

Abstract
Cancer therapies can lead to a broad spectrum of cardiovascular complications. Among these, cardiotoxicities remain of prime concern, but vascular toxicities have emerged as the second most common group. The range of cancer therapies with a vascular toxicity profile and the clinical spectrum of vascular toxic effects are quite broad. Historically, venous thromboembolism has received the greatest attention but, over the past decade, the arterial toxic effects, which can present as acute vasospasm, acute thrombosis and accelerated atherosclerosis, of cancer therapies have gained greater recognition. This Review focuses on these types of cancer therapy-related arterial toxicity, including their mechanisms, and provides an update on venous thromboembolism and pulmonary hypertension associated with cancer therapies. Recommendations for the screening, treatment and prevention of vascular toxic effects of cancer therapies are outlined in the context of available evidence and society guidelines and consensus statements. The shift towards greater awareness of the vascular toxic effects of cancer therapies has further unveiled the urgent needs in this area in terms of defining best clinical practices. Well-designed and well-conducted clinical studies and registries are needed to more precisely define the incidence rates, risk factors, primary and secondary modes of prevention, and best treatment modalities for vascular toxicities related to cancer therapies. These efforts should be complemented by preclinical studies to outline the pathophysiological concepts that can be translated into the clinic and to identify drugs with vascular toxicity potential even before their widespread clinical use.
Cancer therapy is changing, with an ever-expanding armamentarium of therapies and an improving survival across a broad range of cancer types. Concomitantly, the spectrum of cancer therapy-related toxicities has also been transformed through new presentations and with a new level of clinical significance given the increased number of patients with adverse effect-related morbidity and mortality. Among the cardiovascular toxic effects of cancer therapies, cardiac adverse effects remain of great concern. However, vascular toxic effects have emerged as the second most commonly reported cancer therapy-related cardiovascular toxicity; indeed, vascular toxicities were on a par with cardiotoxicity in terms of scientific publications at the height of the era of targeted cancer therapy (Supplementary Figure 1). Importantly, vascular toxicities are the second most common cause of death in patients with cancer undergoing outpatient therapies.
A broad range of cancer therapeutics can induce a variety of vascular toxicities at very different rates (FIG. 1; TABLES 1,2). Most of the attention in this area has historically been focused on venous thromboembolism; however, the new cancer therapies have brought arterial toxicities to the fore. On the basis of the clinical presentation, three main types of cancer therapy-related arterial toxicity can be differentiated: acute vasospasm, acute thrombosis and accelerated atherosclerosis (TABLE 3). These types of vascular toxic effects of cancer therapies are the focus of this Review, and updates on venous thromboembolism and pulmonary hypertension associated with cancer therapies are also provided. The cardiac toxic effects of cancer therapies are discussed in a separate Review in this Issue1.
Fig. 1 |. Spectrum of vascular toxic effects of cancer therapies.
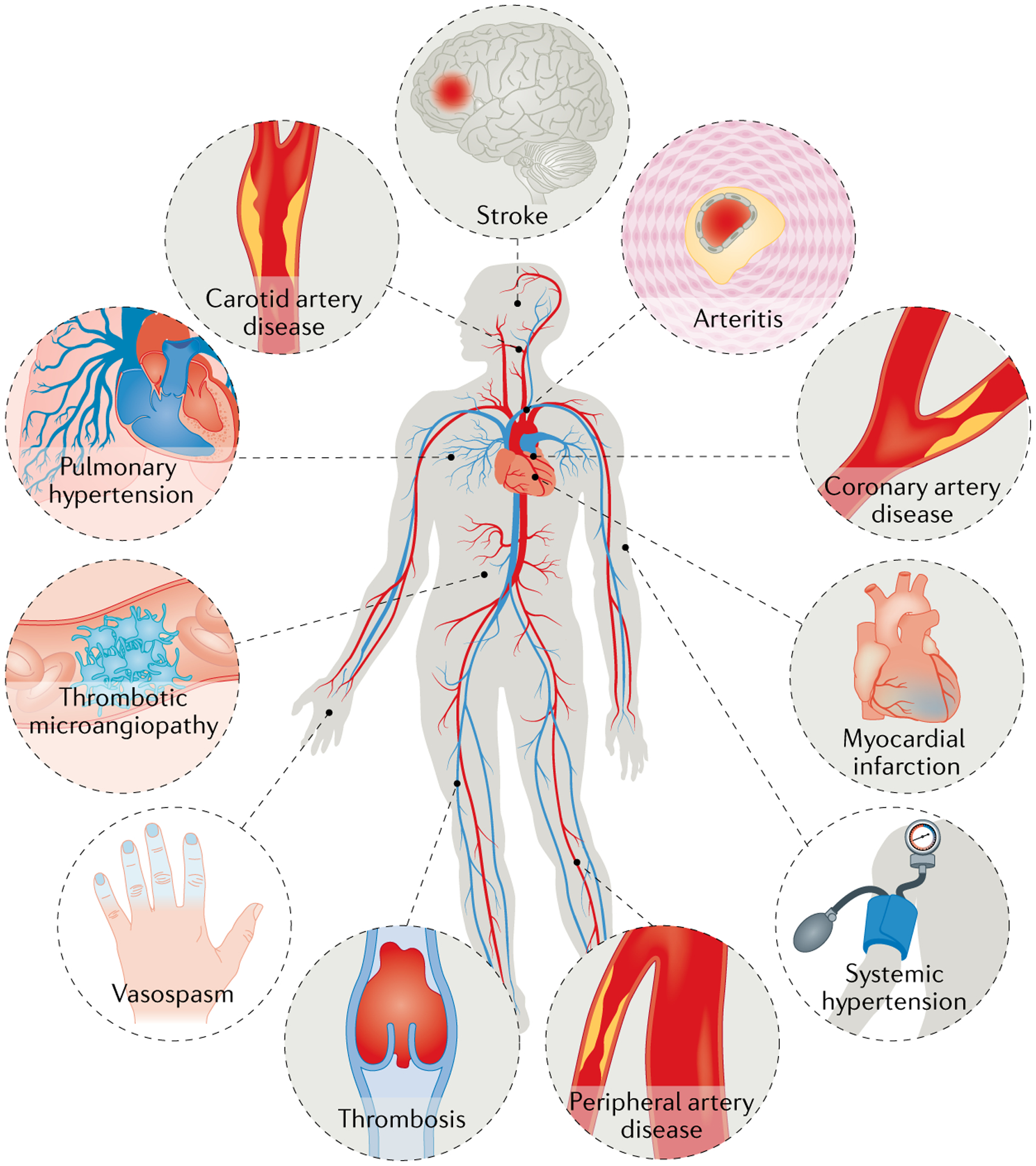
A number of cancer drugs (detailed in TABLES 1,2) can cause various vascular diseases, including coronary artery disease, myocardial infarction, stroke, systemic and pulmonary hypertension, vasospasm and thrombosis.
Table 1 |.
Spectrum of vascular toxic effects of conventional chemotherapies
| Therapy | Cancer therapy indications (label and off-label) | Toxicity | |||||||
|---|---|---|---|---|---|---|---|---|---|
| HTN | Angina | AMI | Raynaud syndrome | Stroke | PAD | Pulmonary HTN | DVT or PE | ||
| Alkylating agents | |||||||||
| Cisplatin | Bladder cancer, breast cancer, cervical cancer, endometrial carcinoma, head and neck cancer, HL, malignant pleural mesothelioma, multiple myeloma, NHL, oesophageal and gastric cancer, osteosarcoma, ovarian cancer, penile cancer, SCLC, testicular cancer | – | ++ | ++ | + | + | + | – | – |
| Cyclophosphamide | ALL, breast cancer, CLL, Ewing sarcoma, HL, multiple myeloma, NHL, SCLC, stem cell transplant condition | – | + | + | – | – | – | + | – |
| Antimetabolites | |||||||||
| 5-Fluorouracil | Anal carcinoma, bladder cancer, breast cancer, cervical cancer, colorectal cancer, gastric cancer, hepatobiliary cancer, oesophageal cancer, pancreatic cancer, squamous cell carcinomas | – | ND | ND | ND | ND | – | – | – |
| Capecitabine | Anal carcinoma, breast cancer, colorectal cancer, fallopian cancer, gastric cancer, hepatobiliary cancer, oesophageal cancer, ovarian cancer, pancreatic cancer, peritoneal cancer, carcinoma of unknown primary | + | ++ | ++ | + | + | – | – | + |
| Gemcitabine | Adenocarcinoma of unknown primary, bladder cancer, breast cancer, cervical cancer, head and neck cancer, hepatobiliary cancer, HL, malignant pleural mesothelioma, NHL, NSCLC, ovarian cancer, pancreatic cancer, sarcomas, SCLC, testicular cancer, uterine cancer | – | + | + | + | – | – | – | – |
| Microtubule-binding agents | |||||||||
| Paclitaxel | Adenocarcinoma of unknown primary, bladder cancer, breast cancer, cervical cancer, head and neck cancers, Kaposi sarcoma, NSCLC, oesophageal and gastric cancer, ovarian cancer, penile cancer, SCLC, soft tissue sarcoma, testicular germ cell tumours, thymoma | + | + | + | – | – | – | – | + |
| Antitumour antibiotics | |||||||||
| Bleomycin | HL, ovarian germ cell cancer, testicular cancer | – | + | + | + | + | – | + | – |
| Vinca alkaloids | |||||||||
| Vincristine | ALL, central nervous system tumours, HL, Ewing sarcoma, gestational trophoblastic tumours, multiple myeloma, NHL, ovarian cancer, primary CNS lymphoma, SCLC, thymoma | ND | ND | ND | ND | – | – | – | – |
| Immunomodulatory drugs | |||||||||
| IFNα2B | Hairy cell leukaemia, Kaposi sarcoma, lymphoma, malignant melanoma | ++ | +++ | ++ | ++ | ++ | ++ | ++ | ++ |
| Lenalidomide | CLL, diffuse large B cell lymphoma, mantle cell lymphoma, multiple myeloma, myelodysplastic syndrome | ++ | ++ | ++ | – | ++ | – | – | +++ |
| Thalidomide | Multiple myeloma, systemic light chain amyloidosis, Waldenström macroglobulinaemia | – | – | – | – | – | – | – | +++ |
Based on data from Micromedex (IBM, NY, USA) and Lexicomp (Wolters Kluwer, Netherlands). –, not reported; +, uncommon (<1%), ++, common (1–10%), +++, very common (>10%). ALL, acute lymphoblastic leukaemia; AMI, acute myocardial infarction; CLL, chronic lymphocytic leukaemia; CNS, central nervous system; DVT, deep-vein thrombosis; HL, Hodgkin lymphoma; HTN, hypertension; ND, frequency not defined; NHL, non-Hodgkin lymphoma; NSCLC, non-small-cell lung cancer; PAD, peripheral artery disease; PE, pulmonary embolism; SCLC, small-cell lung cancer.
Table 2 |.
Spectrum of vascular toxic effects of targeted cancer therapies
| Therapy | Cancer therapy indications (label and off-label) | Toxicity | |||||||
|---|---|---|---|---|---|---|---|---|---|
| HTN | Angina | AMI | Raynaud syndrome | Stroke | PAD | Pulmonary HTN | DVT or PE | ||
| Proteasome inhibitors | |||||||||
| Bortezomib | Follicular lymphoma, mantle cell lymphoma, multiple myeloma, systemic light chain amyloidosis, T cell lymphoma, Waldenström macroglobulinaemia | + | – | ND | – | ND | – | ND | ND |
| Carfilzomib | Multiple myeloma, Waldenström macroglobulinaemia | +++ | +++ | +++ | – | – | – | ++ | – |
| mTOR inhibitors | |||||||||
| Everolimus | Breast cancer, neuroendocrine tumours, RCC | +++ | ++ | + | – | – | – | – | ++ |
| Temsirolimus | RCC | ++ | +++ | – | – | – | – | – | ++ |
| Monoclonal antibodies (target) | |||||||||
| Rituximab (anti-CD20) | Burkitt lymphoma, CLL, CNS lymphoma, HL, NHL, Waldenström macroglobulinaemia | +++ | + | + | – | – | – | – | – |
| Bevacizumab (anti-VEGF–VEGFR2) | Glioblastoma, persistent/recurrent/metastatic cervical cancer, metastatic colorectal cancer, (non-squamous) NSCLC | +++ | ++ | ++ | – | ++ | – | – | +++ |
| Ramucirumab (anti-VEGF–VEGFR2) | Metastatic colorectal cancer, metastatic gastric cancer, metastatic NSCLC | +++ | – | ++ | – | ++ | – | – | – |
| VEGFR fusion molecules | |||||||||
| Aflibercept | Metastatic colorectal cancer | +++ | – | ++ | – | ++ | – | – | ++ |
| Multi-target kinase inhibitors (primary target) | |||||||||
| Vandetanib (VEGFR) | Thyroid cancer | +++ | – | – | – | + | – | – | ++ |
| Axitinib (VEGFR1–VEGFR3) | RCC, thyroid cancer | +++ | + | ++ | – | + | – | – | ++ |
| Lenvatinib (VEGFR1–VEGFR3) | Hepatocellular carcinoma, RCC, thyroid cancer | +++ | – | ++ | – | – | – | – | ++ |
| Pazopanib (VEGFR1–VEGFR3) | RCC, soft tissue carcinoma, thyroid cancer | +++ | +++ | ++ | – | + | – | – | ++ |
| Sorafenib (VEGFR1–VEGFR3) | Angiosarcoma, GIST, hepatocellular cancer, RCC, thyroid cancer | +++ | + | ++ | – | + | – | – | + |
| Sunitinib (VEGFR1–VEGFR3 | GIST, pancreatic neuroendocrine tumours, RCC, soft tissue sarcoma, thyroid cancer | +++ | +++ | + | – | + | – | – | ++ |
| Cabozantinib (VEGFR2) | Hepatocellular carcinoma, RCC, thyroid cancer | +++ | – | ++ | – | ++ | – | – | ++ |
| Regorafenib (VEGFR2) | Colorectal cancer, GIST, hepatocellular carcinoma | +++ | + | + | – | – | – | – | ++ |
| Dasatinib (BCR–ABL1) | GIST, Philadelphia chromosome-positive ALL and CML | – | ++ | – | – | – | – | ++ | + |
| Nilotinib (BCR–ABL1) | GIST, Philadelphia chromosome-positive ALL and CML | ++ | ++ | + | – | ++ | +++ | – | ND |
| Ponatinib (BCR–ABL1) | Philadelphia chromosome-positive ALL and CML | +++ | +++ | +++ | – | ++ | ++ | – | ++ |
| Alectinib (ALK) | NSCLC | – | – | – | – | – | – | – | + |
| Crizotinib (ALK) | NSCLC | – | – | – | – | – | – | – | ++ |
| Dacomitinib (EGFR) | NSCLC | – | ++ | – | – | – | – | – | – |
| Erlotinib (EGFR) | NSCLC, pancreatic cancer | – | +++ | ++a | – | ++a | – | – | +++a |
| Dabrafenib (BRAF) | Melanoma, NSCLC, thyroid cancer | +++ | – | – | – | – | – | – | + |
| Cabozantinib (MET) | Hepatocellular carcinoma, RCC, thyroid cancer | +++ | – | + | – | + | – | – | ++ |
| Crizotinib (MET) | NSCLC | – | – | – | – | – | – | – | ++ |
| Binimetinib (MEK) | Melanoma | ++ | – | – | – | – | – | – | ++b |
| Trametinib (MEK) | Melanoma, NSCLC, thyroid cancer | +++ | – | – | – | – | – | – | ++b |
Based on data from Micromedex (IBM, NY, USA) and Lexicomp (Wolters Kluwer, Netherlands). –, not reported; +, uncommon (<1%); ++, common (1–10%); +++, very common (>10%). ALK, anaplastic lymphoma kinase; ALL, acute lymphoblastic leukaemia; AMI, acute myocardial infarction; CLL, chronic lymphocytic leukaemia; CML, chronic myeloid leukaemia; CNS, central nervous system; DVT, deep-vein thrombosis; EGFR, epidermal growth factor receptor; GIST, gastrointestinal stromal tumours; HL, Hodgkin lymphoma; HTN, hypertension; MEK, MAPK/ERK kinase; mTOR, mechanistic target of rapamycin; ND, frequency not defined; NHL, non-Hodgkin lymphoma; NSCLC, non-small-cell lung cancer; PAD, peripheral artery disease; PE, pulmonary embolism; RCC, renal cell carcinoma; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor.
In combination with gemcitabine.
In combination with a BRAF inhibitor.
Table 3 |.
Overview of the three principal presentations of arterial vascular toxic effects of cancer therapy
| Characteristics | Main presentation | ||
|---|---|---|---|
| Acute vasospasm | Acute thrombosis | Accelerated atherosclerosis | |
| Onset after start of cancer therapy | Days to weeks | Weeks to months | Months to years |
| Reversibility | Very likely | Likely | Very unlikely |
| Primary culprit | Vascular smooth muscle cells | Endothelial cells | Endothelial cells |
| Secondary culprit | Endothelial cells | Platelets | Bone marrow-derived cells, pro-inflammatory cells |
| High levels of circulating endothelial cells | Possible | Yes | Yes |
| Low levels of endothelial progenitor cells | Possible | Yes | Yes |
| Procoagulant microvesicles | No | Yes | No |
| Examples of cancer therapeutics | 5-Fluorouracil, capecitabine, platinum drugs, VEGF inhibitors | Platinum drugs, bleomycin, vinca alkaloids, VEGF inhibitors, ICIs | Nilotinib, ponatinib, cisplatin, VEGF inhibitors |
| Treatment | Nitrates, calcium-channel blockers | Thrombectomy, PTCA and/or stent, DAPT, statin therapy | Revascularization, aspirin, statins, amlodipine, ACE inhibitor, exercise |
| On-therapy screening | Signs and symptoms | Signs and symptoms | Signs and symptoms |
| Vasoreactivity studies, ECG (ST-segment elevation) monitoring | vWF levels, circulating endothelial cell and/or endothelial progenitor cell levels | Ankle–brachial index, cardiac stress test, coronary CT angiography | |
| Pre-therapy screening and prevention | Prophylactic therapy with nitrates and calcium-channel blockers | DAPT, statins | Strict risk factor control, especially lipids, (anti-IL-1β), aspirin, statins and/or other therapies |
| CVD risk stratification: risk factors and/or disease, testing for subclinical ASCVD and/or abnormal vasoreactivity, including endothelial dysfunction | CVD risk stratification: risk factors, testing for subclinical ASCVD, endothelial dysfunction, vWF levels | CVD risk stratification: risk factors, testing for subclinical ASCVD | |
ACE, angiotensin-converting enzyme; ASCDV, atherosclerotic cardiovascular disease; CVD, cardiovascular disease; DAPT, dual antiplatelet therapy; ECG, electrocardiogram; ICI, immune checkpoint inhibitor; PTCA, percutaneous transluminal coronary angioplasty; VEGF, vascular endothelial growth factor; vWF, von Willebrand factor.
Acute arterial vasospasm
Conventional chemotherapies.
The classic examples of chemotherapies that cause acute vasospasm are 5-fluorouracil (5-FU) and its oral prodrug capecitabine, both of which are used in gastrointestinal and gynaecological malignancies in particular2–4 (TABLE 1). A theory for the underlying mechanism is an alteration in vascular smooth muscle cell (VSMC) reactivity, induced by activation of protein kinase C signalling, that leads to dysregulation of calcium handling and to contractile hyperreactivity5. Endothelial dysfunction has a facilitating role in arterial vasospasm6 and experimental studies indicate that 5-FU might also have direct toxic effects on endothelial cells7. Patients with pre-existing endothelial dysfunction, such as those with coronary artery disease (CAD), might be at a higher risk of vasospasm with 5-FU than those without endothelial dysfunction8. Furthermore, thymidine phosphorylase, which catalyses the last step of the conversion of capecitabine to 5-FU, is expressed in atherosclerotic plaques9, thereby potentially leading to higher local concentrations of 5-FU as well as increased exposure and risks. Indeed, CAD, and especially a history of myocardial infarction (MI), is one of the most potent predictors of 5-FU-related cardiotoxicity, increasing the risk by a factor of eight10. The most common presentation of 5-FU-related cardiotoxicity is angina (45% of patients), followed by MI (22%) and arrhythmias (23%); ventricular fibrillation, cardiac arrest and sudden cardiac death have also been described11. Furthermore, Takotsubo cardiomyopathy, heart failure and cardiogenic shock can be seen in <5% of patients11. Profound and prolonged vasoconstriction of the epicardial vessels and/or the coronary microcirculation can conceivably provoke such severe complications, although a direct myocardial toxicity of 5-FU has also been proposed in relation to a decline in cardiac function12.
Paclitaxel therapy has been associated with acute coronary syndromes (ACS), and the prompt resolution of ST-segment elevations with nitroglycerin administration supports the involvement of coronary vasospasm13. CAD can be a concomitant factor in this setting and needs to be evaluated13. Importantly, MI (not only myocardial ischaemia), in some cases with a fatal outcome, has also been reported with paclitaxel therapy14. Furthermore, MI and myocardial ischaemia can be seen up to 14 days after initiation of paclitaxel therapy15. Increased RHO kinase activity in coronary artery VSMCs has been postulated to have a role in paclitaxel-induced coronary vasospasm16. However, another study showed that paclitaxel therapy impairs endothelium-dependent vasorelaxation, which, as mentioned above, might have a contributing role in coronary vasospasm17.
Finally, bleomycin, cisplatin and vinca alkaloids have been associated with endothelial toxicity that can manifest as acute coronary ischaemia without underlying structural disease18,19. These medications are given in combination for patients with testicular cancer, and more than two-thirds of patients can develop angina at any time during chemotherapy. Altered vasoreactivity is also evident in one-third of these patients in the form of Raynaud syndrome, which can precede presentations of acute MI20. Impairment of endothelium-dependent vasodilatation has been shown in patients receiving cisplatin and has been attributed to reductions in AKT–endothelial nitric oxide synthase (eNOS) signalling21.
Targeted therapies.
Vascular endothelial growth factor (VEGF) has an important role in modulating the activity of the nitric oxide (NO) signalling pathway in endothelial cells, even under resting conditions. This critical role is illustrated by the observation that administration of bevacizumab, a monoclonal antibody that blocks VEGFA, at clinically relevant concentrations very specifically and acutely (within 15 min) reduced endothelium-dependent (and NO-dependent) vasorelaxation in healthy volunteers, that is, in the absence of comorbidities and treatments that could alter the response to bevacizumab22. No increase in vascular tone or blood pressure was observed, and a subsequent study confirmed that impairment of endothelium-dependent vasodilatation does not precede the development of hypertension in patients receiving the cancer drug sunitinib, which inhibits multiple receptor tyrosine kinases, including VEGF receptors (VEGFRs)23. In rats, a decrease in endothelium-dependent vasorelaxation without a change in endothelium-independent vasorelaxation was seen after 7 days of treatment with sunitinib at doses higher than those used in clinical practice23. Sunitinib suppressed acetylcholine-induced vasorelaxation similar to a NO antagonist, and the difference in acetylcholine-induced vasorelaxation between control and sunitinib-treated animals disappeared in the presence of the universal NOS inhibitor Nω-nitro-l-arginine methyl ester. These observations support findings from preclinical studies showing that VEGFR inhibition reduces NOS expression and NO availability24. Nevertheless, selective interruption of VEGFR signalling, such as with bevacizumab, probably has a different effect on vascular function than the less selective inhibition with multi-target tyrosine kinase inhibitors (TKIs) such as sunitinib. Indeed, one might argue that the vascular and haemodynamic effects induced by multi-target TKIs do not solely depend on interruption of VEGFR signalling but also on other targeted pathways. Importantly, the vascular alterations seen with VEGF inhibitors extend to the microvasculature. In animal models, impairment of the endothelium-dependent and endothelium-independent vasodilatory response of the coronary microcirculation occurs within a week of sunitinib administration25,26. In patients, sunitinib therapy induced a reduction in coronary flow reserve, which, in the absence of epicardial disease, indicates microvascular impairment27. Likewise, bevacizumab has been shown to impair retinal microvascular function in humans28 and microvascular angina has also been reported29. Other cancer therapeutics associated with a risk of vasospasm to the extent of MI include the TKI sorafenib30,31. A working hypothesis is that sorafenib reduces MAPK/ERK kinase (MEK) activity in VSMCs, which translates into increased activity of the RHO-associated protein kinase (ROCK) pathway, thereby increasing calcium sensitivity in these cells30.
Immunotherapies.
Impairment of endothelium-dependent vasorelaxation has been described in the setting of an acute systemic inflammatory state such as systemic inflammatory response syndrome and sepsis32. Even so, an increase in vasoconstriction and vascular tone does not necessarily occur in patients with systemic inflammatory response syndrome or sepsis33. Likewise, although associated with an inflammatory response, acute vasospasm has not been reported with the use of cancer immunotherapies such as chimeric antigen receptor T cell therapy (CAR T cell therapy), bispecific T cell engager therapy (BiTE therapy) or immune checkpoint inhibitor therapy (ICI therapy)34.
Systemic inflammatory response syndrome.
A widespread inflammatory response that might or might not be associated with infection, characterized by an abnormal temperature (>38 °C or <36 °C) and/or leukocyte count (white blood cells >1,200 per mm3, <4,000 per mm3 or bandaemia ≥10%) and either tachycardia (heart rate >90 bpm) or tachypnoea (respiratory rate >20 breaths per min).
Chimeric antigen receptor T cell therapy.
(CAR T cell therapy). strategy in which T cells harvested from a patient are genetically modified to recognize a specific tumour antigen in an antibody-like fashion, followed by activation of the engineered T cells before administration to the patient. Second-generation and third-generation CAR T cells have improved co-stimulatory domains, and fourth-generation CAR T cells (also known as armoured CAR T cells) express factors that enhance T cell expansion, persistence and anti-humoural activity.
Bispecific T cell engager therapy.
(BiTE therapy). antibody constructs designed to create an immunological synapse between an effector T cell and a tumour cell by simultaneously binding to the T cell-activation molecule CD3 and a tumour-associated antigen, which is CD19 on B cells in the case of blinatumomab (approved for the treatment of B cell acute lymphoblastic leukaemia).
Immune checkpoint inhibitor therapy.
(ICI therapy). Therapy that targets internal T cell inhibitory signals known as immune checkpoints, which control T cell activity in a balance with co-stimulatory signals upon T cell receptor activation following antigen presentation and recognition. Tumours can express ligands for immune checkpoint pathways, such as programmed cell death 1, thereby mediating resistance to T cell-mediated destruction. ICIs can reverse this T cell tolerance towards tumour cells and promote T cell antitumour activity.
Radiation therapy.
Cases of symptomatic coronary vasospasm and variant angina have been reported in patients undergoing radiation therapy for Hodgkin lymphoma35,36. The lack of a response to vasodilatory therapy and the resolution with steroid therapy has led one group to propose that radiation-induced pericarditis might trigger the vasospasm of epicardial coronary arteries36. An alternative mechanism might be radiation-induced vasculitis or arteritis; however, experimental studies in a large-animal model indicated a more direct contribution of radiation to VSMC hyper-reactivity, which can emerge especially when the endothelium is compromised37. Impairment of endothelium-dependent vasorelaxation of the aorta related to a reduction in NO availability has been shown in rabbits38 and the same observations have been made in patients39; strikingly, these consequences can persist for many years40. Notwithstanding studies with opposing results41, available data do suggest that radiation therapy can alter vascular reactivity. This concept is consistent with the overwhelming literature on the negative effect of radiation therapy on endothelial cell function and viability, as reviewed further in the atherosclerosis section below.
Management and prevention.
Management recommendations for cancer therapy-related acute arterial vasospasm are in line with general guideline recommendations (Supplementary Table 1). Vasodilators, such as nitrates and calcium-channel blockers, are mainstay therapy for patients with vasospasm receiving 5-FU, cisplatin, paclitaxel or VEGF inhibitors, among other cancer therapies. Calcium-channel blockers are of greater benefit when microvascular dysfunction is suspected. Often, a particular concern is re-exposure to the cancer therapy, and coverage with vasodilator therapy just before, throughout and for a short time after the treatment course might be advisable. However, this strategy is not always successful, arguing for the involvement of other pathological mechanisms, at least in 5-FU-related cardiotoxicity. Patients who are re-exposed to 5-FU after experiencing cardiotoxicity should undergo electro cardiogram monitoring for the assessment of ischaemic changes and arrhythmias. In these patients and especially in those who experienced cardiac dysfunction with 5-FU therapy, an echocardiography should be repeated after re-exposure even if the patient is completely asymptomatic. In view of the potentially fatal outcomes, the aim should be to detect profound and severe coronary vasoconstriction and any other cardiovascular toxicity as early as possible.
Strategies for the primary prevention of acute arterial vasospasm are not defined for any chemotherapy. Given that ischaemic heart disease is a prominent risk factor for 5-FU-related cardiotoxicity, a thorough clinical history should be obtained for each patient. Whether patients should be screened for subclinical ischaemic heart disease, which parameters to react to and in which format is yet to be defined. For instance, is a history of MI the central and only element that needs to be assessed? Or does a functional (non-invasive, cardiac stress test) or anatomical (coronary CT angiography) test provide better stratification and selection of care? When is the clinical work-up complete? Which patients should be started on vasodilatory therapy even in the absence of (angina) symptoms for the prevention of vasoconstriction? What efforts should be taken to modulate the involved molecular pathways? Another question is the duration of preventive therapy. For most patients, only the time of active cancer therapy requires coverage. However, some patients might have prolonged abnormal vasoreactivity and its manifestations, for example, Raynaud syndrome as well as atypical or typical angina, all of which might require additional specialist care.
Acute arterial thrombosis
Conventional chemotherapies.
Acute thrombosis has classically been described with cisplatin and VEGF inhibitor therapies, which are particularly used in patients with testicular cancer, gynaecological malignancies or renal cell carcinoma42–52. Case reports describing acute coronary thrombosis with cisplatin therapy without the presence of atherosclerotic plaque rupture or clinically significant atherosclerosis suggest superficial erosion of the endothelial monolayer as the underlying mechanism43. Superficial erosion has received increasing attention as a mechanism of ACS over the past 5 years53–56. Blood flow perturbations are an important element in the pathophysiology of superficial erosions because they lead to endothelial activation (and dysfunction), attraction of inflammatory cells such as neutrophils and, ultimately, to endothelial cell apoptosis57,58. Desquamation of endothelial cells leaves von Willebrand factor (vWF) exposed at the level of the subendothelial basement membrane, which is a potent stimulus for platelet activation and aggregation59. Although histological and mechanistic details of superficial erosion have not been defined in patients with cancer, the described sequence might operate in these patients, with some variation(s). For instance, cancer therapeutics, such as cisplatin, have a direct cytotoxic effect on endothelial cells (FIG. 2; TABLE 4). Several cancer therapeutics also have a cytostatic effect, suppressing the migration and proliferation of endothelial cells. Collectively, these effects can lead to the conundrum of the simultaneous induction of endothelial injury and reduction of its repair capacity. Indeed, the proliferation of local resident endothelial cells is an important regenerative mechanism60,61. An additional repair capacity is provided by endothelial progenitor cells (EPCs), although their nature and level of contribution has been debated60–62. Nevertheless, the myelosuppressive effects of chemotherapeutics have to be considered and a reduction in the number of EPCs has been documented in patients receiving cancer therapy63. Although this reduction in EPC numbers is almost always present, arterial thrombosis is not universally seen in patients receiving chemotherapy, pointing towards an individual risk in each patient. However, one has to caution that the real incidence of acute thrombosis in this patient population is unknown because no routine screening is performed and a number of events can remain subclinical. At the other end of the spectrum, simultaneous multivessel thrombosis with acute presentations has been reported with chemotherapy, but this presentation is a rare exception45.
Fig. 2 |. Mechanisms of ischaemia in patients with cancer.
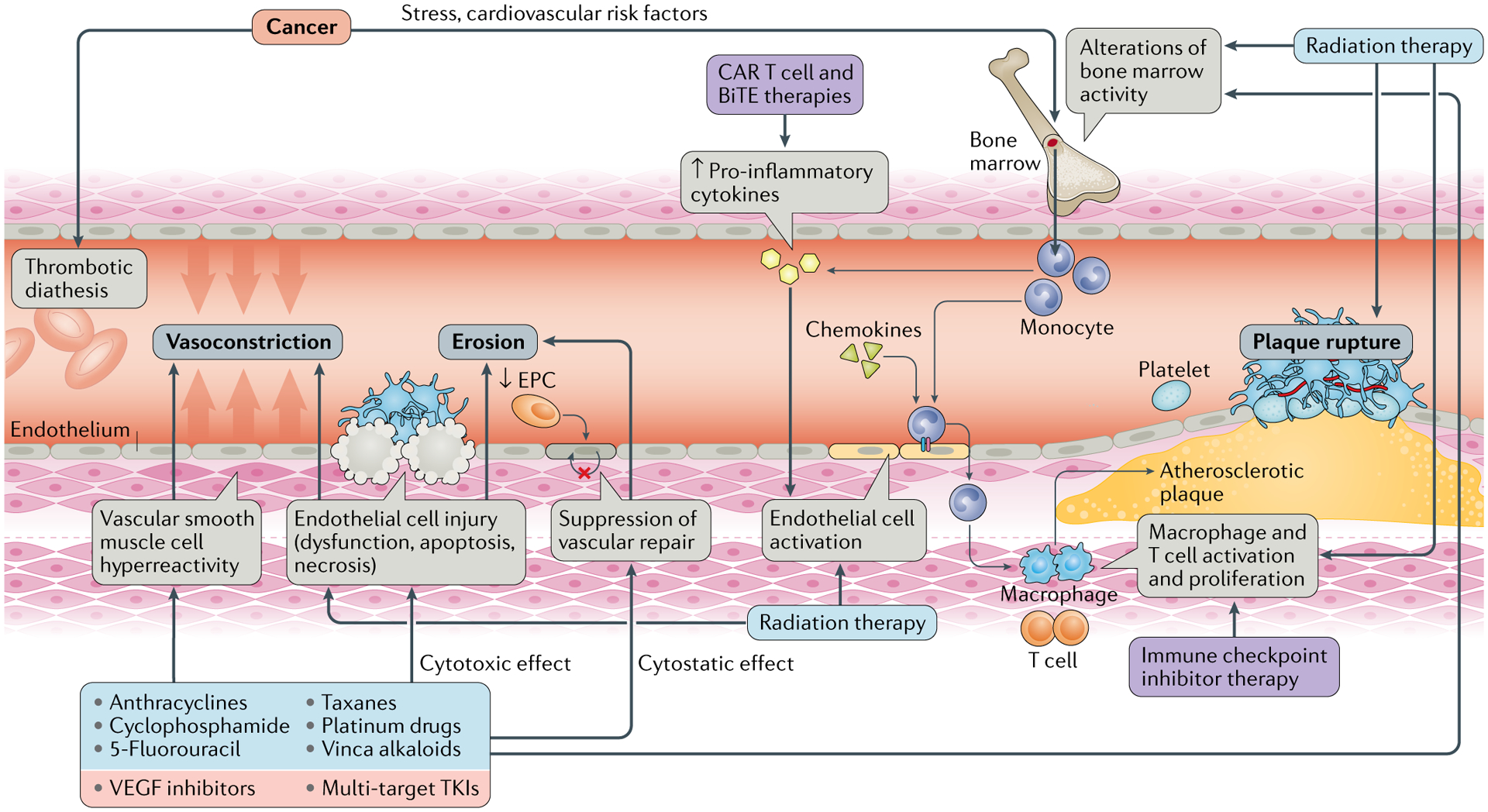
Ischaemia can be precipitated in patients with cancer by various mechanisms, leading to functional or structural alterations in the blood vessels. Increased vasoconstriction can be caused by vascular smooth muscle cell hyper-reactivity, which is classically seen with 5-fluorouracil therapy, and/or endothelial dysfunction, which can be provoked by various and sometimes concomitantly administered chemotherapeutics. Thrombotic occlusion, either partial or complete, can be caused by superficial erosion or by atherosclerotic plaque rupture. Erosion entails the loss of the endothelial monolayer, and multiple chemotherapeutics exert a cytotoxic effect on endothelial cells. This endothelial damage is often coupled with impairment of repair mechanisms, such as inhibition of the proliferation and migration of neighbouring endothelial cells or a reduction in the number of circulating endothelial progenitor cells (EPCs). Atherosclerotic plaque rupture is fostered by plaque inflammation, which also contributes to plaque development and growth and thereby to progressive luminal narrowing. Inflammation is stimulated by increased cytokine levels, for example, as a result of the expansion of some haematopoietic clones through a process called clonal haematopoiesis, or by disinhibition of immune checkpoints on inflammatory cells, mainly the programmed cell death 1–programmed cell death 1 ligand 1 (PD1–PDL1) axis, with immune checkpoint inhibitor therapy. BiTE, bispecific T cell engager; CAR, chimeric antigen receptor; TKI, tyrosine kinase inhibitor; VEGF, vascular endothelial growth factor.
Table 4 |.
Potential mechanisms of endothelial cell toxicity of cancer therapies
| Agent | Mechanism | ||||||
|---|---|---|---|---|---|---|---|
| EC death | EC proliferation and migration | EC-dependent vasodilatation | Oxidative stress | Inflammation | Coagulation disorders, thrombosis | VEGF signalling | |
| Conventional chemotherapies | |||||||
| Anthracyclines | Induction of caspase-mediated apoptosis | Inhibition of EC proliferation | ↓ EC-dependent relaxation | ROS-induced dysregulation of superoxide levels; free radical generation | ND | ↓ ET1 levels | ND |
| Platinum drugs | Direct toxic effect | Inhibition of EC proliferation | ↓ NO levels | Free radical generation | Activation of mononuclear cells and leukocyte–EC interaction | Platelet aggregation; ↑ vWF levels; fibrinolysis (↑ ICAM1 and PAI1 levels) | ND |
| Cyclophosphamide | Direct toxic effect | ND | ↓ ACE levels | ND | ND | Platelet aggregation | ↑ VEGF levels |
| 5-Fluorouracil | Direct toxic effect | ND | ↓ eNOS levels, ↑ EC-independent vasoconstriction via PKC | ND | ↑ Cytokine levels | Promotes prothrombotic state | Possible inhibitory effect |
| Taxanes | Induction of EC apoptosis | Inhibition of EC proliferation, migration and tube formation assay | Impaired nitrate-mediated vasodilatation | ND | ↑ TNF levels | ↑ TNF-induced tissue factor release; ↓ thrombomodulin levels |
↑ VEGF and VCAM1 levels |
| Bleomycin | ↑ FASL levels | ↑ ICAM1 and E-selectin levels | ↓ NO levels | Free radical generation | ↑ Cytokine production; inhibition of NO; anti-inflammatory effect | ND | ND |
| Vinca alkaloids | Induction of caspase-mediated apoptosis | Mitosis-mediated inhibition of EC proliferation | ND | ND | ND | ↓ ET1 levels | ND |
| Immunomodulatory therapies | |||||||
| Methotrexate | Direct toxic effect | ND | ND | ND | ND | Hyperhomocystinaemia | ND |
| Targeted therapies | |||||||
| Nilotinib (BCR–ABL1 inhibitor) | Induction of EC apoptosis | Inhibition | ND | ND | ND | ND | Inhibition |
| Ponatinib (BCR–ABL1 inhibitor) | Induction of EC apoptosis | Inhibition | ND | ND | ND | ND | Inhibition |
| VEGF inhibitors | Induction of EC apoptosis | Inhibition | ↓ NO levels; impaired nitrate-mediated vasodilatation | Free radical generation | ND | ND | Main drug target |
Based on data from REF.115. ACE, angiotensin-converting enzyme; EC, endothelial cell; eNOS, endothelial nitric oxide synthase; ET1, endothelin 1; FASL, FAS ligand; ICAM1, intercellular adhesion molecule 1; ND, not described; NO, nitrogen oxide; PAI1, plasminogen activation inhibitor 1; PKC, protein kinase C; ROS, reactive oxygen species; TNF, tumour necrosis factor; VCAM1, vascular cell adhesion molecule 1; VEGF, vascular endothelial growth factor; vWF, von Willebrand factor.
The individual risk, rate and scope of thrombotic events might depend on a modified Virchow’s triad64. A pre-existing reduction of endothelial and vascular health as a result of cardiovascular disease and risk factors might modify the risk of endothelial and vascular injury with cancer therapy. Blood flow perturbations, such as at flow dividers (bifurcations), inner curvatures of vessels and stenoses, are probably another element influencing the risk of cancer therapy-related vascular toxicity, and their potential role in the pathophysiology of superficial erosion was mentioned above. The third factor is alteration in the prothrombotic and antithrombotic balance, resulting in a prothrombotic state. Cancer cells can express tissue factor and a number of ligands for platelet surface receptors (such as ADP and thromboxane A2) that mediate platelet activation64. Furthermore, tumour-derived VEGFA triggers the release of vWF from endothelial cells, and some types of cancer (especially undifferentiated carcinomas) can express vWF, thereby increasing platelet activation65,66. Activated platelets in turn support tumour growth and metastases. Therefore, a bidirectional relationship exists, known as the ‘platelet–cancer loop’64.
Virchow’s triad.
Concept named after the German pathologist Rudolf Virchow, who, in 1856, described three factors that are critically important in the development of venous thrombosis: stasis, hypercoagulability and endothelial or vascular injury.
Consistent with the concept of a general prothrombotic state related to the underlying cancer is the observation that arterial thromboembolic events (ATEs) are seen in particular in patients with pancreatic, gastric or lung cancer, which is similar to the incidence of venous thromboembolic events (VTEs)67–70. Furthermore, the increased risk of ATEs seems to be highest in the month before and after diagnosis and remains significantly elevated over the first year after diagnosis (FIG. 3). The risk of ATEs is mainly seen with undifferentiated and more advanced cancer disease stages (stages 3 and 4)67. Systemic vWF levels follow a similar trajectory, increasing with tumour stage and early on during cancer therapy such as with cisplatin-based therapies66,71. This observation supports the concept of the double effect of tumour burden and potency of active chemotherapy, generating a substrate that can then precipitate in an acute thrombotic event in areas of disturbed flow dynamics lined by an injured and inadequately repaired endothelial layer.
Fig. 3 |. Risk of arterial thromboembolic events in patients with cancer.
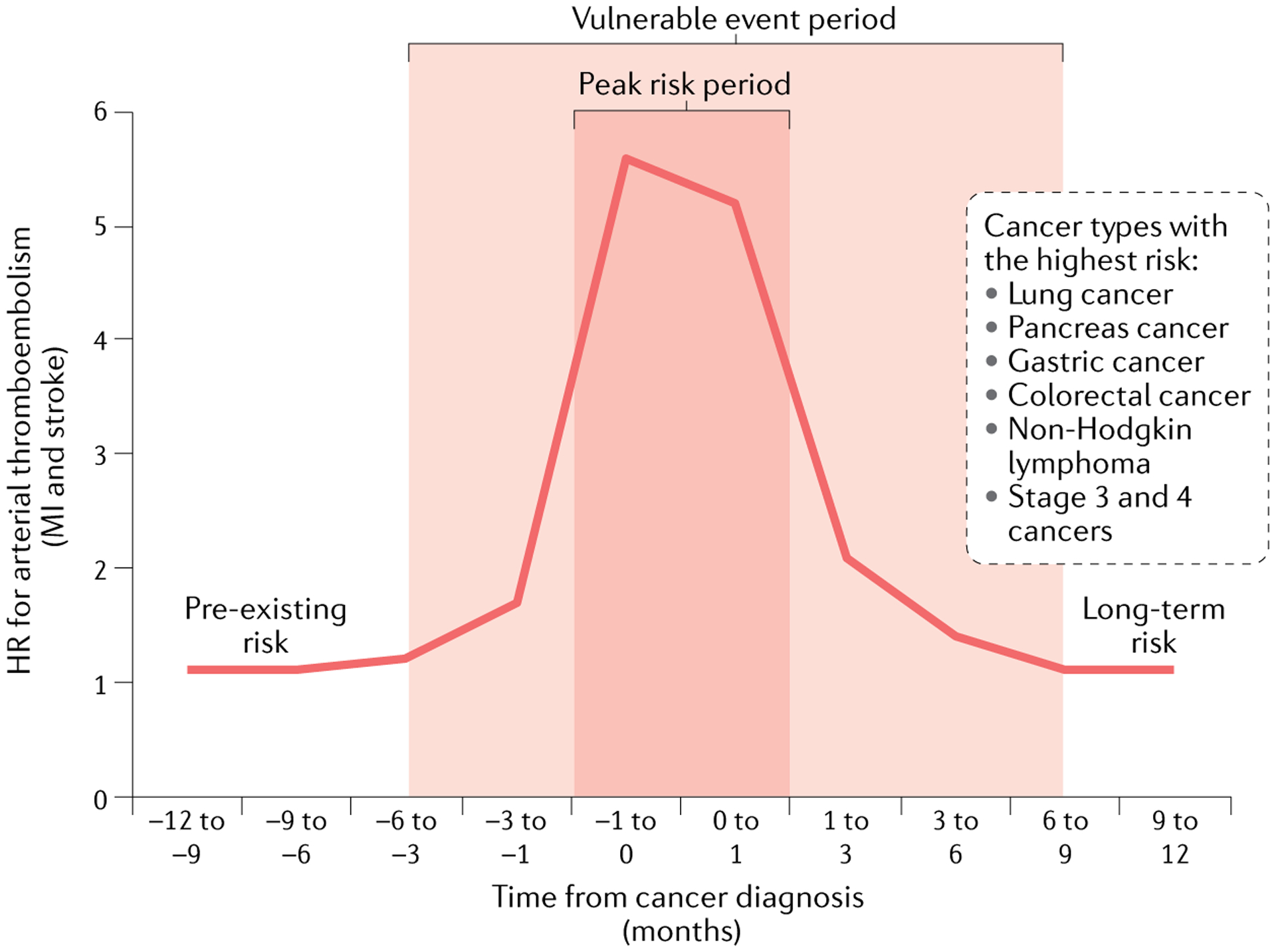
Outline of the risk of arterial thromboembolic events relative to the time of cancer diagnosis in patients in the US Medicare system. A vulnerable and high-risk period of arterial thromboembolic events can be defined as illustrated, particularly for the cancer types listed. Baseline risk factors and long-term risk dynamics remain to be defined. The plot was generated using data from REFS67,253. HR, hazard ratio; MI, myocardial infarction.
Targeted therapies.
In agreement with a general prothrombotic state associated with some cancer types (and contrary to some of the debates in the past), studies suggest that bevacizumab leads to similarly increased risks of ATEs and VTEs72. Documentation of thrombus formation in the arterial system in patients receiving VEGF-inhibitor therapy is conceptually important because ATEs encompass a broad spectrum of clinical presentations and some are more indirect in nature (such as angina, which can be caused not only by thrombosis but also by vasospasm and a haemodynamically significant stenosis)73–75. Mechanistically, bevacizumab, through the formation of immune complexes with its target VEGFA, activates platelets via FcγRIIA receptors76. This activity requires the heparin-binding domain of VEGFA in a manner similar to platelet factor 4 in heparin-induced thrombocytopenia. Indeed, heparin administration increases the deposition of bevacizumab–VEGFA immune complexes on platelets and platelet activation76. In the presence of thrombin (as occurs in hypercoagulable states), the amount of heparin required to trigger platelet activation and aggregation by bevacizumab–VEGFA immune complexes was much lower76. Bevacizumab has also been shown to increase thrombus formation after inferior vena cava obstruction and chemical saphenous vein injury in a murine xenograft model of lung carcinoma77. This effect was due to a shift in the thrombolytic balance via alterations in plasminogen activator inhibitor 1 (PAI1) levels. By binding to VEGFA, bevacizumab neutralizes the inhibitory effect of VEGFA on the expression of PAI1 in tumour cells, thereby increasing PAI1 systemic levels77. These studies illustrate that the prothrombotic effects of bevacizumab extend beyond inhibition of the protective role of VEGF on the endothelium; the consequences of not preventing endothelial dysfunction would include the conversion to a procoagulant state, for example, by increased tissue factor production8,78.
The described effects on the induction of endothelial dysfunction also apply to TKIs targeting VEGFR, although the effects of these therapies are not always as expected and have often been understudied24. Likewise, the direct effects of VEGFR-targeted TKIs on platelets are not always intuitive or consistent. Sunitinib and sorafenib dose-dependently inhibit platelet aggregation, probably via interference with tyrosine kinase signalling in platelets (such as SRC family kinases), possibly contributing to bleeding events79,80. This observation is remarkable because these drugs have also been associated with thrombotic events. The same holds true for the BCR–ABL1 inhibitors dasatinib, nilotinib and ponatinib, which have been associated with ATEs such as MI and stroke in patients with chronic myeloid leukaemia81. Peculiarly, dasatinib and ponatinib inhibit platelet activation and aggregation, but not nilotinib, which has a prothrombotic effect82–86. This finding has important implications not only for the pathophysiology of TKI-induced thromboembolic events, but also for the treatment and prevention of these events because it relates, for instance, to antiplatelet therapy.
Immunotherapies.
Arterial and venous thrombotic events have been reported in patients receiving ICI therapy87–89. Some of these events are temporally related to the initiation of ICI therapy. However, in the absence of any cohort studies, the overall incidence of thromboembolic events with these therapies and whether the incidence is higher than that generally observed in these patient cohorts remains unknown. A link between vascular thrombotic events and vasculitis has been speculated, and the interaction between inflammation and coagulation has been described and termed ‘immunothrombosis’90,91. This relationship is bidirectional, with the release of inflammatory cytokines activating the endothelium towards a procoagulant and platelet-activating phenotype92. The generation of procoagulant microvesicles and tissue factor as well as a decrease in ADMTS13 levels are contributing factors to thrombotic events in the setting of systemic inflammation93. These factors might be more important for CAR T cell (and BiTE) therapies than for ICI therapy.
Radiation therapy.
Although certainly not a common event, acute arterial (even large-vessel) thrombosis in an irradiated segment has been reported in patients, with potentially profound complications such as acute limb ischaemia and stroke94,95. These reports match experimental observations in irradiated canine femoral arteries96. Within just 48 h, endothelial cells showed evidence of severe injury, followed by endothelial denudation and intimal deposition of fibrin. Re-endothelialization was noted within 3 weeks of radiation, but remained incomplete even at 4 months96. However, the radiation dose used in this study (35 Gy) was very high and was delivered in one fraction. Conceivably, endothelial function and viability might already be reduced in some patients at the time of radiation therapy (for example, as a result of pre-existing cardiovascular disease risk factors and atherosclerotic cardiovascular disease (ASCVD) or when combined with chemotherapy with toxic effects on endothelial cells). This pre-existing endothelial damage might lower the resilience of the endothelium to injury and even low radiation doses might lead to the same consequences. In addition, depending on the cardiovascular disease risk factors present and the effects of chemotherapy, reconstitution of the endothelial layer can be further delayed in patients with cancer owing to the suppression of local and bone marrow regenerative capacity97,98.
Management and prevention.
In general, management of acute arterial thrombosis in patients with cancer follows published guidelines (Supplementary Table 2), with the main interventions being anticoagulation, fibrinolysis, antiplatelet therapy, mechanical thrombectomy and treatment of the underlying cause. Treatment of the cancer is expected to decrease the prothrombotic state, but the effect is not instantaneous. If atherosclerotic plaque rupture is present, revascularization strategies are usually pursued, mainly stenting. Manual thrombectomy is no longer a general recommendation for coronary artery lesions in view of higher stroke rates and no decisive benefit on coronary or myocardial ischaemia compared with percutaneous coronary intervention without thrombectomy. Depending on the location and severity of disease, surgery might also be considered. Superficial erosions are usually not revascularized in the absence of any clinically significant stenoses but dual antiplatelet therapy (DAPT) is recommended54. For ACS with documented coronary thrombus, DAPT should be administered for a minimum of 1 year, regardless of the coronary management strategy, that is, even when medical therapy alone is chosen or when undergoing surgery rather than stenting (Supplementary Table 2). Particularly in patients with cancer, this recommendation needs to be balanced with the risk of bleeding. In this context, thrombocytopenia is an important factor to consider, with the Society for Cardiovascular Angiography and Interventions (SCAI) recommendations for triage and platelet cut-off levels indicating platelet counts >50,000 per μl for surgical interventions, >30,000 per μl for percutaneous coronary intervention (PCI) with DAPT and >10,000 per μl for angiography99 (Supplementary Table 7, Supplementary Figure 2).
The recommendations for the duration of DAPT after stenting have been evolving (Supplementary Table 2), and the latest ESC guidelines recommend the universal use of drug-eluting stents (DES) regardless of lesion or patient characteristics100 (Supplementary Table 2). Improvements in the design of DES have markedly reduced the associated risk of thrombosis; in particular, newer-generation DES have a documented lower rate of stent thrombosis than bare-metal stents101. Accordingly, the LEADERS FREE trial102 showed that, in combination with just 1 month of DAPT, a polymer-free biolimus A9-coated stent was superior to a bare-metal stent in terms of safety (stent thrombosis, death and MI) and efficacy (repeat revascularization) in patients at high risk of bleeding (including 15% with anaemia, 15% with anticipated surgery in the next year and 10% with cancer). Although meta-analyses have shown a reduction in both stent thrombosis and stent restenosis with the newer generation of DES than with bare-metal stents in the general patient population, whether these results can be extrapolated to patients with cancer in general is unknown103. Nevertheless, acute stent thrombosis has been reported even after bare-metal stenting in patients with cancer, often shortly after discontinuation of DAPT104. Importantly, prediction scores for the risks of thrombosis and bleeding, such as the DAPT and PRECISE-DAPT scores, have not been validated in patients with cancer (who have a higher risk of both than patients without cancer)105,106. Cancer has been listed as an important risk factor for early stent thrombosis and the most important patient-related risk factor for late stent thrombosis107. The risk of stent thrombosis is further increased in patients with additional comorbidities, including a reduced ejection fraction107. These clinical risk factors for stent thrombosis might not be as easily controllable as technical aspects, which should be managed in a pristine manner in patients with cancer108 (see SCAI recommendations; Supplementary Table 7).
In patients with cancer with an acute coronary event, defining not only the culprit lesion but also the culprit mechanism is paramount. The Universal Definition of MI pursues this concept, and it should be noted that the recommendations provided in this Review apply mainly to type I MI109 (Supplementary Figure 3). Of interest, a 2018 study showed that the majority of MIs in patients with haematological cancers are type II MIs, which might explain why an invasive approach did not translate into better outcomes in these patients, contrary to aspirin, β-blocker and angiotensin-converting enzyme-inhibitor therapy110. The scope of aetiologies for type II MI is very broad, and a number of mechanisms need to be considered in patients with cancer111. Furthermore, the very same patient can be at risk of both type I and type II MI. Therefore, overlap in the types of presentation must be considered and re-defined on an individual basis.
From a preventive standpoint, efforts are directed towards improving endothelial health, which will reduce the risk of all three vascular toxicities associated with cancer therapy. These interventions primarily include statins, angiotensin-converting enzyme inhibitors and exercise24. As shown for patients undergoing treatment with bevacizumab, aspirin can reduce the risk of arterioembolic events in those at highest risk (age >65 years and prior ATE)112. The implications for TKI therapy might be different, as outlined above. However, whether patients receiving TKIs develop thrombosis or bleeding might not only be a function of platelet activity and inhibition but rather the consequence of the effects of VEGFR signalling inhibition on endothelial viability and vascular integrity within the context of the specific histopathological environment113. Similarly, capillary networks are more prone to collapse, with the complications being thrombosis with ischaemia or rupture with bleeding.
At present, no recommendations are available regarding screening for the risk of arterial thrombosis during or before cancer therapy; one reason is the uncertainty about which tests to implement and what parameters to react to and in what manner. However, vWF has emerged as a possible marker. Circulating levels of vWF rise during cisplatin-based chemotherapy in patients with testicular cancer, particularly among those with an ATE71. Indeed, levels of vWF are already higher at baseline among patients with testicular cancer with ATEs114. A clinical fingerprint that can identify patients at high risk of ATEs even before cancer therapy entails at least three of the following five risk factors: BMI >25 kg/m2, current smoking, blood pressure >140/90 mmHg (or treated), hyperlipidaemia (or treated) or elevated fasting plasma glucose levels114. Profiling in this way is intuitively attractive, but the next question is whether this profiling should translate into some form of drug intervention such as statin therapy or DAPT. The confidence level of starting DAPT in patients who are prone to thrombocytopenia and bleeding can be low. Furthermore, the adverse effects of the cancer therapy can evolve and future declines in blood cell counts might need to be anticipated, or prohibitive cytopenias could eventually resolve and/or be bridged by transfusions (although the latter carries its own risks).
Accelerated atherosclerosis
Conventional chemotherapies.
Among the conventional chemotherapies, many can have acute and lasting effects on the vasculature, including those classically associated with cardiotoxicity. A prime example is anthracyclines, which induce (signs of) premature vascular ageing115–117. However, the classic conventional chemotherapy associated with atherosclerosis is cisplatin. As outlined above, cisplatin can induce acute thrombosis as well as acute vasospasm. However, the toxic effects on endothelial cells can last even longer. Studies in long-term cancer survivors have shown evidence of endothelial cell activation and dysfunction as well as elevated levels of circulating endothelial cells two decades after termination of the cancer therapy118. Similarly, circulating cisplatin levels can remain detectable over this time frame119. Of note, survivors of testicular cancer treated with cisplatin-based therapies have an up to sevenfold higher than expected (on average, an approximately twofold higher) risk of CAD120–124, and mediastinal radiation seems to have an additive effect on the risk of CAD125,126. Importantly, although these patients receive other agents with toxic effects on endothelial cells, such as bleomycin, etoposide and vinca alkaloids, the impression is that platinum drugs are still the main culprit125. The cumulative platinum dose seems to be important for both general and cardiovascular long-term toxicity127. The risk of cardiovascular disease might be particularly high with doses of platinum >3,000 mg (REF.128). However, intermediate and high risks of cardiovascular disease can already be seen in patients exposed to cisplatin doses above a much lower threshold of 850 mg (REF.129). Further to consider, survivors of testicular cancer have metabolic derangements that confer a cardiovascular risk130; about 20% of patients have hypertension and 33% patients have hyperlipidaemia127. Importantly, cisplatin itself does not have a long-term effect on the blood lipid profile131. Not unexpectedly, for survivors of testicular cancer, discussions have emerged similar to the debates on androgen-deprivation therapy for patients with prostate cancer132–134.
Targeted therapies.
As outlined above, VEGF signalling inhibition negatively affects NO production and thereby might contribute to atherosclerosis. Indeed, pan-VEGFR inhibition has been shown to lead to accelerated atherosclerosis in a rodent model albeit without an increase in plaque vulnerability135. These findings are interesting because VEGF administration and its inhibition have been linked not only to the function of endothelial cells but also to endothelial cell proliferation and plaque neovascularization136. Importantly, plaque neovascularization contributes not only to plaque progression but also to plaque inflammation and vulnerability136. Therefore, antiangiogenic VEGF-inhibitor therapy would be expected to have an overall stabilizing effect and, indeed, bevacizumab therapy reduced the neovascularization and growth of established plaques in another experimental study24. The observation that atherosclerosis shows angiogenesis-dependent and angiogenesis-independent phases might provide some explanation for the apparently conflicting results136. At least depending on the stage of atherosclerosis, the effects of VEGF inhibition can vary and VEGF inhibition might not always be synonymous with angiogenesis inhibition24,137–141. Along these lines, the effects of VEGFR inhibition with a TKI might differ considerably from the effects of VEGF neutralization with bevacizumab. Furthermore, the vascular effects of VEGF and VEGF signalling inhibition might depend on local VEGF concentrations even when accounting for other factors. Low VEGF levels are necessary for vascular homeostasis, whereas high concentrations result in proliferative effects. Accordingly, the ultimate effects of VEGF inhibition can be harmful or therapeutic, depending on the host milieu, and can therefore be very difficult to predict.
The possibility of accelerated atherosclerosis as an adverse effect of cancer therapy has received attention in particular with regards to the use of the two TKIs targeting BCR–ABL1, nilotinib and ponatinib. Profound and progressive peripheral artery disease was first recognized with nilotinib142–153. An even more aggressive phenotype was then seen with ponatinib, leading to a temporary suspension of sales154. Meta-analyses confirmed the increased risk of cardiovascular disease with nilotinib and ponatinib therapies155,156. Expressed in event rates per exposure time, the risk of MI is more than twofold higher with nilotinib than with rofecoxib (Vioxx), a cyclooxygenase 2 inhibitor that was removed from the market because of an unacceptably high risk of cardiovascular disease157. Another interesting aspect that has emerged is a potential risk of cardiovascular disease even with the BCR–ABL1 TKI dasatinib, although on a lower scale.
These clinical observations support in vitro findings: ponatinib, nilotinib and dasatinib (in decreasing order of potency) were the three TKIs targeting BCR–ABL1 with notable effects on endothelial cells158. These effects included alteration of the expression of numerous genes, suppression of endothelial function and even cell toxicity. Molecular and mechanistic overlap has been difficult to identify, but nilotinib has been shown to downregulate VEGFR2 expression159; therefore, nilotinib and ponatinib share inhibition of the VEGF signalling pathway as a common mechanism159,160. In experimental models, the progressive nature of atherosclerosis could be replicated, at least in part, with nilotinib and ponatinib treatment161. Interestingly, ponatinib therapy did not increase but rather reduced atherosclerotic plaque volume. Nevertheless, both nilotinib and ponatinib therapies led to changes that were consistent with increased atherosclerotic plaque vulnerability161.
Given that the population of patients with chronic myeloid leukaemia, in whom these drugs are primarily used, includes mainly elderly individuals who are at high risk of ASCVD, whether the clinical events noticed were truly attributable to the TKIs, the target population or the combination of the two has been a matter of debate. A carefully conducted study showed that patients receiving nilotinib had a much higher adverse vascular event rate than the control cohort of patients who were matched by age and cardiovascular risk factors159. The additional observation that general prediction models of the risk of ASCVD could stratify patients undergoing nilotinib therapy for cardiovascular events has several implications: it indicates that the underlying process of vascular toxicity in these patients is indeed atherosclerosis rather than, for instance, vasculitis, that the patients have pre-existing risk factors and that, possibly, the interaction of these risk factors with the cancer therapeutic leads to the acceleration of the disease process151,162–165. Regarding the pre-existing risk of ASCVD, a preliminary finding is the higher incidence of age-related clonal haematopoiesis mutations in patients receiving nilotinib who develop ASCVD than in those who did not (two-thirds versus one-third)159; the specific link between nilotinib therapy and these mutations is not known. Likewise, if any such correlation exists with ponatinib remains unknown. If and to what extent these drugs affect the bone marrow cell population is yet to be defined.
These considerations are intriguing in view of the discovery of age-related clonal haematopoiesis or clonal haematopoiesis of indeterminate potential (CHIP) mutations as a shared pathway to ASCVD and haematological malignancies166–168. Bone marrow cells, like any other cells in the body, can undergo somatic mutations. The likelihood of mutations increases with time and, therefore, with the increasing age of the individual. DNMT3 and TET2 are the most commonly affected genes, and their mutations result in a loss of function, consistent with their role as tumour-suppressor genes169. Alterations in DNMT3A, which encodes DNA methyltransferase 3A (DNMT3A), have been reported in patients with various haematological malignancies, including acute myeloid leukaemia (AML). TET2 mutations also occur in AML as well as in other myeloproliferative disorders and myelodysplastic syndrome170. A study in mice has shown that TET2-deficiency increases pro-IL-1β expression (via epigenetic mechanisms) and pro-IL-1β processing (via modulation of NLRP3 inflammasome expression and activity) in bone marrow-derived macrophages171. These cells infiltrate the vasculature and contribute to a much more pronounced inflammatory phenotype of atherosclerotic plaques (FIG. 4). This mechanism might explain the striking observations of population-based studies showing an eightfold increased risk of MI in patients with TET2 mutations compared with those without this genetic fingerprint169. Conversely, IL-1β-directed therapy with a monoclonal antibody has been shown to decrease recurrent acute ischaemic events in patients with previous ACS, including MI, in addition to decreasing the risk of lung cancer172,173.
Fig. 4 |. Pathophysiological processes contributing to atherosclerosis in patients with cancer.
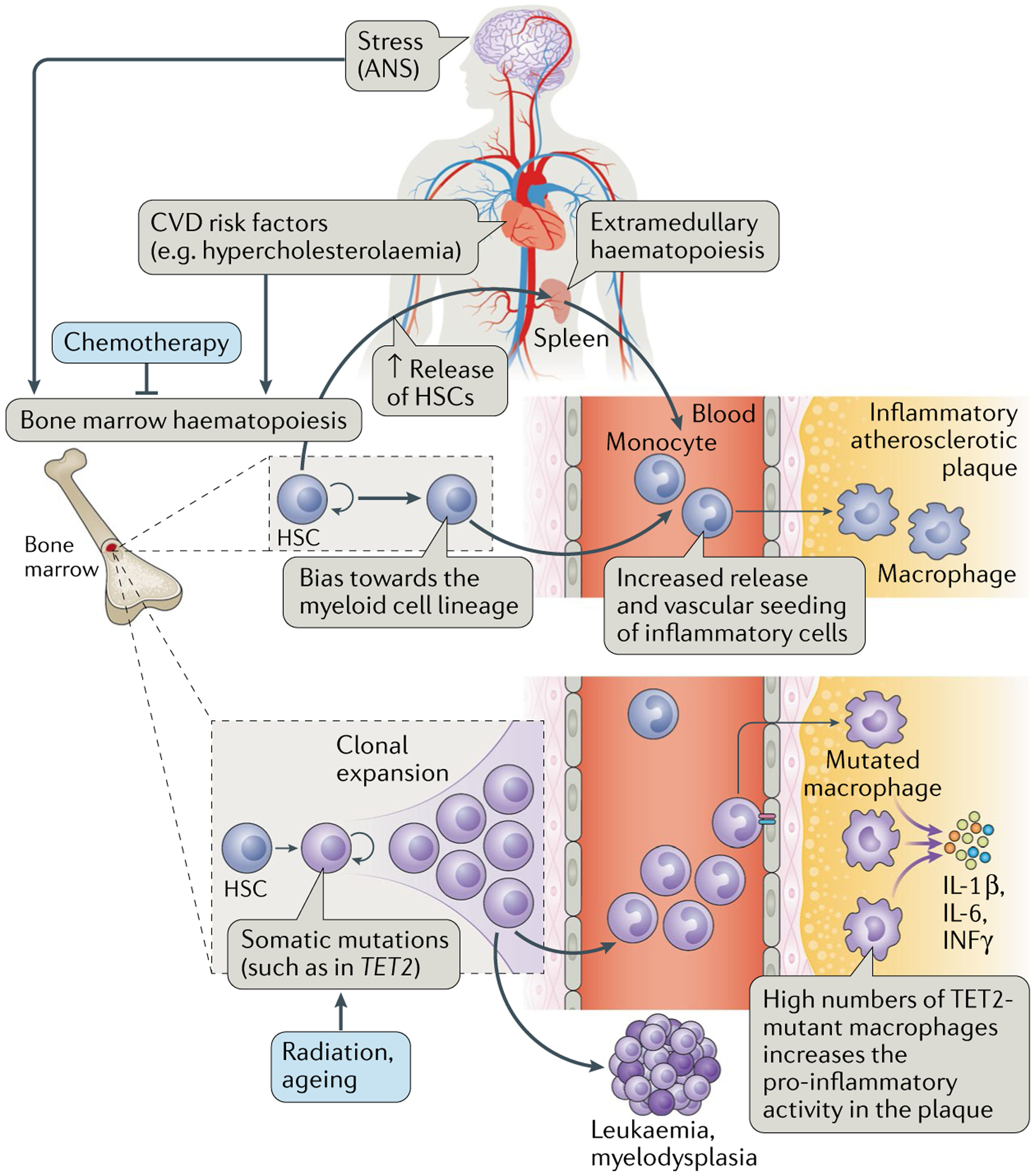
Somatic mutations can lead to alterations in the haematopoietic stem cell (HSC) clones in the bone marrow, such as mutations in TET2, which, through a process termed clonal haematopoiesis, ultimately leads to the generation of a macrophage pool with greater inflammatory activity. In addition, stress, via β3-adrenergic receptor activation, leads to activation of HSCs and increased proliferation. These cells circulate in the blood and seed into the spleen, where they proliferate and continue to mature. The end effect is an increased infiltration of inflammatory cells into atherosclerotic plaques. Both of these processes — stress as well as somatic mutations and clonal haematopoiesis — can also contribute to and result in malignancies. Cancer therapies can affect the bone marrow, for example, leading to bone marrow suppression as well as mutations and derangements of bone marrow cells. ANS, autonomic nervous system; CVD cardiovascular disease.
Patients with myelodysplastic syndrome (MDS) have long been known to have a higher risk of cardiovascular disease174. This high risk is, at least in part, due to acute thrombotic events in the arterial and venous circulation, especially among those with JAK2 mutations175. Advanced atherosclerosis and microvascular dysfunction have also been seen in these patients176,177. The adaptor protein LNK (also known as SH2B adapter protein 3) operates within the negative feedback loop to JAK2, and LNK loss of function increases the risk of MDS178. Fascinatingly, LNK has also been shown to limit myelopoiesis and platelet activation in hypercholesterolaemia, and LNK loss of function would therefore also predispose to the progression and complication of ASCVD179,180.
An important additional aspect is that the bone marrow is influenced by various factors, including the autonomic nervous system. In MDS and AML, β2-adrenergic receptor activation in the bone marrow contributes to disease progression at the cost of reduced activity in the β3-adrenergic receptor-related regenerative niche181. The bone marrow regenerative niche is also activated by exposure to cardiometabolic risk factors, such as hyperlipidaemia, and shows higher activity in patients with ASCVD in general than in healthy individuals182–187. In the setting of stress exposure, as seen for instance with MI183–187, β3-adrenergic receptor stimulation of the bone marrow leads to the release of haematopoietic and myeloid progenitor cells that seed in the spleen, feed extramedullary haematopoiesis and eventually enrich the inflammatory cell population in atherosclerotic plaques as monocytes and macrophages183–187. This finding provides one explanation to the intriguing clinical observation of an increased risk of recurrent MI early after an MI. If and to what degree (towards greater or lower activity) this axis is altered in patients with cancer in general as well as in patients with haematological malignancies is yet to be determined. Of note, in the seminal studies dissecting the effect of chronic stress on the bone marrow and ASCVD, 5-FU treatment was found to lead to an impressive suppression of the stimulating effects of the autonomic nervous system on the bone marrow, with a prominent rebound thereafter185.
Immunotherapies.
The cytokine release in the setting of CAR T cell (and BiTE) therapy has the theoretic potential to influence the clinical course of patients with ASCVD. Inflammation is an important parameter for the biological activity of atherosclerotic plaques and can be fostered by other concomitant acute or chronic inflammatory conditions in the body. For instance, the risk of acute MI increases at times of acute infections, then slowly declines188. Various cytokines, such as IFNγ and IL-6, have been shown to accelerate atherosclerosis189–191. However, these latter observations were made under very different conditions. Patients undergoing CAR T cell therapy often do so as a last resort and have therefore been exposed to several previous cancer treatments. In addition, suppressive effects of preconditioning interventions might alter the vascular effect of the inflammatory response occurring with CAR T cell therapy, although this hypothesis is yet to be tested, for example, by comparing with BiTE therapy, which does not require preconditioning192.
Overall more predictable seem to be the effects of inhibition of immune checkpoints, which are broader and longer lasting. Under inflammatory conditions, the absence or blockade of programmed cell death 1 ligand 1 (PDL1), PDL2 or programmed cell death 1 (PD1) results in enhanced T cell responses and acceleration or exacerbation of disease. For instance, PDL1 is detectable in cardiac allografts but not in coronary arteries or in the myocardium of normal hearts193. In transplanted hearts, PDL1 expression was especially notable in the VSMCs of coronary arteries affected by transplant vasculopathy and increased in these cells in response to IFNγ193. Importantly, treatment with an anti-PDL1 monoclonal antibody increased transplant vasculopathy in a dose-dependent manner193, in conjucntion with an increased number of infiltrating CD4+ T cells and CD8+ T cells, most of which centring around coronary arteries.
Further in keeping with the theory that PD1 is critical for maintaining the immune-privileged territory of the vasculature is the observation that inhibition of PD1 signalling leads to fulminant vasculitis in a chimeric mouse model194. Importantly, such a reaction occurs whether inflammatory cells are reconstituted from healthy volunteers or patients with giant cell arteritis (GCA). In GCA, dendritic cells express low levels of PDL1 and thereby provide insufficient negative counterbalance to PD1+ effector T cells195. The T cells then become more infiltrative and more active, leading to increased cytokine production and ultimately profound alternation of the normal vascular structure. Progressive luminal narrowing and occlusion are most important for clinical manifestations such as ischaemic optic neuropathy, which can lead to blindness195. The outgrowth of myofibroblasts is crucial in this context and was found to be accelerated by PD1–PDL1 inhibition, as was the extent of neoangiogenesis in GCA vessels. Accordingly, the effects of immune checkpoint inhibition can be profound in patients in whom the immunoprivilege of the vasculature is broken. Intriguingly, emerging vasculitis has been reported even in patients with no such clinical history with cytotoxic T lymphocyte antigen 4 (CTLA4) and PDL1 inhibitor therapy196–200. The case reports usually involve large-to-medium-sized arteries, although small-vessel vasculitis and vasculitic neuropathy have also been reported201–203.
Reports on the dynamics of ASCVD in patients receiving ICIs are scarce. In experimental studies in atherosclerosis-prone Ldlr−/− mice, genetic deficiency of PDL1, PDL2 or PD1 increased the size and inflammatory cell infiltrate of atherosclerotic plaques, including macrophages and T cell populations204,205. An exaggerated immune response was noted, that is, T cells were more reactive (including a higher production of pro-inflammatory cytokines) and more proliferative to antigens and oxidized LDL, presumably as a consequence of removal of restraints within them or in PDL1-deficient or PDL2-deficient antigen-presenting cells. Further in line with the theory of removal of restraints, one of the most striking observations was the prominent increase in CD8+ T cells, otherwise present in only very low numbers in atherosclerotic plaques204,205. Furthermore, PD1-deficient CD8+ T cells showed greater cytotoxic activity against resident cells, that is, VSMCs and endothelial cells. This cytotoxic activity is important because depletion of VSMCs weakens the fibrous caps of atherosclerotic plaques, and the apoptosis of endothelial cells leads to plaque erosion. Most importantly, treatment with an anti-PD1 monoclonal antibody did not increase plaque size but increased the abundance and activity of CD4+ T cells and CD8+ T cells204,205. This finding is likewise in keeping with a shift towards a more inflammatory and vulnerable phenotype of atherosclerosis. Of further note, these changes occurred despite the expansion and preserved activity of regulatory T cells, which have a presumed protective function206.
Radiation therapy.
Several studies have indicated that coronary and other vascular diseases are among (if not) the most common cardiovascular diseases in patients who receive radiation therapy, and most patients who develop vascular events have at least one cardiovascular risk factor207. The vascular territories exposed to radiation therapy or in the proximity of radiation fields include the carotid and intracranial arteries in head and neck tumours, the coronary arteries in lymphoma, breast, lung, oesophageal and gastric cancers, and the aorta, renal, intestinal and peripheral arteries in lymphoma, intestinal and testicular cancers208. Unique features are media disruption, fibrosis and atrophy as well as adventitial thickening and fibrosis209,210. By contrast, intimal plaques might be rather similar in appearance to those seen in patients not exposed to radiation therapy, although possibly less proliferative and more fibrocalcific or fibrofatty209–211. All these autopsy studies outlined the difficulties in clearly differentiating radiation-induced atherosclerosis from conventional atherosclerosis, especially in elderly patients and in those with cardiovascular risk factors208. Therefore, only cases with atherosclerosis that is out of proportion in severity and/or location to what would be expected have been considered to be causally related to radiation. However, the disease process can conceivably overlap and radiation might have a contributing role, adding to and aggravating the very processes that lead to atherosclerosis212. Induction of oxidative stress and activation of nuclear factor-κB are common elements, as is endothelial activation and dysfunction, which are the initial phases of ASCVD213. Of note, sustained inflammation and a link to the inflammasome–interleukin system have been shown in long-term survivors of cancer after radiation therapy214. Furthermore, in patients with breast cancer, the risk of acute coronary events increased with greater exposure to chest radiation and the presence of traditional cardiovascular risk factors215. Experimental work has indicated that the acute effects of radiation reflect endothelial cell apoptosis and the chronic effects reflect endothelial cell senescence216. Progenitor cells seem to be even more sensitive to cell death than well-differentiated cells, but radiation doses of ≥10 Gy are required to induce a sizeable effect. At the same dose, mature endothelial cells undergo senescence and, similar to fibroblasts, these cells display a secretory phenotype with the production of cytokines important for the induction of the inflammatory ASCVD phenotype. In rodent models of atherosclerosis, radiation doses of 2–8 Gy increase the number and size of atherosclerotic plaques217. These lesions show comparatively more macrophages and thrombotic features (and less collagen) than non-irradiated lesions, especially at higher radiation doses. After exposure to a single dose of 14 Gy or 20 Gy delivered in fractions of 2 Gy, carotid arteries of Apoe−/− mice show profound inflammation, thrombosis and intraplaque haemorrhage218,219. Neither atorvastatin nor clopidogrel was therapeutic220, whereas high-dose aspirin increased collagen content and decreased adhesion molecule expression without reducing the overall disease burden221. These findings beget the hypothesis that radiation-induced and conventional atherosclerosis might not be identical processes, although the experimental context in which these results were obtained needs to be considered222.
Management and prevention.
Patients with cancer who have accelerated atherosclerosis should be treated according to current society guidelines (Supplementary Table 3), regardless of the cancer therapy received. Optimal medical therapy is the very foundation of treatment, directed at cardiovascular risk factor control, stabilization of atherosclerotic plaques and reduction of further growth (or, less likely, regression) of stenoses. These patients require serial surveillance to define the dynamics of progression of disease and the possible eventual need for percutaneous or surgical intervention. In the non-acute setting in the general population, revascularizations mainly serve the purpose of symptom and quality-of-life improvement. However, some studies indicate that interventions in the setting of a large burden of myocardial ischaemia are also prognostically relevant223. Additionally, progressive luminal obstruction, unhalted by other efforts, is likely to require intervention to maintain patency and perfusion. This situation is illustrated in the accelerated nature of peripheral artery disease in patients receiving nilotinib, which can lead to limb ischaemia and limb loss. Given the predisposition of the peripheral circulation to atherosclerosis, serial ankle–brachial indices have been suggested as a mode of surveillance. Details about the optimal time intervals for screening, the duration of follow-up and the cut-off points to designate a clinically significant change have not been defined. Extrapolating from longitudinal cohort studies, a drop in the ankle–brachial index of ≥0.02 within 6 months could be deemed clinically significant. The inter-test variability needs to be considered as well as the possibility that progression might not be linear. Given that the development of severe arterial occlusive disease showed a binary prediction pattern by the ESC score, that is, the risk was essentially confined to those with a score ≥5, starting serial surveillance efforts in this group of patients would be reasonable. This surveillance would need to be over years and even after completion of the cancer therapy regimen (Supplementary Figure 4).
Long-term surveillance is also a major requirement after radiation therapy, tailored on the basis of concomitant risk factors. For instance, whereas the risk of MI does not increase significantly before 15–20 years of follow-up in survivors of childhood cancer, an increased risk of ACS can become apparent in adult patients with breast cancer within 5–10 years of chest radiation therapy215. Patients with breast cancer with additional risk factors for cardiovascular disease are at a higher risk of ACS after radiation therapy, and those with a history of ischaemic heart disease and MI are at the highest risk215. In this and other studies, a linear relationship between mean heart radiation dose and the rate of major coronary events was observed, starting a 2–4 Gy with an increase in risk of 7.4% per Gy (REF.224). Therefore, reductions in radiation exposure are important, and can be accomplished by several strategies, including breath holding, prone positioning and proton beam therapy225–228.
With regard to surveillance studies, coronary artery calcium scanning and coronary CT angiography have been considered to assess the structural burden of CAD in patients after chest radiation therapy229 (Supplementary Figure 5). However, which parameters would require action and what type of action remain uncertain. Non-invasive stress tests provide more guidance on functional significance, and their combination with an exercise oxygen consumption study might be particularly useful because this test can also indicate the disease burden induced by the pulmonary adverse effects of chest radiation. This information is important for treatment planning in these patients in general and in cases of eventual open-heart surgery. Acute mortality is low, but these patients have a risk of early right ventricular and pulmonary dysfunction230. The routine use of the internal mammary artery for coronary artery bypass graft surgery might not be possible and severe inflammatory changes of the pericardium might complicate surgery. By contrast, the outcomes with PCI are good, both acutely and long term, irrespective of whether radiation therapy was performed before or after PCI231–233.
Finally, the dynamics of ASCVD are putatively increased in patients receiving immunotherapy given that inflammation accelerates atherosclerosis. Closer surveillance from a vascular disease standpoint is recommendable for patients receiving ICI, especially those receiving long-term therapy, those with previous exposure to other vascular toxic drugs and those with (pre-)existing ASCVD. Those with a recent acute ischaemic event should not be treated with ICIs (nor any other chemotherapy with cardiovascular toxicity) until the acute injury phase is resolved. This point is important with regard to both the expression of PDL1 in the myocardium secondary to the injury as well as the likely infiltration of inflammatory cells at both epicardial and myocardial levels234. Expression levels of PDL1 over time after MI are unknown, but healing can take up to 90 days depending on the extent of MI235.
Venous thromboembolic disease update
Historically, more attention has been given to venous thromboembolic disease induced by cancer therapies, not least because of the well-known increased incidence of VTEs in patients with cancer68. Compared with patients without cancer, those with cancer have a fourfold to sevenfold higher risk of VTE236. The risk is greatest in the first months after cancer diagnosis and in those with more advanced (metastatic) cancers69. Pancreatic and brain cancers have consistently been ranked as conferring the highest risk of VTE across multiple studies. Taken together, these observations suggest that cancers with the worst prognosis are often those with the highest risk of VTEs68. This observation might explain, at least in part, the worse prognosis of patients with cancer and VTEs (threefold higher compared with patients with cancer without VTE and eightfold higher compared with patients with VTE and without cancer)68. Challenges in the management of VTEs are the risk of bleeding and the risk of recurrent VTEs. Risk-prediction scores have been developed to predict thrombotic risk and both initial and recurrent VTE237 (Supplementary Table 4). However, no risk calculator for bleeding risk is available and therefore no net-gain calculator integrating the risks of both thrombosis and bleeding is currently available. Differences in the thrombotic and bleeding risks are also influenced by the specific type of cancer therapy, which is often not considered (such as higher risk of bleeding than risk of thrombosis with VEGF-inhibitor therapy)238,239.
Among the thrombotic risk-prediction scores, the Khorana score is the most widely tested, externally validated and endorsed in society guidelines. The 2019 National Comprehensive Cancer Network (NCCN) guidelines state that ambulatory patients with cancer with a Khorana score of ≥3 could be considered for primary prophylactic anticoagulation240. This concept is supported by subgroup analyses from two large randomized trials (PROTECHT and SAVE-ONCO)241,242 on low-molecular-weight heparin (LMWH) for the primary prevention of thromboembolic events in patients with cancer. Studies on the use of apixaban (AVERT trial)243 and rivaroxaban (CASSINI trial)244 for the primary prophylaxis of VTEs in patients with cancer with a Khorana score of ≥2 have been published in 2019. Apixaban decreased the risk of VTE by 60% at the cost of a twofold higher risk of bleeding, whereas rivaroxaban was similarly effective only in the on-treatment analyses at a similarly increased (although not significant) risk of bleeding. How these results will influence the treatment recommendations remains to be seen. For the time being, only patients with multiple myeloma receiving immunomodulatory therapy are recommended to receive primary prophylaxis with either aspirin, LMWH or warfarin. In patients with cancer and VTE, guideline recommendations for treatment vary by society (Supplementary Table 4). The latest is the 2019 NCCN guideline, which now lists rivaroxaban as a viable option for monotherapy as an alternative to dalteparin (category 1) and also apixaban for patients who refuse or cannot take LMWH, for example, owing to heparin-induced thrombocytopenia240. Combination therapy with LMWH or unfractionated heparin for 5–10 days, followed by edoxaban (or dabigatran) is another viable option. LMWH had been the preferred choice given its greater efficacy in preventing recurrent VTE compared with that of warfarin. Non-vitamin K antagonist oral anticoagulants can be as effective as LMWH but have a higher risk of major bleeding, and especially upper gastrointestinal bleeding (and possibly also genitourinary bleeding) as seen in the Hokusai VTE Cancer trial245. For this reason, the International Society on Thrombosis and Haemostasis Guidance Statement suggests the use of specific direct oral anticoagulants (edoxaban and rivaroxaban) for patients with cancer and an acute diagnosis of VTE who have a low risk of bleeding and no drug–drug interactions with current systemic therapy, and LMWHs for patients with cancer with an acute diagnosis of VTE and a high risk of bleeding, including patients with luminal gastrointestinal cancers with an intact primary, patients with cancers at risk of bleeding from the genitourinary tract, bladder or nephrostomy tubes, or patients with active gastrointestinal mucosal abnormalities such as duodenal ulcers, gastritis, oesophagitis or colitis246.
Treatment with the same anticoagulant should be pursued for 3 months, and overall treatment should continue as long as the cancer is active, under active treatment or if risk factors for recurrence persist. Patients who develop recurrent VTE while receiving anticoagulation are generally switched to LMWH if they are not already receiving it; otherwise, the dose of LMWH is increased by 25% (anti-factor Xa levels and heparin-induced thrombocytopenia should be considered). For patients with thrombocytopenia, the NCCN guidelines recommend enoxaparin as the only agent at full dose for platelet counts >50,000 per μl, at half dose for platelet counts of 25,000–50,000 per μl and withholding of the drug or combination with platelet transfusion for platelet counts <25,000 per μl (REF.240). Cost considerations are an important aspect because different insurance plans might cover one but not other anticoagulants.
Pulmonary hypertension update
A unique form of pulmonary hypertension was recognized on the basis of the clustering of nine cases in the French PH registry247. All but one patient had received dasatinib for >2 years, and all but two had received a dose of ≥100 mg per day. On right heart catheterization, the pulmonary capillary wedge pressure was normal in all patients and all but one patient did not respond to vasodilatory therapy. When dasatinib therapy was discontinued, pulmonary arterial pressures dropped but, as longer-term follow-up data (average 24 months) showed, one-third of patients still had elevated pulmonary arterial resistance and pressures. Mechanistically, experimental studies have shown that dasatinib therapy leads to endothelial injury and that structural rather than functional alterations of the pulmonary vasculature can evolve248. Whether endothelin-receptor antagonists should be the preferred treatment strategy for dasatinib-induced hypertension or whether general treatment guidelines (Supplementary Table 5) should be followed is unknown. Universal screening has not been endorsed, but patients developing dyspnoea while receiving cancer therapy or signs and symptoms of right heart failure need to be evaluated by echocardiography and, if confirmed, by right heart catheterization249,250. Further stratification for management depends on test results and symptom status. An important update is the recommendation of the 6th World Symposium on Pulmonary Hypertension to decrease the cut-off level for pulmonary arterial hypertension from a resting mean pulmonary arterial pressure (PAP) of 25 mmHg to 20 mmHg (REF.251). The argument in favour is that this definition is no longer arbitrary but instead represents the 97.5 percentile of the upper limit of normal, that is, 2 standard deviations above the mean PAP in normal individuals. Considering an increase in cardiac output or pulmonary arterial wedge pressure, the task force also proposed to include a pulmonary vascular resistance of ≥3 Wood units in the definition of all forms of precapillary pulmonary hypertension associated with a mean PAP >20 mmHg. Other cancer therapies that can lead to pulmonary hypertension encompass those that induce pulmonary toxicity and fibrosis, including bleomycin, TKIs targeting the epidermal growth factor receptor and AKT, and radiation therapy; these patients require proper pulmonary follow-up.
Future directions
Studies and registries are needed to answer several of the questions raised in this Review. A more precise estimate of the incidence of the various arterial toxicity events with cancer therapies is needed. Risk factors and risk groups, if possible, need to be defined. These definitions would facilitate screening efforts before, during and after therapy. The specific modes and frequencies of these screenings also need to be defined along with which parameters at which cut-off levels should trigger a (clinical) response. Primary and secondary prevention efforts should follow the pathophysiology involved, and appropriate management of vascular disease should enable the best-possible cancer therapy to continue and be completed. Finally, novel insight into the pathophysiology of vascular disease and the vascular nature of cardiotoxicity might also be gained252.
Conclusions
Arterial vascular toxic effects of cancer therapies can present as acute vasospasm, acute thrombosis and accelerated atherosclerosis. The management of cancer therapy-related vascular toxicities is directed towards the underlying pathological mechanism and, therefore, defining the mechanisms is a central element. The best modes of pre-therapy screening, surveillance and prevention are yet to be defined. Well-conducted research is needed to define more precisely the risk, risk factors and risk management of the vascular toxic effects of cancer therapies. Future studies are also needed to provide more insight into the pathophysiology of vascular disease and the vascular nature of cardiotoxicity with cancer therapies.
Supplementary Material
Key points.
Vascular toxic effects of cancer therapies include arterial and venous events and affect the systemic and pulmonary circulations.
Cancer therapy-related arterial toxicities can present as acute vasospasm, acute thrombosis and accelerated atherosclerosis.
The management of cancer therapy-related vascular toxicities is directed towards the underlying pathological mechanism; therefore, defining the underlying mechanism is a central element.
The best modes of pre-therapy screening, surveillance and prevention are yet to be defined.
Clinical studies and registries are needed to define more precisely the risk, risk factors and risk management of the vascular toxic effects of cancer therapies.
Experimental studies should provide insight into the pathophysiological mechanisms of cardiovascular toxic effects of cancer therapies, which might also lead to an improved understanding of cardiovascular diseases in general.
Acknowledgements
The author received and receives support from the NIH (HL116952 and CA233610) and the Miami Heart Research Institute/Florida Heart Research Foundation.
Footnotes
Competing interests
The author declares no competing interests.
Supplementary information
Supplementary information is available for this paper at https://doi.org/10.1038/s41569-020-0347-2.
References
- 1.Herrmann J Adverse cardiac effects of cancer therapies: cardiotoxicity and arrhythmia. Nat. Rev. Cardiol 10.1038/s41569-020-0348-1 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kleiman NS, Lehane DE, Geyer CE Jr., Pratt CM & Young JB Prinzmetal’s angina during 5-fluorouracil chemotherapy. Am. J. Med 82, 566–568 (1987). [DOI] [PubMed] [Google Scholar]
- 3.Collins C & Weiden PL Cardiotoxicity of 5-fluorouracil. Cancer Treat. Rep 71, 733–736 (1987). [PubMed] [Google Scholar]
- 4.Schnetzler B, Popova N, Collao Lamb C & Sappino AP Coronary spasm induced by capecitabine. Ann. Oncol 12, 723–724 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Mosseri M, Fingert HJ, Varticovski L, Chokshi S & Isner JM In vitro evidence that myocardial ischemia resulting from 5-fluorouracil chemotherapy is due to protein kinase C-mediated vasoconstriction of vascular smooth muscle. Cancer Res. 53, 3028–3033 (1993). [PubMed] [Google Scholar]
- 6.Lanza GA, Careri G & Crea F Mechanisms of coronary artery spasm. Circulation 124, 1774–1782 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Cwikiel M, Eskilsson J, Wieslander JB, Stjernquist U & Albertsson M The appearance of endothelium in small arteries after treatment with 5-fluorouracil. An electron microscopic study of late effects in rabbits. Scanning Microsc. 10, 805–818; discussion 819 (1996). [PubMed] [Google Scholar]
- 8.Herrmann J & Lerman A The endothelium: dysfunction and beyond. J. Nucl. Cardiol 8, 197–206 (2001). [DOI] [PubMed] [Google Scholar]
- 9.Boyle JJ et al. Expression of angiogenic factor thymidine phosphorylase and angiogenesis in human atherosclerosis. J. Pathol 192, 234–242 (2000). [DOI] [PubMed] [Google Scholar]
- 10.Meyer CC, Calis KA, Burke LB, Walawander CA & Grasela TH Symptomatic cardiotoxicity associated with 5-fluorouracil. Pharmacotherapy 17, 729–736 (1997). [PubMed] [Google Scholar]
- 11.Saif MW, Shah MM & Shah AR Fluoropyrimidine-associated cardiotoxicity: revisited. Expert Opin. Drug Saf 8, 191–202 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Sara JD et al. 5-fluorouracil and cardiotoxicity: a review. Ther. Adv. Med. Oncol 10, 1758835918780140 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gemici G, Cincin A, Degertekin M & Oktay A Paclitaxel-induced ST-segment elevations. Clin. Cardiol 32, E94–E96 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nguyen-Ho P, Kleiman NS & Verani MS Acute myocardial infarction and cardiac arrest in a patient receiving paclitaxel. Can. J. Cardiol 19, 300–302 (2003). [PubMed] [Google Scholar]
- 15.Arbuck SG et al. A reassessment of cardiac toxicity associated with Taxol. J. Natl. Cancer Inst. Monogr 15, 117–130 (1993). [PubMed] [Google Scholar]
- 16.Shiroto T et al. Role of Rho-kinase in the pathogenesis of coronary hyperconstricting responses induced by drug-eluting stents in pigs in vivo. J. Am. Coll. Cardiol 54, 2321–2329 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Vassilakopoulou M et al. Paclitaxel chemotherapy and vascular toxicity as assessed by flow-mediated and nitrate-mediated vasodilatation. Vasc. Pharmacol 53, 115–121 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Doll DC et al. Acute vascular ischemic events after cisplatin-based combination chemotherapy for germ-cell tumors of the testis. Ann. Intern. Med 105, 48–51 (1986). [DOI] [PubMed] [Google Scholar]
- 19.Taniguchi T, Nakamura T & Sawada T Arterial stiffness, endothelial dysfunction and recurrent angina post-chemotherapy. QJM 108, 653–655 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Stefenelli T, Kuzmits R, Ulrich W & Glogar D Acute vascular toxicity after combination chemotherapy with cisplatin, vinblastine, and bleomycin for testicular cancer. Eur. Heart J 9, 552–556 (1988). [DOI] [PubMed] [Google Scholar]
- 21.Sekijima T et al. Impact of platinum-based chemotherapy on the progression of atherosclerosis. Climacteric 14, 31–40 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Thijs AM et al. Role of endogenous vascular endothelial growth factor in endothelium-dependent vasodilation in humans. Hypertension 61, 1060–1065 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Thijs AM et al. Impaired endothelium-dependent vasodilation does not initiate the development of sunitinib-associated hypertension. J. Hypertens 33, 2075–2082 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Touyz RM, Herrmann SMS & Herrmann J Vascular toxicities with VEGF inhibitor therapies-focus on hypertension and arterial thrombotic events. J. Am. Soc. Hypertens 12, 409–425 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kappers MH et al. Sunitinib-induced systemic vasoconstriction in swine is endothelin mediated and does not involve nitric oxide or oxidative stress. Hypertension 59, 151–157 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Kappers MH et al. Hypertension induced by the tyrosine kinase inhibitor sunitinib is associated with increased circulating endothelin-1 levels. Hypertension 56, 675–681 (2010). [DOI] [PubMed] [Google Scholar]
- 27.Sen F et al. Impaired coronary flow reserve in metastatic cancer patients treated with sunitinib. J. BUON 18, 775–781 (2013). [PubMed] [Google Scholar]
- 28.Reimann M et al. Anti-vascular endothelial growth factor therapy impairs endothelial function of retinal microcirculation in colon cancer patients – an observational study. Exp. Transl. Stroke Med 5, 7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katoh M et al. Bevacizumab-related microvascular angina and its management with Nicorandil. Int. Heart J 58, 803–805 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Arima Y et al. Sorafenib-induced acute myocardial infarction due to coronary artery spasm. J. Cardiol 54, 512–515 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Naib T, Steingart RM & Chen CL Sorafenib-associated multivessel coronary artery vasospasm. Herz 36, 348–351 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Hingorani AD et al. Acute systemic inflammation impairs endothelium-dependent dilatation in humans. Circulation 102, 994–999 (2000). [DOI] [PubMed] [Google Scholar]
- 33.Pleiner J et al. Simvastatin prevents vascular hyporeactivity during inflammation. Circulation 110, 3349–3354 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Levy B et al. Vascular hyporesponsiveness to vasopressors in septic shock: from bench to bedside. Intensive Care Med. 36, 2019–2029 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Miller DD, Waters DD, Dangoisse V & David PR Symptomatic coronary artery spasm following radiotherapy for Hodgkin’s disease. Chest 83, 284–285 (1983). [DOI] [PubMed] [Google Scholar]
- 36.Yahalom J, Hasin Y & Fuks Z Acute myocardial infarction with normal coronary arteriogram after mantle field radiation therapy for Hodgkin’s disease. Cancer 52, 637–641 (1983). [DOI] [PubMed] [Google Scholar]
- 37.Egashira S et al. Mechanisms of ergonovine-induced hyperconstriction of coronary artery after x-ray irradiation in pigs. Basic Res. Cardiol 90, 167–175 (1995). [DOI] [PubMed] [Google Scholar]
- 38.Soloviev AI et al. Mechanisms of endothelial dysfunction after ionized radiation: selective impairment of the nitric oxide component of endothelium-dependent vasodilation. Br. J. Pharmacol 138, 837–844 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sugihara T et al. Preferential impairment of nitric oxide-mediated endothelium-dependent relaxation in human cervical arteries after irradiation. Circulation 100, 635–641 (1999). [DOI] [PubMed] [Google Scholar]
- 40.Beckman JA, Thakore A, Kalinowski BH, Harris JR & Creager MA Radiation therapy impairs endothelium-dependent vasodilation in humans. J. Am. Coll. Cardiol 37, 761–765 (2001). [DOI] [PubMed] [Google Scholar]
- 41.Levesque L et al. Effects of radiation therapy on vascular responsiveness. J. Cardiovasc. Pharmacol 37, 381–393 (2001). [DOI] [PubMed] [Google Scholar]
- 42.Centola M et al. A rare case of large intracoronary thrombosis in advanced breast cancer patient treated with epirubicin and cisplatin. J. Cardiovasc. Med 17 (Suppl. 2), e241–e243 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Ito D et al. Primary percutaneous coronary intervention and intravascular ultrasound imaging for coronary thrombosis after cisplatin-based chemotherapy. Heart Vessels 27, 634–638 (2012). [DOI] [PubMed] [Google Scholar]
- 44.Jafri M & Protheroe A Cisplatin-associated thrombosis. Anticancer Drugs 19, 927–929 (2008). [DOI] [PubMed] [Google Scholar]
- 45.Karabay KO, Yildiz O & Aytekin V Multiple coronary thrombi with cisplatin. J. Invasive Cardiol 26, E18–E20 (2014). [PubMed] [Google Scholar]
- 46.Karavelioglu Y, Ekicibasi E, Tanalp AC, Karapinar H & Aung SM Worm-like thrombus in left main coronary artery after cytostatic treatment. Blood Coagul. Fibrinolysis 21, 491–493 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Morlese JF, Jeswani T, Beal I, Wylie P & Bell J Acute ventricular and aortic thrombosis post chemotherapy. Br. J. Radiol 80, e75–e77 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Pretnar-Oblak J, Zaletel M, Jagodic M & Zaletel M Thrombosis of internal carotid artery after cisplatin-based chemotherapy. Eur. Neurol 57, 109–110 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Rishi A & Ghoshal S Acute multiple arterial thrombosis after cisplatin in base of tongue carcinoma: case report. Head Neck 35, E269–E271 (2013). [DOI] [PubMed] [Google Scholar]
- 50.Kawano N et al. Two cases of acute myocardial infarction during combined chemotherapy in young patients with testicular cancer. J. Cardiol. Cases 7, e176–e180 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ozkan TA, Aydin U, Ay D & Cebeci IO Cisplatin and bleomycin-induced acute peripheral-vascular stenosis in patient with testicular cancer. Urol. Ann 8, 483–485 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moore RA et al. High incidence of thromboembolic events in patients treated with cisplatin-based chemotherapy: a large retrospective analysis. J. Clin. Oncol 29, 3466–3473 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Crea F & Libby P Acute coronary syndromes: the way forward from mechanisms to precision treatment. Circulation 136, 1155–1166 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Partida RA, Libby P, Crea F & Jang IK Plaque erosion: a new in vivo diagnosis and a potential major shift in the management of patients with acute coronary syndromes. Eur. Heart J 39, 2070–2076 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Quillard T et al. TLR2 and neutrophils potentiate endothelial stress, apoptosis and detachment: implications for superficial erosion. Eur. Heart J 36, 1394–1404 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Libby P, Pasterkamp G, Crea F & Jang IK Reassessing the mechanisms of acute coronary syndromes. Circ. Res 124, 150–160 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Folco EJ et al. Neutrophil extracellular traps induce endothelial cell activation and tissue factor production through interleukin-1α and cathepsin G. Arterioscler. Thromb. Vasc. Biol 38, 1901–1912 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Franck G et al. Flow perturbation mediates neutrophil recruitment and potentiates endothelial injury via TLR2 in mice: implications for superficial erosion. Circ. Res 121, 31–42 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Libby P & Croce K Visualizing the vascular glue “shearing” the barriers to atherosclerotic plaque imaging. JACC Cardiovasc. Imaging 3, 956–958 (2010). [DOI] [PubMed] [Google Scholar]
- 60.Hagensen MK et al. Circulating endothelial progenitor cells do not contribute to regeneration of endothelium after murine arterial injury. Cardiovasc. Res 93, 223–231 (2012). [DOI] [PubMed] [Google Scholar]
- 61.Padfield GJ, Newby DE & Mills NL Understanding the role of endothelial progenitor cells in percutaneous coronary intervention. J. Am. Coll. Cardiol 55, 1553–1565 (2010). [DOI] [PubMed] [Google Scholar]
- 62.Douglas G et al. Endothelial cell repopulation after stenting determines in-stent neointima formation: effects of bare-metal vs. drug-eluting stents and genetic endothelial cell modification. Eur. Heart J 34, 3378–3388 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ramcharan KS, Lip GY, Stonelake PS & Blann AD Effect of standard chemotherapy and antiangiogenic therapy on plasma markers and endothelial cells in colorectal cancer. Br. J. Cancer 111, 1742–1749 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Oren O & Herrmann J Arterial events in cancer patients-the case of acute coronary thrombosis. J. Thorac. Dis 10, S4367–S4385 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bauer AT et al. von Willebrand factor fibers promote cancer-associated platelet aggregation in malignant melanoma of mice and humans. Blood 125, 3153–3163 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang AJ et al. Cancer cell-derived von Willebrand factor enhanced metastasis of gastric adenocarcinoma. Oncogenesis 7, 12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Navi BB et al. Risk of arterial thromboembolism in patients with cancer. J. Am. Coll. Cardiol 70, 926–938 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Timp JF, Braekkan SK, Versteeg HH & Cannegieter SC Epidemiology of cancer-associated venous thrombosis. Blood 122, 1712–1723 (2013). [DOI] [PubMed] [Google Scholar]
- 69.Chew HK, Wun T, Harvey D, Zhou H & White RH Incidence of venous thromboembolism and its effect on survival among patients with common cancers. Arch. Intern. Med 166, 458–464 (2006). [DOI] [PubMed] [Google Scholar]
- 70.Blann AD & Dunmore S Arterial and venous thrombosis in cancer patients. Cardiol. Res. Pract 2011, 394740 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dieckmann KP, Struss WJ & Budde U Evidence for acute vascular toxicity of cisplatin-based chemotherapy in patients with germ cell tumour. Anticancer Res. 31, 4501–4505 (2011). [PubMed] [Google Scholar]
- 72.Patel JN et al. Bevacizumab and the risk of arterial and venous thromboembolism in patients with metastatic, castration-resistant prostate cancer treated on Cancer and Leukemia Group B (CALGB) 90401 (Alliance). Cancer 121, 1025–1031 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Suzuki K, Yanagimura T, Ohashi K, Kagamu H & Takada T Bevacizumab-induced aortic arterial thrombosis. Intern. Med 57, 2987–2990 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lamba G, Deol R, Shah D, Sahni R & Malhotra BK Sunitinib and thrombosis. J. Gastrointest. Cancer 43 (Suppl. 1), S128–S130 (2012). [DOI] [PubMed] [Google Scholar]
- 75.Grieco A, Lombardo A & Biolato M Ventricular thrombosis during sorafenib therapy for advanced hepatocellular carcinoma. Eur. J. Gastroenterol. Hepatol 25, 1001–1002 (2013). [DOI] [PubMed] [Google Scholar]
- 76.Meyer T et al. Bevacizumab immune complexes activate platelets and induce thrombosis in FCGR2A transgenic mice. J. Thromb. Haemost 7, 171–181 (2009). [DOI] [PubMed] [Google Scholar]
- 77.Chen N et al. Bevacizumab promotes venous thromboembolism through the induction of PAI-1 in a mouse xenograft model of human lung carcinoma. Mol. Cancer 14, 140 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kilickap S, Abali H & Celik I Bevacizumab, bleeding, thrombosis, and warfarin. J. Clin. Oncol 21, 3542 (2003). [DOI] [PubMed] [Google Scholar]
- 79.Sabrkhany S et al. Sunitinib uptake inhibits platelet function in cancer patients. Eur. J. Cancer 66, 47–54 (2016). [DOI] [PubMed] [Google Scholar]
- 80.Walraven M et al. Platelet function is disturbed by the angiogenesis inhibitors sunitinib and sorafenib, but unaffected by bevacizumab. Angiogenesis 21, 325–334 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Herrmann J Tyrosine kinase inhibitors and vascular toxicity: impetus for a classification system? Curr. Oncol. Rep 18, 33 (2016). [DOI] [PubMed] [Google Scholar]
- 82.Gratacap MP et al. The new tyrosine-kinase inhibitor and anticancer drug dasatinib reversibly affects platelet activation in vitro and in vivo. Blood 114, 1884–1892 (2009). [DOI] [PubMed] [Google Scholar]
- 83.Quintas-Cardama A, Han X, Kantarjian H & Cortes J Tyrosine kinase inhibitor-induced platelet dysfunction in patients with chronic myeloid leukemia. Blood 114, 261–263 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Alhawiti N et al. The tyrosine kinase inhibitor, nilotinib potentiates a prothrombotic state. Thromb. Res 145, 54–64 (2016). [DOI] [PubMed] [Google Scholar]
- 85.Loren CP et al. The BCR-ABL inhibitor ponatinib inhibits platelet immunoreceptor tyrosine-based activation motif (ITAM) signaling, platelet activation and aggregate formation under shear. Thromb. Res 135, 155–160 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Neelakantan P et al. Platelet dysfunction associated with ponatinib, a new pan BCR-ABL inhibitor with efficacy for chronic myeloid leukemia resistant to multiple tyrosine kinase inhibitor therapy. Haematologica 97, 1444 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tsukamoto J et al. Thromboembolic events related to treatment with checkpoint inhibitors: report of two cases. Case Rep. Oncol 11, 648–653 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kunimasa K et al. Pembrolizumab-induced acute thrombosis: a case report. Medicine 97, e10772 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Boutros C et al. Arterial thrombosis and anti-PD-1 blockade. Eur. J. Cancer 91, 164–166 (2018). [DOI] [PubMed] [Google Scholar]
- 90.Lipinska-Gediga M Coagulopathy in sepsis – a new look at an old problem. Anaesthesiol. Intensive Ther 48, 352–359 (2016). [DOI] [PubMed] [Google Scholar]
- 91.Budnik I & Brill A Immune factors in deep vein thrombosis initiation. Trends Immunol. 39, 610–623 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Branchford BR & Carpenter SL The role of inflammation in venous thromboembolism. Front. Pediatr 6, 142 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bockmeyer CL et al. Inflammation-associated ADAMTS13 deficiency promotes formation of ultra-large von Willebrand factor. Haematologica 93, 137–140 (2008). [DOI] [PubMed] [Google Scholar]
- 94.Higgins JN, Wlodarczyk Z, Platts AD & Hamilton G Radiation-induced acute femoral artery thrombosis treated by thrombolysis. Br. J. Surg 79, 909–910 (1992). [DOI] [PubMed] [Google Scholar]
- 95.Reed R & Sadiq S Acute carotid artery thrombosis after neck irradiation. J. Ultrasound Med 13, 641–644 (1994). [DOI] [PubMed] [Google Scholar]
- 96.Fonkalsrud EW, Sanchez M, Zerubavel R & Mahoney A Serial changes in arterial structure following radiation therapy. Surg. Gynecol. Obstet 145, 395–400 (1977). [PubMed] [Google Scholar]
- 97.Schmidt-Lucke C et al. Reduced number of circulating endothelial progenitor cells predicts future cardiovascular events: proof of concept for the clinical importance of endogenous vascular repair. Circulation 111, 2981–2987 (2005). [DOI] [PubMed] [Google Scholar]
- 98.Vasa M et al. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ. Res 89, E1–7 (2001). [DOI] [PubMed] [Google Scholar]
- 99.Iliescu CA et al. SCAI Expert Consensus Statement: evaluation, management, and special considerations of cardio-oncology patients in the cardiac catheterization laboratory (endorsed by the Cardiological Society of India, and Sociedad Latino Americana de Cardiologia Intervencionista). Catheter. Cardiovasc. Interv 87, E202–E223 (2016). [DOI] [PubMed] [Google Scholar]
- 100.Neumann FJ et al. 2018 ESC/EACTS Guidelines on myocardial revascularization. Eur. Heart J 40, 87–165 (2019). [DOI] [PubMed] [Google Scholar]
- 101.Gogas BD, McDaniel M, Samady H & King SB III. Novel drug-eluting stents for coronary revascularization. Trends Cardiovasc. Med 24, 305–313 (2014). [DOI] [PubMed] [Google Scholar]
- 102.Urban P et al. Polymer-free drug-coated coronary stents in patients at high bleeding risk. N. Engl. J. Med 373, 2038–2047 (2015). [DOI] [PubMed] [Google Scholar]
- 103.Palmerini T et al. Stent thrombosis with drug-eluting and bare-metal stents: evidence from a comprehensive network meta-analysis. Lancet 379, 1393–1402 (2012). [DOI] [PubMed] [Google Scholar]
- 104.Smith SC, Winters KJ & Lasala JM Stent thrombosis in a patient receiving chemotherapy. Cathet. Cardiovasc. Diagn 40, 383–386 (1997). [DOI] [PubMed] [Google Scholar]
- 105.Yeh RW et al. Development and validation of a prediction rule for benefit and harm of dual antiplatelet therapy beyond 1 year after percutaneous coronary intervention. JAMA 315, 1735–1749 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Costa F et al. Derivation and validation of the predicting bleeding complications in patients undergoing stent implantation and subsequent dual antiplatelet therapy (PRECISE-DAPT) score: a pooled analysis of individual-patient datasets from clinical trials. Lancet 389, 1025–1034 (2017). [DOI] [PubMed] [Google Scholar]
- 107.Gori T et al. Predictors of stent thrombosis and their implications for clinical practice. Nat. Rev. Cardiol 16, 243–256 (2019). [DOI] [PubMed] [Google Scholar]
- 108.Iliescu C et al. SCAI Expert Consensus Statement: evaluation, management, and special considerations of cardio-oncology patients in the cardiac catheterization laboratory (endorsed by the Cardiological Society of India, and Sociedad Latino Americana de Cardiologia Intervencionista). Catheter. Cardiovasc. Interv 87, 895–899 (2016). [DOI] [PubMed] [Google Scholar]
- 109.Thygesen K et al. Fourth universal definition of myocardial infarction (2018). J. Am. Coll. Cardiol 72, 2231–2264 (2018). [DOI] [PubMed] [Google Scholar]
- 110.Park JY et al. Acute coronary syndromes in patients with active hematologic malignancies - Incidence, management, and outcomes. Int. J. Cardiol 275, 6–12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.McCarthy CP, Vaduganathan M & Januzzi JL Jr. Type 2 myocardial infarction-diagnosis, prognosis, and treatment. JAMA 320, 433–434 (2018). [DOI] [PubMed] [Google Scholar]
- 112.Scappaticci FA et al. Arterial thromboembolic events in patients with metastatic carcinoma treated with chemotherapy and bevacizumab. J. Natl Cancer Inst 99, 1232–1239 (2007). [DOI] [PubMed] [Google Scholar]
- 113.Lee S et al. Autocrine VEGF signaling is required for vascular homeostasis. Cell 130, 691–703 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lubberts S et al. Vascular fingerprint and vascular damage markers associated with vascular events in testicular cancer patients during and after chemotherapy. Eur. J. Cancer 63, 180–188 (2016). [DOI] [PubMed] [Google Scholar]
- 115.Soultati A et al. Endothelial vascular toxicity from chemotherapeutic agents: preclinical evidence and clinical implications. Cancer Treat. Rev 38, 473–483 (2012). [DOI] [PubMed] [Google Scholar]
- 116.Jang WJ, Choi DY & Jeon IS Vascular endothelial dysfunction after anthracyclines treatment in children with acute lymphoblastic leukemia. Korean J. Pediatr 56, 130–134 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Drafts BC et al. Low to moderate dose anthracycline-based chemotherapy is associated with early noninvasive imaging evidence of subclinical cardiovascular disease. JACC Cardiovasc. Imaging 6, 877–885 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Vaughn DJ, Palmer SC, Carver JR, Jacobs LA & Mohler ER Cardiovascular risk in long-term survivors of testicular cancer. Cancer 112, 1949–1953 (2008). [DOI] [PubMed] [Google Scholar]
- 119.Gietema JA et al. Circulating plasma platinum more than 10 years after cisplatin treatment for testicular cancer. Lancet 355, 1075–1076 (2000). [DOI] [PubMed] [Google Scholar]
- 120.Meinardi MT et al. Cardiovascular morbidity in long-term survivors of metastatic testicular cancer. J. Clin. Oncol 18, 1725–1732 (2000). [DOI] [PubMed] [Google Scholar]
- 121.van den Belt-Dusebout AW et al. Long-term risk of cardiovascular disease in 5-year survivors of testicular cancer. J. Clin. Oncol 24, 467–475 (2006). [DOI] [PubMed] [Google Scholar]
- 122.van den Belt-Dusebout AW et al. Treatment-specific risks of second malignancies and cardiovascular disease in 5-year survivors of testicular cancer. J. Clin. Oncol 25, 4370–4378 (2007). [DOI] [PubMed] [Google Scholar]
- 123.Haugnes HS et al. Cardiovascular risk factors and morbidity in long-term survivors of testicular cancer: a 20-year follow-up study. J. Clin. Oncol 28, 4649–4657 (2010). [DOI] [PubMed] [Google Scholar]
- 124.Huddart RA et al. Cardiovascular disease as a long-term complication of treatment for testicular cancer. J. Clin. Oncol 21, 1513–1523 (2003). [DOI] [PubMed] [Google Scholar]
- 125.Gugic J, Zaletel LZ & Oblak I Treatment-related cardiovascular toxicity in long-term survivors of testicular cancer. Radiol. Oncol 51, 221–227 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Haugnes HS, Oldenburg J & Bremnes RM Pulmonary and cardiovascular toxicity in long-term testicular cancer survivors. Urol. Oncol 33, 399–406 (2015). [DOI] [PubMed] [Google Scholar]
- 127.Bokemeyer C, Berger CC, Kuczyk MA & Schmoll HJ Evaluation of long-term toxicity after chemotherapy for testicular cancer. J. Clin. Oncol 14, 2923–2932 (1996). [DOI] [PubMed] [Google Scholar]
- 128.Ishioka J et al. Cardiovascular events in survivors of high-dose chemotherapy for germ cell tumors. Int. J. Urol 15, 642–645 (2008). [DOI] [PubMed] [Google Scholar]
- 129.Haugnes HS et al. Predicted cardiovascular mortality and reported cardiovascular morbidity in testicular cancer survivors. J. Cancer Surviv 2, 128–137 (2008). [DOI] [PubMed] [Google Scholar]
- 130.Gietema JA et al. Long-term follow-up of cardiovascular risk factors in patients given chemotherapy for disseminated nonseminomatous testicular cancer. Ann. Intern. Med 116, 709–715 (1992). [DOI] [PubMed] [Google Scholar]
- 131.Koc G, Divrik TR, Unlu N, Bulut V & Zorlu F Does cisplatin-based chemotherapy effect on blood lipid levels of patients with germ cell testicular tumor in long-term follow-up? Int. Urol. Nephrol 43, 1095–1100 (2011). [DOI] [PubMed] [Google Scholar]
- 132.Levine GN et al. Androgen-deprivation therapy in prostate cancer and cardiovascular risk: a science advisory from the American Heart Association, American Cancer Society, and American Urological Association: endorsed by the American Society for Radiation Oncology. Circulation 121, 833–840 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bosco C et al. Quantifying observational evidence for risk of fatal and nonfatal cardiovascular disease following androgen deprivation therapy for prostate cancer: a meta-analysis. Eur. Urol 68, 386–396 (2015). [DOI] [PubMed] [Google Scholar]
- 134.Keating NL Type of androgen deprivation therapy and risk of cardiovascular disease. Eur. Urol 72, 929–930 (2017). [DOI] [PubMed] [Google Scholar]
- 135.Winnik S et al. Systemic VEGF inhibition accelerates experimental atherosclerosis and disrupts endothelial homeostasis – implications for cardiovascular safety. Int. J. Cardiol 168, 2453–2461 (2013). [DOI] [PubMed] [Google Scholar]
- 136.Herrmann J, Lerman LO, Mukhopadhyay D, Napoli C & Lerman A Angiogenesis in atherogenesis. Arterioscler. Thromb. Vasc. Biol 26, 1948–1957 (2006). [DOI] [PubMed] [Google Scholar]
- 137.Celletti FL et al. Vascular endothelial growth factor enhances atherosclerotic plaque progression. Nat. Med 7, 425–429 (2001). [DOI] [PubMed] [Google Scholar]
- 138.Zhao Q et al. Vascular endothelial growth factor is necessary in the development of arteriosclerosis by recruiting/activating monocytes in a rat model of long-term inhibition of nitric oxide synthesis. Circulation 105, 1110–1115 (2002). [DOI] [PubMed] [Google Scholar]
- 139.Lemstrom KB et al. Vascular endothelial growth factor enhances cardiac allograft arteriosclerosis. Circulation 105, 2524–2530 (2002). [DOI] [PubMed] [Google Scholar]
- 140.Moulton KS et al. Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Circulation 99, 1726–1732 (1999). [DOI] [PubMed] [Google Scholar]
- 141.Moulton KS et al. Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proc. Natl Acad. Sci. USA 100, 4736–4741 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Aichberger KJ et al. Progressive peripheral arterial occlusive disease and other vascular events during nilotinib therapy in CML. Am. J. Hematol 86, 533–539 (2011). [DOI] [PubMed] [Google Scholar]
- 143.Le Coutre P et al. Severe peripheral arterial disease during nilotinib therapy. J. Natl Cancer Inst 103, 1347–1348 (2011). [DOI] [PubMed] [Google Scholar]
- 144.Tefferi A & Letendre L Nilotinib treatment-associated peripheral artery disease and sudden death: yet another reason to stick to imatinib as front-line therapy for chronic myelogenous leukemia. Am. J. Hematol 86, 610–611 (2011). [DOI] [PubMed] [Google Scholar]
- 145.Breccia M, Efficace F & Alimena G Progressive arterial occlusive disease (PAOD) and pulmonary arterial hypertension (PAH) as new adverse events of second generation TKIs in CML treatment: who’s afraid of the big bad wolf? Leuk. Res 36, 813–814 (2012). [DOI] [PubMed] [Google Scholar]
- 146.Giles FJ et al. Rates of peripheral arterial occlusive disease in patients with chronic myeloid leukemia in the chronic phase treated with imatinib, nilotinib, or non-tyrosine kinase therapy: a retrospective cohort analysis. Leukemia 27, 1310–1315 (2013). [DOI] [PubMed] [Google Scholar]
- 147.Kim TD et al. Peripheral artery occlusive disease in chronic phase chronic myeloid leukemia patients treated with nilotinib or imatinib. Leukemia 27, 1316–1321 (2013). [DOI] [PubMed] [Google Scholar]
- 148.Levato L et al. Progressive peripheral arterial occlusive disease and other vascular events during nilotinib therapy in chronic myeloid leukemia: a single institution study. Eur. J. Haematol 90, 531–532 (2013). [DOI] [PubMed] [Google Scholar]
- 149.Tefferi A Nilotinib treatment-associated accelerated atherosclerosis: when is the risk justified? Leukemia 27, 1939–1940 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Maurizot A et al. Rapid clinical improvement of peripheral artery occlusive disease symptoms after nilotinib discontinuation despite persisting vascular occlusion. Blood Cancer J. 4, e247 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Breccia M, Molica M, Zacheo I, Serrao A & Alimena G Application of systematic coronary risk evaluation chart to identify chronic myeloid leukemia patients at risk of cardiovascular diseases during nilotinib treatment. Ann. Hematol 94, 393–397 (2015). [DOI] [PubMed] [Google Scholar]
- 152.Mirault T, Rea D, Azarine A & Messas E Rapid onset of peripheral artery disease in a chronic myeloid leukemia patient without prior arterial disorder: direct relationship with nilotinib exposure and clinical outcome. Eur. J. Haematol 94, 363–367 (2015). [DOI] [PubMed] [Google Scholar]
- 153.Valent P et al. Vascular safety issues in CML patients treated with BCR/ABL1 kinase inhibitors. Blood 125, 901–906 (2015). [DOI] [PubMed] [Google Scholar]
- 154.Herrmann J et al. Complicated and advanced atherosclerosis in a young woman with Philadelphia chromosome-positive acute lymphoblastic leukemia: success and challenges of BCR/ABL1-targeted cancer therapy. Mayo Clin. Proc 90, 1167–1168 (2015). [DOI] [PubMed] [Google Scholar]
- 155.Chai-Adisaksopha C, Lam W & Hillis C Major arterial events in patients with chronic myeloid leukemia treated with tyrosine kinase inhibitors: a meta-analysis. Leuk. Lymphoma 57, 1300–1310 (2016). [DOI] [PubMed] [Google Scholar]
- 156.Douxfils J et al. Association between BCR-ABL tyrosine kinase inhibitors for chronic myeloid leukemia and cardiovascular events, major molecular response, and overall survival: a systematic review and meta-analysis. JAMA Oncol. 2, 625–632 (2016). [DOI] [PubMed] [Google Scholar]
- 157.Dahlen T et al. Cardiovascular events associated with use of tyrosine kinase inhibitors in chronic myeloid leukemia: a population-based cohort study. Ann. Intern. Med 165, 161–166 (2016). [DOI] [PubMed] [Google Scholar]
- 158.Gover-Proaktor A et al. Bosutinib, dasatinib, imatinib, nilotinib, and ponatinib differentially affect the vascular molecular pathways and functionality of human endothelial cells. Leuk. Lymphoma 60, 189–199 (2019). [DOI] [PubMed] [Google Scholar]
- 159.Hadzijusufovic E et al. Nilotinib-induced vasculopathy: identification of vascular endothelial cells as a primary target site. Leukemia 31, 2388–2397 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Gover-Proaktor A et al. Ponatinib reduces viability, migration, and functionality of human endothelial cells. Leuk. Lymphoma 58, 1455–1467 (2017). [DOI] [PubMed] [Google Scholar]
- 161.Pouwer MG et al. The BCR-ABL1 inhibitors imatinib and ponatinib decrease plasma cholesterol and atherosclerosis, and nilotinib and ponatinib activate coagulation in a translational mouse model. Front. Cardiovasc. Med 5, 55 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Bocchia M et al. Genetic predisposition and induced pro-inflammatory/pro-oxidative status may play a role in increased atherothrombotic events in nilotinib treated chronic myeloid leukemia patients. Oncotarget 7, 72311–72321 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Breccia M et al. Proposal for a tailored stratification at baseline and monitoring of cardiovascular effects during follow-up in chronic phase chronic myeloid leukemia patients treated with nilotinib frontline. Crit. Rev. Oncol. Hematol 107, 190–198 (2016). [DOI] [PubMed] [Google Scholar]
- 164.Aghel N, Lipton JH, Atenafu EG, Kim DDH & Delgado DH Cardiovascular events after exposure to nilotinib in chronic myeloid leukemia: long-term follow-up. Clin. Lymphoma Myeloma Leuk 17, 870–878.e1 (2017). [DOI] [PubMed] [Google Scholar]
- 165.Fujioka I et al. Features of vascular adverse events in Japanese patients with chronic myeloid leukemia treated with tyrosine kinase inhibitors: a retrospective study of the CML cooperative study group database. Ann. Hematol 97, 2081–2088 (2018). [DOI] [PubMed] [Google Scholar]
- 166.Fuster JJ & Walsh K Somatic mutations and clonal hematopoiesis: unexpected potential new drivers of age-related cardiovascular disease. Circ. Res 122, 523–532 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Natarajan P, Jaiswal S & Kathiresan S Clonal hematopoiesis: somatic mutations in blood cells and atherosclerosis. Circ. Genom. Precis. Med 11, e001926 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ebert BL & Libby P Clonal hematopoiesis confers predisposition to both cardiovascular disease and cancer: a newly recognized link between two major killers. Ann. Intern. Med 169, 116–117 (2018). [DOI] [PubMed] [Google Scholar]
- 169.Jaiswal S et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med 377, 111–121 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Cimmino L et al. Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell 170, 1079–1095.e20 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Fuster JJ et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 355, 842–847 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ridker PM et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med 377, 1119–1131 (2017). [DOI] [PubMed] [Google Scholar]
- 173.Ridker PM et al. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 390, 1833–1842 (2017). [DOI] [PubMed] [Google Scholar]
- 174.Brunner AM et al. Risk and timing of cardiovascular death among patients with myelodysplastic syndromes. Blood Adv. 1, 2032–2040 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Carobbio A et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an international study of 891 patients. Blood 117, 5857–5859 (2011). [DOI] [PubMed] [Google Scholar]
- 176.Cucuianu A et al. Arterial stenosis and atherothrombotic events in polycythemia vera and essential thrombocythemia. Rom. J. Intern. Med 44, 397–406 (2006). [PubMed] [Google Scholar]
- 177.Vianello F et al. Coronary microvascular dysfunction due to essential thrombocythemia and policythemia vera: the missing piece in the puzzle of their increased cardiovascular risk? Am. J. Hematol 90, 109–113 (2015). [DOI] [PubMed] [Google Scholar]
- 178.Maslah N, Cassinat B, Verger E, Kiladjian JJ & Velazquez L The role of LNK/SH2B3 genetic alterations in myeloproliferative neoplasms and other hematological disorders. Leukemia 31, 1661–1670 (2017). [DOI] [PubMed] [Google Scholar]
- 179.Wang W et al. LNK/SH2B3 loss of function promotes atherosclerosis and thrombosis. Circ. Res 119, e91–e103 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Swirski FK & Nahrendorf M Bone marrow takes center stage in cardiovascular disease. Circ. Res 119, 701–703 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hanoun M, Maryanovich M, Arnal-Estape A & Frenette PS Neural regulation of hematopoiesis, inflammation, and cancer. Neuron 86, 360–373 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Seijkens T et al. Hypercholesterolemia-induced priming of hematopoietic stem and progenitor cells aggravates atherosclerosis. FASEB J. 28, 2202–2213 (2014). [DOI] [PubMed] [Google Scholar]
- 183.Dutta P et al. Myocardial infarction accelerates atherosclerosis. Nature 487, 325–329 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Dutta P et al. Myocardial infarction activates CCR2+ hematopoietic stem and progenitor cells. Cell Stem Cell 16, 477–487 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Heidt T et al. Chronic variable stress activates hematopoietic stem cells. Nat. Med 20, 754–758 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Hoogeveen RM et al. Monocyte and haematopoietic progenitor reprogramming as common mechanism underlying chronic inflammatory and cardiovascular diseases. Eur. Heart J 39, 3521–3527 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.van der Valk FM et al. Increased haematopoietic activity in patients with atherosclerosis. Eur. Heart J 38, 425–432 (2017). [DOI] [PubMed] [Google Scholar]
- 188.Musher DM, Abers MS & Corrales-Medina VF Acute infection and myocardial infarction. N. Engl. J. Med 380, 171–176 (2019). [DOI] [PubMed] [Google Scholar]
- 189.Gotsman I & Lichtman AH Targeting interferon-γ to treat atherosclerosis. Circ. Res 101, 333–334 (2007). [DOI] [PubMed] [Google Scholar]
- 190.Akita K et al. An interleukin-6 receptor antibody suppresses atherosclerosis in atherogenic mice. Front. Cardiovasc. Med 4, 84 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Schuett H et al. Transsignaling of interleukin-6 crucially contributes to atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol 32, 281–290 (2012). [DOI] [PubMed] [Google Scholar]
- 192.Slaney CY, Wang P, Darcy PK & Kershaw MH CARs versus BiTEs: a comparison between T cell-redirection strategies for cancer treatment. Cancer Discov. 8, 924–934 (2018). [DOI] [PubMed] [Google Scholar]
- 193.Koga N et al. Blockade of the interaction between PD-1 and PD-L1 accelerates graft arterial disease in cardiac allografts. Arterioscler. Thromb. Vasc. Biol 24 2057–2062 (2004). [DOI] [PubMed] [Google Scholar]
- 194.Zhang H et al. Immunoinhibitory checkpoint deficiency in medium and large vessel vasculitis. Proc. Natl Acad. Sci. USA 114, E970–E979 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Weyand CM, Berry GJ & Goronzy JJ The immunoinhibitory PD-1/PD-L1 pathway in inflammatory blood vessel disease. J. Leukoc. Biol 103, 565–575 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Goldstein BL, Gedmintas L & Todd DJ Drug-associated polymyalgia rheumatica/giant cell arteritis occurring in two patients after treatment with ipilimumab, an antagonist of CTLA-4. Arthritis Rheumatol. 66, 768–769 (2014). [DOI] [PubMed] [Google Scholar]
- 197.Padda A et al. Ipilimumab induced digital vasculitis. J. Immunother. Cancer 6, 12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Kang A, Yuen M & Lee DJ Nivolumab-induced systemic vasculitis. JAAD Case Rep. 4, 606–608 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Laubli H et al. Cerebral vasculitis mimicking intracranial metastatic progression of lung cancer during PD-1 blockade. J. Immunother. Cancer 5, 46 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Loricera J et al. Subclinical aortitis after starting nivolumab in a patient with metastatic melanoma. A case of drug-associated aortitis? Clin. Exp. Rheumatol 36 (Suppl. 111), 171 (2018). [PubMed] [Google Scholar]
- 201.Daxini A, Cronin K & Sreih AG Vasculitis associated with immune checkpoint inhibitors-a systematic review. Clin. Rheumatol 37, 2579–2584 (2018). [DOI] [PubMed] [Google Scholar]
- 202.Castillo B, Gibbs J, Brohl AS & Seminario-Vidal L Checkpoint inhibitor-associated cutaneous small vessel vasculitis. JAAD Case Rep. 4, 675–677 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Aya F et al. Vasculitic neuropathy induced by pembrolizumab. Ann. Oncol 28, 433–434 (2017). [DOI] [PubMed] [Google Scholar]
- 204.Bu DX et al. Impairment of the programmed cell death-1 pathway increases atherosclerotic lesion development and inflammation. Arterioscler. Thromb. Vasc. Biol 31, 1100–1107 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Gotsman I et al. Proatherogenic immune responses are regulated by the PD-1/PD-L pathway in mice. J. Clin. Invest 117, 2974–2982 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Cochain C et al. Programmed cell death-1 deficiency exacerbates T cell activation and atherogenesis despite expansion of regulatory T cells in atherosclerosis-prone mice. PLOS ONE 9, e93280 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Hull MC, Morris CG, Pepine CJ & Mendenhall NP Valvular dysfunction and carotid, subclavian, and coronary artery disease in survivors of hodgkin lymphoma treated with radiation therapy. JAMA 290, 2831–2837 (2003). [DOI] [PubMed] [Google Scholar]
- 208.Patel DA et al. Clinical manifestations of noncoronary atherosclerotic vascular disease after moderate dose irradiation. Cancer 106, 718–725 (2006). [DOI] [PubMed] [Google Scholar]
- 209.Brosius FC 3rd, Waller BF & Roberts WC Radiation heart disease. Analysis of 16 young (aged 15 to 33 years) necropsy patients who received over 3,500 rads to the heart. Am. J. Med 70, 519–530 (1981). [DOI] [PubMed] [Google Scholar]
- 210.Virmani R, Farb A, Carter AJ & Jones RM Pathology of radiation-induced coronary artery disease in human and pig. Cardiovasc. Radiat. Med 1, 98–101 (1999). [DOI] [PubMed] [Google Scholar]
- 211.Veinot JP & Edwards WD Pathology of radiation-induced heart disease: a surgical and autopsy study of 27 cases. Hum. Pathol 27, 766–773 (1996). [DOI] [PubMed] [Google Scholar]
- 212.Schultz-Hector S & Trott KR Radiation-induced cardiovascular diseases: is the epidemiologic evidence compatible with the radiobiologic data? Int. J. Radiat. Oncol. Biol. Phys 67, 10–18 (2007). [DOI] [PubMed] [Google Scholar]
- 213.Halle M et al. Sustained inflammation due to nuclear factor-kappa B activation in irradiated human arteries. J. Am. Coll. Cardiol 55, 1227–1236 (2010). [DOI] [PubMed] [Google Scholar]
- 214.Christersdottir T et al. Prevention of radiotherapy-induced arterial inflammation by interleukin-1 blockade. Eur. Heart. J 40, 2495–2503 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Darby SC et al. Risk of ischemic heart disease in women after radiotherapy for breast cancer. N. Engl. J. Med 368, 987–998 (2013). [DOI] [PubMed] [Google Scholar]
- 216.Venkatesulu BP et al. Radiation-induced endothelial vascular injury: a review of possible mechanisms. JACC Basic Transl. Sci 3, 563–572 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Stewart FA, Seemann I, Hoving S & Russell NS Understanding radiation-induced cardiovascular damage and strategies for intervention. Clin. Oncol 25, 617–624 (2013). [DOI] [PubMed] [Google Scholar]
- 218.Stewart FA et al. Ionizing radiation accelerates the development of atherosclerotic lesions in ApoE−/− mice and predisposes to an inflammatory plaque phenotype prone to hemorrhage. Am. J. Pathol 168, 649–658 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Hoving S et al. Single-dose and fractionated irradiation promote initiation and progression of atherosclerosis and induce an inflammatory plaque phenotype in ApoE−/− mice. Int. J. Radiat. Oncol. Biol. Phys 71, 848–857 (2008). [DOI] [PubMed] [Google Scholar]
- 220.Hoving S et al. Anti-inflammatory and anti-thrombotic intervention strategies using atorvastatin, clopidogrel and knock-down of CD40L do not modify radiation-induced atherosclerosis in ApoE null mice. Radiother. Oncol 101, 100–108 (2011). [DOI] [PubMed] [Google Scholar]
- 221.Hoving S et al. NO-donating aspirin and aspirin partially inhibit age-related atherosclerosis but not radiation-induced atherosclerosis in ApoE null mice. PLOS ONE 5, e12874 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Hoving S et al. Irradiation induces different inflammatory and thrombotic responses in carotid arteries of wildtype C57BL/6J and atherosclerosis-prone ApoE−/− mice. Radiother. Oncol 105, 365–370 (2012). [DOI] [PubMed] [Google Scholar]
- 223.Hachamovitch R, Hayes SW, Friedman JD, Cohen I & Berman DS Comparison of the short-term survival benefit associated with revascularization compared with medical therapy in patients with no prior coronary artery disease undergoing stress myocardial perfusion single photon emission computed tomography. Circulation 107, 2900–2907 (2003). [DOI] [PubMed] [Google Scholar]
- 224.Jacobse JN et al. Radiation dose-response for risk of myocardial infarction in breast cancer survivors. Int. J. Radiat. Oncol. Biol. Phys 103, 595–604 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.James M et al. Ischaemic heart disease following conventional and hypofractionated radiation treatment in a contemporary breast cancer series. J. Med. Imaging Radiat. Oncol 62, 425–431 (2018). [DOI] [PubMed] [Google Scholar]
- 226.Jagsi R, Griffith KA, Koelling T, Roberts R & Pierce LJ Rates of myocardial infarction and coronary artery disease and risk factors in patients treated with radiation therapy for early-stage breast cancer. Cancer 109, 650–657 (2007). [DOI] [PubMed] [Google Scholar]
- 227.Constine LS, Schwartz RG, Savage DE, King V & Muhs A Cardiac function, perfusion, and morbidity in irradiated long-term survivors of Hodgkin’s disease. Int. J. Radiat. Oncol. Biol. Phys 39, 897–906 (1997). [DOI] [PubMed] [Google Scholar]
- 228.Glanzmann C, Kaufmann P, Jenni R, Hess OM & Huguenin P Cardiac risk after mediastinal irradiation for Hodgkin’s disease. Radiother. Oncol 46, 51–62 (1998). [DOI] [PubMed] [Google Scholar]
- 229.Apter S et al. Cardiovascular calcifications after radiation therapy for Hodgkin lymphoma: computed tomography detection and clinical correlation. Coron. Artery Dis 17, 145–151 (2006). [DOI] [PubMed] [Google Scholar]
- 230.Hicks GL Jr. Coronary artery operation in radiation-associated atherosclerosis: long-term follow-up. Ann. Thorac. Surg 53, 670–674 (1992). [DOI] [PubMed] [Google Scholar]
- 231.Fender EA et al. Coronary artery bypass grafting in patients treated with thoracic radiation: a case-control study. Open Heart 5, e000766 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Fender EA et al. Percutaneous revascularization in patients treated with thoracic radiation for cancer. Am. Heart J 187, 98–103 (2017). [DOI] [PubMed] [Google Scholar]
- 233.Liang JJ et al. Outcomes after percutaneous coronary intervention with stents in patients treated with thoracic external beam radiation for cancer. JACC Cardiovasc. Interv 7, 1412–1420 (2014). [DOI] [PubMed] [Google Scholar]
- 234.Baban B, Liu JY, Qin X, Weintraub NL & Mozaffari MS Upregulation of programmed death-1 and its ligand in cardiac injury models: interaction with GADD153. PLOS ONE 10, e0124059 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Arai AE Healing after myocardial infarction: a loosely defined process. JACC Cardiovasc. Imaging 8, 680–683 (2015). [DOI] [PubMed] [Google Scholar]
- 236.Khorana AA, Carrier M, Garcia DA & Lee AY Guidance for the prevention and treatment of cancer-associated venous thromboembolism. J. Thromb. Thrombolysis 41, 81–91 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.van Es N et al. Comparison of risk prediction scores for venous thromboembolism in cancer patients: a prospective cohort study. Haematologica 102, 1494–1501 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Totzeck M, Mincu RI & Rassaf T Cardiovascular adverse events in patients with cancer treated with bevacizumab: a meta-analysis of more than 20 000 patients. J. Am. Heart. Assoc 6, e006278 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Totzeck M, Mincu RI, Mrotzek S, Schadendorf D & Rassaf T Cardiovascular diseases in patients receiving small molecules with anti-vascular endothelial growth factor activity: a meta-analysis of approximately 29,000 cancer patients. Eur. J. Prev. Cardiol 25, 482–494 (2018). [DOI] [PubMed] [Google Scholar]
- 240.Streiff MB NCCN guidelines. Cancer-associated venous thromboembolic disease. NCCN https://www.nccn.org/professionals/physician_gls/default.aspx#supportive (2019). [DOI] [PubMed] [Google Scholar]
- 241.Agnelli G et al. Nadroparin for the prevention of thromboembolic events in ambulatory patients with metastatic or locally advanced solid cancer receiving chemotherapy: a randomised, placebo-controlled, double-blind study. Lancet Oncol. 10, 943–949 (2009). [DOI] [PubMed] [Google Scholar]
- 242.Verso M, Agnelli G, Barni S, Gasparini G & LaBianca R A modified Khorana risk assessment score for venous thromboembolism in cancer patients receiving chemotherapy: the Protecht score. Intern. Emerg. Med 7, 291–292 (2012). [DOI] [PubMed] [Google Scholar]
- 243.Carrier M et al. Apixaban to prevent venous thromboembolism in patients with cancer. N. Engl. J. Med 380, 711–719 (2019). [DOI] [PubMed] [Google Scholar]
- 244.Khorana AA et al. Rivaroxaban for thromboprophylaxis in high-risk ambulatory patients with cancer. N. Engl. J. Med 380, 720–728 (2019). [DOI] [PubMed] [Google Scholar]
- 245.Raskob GE et al. Edoxaban for the treatment of cancer-associated venous thromboembolism. N. Engl. J. Med 378, 615–624 (2018). [DOI] [PubMed] [Google Scholar]
- 246.Khorana AA et al. Role of direct oral anticoagulants in the treatment of cancer-associated venous thromboembolism: guidance from the SSC of the ISTH. J. Thromb. Haemost 16, 1891–1894 (2018). [DOI] [PubMed] [Google Scholar]
- 247.Montani D et al. Pulmonary arterial hypertension in patients treated by dasatinib. Circulation 125, 2128–2137 (2012). [DOI] [PubMed] [Google Scholar]
- 248.Guignabert C et al. Dasatinib induces lung vascular toxicity and predisposes to pulmonary hypertension. J. Clin. Invest 126, 3207–3218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Weatherald J, Chaumais MC & Montani D Pulmonary arterial hypertension induced by tyrosine kinase inhibitors. Curr. Opin. Pulm. Med 23, 392–397 (2017). [DOI] [PubMed] [Google Scholar]
- 250.Weatherald J et al. Long-term outcomes of dasatinib-induced pulmonary arterial hypertension: a population-based study. Eur. Respir. J 50 (2017). [DOI] [PubMed] [Google Scholar]
- 251.Simonneau G et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J 53, 1801913 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Herrmann J et al. Vascular toxicities of cancer therapies: the old and the new – an evolving avenue. Circulation 133, 1272–1289 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Navi BB et al. Arterial thromboembolic events preceding the diagnosis of cancer in older persons. Blood 133, 781–789 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.