

Abstract
Obese asthmatics tend to have severe, poorly controlled disease and exhibit methacholine hyperresponsiveness manifesting in proximal airway narrowing and distal lung tissue collapsibility. Substantial weight loss in obese asthmatics or in mouse models of the condition decreases methacholine hyperresponsiveness. Ketone bodies are rapidly elevated during weight loss, coinciding with or preceding relief from asthma-related comorbidities. As ketone bodies may exert numerous potentially therapeutic effects, augmenting their systemic concentrations is being targeted for the treatment of several conditions. Circulating ketone body levels can be increased by feeding a ketogenic diet or by providing a ketone ester dietary supplement, which we hypothesized would exert protective effects in mouse models of inherent obese asthma. Weight loss induced by feeding a low-fat diet to mice previously fed a high-fat diet was preceded by increased urine and blood levels of the ketone body β-hydroxybutyrate (BHB). Feeding a ketogenic diet for 3 wk to high-fat diet-fed obese mice or genetically obese db/db mice increased BHB concentrations and decreased methacholine hyperresponsiveness without substantially decreasing body weight. Acute ketone ester administration decreased methacholine responsiveness of normal mice, and dietary ketone ester supplementation of high-fat diet-fed mice decreased methacholine hyperresponsiveness. Ketone ester supplementation also transiently induced an “antiobesogenic” gut microbiome with a decreased Fermicutes/Bacteroidetes ratio. Dietary interventions to increase systemic BHB concentrations could provide symptom relief for obese asthmatics without the need for the substantial weight loss required of patients to elicit benefits to their asthma through bariatric surgery or other diet or lifestyle alterations.
Keywords: asthma, β-hydroxybutyrate, ketogenic diet, mice, obesity
INTRODUCTION
Asthma is a common, chronic pulmonary disorder that while diverse in presentation typically involves a complex interaction of airway reactivity and inflammation (1). Obesity is highly prevalent among asthmatics (2). Whereas about one-third of the U.S population is obese, the prevalence of obesity among asthmatics is ≥50% (3, 4), and asthma is among the most important comorbidities of obesity (5). Obese asthmatics respond poorly to standard therapy (6) and are hospitalized for their asthma at an increased rate relative to normal weight asthmatics (7). Obesity alters responsiveness to bronchoconstricting agents, making both humans and mice hyperresponsive to the most common clinically used asthma diagnostic, methacholine (8–11). Metabolic reprogramming, oxidative stress, and altered immune status all contribute to obese asthma and are considered mediators of lung dysfunction (2, 12–14). Obese asthma exists as phenotypes termed “inherent” and “allergic,” both of which can be effectively modeled in mice. Whereas allergic asthma typically manifests in large airway hyperresponsiveness reactivity (AHR), obese asthma involves both proximal and distal lung compartments (15–17). Animal models suggest that obesity causes an inherent asthma-like phenotype involving proximal and distal lung compartments (18) through innate immune pathways, with the same mediators (e.g., TNF, IL-1β, and IL-17A) augmenting pathologic outcomes in obese allergic asthma (19–23). Altered immune responses and changes in pulmonary structure and function are evident in obese asthma patients, with TNF, macrophages (21), adipokines (24–26), and altered cellular metabolism (especially mitochondrial metabolism) (27–34) emerging as important modulators. Elevated markers of oxidative stress are found in obese asthmatics and are believed to contribute to the pathogenesis of obese asthma (35, 36).
Obesity is fundamentally a state of altered metabolism (37). In obese asthmatics, adipose tissue preferentially stores fatty acids instead of liberating them (38) and is more proinflammatory than adipose tissue from obese nonasthmatics (39). Weight loss reduces several comorbidities associated with obesity (40–42), including the hyperresponsiveness to inhaled methacholine manifest in inherent and allergic asthma (8, 11, 12, 18, 43–45). In humans, 5–10% body weight loss is typically required to promote benefit (defined as a minimal clinically important difference) in obese asthmatics (46), whereas body weight loss in mouse models of obese asthma is typically much more substantial, approaching the weights of normal-weight mice (18). However, reducing caloric intake, increasing energy expenditure, and pharmacological interventions as means of weight loss in human populations have limited long-term success due to poor long-term compliance, a lack of desirability, and high cost that limit their success as treatments for obese asthma. Bariatric surgery is the most effective weight loss intervention in the morbidly obese and is also an effective therapy for the comorbidities of obesity (11, 40–43, 47–49). Ketone bodies are elevated during weight loss (50–52), including for a brief period following bariatric surgery (53), and can modulate several of the key pathological processes involved in obese asthma (54–56). Despite our reports on the benefits of bariatric surgery, especially for nonallergic obese asthmatics (6, 8, 11, 39, 43, 47, 57–60), the limited success of this intervention on lung function in obese allergic asthmatics, and the poor long-term weight loss achieved by lifestyle alterations, highlight the continued need for specific treatments for obese allergic asthma.
During weight loss, fatty acids mobilized from adipose tissue are catabolized in the liver to the ketone bodies acetoacetate (AcAc) and β-hydroxybutyrate (BHB). Whereas circulating ketone body concentrations are normally repressed after eating, they become elevated during events that promote weight loss, including calorie restriction and exercise (51). Ketone bodies provide an energy source that makes cells less reliant on glycolysis (61). Ketone bodies also function as antioxidants (54, 55) and exert anti-inflammatory effects, including inhibition of the NLRP3 inflammasome and subsequent IL-1β production (56, 62), which are implicated in the pathogenesis of allergic asthma and obese asthma in mice fed a high-fat diet (20, 56, 62, 63). In addition, elevated BHB levels induced through alternate day caloric restriction are correlated with reductions in oxidative stress and inflammation, along with improved clinical findings, in overweight asthmatic subjects (64). Importantly, augmenting ketone bodies in humans is well tolerated (65).
Despite the strong connections between the mechanisms underlying obese asthma and the beneficial effects of ketone bodies, their potential to be used therapeutically in obese asthma has not been evaluated. We hypothesized that since weight loss elicits increases in ketone bodies that can exert significant anti-inflammatory, redox-regulating, and metabolic effects, ketone bodies could be relevant tools for treating obese inherent asthma. Our objective was to evaluate the effectiveness of augmenting ketone body concentrations on diminishing pathological features of obese asthma in mouse models and to provide mechanistic insight into the means by which benefit may be induced. Further understanding the efficacy and mechanisms of ketone bodies could provide new pharmacological targets for treatment or prevention of obese asthma, as could be addressed in subsequent clinical trials.
MATERIALS AND METHODS
Study Approval
Animal experiments were reviewed and approved by the University of Vermont’s Institutional Animal Care and Use Committee (protocol nos. 16-041, 18-023, and PROTO202000195), in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals prepared by the Institute of Laboratory Animal Resources, National Research Council, and published by the National Academy Press (revised 2011). Studies involving potentially hazardous materials were reviewed and approved by the University of Vermont’s Institutional Biosafety Committee (protocol nos. 09-018 and REG201900052).
Mice
Wild-type C57BL/6J mice (stock no. 000664) were purchased from The Jackson Laboratory (Bar Harbor, ME). Leprdb/+ mice on the C57BL/6J background purchased from The Jackson Laboratory (stock number 000697) were bred at the University of Vermont. Age-matched, female, normal-weight Lepr+/+ and obese Leprdb/db offspring were used for studies. All animals were maintained on 12-h light-dark cycle and were provided chow (∼15% kcal from fat; TestDiet, St. Louis, MO) and water ad libitum in an American Association for the Accreditation of Laboratory Animal Care-accredited facility. Low-fat diet (D12450B), high-fat diet (D12492), and ketogenic diet (D03022101) were purchased from Research Diets (New Brunswick, NJ) and stored frozen. Food in cages was replaced twice each week. Mice were euthanized with sodium pentobarbital (150 mg/kg by intraperitoneal injection; Wilcox Pharmacy, Rutland, VT).
High-Fat Diet Model of Obese Inherent Asthma and Diet-Induced Weight Loss
Twenty-one-week-old male C57BL/6J mice were purchased from The Jackson Laboratory having been maintained on food containing 10% fat (low-fat diet; LFD) or 60% fat (high-fat diet; HFD) from 6 wk of age. Mice were acclimated at the University of Vermont for a week following shipping and were maintained on their same food or subjected to dietary weight loss regimens. Obese mice were switched from the HFD to the LFD (HFDtoLFD), periodically assessed for body weight and ketone concentrations in urine or blood, and maintained on the LFD for a time sufficient to induce significant and substantial weight loss compared with the matched HFD mice that continued to gain weight, which was typically 2–3 wk and making the weight of the HFDtoLFD mice indistinguishable from the LFD-fed mice.
Genetic Model of Obese Inherent Asthma
Twenty-week-old male and female Leprdb/db mice and their lean control littermates were maintained on normal chow (NC) and then randomly divided into groups to be switched to the ketogenic diet (KD) or kept on NC. The mice were maintained on these diets for 3 wk until they were assessed for pulmonary responsiveness to inhaled methacholine.
Ketone Ester Supplementation of Mouse Diets
Twenty-week-old male C57BL/6J mice were purchased from The Jackson Laboratory having been maintained on LFD or HFD from 6 wk of age. After 1 wk of acclimation at the University of Vermont, the mice were switched to HFD or LFD containing 20% wt/vol water for 1 wk. Some of the obese mice were then switched to HFD containing 20% w/v ketone ester (KE; HVMN Inc., San Francisco, CA) and maintained on these diets for 3 wk until they were assessed for pulmonary responsiveness to inhaled methacholine. The ketone ester contains 25 g of pure ketones per 65 mL and less than 2% stevia leaf extract, flavorings, and preservatives.
Ketone Ester Gavage
Twenty-week-old C57BL/6J mice were gavaged with 200 µl of KE, and BHB levels were measured in blood collected from the submandibular vein using a ketone meter (Keto-Mojo, Napa, CA). Mice gavaged with 200 µl of KE or water were rested for a half hour then were assessed for acute effects of KE on pulmonary responsiveness to inhaled methacholine (∼1 h following water or KE gavage, accounting for time needed for mice to reach a steady anesthetic plane and be baselined on the flexiVent).
Assessment of Pulmonary Responsiveness to Methacholine
Responsiveness to inhaled methacholine was assessed in closed-chested mice. The mice were anesthetized with intraperitoneal sodium pentobarbital (90 mg/kg), the trachea was cannulated with a blunted 18 g needle, and the mice were connected to a flexiVent computer controlled small animal ventilator (SCIREQ, Inc., Montreal, Canada). The mice were ventilated at 200 breaths/min with a 0.25-mL tidal volume and 3-cmH2O positive end-expiratory pressure (PEEP). Next, the mice were paralyzed with an intraperitoneal injection of pancuronium bromide (0.8 µg/kg). The mice were stabilized over ∼10 min of regular ventilation at a PEEP of 3 cmH2O. A standard lung volume history was then established by delivering two total lung capacity maneuvers to a pressure limit of 25 cmH2O and holding for 3 s. Next, two baseline measurements of respiratory input impedance (Zrs) were obtained from 2-s multifrequency oscillations at PEEP = 3 cmH2O. This was followed by an inhalation of aerosolized PBS (control) for 10 s, achieved by an in-line piezo electric nebulizer (Aeroneb, Aerogen, Galway, Ireland). Zrs was then measured every 10 s for 3 min (18 measurements of Zrs in total). This complete sequence of maneuvers and measurements was then repeated for aerosol exposures to saline and three ascending doses of aerosolized methacholine (12.5, 25, and 50 mg/mL). Data were fit to the constant phase model of the lung (66) to provide values reflecting airway resistance (RN), tissue damping (G), and tissue elastance (H). Individual data points were excluded when the coefficient of determination to the constant phase model was below 0.85 or when values were below baseline levels. All data points for a particular dose of methacholine in an individual mouse were excluded when >50% or more of individual data points (>9 of 18) were excluded. The mean values ± SE of RN, G, and H in each of the mouse groups, at each incremental methacholine dose, are reported.
Serum Collection and Analysis
Following euthanasia at the completion of flexiVent analysis, ∼300 μL of blood were collected via cardiac puncture from the right ventricle using a 25-g needle attached to a 1-mL syringe, transferred into serum separator tubes (Becton Dickinson, Franklin Lanes, NJ) and centrifuged, and serum was kept frozen at −80°C. Serum was used to determine the concentration of beta-hydroxybutyrate (BHB) (BHB colorimetric assay kit no. 700190; Cayman Chemical), 8-isoprostane (8-isoprostane ELISA kit; Cayman Chemical), and l-lactate [l-lactate assay kit II (Eton Bioscience) following deproteination using StrataClean resin (Agilent, Santa Clara, CA)].
Bronchoalveolar Lavage Collection and Processing
Anesthetized mice, following pulmonary function assessment in some studies, were lavaged through an 18-gauge tracheal cannula with 1 mL of room temperature DPBS (Sigma-Aldrich, St. Louis, MO). Cells were manually counted immediately in white blood cell stain (0.2 mg/mL crystal violet in 2% acetic acid) using a hemocytometer, the lavage fluid was centrifuged at 400 g for 10 min at room temperature, and cell-free supernatants were frozen at −80°C for analysis. Cell pellets were resuspended in saline and mounted on slides by cytospin (100,000 cells per slide) for hematoxylin and eosin (H&E) staining and differential analysis.
Urine Collection and Analysis
Urine was collected from mice by scruffing them over parafilm and waiting for voluntary urination. ∼50 to 100 μL of urine was collected from each mouse and transferred to a tube with a pipette and frozen at −80°C until analysis. Urine was later used to determine the concentration of BHB using a commercial colorimetric assay (BHB colorimetric assay kit no. 700190, Cayman Chemical).
Quantitative RT-PCR
Following the collection of BAL fluid, lungs were dissected, ground to a fine powder using a liquid nitrogen-chilled mortar and pestle, and stored at −80°C until analysis. Total RNA was extracted from an aliquot of the snap-frozen and pulverized lung lobes using the PrepEase RNA Isolation kit (USB Corp., Cleveland, OH) and reverse transcribed to cDNA using the iScript kit (Bio-Rad, Hercules, CA). Primers were designed for mouse genes using NCBI Primer-BLAST (RRID:SCR_003095) and synthesized by Integrated DNA Technologies (Coralville, IA). Quantitative RT-PCR was performed using SYBR Green Supermix on a CFX96 or Chromo4 thermocycler using CFX Manager or Opticon Monitor software (Bio-Rad) and normalized to Rpl13a using the ΔΔCT method (Table 1).
Table 1.
Primers used for RT-qPCR analysis of lung gene expression
| Gene | Name | Forward Primer | Reverse Primer |
|---|---|---|---|
| Acta2 | Actin alpha 2 | TGTGCTGGACTCTGGAGATG | GAAGGAATAGCCACGCTCAG |
| Actg | Actin gamma | GGATCGGTGGCTCCATTCTG | TGAGGTGTGTACATTTGCCAG |
| Adipoq | Adiponectin | TGTTCCTCTTAATCCTGCCCA | CCAACCTGCACAAGTTCCCTT |
| Ccl2 | Chemokine (C-C motif) ligand 2 | TCCCTGTCATGCTTCTGGGC | GTTCTGATCTCATTTGGTTCCGATCC |
| Clca1 | Chloride channel accessory 1 | AAGCAAACCACTCCCATGAC | TGCGAAAGCATCAACAAGAC |
| Col1a1 | Collagen type I alpha I | GAGCGGAGAGTACTGGATCG | GTTCGGGCTGATGTACCAGT |
| Il1b | Interleukin 1 beta | GCCCATCCTCTGTGACTCAT | AGGCCACAGGTATTTTGTCG |
| Il6 | Interleukin 6 | CCGGAGAGGAGACTTCACAG | GAGCATTGGAAATTGGGGTA |
| Il17a | Interleukin 17 A | CTGCTGAGCCTGGCGGCTAC | GGCGGCACTGAGCTTCCCAG |
| Muc5ac | Mucin 5 subtype AC | CCATGCAGAGTCCTCAGAACAA | TTACTGGAAAGGCCCAAGCA |
| Muc5b | Mucin 5 subtype B | ACACATGCACCTGCCTCTCTGA | TCCATGGAGTACTTGGATATTC |
| Myh11 | Myosin heavy polypeptide 11 | CTCTGGCCTCTTCTGTGTGG | TCTTTCTTGCCCTTGTGGGA |
| Myod | Myogenic differentiation 1 | GCCGCCTGAGCAAAGTGAATG | CAGCGGTCCAGGTGCGTAGAAG |
| Myog | Myogenin | CTACAGGCCTTGCTCAGCTC | ACGATGGACGTAAGGGAGTG |
| Nlrp3 | NRL family pyrin domain containing 3 | ATGCTGCTTCGACATCTCCT | AACCAATGCGAGATCCTGAC |
| Rpl13a | Ribosomal protein L13a | CCCTCCACCCTATGACAAGA | CTGCCTGTTTCCGTAACCTC |
| Saa3 | Serum amyloid A3 | CAGGATGAAGCCTTCCATTG | CATGACTGGGAACAACAGGA |
| Tagln | Transgelin | GCGGCCTTTAAACCCCTCA | CTCCACTAGTCGCTCCTCCA |
| Tjp1 | Tight junction protein 1 | CCACCTCTGTCCAGCTCTTC | CACCGGAGTGATGGTTTTCT |
| Tnfa | Tumor necrosis factor | TCCCAGGTTCTCTTCAAGGGA | GGTGAGGAGCACGTAGTCGC |
| Vim | Vimentin | TGCTTCAAGACTCGGTGGAC | AAGCGCACCTTGTCGATGTA |
Measurement of Fecal Bacterial Diversity
From fecal pellets collected at the time of euthanasia, bacterial DNA was extracted (Qiagen) and subjected to quantitative PCR analysis of the 16S ribosomal RNA (rRNA) present in all eubacteria as well as the 16S rRNA present specifically in the flora of the mouse gastrointestinal tract, including the phyla Bacteroidetes (Bacteroides, mouse intestinal Bacteroides), Fermicutes (Clostridium perfiringens, Eubacterium rectale/Clostridium coccoides, and Lactobacillus/Lactococcus), and Proteobacteria (Enterobacteriaceae, Salmonella, and Helicobacter pylori) using primers described previously (67). Standards were generated from the pooled products of mixed mouse fecal samples following 20 rounds of PCR, which were subsequently used to establish standard curves for each of the primer reactions over a range of 4-fold dilutions between 1:100 and 1:400,000. Cycle threshold values for each PCR reaction were applied to the respective standard curve, run on each plate on a Bio-Rad CFX96 or Chromo4 96-well quantitative real-time PCR detection system. PCR product abundance was log transformed and the average abundance in the low-fat diet group was used to normalize relative abundance from each animal, values that were then used to calculate the means ± SE for each bacterial 16S rRNA in each mouse group. The average relative abundance of specific 16S rRNAs of each phyla in each mouse were used to calculate the overall Bacteroidetes, Fermicutes, and Proteobacteria abundance, from which the Fermicutes:Bacteroidetes ratios were calculated.
Data Acquisition, Data Availability, and Statistical Analysis
All experiments involved multiple mice per group and were replicated. The data sets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request. Data were analyzed by two-tailed unpaired t test or one-way or two-way ANOVA and Bonferroni, Tukey, or Dunnett’s multiple comparisons post hoc test using GraphPad Prism 9.1.2 for Windows (GraphPad Software, Inc., La Jolla, CA; RRID:SCR_002798). Data are presented as means ± SE. P < 0.05 in the t test or multiple comparisons post hoc test was considered statistically significant. Significance levels of the tested comparisons are indicated in the figure legends.
RESULTS
Elevated Ketone Body Concentration Precedes Diet-Induced Weight Loss in Obese Mice Previously Fed a High-Fat Diet
The high-fat diet-induced obesity model most closely replicates the findings of weight gain and the development of metabolic syndrome common in obese humans (68, 69). We have previously reported that diet-induced weight loss in obese mice previously fed a high-fat diet decreases methacholine hyperresponsiveness (18) and promotes visceral adipose tissue homeostasis (70). To examine the temporal relationships between body weight and BHB levels, C57BL/6J mice were maintained on low-fat diet (LFD) or high-fat diet (HFD) until the latter were markedly obese. Then, a cohort of the high-fat diet-fed mice were provided the low-fat diet for 3 wk (HFDtoLFD) while other mice were maintained on their original diets. Whereas body weights of the HFDtoLFD mice did not differ significantly from the HFD group until day 9 (Fig. 1A), urinary BHB levels were significantly elevated on days 2 and 7 in the HFDtoLFD mice (Fig. 1B), with levels declining to near baseline at day 9 and beyond. At the end of the study, visceral adipose tissue (VAT) weight was significantly decreased in the HFDtoLFD mice to levels equivalent to those of the LFD mice (Fig. 1C), whereas serum BHB concentrations were the same across the groups at the end of the study (means ± SD for LFD = 0.25 ± 0.01 mM, HFD = 0.27 ± 0.02 mM, and HFDtoLFD = 0.27 ± 0.02 mM; P = 0.29). In a separate cohort of HFDtoLFD mice, blood BHB levels were significantly elevated over baseline on days 1, 2, and 3 after switching from the high-fat to the low-fat diet and, although still significantly elevated on day 6, were returning to baseline levels, which were low again on day 9 (Fig. 1D).
Figure 1.
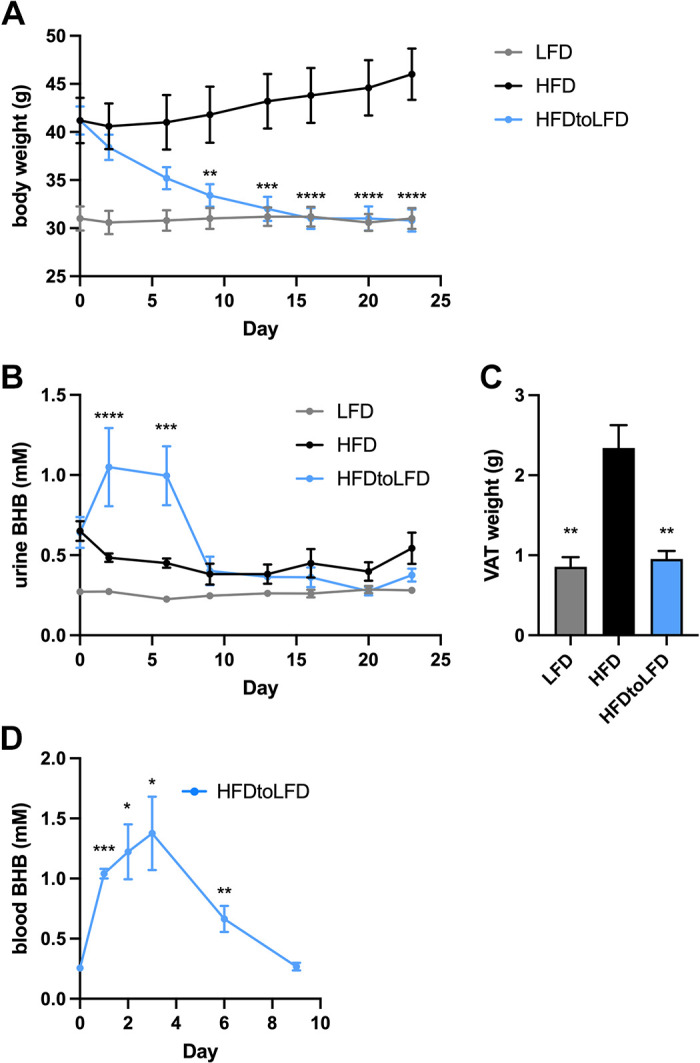
High-fat diet-induced obese mice switched to a low-fat diet produce β-hydroxybutyrate preceding weight loss. Mice were fed a low-fat diet (LFD) or a high-fat diet (HFD) for 16 wk and then either maintained on the same diets or switched (day 0) from the HFD to a LFD (HFDtoLFD) for 3 additional weeks. Body weight (A), urine β-hydroxybutyrate (BHB) (B), and visceral adipose tissue (VAT) weight at day 23 (C) were measured. In a separate cohort of HFDtoLFD mice, blood β-hydroxybutyrate (BHB) was measured daily (D); n = 5 mice/group. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001, comparing HFDtoLFD and HFD (A and B), compared with HFD group (C), or compared with day 0 (D).
Feeding a Ketogenic Diet to Diet-Induced Obese Mice Modestly Decreases Body Weight but Substantially Increases Serum BHB Concentrations and Decreases Methacholine Hyperresponsiveness
A ketogenic diet is one in which the fat content is elevated beyond that in the HFD at the expense of reducing carbohydrates and in which circulating ketone body levels are elevated. Increasing dietary fat content causes “therapeutic ketosis” in the absence of fasting and induces a unique metabolic state in mice that results in weight loss of similar magnitude to that of caloric restriction (71). As elevations in ketone body concentrations precede diet-induced weight loss, we examined whether providing a high-fat (80% of calories), low-carbohydrate (<0.1% of calories) ketogenic diet (KD) to obese mice previously fed a high-fat diet would attenuate methacholine hyperresponsiveness to a magnitude similar to that elicited by feeding a low-fat diet (18). Mice were maintained on LFD or HFD until the HFD-fed mice were significantly and substantially obese. Mice were then maintained on these diets, or a cohort of the HFD-fed mice were either provided the LFD (HFDtoLFD) or the KD (HFDtoKD) for 3 wk. Whereas feeding the LFD to HFD-induced obese mice substantially decreased body weight to levels similar to the LFD mice, obese mice fed the KD only modestly decreased body weight (Fig. 2A) and had significantly elevated levels of serum BHB (Fig. 2B) compared with the HFD-fed mice. Bronchoalveolar lavage cellularity was not significantly different between the groups (Fig. 2C), and all cells were identified as alveolar macrophages by H&E staining. Assessment of inhaled methacholine responsiveness, a defining criterion of obesity-associated asthma (12, 18, 47), showed significantly decreased central airway (Newtonian) resistance (RN), tissue damping (G), and tissue elastance (H) in mice switched from the HFD to either LFD or KD (Fig. 2, D and E).
Figure 2.

High-fat diet-induced obese mice switched to low-fat or ketogenic diets exhibit decreased methacholine hyperresponsiveness. Mice were fed a low-fat diet (LFD) or a high-fat diet (HFD) for 16 wk and then either maintained on the same diets or switched from the HFD to a LFD (HFDtoLFD) or ketogenic diet (HFDtoKD) for 3 additional weeks. Body weight (A), serum β-hydroxybutyrate (BHB) (B), and total bronchoalveolar lavage (BAL) cells (C) were measured. Airway resistance (RN) (D), tissue damping (G) (E), and tissue elastance (H) (F) were measured; n = 8–10 mice/group. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001, compared with indicated group.
Feeding a Ketogenic Diet to Genetically Obese Mice Substantially Increases Serum BHB Concentrations and Decreases Methacholine Hyperresponsiveness of the Central Airways
The leptin “resistant” db/db mice on a C57BL/6J background are a genetic model of hyperphagic obesity in which mice are deficient in the long form of the leptin receptor (ObRb), conferring impaired signaling along the canonical pathway in response to leptin (72), much like what is observed functionally in obese humans and accompanied by other manifestations of metabolic syndrome. Studies in db/db mice demonstrate obesity-associated defects in both the innate and adaptive immune responses (73) and inherent methacholine hyperresponsiveness when obese (74). Wild-type (WT) and homozygous Leprdb/db (DbDb) mice were maintained on normal chow, and then cohorts from each strain were switched to KD or maintained on normal chow for 3 wk. WT mice remained normal weight on either food, whereas the DbDb mice were markedly obese and slightly increased their weight on the KD (Fig. 3A). In both WT and DbDb mice, feeding the KD significantly and substantially increased serum BHB levels at the end of the study (Fig. 3B), whereas there was no effect of the type of food consumed on BAL cells (Fig. 3C), all of which were alveolar macrophages. DbDb mice exhibited profound methacholine hyperresponsiveness, with significant increases in central airway resistance and tissue elastance (Fig. 3, D–F). DbDb mice fed the KD showed significantly decreased central airway resistance in response to the highest dose of inhaled methacholine compared with DbDb mice fed normal chow.
Figure 3.

Genetically obese mice fed a ketogenic exhibit decreased methacholine hyperresponsiveness. Wild-type (WT) or Leprdb/db (DbDb) mice were maintained on a normal diet (Chow) or switched to a ketogenic diet (KD) for 3 wk. Body weight was measured pre- and postswitching diets (A). Serum β-hydroxybutyrate (BHB) (B), total bronchoalveolar lavage (BAL) cells (C), airway resistance (RN) (D), tissue damping (G) (E), and tissue elastance (H) (F) were measured at the end of the study; n = 8–9 mice/group. *P ≤ 0.05, **P ≤ 0.01, ****P ≤ 0.0001, compared with indicated group.
Substantially Elevating Serum BHB Levels Decreases Methacholine Responsiveness
Since ketone ester supplementation in the food decreased methacholine hyperresponsiveness in diet-induced and genetic models of obesity while slightly, albeit significantly, increasing serum BHB levels at the end of the studies, we sought to determine the impact of substantially elevated circulating BHB levels during methacholine challenge. Mice fed normal chow were administered by oral gavage 200 μL of water or an equal volume containing 80 mg ketone ester. Blood BHB levels were substantially elevated above 7 mM at 30 min and 1 h and then began to drop by 2 h, with levels falling to baseline by 6 h (Fig. 4A). A separate cohort of mice were administered water or KE by gavage then assessed for methacholine responsiveness 1 h later, at the time of peak circulating BHB levels. Compared with the water control mice, mice acutely administered KE displayed significantly decreased methacholine responsiveness, especially in tissue damping and tissue elastance (Fig. 4, B–D). As BHB has been reported to exert antioxidant properties (54, 55, 75, 76), serum levels of 8-isoprostanes, a stable product of lipid peroxidation elevated in exhaled breath of obese asthmatics (14), were measured. The levels of serum 8-isoprostanes present in the mice administered ketone ester by gavage approximately 1 h earlier were significantly less than those in mice administered water (Fig. 4E). Since BHB can be used as an energetic substrate to provide ATP as an alternative to utilizing dietary carbohydrates (e.g., glucose) (77, 78), and since glucose metabolism generates lactate that has been implicated as a driver of asthma pathogenesis (79–81), serum lactate was also measured. In mice administered ketone ester, the serum lactate levels were significantly and substantially decreased (Fig. 4F).
Figure 4.

Ketone ester administration acutely decreases methacholine responsiveness. Mice were administered 200 μL of water (H2O) or ketone ester (KE; ∼4 g/kg) by oral gavage. Blood β-hydroxybutyrate (BHB) (A) was measured before or at several time points following KE administration; n = 5 mice/group. Airway resistance (RN) (B), tissue damping (G) (C), and tissue elastance (H) (C) were measured 1 h following water or ketone ester gavage. Serum 8-isoprostane (E) and lactate (F) were measured; n = 6 mice/group. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001, compared with time 0 by ANOVA (A) or the water group by t test (E and F). *P ≤ 0.05, **P ≤ 0.01, compared with water group at the same dose of methacholine (B–D).
Ketone Ester Supplementation Decreases the Methacholine Hyperresponsiveness of HFD-Induced Obese Mice
Ketone esters are a dietary supplement approved for human use that transiently increase circulating BHB levels (55). C57BL/6J mice were maintained on LFD or HFD until the HFD-fed mice were obese, and then mice were continued on the same food or a cohort of the obese mice were administered a HFD containing 20% by weight ketone ester (HFD + KE) for 3 wk. The body weight of the mice at the end of the feeding regimens was elevated in both the HFD and HFD + KE groups, although there was a trend toward a decreased weight in the HFD + KE mice compared with the HFD mice that did not reach significance (Fig. 5A). Whereas serum BHB levels were slightly elevated in the HFD-fed mice compared with the LFD-fed mice, the levels were further substantially and significantly elevated in the HFD + KE mice (Fig. 5B), The BAL alveolar macrophage numbers were not different between the groups (Fig. 5C). The HFD-fed mice exhibited methacholine hyperresponsiveness, particularly in central airway resistance and tissue damping, whereas the HFD + KE mice exhibited significantly attenuated methacholine responsiveness in both parameters RN and G (Fig. 5, D–F). Additionally, whereas serum 8-isoprostane levels were not different between the groups (Fig. 5G), serum lactate levels in mice fed the HFD + KE were significantly decreased compared with HFD-fed mice (Fig. 5H). Finally, lung expression of several genes of potential relevance to the mechanism whereby ketone ester feeding may elicit an inhibitory effect on methacholine hyperresponsiveness was assessed. Lung expression of adipogenic (Adipoq), epithelial (Muc5ac and Muc5b), mesenchymal (Col1a1 and Actg), muscle (Myh11, Myod, and Myog), and obesity-associated proinflammatory genes (Ccl2, Tnfa, Il6, IL17a, Saa3, and Nlrp3) was not significantly different between LFD and HFD, and none was not significantly different between HFD and HFD + KE (Fig. 5I). Expression of the epithelial gene chloride channel accessory 1 (Clca1), the smooth muscle and mesenchymal genes actin alpha 2 (Acta2), vimentin (Vim), and transgelin (Tagln), as well as the proinflammatory cytokine IL-1β (Il1b) was significantly increased in the lungs of HFD-fed mice compared with LFD-fed mice. However, expression of these genes was not significantly decreased in the HFD + KE lungs compared with those from HFD-fed mice (Fig. 5I). Expression in the lung of tight junction protein 1 (Tjp1) was significantly elevated in the HFD + KE group compared with the HFD group (Fig. 5I).
Figure 5.

Ketone ester supplementation decreases methacholine hyperresponsiveness in high-fat diet-induced obese mice. Mice were maintained on a low-fat diet (LFD), a high-fat diet (HFD), or a high-fat diet containing 20% (by weight) ketone ester (HFD + KE) for 3 wk. Body weight (A), serum β-hydroxybutyrate (BHB) (B), and total bronchoalveolar lavage (BAL) cells (C) were measured. Airway resistance (RN) (D), tissue damping (G) (E), and tissue elastance (H) (F) were measured. Serum 8-isoprostane (G), serum lactate (H), and lung gene expression were measured and presented as expression relative to the mean of the LFD group, scaling from 0 to 3 (I); n = 8–10 mice/group.. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001, compared with indicated group (A–H) or HFD group (I).
Dietary Ketone Ester Supplementation Transiently Promotes an Antiobesogenic Gut Microbiome
We have previously reported that diet-induced weight loss in obese mice previously fed a high-fat diet promotes an antiobesogenic gut microbiome characterized by decreases in the elevated fecal Firmicutes/Bacteroidetes ratio present in obesity (18). To examine the impact ketone ester supplementation has on the fecal microbiome, normal chow-fed C57BL/6 mice were fed normal chow containing 20% water or KE (by weight) for 15 days then fed normal chow for an additional 15 days. Whereas mice fed the normal chow (even with 20% water) increased body weight steadily over the course of the study, mice fed chow containing KE displayed a transient decrease in body weight followed by a similar slope of body weight increase after day 5 of the KE feeding and throughout the remainder of the study (Fig. 6A). Whereas there were no differences in the overall abundance of total eubacterial 16S rRNA in the feces (Fig. 6B), there were significantly elevated abundances of Bacteroidetes in the KE-fed mice at 15 days that remained following an additional 15 days of feeding normal chow (Fig. 6C). In mice fed KE for 15 days then fed normal chow, fecal Fermicutes levels were significantly elevated (Fig. 6D), whereas there were no differences in the abundance of Proteobacteria between the groups (Fig. 6E). At the end of the first 15-day KE-feeding period, the calculated fecal Firmicutes/Bacteroidetes ratio was substantially decreased relative to the chow-fed mice, an effect that was no longer present on day 30, after 15 days of again feeding normal chow (Fig. 6F).
Figure 6.
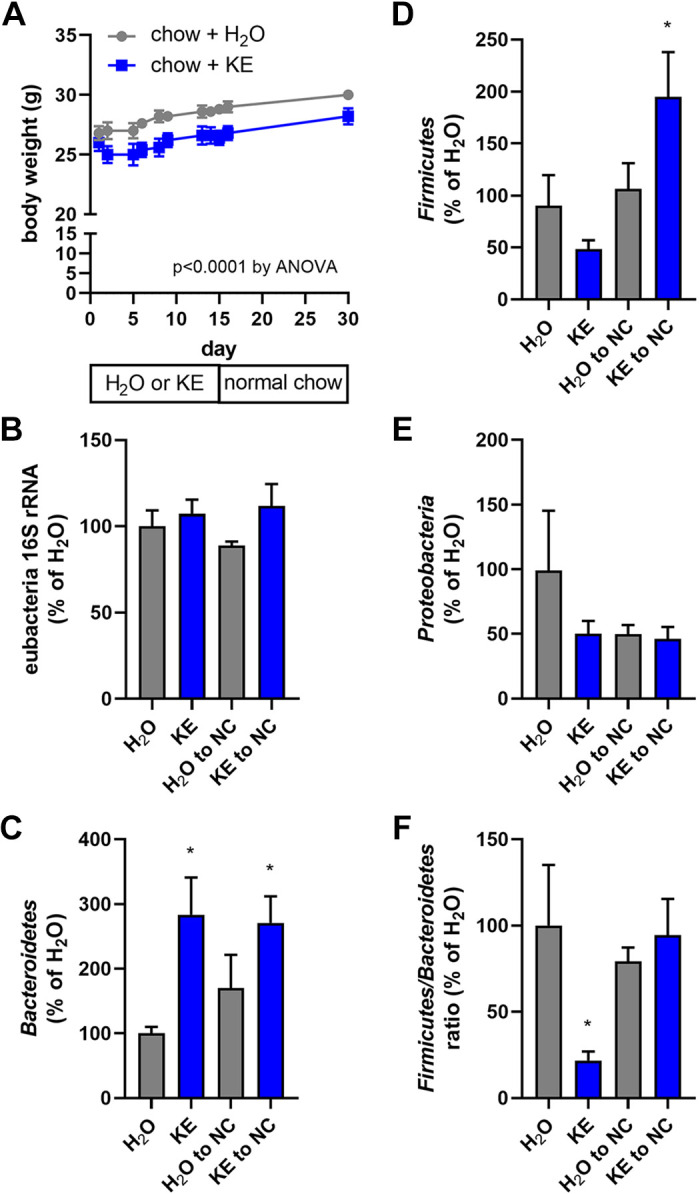
Ketone ester feeding promotes a transient anti-obesogenic gut microbiome. Mice were maintained on normal chow until day 0 and then maintained on chow containing 20% (by weight) water (H2O) or 20% ketone ester (KE) for 15 days and then switched back to normal chow (NC) until day 30. Body weight was measured on several occasions throughout the 30-day feeding regimen (A). Fecal pellet total eubacterial 16S rRNA (B) and phlya-specific 16S rRNA for Bacteroidetes (C), Firmicutes (D), and Proteobacteria (E) DNA content was quantitated on days 15 and 30, and expressed relative to that found in the chow + H2O fed mice at day 15. The Firmicutes/Bacteroidetes ratio, relative to that found in the chow + H2O fed mice at day 15, was calculated (F); n = 5 mice/group. *P ≤ 0.05, compared with indicated group.
DISCUSSION
Given the rapidly worsening global epidemics of asthma (82) and obesity (42), and the heterogeneity of these conditions (83, 84), there is a pressing need to understand the efficacy of and mechanisms through which mediators associated with the beneficial effects of weight loss, which may provide novel insight into the treatment of obese asthma not afforded by conventional approaches. Our studies demonstrate that dietary treatments inducing acute (ketone ester gavage), intermediate duration (low-fat diet-induced weight loss), or protracted (ketogenic diet or ketone ester supplementation) increases in the ketone body β-hydroxybutyrate decrease methacholine responsiveness and obesity-associated methacholine hyperresponsiveness. Whereas the physiological mechanisms underlying the methacholine hyperresponsiveness in obese asthma are uncertain and remain the subject of investigation (85), both large airway and peripheral lung dysfunction are present in obese asthma (15–17), and our interventions show that increased ketone body concentrations benefit both the proximal and distal airways. First, ketone bodies decreased the parameter RN that provides a measure of the flow resistance of the entire airway tree. RN is more sensitive to contraction of the central rather than the peripheral airways (86). Second, the interventions also decreased tissue damping, G, and tissue elastance, H. Both G and H are increased by the development of heterogeneous ventilation to the distal reaches of the lung due to variations in airway narrowing, and also by derecruitment of lung units (86). Both ventilation heterogeneity and derecruitment are events that manifest predominately in the periphery of the lung and that are particularly sensitive to contraction of peripheral airways (87). However, the central airways and the lung periphery are not mechanically independent because the parenchymal tissue is tethered to the airways (85). It is therefore compelling to speculate that a primary effect of ketones may be on bronchial smooth muscle cells, attenuating their capacity to contract in response to methacholine and thereby affecting the changes in all three mechanics parameters: RN, G, and H. Based on the results reported herein, we posit that providing elevated levels of ketone bodies in the setting of obesity could provide benefit (53) to intrinsic asthma and may do so through a number of direct and indirect effects on lung physiology throughout the proximal airways and distal airspaces that modulate inhaled methacholine hyperresponsiveness.
Intriguingly, in obese subjects undergoing bariatric surgery, ketone body levels in the circulation rise very rapidly, even before substantial weight loss is realized (88), coinciding with relief from many asthma-associated comorbidities. Similarly, our high-fat diet to low-fat diet weight loss model induced rapid and transient elevations, over the course of 6 days, in systemic ketone body concentrations that preceded and accompanied substantial weight loss. Healthy weight loss induces the mobilization of fat from adipose tissue that is subsequently catabolized in the liver through β-oxidation to form the ketone bodies β-hydroxybutyrate (BHB) and acetoacetate (AcAc), which then enter the circulation and can be utilized by cells throughout the body. Ketone bodies exert their metabolic functions within mitochondria through their entry into the tricarboxylic acid cycle to generate 1 GTP and 11 ATP molecules per acetyl group oxidized (77). Ketones also decrease circulating levels of glucose and the rate of glycolysis, even in states where carbohydrates are abundant (89–91). As an increased rate of glycolysis can promote inflammation (92, 93), it is possible that some of the beneficial effects of ketones observed in our studies are derived from their ability to provide an alternative fuel source to glucose and promote oxidative phosphorylation. The consequences would be a decreased production of lactate, which has been implicated as a pathogenic driver of asthma (79–81).
In our acute studies of ketone ester supplementation that decreased methacholine-induced tissue damping and tissue elastance, circulating lactate concentrations were significantly decreased. In addition, compared with mice fed a high-fat diet, the fed a high-fat diet-fed mice supplemented with ketone ester had lower levels of serum lactate. These results show less utilization of glucose as a cellular fuel when ketone body concentrations are elevated. Ketone bodies also perform additional functions systemically in concert with and independent of their ability to function as a cellular fuel (50, 51, 61). Ketone bodies have redox-modulating and anti-inflammatory functions. Airway mitochondria are altered in animals fed a high-fat diet (32), and mitochondrial reactive oxygen species are involved in the complications of obesity (29, 33, 94). Furthermore, increased transcription and release of cytokines (95, 96) are a consequence of signaling through redox-sensitive pathways (97, 98). Ketones decrease ROS production by acting directly as antioxidants and by inducing the expression of genes associated with resistance to oxidative stress (54, 55, 75, 76). Increased methacholine reactivity is observed in allergen-challenged obese mice (99–102) and elevated levels of oxidative stress in the airways of obese asthmatics increase proportionally with BMI (13). Acute ketone ester supplementation significantly decreased circulating levels of 8-isoprostanes, a biomarker of lipid peroxidation elevated in obesity (103). In overweight asthmatics, the improved clinical findings and decreases in oxidative stress in subjects practicing alternate day calorie restriction are associated with elevated circulating levels of ketone bodies (BHB) (64).
In contrast to their association with ketoacidosis, a pathogenic state that can come about in the setting of diabetes and alcohol consumption (104) in which circulating ketone levels are well above those achieved during the feeding of a ketogenic diet or starvation (51), elevating ketone bodies is safe both in animal models of disease and in human subjects (65, 90, 91, 105–110). A ketogenic diet contains sufficient protein, reduced carbohydrates, and an abundance of fat that serves as a substrate for ketone body formation (71). In our hands, feeding a ketogenic diet for 10 days to mice previously fed a high-fat diet augmented circulating BHB levels but not to remarkably high concentrations. Other methods to rapidly increase the levels of circulating ketone bodies include the feeding of ketone esters that provide long-lived and high levels of ketone bodies. Ketone esters are generally regarded as safe (meaning they can be considered a dietary supplement) and have shown benefits to elite athletes and in patients with chronic disease (65, 90, 91, 105, 106). As used in our studies, the ketone ester (R)-1,3-beta hydroxybutyrate (R)-1,3-butanediol (91, 105, 106) augments circulating BHB levels and was provided by gavage akin to how it is acutely consumed as a supplement, or incorporated into mouse food at 20% of weight (and approximately calories) to promote protracted consumption. This ketone ester supplementation strategy could be optimized as an approach to promote a state of “therapeutic ketosis” similar to that achieved through the feeding of a ketogenic diet or fasting, without any caloric deficit or the need for substantial lifestyle modification.
Ketone bodies can also function through the cell surface receptors hydroxycarboxylic acid receptor 2 (HCAR2/GPR109a) and free fatty acid receptor 3 (FFAR3/GPR41) (50, 51, 111, 112). Additionally, ketone bodies have also been reported to function as class-I histone deacetylase (HDAC) inhibitors (50, 51, 76), and to induce β-hydroxybutyrylation of histone H3 lysines (113) to influence gene expression. We detected increased expression of the smooth muscle-associated genes, Tagln (transgelin/SM-22 alpha) and Acta2 (actin alpha 2), in the lungs of methacholine-hyperresponsive, HFD-fed obese mice, which appeared to be decreased in obese mice fed HFD containing ketone ester. Acta2 encodes alpha smooth muscle actin, necessary for agonist-induced contraction and elevated in asthmatics (114). Tagln encodes an actin-crosslinking protein, which although not affecting the rate of actin filament propulsion is also elevated in asthmatic airways (114). In mice fed high-fat diet supplemented with ketone ester, lung expression of the tight junction protein Tjp1 was significantly increased, which may augment airway barrier integrity and limit access of inhaled methacholine to the underlying smooth muscle, thereby decreasing responsiveness. It is possible that BHB affects bronchial smooth muscle responses to methacholine, which could inhibit contraction at many distinct cellular levels. Expression of the gene encoding IL-1β was significantly elevated in the lungs of high-fat diet-fed mice and was not as highly expressed in obese mice provided ketone ester supplementation. BHB inhibits NLRP3 inflammasome-mediated IL-1β processing (56, 62), and coupled with decreased lung Il1b expression could translate into substantially decreased bioactive IL-1β, a cytokine that is a cause of elevated glycolysis and accompanying pathology in asthma (80, 115, 116).
Our results provide innovative first steps toward a safe, efficacious, and cost-effective strategy that may uniquely target obese asthma. However, these results may also be translatable to allergic asthma in which a more robust inflammatory response is present. While it is widely-accepted that 5–10% weight loss is required to achieve improvement in obese asthmatics (46), the modest reductions in weight elicited by feeding a ketogenic diet or providing ketone ester supplementation implicate effects besides weight loss for which BHB may be responsible. While a transient “antiobesogenic” gut microbiota was achieved following dietary supplementation with the ketone ester, weight gains over time were similar between the treated and untreated groups. The decreased Fermicutes/Bacteroidetes ratio present during ketone ester supplementation may also provide short-chain fatty acids, such as butyrate, which has been reported to elicit therapeutic effects in asthma (117). Butyrate may even be a substrate for conversion into BHB endogenously (118), suggesting that some of the beneficial effects of butyrate may be due to BHB. Intriguingly, medium-chain triglyceride supplementation is a therapeutic arm of The PrecISE Network, a National Heart, Lung, and Blood Institute-sponsored trial providing biomarker-based investigational interventions to patients with specific endotypes of severe asthma (119). Medium-chain triglycerides are preferentially catabolized to form ketone bodies, and BHB levels are a target, although not necessarily a mediator, for achieving potentially therapeutic levels of supplementation in this intervention arm targeted at high-arginine metabolic severe asthma (119). Certainly, metabolic therapy for obese asthma is a potential complement or alternative to other conventional approaches for the treatment of these patients.
DATA AVAILABILITY
The data that support this study are available from the author on reasonable request.
GRANTS
This work was supported by NIH National Heart, Lung, and Blood Institute Grants R01HL133920 (to M.E.P.) and R01HL142081 (to M.E.P.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.M.M. and M.E.P. conceived and designed research; M.M.M., L.F.R., C.J.W., M.L.T.B., J.L.A., and M.E.P. performed experiments; M.M.M., J.L.A., and M.E.P. analyzed data; M.M.M. and M.E.P. interpreted results of experiments; M.E.P. prepared figures; M.E.P. drafted manuscript; M.M.M., L.F.R., C.J.W., M.L.T.B., and M.E.P. edited and revised manuscript; M.M.M., L.F.R., C.J.W., M.L.T.B., J.L.A., and M.E.P. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Nirav Daphtary, Minara Aliyeva, and Dr. Jason H. T. Bates for technical and computational support in the generation and interpretation of flexiVent data. We thank Olivia Johnson and Anne E. Dixon for helpful discussions.
REFERENCES
- 1.US Department of Health and Human Services. 2020 Focused Updates to the Asthma Management Guidelines. A Report from the National Asthma Education and Prevention Program Coordinating Committee Expert Panel Working Group National Heart, Lung, and Blood Institute, National Institutes of Health, US Department of Health and Human Services. https://www.nhlbi.nih.gov/health-topics/asthma-management-guidelines-2020-updates [2021 Jul 7].
- 2.Dixon AE, Poynter ME. Mechanisms of asthma in obesity. pleiotropic aspects of obesity produce distinct asthma phenotypes. Am J Respir Cell Mol Biol 54: 601–608, 2016. doi: 10.1165/rcmb.2016-0017PS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schatz M, Hsu JW, Zeiger RS, Chen W, Dorenbaum A, Chipps BE, Haselkorn T. Phenotypes determined by cluster analysis in severe or difficult-to-treat asthma. J Allergy Clin Immunol 133: 1549–1556, 2014. doi: 10.1016/j.jaci.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 4.Schatz M, Zeiger RS, Zhang F, Chen W, Yang SJ, Camargo CA Jr.. Overweight/obesity and risk of seasonal asthma exacerbations. J Allergy Clin Immunol Pract 1: 618–622, 2013. doi: 10.1016/j.jaip.2013.07.009. [DOI] [PubMed] [Google Scholar]
- 5.Beuther DA, Sutherland ER. Overweight, obesity, and incident asthma: a meta-analysis of prospective epidemiologic studies. Am J Respir Crit Care Med 175: 661–666, 2007. doi: 10.1164/rccm.200611-1717OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dixon A. The treatment of asthma in obesity. Expert Rev Respir Med 6: 331–340, 2012. doi: 10.1586/ers.12.22. [DOI] [PubMed] [Google Scholar]
- 7.Vortmann M, Eisner MD. BMI and health status among adults with asthma. Obesity (Silver Spring) 16: 146–152, 2008. doi: 10.1038/oby.2007.7. [DOI] [PubMed] [Google Scholar]
- 8.Sideleva O, Dixon A. The many faces of asthma in obesity. J Cell Biochem 115: 421–426, 2014. doi: 10.1002/jcb.24678. [DOI] [PubMed] [Google Scholar]
- 9.Wenzel SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med 18: 716–725, 2012. doi: 10.1038/nm.2678. [DOI] [PubMed] [Google Scholar]
- 10.Shore SA. Obesity and asthma: lessons from animal models. J Appl Physiol (1985) 102: 516–528, 2007. doi: 10.1152/japplphysiol.00847.2006. [DOI] [PubMed] [Google Scholar]
- 11.Sideleva O, Black K, Dixon AE. Effects of obesity and weight loss on airway physiology and inflammation in asthma. Pulm Pharmacol Ther 26: 455–458, 2013. doi: 10.1016/j.pupt.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ather JL, Poynter ME, Dixon AE. Immunological characteristics and management considerations in obese patients with asthma. Expert Rev Clin Immunol 11: 793–803, 2015. doi: 10.1586/1744666X.2015.1040394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holguin F, Fitzpatrick A. Obesity, asthma, and oxidative stress. J Appl Physiol (1985) 108: 754–759, 2010. doi: 10.1152/japplphysiol.00702.2009. [DOI] [PubMed] [Google Scholar]
- 14.Komakula S, Khatri S, Mermis J, Savill S, Haque S, Rojas M, Brown L, Teague GW, Holguin F. Body mass index is associated with reduced exhaled nitric oxide and higher exhaled 8-isoprostanes in asthmatics. Respir Res 8: 32, 2007. doi: 10.1186/1465-9921-8-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bates JH, Peters U, Daphtary N, MacLean ES, Hodgdon K, Kaminsky DA, Bhatawadekar S, Dixon AE. Altered airway mechanics in the context of obesity and asthma. J Appl Physiol (1985) 130: 36–47, 2021. doi: 10.1152/japplphysiol.00666.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhatawadekar SA, Peters U, Walsh RR, Daphtary N, MacLean ES, Mori V, Hodgdon K, Kinsey CM, Kaminsky DA, Bates JH, Dixon AE. Central airway collapse is related to obesity independent of asthma phenotype. Respirology 26: 334–341, 2021. doi: 10.1111/resp.14005. [DOI] [PubMed] [Google Scholar]
- 17.Dixon AE, Peters U, Walsh R, Daphtary N, MacLean ES, Hodgdon K, Kaminsky DA, Bates JH. Physiological signature of late-onset nonallergic asthma of obesity. ERJ Open Res 6: 00049-2020, 2020. doi: 10.1183/23120541.00049-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ather JL, Chung M, Hoyt LR, Randall MJ, Georgsdottir A, Daphtary NA, Aliyeva MI, Suratt BT, Bates JH, Irvin CG, Russell SR, Forgione PM, Dixon AE, Poynter ME. Weight loss decreases inherent and allergic methacholine hyperresponsiveness in mouse models of diet-induced obese asthma. Am J Respir Cell Mol Biol 55: 176–187, 2016. doi: 10.1165/rcmb.2016-0070OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mathews JA, Wurmbrand AP, Ribeiro L, Neto FL, Shore SA. Induction of IL-17A precedes development of airway hyperresponsiveness during diet-induced obesity and correlates with complement factor D. Front Immunol 5: 440, 2014. doi: 10.3389/fimmu.2014.00440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim HY, Lee HJ, Chang YJ, Pichavant M, Shore SA, Fitzgerald KA, Iwakura Y, Israel E, Bolger K, Faul J, DeKruyff RH, Umetsu DT. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat Med 20: 54–61, 2014. doi: 10.1038/nm.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim JY, Sohn JH, Lee JH, Park JW. Obesity increases airway hyperresponsiveness via the TNF-alpha pathway and treating obesity induces recovery. PLoS One 10: e0116540, 2015. doi: 10.1371/journal.pone.0116540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Williams AS, Chen L, Kasahara DI, Si H, Wurmbrand AP, Shore SA. Obesity and airway responsiveness: role of TNFR2. Pulm Pharmacol Ther 26: 444–454, 2013. doi: 10.1016/j.pupt.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Williams AS, Mathews JA, Kasahara DI, Wurmbrand AP, Chen L, Shore SA. Innate and ozone-induced airway hyperresponsiveness in obese mice: role of TNF-α. Am J Physiol Lung Cell Mol Physiol 308: L1168–L1177, 2015. doi: 10.1152/ajplung.00393.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shore SA, Schwartzman IN, Mellema MS, Flynt L, Imrich A, Johnston RA. Effect of leptin on allergic airway responses in mice. J Allergy Clin Immunol 115: 103–109, 2005. doi: 10.1016/j.jaci.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 25.Shore SA, Terry RD, Flynt L, Xu A, Hug C. Adiponectin attenuates allergen-induced airway inflammation and hyperresponsiveness in mice. J Allergy Clin Immunol 118: 389–395, 2006. doi: 10.1016/j.jaci.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 26.Williams AS, Kasahara DI, Verbout NG, Fedulov AV, Zhu M, Si H, Wurmbrand AP, Hug C, Ranscht B, Shore SA. Role of the adiponectin binding protein, T-cadherin (Cdh13), in allergic airways responses in mice. PLoS One 7: e41088, 2012. doi: 10.1371/journal.pone.0041088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aguilera-Aguirre L, Bacsi A, Saavedra-Molina A, Kurosky A, Sur S, Boldogh I. Mitochondrial dysfunction increases allergic airway inflammation. J Immunol 183: 5379–5387, 2009. doi: 10.4049/jimmunol.0900228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahangari F, Sood A, Ma B, Takyar S, Schuyler M, Qualls C, Dela Cruz CS, Chupp GL, Lee CG, Elias JA. Chitinase 3-like-1 Regulates both visceral fat accumulation and asthma-like Th2 inflammation. Am J Respir Crit Care Med 191: 746–757, 2015. doi: 10.1164/rccm.201405-0796OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arruda AP, Pers BM, Parlakgul G, Guney E, Inouye K, Hotamisligil GS. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat Med 20: 1427–1435, 2014. doi: 10.1038/nm.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flaquer A, Heinzmann A, Rospleszcz S, Mailaparambil B, Dietrich H, Strauch K, Grychtol R. Association study of mitochondrial genetic polymorphisms in asthmatic children. Mitochondrion 14: 49–53, 2014. doi: 10.1016/j.mito.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 31.Jaffer OA, Carter AB, Sanders PN, Dibbern ME, Winters CJ, Murthy S, Ryan AJ, Rokita AG, Prasad AM, Zabner J, Kline JN, Grumbach IM, Anderson ME. Mitochondrial-targeted antioxidant therapy decreases transforming growth factor-beta-mediated collagen production in a murine asthma model. Am J Respir Cell Mol Biol 52: 106–115, 2015. doi: 10.1165/rcmb.2013-0519OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leishangthem GD, Mabalirajan U, Singh VP, Agrawal A, Ghosh B, Dinda AK. Ultrastructural changes of airway in murine models of allergy and diet-induced metabolic syndrome. ISRN Allergy 2013: 261297, 2013. 261297, 2013. doi: 10.1155/2013/261297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taddeo EP, Laker RC, Breen DS, Akhtar YN, Kenwood BM, Liao JA, Zhang M, Fazakerley DJ, Tomsig JL, Harris TE, Keller SR, Chow JD, Lynch KR, Chokki M, Molkentin JD, Turner N, James DE, Yan Z, Hoehn KL. Opening of the mitochondrial permeability transition pore links mitochondrial dysfunction to insulin resistance in skeletal muscle. Mol Metab 3: 124–134, 2014. doi: 10.1016/j.molmet.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zifa E, Daniil Z, Skoumi E, Stavrou M, Papadimitriou K, Terzenidou M, Kostikas K, Bagiatis V, Gourgoulianis KI, Mamuris Z. Mitochondrial genetic background plays a role in increasing risk to asthma. Mol Biol Rep 39: 4697–4708, 2012. doi: 10.1007/s11033-011-1262-8. [DOI] [PubMed] [Google Scholar]
- 35.Liu X, Lin R, Zhao B, Guan R, Li T, Jin R. Correlation between oxidative stress and the NF-kappaB signaling pathway in the pulmonary tissues of obese asthmatic mice. Mol Med Rep 13: 1127–1134, 2016. doi: 10.3892/mmr.2015.4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Manna P, Jain SK. Obesity, oxidative stress, adipose tissue dysfunction, and the associated health risks: causes and therapeutic strategies. Metab Syndr Relat Disord 13: 423–444, 2015. doi: 10.1089/met.2015.0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest 121: 2111–2117, 2011. doi: 10.1172/JCI57132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Virtue S, Vidal-Puig A. Adipose tissue expandability, lipotoxicity and the Metabolic Syndrome–an allostatic perspective. Biochim Biophys Acta 1801: 338–349, 2010. doi: 10.1016/j.bbalip.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 39.Sideleva O, Suratt BT, Black KE, Tharp WG, Pratley RE, Forgione P, Dienz O, Irvin CG, Dixon AE. Obesity and asthma: an inflammatory disease of adipose tissue not the airway. Am J Respir Crit Care Med 186: 598–605, 2012. doi: 10.1164/rccm.201203-0573OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buchwald H, Avidor Y, Braunwald E, Jensen MD, Pories W, Fahrbach K, Schoelles K. Bariatric surgery: a systematic review and meta-analysis. JAMA 292: 1724–1737, 2004. doi: 10.1001/jama.292.14.1724. [DOI] [PubMed] [Google Scholar]
- 41.Saber AA, Elgamal MH, McLeod MK. Bariatric surgery: the past, present, and future. Obes Surg 18: 121–128, 2008. doi: 10.1007/s11695-007-9308-7. [DOI] [PubMed] [Google Scholar]
- 42.Switzer NJ, Mangat HS, Karmali S,. Current trends in obesity: body composition assessment, weight regulation, and emerging techniques in managing severe obesity. J Intervent Gastroenterol 3: 34–36, 2013. doi: 10.7178/jig.106. [DOI] [Google Scholar]
- 43.Pradeepan S, Garrison G, Dixon AE. Obesity in asthma: approaches to treatment. Curr Allergy Asthma Rep 13: 434–442, 2013. doi: 10.1007/s11882-013-0354-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Al-Alwan A, Bates JH, Chapman DG, Kaminsky DA, DeSarno MJ, Irvin CG, Dixon AE. The nonallergic asthma of obesity. A matter of distal lung compliance. Am J Respir Crit Care Med 189: 1494–1502, 2014. doi: 10.1164/rccm.201401-0178OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chapman DG, Irvin CG, Kaminsky DA, Forgione PM, Bates JH, Dixon AE. Influence of distinct asthma phenotypes on lung function following weight loss in the obese. Respirology 19: 1170–1177, 2014. doi: 10.1111/resp.12368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nyenhuis SM, Dixon AE, Ma J. Impact of lifestyle interventions targeting healthy diet, physical activity, and weight loss on asthma in adults: what is the evidence? J Allergy Clin Immunol Pract 6: 751–763, 2018. doi: 10.1016/j.jaip.2017.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dixon AE, Pratley RE, Forgione PM, Kaminsky DA, Whittaker-Leclair LA, Griffes LA, Garudathri J, Raymond D, Poynter ME, Bunn JY, Irvin CG. Effects of obesity and bariatric surgery on airway hyperresponsiveness, asthma control, and inflammation. J Allergy Clin Immunol 128: 508–515, 2011. doi: 10.1016/j.jaci.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gan SS, Talbot ML, Jorgensen JO. Efficacy of surgery in the management of obesity-related type 2 diabetes mellitus. ANZ J Surg 77: 958–962, 2007. doi: 10.1111/j.1445-2197.2007.04290.x. [DOI] [PubMed] [Google Scholar]
- 49.Goldfine AB, Shoelson SE, Aguirre V. Expansion and contraction: treating diabetes with bariatric surgery. Nat Med 15: 616–617, 2009. doi: 10.1038/nm0609-616. [DOI] [PubMed] [Google Scholar]
- 50.Newman JC, Verdin E. beta-hydroxybutyrate: much more than a metabolite. Diabetes Res Clin Pract 106: 173–181, 2014. doi: 10.1016/j.diabres.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Newman JC, Verdin E. Ketone bodies as signaling metabolites. Trends Endocrinol Metab 25: 42–52, 2014. doi: 10.1016/j.tem.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Soeters MR, Sauerwein HP, Faas L, Smeenge M, Duran M, Wanders RJ, Ruiter AF, Ackermans MT, Fliers E, Houten SM, Serlie MJ. Effects of insulin on ketogenesis following fasting in lean and obese men. Obesity 17: 1326–1331, 2009. doi: 10.1038/oby.2008.678. [DOI] [PubMed] [Google Scholar]
- 53.Balasse EO, Fery F. Ketone body production and disposal: effects of fasting, diabetes, and exercise. Diabetes Metab Rev 5: 247–270, 1989. doi: 10.1002/dmr.5610050304. [DOI] [PubMed] [Google Scholar]
- 54.Cheng B, Lu H, Bai B, Chen J. d-beta-Hydroxybutyrate inhibited the apoptosis of PC12 cells induced by H2O2 via inhibiting oxidative stress. Neurochem Int 62: 620–625, 2013. doi: 10.1016/j.neuint.2012.09.011. [DOI] [PubMed] [Google Scholar]
- 55.Haces ML, Hernandez-Fonseca K, Medina-Campos ON, Montiel T, Pedraza-Chaverri J, Massieu L. Antioxidant capacity contributes to protection of ketone bodies against oxidative damage induced during hypoglycemic conditions. Exp Neurol 211: 85–96, 2008. doi: 10.1016/j.expneurol.2007.12.029. [DOI] [PubMed] [Google Scholar]
- 56.Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, D’Agostino D, Planavsky N, Lupfer C, Kanneganti TD, Kang S, Horvath TL, Fahmy TM, Crawford PA, Biragyn A, Alnemri E, Dixit VD. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med 21: 263–269, 2015. doi: 10.1038/nm.3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dixon AE, Holguin F, Sood A, Salome CM, Pratley RE, Beuther DA, Celedon JC, Shore SA, American Thoracic Society Ad Hoc Subcommittee on Obesity and Lung Disease. An official American Thoracic Society Workshop report: obesity and asthma. Proc Am Thorac Soc 7: 325–335, 2010. doi: 10.1513/pats.200903-013ST. [DOI] [PubMed] [Google Scholar]
- 58.Lugogo NL, Kraft M, Dixon AE. Does obesity produce a distinct asthma phenotype? J Appl Physiol (1985) 108: 729–734, 2010. doi: 10.1152/japplphysiol.00845.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.O’Donnell CP, Holguin F, Dixon AE. Pulmonary physiology and pathophysiology in obesity. J Appl Physiol (1985) 108: 197–198, 2010. doi: 10.1152/japplphysiol.01208.2009. [DOI] [PubMed] [Google Scholar]
- 60.Raviv S, Dixon AE, Kalhan R, Shade D, Smith LJ. Effect of obesity on asthma phenotype is dependent upon asthma severity. J Asthma 48: 98–104, 2011. doi: 10.3109/02770903.2010.534220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vidali S, Aminzadeh S, Lambert B, Rutherford T, Sperl W, Kofler B, Feichtinger RG. Mitochondria: the ketogenic diet–a metabolism-based therapy. Int J Biochem Cell Biol 63: 55–59, 2015. doi: 10.1016/j.biocel.2015.01.022. [DOI] [PubMed] [Google Scholar]
- 62.Goldberg EL, Asher JL, Molony RD, Shaw AC, Zeiss CJ, Wang C, Morozova-Roche LA, Herzog RI, Iwasaki A, Dixit VD. Beta-hydroxybutyrate deactivates neutrophil NLRP3 inflammasome to relieve gout flares. Cell Rep 18: 2077–2087, 2017. doi: 10.1016/j.celrep.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fu SP, Li SN, Wang JF, Li Y, Xie SS, Xue WJ, Liu HM, Huang BX, Lv QK, Lei LC, Liu GW, Wang W, Liu JX. BHBA suppresses LPS-induced inflammation in BV-2 cells by inhibiting NF-kappaB activation. Mediators Inflamm 2014: 983401, 2014., doi: 10.1155/2014/983401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Johnson JB, Summer W, Cutler RG, Martin B, Hyun DH, Dixit VD, Pearson M, Nassar M, Telljohann R, Tellejohan R, Maudsley S, Carlson O, John S, Laub DR, Mattson MP. Alternate day calorie restriction improves clinical findings and reduces markers of oxidative stress and inflammation in overweight adults with moderate asthma. Free Radic Biol Med 42: 665–674, 2007. [Erratum in Free Radic Biol Med 43:1348, 2007]. doi: 10.1016/j.freeradbiomed.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hashim SA, VanItallie TB. Ketone body therapy: from the ketogenic diet to the oral administration of ketone ester. J Lipid Res 55: 1818–1826, 2014. doi: 10.1194/jlr.R046599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bates JH, Irvin CG, Farre R, Hantos Z. Oscillation mechanics of the respiratory system. Compr Physiol 1: 1233–1272, 2011. doi: 10.1002/cphy.c100058. [DOI] [PubMed] [Google Scholar]
- 67.Barman M, Unold D, Shifley K, Amir E, Hung K, Bos N, Salzman N. Enteric salmonellosis disrupts the microbial ecology of the murine gastrointestinal tract. Infect Immun 76: 907–915, 2008. doi: 10.1128/IAI.01432-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.West DB, Boozer CN, Moody DL, Atkinson RL. Dietary obesity in nine inbred mouse strains. Am J Physiol Regul Integr Comp Physiol 262: R1025–R1032, 1992. doi: 10.1152/ajpregu.1992.262.6.R1025. [DOI] [PubMed] [Google Scholar]
- 69.Lutz TA, Woods SC. Overview of animal models of obesity. Curr Protocol Pharmacol 58: 5.61.1–5.61.18. 2012. doi: 10.1002/0471141755.ph0561s58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ather JL, Van Der Vliet KE, Mank MM, Reed LF, Dixon AE, Poynter ME. Obese adipose tissue modulates proinflammatory responses of mouse airway epithelial cells. Am J Physiol Regul Integr Comp Physiol 321: R79–R90, 2021. doi: 10.1152/ajpregu.00316.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kennedy AR, Pissios P, Otu H, Roberson R, Xue B, Asakura K, Furukawa N, Marino FE, Liu FF, Kahn BB, Libermann TA, Maratos-Flier E. A high-fat, ketogenic diet induces a unique metabolic state in mice. Am J Physiol Endocrinol Metab 292: E1724–E1739, 2007. [Erratum in Am J Physiol Endocrinol Metab 293: E1846, 2007]. doi: 10.1152/ajpendo.00717.2006. [DOI] [PubMed] [Google Scholar]
- 72.Hummel KP, Dickie MM, Coleman DL. Diabetes, a new mutation in the mouse. Science 153: 1127–1128, 1966. doi: 10.1126/science.153.3740.1127. [DOI] [PubMed] [Google Scholar]
- 73.Ubags ND, Burg E, Antkowiak M, Wallace AM, Dilli E, Bement J, Wargo MJ, Poynter ME, Wouters EF, Suratt BT. A comparative study of lung host defense in murine obesity models. insights into neutrophil function. Am J Respir Cell Mol Biol 55: 188–200, 2016. doi: 10.1165/rcmb.2016-0042OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shore SA. Obesity, airway hyperresponsiveness, and inflammation. J Appl Physiol (1985) 108: 735–743, 2010. doi: 10.1152/japplphysiol.00749.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Julio-Amilpas A, Montiel T, Soto-Tinoco E, Geronimo-Olvera C, Massieu L. Protection of hypoglycemia-induced neuronal death by beta-hydroxybutyrate involves the preservation of energy levels and decreased production of reactive oxygen species. J Cereb Blood Flow Metab 35: 851–860, 2015. doi: 10.1038/jcbfm.2015.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD, Newgard CB, Farese RV Jr, de Cabo R, Ulrich S, Akassoglou K, Verdin E. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 339: 211–214, 2013. doi: 10.1126/science.1227166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Laffel L. Ketone bodies: a review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab Res Rev 15: 412–426, 1999. doi:. [DOI] [PubMed] [Google Scholar]
- 78.Owen OE, Morgan AP, Kemp HG, Sullivan JM, Herrera MG, Cahill GF Jr.. Brain metabolism during fasting. J Clin Invest 46: 1589–1595, 1967. doi: 10.1172/JCI105650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Manuel AM, van de Wetering C, MacPherson M, Erickson C, Murray C, Aboushousha R, van der Velden J, Dixon AE, Poynter ME, Irvin CG, Taatjes DJ, van der Vliet A, Anathy V, Janssen-Heininger YM. Dysregulation of pyruvate kinase M2 promotes inflammation in a mouse model of obese allergic asthma. Am J Respir Cell Mol Biol 64: 709–721, 2021. doi: 10.1165/rcmb.2020-0512OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Qian X, Aboushousha R, van de Wetering C, Chia SB, Amiel E, Schneider RW, van der Velden JL, Lahue KG, Hoagland DA, Casey DT, Daphtary N, Ather JL, Randall MJ, Aliyeva M, Black KE, Chapman DG, Lundblad LK, McMillan DH, Dixon AE, Anathy V, Irvin CG, Poynter ME, Wouters EF, Vacek PM, Henket M, Schleich F, Louis R, van der Vliet A, Janssen-Heininger YM. IL-1/inhibitory kappaB kinase epsilon-induced glycolysis augment epithelial effector function and promote allergic airways disease. J Allergy Clin Immunol 142: 435–450, 2018. e410 doi: 10.1016/j.jaci.2017.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.van de Wetering C, Aboushousha R, Manuel AM, Chia SB, Erickson C, MacPherson MB, van der Velden JL, Anathy V, Dixon AE, Irvin CG, Poynter ME, van der Vliet A, Wouters EF, Reynaert NL, Janssen-Heininger YM. Pyruvate kinase M2 promotes expression of proinflammatory mediators in house dust mite-induced allergic airways disease. J Immunol 204: 763–774, 2020. doi: 10.4049/jimmunol.1901086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Anandan C, Nurmatov U, van Schayck OC, Sheikh A. Is the prevalence of asthma declining? Systematic review of epidemiological studies. Allergy 65: 152–167, 2010. doi: 10.1111/j.1398-9995.2009.02244.x. [DOI] [PubMed] [Google Scholar]
- 83.Moore WC, Bleecker ER, Curran-Everett D, Erzurum SC, Ameredes BT, Bacharier L, Calhoun WJ, Castro M, Chung KF, Clark MP, Dweik RA, Fitzpatrick AM, Gaston B, Hew M, Hussain I, Jarjour NN, Israel E, Levy BD, Murphy JR, Peters SP, Teague WG, Meyers DA, Busse WW, Wenzel SE. Characterization of the severe asthma phenotype by the National Heart, Lung, and Blood Institute’s Severe Asthma Research Program. J Allergy Clin Immunol 119: 405–413, 2007. doi: 10.1016/j.jaci.2006.11.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Moore WC, Meyers DA, Wenzel SE, Teague WG, Li H, Li X, D’Agostino R Jr, Castro M, Curran-Everett D, Fitzpatrick AM, Gaston B, Jarjour NN, Sorkness R, Calhoun WJ, Chung KF, Comhair SA, Dweik RA, Israel E, Peters SP, Busse WW, Erzurum SC, Bleecker ER, National Heart, Lung, and Blood Institute’s Severe Asthma Research Program. Identification of asthma phenotypes using cluster analysis in the Severe Asthma Research Program. Am J Respir Crit Care Med 181: 315–323, 2010. doi: 10.1164/rccm.200906-0896OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bates JH. Physiological mechanisms of airway hyperresponsiveness in obese asthma. Am J Respir Cell Mol Biol 54: 618–623, 2016. doi: 10.1165/rcmb.2016-0019PS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bates JH. Constant phase model of impedance. In: Lung Mechanics an Inverse Modeling Approach. Cambridge, UK: Cambridge University Press, 2009. [Google Scholar]
- 87.Wagers S, Lundblad LK, Ekman M, Irvin CG, Bates JH. The allergic mouse model of asthma: normal smooth muscle in an abnormal lung? J Appl Physiol (1985) 96: 2019–2027, 2004. doi: 10.1152/japplphysiol.00924.2003. [DOI] [PubMed] [Google Scholar]
- 88.Aberle J, Reining F, Dannheim V, Flitsch J, Klinge A, Mann O. Metformin after bariatric surgery–an acid problem. Exp Clin Endocrinol Diabetes 120: 152–153, 2012. doi: 10.1055/s-0031-1285911. [DOI] [PubMed] [Google Scholar]
- 89.Lund TM, Ploug KB, Iversen A, Jensen AA, Jansen-Olesen I. The metabolic impact of beta-hydroxybutyrate on neurotransmission: Reduced glycolysis mediates changes in calcium responses and KATP channel receptor sensitivity. J Neurochem 132: 520–531, 2015. doi: 10.1111/jnc.12975. [DOI] [PubMed] [Google Scholar]
- 90.Kesl SL, Poff AM, Ward NP, Fiorelli TN, Ari C, Van Putten AJ, Sherwood JW, Arnold P, D’Agostino DP. Effects of exogenous ketone supplementation on blood ketone, glucose, triglyceride, and lipoprotein levels in Sprague-Dawley rats. Nutr Metab (Lond) 13: 9, 2016. doi: 10.1186/s12986-016-0069-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cox PJ, Kirk T, Ashmore T, Willerton K, Evans R, Smith A, Murray AJ, Stubbs B, West J, McLure SW, King MT, Dodd MS, Holloway C, Neubauer S, Drawer S, Veech RL, Griffin JL, Clarke K. Nutritional ketosis alters fuel preference and thereby endurance performance in athletes. Cell Metab 24: 256–268, 2016. doi: 10.1016/j.cmet.2016.07.010. [DOI] [PubMed] [Google Scholar]
- 92.Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, Redmann V, Freitas TC, Blagih J, van der Windt GJ, Artyomov MN, Jones RG, Pearce EL, Pearce EJ. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat Immunol 15: 323–332, 2014. doi: 10.1038/ni.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, Pearce EJ. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115: 4742–4749, 2010. doi: 10.1182/blood-2009-10-249540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Carvajal K, Balderas-Villalobos J, Bello-Sanchez MD, Phillips-Farfan B, Molina-Munoz T, Aldana-Quintero H, Gomez-Viquez NL. Ca(2+) mishandling and cardiac dysfunction in obesity and insulin resistance: role of oxidative stress. Cell Calcium 56: 408–415, 2014. doi: 10.1016/j.ceca.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 95.Lee JH, Kagan E. Role of nicotinamide adenine dinucleotide phosphate oxidase in mediating vesicant-induced interleukin-6 secretion in human airway epithelial cells. Am J Respir Cell Mol Biol 50: 713–722, 2014. doi: 10.1165/rcmb.2012-0527OC. [DOI] [PubMed] [Google Scholar]
- 96.Pelaia G, Cuda G, Vatrella A, Gallelli L, Fratto D, Gioffre V, D’Agostino B, Caputi M, Maselli R, Rossi F, Costanzo FS, Marsico SA. Effects of hydrogen peroxide on MAPK activation, IL-8 production and cell viability in primary cultures of human bronchial epithelial cells. J Cell Biochem 93: 142–152, 2004. doi: 10.1002/jcb.20124. [DOI] [PubMed] [Google Scholar]
- 97.Rahman I, Adcock IM. Oxidative stress and redox regulation of lung inflammation in COPD. Eur Respir J 28: 219–242, 2006. doi: 10.1183/09031936.06.00053805. [DOI] [PubMed] [Google Scholar]
- 98.Chiarella SE, Soberanes S, Urich D, Morales-Nebreda L, Nigdelioglu R, Green D, Young JB, Gonzalez A, Rosario C, Misharin AV, Ghio AJ, Wunderink RG, Donnelly HK, Radigan KA, Perlman H, Chandel NS, Budinger GR, Mutlu GM. beta(2)-Adrenergic agonists augment air pollution-induced IL-6 release and thrombosis. J Clin Invest 124: 2935–2946, 2014. doi: 10.1172/JCI75157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Johnston RA, Zhu M, Rivera-Sanchez YM, Lu FL, Theman TA, Flynt L, Shore SA. Allergic airway responses in obese mice. Am J Respir Crit Care Med 176: 650–658, 2007. doi: 10.1164/rccm.200702-323OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dahm PH, Richards JB, Karmouty-Quintana H, Cromar KR, Sur S, Price RE, Malik F, Spencer CY, Barreno RX, Hashmi SS, Blackburn MR, Haque IU, Johnston RA. Effect of antigen sensitization and challenge on oscillatory mechanics of the lung and pulmonary inflammation in obese carboxypeptidase E-deficient mice. Am J Physiol Regul Integr Comp Physiol 307: R621–R633, 2014. doi: 10.1152/ajpregu.00205.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Calixto MC, Lintomen L, Schenka A, Saad MJ, Zanesco A, Antunes E. Obesity enhances eosinophilic inflammation in a murine model of allergic asthma. Br J Pharmacol 159: 617–625, 2010. doi: 10.1111/j.1476-5381.2009.00560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lintomen L, Calixto MC, Schenka A, Antunes E. Allergen-induced bone marrow eosinophilopoiesis and airways eosinophilic inflammation in leptin-deficient ob/ob mice. Obesity (Silver Spring) 20: 1959–1965, 2012. doi: 10.1038/oby.2012.93. [DOI] [PubMed] [Google Scholar]
- 103.Duchene B, Caffry S, Kaminsky DA, Que LG, Poynter ME, Dixon AE. Functional significance of 8-isoprostanes in sinonasal disease and asthma. Respir Med 185: 106506, 2021. doi: 10.1016/j.rmed.2021.106506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nyenwe EA, Kitabchi AE. The evolution of diabetic ketoacidosis: an update of its etiology, pathogenesis and management. Metab Clin Expe 65: 507–521, 2016. doi: 10.1016/j.metabol.2015.12.007. [DOI] [PubMed] [Google Scholar]
- 105.Veech RL. Ketone ester effects on metabolism and transcription. J Lipid Res 55: 2004–2006, 2014. doi: 10.1194/jlr.R046292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Clarke K, Tchabanenko K, Pawlosky R, Carter E, Todd King M, Musa-Veloso K, Ho M, Roberts A, Robertson J, Vanitallie TB, Veech RL. Kinetics, safety and tolerability of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate in healthy adult subjects. Regul Toxicol Pharmacol 63: 401–408, 2012. doi: 10.1016/j.yrtph.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kashiwaya Y, Takeshima T, Mori N, Nakashima K, Clarke K, Veech RL. D-beta-hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proc Natl Acad Sci U S A 97: 5440–5444, 2000. doi: 10.1073/pnas.97.10.5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Poplawski MM, Mastaitis JW, Isoda F, Grosjean F, Zheng F, Mobbs CV. Reversal of diabetic nephropathy by a ketogenic diet. PLoS One 6: e18604, 2011. doi: 10.1371/journal.pone.0018604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Masuda R, Monahan JW, Kashiwaya Y. D-beta-hydroxybutyrate is neuroprotective against hypoxia in serum-free hippocampal primary cultures. J Neurosci Res 80: 501–509, 2005. doi: 10.1002/jnr.20464. [DOI] [PubMed] [Google Scholar]
- 110.Meidenbauer JJ, Roberts MF. Reduced glucose utilization underlies seizure protection with dietary therapy in epileptic EL mice. Epilepsy Behav 39: 48–54, 2014. doi: 10.1016/j.yebeh.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Graff EC, Fang H, Wanders D, Judd RL. Anti-inflammatory effects of the hydroxycarboxylic acid receptor 2. Metabolism 65: 102–113, 2016. doi: 10.1016/j.metabol.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 112.Kimura I, Inoue D, Maeda T, Hara T, Ichimura A, Miyauchi S, Kobayashi M, Hirasawa A, Tsujimoto G. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41). Proc Natl Acad Sci U S A 108: 8030–8035, 2011. doi: 10.1073/pnas.1016088108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Xie Z, Zhang D, Chung D, Tang Z, Huang H, Dai L, Qi S, Li J, Colak G, Chen Y, Xia C, Peng C, Ruan H, Kirkey M, Wang D, Jensen LM, Kwon OK, Lee S, Pletcher SD, Tan M, Lombard DB, White KP, Zhao H, Li J, Roeder RG, Yang X, Zhao Y. Metabolic regulation of gene expression by histone lysine beta-hydroxybutyrylation. Mol Cell 62: 194–206, 2016. doi: 10.1016/j.molcel.2016.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Leguillette R, Laviolette M, Bergeron C, Zitouni N, Kogut P, Solway J, Kachmar L, Hamid Q, Lauzon AM. Myosin, transgelin, and myosin light chain kinase: expression and function in asthma. Am J Respir Crit Care Med 179: 194–204, 2009. doi: 10.1164/rccm.200609-1367OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Aboushousha R, Elko E, Chia SB, Manuel AM, van de Wetering C, van der Velden J, MacPherson M, Erickson C, Reisz JA, D’Alessandro A, Wouters EF, Reynaert NL, Lam YW, Anathy V, van der Vliet A, Seward DJ, Ymw JH. Glutathionylation chemistry promotes interleukin-1 beta-mediated glycolytic reprogramming and pro-inflammatory signaling in lung epithelial cells. FASEB J 35: e21525, 2021. doi: 10.1096/fj.202002687rr. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.van de Wetering C, Manuel AM, Sharafi M, Aboushousha R, Qian X, Erickson C, MacPherson M, Chan G, Adcock IM, ZounematKermani N, Schleich F, Louis R, Bohrnsen E, D’Alessandro A, Wouters EF, Reynaert NL, Li J, Wolf CR, Henderson CJ, Lundblad LK, Poynter ME, Dixon AE, Irvin CG, van der Vliet A, van der Velden JL, Janssen-Heininger YM. Glutathione-S-transferase P promotes glycolysis in asthma in association with oxidation of pyruvate kinase M2. Redox Biol 47: 102160, 2021. doi: 10.1016/j.redox.2021.102160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Marsland BJ, Trompette A, Gollwitzer ES. The gut-lung axis in respiratory disease. Ann Am Thorac Soc 12: S150–156, 2015. doi: 10.1513/annalsats.201503-133aw. [DOI] [PubMed] [Google Scholar]
- 118.Herrick KJ, Hippen AR, Kalscheur KF, Schingoethe DJ, Casper DP, Moreland SC, van Eys JE. Single-dose infusion of sodium butyrate, but not lactose, increases plasma beta-hydroxybutyrate and insulin in lactating dairy cows. J Dairy Sci 100: 757–768, 2017. doi: 10.3168/jds.2016-11634. [DOI] [PubMed] [Google Scholar]
- 119.Israel E, Denlinger LC, Bacharier LB, LaVange LM, Moore WC, Peters MC, et al. PrecISE: Precision Medicine in Severe Asthma: an adaptive platform trial with biomarker ascertainment. J Allergy Clin Immunol 147: 1594–1601, 2021. doi: 10.1016/j.jaci.2021.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support this study are available from the author on reasonable request.