even mechanical circulatory support, including extracorporeal membrane oxygenation, as a bridge to recovery, as has been shown in patients who developed fulminant myocarditis with cyclophosphamide and ICIs187,188.
For early detection of cancer therapy-related myocarditis, the standard 12-lead ECG can be very effective. A declining R-wave amplitude (low voltage) can indicate progressive pericardial effusion and loss of myocardial mass, as occurring in haemorrhagic myopericarditis induced by cyclophosphamide. Other ECG indicators of myocardial inflammation include PR interval prolongation, heart block, bradycardia, ventricular ectopy and VT. Biomarkers have a supporting role, and the classic indicator of myocarditis is a protracted period of markedly elevated circulating cTn levels189. In a retrospective series of 35 patients with ICI-induced myocarditis, circulating cTn and BNP levels were elevated in 33 individuals (94%)190. However, ECG and cTn levels were an integral element in the diagnostic inclusion of patients in this study, which might explain the high levels of these markers. Of note, in another study on ICI-induced myocarditis and cardiomyopathy, circulating BNP levels were elevated in all patients but cTn levels were elevated in only 46%177. Coronary angiography is usually performed to exclude CAD leading to MI as the main differential diagnosis. In cases resembling culprit-lesion acute coronary syndrome, cardiac MRI is very valuable to identify acute MI with resolution of epicardial culprit-vessel lesion, acute myocarditis, Takotsubo cardiomyopathy or other cardiomyopathies191. Cardiac positron emission tomography might have a complementary role192.
In the aforementioned study involving 35 patients with ICI-related myocarditis, dyspnoea and oxygen requirement were two differentiating clinical features between those who developed major adverse cardiac events (MACE; including haemodynamically relevant heart block, cardiac arrest, cardiogenic shock and cardiac-related death) and those who did not190. Nearly 50% of all patients experienced MACE, with a mortality of 17%. These patients did not receive steroids as quickly or at as high a dosage as patients without MACE. LVEF was normal in 38% of patients with MACE and in nearly 50% of the patients in the overall cohort.
Immunosuppressive therapies have been recommended for all acutely life-threatening scenarios; that is, with confirmed myocarditis and VT or ventricular fibrillation. Immunosuppressive therapies should also be considered for any other potentially life-threatening presentation, such as advanced conduction disease or heart block owing to presumed myocarditis, pericarditis with cardiac tamponade and acute MI with coronary vasculitis on angiography. Some patients might require a quick escalation of immunosuppressive therapy by including immunoglobulin, antithymocyte globulin, infliximab (if HF is not present), mycophenolate mofetil or tacrolimus193. Plasmapheresis has also been implemented, with the goal of accelerating removal of the contributing drug (as well as any potential circulating autoantibodies). This approach is important with ICIs because their half-lives are extremely long: 14.5 days for ipilimumab, 25.0 days for pembrolizumab, 26.7 days for nivolumab and 27.0 days for atezolizumab. Finally, the CTLA4 agonist abatacept might be used in cases of steroid-refractory myocarditis194. Importantly, the clinical course of ICI-induced myocarditis can be so fulminant that mechanical support such as extracorporeal membrane oxygenation can become necessary and life-saving while all other measures are continued188. Overall, the hard evidence available at present is insufficient to support any of the anecdotal, albeit reasonable, strategies outlined above, and more evidence-based guidance in this area is needed. Current consensus recommendations by oncology societies are listed in Supplementary Table 2.
Arrhythmias related to cancer therapy
Several rhythm abnormalities can be seen in patients with cancer as they undergo therapy owing to several potential drug–drug interactions, metabolic and electrolyte derangements, and evolving toxic effects. In general, cancer therapy-related arrhythmias can be differentiated into bradycardia and tachycardia, with atrial fibrillation (AF) emerging as an important complication (TABLE 4). The real incidence of cancer therapy-induced arrhythmias is likely to be underestimated because routine cardiac monitoring is often not performed or includes only non-continuous 12-lead ECGs.
Table 4 |.
Types of arrhythmia reported with the use of cancer therapies
| Therapy class | Agent (target) | AF | SVT | Bradycardia | AV block | QTc prolongation | TdP | VT/VF | SCD |
|---|---|---|---|---|---|---|---|---|---|
| Miscellaneous | Arsenic trioxide | ++ | ++ | – | + | +++ | ++ | + | + |
| Alkylating agents | Anthracyclines; acute | ND | ND | ND | ND | ND | – | ND | ND |
| Busulfan | ND | ND | – | ND | – | – | – | ND | |
| Cyclophosphamide | ND | ND | – | ND | ND | – | ND | – | |
| Ifosfamide | ND | – | ND | – | – | – | ND | ND | |
| Melphalan | ND | ND | – | – | – | – | ND | ND | |
| Antimetabolites | 5-Fluorouracil | ND | ND | ND | ND | ND | – | ND | ND |
| Capecitabine | ++ | – | ++ | – | + | – | – | + | |
| Clofarabine | ND | ND | ND | – | – | – | – | – | |
| Cytarabine | ND | – | ND | – | – | – | – | – | |
| Gemcitabine | + | + | – | – | – | – | – | – | |
| Microtubule-binding agents | Paclitaxel | + | + | ++ | + | – | – | + | – |
| Platinum-based drugs | Cisplatin | + | + | + | + | – | – | + | – |
| Immunomodulatory drugs | Lenalidomide | ND | ND | ND | – | – | – | – | – |
| Thalidomide | + | + | + | – | – | – | – | – | |
| Proteasome inhibitors | Bortezomib | ND | – | ND | ND | ND | ND | ND | ND |
| Carfilzomib | ND | ND | ND | ND | – | – | – | ND | |
| HDAC inhibitors | Romidepsin | + | ++ | – | – | ++ | + | ++ | + |
| Panobinostat | – | – | – | – | ++ | – | – | – | |
| Vorinostat | – | – | ND | – | ++ | – | – | – | |
| CDK4/CDK6 inhibitors | Ribociclib | – | – | – | – | ++ | – | – | – |
| mTOR inhibitors | Everolimus | ++ | – | – | – | – | – | – | – |
| Monoclonal antibodies | Alemtuzumab (anti-CD52) | ++ | – | ++ | – | – | – | + | + |
| Cetuximab (anti-EGFR/HER1) | + | – | + | – | – | – | + | + | |
| Necitumumab (anti-EGFR/HER1) | – | + | – | – | – | – | – | ++ | |
| Pertuzumab (anti-EGFR/HER1) | + | + | + | – | – | – | + | + | |
| Rituximab (anti-CD20) | + | + | + | + | + | + | + | + | |
| Trastuzumab (anti-HER2/ERBB2) | ++ | ++ | + | – | – | – | + | – | |
| Multi-target kinase inhibitors | Osimertinib (EGFR/HER1) | – | – | – | – | ++ | – | – | – |
| Lapatinib (HER2/ERBB2) | + | + | – | – | + | – | – | – | |
| Lenvatinib (VEGFR) | – | – | – | – | ++ | – | – | – | |
| Pazopanib (VEGFR) | – | – | +++ | – | ++ | – | – | – | |
| Sorafenib (VEGFR) | + | – | + | + | + | + | – | – | |
| Sunitinib (VEGFR) | – | – | + | – | + | + | – | – | |
| Vandetanib (VEGFR) | – | – | – | – | +++ | – | + | + | |
| Bosutinib (BCR–ABL1) | – | – | + | – | ++ | – | – | – | |
| Dasatinib (BCR–ABL1) | + | + | – | – | + | – | + | + | |
| Imatinib (BCR–ABL1) | + | + | – | – | – | – | – | – | |
| Nilotinib (BCR–ABL1) | ++ | – | ++ | ++ | ++ | – | – | + | |
| Ponatinib (BCR–ABL1) | ++ | + | + | + | + | – | + | ||
| Ibrutinib (BTK) | +++ | – | – | – | – | – | + | + | |
| Alectinib (ALK) | – | – | +++ | – | + | – | – | – | |
| Ceritinib (ALK) | – | – | + | – | ++ | – | – | – | |
| Crizotinib (ALK) | – | – | +++ | – | + | – | – | – | |
| Brigatinib (ALK) | – | – | ++ | – | – | – | – | – | |
| Lorlatinib (ALK) | – | – | – | + | – | – | – | – | |
| Encorafenib (BRAF) | – | – | – | – | + | – | – | – | |
| Vemurafenib (BRAF) | ++ | + | + | – | +++ | + | – | – | |
| Gilteritinib (FTL3) | – | – | – | – | ++ | – | – | – | |
| Trametinib (MEK) | – | – | ++ | – | ++ | – | – | – | |
| Ruxolitinib (JAK) | – | – | + | – | + | – | – | – | |
| Immune checkpoint inhibitors | Ipilimumab (anti-CTLA4) | + | – | + | + | – | – | + | + |
| Nivolumab (anti-PD1) | + | – | + | + | – | – | + | + | |
| Pembrolizumab (anti-PD1) | + | – | + | + | – | – | + | + | |
| CAR T cell therapy | Tisagenlecleucel (anti-CD19) | ++ | ++ | – | – | – | – | – | – |
Frequency not always defined for the individual entities, but when available: –, not reported or very few data available; +, uncommon (<1%); ++, common (1–10%); +++, very common (>10%). AF, atrial fibrillation; AV, atrioventricular; CAR, chimeric antigen receptor; CDK, cyclin-dependent kinase; CTLA4, cytotoxic T lymphocyte antigen 4; EGFR, epidermal growth factor receptor; HDAC, histone deacetylase; JAK, Janus kinase; MEK, MAPK/ERK kinase; mTOR, mechanistic target of rapamycin; ND, frequency not defined; PD1, programmed cell death 1; QTc, corrected QT interval; SCD, sudden cardiac death; SVT, supraventricular tachycardia; TdP, torsades de pointes; VEGFR, vascular endothelial growth factor receptor; VF, ventricular fibrillation; VT, ventricular tachycardia.
Bradycardia
Conventional chemotherapies.
Cardiac arrhythmias were first noted to occur with paclitaxel, more specifically its Kolliphor EL (formerly known as Cremophor EL) formulation, when continuous cardiac monitoring was used to assess hypersensitivity reactions. These paclitaxel-induced arrhythmias are mainly episodes of asymptomatic bradycardia occurring in nearly 30% of patients195. All other arrhythmias are rare: heart block in 0.1% of patients, supraventricular tachycardia including AF and flutter in 0.2%, and VT and ventricular fibrillation in 0.3%. Most of these arrhythmias are noted with the first or second cycle of paclitaxel therapy, sometimes even within the first 24 h (REF.196). Typically, these episodes are self-limiting and resolve in 48–72 h after discontinuation of therapy, although brief episodes of supraventricular tachycardia and premature ventricular contractions can persist for up to 1–2 weeks. The mechanisms of paclitaxel-induced arrhythmias are not precisely defined; for example, whether these phenomena are inherent to paclitaxel or the vehicle used in the Kolliphor EL formulation is not clear.
Another chemotherapeutic classically associated with bradycardia is thalidomide, affecting as many as 50% of all patients with multiple myeloma who were treated with this medication. Elderly patients and those with comorbidities or receiving combination therapies with β-blockers, calcium-channel blockers, digoxin and antiarrhythmic drugs, or exposed to doxorubicin or cyclophosphamide and/or chest radiation therapy are at higher risk of bradycardia. Over-reactivity of the parasympathetic nervous system and thalidomide-induced hypothyroidism have been discussed as potential mechanisms197.
Targeted cancer therapies.
Sinus bradycardia can be seen with various TKIs (TABLE 4). Among VEGF pathway inhibitors, sinus bradycardia is most common with pazopanib (2–19%) but in a 2018 phase II study198, grade 4 bradycardia events were reported in 3% of patients with glioblastoma who were receiving bevacizumab in combination with the histone deacetylase (HDAC) inhibitor vorinostat. As in any other patients, other causes of sinus bradycardia in patients with cancer need to be excluded; for instance, sunitinib therapy can cause hypothyroidism, thereby leading to bradycardia199.
The other class of TKIs that has been associated with risk of bradycardia is ALK inhibitors (BOX 2). Sinus bradycardia has been reported in up to 15% of patients treated with crizotinib and up to 4% of patients treated with ceritinib200. Sinoatrial arrest and asystole have been reported with therapy with ibrutinib, an inhibitor of the tyrosine-protein kinase BTK, in addition to its well-known association with AF and ventricular arrhythmias (see later)201.
Immunotherapies.
In patients receiving ICI therapy, bradycardia can be seen in the setting of high degrees of atrioventricular (AV) block178,202. This AV block is secondary to inflammatory infiltration of the myocardium (that is, ICI-associated myocarditis), which can include the AV nodal area and the conduction system in the septum. The extent of AV block can warrant pacemaker implantation, even permanent devices if no resolution occurs in the setting of evolving fibrosis. On the basis of a 2018 systematic review, 10% of the cardiotoxicity events associated with ICI therapy were AV block or conduction disease, which leads to death in 50% of these patients176.
Radiation therapy.
Bradycardia can develop in patients after radiation therapy as a result of radiation injury and fibrosis in the heart with involvement of the conduction system, including the AV nodal area, the AV and His bundle and bundle branches203–205. In addition, accelerated CAD that affects the sinoatrial artery and the AV nodal branch can contribute to this presentation206. Calcifications of the aortomitral curtain and extensive calcification of the mitral annulus are indicative of the risk of bradycardia207,208.
Management and prevention.
Patients who are at risk of bradycardia with cancer therapy are not well defined but might present with any of the following elements: evidence of pre-existing cardiac conduction abnormalities (bundle branch or AV block); requirement of negatively dromotropic and chronotropic medications (β-blockers, digoxin and calcium-channel blockers) or antiarrhythmic agents; or poor tolerance of bradycardias. These patients often have underlying ischaemic heart disease, cardiomyopathy or HF. In these patients, chemotherapeutics that have been associated with bradycardia, such as crizotinib, paclitaxel, pazopanib and thalidomide, should be used very carefully, with proper monitoring of heart rate and blood pressure. In the event of symptomatic bradycardia, any potentially contributing medications are to be stopped until its resolution. Thereafter, the risks and benefits of resuming any bradycardia-associated cancer therapy and cardiovascular medications alone or in combination with dose reduction and with or without pacemaker implantation as per the guidelines need to be determined (Supplementary Table 5). Any life-threatening bradycardia requires discontinuation of the cancer therapy unless concurrent medications associated with bradycardia can be discontinued or adjusted in dose (such as a β-blocker and calcium-channel blocker) to allow resumption of the cancer therapy with frequent monitoring (with or without pacemaker support). Electrolytes, especially serum K+ levels, and renal and thyroid function should be checked. In patients receiving ICI therapy, the development of new conduction disease should prompt an evaluation for the presence of myocarditis.
QTc prolongation and VT
The risk of corrected QT interval (QTc) prolongation has been attributed to the effects of anticancer drugs on the inward current (increase) and outward current (decrease), leading to prolongation of the ventricular action potential, especially the repolarization period. Repolarization is driven by two delayed rectifier K+ current subtypes, IKr (rapid) and IKs (slow), and most drug-induced QTc prolongations are related to blockade of IKr, which is carried by K+ voltage-gated channel subfamily H member 2, commonly known as the hERG channel (encoded by KCNH2). However, hERG channel blockade might not always translate into QTc prolongation and might not be the sole mechanism.
Conventional chemotherapies.
Arsenic trioxide is a classic agent among conventional chemotherapeutics with the potential to induce QTc prolongation, with up to one third of patients who receive this medication experiencing an increase in the QTc of 30–60 ms from baseline and one third experiencing an increase of >60 ms (REF.209). QTc prolongations of >500 ms can be seen in as many as 65% of patients receiving arsenic trioxide with the use of the Bazett rate correction formula, but in only 24–32% of patients if alternative formulas, such as the Fridericia formula, are used210. The latter is preferred in patients with cancer because this formula is associated with less overcorrection at higher heart rates and leads to fewer unnecessary cancer treatment interruptions.
Torsades de pointes is usually not seen with arsenic trioxide therapy unless other contributing factors, such as electrolyte abnormalities, are present210. Sudden cardiac death has been reported but is extremely rare. Mechanistically, arsenic trioxide can block both IKr and IKs but activates the ATP-dependent K+ current IK-ATP (REF.211). Other conventional chemotherapeutics with the potential to induce QTc prolongation include oxaliplatin, which increases the inward Na+ current211. For drugs such as paclitaxel, docetaxel and 5-FU, induction of myocardial ischaemia might be another potential mechanism leading to QTc prolongation.
Targeted cancer therapies.
TKIs have a heterogeneous effect on QTc: on average, a 15-ms increase from baseline but notably more for sunitinib, lapatinib, nilotinib and vandetanib (on average, increases of 22.4, 23.4, 25.8 and 36.4 ms, respectively, from baseline)197. Although the incidences can differ considerably, in patients receiving TKIs, including dasatinib, nilotinib, pazopanib and sunitinib, QTc prolongations to 500 ms were noted in <5% of patients and ventricular arrhythmia and sudden cardiac death were noted in <1% of patients212. Vandetanib is the drug with the most robust evidence by the number of studies, and the incidence of all-grade or high-grade QTc prolongation with vandetanib is 16.4% and 3.7%, respectively, among patients with non-thyroid cancer and 18% and 12%, respectively, among patients with thyroid cancer, who have longer durations of treatment197.
A systematic, registry-based study published in 2018 confirmed the reports on the risk of ventricular arrhythmias with ibrutinib213–218. On the basis of the Naranjo Adverse Drug Reaction Probability Scale score, the association of ibrutinib with ventricular arrhythmias was deemed to be at least probable, and overall a more than 10 times higher than expected rate of VT was observed. Of note, VT and ventricular fibrillation, even polymorphic VT, has been reported with ibrutinib treatment even in the presence of a normal QTc218. AF remained a predictor of VT in the adjusted analyses. In mice, atrial and ventricular arrhythmias were seen even after a single dose of ibrutinib, and high serum concentrations of ibrutinib, rather than chronicity of the treatment, seems to be the determining factor219.
HDAC inhibitors (BOX 2) have also been associated with QTc prolongation and arrhythmias197. QTc prolongation can be seen in 10% of patients receiving the HDAC inhibitor romidepsin, supraventricular tachycardia can be seen in 38%, VT can be seen in 14%, atrial ectopy can be seen in 65% and ventricular ectopy can be seen in 38%. Both QTc prolongation and arrhythmias usually resolve before the next cycle of therapy. However, cases of sudden cardiac death have been reported with the use of romidepsin, underscoring the need for vigilance. QTc prolongation has also been reported in 10% of patients receiving dacinostat, 6.3–28.0% of patients receiving panobinostat and 3.5–6.0% of patients receiving vorinostat212. These observations are consistent with a class effect, and blockade of the hERG channel by HDAC inhibitors has been proposed as a mechanistic explanation. The risk of QTc prolongation increases as a function of peak dose; that is, it is highest with short bolus administrations. The risk of torsades de pointes is higher in women, elderly patients and patients with bradyarrhythmias, electrolyte abnormalities, structural heart diseases or baseline QTc prolongation.
Inhibitors of the cyclin-dependent kinases CDK4 and CDK6 (BOX 2) are another class of drug that has been associated with QTc-prolonging potential, albeit with great variations in the associated risk220. Ribociclib is the drug associated with the highest risk, and QTc prolongation is seen in a concentration-dependent manner, usually within the first 4 weeks of treatment, and the ECG changes are reversible with therapy interruption. In clinical trials, 6% of patients with advanced or metastatic breast cancer who were treated with ribociclib (in combination with an aromatase inhibitor or fulvestrant) had a >60-ms increase in QTc from baseline, and 1% had a QTc of >500 ms (REF.221). No cases of torsades de pointes were reported but one sudden cardiac death (0.3%) occurred in a patient with concomitant hypokalaemia222. Ribociclib should not be combined with tamoxifen given the nearly threefold higher incidence of QTc increase (by >60 ms) than that seen with single therapy. Palbociclib and abemaciclib do not seem to lead to clinically significant (increase of >60 ms or duration >500 ms) QTc prolongations223.
Immunotherapies.
In patients receiving ICIs, ventricular arrhythmias might be a result of the inflammatory infiltration of the myocardium178. Ventricular arrhythmias are seen in 5–10% of patients receiving ICIs and are associated with 40% mortality176. Similarly to new-onset conduction disease, ventricular arrhythmias indicate a more complicated clinical course and should prompt investigations into the presence of myocarditis. Ventricular arrhythmias could also conceivably be seen with other forms of cardiomyopathy reported with ICI therapy.
Radiation therapy.
Despite the ample reports on cardiac fibrosis in patients who underwent radiation therapy involving the chest, reports of ventricular arrhythmia are scarce. Indeed, cardiac radiation therapy is being explored as an alternative to invasive ventricular ablation in the treatment of ventricular arrhythmia224. In survivors of childhood cancer, some studies indicate a 3–5% incidence of VT, but incidence rates differ by treatment: 4% among those treated with chest radiation therapy and 8% among those treated with both chest radiation therapy and anthracyclines225. Importantly, ventricular arrhythmias are not restricted to patients with cardiac dysfunction and can be noted even in those with preserved LVEF225.
Management and prevention.
Patients with cancer who have ECG abnormalities, impaired exercise capacity or cardiovascular diseases at baseline should be assumed to be more susceptible to cancer therapy-induced arrhythmias, as are those undergoing treatment regimens with known cardiotoxicity potential. Therefore, as a general rule, comorbidities that could represent a possible arrhythmogenic substrate should be identified and treated aggressively before and during cancer therapy. Early identification and appropriate management of cardiac ischaemia, dysfunction and remodelling is also likely to be the best strategy to modulate the arrhythmogenic substrate and improve outcomes in patients with cancer therapy-induced arrhythmias. These recommendations hold true for QTc prolongation and related ventricular arrhythmias.
Crizotinib, dasatinib, lapatinib, nilotinib, pazopanib, sorafenib, sunitinib, vandetanib and vemurafenib should be administered with caution in patients with pre-existing QTc prolongation or QTc prolongation-related risk factors including medications and drug–drug interactions. As illustrated for several TKIs, such as vandetanib, electrolyte levels should be corrected before initiation of cancer therapy (the goal value for serum K+ levels is 4 mEq/l to the upper limit of normal and for serum Mg2+ and serum Ca2+ levels within the normal range) and should be monitored along with serial ECGs, as outlined earlier (at baseline, at 2–4 weeks, at 8–12 weeks and every 3 months thereafter). The same frequency of monitoring is required after dose reductions or therapy interruptions of more than 2 weeks. Importantly, vandetanib has a half-life of 19 days and, therefore, any adverse reactions can resolve slowly. The upper limit for QTc with vandetanib therapy is 450 ms before the start of therapy and 500 ms during therapy. If these thresholds are surpassed, therapy should be stopped and might be resumed at a reduced dose. With nilotinib therapy, ECGs should be taken 7 days after initiation or change of therapy, and any QTc >480 ms requires a temporary cessation of therapy (or permanent cessation if QTc prolongation is recurrent after measures have been taken) until QTc is 450–480 ms (then therapy should be resumed at half the dose) or <450 ms (then therapy should be resumed at the full dose). Any grade 4 (that is, life-threatening) QTc event also precludes any further cancer therapy. Ventricular arrhythmias should be managed as usual according to clinical guidelines226 (Supplementary Table 6).
Atrial fibrillation
AF in patients with cancer has been reported for more than half a century, initially as a consequence of neoplastic infiltration or mechanical pressure on the heart or as a complication of oncological thoracic surgery or medical therapy. Subsequently, a bidirectional and more so multifactorial association between cancer and AF was recognized. For instance, the Women’s Health Study227 showed that the risk of cancer was threefold higher in the first 3 months after diagnosis of AF and remained elevated by 42% beyond the first year. Conversely, the risk of AF was found to be increased by 20% in the first 3 months after cancer diagnosis227. The exact mechanisms underlying this association are not defined, in particular, how AF begets cancer. One possible explanation is that anticoagulant use for treatment of AF unmasks the presence of malignancies by the induction of bleeding events in the tumour228. Cancer antigen 125 (also known as MUC16) is not only a marker for tumours, such as in ovarian cancer, but is also a predictor of AF in postmenopausal women229. Shared risk factors in AF and cancer include obesity and inflammation. Whether AF in patients with cancer can occur without any underlying substrate and predisposition is not fully clear. Patients with cancer are usually not fully characterized in terms of atrial filling pressures and pre-existing remodelling dynamics. General precipitating and aggravating factors for AF in patients with cancer include cardiac masses or infiltration, sympathetic stressors, acute and chronic inflammation, pericarditis, mediastinal irradiation, surgery, bone marrow transplantation and chemotherapy230.
Conventional chemotherapy.
As outlined in TABLE 4, AF has been reported with numerous traditional chemotherapies, especially with melphalan and paclitaxel. The precise mechanisms of melphalan-related supraventricular tachycardia are not known. AF is seen in 8% of patients receiving melphalan, and is more common in elderly patients and in those with reduced renal function or hypertension231. Left atrial enlargement and particularly a history of AF are predictive of the risk of AF in patients receiving melphalan231. A history of cardiovascular disease has also been considered to be a risk factor for AF in patients receiving paclitaxel, but AF can occur in the absence of risk factors for AF. The incidence of paclitaxel-related AF is <2%232.
Targeted cancer therapy.
A renaissance of the topic of AF and cancer has occurred with the use of the TKI ibrutinib233. Incidence rates of AF with the use of ibrutinib range from 3% to 16% (on average 8%) according to a systematic review published in 2017 (REFS234,235). A risk prediction model for AF in patients with chronic lymphocytic leukaemia was developed at the Mayo Clinic and externally validated233. The variables included in this model include age (<65 years, 65–74 years and >74 years), sex, valvular heart disease and hypertension233. The score categories are 0–1, 2–3, 4 and 5+, and each step up in category corresponds to a doubling of the risk of AF (zero, twofold, fourfold and eightfold increase). The predictiveness of this model for AF in patients receiving ibrutinib was confirmed, as was the Framingham model and a model by Visentin et al. (the latter potentially performing the best)236,237. Other studies are in general agreement with the concept that patients who develop AF with exposure to ibrutinib have either a history of AF or predisposition for AF238,239. The underlying mechanisms are not clear but might be related to suppression of phosphoinositide 3-kinase (PI3K)–AKT pathway activity in cardiomyocytes201,240. The PI3K–AKT pathway is regulated by BTK and the tyrosine-protein kinase TEC, both targets of ibrutinib201,241. The second-generation BTK inhibitor acalabrutinib neither increases the risk of AF and bleeding nor inhibits TEC and SRC family members as ibrutinib does242,243.
Immunotherapy.
Inflammation and AF have been linked but with debated causality (direct, indirect or not at all)244–246. Nevertheless, patients with higher levels of C-reactive protein (CRP) in the plasma have more AF episodes, and baseline plasma CRP levels are predictive of future risk of AF247. Not surprisingly, new-onset AF has been seen in patients receiving CAR T cell therapy, even in very young patients and those without a history of AF123,248. AF has also been reported with ICI therapy249,250. AF in this setting could be caused by the induction of pericarditis or cardiomyopathy. Other conditions induced by ICIs that can contribute to AF include, for instance, thyroiditis.
Radiation therapy.
AF can develop in the setting of pericarditis or as a consequence of the development of restrictive cardiomyopathy in patients who have under-gone radiation therapy. Nevertheless, a history of HF is less common in patients with cancer who have under-gone chest radiation therapy and develop AF than in the general AF population251. Some studies even concluded that the risk of AF is not overall higher in this patient population than in the general population. As with VT, radiation therapy is now being tested as a non-invasive ablative strategy for AF treatment252.
Management and prevention.
The principles and goals of the management of AF in patients with cancer are generally the same as those in the general population (Supplementary Table 7), albeit with some important differences (FIG. 3). The first goal is to control heart rates with a lenient target with the use of β-blockers, Ca2+ channel blockers and digoxin. If this strategy does not suffice and patients remain symptomatic, for example, with palpitations, dyspnoea and effort intolerance, antiarrhythmic drugs can be used. In patients with cancer who are actively receiving cancer therapy, various drug–drug interactions can be a complicating factor for any such interventions. This complication particularly exists for multitargeted TKIs.
Fig. 3 |. Main elements in the treatment of patients with cancer and atrial fibrillation.

In patients with cancer, predisposing conditions for atrial fibrillation should be identified and addressed if possible. These include common risk factors for atrial fibrillation such as old age (>60 years), valvular heart disease, hypertension, obstructive sleep apnoea, chronic kidney disease, diabetes mellitus and smoking. Cancer therapies that have been associated with the risk of atrial fibrillation are listed in TABLE 4 and include chemical compounds such as melphalan, targeted agents such as the tyrosine kinase inhibitor (TKI) ibrutinib and immunotherapies that increase inflammation and cytokine production such as immune checkpoint inhibitor and chimeric antigen receptor (CAR) T cell therapies. Various other factors are important in patients with cancer, including metabolic (such as hyperthyroidism) and electrolyte abnormalities, autonomic nervous system stimulation (pain or stress), cardiac infiltration or pericarditis and/or pericardial effusion. These predisposing factors can contribute to morbidity and death in patients with cancer. Atrial fibrillation symptoms include palpitations, chest discomfort and dyspnoea. Atrial fibrillation can lead to thromboembolism, myocardial ischaemia and heart failure. To reduce symptoms and the risk of complications, the decisions have to be made whether interventions should be pursued and what they should be. However, risk scores to guide decisions regarding anticoagulation have not been validated for patients with cancer. Similarly, results from landmark randomized clinical trials (RCTs) involving patients with atrial fibrillation cannot be easily translated to patients with cancer. ACE, angiotensin-converting enzyme; ARB, angiotensin-receptor blocker; LAA, left atrial appendage; NOAC, non-vitamin K antagonist oral anticoagulant; VKA, vitamin K antagonist.
An illustrative example in this regard is ibrutinib, which can increase the plasma levels of amiodarone, carvedilol, digoxin, diltiazem and verapamil253. Conversely, amiodarone and the calcium-channel blockers diltiazem and verapamil can increase the plasma levels of ibrutinib severalfold by interfering with the hepatic metabolism of ibrutinib through inhibition of cytochrome P450 3A4 (CYP3A4)253. Therefore, the β-blockers atenolol and metoprolol should be used as first-line agents253. Class Ib (mexiletine) and class Ic (flecainide and propafenone) drugs and sotalol might be valid choices for antiarrhythmic drug therapy, depending on cardiovascular comorbidities. If needed in patients with HF, amiodarone and digoxin should be used very carefully (as substrates of P-glycoprotein, which is inhibited by ibrutinib; amiodarone is also an inhibitor of P-glycoprotein and a major CYP3A substrate); dronedarone should not be used (as a moderate CYP3A4 inhibitor and a major CYP3A4 substrate). More than two thirds of patients with AF who are receiving ibrutinib might not experience long-term success with cardioversion, suggesting that antiarrhythmic therapy should be started even when cardioversion is considered239. The role of ablation in patients with cancer and AF is currently not defined, especially as a first-line therapy to avoid potentially fatal drug–drug interactions. If at all possible, ibrutinib therapy should not be discontinued but instead the dose should be reduced because no significant difference in the rate of AF resolution with the two strategies has been observed, whereas ibrutinib therapy discontinuation leads to a significantly (about twofold) higher risk of cancer progression239. A systematic review did not find a relationship between ibrutinib dose and the occurrence of either AF or bleeding235.
Anticoagulation therapy in patients with cancer can be problematic in general and especially in patients receiving ibrutinib because they are predisposed to bleeding (60% incidence in single-group studies, 44% in randomized clinical trials, with high-grade haemorrhage in up to 7% of patients)254. Ibrutinib has a unique antiplatelet effect, inhibiting mainly von Willebrand factor and collagen-mediated platelet activation (in addition to fibrinogen-activated platelet activation), which could be very effective in the setting of atherosclerotic plaque rupture255,256. Importantly, these activation pathways are distinct from those inhibited by aspirin (cyclooxygenase) and thienopyridines (ADP receptor), and combination therapy would lead to a profoundly additive effect and bleeding risk; therefore, this strategy is not recommended. The combination of any antiplatelet agent with anticoagulation therapy increases the risk of bleeding by default. In terms of drug–drug interactions, the adverse potential is not deemed very high for the combination of ibrutinib and warfarin, which together with the option of warfarin reversal has favoured the use of warfarin in patients receiving ibrutinib therapy241. However, wide fluctuations in the international normalized ratio can be seen in patients receiving ibrutinib and warfarin, and although warfarin was allowed initially in clinical trials, the trial criteria were later amended to exclude patients receiving warfarin because of excessive bleeding events256. Low-molecular-weight heparin has therefore often been a preferred choice for anticoagulation in these patients (especially in those with normal renal function). Nevertheless, the costs and discomfort of the injections remain major disadvantages of using low-molecular-weight heparin, especially considering the chronicity of this treatment. Direct oral anticoagulants have emerged as an attractive alternative, even in patients with cancer and AF257–262. Given the inhibitory action on P-glycoprotein, ibrutinib has the potential to increase the serum levels of all direct oral anticoagulants, especially dabigatran and edoxaban and, to a lesser degree, apixaban and rivaroxaban263. Indeed, no grade 3 bleeding events were noted in 18 patients who developed AF with ibrutinib therapy, seven of whom were treated with apixaban264. Anticoagulation therapy in patients receiving ibrutinib should be for the duration of the increased risk of AF, which is certainly for the duration of ibrutinib therapy. However, these patients might be at high risk of AF regardless of receiving ibrutinib233,238. For patients with malignancies in general, an approach that is based on the stage within the continuum of cancer care might be advisable (BOX 5).
Box 5 |. Anticoagulation strategies in patients with cancer and atrial fibrillation.
In the absence of clinical trial data, safety concerns guide decision-making regarding the anticoagulation regimen for patients with cancer and atrial fibrillation, assuming that all strategies have equal efficacy to prevent thromboembolism. On the basis of the factors outlined in the table, a reasonable approach would be to use either vitamin K antagonists (VKAs) or non-vitamin K antagonist oral anticoagulants (NOACs) before and after active cancer therapy, when steady-state conditions are reached (that is, no major changes in drug regimen, renal and liver function, and blood counts and coagulation status are expected). During active cancer therapy, low-molecular-weight heparin (LMWH) might be the preferred choice.
| Anticoagulant regimen | Preferred timing with respect to cancer therapy | Drugs and dosing | Reversibility | Drug-drug interactions | Reduced renal function | Reduced liver function | Cost | Comments |
|---|---|---|---|---|---|---|---|---|
| VKAs | Before and after | Coumadin, dosing according to INR | Vitamin K, fresh frozen plasma or prothrombin complex concentrate | +++ | Preferred if severe end-stage renal disease without haemodialysis | Not required | Low | Inconvenience owing to the need for recurrent INR checks |
| LMWH | During | Enoxaparin 1 mg/kg subcutaneously twice daily; dalteparin 200 U/kg subcutaneously daily | Protamine (but unlike with unfractionated heparin, it does not completely abolish the anti-factor Xa activity of LMWH) | + | Caution if eGFR <30 ml/min; monitor factor Xa levels | Not required | High | Heparin-induced thrombocytopenia; discomfort with injections; challenging long-term treatment |
| NOACs | Before and after | Rivaroxaban 20 mg orally daily; endoxaban 60 mg orally daily; dabigatran 150 mg orally twice daily; apixaban 5 mg orally twice daily | Idarucizumab (Praxbind) for dabigatran; andexanet alfa (Andexxa), if available, for apixaban or rivaroxaban; or four-factor prothrombin complex concentrate for all other NOACs | +++ | Reduce rivaroxaban dosage to 15 mg daily; reduce endoxaban dosage to 30 mg daily if eGFR is 15–50 ml/min; reduce dabigatran dosage to 75 mg twice daily if eGFR is 15–30 ml/min; reduce apixaban dosage to 2.5 mg twice daily if serum creatinine level is ≥1.5 mg/dl and either age ≥80 years or weight ≤60 kg | Not recommended with moderate-to-severe (rivaroxaban and endoxaban) or severe (apixaban) liver dysfunction (Child-Pugh class B/C and class C) | High | Lack of ample experience and publications in patients with cancer; concerns for use in patients with gastrointestinal (and genitourinary) tract lesions |
egFR, estimated glomerular filtration rate; INR, international normalized ratio.
Importantly, when choosing the anticoagulation strategy, the CHA2DS2-VASc score seems to perform the same in patients with cancer as in patients without cancer for those with baseline AF265–267. However, the CHA2DS2-VASc score does not account for cancer-induced hypercoagulability and does not perform as well for patients who newly develop AF during cancer therapy268–270. With regard to bleeding risk prediction, differences in patients with cancer are also not included in the HAS-BLED score and, for this reason, this score might not perform ideally in patients with cancer either265. These difficulties might explain why, at least in the USA, patients with cancer take medications for anticoagulation at a much lower rate than patients without cancer, despite deriving the same benefit271. Intriguingly, involvement of a cardiologist markedly improved this management aspect271.
In terms of primary prevention, how to identify patients with cancer at high risk of AF accurately and whether they should be prophylactically treated with antiarrhythmic drugs is currently unknown. These are pertinent questions especially for those patients with cancer whose treatment course can be greatly affected in a negative manner by the development of AF (for example, those undergoing bone marrow transplantation or CAR T cell therapy or responders to long-term therapy with ibrutinib).
Conclusions
Cancer therapy has evolved remarkably over the decades, from chemical therapeutics to targeted molecular therapies and, most recently, immunotherapies. With these developments, the cardiovascular toxicity profile of cancer therapeutics is broadening. Although familiarity with the old concepts and management recommendations has to remain, one has to be attentive to new concepts and discoveries in cardio-oncology. This multidisciplinary area will gain importance in the years to come, with the ageing of the general population and the consequent increase in the incidence and prevalence of both cancer and cardiovascular diseases. Although this Review focuses on the active therapy phase in patients with cancer, the continuum of care should not be forgotten. Some patients will need to be evaluated before and continued to be followed up for cardiovascular risks after exposure to the therapies discussed in this Review. In addition to cardiotoxicity, vascular toxicity and arrhythmias associated with cancer therapies are important topic areas for every cardiologist to know given the potential for fatal outcomes. Finally, in view of the improving survival rates in patients with cancer, how cardiovascular diseases and cardiovascular toxicities of cancer therapies are managed in these patients will become increasingly important.
Supplementary Material
Key points.
Cancer therapy has evolved from the administration of chemical compounds and radiation therapy to the use of targeted agents and immunotherapies.
Along with these developments, the cardiovascular toxicity spectrum of cancer therapies has been changing but cardiac toxicity remains of greatest concern.
Inflammatory and immune mechanisms have to be taken into account when considering cardiotoxicity in patients receiving immune checkpoint inhibitor or chimeric antigen receptor T cell therapies.
With the newer cancer therapies, atrial fibrillation is emerging as the most relevant and practically challenging arrhythmia in patients with cancer.
Corrected QT interval prolongation, ventricular arrhythmias and cardiac arrest can also occur with many of the newer targeted agents.
Acknowledgements
The author receives support from the US National Institutes of Health (HL116952 and CA233610) and the Miami Heart Research Institute/Florida Heart Research Foundation.
Footnotes
Competing interests
The author declares no competing interests.
Supplementary information
Supplementary information is available for this paper at https://doi.org/10.1038/s41569-020-0348-1.
References
- 1.Global Burden of Disease Cancer Collaboration. The global burden of cancer 2013. JAMA Oncol. 1, 505–527 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bluethmann SM, Mariotto AB & Rowland JH Anticipating the “silver tsunami”: prevalence trajectories and comorbidity burden among older cancer survivors in the United States. Cancer Epidemiol. Biomarkers Prev 25, 1029–1036 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ewer MS & Ewer SM Cardiotoxicity of anticancer treatments: what the cardiologist needs to know. Nat. Rev. Cardiol 7, 564–575 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Ewer MS & Ewer SM Cardiotoxicity of anticancer treatments. Nat. Rev. Cardiol 12, 547–558 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Herrmann J Vascular toxic effects of cancer therapies. Nat. Rev. Cardiol 10.1038/s41569-020-0347-2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gianni L et al. Anthracycline cardiotoxicity: from bench to bedside. J. Clin. Oncol 26, 3777–3784 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herrmann J et al. Evaluation and management of patients with heart disease and cancer: cardio-oncology. Mayo Clin. Proc 89, 1287–1306 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bristow MR et al. Early anthracycline cardiotoxicity. Am. J. Med 65, 823–832 (1978). [DOI] [PubMed] [Google Scholar]
- 9.Bristow MR, Billingham ME, Mason JW & Daniels JR Clinical spectrum of anthracycline antibiotic cardiotoxicity. Cancer Treat. Rep 62, 873–879 (1978). [PubMed] [Google Scholar]
- 10.Ferrans VJ Overview of cardiac pathology in relation to anthracycline cardiotoxicity. Cancer Treat. Rep 62, 955–961 (1978). [PubMed] [Google Scholar]
- 11.Berry GJ & Jorden M Pathology of radiation and anthracycline cardiotoxicity. Pediatr. Blood Cancer 44, 630–637 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Ewer MS et al. A comparison of cardiac biopsy grades and ejection fraction estimations in patients receiving Adriamycin. J. Clin. Oncol 2, 112–117 (1984). [DOI] [PubMed] [Google Scholar]
- 13.Nousiainen T, Jantunen E, Vanninen E & Hartikainen J Early decline in left ventricular ejection fraction predicts doxorubicin cardiotoxicity in lymphoma patients. Br. J. Cancer 86, 1697–1700 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Felker GM et al. Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy. N. Engl. J. Med 342, 1077–1084 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Mazur M et al. Burden of cardiac arrhythmias in patients with anthracycline-related cardiomyopathy. JACC Clin. Electrophysiol 3, 139–150 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Kremer LC, van der Pal HJ, Offringa M, van Dalen EC & Voute PA Frequency and risk factors of subclinical cardiotoxicity after anthracycline therapy in children: a systematic review. Ann. Oncol 13, 819–829 (2002). [DOI] [PubMed] [Google Scholar]
- 17.Zhang S et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med 18, 1639–1642 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Chen B, Peng X, Pentassuglia L, Lim CC & Sawyer DB Molecular and cellular mechanisms of anthracycline cardiotoxicity. Cardiovasc. Toxicol 7, 114–121 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Varga ZV, Ferdinandy P, Liaudet L & Pacher P Drug-induced mitochondrial dysfunction and cardiotoxicity. Am. J. Physiol. Heart Circ. Physiol 309, H1453–H1467 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ichikawa Y et al. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Invest 124, 617–630 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lebrecht D, Setzer B, Ketelsen UP, Haberstroh J & Walker UA Time-dependent and tissue-specific accumulation of mtDNA and respiratory chain defects in chronic doxorubicin cardiomyopathy. Circulation 108, 2423–2429 (2003). [DOI] [PubMed] [Google Scholar]
- 22.De Angelis A et al. Anthracycline cardiomyopathy is mediated by depletion of the cardiac stem cell pool and is rescued by restoration of progenitor cell function. Circulation 121, 276–292 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Piegari E et al. Doxorubicin induces senescence and impairs function of human cardiac progenitor cells. Basic Res. Cardiol 108, 334 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Ali MK, Ewer MS, Gibbs HR, Swafford J & Graff KL Late doxorubicin-associated cardiotoxicity in children. The possible role of intercurrent viral infection. Cancer 74, 182–188 (1994). [DOI] [PubMed] [Google Scholar]
- 25.Carter P et al. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc. Natl Acad. Sci. USA 89, 4285–4289 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moasser MM The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 26, 6469–6487 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moasser MM & Krop IE The evolving landscape of HER2 targeting in breast cancer. JAMA Oncol. 1, 1154–1161 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Slamon DJ et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med 344, 783–792 (2001). [DOI] [PubMed] [Google Scholar]
- 29.Farolfi A et al. Trastuzumab-induced cardiotoxicity in early breast cancer patients: a retrospective study of possible risk and protective factors. Heart 99, 634–639 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Nowsheen S et al. Trastuzumab in female breast cancer patients with reduced left ventricular ejection fraction. J. Am. Heart Assoc 7, e008637 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ewer MS & Lippman SM Type II chemotherapy-related cardiac dysfunction: time to recognize a new entity. J. Clin. Oncol 23, 2900–2902 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Ewer MS et al. Reversibility of trastuzumab-related cardiotoxicity: new insights based on clinical course and response to medical treatment. J. Clin. Oncol 23, 7820–7826 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Tan TC et al. Time trends of left ventricular ejection fraction and myocardial deformation indices in a cohort of women with breast cancer treated with anthracyclines, taxanes, and trastuzumab. J. Am. Soc. Echocardiogr 28, 509–514 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Yu AF et al. Trastuzumab interruption and treatment-induced cardiotoxicity in early HER2-positive breast cancer. Breast Cancer Res. Treat 149, 489–495 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guarneri V et al. Long-term cardiac tolerability of trastuzumab in metastatic breast cancer: the M.D. Anderson Cancer Center experience. J. Clin. Oncol 24, 4107–4115 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Cardinale D et al. Trastuzumab-induced cardiotoxicity: clinical and prognostic implications of troponin I evaluation. J. Clin. Oncol 28, 3910–3916 (2010). [DOI] [PubMed] [Google Scholar]
- 37.ElZarrad MK et al. Trastuzumab alters the expression of genes essential for cardiac function and induces ultrastructural changes of cardiomyocytes in mice. PLoS One 8, e79543 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Azambuja E et al. Trastuzumab-associated cardiac events at 8 years of median follow-up in the Herceptin Adjuvant trial (BIG 1–01). J. Clin. Oncol 32, 2159–2165 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Chen J et al. Incidence of heart failure or cardiomyopathy after adjuvant trastuzumab therapy for breast cancer. J. Am. Coll. Cardiol 60, 2504–2512 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Bowles EJ et al. Risk of heart failure in breast cancer patients after anthracycline and trastuzumab treatment: a retrospective cohort study. J. Natl Cancer Inst 104, 1293–1305 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baselga J et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N. Engl. J. Med 366, 109–119 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Swain SM et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N. Engl. J. Med 372, 724–734 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Piccart-Gebhart M et al. Adjuvant lapatinib and trastuzumab for early human epidermal growth factor receptor 2-positive breast cancer: results from the randomized phase III Adjuvant Lapatinib and/or Trastuzumab Treatment Optimization trial. J. Clin. Oncol 34, 1034–1042 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Watanabe H et al. Congestive heart failure during osimertinib treatment for epidermal growth factor receptor (EGFR)-mutant non-small cell lung cancer (NSCLC). Intern. Med 56, 2195–2197 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bhullar KS et al. Kinase-targeted cancer therapies: progress, challenges and future directions. Mol. Cancer 17, 48 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thompson PA, Kantarjian HM & Cortes JE Diagnosis and treatment of chronic myeloid leukemia in 2015. Mayo Clin. Proc 90, 1440–1454 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kerkela R et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat. Med 12, 908–916 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Herman EH et al. A multifaceted evaluation of imatinib-induced cardiotoxicity in the rat. Toxicol. Pathol 39, 1091–1106 (2011). [DOI] [PubMed] [Google Scholar]
- 49.Wolf A et al. Imatinib does not induce cardiotoxicity at clinically relevant concentrations in preclinical studies. Leuk. Res 34, 1180–1188 (2010). [DOI] [PubMed] [Google Scholar]
- 50.Ribeiro AL et al. An evaluation of the cardiotoxicity of imatinib mesylate. Leuk. Res 32, 1809–1814 (2008). [DOI] [PubMed] [Google Scholar]
- 51.Estabragh ZR et al. A prospective evaluation of cardiac function in patients with chronic myeloid leukaemia treated with imatinib. Leuk. Res 35, 49–51 (2011). [DOI] [PubMed] [Google Scholar]
- 52.Force T, Krause DS & Van Etten RA Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat. Rev. Cancer 7, 332–344 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Greineder CF, Kohnstamm S & Ky B Heart failure associated with sunitinib: lessons learned from animal models. Curr. Hypertens. Rep 13, 436–441 (2011). [DOI] [PubMed] [Google Scholar]
- 54.Hasinoff BB, Patel D & O’Hara KA Mechanisms of myocyte cytotoxicity induced by the multiple receptor tyrosine kinase inhibitor sunitinib. Mol. Pharmacol 74, 1722–1728 (2008). [DOI] [PubMed] [Google Scholar]
- 55.Hasinoff BB & Patel D The lack of target specificity of small molecule anticancer kinase inhibitors is correlated with their ability to damage myocytes in vitro. Toxicol. Appl. Pharmacol 249, 132–139 (2010). [DOI] [PubMed] [Google Scholar]
- 56.Hasinoff BB The cardiotoxicity and myocyte damage caused by small molecule anticancer tyrosine kinase inhibitors is correlated with lack of target specificity. Toxicol. Appl. Pharmacol 244, 190–195 (2010). [DOI] [PubMed] [Google Scholar]
- 57.Will Y et al. Effect of the multitargeted tyrosine kinase inhibitors imatinib, dasatinib, sunitinib, and sorafenib on mitochondrial function in isolated rat heart mitochondria and H9c2 cells. Toxicol. Sci 106, 153–161 (2008). [DOI] [PubMed] [Google Scholar]
- 58.Jacob F et al. Analysis of tyrosine kinase inhibitor-mediated decline in contractile force in rat engineered heart tissue. PLoS One 11, e0145937 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lamore SD et al. Deconvoluting kinase inhibitor induced cardiotoxicity. Toxicol. Sci 158, 213–226 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stuhlmiller TJ et al. Kinome and transcriptome profiling reveal broad and distinct activities of erlotinib, sunitinib, and sorafenib in the mouse heart and suggest cardiotoxicity from combined signal transducer and activator of transcription and epidermal growth factor receptor inhibition. J. Am. Heart Assoc 6, e006635 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sharma A et al. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci. Transl Med 9, eaaf2584 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shim JV et al. Mechanistic systems modeling to improve understanding and prediction of cardiotoxicity caused by targeted cancer therapeutics. Front. Physiol 8, 651 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brown SA, Sandhu N & Herrmann J Systems biology approaches to adverse drug effects: the example of cardio-oncology. Nat. Rev. Clin. Oncol 12, 718–731 (2015). [DOI] [PubMed] [Google Scholar]
- 64.Brown SA, Nhola L & Herrmann J Cardiovascular toxicities of small molecule tyrosine kinase inhibitors: an opportunity for systems-based approaches. Clin. Pharmacol. Ther 101, 65–80 (2017). [DOI] [PubMed] [Google Scholar]
- 65.Plana JC et al. Expert consensus for multimodality imaging evaluation of adult patients during and after cancer therapy: a report from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J. Am. Soc. Echocardiogr 27, 911–939 (2014). [DOI] [PubMed] [Google Scholar]
- 66.Cardinale D et al. Prognostic value of troponin I in cardiac risk stratification of cancer patients undergoing high-dose chemotherapy. Circulation 109, 2749–2754 (2004). [DOI] [PubMed] [Google Scholar]
- 67.Negishi T, Thavendiranathan P, Negishi K & Marwick TH Rationale and design of the strain surveillance of chemotherapy for improving cardiovascular outcomes: the SUCCOUR trial. JACC Cardiovasc. Imaging 11, 1098–1105 (2018). [DOI] [PubMed] [Google Scholar]
- 68.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03879629 (2019) [DOI] [PubMed]
- 69.Yancy CW et al. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. J. Am. Coll. Cardiol 70, 776–803 (2017). [DOI] [PubMed] [Google Scholar]
- 70.Yancy CW et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 128, e240–e327 (2013). [DOI] [PubMed] [Google Scholar]
- 71.Curigliano G et al. Cardiovascular toxicity induced by chemotherapy, targeted agents and radiotherapy: ESMO clinical practice guidelines. Ann. Oncol 23, vii155–vii166 (2012). [DOI] [PubMed] [Google Scholar]
- 72.Russell RR et al. The role and clinical effectiveness of multimodality imaging in the management of cardiac complications of cancer and cancer therapy. J. Nucl. Cardiol 23, 856–884 (2016). [DOI] [PubMed] [Google Scholar]
- 73.Chang HM, Okwuosa TM, Scarabelli T, Moudgil R & Yeh ETH Cardiovascular complications of cancer therapy: best practices in diagnosis, prevention, and management: part 2. J. Am. Coll. Cardiol 70, 2552–2565 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Alexander J et al. Serial assessment of doxorubicin cardiotoxicity with quantitative radionuclide angiocardiography. N. Engl. J. Med 300, 278–283 (1979). [DOI] [PubMed] [Google Scholar]
- 75.Avila MS et al. Carvedilol for prevention of chemotherapy-related cardiotoxicity: the CECCY trial. J. Am. Coll. Cardiol 71, 2281–2290 (2018). [DOI] [PubMed] [Google Scholar]
- 76.Urbanek K et al. Cardioprotection by targeting the pool of resident and extracardiac progenitors. Curr. Drug Targets 16, 884–894 (2015). [DOI] [PubMed] [Google Scholar]
- 77.Hamed S et al. Erythropoietin improves myocardial performance in doxorubicin-induced cardiomyopathy. Eur. Heart J 27, 1876–1883 (2006). [DOI] [PubMed] [Google Scholar]
- 78.Hoch M et al. Erythropoietin preserves the endothelial differentiation capacity of cardiac progenitor cells and reduces heart failure during anticancer therapies. Cell Stem Cell 9, 131–143 (2011). [DOI] [PubMed] [Google Scholar]
- 79.Burridge PW et al. Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat. Med 22, 547–556 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen IY, Matsa E & Wu JC Induced pluripotent stem cells: at the heart of cardiovascular precision medicine. Nat. Rev. Cardiol 13, 333–349 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pituskin E et al. Multidisciplinary approach to novel therapies in cardio-oncology research (MANTICORE 101-Breast): a randomized trial for the prevention of trastuzumab-associated cardiotoxicity. J. Clin. Oncol 35, 870–877 (2017). [DOI] [PubMed] [Google Scholar]
- 82.Boekhout AH et al. Angiotensin II-receptor inhibition with candesartan to prevent trastuzumab-related cardiotoxic effects in patients with early breast cancer: a randomized clinical trial. JAMA Oncol. 2, 1030–1037 (2016). [DOI] [PubMed] [Google Scholar]
- 83.Nowsheen S et al. Trastuzumab in female breast cancer patients with reduced left ventricular ejection fraction. J. Am. Heart Assoc 7, e008637 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Crone SA et al. ErbB2 is essential in the prevention of dilated cardiomyopathy. Nat. Med 8, 459–465 (2002). [DOI] [PubMed] [Google Scholar]
- 85.Herrmann J, Herrmann SM & Haddad TC New-onset heart failure in association with severe hypertension during trastuzumab therapy. Mayo Clin. Proc 89, 1734–1739 (2014). [DOI] [PubMed] [Google Scholar]
- 86.Gilchrist SC et al. Cardio-oncology rehabilitation to manage cardiovascular outcomes in cancer patients and survivors: a scientific statement from the American Heart Association. Circulation 139, e997–e1012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Squires RW, Shultz AM & Herrmann J Exercise training and cardiovascular health in cancer patients. Curr. Oncol. Rep 20, 27 (2018). [DOI] [PubMed] [Google Scholar]
- 88.Cerny J, Hassan A, Smith C & Piperdi B Coronary vasospasm with myocardial stunning in a patient with colon cancer receiving adjuvant chemotherapy with FOLFOX regimen. Clin. Colorectal Cancer 8, 55–58 (2009). [DOI] [PubMed] [Google Scholar]
- 89.Basselin C et al. 5-Fluorouracil-induced Tako-Tsubo-like syndrome. Pharmacotherapy 31, 226 (2011). [DOI] [PubMed] [Google Scholar]
- 90.Gianni M, Dentali F & Lonn E 5 flourouracil-induced apical ballooning syndrome: a case report. Blood Coagul. Fibrinolysis 20, 306–308 (2009). [DOI] [PubMed] [Google Scholar]
- 91.Grunwald MR, Howie L & Diaz LA Jr. Takotsubo cardiomyopathy and fluorouracil: case report and review of the literature. J. Clin. Oncol 30, e11–e14 (2012). [DOI] [PubMed] [Google Scholar]
- 92.Kobayashi N et al. A case of takotsubo cardiomyopathy during 5-fluorouracil treatment for rectal adenocarcinoma. J. Nippon Med. Sch 76, 27–33 (2009). [DOI] [PubMed] [Google Scholar]
- 93.Ozturk MA, Ozveren O, Cinar V, Erdik B & Oyan B Takotsubo syndrome: an underdiagnosed complication of 5-fluorouracil mimicking acute myocardial infarction. Blood Coagul. Fibrinolysis 24, 90–94 (2013). [DOI] [PubMed] [Google Scholar]
- 94.S YH, Tornvall P, Tornerud M & Henareh L Capecitabine caused cardiogenic shock through induction of global takotsubo syndrome. Cardiovasc. Revasc. Med 14, 57–61 (2013). [DOI] [PubMed] [Google Scholar]
- 95.Stewart T, Pavlakis N & Ward M Cardiotoxicity with 5-fluorouracil and capecitabine: more than just vasospastic angina. Intern. Med. J 40, 303–307 (2010). [DOI] [PubMed] [Google Scholar]
- 96.Dechant C et al. Acute reversible heart failure caused by coronary vasoconstriction due to continuous 5-fluorouracil combination chemotherapy. Case Rep. Oncol 5, 296–301 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tsibiribi P et al. Cardiac lesions induced by 5-fluorouracil in the rabbit. Hum. Exp. Toxicol 25, 305–309 (2006). [DOI] [PubMed] [Google Scholar]
- 98.Martin M et al. Lethal cardiac toxicity after cisplatin and 5-fluorouracil chemotherapy. Report of a case with necropsy study. Am. J. Clin. Oncol 12, 229–234 (1989). [DOI] [PubMed] [Google Scholar]
- 99.Eskandari MR, Moghaddam F, Shahraki J & Pourahmad J A comparison of cardiomyocyte cytotoxic mechanisms for 5-fluorouracil and its pro-drug capecitabine. Xenobiotica 45, 79–87 (2015). [DOI] [PubMed] [Google Scholar]
- 100.Focaccetti C et al. Effects of 5-fluorouracil on morphology, cell cycle, proliferation, apoptosis, autophagy and ROS production in endothelial cells and cardiomyocytes. PLoS One 10, e0115686 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lamberti M et al. A mechanistic study on the cardiotoxicity of 5-fluorouracil in vitro and clinical and occupational perspectives. Toxicol. Lett 227, 151–156 (2014). [DOI] [PubMed] [Google Scholar]
- 102.Lischke J, Lang C, Sawodny O & Feuer R Impairment of energy metabolism in cardiomyocytes caused by 5-FU catabolites can be compensated by administration of amino acids. Conf. Proc. IEEE Eng. Med. Biol. Soc 2015, 5363–5366 (2015). [DOI] [PubMed] [Google Scholar]
- 103.Polk A, Vistisen K, Vaage-Nilsen M & Nielsen DL A systematic review of the pathophysiology of 5-fluorouracil-induced cardiotoxicity. BMC Pharmacol. Toxicol 15, 47 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sara JD et al. 5-fluorouracil and cardiotoxicity: a review. Ther. Adv. Med. Oncol 10, 1758835918780140 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Arellano M, Malet-Martino M, Martino R & Gires P The anti-cancer drug 5-fluorouracil is metabolized by the isolated perfused rat liver and in rats into highly toxic fluoroacetate. Br. J. Cancer 77, 79–86 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Matsubara I, Kamiya J & Imai S Cardiotoxic effects of 5-fluorouracil in the guinea pig. Jpn. J. Pharmacol 30, 871–879 (1980). [DOI] [PubMed] [Google Scholar]
- 107.Diasio RB The role of dihydropyrimidine dehydrogenase (DPD) modulation in 5-FU pharmacology. Oncology 12, 23–27 (1998). [PubMed] [Google Scholar]
- 108.Papanastasopoulos P & Stebbing J Molecular basis of 5-fluorouracil-related toxicity: lessons from clinical practice. Anticancer Res. 34, 1531–1535 (2014). [PubMed] [Google Scholar]
- 109.Franco TH, Khan A, Joshi V & Thomas B Takotsubo cardiomyopathy in two men receiving bevacizumab for metastatic cancer. Ther. Clin. Risk Manag 4, 1367–1370 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Numico G et al. Takotsubo syndrome in a patient treated with sunitinib for renal cancer. J. Clin. Oncol 30, e218–e220 (2012). [DOI] [PubMed] [Google Scholar]
- 111.Touyz RM & Herrmann J Cardiotoxicity with vascular endothelial growth factor inhibitor therapy. NPJ Precis. Oncol 2, 13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Baffert F et al. Cellular changes in normal blood capillaries undergoing regression after inhibition of VEGF signaling. Am. J. Physiol. Heart Circ. Physiol 290, H547–H559 (2006). [DOI] [PubMed] [Google Scholar]
- 113.Kamba T et al. VEGF-dependent plasticity of fenestrated capillaries in the normal adult microvasculature. Am. J. Physiol. Heart Circ. Physiol 290, H560–H576 (2006). [DOI] [PubMed] [Google Scholar]
- 114.Lazarus A & Keshet E Vascular endothelial growth factor and vascular homeostasis. Proc. Am. Thorac. Soc 8, 508–511 (2011). [DOI] [PubMed] [Google Scholar]
- 115.Maharaj AS & D’Amore PA Roles for VEGF in the adult. Microvasc. Res 74, 100–113 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.June CH & Sadelain M Chimeric antigen receptor therapy. N. Engl. J. Med 379, 64–73 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Brudno JN & Kochenderfer JN Chimeric antigen receptor T-cell therapies for lymphoma. Nat. Rev. Clin. Oncol 15, 31–46 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Brichard VG & Godechal Q MAGE-A3-specific anticancer immunotherapy in the clinical practice. Oncoimmunology 2, e25995 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Linette GP et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 122, 863–871 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cameron BJ et al. Identification of a titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci. Transl Med 5, 197ra103 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Morgan RA et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther 18, 843–851 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ahmed N et al. Human epidermal growth factor receptor 2 (HER2) -specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J. Clin. Oncol 33, 1688–1696 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Brudno JN & Kochenderfer JN Toxicities of chimeric antigen receptor T cells: recognition and management. Blood 127, 3321–3330 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bonifant CL, Jackson HJ, Brentjens RJ & Curran KJ Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 3, 16011 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Neelapu SS et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat. Rev. Clin. Oncol 15, 47–62 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sato R & Nasu M A review of sepsis-induced cardiomyopathy. J. Intensive Care 3, 48 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Krishnagopalan S, Kumar A, Parrillo JE & Kumar A Myocardial dysfunction in the patient with sepsis. Curr. Opin. Crit. Care 8, 376–388 (2002). [DOI] [PubMed] [Google Scholar]
- 128.Vallabhajosyula S et al. New-onset heart failure and mortality in hospital survivors of sepsis-related left ventricular dysfunction. Shock 49, 144–149 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Court O, Kumar A, Parrillo JE & Kumar A Clinical review: myocardial depression in sepsis and septic shock. Crit. Care 6, 500–508 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Morelli A et al. Effect of heart rate control with esmolol on hemodynamic and clinical outcomes in patients with septic shock: a randomized clinical trial. JAMA 310, 1683–1691 (2013). [DOI] [PubMed] [Google Scholar]
- 131.Kumar A et al. Cardiovascular response to dobutamine stress predicts outcome in severe sepsis and septic shock. Crit. Care 12, R35 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Przepiorka D et al. FDA approval: blinatumomab. Clin. Cancer Res 21, 4035–4039 (2015). [DOI] [PubMed] [Google Scholar]
- 133.Slaney CY, Wang P, Darcy PK & Kershaw MH CARs versus BiTEs: a comparison between T cell-redirection strategies for cancer treatment. Cancer Discov. 8, 924–934 (2018). [DOI] [PubMed] [Google Scholar]
- 134.Pardoll DM The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Boutros C et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat. Rev. Clin. Oncol 13, 473–486 (2016). [DOI] [PubMed] [Google Scholar]
- 136.Sury K, Perazella MA & Shirali AC Cardiorenal complications of immune checkpoint inhibitors. Nat. Rev. Nephrol 14, 571–588 (2018). [DOI] [PubMed] [Google Scholar]
- 137.Yang S & Asnani A Cardiotoxicities associated with immune checkpoint inhibitors. Curr. Probl. Cancer 42, 422–432 (2018). [DOI] [PubMed] [Google Scholar]
- 138.Ederhy S et al. Takotsubo-like syndrome in cancer patients treated with immune checkpoint inhibitors. JACC Cardiovasc. Imaging 11, 1187–1190 (2018). [DOI] [PubMed] [Google Scholar]
- 139.Giza DE et al. Stress-induced cardiomyopathy in cancer patients. Am. J. Cardiol 120, 2284–2288 (2017). [DOI] [PubMed] [Google Scholar]
- 140.Ogawa Y Paradigm shift in radiation biology/radiation oncology-exploitation of the “H2O2 effect” for radiotherapy using low-LET (linear energy transfer) radiation such as X-rays and high-energy electrons. Cancers 8, 28 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Saiki H et al. Risk of heart failure with preserved ejection fraction in older women after contemporary radiotherapy for breast cancer. Circulation 135, 1388–1396 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Heselich A et al. High LET radiation shows no major cellular and functional effects on primary cardiomyocytes in vitro. Life Sci. Space Res 16, 93–100 (2018). [DOI] [PubMed] [Google Scholar]
- 143.Hughson RL, Helm A & Durante M Heart in space: effect of the extraterrestrial environment on the cardiovascular system. Nat. Rev. Cardiol 15, 167–180 (2018). [DOI] [PubMed] [Google Scholar]
- 144.Fajardo LF & Stewart JR Experimental radiation-induced heart disease. I. Light microscopic studies. Am. J. Pathol 59, 299–316 (1970). [PMC free article] [PubMed] [Google Scholar]
- 145.Khan MY Radiation-induced cardiomyopathy. I. An electron microscopic study of cardiac muscle cells. Am. J. Pathol 73, 131–146 (1973). [PMC free article] [PubMed] [Google Scholar]
- 146.Fajardo LF & Stewart JR Capillary injury preceding radiation-induced myocardial fibrosis. Radiology 101, 429–433 (1971). [DOI] [PubMed] [Google Scholar]
- 147.Stewart FA, Seemann I, Hoving S & Russell NS Understanding radiation-induced cardiovascular damage and strategies for intervention. Clin. Oncol 25, 617–624 (2013). [DOI] [PubMed] [Google Scholar]
- 148.Cuomo JR et al. How to prevent and manage radiation-induced coronary artery disease. Heart 104, 1647–1653 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Taunk NK, Haffty BG, Kostis JB & Goyal S Radiation-induced heart disease: pathologic abnormalities and putative mechanisms. Front. Oncol 5, 39 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Cosset JM et al. Pericarditis and myocardial infarctions after Hodgkin’s disease therapy. Int. J. Radiat. Oncol. Biol. Phys 21, 447–449 (1991). [DOI] [PubMed] [Google Scholar]
- 151.Jensen SA & Sorensen JB Risk factors and prevention of cardiotoxicity induced by 5-fluorouracil or capecitabine. Cancer Chemother. Pharmacol 58, 487–493 (2006). [DOI] [PubMed] [Google Scholar]
- 152.Meyer CC, Calis KA, Burke LB, Walawander CA & Grasela TH Symptomatic cardiotoxicity associated with 5-fluorouracil. Pharmacotherapy 17, 729–736 (1997). [PubMed] [Google Scholar]
- 153.Clasen SC et al. Fluoropyrimidine-induced cardiac toxicity: challenging the current paradigm. J. Gastrointest. Oncol 8, 970–979 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ma WW et al. Emergency use of uridine triacetate for the prevention and treatment of life-threatening 5-fluorouracil and capecitabine toxicity. Cancer 123, 345–356 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Truitt R et al. Increased afterload augments sunitinib-induced cardiotoxicity in an engineered cardiac microtissue model. JACC Basic Transl Sci. 3, 265–276 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Touyz RM, Herrmann SMS & Herrmann J Vascular toxicities with VEGF inhibitor therapies-focus on hypertension and arterial thrombotic events. J. Am. Soc. Hypertens 12, 409–425 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Chu TF et al. Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet 370, 2011–2019 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Di Lorenzo G et al. Cardiovascular toxicity following sunitinib therapy in metastatic renal cell carcinoma: a multicenter analysis. Ann. Oncol 20, 1535–1542 (2009). [DOI] [PubMed] [Google Scholar]
- 159.Abdel-Qadir H, Ethier JL, Lee DS, Thavendiranathan P & Amir E Cardiovascular toxicity of angiogenesis inhibitors in treatment of malignancy: a systematic review and meta-analysis. Cancer Treat. Rev 53, 120–127 (2017). [DOI] [PubMed] [Google Scholar]
- 160.Annane D et al. A global perspective on vasoactive agents in shock. Intensive Care Med. 44, 833–846 (2018). [DOI] [PubMed] [Google Scholar]
- 161.Donis N, Oury C, Moonen M & Lancellotti P Treating cardiovascular complications of radiotherapy: a role for new pharmacotherapies. Expert Opin. Pharmacother 19, 431–442 (2018). [DOI] [PubMed] [Google Scholar]
- 162.Lancellotti P et al. Expert consensus for multi-modality imaging evaluation of cardiovascular complications of radiotherapy in adults: a report from the European Association of Cardiovascular Imaging and the American Society of Echocardiography. J. Am. Soc. Echocardiogr 26, 1013–1032 (2013). [DOI] [PubMed] [Google Scholar]
- 163.Iliescu CA et al. SCAI expert consensus statement: evaluation, management, and special considerations of cardio-oncology patients in the cardiac catheterization laboratory (endorsed by the Cardiological Society of India, and Sociedad Latino Americana de Cardiologia Intervencionista). Catheter. Cardiovasc. Interv 87, E202–E223 (2016). [DOI] [PubMed] [Google Scholar]
- 164.Appelbaum F et al. Acute lethal carditis caused by high-dose combination chemotherapy. A unique clinical and pathological entity. Lancet 1, 58–62 (1976). [DOI] [PubMed] [Google Scholar]
- 165.O’Connell TX & Berenbaum MC Cardiac and pulmonary effects of high doses of cyclophosphamide and isophosphamide. Cancer Res. 34, 1586–1591 (1974). [PubMed] [Google Scholar]
- 166.Dhesi S et al. Cyclophosphamide-induced cardiomyopathy: a case report, review, and recommendations for management. J. Investig. Med. High Impact Case Rep 1, 2324709613480346 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Bianchini G & Gianni L The immune system and response to HER2-targeted treatment in breast cancer. Lancet Oncol. 15, e58–e68 (2014). [DOI] [PubMed] [Google Scholar]
- 168.Yousif NG & Al-Amran FG Novel Toll-like receptor-4 deficiency attenuates trastuzumab (Herceptin) induced cardiac injury in mice. BMC Cardiovasc. Disord 11, 62 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Asawaeer M, Barton D, Radio S & Chatzizisis YS Tyrosine kinase inhibitor-induced acute myocarditis, myositis, and cardiogenic shock. Methodist Debakey Cardiovasc. J 14, e5–e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Palmieri DJ & Carlino MS Immune checkpoint inhibitor toxicity. Curr. Oncol. Rep 20, 72 (2018). [DOI] [PubMed] [Google Scholar]
- 171.Puzanov I et al. Managing toxicities associated with immune checkpoint inhibitors: consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J. Immunother. Cancer 5, 95 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Spain L, Diem S & Larkin J Management of toxicities of immune checkpoint inhibitors. Cancer Treat. Rev 44, 51–60 (2016). [DOI] [PubMed] [Google Scholar]
- 173.Eigentler TK et al. Diagnosis, monitoring and management of immune-related adverse drug reactions of anti-PD-1 antibody therapy. Cancer Treat. Rev 45, 7–18 (2016). [DOI] [PubMed] [Google Scholar]
- 174.Wang DY et al. Fatal toxic effects associated with immune checkpoint inhibitors: a systematic review and meta-analysis. JAMA Oncol. 4, 1721–1728 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lyon AR, Yousaf N, Battisti NML, Moslehi J & Larkin J Immune checkpoint inhibitors and cardiovascular toxicity. Lancet Oncol. 19, e447–e458 (2018). [DOI] [PubMed] [Google Scholar]
- 176.Mir H et al. Cardiac complications associated with checkpoint inhibition: a systematic review of the literature in an important emerging area. Can. J. Cardiol 34, 1059–1068 (2018). [DOI] [PubMed] [Google Scholar]
- 177.Escudier M et al. Clinical features, management, and outcomes of immune checkpoint inhibitor-related cardiotoxicity. Circulation 136, 2085–2087 (2017). [DOI] [PubMed] [Google Scholar]
- 178.Johnson DB et al. Fulminant myocarditis with combination immune checkpoint blockade. N. Engl. J. Med 375, 1749–1755 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Nishimura H et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 291, 319–322 (2001). [DOI] [PubMed] [Google Scholar]
- 180.Okazaki T et al. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat. Med 9, 1477–1483 (2003). [DOI] [PubMed] [Google Scholar]
- 181.Grabie N et al. Endothelial programmed death-1 ligand 1 (PD-L1) regulates CD8+ T-cell mediated injury in the heart. Circulation 116, 2062–2071 (2007). [DOI] [PubMed] [Google Scholar]
- 182.Lichtman AH The heart of the matter: protection of the myocardium from T cells. J. Autoimmun 45, 90–96 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Rodig N et al. Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur. J. Immunol 33, 3117–3126 (2003). [DOI] [PubMed] [Google Scholar]
- 184.Tarrio ML, Grabie N, Bu DX, Sharpe AH & Lichtman AH PD-1 protects against inflammation and myocyte damage in T cell-mediated myocarditis. J. Immunol 188, 4876–4884 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Baban B, Liu JY, Qin X, Weintraub NL & Mozaffari MS Upregulation of programmed death-1 and Its ligand in cardiac injury models: interaction with GADD153. PLoS One 10, e0124059 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Varricchi G, Galdiero MR & Tocchetti CG Cardiac toxicity of immune checkpoint inhibitors: cardio-oncology meets immunology. Circulation 136, 1989–1992 (2017). [DOI] [PubMed] [Google Scholar]
- 187.Freilich M et al. Recovery from anthracycline cardiomyopathy after long-term support with a continuous flow left ventricular assist device. J. Heart Lung Transpl 28, 101–103 (2009). [DOI] [PubMed] [Google Scholar]
- 188.Arangalage D et al. Survival after fulminant myocarditis induced by immune-checkpoint inhibitors. Ann. Intern. Med 167, 683–684 (2017). [DOI] [PubMed] [Google Scholar]
- 189.Mahajan VS & Jarolim P How to interpret elevated cardiac troponin levels. Circulation 124, 2350–2354 (2011). [DOI] [PubMed] [Google Scholar]
- 190.Mahmood SS et al. Myocarditis in patients treated with immune checkpoint inhibitors. J. Am. Coll. Cardiol 71, 1755–1764 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.O’Regan DP & Cook SA Myocarditis or myocardial infarction? MRI can help. Heart 97, 1283 (2011). [DOI] [PubMed] [Google Scholar]
- 192.Miller EJ & Culver DA Establishing an evidence-based method to diagnose cardiac sarcoidosis: the complementary use of cardiac magnetic resonance imaging and FDG-PET. Circ. Cardiovasc. Imaging 11, e007408 (2018). [DOI] [PubMed] [Google Scholar]
- 193.Wang DY, Okoye GD, Neilan TG, Johnson DB & Moslehi JJ Cardiovascular toxicities associated with cancer immunotherapies. Curr. Cardiol. Rep 19, 21 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Salem JE et al. Abatacept for severe immune checkpoint inhibitor-associated myocarditis. N. Engl. J. Med 380, 2377–2379 (2019). [DOI] [PubMed] [Google Scholar]
- 195.Arbuck SG et al. A reassessment of cardiac toxicity associated with Taxol. J. Natl Cancer Inst. Monogr 15, 117–130 (1993). [PubMed] [Google Scholar]
- 196.Pai VB & Nahata MC Cardiotoxicity of chemotherapeutic agents: incidence, treatment and prevention. Drug Saf 22, 263–302 (2000). [DOI] [PubMed] [Google Scholar]
- 197.Tamargo J, Caballero R & Delpon E Cancer chemotherapy and cardiac arrhythmias: a review. Drug Saf. 38, 129–152 (2015). [DOI] [PubMed] [Google Scholar]
- 198.Ghiaseddin A et al. Phase II study of bevacizumab and vorinostat for patients with recurrent World Health Organization grade 4 malignant glioma. Oncologist 23, 157–e121 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Lele AV, Clutter S, Price E & De Ruyter ML Severe hypothyroidism presenting as myxedema coma in the postoperative period in a patient taking sunitinib: case report and review of literature. J. Clin. Anesth 25, 47–51 (2013). [DOI] [PubMed] [Google Scholar]
- 200.Herrmann J Tyrosine kinase inhibitors and vascular toxicity: impetus for a classification system? Curr. Oncol. Rep 18, 33 (2016). [DOI] [PubMed] [Google Scholar]
- 201.Mathur K, Saini A, Ellenbogen KA & Shepard RK Profound sinoatrial arrest associated with Ibrutinib. Case Rep. Oncol. Med 2017, 7304021 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Cooper LT Jr. Myocarditis. N. Engl. J. Med 360, 1526–1538 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Kaplan BM, Miller AJ, Bharati S, Lev M & Martin Grais I Complete AV block following mediastinal radiation therapy: electrocardiographic and pathologic correlation and review of the world literature. J. Interv. Card. Electrophysiol 1, 175–188 (1997). [DOI] [PubMed] [Google Scholar]
- 204.Orzan F et al. Associated cardiac lesions in patients with radiation-induced complete heart block. Int. J. Cardiol 39, 151–156 (1993). [DOI] [PubMed] [Google Scholar]
- 205.Tzivoni D, Ratzkowski E, Biran S, Brook JG & Stern S Complete heart block following therapeutic irradiation of the left side of the chest. Chest 71, 231–234 (1977). [DOI] [PubMed] [Google Scholar]
- 206.Cohen SI, Bharati S, Glass J & Lev M Radiotherapy as a cause of complete atrioventricular block in Hodgkin’s disease. An electrophysiological-pathological correlation. Arch. Intern. Med 141, 676–679 (1981). [PubMed] [Google Scholar]
- 207.Santoro F et al. Late calcification of the mitral-aortic junction causing transient complete atrio-ventricular block after mediastinal radiation of Hodgkin lymphoma: multimodal visualization. Int. J. Cardiol 155, e49–e50 (2012). [DOI] [PubMed] [Google Scholar]
- 208.Nair CK et al. Conduction defects and mitral annulus calcification. Br. Heart J 44, 162–167 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Barbey JT, Pezzullo JC & Soignet SL Effect of arsenic trioxide on QT interval in patients with advanced malignancies. J. Clin. Oncol 21, 3609–3615 (2003). [DOI] [PubMed] [Google Scholar]
- 210.Roboz GJ et al. Prevalence, management, and clinical consequences of QT interval prolongation during treatment with arsenic trioxide. J. Clin. Oncol 32, 3723–3728 (2014). [DOI] [PubMed] [Google Scholar]
- 211.Duan J et al. Anticancer drugs-related QTc prolongation, torsade de pointes and sudden death: current evidence and future research perspectives. Oncotarget 9, 25738–25749 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Porta-Sanchez A et al. Incidence, diagnosis, and management of QT prolongation induced by cancer therapies: a systematic review. J. Am. Heart Assoc 6, e007724 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Cheng C, Woronow D, Nayernama A, Wroblewski T & Jones SC Ibrutinib-associated ventricular arrhythmia in the FDA adverse event reporting system. Leuk. Lymphoma 59, 3016–3017 (2018). [DOI] [PubMed] [Google Scholar]
- 214.Tomcsanyi J, Matrai Z & Tomcsanyi K Ventricular tachycardia caused by ibrutinib. J. Emerg. Med 53, e27 (2017). [DOI] [PubMed] [Google Scholar]
- 215.Lampson BL et al. Ventricular arrhythmias and sudden death in patients taking ibrutinib. Blood 129, 2581–2584 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Beyer A, Ganti B, Majkrzak A & Theyyunni N A perfect storm: tyrosine kinase inhibitor-associated polymorphic ventricular tachycardia. J. Emerg. Med 52, e123–e127 (2017). [DOI] [PubMed] [Google Scholar]
- 217.Wallace N, Wong E, Cooper D & Chao H A case of new-onset cardiomyopathy and ventricular tachycardia in a patient receiving ibrutinib for relapsed mantle cell lymphoma. Clin. Case Rep 4, 1120–1121 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Tomcsanyi J, Nenyei Z, Matrai Z & Bozsik B Ibrutinib, an approved tyrosine kinase inhibitor as a potential cause of recurrent polymorphic ventricular tachycardia. JACC Clin. Electrophysiol 2, 847–849 (2016). [DOI] [PubMed] [Google Scholar]
- 219.Tuomi JM, Xenocostas A & Jones DL Increased susceptibility for atrial and ventricular cardiac arrhythmias in mice treated with a single high dose of ibrutinib. Can. J. Cardiol 34, 337–341 (2018). [DOI] [PubMed] [Google Scholar]
- 220.Thill M & Schmidt M Management of adverse events during cyclin-dependent kinase 4/6 (CDK4/6) inhibitor-based treatment in breast cancer. Ther. Adv. Med. Oncol 10, 1758835918793326 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.US Food and Drug Administration. Package insert Kisqali (ribociclib) tablets prescribing information (FDA, 2018). [Google Scholar]
- 222.Shah A et al. FDA approval: ribociclib for the treatment of postmenopausal women with hormone receptor-positive, HER2-negative advanced or metastatic breast cancer. Clin. Cancer Res 24, 2999–3004 (2018). [DOI] [PubMed] [Google Scholar]
- 223.Bellet M et al. Palbociclib and ribociclib in breast cancer: consensus workshop on the management of concomitant medication. Ther. Adv. Med. Oncol 11, 1758835919833867 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Cuculich PS et al. Noninvasive cardiac radiation for ablation of ventricular tachycardia. N. Engl. J. Med 377, 2325–2336 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Larsen RL et al. Electrocardiographic changes and arrhythmias after cancer therapy in children and young adults. Am. J. Cardiol 70, 73–77 (1992). [DOI] [PubMed] [Google Scholar]
- 226.Al-Khatib SM et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J. Am. Coll. Cardiol 72, e91–e220 (2018). [DOI] [PubMed] [Google Scholar]
- 227.Conen D et al. Risk of malignant cancer among women with new-onset atrial fibrillation. JAMA Cardiol. 1, 389–396 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Wallis CJD et al. Association between use of antithrombotic medication and hematuria-related complications. JAMA 318, 1260–1271 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Sekiguchi H et al. Cancer antigen-125 plasma level as a biomarker of new-onset atrial fibrillation in postmenopausal women. Heart 103, 1368–1373 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Farmakis D, Parissis J & Filippatos G Insights into onco-cardiology: atrial fibrillation in cancer. J. Am. Coll. Cardiol 63, 945–953 (2014). [DOI] [PubMed] [Google Scholar]
- 231.Feliz V et al. Melphalan-induced supraventricular tachycardia: incidence and risk factors. Clin. Cardiol 34, 356–359 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Zhao D et al. Atrial fibrillation following treatment with paclitaxel: a case report. Biomed. Rep 9, 540–544 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Shanafelt TD et al. Atrial fibrillation in patients with chronic lymphocytic leukemia (CLL). Leuk. Lymphoma 58, 1630–1639 (2017). [DOI] [PubMed] [Google Scholar]
- 234.Leong DP et al. The risk of atrial fibrillation with ibrutinib use: a systematic review and meta-analysis. Blood 128, 138–140 (2016). [DOI] [PubMed] [Google Scholar]
- 235.Yun S, Vincelette ND, Acharya U & Abraham I Risk of atrial fibrillation and bleeding diathesis associated with ibrutinib treatment: a systematic review and pooled analysis of four randomized controlled trials. Clin. Lymphoma Myeloma Leuk 17, 31–37 e13 (2017). [DOI] [PubMed] [Google Scholar]
- 236.Visentin A et al. A scoring system to predict the risk of atrial fibrillation in chronic lymphocytic leukemia. Hematol. Oncol 37, 508–512 (2019). [DOI] [PubMed] [Google Scholar]
- 237.Archibald W et al. Atrial fibrillation (AF) in patients with CLL treated with ibrutinib: assessing prediction models and clinical outcomes. J. Clin. Oncol 37, 7522–7522 (2019). [Google Scholar]
- 238.Wiczer TE et al. Cumulative incidence, risk factors, and management of atrial fibrillation in patients receiving ibrutinib. Blood Adv. 1, 1739–1748 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Thompson PA et al. Atrial fibrillation in CLL patients treated with ibrutinib. An international retrospective study. Br. J. Haematol 175, 462–466 (2016). [DOI] [PubMed] [Google Scholar]
- 240.Pretorius L et al. Reduced phosphoinositide 3-kinase (p110alpha) activation increases the susceptibility to atrial fibrillation. Am. J. Pathol 175, 998–1009 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Ganatra S et al. Ibrutinib-associated atrial fibrillation. JACC Clin. Electrophysiol 4, 1491–1500 (2018). [DOI] [PubMed] [Google Scholar]
- 242.Patel V et al. Comparison of acalabrutinib, a selective bruton tyrosine kinase inhibitor, with Ibrutinib in chronic lymphocytic leukemia cells. Clin. Cancer Res 23, 3734–3743 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Owen C, Berinstein NL, Christofides A & Sehn LH Review of Bruton tyrosine kinase inhibitors for the treatment of relapsed or refractory mantle cell lymphoma. Curr. Oncol 26, e233–e240 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Boos CJ, Anderson RA & Lip GY Is atrial fibrillation an inflammatory disorder? Eur. Heart J 27, 136–149 (2006). [DOI] [PubMed] [Google Scholar]
- 245.Pastori D et al. Inflammation and the risk of atrial high-rate episodes (AHREs) in patients with cardiac implantable electronic devices. Clin. Res. Cardiol 107, 772–777 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Hu YF, Chen YJ, Lin YJ & Chen SA Inflammation and the pathogenesis of atrial fibrillation. Nat. Rev. Cardiol 12, 230–243 (2015). [DOI] [PubMed] [Google Scholar]
- 247.Aviles RJ et al. Inflammation as a risk factor for atrial fibrillation. Circulation 108, 3006–3010 (2003). [DOI] [PubMed] [Google Scholar]
- 248.Davila ML et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl Med 6, 224ra225 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Palla AR, Kennedy D, Mosharraf H & Doll D Autoimmune hemolytic anemia as a complication of nivolumab therapy. Case Rep. Oncol 9, 691–697 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Dein E et al. Two cases of sinusitis induced by immune checkpoint inhibition. J. Immunother 40, 312–314 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Vaidya V et al. Atrial fibrillation after thoracic radiotherapy for cancer: examining differences in clinical characteristics at time of diagnosis compared with the general population. J. Am. Coll. Cardiol 65, A318 (2015). [Google Scholar]
- 252.Zei PC & Soltys S Ablative radiotherapy as a noninvasive alternative to catheter ablation for cardiac arrhythmias. Curr. Cardiol. Rep 19, 79 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Vrontikis A et al. Proposed algorithm for managing Ibrutinib-related atrial fibrillation. Oncology 30, 970–974, 980–971, C973 (2016). [PubMed] [Google Scholar]
- 254.Aguilar C Ibrutinib-related bleeding: pathogenesis, clinical implications and management. Blood Coagul. Fibrinolysis 29, 481–487 (2018). [DOI] [PubMed] [Google Scholar]
- 255.Busygina K et al. Oral Bruton tyrosine kinase inhibitors selectively block atherosclerotic plaque-triggered thrombus formation in humans. Blood 131, 2605–2616 (2018). [DOI] [PubMed] [Google Scholar]
- 256.Shatzel JJ et al. Ibrutinib-associated bleeding: pathogenesis, management and risk reduction strategies. J. Thromb. Haemost 15, 835–847 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Laube ES et al. Rivaroxaban for stroke prevention in patients with nonvalvular atrial fibrillation and active cancer. Am. J. Cardiol 120, 213–217 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Melloni C et al. Efficacy and safety of apixaban versus warfarin in patients with atrial fibrillation and a history of cancer: insights from the Aristotle trial. Am. J. Med 130, 1440–1448.e1 (2017). [DOI] [PubMed] [Google Scholar]
- 259.Russo V et al. Use of non-vitamin k antagonist oral anticoagulants in atrial fibrillation patients with malignancy: clinical practice experience in a single institution and literature review. Semin. Thromb. Hemost 44, 370–376 (2018). [DOI] [PubMed] [Google Scholar]
- 260.Shah S et al. Comparative effectiveness of direct oral anticoagulants and warfarin in patients with cancer and atrial fibrillation. Blood Adv. 2, 200–209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Vedovati MC et al. Patients with cancer and atrial fibrillation treated with doacs: a prospective cohort study. Int. J. Cardiol 269, 152–157 (2018). [DOI] [PubMed] [Google Scholar]
- 262.Xiang E et al. Anticoagulation prescribing patterns in patients with cancer. J. Thromb. Thrombolysis 45, 89–98 (2018). [DOI] [PubMed] [Google Scholar]
- 263.Mulligan SP, Ward CM, Whalley D & Hilmer SN Atrial fibrillation, anticoagulant stroke prophylaxis and bleeding risk with ibrutinib therapy for chronic lymphocytic leukaemia and lymphoproliferative disorders. Br. J. Haematol 175, 359–364 (2016). [DOI] [PubMed] [Google Scholar]
- 264.Ahn IE et al. Depth and durability of response to ibrutinib in CLL: 5-year follow-up of a phase 2 study. Blood 131, 2357–2366 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Lee YJ et al. Bleeding risk and major adverse events in patients with cancer on oral anticoagulation therapy. Int. J. Cardiol 203, 372–378 (2016). [DOI] [PubMed] [Google Scholar]
- 266.Friberg L, Skeppholm M & Terent A Benefit of anticoagulation unlikely in patients with atrial fibrillation and a CHA2DS2-VASc score of 1. J. Am. Coll. Cardiol 65, 225–232 (2015). [DOI] [PubMed] [Google Scholar]
- 267.Hu YF et al. Incident thromboembolism and heart failure associated with new-onset atrial fibrillation in cancer patients. Int. J. Cardiol 165, 355–357 (2013). [DOI] [PubMed] [Google Scholar]
- 268.D’Souza M et al. CHA2DS2-VASc score and risk of thromboembolism and bleeding in patients with atrial fibrillation and recent cancer. Eur. J. Prev. Cardiol 25, 651–658 (2018). [DOI] [PubMed] [Google Scholar]
- 269.Hu WS & Lin CL Comparison of CHA2DS2-VASc, CHADS2 and HATCH scores for the prediction of new-onset atrial fibrillation in cancer patients: a nationwide cohort study of 760,339 study participants with competing risk analysis. Atherosclerosis 266, 205–211 (2017). [DOI] [PubMed] [Google Scholar]
- 270.Hu WS & Lin CL Impact of atrial fibrillation on the development of ischemic stroke among cancer patients classified by CHA2DS2-VASc score-a nationwide cohort study. Oncotarget 9, 7623–7630 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.O’Neal WT et al. Provider specialty, anticoagulation, and stroke risk in patients with atrial fibrillation and cancer. J. Am. Coll. Cardiol 72, 1913–1922 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.