

Keywords: bile acid, FXR, motility, secretion, TGR5
Abstract
Bile acids (BAs) are known to be important regulators of intestinal motility and epithelial fluid and electrolyte transport. Over the past two decades, significant advances in identifying and characterizing the receptors, transporters, and ion channels targeted by BAs have led to exciting new insights into the molecular mechanisms involved in these processes. Our appreciation of BAs, their receptors, and BA-modulated ion channels as potential targets for the development of new approaches to treat intestinal motility and transport disorders is increasing. In the current review, we aim to summarize recent advances in our knowledge of the different BA receptors and BA-modulated ion channels present in the gastrointestinal system. We discuss how they regulate motility and epithelial transport, their roles in pathogenesis, and their therapeutic potential in a range of gastrointestinal diseases.
INTRODUCTION
Bile acids (BAs) are amphipathic steroidal molecules found as a major component of bile. BAs are synthesized in hepatocytes, stored in the gall bladder, and released into the small intestine in response to the ingestion of a meal. Classically, BAs are known for their roles in digestion through facilitating the solubilization of fatty acids, stimulating bile flow and intestinal motility, regulating glucose levels, and promoting cholesterol secretion and catabolism (1). BAs modulate their own synthesis, transport, and detoxification and aid in the maintenance of a healthy gut microbiome. Primary BAs, such as cholic acid (CA) and chenodeoxycholic acid (CDCA), are synthesized in the liver and the vast majority are conjugated (e.g., tauro-CA or glyco-CA) before their release into the ileum. In the terminal ileum, ∼95% of released BAs are reabsorbed through the ileal BA transporter, IBAT (also known as apical sodium-dependent BA transporter, ASBT). BAs that are not reabsorbed in the ileum enter the colon where they undergo structural modifications catalyzed by anaerobic bacterial enzymes, such as 7α-dehydroxylase, to become secondary BAs (e.g., deoxycholic acid, DCA and lithocholic acid, LCA) (2).
Normally, the BAs are reabsorbed in the terminal ileum and are recycled back to the liver via the enterohepatic circulation. However, disruptions to the enterohepatic circulation, whether due to increased BA synthesis or defective re-uptake in the small intestine, results in BA accumulation in the colon. When present at physiological levels, BAs act as important signaling molecules that exert their actions through specific receptors located on the cell surface and in the nucleus. However, given their amphipathic nature and detergent-like properties, increased levels of BAs in the colon can have deleterious effects on the lipid composition of cellular membranes in this region and exert cytotoxic and membranolytic effects (3, 4). Given the detergent-like properties of BAs, caution must be taken when interpreting studies where high concentrations of exogenous BAs have been used. Unless specifically assessed, any observed effects may be attributable to deleterious effects of BAs on the lipid composition of cellular membranes, receptor-mediated effects, or a combination of the two. In this review, we aim to summarize recent advances in the field of BA signaling in the gut with a particular emphasis on how they regulate transport function and motility, and consideration of the effects of physiological versus pathophysiological concentrations of BAs.
BILE ACID RECEPTORS
The discovery that BAs can act as transmitters and mediate cellular signaling processes has been a significant advancement to our understanding of how they exert their functional effects. BAs selectively activate a broad range of G protein-coupled receptors (GPCRs), nuclear receptors, and ion channels. Strategies that indirectly alter their actions at these sites are already used to treat a range of conditions. For example, modulation of luminal BA levels, either through sequestration or altered transport, is currently the main therapeutic approach to treat BA-associated disorders, such as cholestatic itch or gallstones. However, the potential for directly targeting BA-activated receptors and ion channels now offers additional opportunities for therapy. This review provides a summary of GPCRs, nuclear receptors, and ion channels that are influenced by BAs. It aims to contextualize how the distribution, functions, and regulation of BA receptors relate to gastrointestinal (GI) physiology and how their dysregulation is implicated in digestive disease and disorders.
BILE ACID INTERACTION WITH G PROTEIN-COUPLED RECEPTORS
GPCRs are dynamic signaling proteins that are mainly located on the cell surface. They enable cells to detect and respond to a diverse array of extracellular ligands and stimuli. It is now known that BAs can activate several distinct GPCRs, including established bona fide BA receptors (Takeda G protein-coupled receptor 5, TGR5), receptors that interact with BAs in addition to their primary orthosteric agonists (e.g., muscarinic receptors), and putative BA receptors (e.g., Mrgprs). A summary of the current findings is provided in Table 1, with a more detailed description listed in the following sections.
Table 1.
Effects of BAs on G protein-coupled receptors and nuclear receptors
| Receptor | Bile Acid | Concentration | Tissue Preparation | Response | Effect | Solvent | Refs. |
|---|---|---|---|---|---|---|---|
| G protein-coupled receptors | |||||||
| TGR5 (GPR130, GpBAR1) | DCA OA |
1–100 µM 100 µM |
Proximal colon from tgr5-WT and KO mice | ↑ WT KO only 100 µM at DCA |
5-HT and CGRP from EC cells to stimulate peristaltic reflex | 0.1% Ethanol, distilled water | (5) |
| DCA TLCA CCDC |
100 µM | DRG neurons innervating colon (mouse) | ↑ ↑ ↑ |
Increased intracellular Ca2+ in subpopulations of neurons (%) DCA ∼20%, TLCA∼ 25%, CCDC∼30% |
NA | (6) | |
| CCDC | 100 µM, 100 µL rectal enema | Spinal cord (mouse) | ↑ | Phosphorylated MAP-kinase-ERK-1/2 immunoreactivity (pERK-IR) enhanced in response to colorectal distention | Saline | ||
| INT-777 | 10–100 µM | Proximal colon (mouse) | ↑ | Increased transepithelial resistance and reduced short circuit current, reduced with TTX or neuron-free preparation | DMSO | (7) | |
| UDCA | 3–60 µM | - | Weak TGR5 agonist | ||||
| LCA, DCA | 10 and 30 µM | Enteroendocrine cell line, STC-1 | ↑ | Promote GLP-1 secretion | DMSO | (8) | |
| Cells transfected withTGR5 siRNA | - | Reduced LCA responses at 10 and 30 µM | |||||
| Cells transfected with TGR5 cDNA | ↑ | Increased LCA responses at 30 µM | |||||
| Range of conjugated & unconjugated BAs | Concentration curve | COS-7 monkey kidney cells expressing human or rat TGR5 | ↑ | Increased cAMP production rank order of potency LCA>DCA>CDCA UDCA and CA ∼20% efficacy at 10 µM | Modified Krebs-Ringer buffer | (9) | |
| UDCA | 10 mM | -UDCA | GLP-1 and PYY release | ||||
| BA mixture | Total BA ∼9 mM, 1 mM each | In vivo intraluminal tgr5-WT and KO mice | BA mix: ↑ WT - KO |
Isotonic saline | |||
| LCA RO5527239 (TGR5 agonist) | Concentration curve | CHO cells transfected with TGR5 | ↑ | Increased cAMP production EC50 = 457 and 3.6 nM EMax∼110% both |
NA | (10) | |
| DCA RO5527239 | Concentration curve | STC-1 | ↑ | Increased cAMP production EC50 = 1.59 µM and 321 nM |
|||
| GLP-1 release EC50 = 11 µM and 321 nM |
|||||||
| MrgprX4 | DCA, UDCA CDCA, CA, OA | 2–100 µM | HEK293 cells | ↑ | Increased [Ca2]+i | Water, DMSO, or 0.1 M NaOHNA | (11) |
| DCA UDCA |
10 µM 100 µM |
DRG from +X4 humanized mice | ↑ | ∼5%–6% of +X4 sensory neurons activated by both | |||
| DCA, t-DCA, UDCA CDCA | 1–2 mM | In vivo +X4 humanized mice | ↑ | Increased cholestatic itch | |||
| Muscarinic M2 | t-CDCA, g-DCA, t-DCA, t-CA | 100 µM | Ventricular neonatal rat myocytes | ↑ | Reduced contraction (Gi coupled) (role in TGR5 mediated release of cAMP found to be independent of contraction response) | L-15 media | (12) |
| TCA | 0–100 µM | Ventricular neonatal rat myocytes | Partial agonist | Radioligand binding, Kd = 17 µM |
DMSO HBSS | (13) | |
| Reduced cAMP production (30% of carbachol response) | |||||||
| Reduced contraction (0.2 and 1 mM) | |||||||
| Muscarinic M3 | CDCA, LCA, g-LCA, t-LCA, g-DCA, t-DCA, UDCA | 30 µM | Ex vivo, precision-cut lung slices | ↓ | Inhibited acetylcholine contractile responses t-LCA IC50 = 3.2 µM | NA | (14) |
| S1PR2 | t-CA, t-DCA, g-DCA, g-CA, t-UDCA | 50–100 µM | Primary rat hepatocytes | ↑ | Activation of ERK1/2 and AKT | DMSO | (15) |
| t-CA | 100 µM | MLE | Activation of ERK1/2 and AKT Cell proliferation and migration |
PBS | (16) | ||
| FPR | CDCA | 25–400 µM | Human monocytes | ↓ | Inhibited monocyte chemotaxis to fMLP IC50 = 100 µM | Ethanol | (17) |
| Human monocytes | Inhibited monocyte Ca flux by fMLP | ||||||
| EFTR cell | Inhibited cell migration by fMLP IC50 = 125 µM | ||||||
| EFTR cell | Inhibited Ca2+ flux by fMLP IC50 = 50 µM | ||||||
| Human monocytes EFTR cell | Radioligand fMLP binding IC50 = 140 µM |
||||||
| DCA | 25–200 µM | Human monocytes and neutrophils | ↓ | Inhibited fMLP induced chemotaxis IC50 = 100 µM | PBS | (18) | |
| 25 µM | Inhibited fMLP induced Ca2+ mobilization | ||||||
| 5–200 µM | Radioligand fMLP binding | ||||||
| Nuclear receptors | |||||||
| FXR | CDCA>DCA and LCA | 50 µM concentration curve for CDCA | CV-1 cells with rat FXR HepG2 cells with human FXR |
↑ | Transactivation of FXR with luciferase reporter CDCA EC50 = 50 µM and 10 µM rat and human FXR |
Ethanol | (19) |
| CDCA>DCA and LCA | 100 µM | CV-1 cells | ↑ | Reporter gene assay. Increased CAT activity |
NA | (20) | |
| ↑ | |||||||
| CDCA, g-CDCA, t-CDCA | FRET ligand sensing assay Increased relative fluorescence CDCA EC50 = 4.5 µM g- and t-CDCA EC50 = 10 µM |
||||||
| CA GW4062 (FXR agonist) | 200 mg/kg 100 mg/kg |
Mice treated in vivo 72 h before to tissue harvest | Ileum, jejunum and duodenum ↑ Liver- |
FGF-15 mRNA present in ileum, jejunum and duodenum | NA | (21, 22) | |
| CDCA | Concentration curve | Human ileum and colon mucosal biopsies | Ileum ↑ Colon– |
FGF19 expression EC50 = 20 µM |
|||
| CA DCA LCA |
50 µM | 6-h incubation | Comparison with matched con. CDCA | 80% of CDCA response 40% 4% |
|||
| OCA | 20 µM | 5 times greater than CDCA response | |||||
| PXR | LCA 3-keto-LCA |
Concentration curve | CV-1 cells with human PXR | ↑ ↑ |
Scintillation proximity binding assay IC50 = 9 and 15 µM |
NA | (23) |
| LCA | 100 µM | CV-1 cells with human or mouse PXR and reporter plasmid | ↑ (Human) | Reporter gene assay. Increased CAT activity | |||
| 3-keto-LCA | (Cyp3a23)2-tk-CAT | ↑ | |||||
| CDCA | - | ||||||
| CA | - | ||||||
| DCA | - | ||||||
| CDCA DCA LCA |
100 µM | CV-1 cells with human and rodent PXR and reporter plasmids | ↑ ↑ ↑ |
Reporter gene activity and CYP3A4 reporter gene activity | NA | (24) | |
| LCA | 8 mg/day for 4 days, oral gavage | PXR-WT and KO mice in vivo | ↑ WT -KO |
Northern blot analysis CYP3A11 mRNA | |||
| VDR | LCA 3-keto-LCA |
Concentration curve | HEK293 cells transfected with VDR | ↑ | GAL4-receptor luciferase assay EC50 = 8 and 3 µM |
NA | (25) |
| LCA 3-keto-LCA |
Monkey kidney COS-7 | ↑ | Competitive binding assay [3H]1,25(OH)2D3 Ki = 29 and 8 µM |
||||
| CA, CDCA | |||||||
| LCA | 0.8 mmol/kg, oral gavage | Ileum tissue of Vdr WT and KO | ↑ WT -KO |
Induces mRNA expression of Cyp24a1 | Ethanol and corn oil | (26) | |
CA, cholic acid; CDCA, chenodeoxycholic acid; DCA, deoxycholic acid; LCA, lithocholic acid. 3-Keto-LCA is the major metabolite of LCA in rats. UDCA, ursodeoxycholic acid; t-, tauro-conjugated; g-, glyco-conjugated bile acid. Cell lines: rat basophil leukemia cell line transfected with FPR EFTR cell; mouse large cholangiocytes MLE; monkey kidney CV-1 cells; human hepatoma HepG2; enteroendocrine cell line, STC-1. Responses: activation, ↑; inhibition, ↓; no change, -. Solvents: NA, no information available; DMSO stock, dimethyl sulfoxide (DMSO).
Takeda G Protein-Coupled Receptor 5
TGR5, also known as bile acid receptor, GPBA, GpBAR1, or M-BAR1 is activated by both primary and secondary BAs. TGR5 is Gs-coupled meaning activation of the receptor stimulates adenylyl cyclase to produce cAMP. TGR5 is expressed by a diverse range of cell types in tissues including, but not limited to, gastrointestinal, liver, brown adipose, skeletal muscle, and placental. Within the gastrointestinal tract, TGR5 is expressed by epithelia, enteroendocrine cell subsets, enteric neurons, enteric glia, smooth muscle, monocytes/macrophages, and extrinsic afferents (6, 27). The roles of TGR5 in intestinal motility and secretion will be discussed in detail throughout this review. TGR5 has additional signaling roles in many other gastrointestinal processes that have not been discussed here including inflammation and the development of cancers, and visceral pain (6, 28).
Mas-Related G Protein-Coupled Receptor X4
Mrgprs are a family of orphan GPCRs consisting of more than 50 members. There are several subfamilies, with mouse and human receptors characterized separately. For example, sensory neurons in the dorsal root ganglia (DRG) of mice express MRGPRA1, whereas the ortholog Mas-related G protein-coupled receptor X4 (MRGPRX4) is expressed in human tissues (29). Their contributions to sensation have not been fully characterized, but roles in nociception and itch have been identified. In patients with the gall bladder condition, primary biliary cholangitis, elevated levels of blood BAs are known to cause the symptom of cholestatic itch and BA sequestrants or IBAT inhibitors are used to treat the condition. However, when studying BA activation of the MRGPR family of receptors, interpretation of results may be complicated by differences in the responses of the mouse and human orthologs. For example, recent evidence from in vivo studies in transgenic mice indicates that BA-induced itch is not dependent on the expression of several Mrgprs. Mice lacking a cluster of Mrgpr genes (Mrgpr-cluster–/–) exhibit itch in response to DCA in a similar manner to control mice (30). HEK cells transfected with the mouse ortholog MRGPRA1 do not respond to BAs as measured by Ca2+ signaling. In contrast, cells expressing the human MRGPRX4 are activated by BAs, and this is the only human receptor to respond (11). Similarly, mice expressing the humanized receptor demonstrate a greater itch response following acute injection of DCA or ursodeoxycholic acid (UDCA) compared with control mice. This same study showed that control mice responded comparably to both BAs, and this is evidence that a Mrgpr-mediated response may not involve activation of TGR5, since UDCA is a poor agonist for the latter receptor (11). It should be noted that longer administration of UDCA is used to treat pruritus associated with cholestasis, with suggested benefits such as stimulation of biliary BA excretion, replacement of the toxic endogenous BA pool, and stabilization of plasma membranes and cytoprotection of cholangiocytes (31).
In the context of the digestive system, the expression of Mrgprs has been described predominantly in primary afferent DRG neurons innervating the intestine. However, some studies have characterized mRNA expression in healthy or inflamed GI tissues. More specifically, expressions of MrgA4, MrgB2, MrgB8, MrgE, and MrgF have been demonstrated by immunohistochemistry in neurochemically defined subsets of enteric neurons (32–35). Activation of Mrgprs in cells such as mast cells could in turn activate closely associated primary afferent and sensory neurons through release of mediators including histamine and proteases. Mrgprs can interact with other cell surface receptors, such as transient receptor potential (TRP) channels to promote cellular responses (36). The relative importance of these interactions in the context of other BA-activated receptors and ion channels is not known. There are populations of primary afferent neurons innervating the gut wall that coexpress Mrgprs and TGR5. Given that both receptors are involved in histamine-independent itch pathways in the skin, this has led some researchers to postulate their function as a visceral “irritant sensing system,” suggesting their contribution to pain and symptomology in irritable bowel syndrome (IBS) (6). In a recent study in mice by Castro et al., chronic visceral hypersensitivity (CVH) responses to colorectal distention were induced by intraluminal application of DCA, and agonists of MRGPRA3 and MRGPRC11. Whether DCA was acting on Mrgpr receptors was not studied here. Although the expression profiles of TGR5, MRGPRA3, and MRGPRC11 were identified in mouse sensory DRG neurons, expression of MRGPRA1 was not mentioned. The same study examined the response profiles of human DRG neurons. Although they found that cells responded to TGR5-preferring agonists they did not study receptor specificity for these responses and this may be due to limitations in the pharmacological tools currently available. The expression profiles of MRGPRX4 were also not mentioned (6).
Muscarinic Acetylcholine Receptors
Some BAs can interact with muscarinic receptors to influence cholinergic signaling. This is likely the result of structural similarities between some BAs and acetylcholine (ACh), as has been identified for lithocholic acid conjugates (37, 38). Secondary BAs, including taurolithocholic acid (t-LCA), block cholinergic-induced contractions of mouse and human airway smooth muscle evoked by either exogenous ACh or by electrical stimulation of parasympathetic nerve terminals, possibly by acting as antagonists of M3 muscarinic receptors (14). Urso et al. found that this was not through specific activation of TGR5, as BAs continued to relax preconstricted smooth muscle tissues from tgr5−/− mice, and selective TGR5 small molecule agonists did not relax tissue from wild-type animals. A role for BAs in muscarinic signaling is further supported by studies in cardiomyocytes, where taurocholate inhibited cAMP-production in a pertussis toxin-sensitive manner to limit contraction. This response was prevented upon pharmacological inhibition or siRNA knockdown of the M2 muscarinic receptor (12, 13). Muscarinic receptors have prominent roles in gastrointestinal motility and secretion. Therefore, it is surprising that not more is known about the implications of BAs interacting with muscarinic receptors in these processes.
Sphingosine-1-Phosphate Receptor 2
Sphingosine-1-phosphate receptor 2 (S1PR2) is one of a series of receptors that respond to the bioactive lipid mediator, lysosphingolipid (S1P). S1PRs are ubiquitously expressed in different tissues and this is reflected by their roles in many processes including angiogenesis, lymphangiogenesis, cell proliferation and motility, immune cell function, and lipid metabolism (39). S1PR2 also has a role as a BA receptor (40). In GPCR screening assays, rodent hepatocytes responded to BAs through extracellular signal-regulated kinase (ERK)1/2 and protein kinase B/Akt signaling. These BA-mediated responses were reduced by an S1PR2-selective antagonist, knockdown of the receptor, and using cells from S1PR2 knockout mice (15). Furthermore, the proliferation of cholangiocytes and onset of liver fibrosis in response to experimental bile duct ligation were attenuated in S1PR2-deficient mice, consistent with the proposed interaction of S1PR2 and BAs (16).
Formyl Peptide Receptors
Formyl peptide receptors (FPRs) are a group of GPCRs involved in innate immune responses. N-Formyl-methionyl-leucyl-phenylalanine (fMLP), a major chemotactic factor, is an agonist with different binding affinities for the three FPRs. fMLP binds with high- and low affinities to formyl peptide receptor (FPR) and formyl peptide receptor-like type-1 (FPRL-1), respectively. However, formyl peptide receptor-like type-2 (FPRL-2) does not bind to N-formyl peptides. Roles for FPRs in BA-induced signaling have been demonstrated in studies on human monocytes and using human embryonic kidney cells (HEK-293) transfected with FPR and FPRL-1. In these cells, pretreatment with CDCA and DCA inhibited fMLP binding to its receptors and associated elevations in intracellular Ca2+ (17, 18). In these same studies, cell migration stimulated by fMLP was inhibited by CDCA and DCA treatment (17, 18). Although these studies did not assess whether TGR5 mediates BA-dependent inhibition of chemotaxis and calcium signaling in these cells, computational modeling experiments have demonstrated antagonist binding sites for DCA on the FPR (41), supporting the idea that direct inhibition of these BA receptors is involved. Intestinal microbiota are known to produce fMLP, and recent studies of interactions between the microbiome, FPRs, and gut motility suggest that FPR-expressing enteric neurons are important mediators of probiotic-induced gut motility (42). It is therefore plausible that BAs could modulate gastrointestinal physiology through actions on neuronal FPRs. However, direct experimental evidence for such interactions is currently lacking. Furthermore, since FPRs are also expressed by intestinal neutrophils with different expression patterns noted for cells isolated from tissues of patients with Crohn’s disease (43), BA/FPR interactions on intestinal immune cells could have important implications for pathogenesis of inflammatory bowel diseases (IBDs).
NUCLEAR RECEPTORS FOR BILE ACIDS
Nuclear receptors are a family of ligand-activated transcription factors, comprised of a ligand-binding domain and a DNA-binding domain to promote or suppress transcription. Activators for these receptors include steroid hormones such as estrogen and progesterone and lipid-soluble mediators such as retinoic acid and vitamin D. In this way they allow factors such as hormones or nutrients to modulate gene expression (44). BAs are ligands for several nuclear receptors with roles in glucose, lipid, and energy metabolism (45). These receptors regulate BA levels in the enterohepatic circulation and mediate processes in inflammation. They represent novel targets for a range of therapies for metabolic, cholestatic liver, and inflammatory bowel diseases (46, 47).
Farnesoid X Receptor
Farnesol was originally thought to be the cognate ligand for farnesoid X receptor (FXR), but this agonist only activates the receptor at supraphysiological concentrations (48). In contrast, conjugated and unconjugated BAs activate FXR at physiological levels with CDCA being one of the most potent agonists (19, 20, 49). FXR is highly expressed in liver and intestinal tissues and has significant roles in mediating both metabolic processes and BA homeostasis. Studies of FXR knockout mice provide evidence for the roles of FXR-dependent BA metabolism, as these mice have elevated levels of circulating BAs and they excrete fewer BAs in their fecal matter (50). In hepatocytes, FXR activation limits the production of primary BAs by various pathways that include downregulating the rate-limiting enzyme for BA synthesis. It also stimulates the secretion of BA from the liver into the gall bladder and limits hepatic reuptake. In intestinal cells, FXR activation promotes downstream pathways that lead to intracellular transport of BAs and reduce BA production in hepatocytes (45, 51). In human ileal enterocytes, BA-mediated activation of FXR stimulates transcription of the hormone fibroblast growth factor 19 (FGF-19, mouse ortholog FGF-15) (21, 22). FGF-19/FGF-15 then activates a downstream pathway in hepatocytes, ultimately inhibiting the production of BAs (45). Maintaining BA homeostasis is important for liver function but when improperly regulated can lead to a range of digestive diseases. For example, BA-diarrhea (BAD) is thought to be caused by alterations in enterohepatic circulation. Given the roles of FXR in stimulating these processes, FXR agonists have been successfully tested for their therapeutic potential to treat this condition (51, 52). The therapeutic potential may extend beyond regulating the enterohepatic circulation of BAs since activation of FXR has been shown to promote antisecretory effects in colonic epithelial cells (53). These additional roles are discussed in more detail later in this review (see bile acids and intestinal transport of electrolytes and fluid). In the liver, FXR activation also limits glucogenesis and lipogenesis, therefore, targeting FXR has potential as a therapeutic strategy to treat a range of metabolic conditions, such as nonalcoholic steatohepatitis (54, 55).
Pregnane X Receptor
A range of lipophilic chemicals bind to Pregnane X receptor (PXR) including xenobiotics, endogenous hormones, and BAs (56). PXR is responsible for the metabolism, detoxification, and elimination of such agents. It is highly expressed in the liver and small intestine (24). PXR activation enhances the production of enzymes responsible for BA metabolism/detoxification, represses enzymes involved in BA synthesis, and alters the expression of BA transport proteins (23, 24, 57). LCA also activates PXR (24, 57). Along with FXR, targeting PXR has been proposed as a possible IBD treatment, particularly for patients with Crohn’s disease associated with BA-diarrhea [BAD (58)]. In patients with BAD, the activities of enzymes that catalyze BA metabolism are reduced, enzymes that facilitate BA synthesis are elevated and hence the balance of BAs levels is dysregulated. These studies have postulated that PXR or FXR may be deactivated in these patients (59, 60). Dextran sulfate sodium (DSS)-induced colitis is more severe in PXR knockout mice when compared with the wild-type control, and activation of PXR can alleviate the development of inflammation in wild-type mice (61).
Vitamin D Receptor
Typically, vitamin D receptor (VDR) responds to the active form of vitamin D, 1α,25-dihydroxyvitamin D3 1,25(OH)2D3. The receptor is expressed in the intestine and kidneys where it mediates a range of processes such as calcium homeostasis, cellular differentiation, and immunity.
LCA is a metabolite of CDCA. Although the primary BAs, CDCA and CA, do not activate VDR, LCA is a potent VDR agonist (25). Oral administration of LCA to mice can induce the expression of some of the same targets genes as VDR activation by 1,25(OH)2D3 (26). Although clarity on the physiological relevance for LCA activation of VDR is lacking, it is thought to have a protective role by mediating the elimination and breakdown of LCA, given the cytotoxic and pro-oncogenic nature of the BA (62, 63). Since, intestinal bacteria metabolize CDCA to LCA, it is hypothesized that this provides a mechanism for intestinal bacteria to regulate host VDR function. Recent evidence suggests that the LCA-VDR interactions may have protective roles for the intestinal epithelial barrier (64). This is supported by observations that VDR knockout mice develop dextran sulfate sodium (DSS) colitis more quickly and robustly than wild-type animals (65), and in patients with Crohn’s disease or ulcerative colitis colonic epithelial VDR expression is reduced (66).
LCA acts on FXR, PXR, and VDR to modulate BA metabolism, and the downstream effects of these interactions regulate the activities of the cytochrome P450 (CYP) family of enzymes (45). These enzymes protect the body from a variety of lipophilic chemicals, such as BAs, and their actions are controlled at a transcriptional level by many different signaling molecules. Activation of VDR, by either LCA or 1,25(OH)2D3, induces transcription of the CYP3A4 gene (25). The CYP3A4 enzyme assists in the detoxification of LCA and other BAs by catalyzing their hydroxylation. Hydroxylation of BAs means they are more hydrophilic, facilitating their excretion in fecal matter or urine. The importance of this interaction is demonstrated in VDR knockout mice, where intestinal, circulating, and fecal BA pools are all dysregulated (65, 67). Insertion of the CYP3A4 gene into these mutant mice improves the BA pool size so that the relative levels of BAs in liver and intestine homogenates return to levels comparable with those of wild-type mice. It also reduces the effects of LCA-induced toxicity by stimulating LCA metabolism (68).
Constitutive Androstane Receptor
Constitutive androstane receptor (CAR) is expressed in the liver and intestines where it mediates the detoxification of phenobarbital-like inducers (69). Although endogenous activators for CAR are not well characterized, both androstenol and androstanol have been identified as repressive endogenous ligands for constitutive activity. BAs are unlikely to directly activate CAR (70) but indirect activation has been suggested in LCA-induced liver toxicity models. CAR knockout mice develop more severe damage compared with wild-type controls, highlighting that LCA detoxification could not occur in these mice (71). CAR expression is reduced in the colon from mice with DSS-induced colitis. Similarly, mucosal intestinal biopsies from patients with Crohns’ disease and ulcerative colitis also have reduced CAR expression when compared with healthy individuals (72).
FXR, PXR, and CAR regulate overlapping target genes in BA metabolism. To investigate how they coordinate their activities, several studies have used double knockout mice for different combinations of these receptors. They suggest that the receptors jointly protect against BA-induced liver toxicity (71, 73, 74). The gut microbiome regulates the function and expression of CAR and PXR receptors by stimulating the conversion of primary BAs to secondary BAs. Tissues from germ-free (GF) mice express significantly lower mRNA levels for CAR, PXR, and FXR as well as for a range of cytochrome P450 enzymes relative to mice housed under specific pathogen-free (SPF) conditions (75). In conventional mice, daily administration of CAR and PXR agonists changes the biodiversity of their microbiome by reducing two taxa in the Bifidobacterium genus (76). Again, this highlights the potential interplay between the gut microbiome, the regulation of BAs levels, the expression this group of receptors, and their activation by BAs.
BILE ACIDS AS MODIFIERS OF ION CHANNEL ACTIVITY
BAs can also exert their effects through interactions with ion channels. Specific ionotropic receptors for BAs have not been identified to date. However, there is good evidence that BAs can modulate the functions of several members of the epithelial sodium channel/degenerin (ENaC/DEG) family. This interaction may be physiologically relevant in tissues where BAs have access to these channels, such as in the intestine and bile ducts. The regulation of these channels by hormones and local factors is highly complex and involves the regulation of channel expression, trafficking, and open probability. Several members of the ENaC/DEG family of ion channels can qualify as ligand-gated ion channels. These include the acid sensing ion channels (ASICs) and the FMRFamide-activated Na+ channel (FaNaC) that are activated by protons and the tetrapeptide FMRFamide, respectively. For other members of this family, no specific activating ligands have been described with the possible exception of BA-sensitive ion channel (BASIC). A summary of the actions of BAs on ion channels and ionotropic receptors is provided in Table 2.
Table 2.
Effects of bile acids on ion channels and ionotropic receptors
| Ion Channel | Bile Acid | Effect: Stimulation ↑ Inhibition ↓ | Concentration Used or EC50/IC50, if Available | Solvent | Cells Expression System | Refs. |
|---|---|---|---|---|---|---|
| Epithelial sodium channel/degenerin ion channel family | ||||||
| BASICrat | g-CDCA, t-CDCA g-HDCA, t-HDCA |
↑↑↑ | 1–1.5 mM g-HDCA: EC50 ∼ 2 mM |
NA | X. laevis oocytes | (77, 78) |
| t-CA, t-βMCA | ↑ | |||||
| UDCA | ↑↑↑ | EC50 ∼ 2.5 mM | ||||
| t-UDCA, t-HDCA | ↑↑↑ | 1–2 mM t-UDCA: EC50 ∼ 2.7 mM |
NA | HEK293 cells | (79) | |
| t-CA, t-LCA t-DCA, t-CDCA |
↑↑ | |||||
| t-CDCA, t-HDCA t-UDCA |
↑↑ | 500 µM–1 mM | NA | Normal rat cholangiocytes (NRC) | (80) | |
| BASIChuman | t-DCA | ↑↑↑ | 500 µM–1 mM | Bath solution | X. laevis oocytes | (77, 81, 82) |
| t-CDCA | ↑↑↑ | NA | ||||
| t-CA, t-HDCA | ↑ | |||||
| t-DCA, t-CDCA | ↑↑↑ | 1–2 mM | NA | HEK293 cells | (79) | |
| t-CA, t-UDCA t-HDCA, t-LCA |
↑ | |||||
| BASICmouse | t-CA, t-UDCA, t-HDCA t-LCA, t-DCA, t-CDCA |
↑↑ | 1–2 mM t-UDCA: EC50 ∼ 4.5 mM |
NA | HEK293 cells | (79) |
| αβγENaCrat | CA | ↑↑ | 1 mM t-DCA: EC50 ∼ 260 µM t-CA: EC50 ∼ 410 µM |
DMSO stock | X. laevis oocytes | (83) |
| t-DCA | ↑↑↑ | Bath solution | ||||
| g-CA, t-CA | ↑↑ | |||||
| t-CDCA, t-UDCA | ↑ | |||||
| t-βMCA, t-HDCA | No effect | |||||
| αβγENaChuman | t-DCA | ↑↑↑ | 250 µM | Bath solution | X. laevis oocytes | (84) |
| t-CA, t-CDCA | ↑↑ | |||||
| CA, DCA | ↑ | |||||
| CDCA | ↓ | |||||
| δβγENaChuman | CA, t-CA, DCA, t-DCA CDCA, t-CDCA | ↑↑↑ | ||||
| αβγENaCmouse | DCA, t-CA | ↑↑↑ | 1 mM DCA: EC50 ∼ 80 µM t-CA: EC50 ∼ 150 µM |
H2O stock | X. laevis oocytes | (85) |
| g-CA | ↑↑ | |||||
| CA, t-LCA, UCA | ↑↑ | DMSO stock | ||||
| CDCA, t-CDCA 12βHICA | ↑ | |||||
| HDCA | ↓ | H2O stock | ||||
| t-HDCA | ↓↓ | |||||
| t-CA | ↑↑ | 1 mM t-CA: EC50 ∼ 200 µM |
H2O stock |
mpkCCD14 cells | ||
| t-HDCA | ↓ | |||||
| ENaChuman | UDCA | ↓↓ | 500 µM | NA | NuLi-1 and CuFi-1 airway epithelial cells | (86) |
| ASIC1ahuman | t-CDCA | ↑↑↑ | 500 µM t-DCA: EC50 ∼ 150 µM |
Bath solution | X. laevis oocytes | (87) |
| t-DCA | ↑↑ | |||||
| t-CA | ↑ | |||||
| Other ion channels and ionotropic receptors | ||||||
| BKCarabbit | CA, DCA, LCA, t-LCA | ↑↑↑ | 100 µM | DMSO ethanol stock | Smooth muscle cells | (88) |
| NMDAmouse | CA, g-CA, t-CA | ↓↓↓ | IC50 ∼ 500-700 µM | NA | Hypothalamic neurons | (89, 90) |
| DCA, t-DCA CDCA, t-CDCA |
IC50 ∼ 60-70 µM | |||||
| g-CDCA | IC50 ∼ 130 µM | |||||
| GABAAmouse | CA, g-CA, t-CA | ↓↓↓ | IC50 ∼ 500–700 µM | |||
| DCA, t-DCA | IC50 ∼ 150 µM | |||||
| CDCA, g-CDCA t-CDCA |
IC50 ∼ 70–80 µM | |||||
| UDCA | IC50 ∼ 70–100 µM | |||||
| GABAA
α1ratβ1ratγ2Lmouse α1ratβ2ratγ2Lmouse α1ratβ3humanγ2Lmouse α1ratβ3human |
UDCA | ↓↓↓ |
IC50 ∼ 70–100 µM |
NA | HEK293 cells | (90) |
| β3human
β3humanγ2Lmouse |
IC50 ∼ 7 µM IC50 ∼ 200 µM |
|||||
| NMDA GluN1rat/2Arat GluN1rat/2Brat |
CA, g-CA, t-CA CDCA, g-CDCA t-CDCA |
↓ | 100 µM | NA | X. laevis oocytes | (91) |
| GluN1rat/2Drat | CA, g-CA, t-CA | ↓ |
100 µM t-CDCA: IC50 ∼ 180 µM |
|||
| CDCA, g-CDCA t-CDCA |
↓↓ | |||||
| GluN1rat/3Bmouse | CA, g-CA, t-CA | ↓ | 100 µM t-CDCA: IC50 ∼ 40–80 µM |
|||
| CDCA, g-CDCA t-CDCA |
↓↓↓ | |||||
| P2X4human | CA, t-CA, CDCA | No effect |
250–500 µM t-DCA: IC50 ∼ 160 µM |
Bath solution | X. laevis oocytes | (92) |
| t-CDCA | ↓↓ | |||||
| DCA, t-DCA | ↓↓↓ | |||||
| P2X2rat | t-CA | No effect | 20 µM t-HDCA: IC50 ∼ 11 µM |
H2O stock | X. laevis oocytes | (93) |
| t-UDCA, t-DCA | ↓ | |||||
| t-CDCA | ↓↓ | |||||
| t-LCA, HDCA t-HDCA, g-HDCA |
↓↓↓ | |||||
| P2X1rat | t-HDCA | No effect | 20 µM | |||
| t-LCA | ↓ | 200 µM | ||||
| P2X3rat | t-HDCA | No effect | 20 µM | |||
| P2X7rat | t-LCA | 200 µM | ||||
| P2X4rat | t-LCA | ↑ | 200 µM | |||
| t-HDCA | No effect | 20 µM | ||||
| P2X2human | t-LCA | ↑↑ | 20 µM | |||
| t-HDCA | No effect | |||||
CA, cholic acid; CDCA, chenodeoxycholic acid; DCA, deoxycholic acid; HDCA, hyodeoxycholic acid; LCA, lithocholic acid; UCA, ursocholic acid; UDCA, ursodeoxycholic acid; 12βHICA, 12β-hydroxyisocholic acid; βMCA, β-muricholic acid; t-, tauro-conjugated; g-, glyco-conjugated bile acid. Solvents: NA, no information available; bath solution, bile acids/bile salts were directly dissolved in bath solution to obtain the indicated final concentrations; DMSO stock, dimethyl sulfoxide (DMSO) stock solutions were prepared with bile acids at a concentration of 100 mM; H2O stock, aqueous stock solutions were prepared with bile acid concentrations ranging from 20 to 100 mM; DMSO/ethanol stock, concentrated dimethyl sulfoxide (DMSO) stock solutions were prepared with bile acids at a concentration of 333 mM and then further diluted 1/10 in 95% ethanol.
BA-Sensitive Ion Channel
BA-sensitive ion channel (BASIC) is a member of the ENaC/DEG ion channel family (previously known as intestinal Na+ channel (INaC) in humans or brain-liver-intestine Na+ channel (BLINaC) in rodents) (94, 95). BASIC is expressed on the apical membrane of rat cholangiocytes (77, 80). It shows very low basal activity in heterologous expression systems, but is strongly activated by submillimolar concentrations of BAs (77–82). Based on this observation, it is possible that BAs may act as endogenous ligands for the channel in tissues where they are present at high concentrations, such as the bile ducts and the small intestine. However, the stimulatory effect of BA on BASIC does not seem to be as potent and as specific as the stimulatory effect of ligands on classical ionotropic receptors. Moreover, a modulatory effect of BAs on ion channel activity does not seem to be limited to BASIC.
Epithelial Sodium Channel
Epithelial sodium channel (ENaC) is expressed in the apical membrane of epithelia in a range of tissues, including the distal colon and the biliary ducts (96). It mediates transepithelial sodium absorption and therefore regulates fluid volume (97–100). Recent evidence suggests that BA sensitivity is not a unique feature of BASIC but may be a common feature of channels belonging to the ENaC/DEG family. Similar or lower concentrations of BAs needed to activate BASIC can stimulate the rat, mouse, and human ENaC (83–85). Generally, BAs activate ENaC with varying abilities, but some also inhibit the channel (84–86).
It is likely that exposure of ENaC to changing levels of BAs in cholangiocytes and distal colon may modify the function of the channel under physiological conditions. In kidney and distal colon, ENaC plays an important role in mediating sodium absorption. In these tissues, ENaC is mainly stimulated by the mineralocorticoid aldosterone but can also be regulated by additional mediators and local mechanisms (101–104).
Despite the location of ENaC on the apical membrane, BAs may also affect the channel through interactions originating from the basal surface. Preliminary studies demonstrated that LCA applied to the serosal side of the mouse distal colon had a sustained stimulatory effect on ENaC-mediated transepithelial Na+ transport. This is reminiscent of the stimulatory effect of aldosterone (105). The delayed and sustained stimulatory effect of LCA on ENaC may be due to transcriptional regulation involving a nuclear receptor, such as FXR. A dual stimulatory effect of BAs on ENaC from the apical and basolateral side may serve as a compensatory mechanism in the colon to limit fluid loss during diarrhea resulting from impaired ileal BA absorption (for a more detailed discussion of the effects of BA on intestinal transport see bile acids and intestinal transport of electrolytes and fluid).
Human Acid-Sensing Ion Channel ASIC1a
ASIC1a is another member of the ENaC/DEG family of ion channels. Although BAs do not directly activate ASIC1a, they positively modulate proton-activated ASIC1a currents (87). BAs may not act as classical ligands on the orthosteric site of the channel, but rather modulate the function of already active ASIC1a channels by interacting with a topographically distinct, allosteric site. Using site-directed mutagenesis and computer simulation it has been shown that BAs may directly interact with the transmembrane domains of ENaC/DEG ion channels in the so-called “degenerin region” known to be functionally important for channel gating (81, 84, 87). BA binding to the transmembrane domains of the channel probably stabilizes the channel open conformation leading to increased channel activity. Several ASICs are responsible for distinct proton-gated currents in neurons regulating gastrointestinal function (106). Thus, it is tempting to speculate that modulation of ASICs by BAs may contribute to gastrointestinal physiology and pathophysiology.
Other Ion Channels and Ionotropic Receptors
BA may also modulate other ion channels and ionotropic receptors. For example, BAs have been shown to modify the function of large-conductance Ca2+- and voltage-activated K+ (BK) channels via distinct channel subunits and sites (88), they also inhibit currents mediated by ionotropic NMDA and GABAA receptors (89–91). Recently, it has been demonstrated that the human purinergic receptor P2X4, an ATP-gated nonselective cation channel, is inhibited by unconjugated or tauro-conjugated DCA (92). The inhibitory effect of BAs is not limited to P2X4 and has also been reported for the related P2X2 receptor (93). A comparison of the crystal structure of P2X4 revealed a high degree of similarity in topology and architecture with that of ASIC1 (107), suggesting structural similarities of their BA binding sites. Indeed, tauro-DCA probably interacts with the transmembrane domains of P2X4 thereby stabilizing its closed state (92). P2X4 is abundantly expressed in hepatocytes and cholangiocytes (108, 109) and P2X2 is present in gall bladder and enteric neurons (110, 111). Therefore, BAs may potentially play a role as local modulators of purinoceptors in these systems.
BILE ACIDS AND INTESTINAL TRANSPORT OF ELECTROLYTES AND FLUID
Bile Acids Modulate Intestinal Fluid and Electrolyte Transport in Pathophysiology
Intestinal fluid absorption is critical for effective homeostatic control of whole body fluid and electrolyte balance. Na+ absorption drives fluid uptake across small intestinal villi and surface cells of the colon, whereas Cl− secretion drives fluid secretion occurring mainly from the crypts. Three main mechanisms drive Na+ absorption along the gut as follows: while digestion is occurring, small intestinal Na+-nutrient co-transporters predominate; during the interdigestive period, Na+/H+ exchange predominates along the entire length of the intestine; and electrogenic Na+ absorption is the primary mechanism driving fluid absorption in the distal colon (112). Depending on physiological versus pathophysiological conditions, BAs can activate both proabsorptive and prosecretory pathways to influence ion flux and water retention.
There has long been an association between increased luminal BA levels and the onset of diarrhea in humans. However, the roles of BAs in fluid dysregulation were not recognized until much later than their effects on intestinal motility (see bile acids and intestinal motility). The first preclinical demonstration of such actions came in 1966, where perfusion of DCA, at a concentration of 100 µM, to the rat jejunum inhibited intestinal fluid absorption (113). Based on observations in patients with ileitis and increased BA turnover, Hofmann (114) proposed that the increased levels of luminal BAs were causing the clinical symptoms of diarrhea. This hypothesis was subsequently supported by clinical studies showing that direct administration of dihydroxy BAs to the colorectum induced diarrhea in healthy individuals (115), whereas infusion of conjugated and unconjugated BAs inhibited jejunal water absorption (116).
Typically, only dihydroxy BAs (e.g., DCA and CDCA) alter epithelial transport processes and, in humans, diarrhea only occurs when BAs are present at pathophysiological levels (see Table 3). In the small intestine where their uptake to enterohepatic circulation can occur via IBAT, conjugates of DCA and CDCA exert similar actions to their nonconjugated counterparts but with lower potency. However, in the colon there is a lack of apical uptake pathways and therefore luminal conjugated BAs cannot enter the epithelium unless the barrier becomes compromised, as occurs in conditions of intestinal inflammation. Conjugated BAs can then gain access to the basolateral domain and enter the epithelial cell via basolateral transporters to stimulate increases in free cytosolic Ca2 (for more details see Molecular Mechanisms of Bile Acid-Induced Fluid Transport), thereby activating intracellular mechanisms that promote fluid secretion and inhibit absorption (133, 134). Interestingly, unlike the other dihydroxy BAs, UDCA does not stimulate fluid secretion in the colon and appears to exert antisecretory actions when it is present at sufficiently high levels (135).
Table 3.
Effects of bile acids on gastrointestinal motility
| GI Region/Application Method/ Measurement | Animal Species | Bile Acid | Concentration Used | Effect: | Refs. |
|---|---|---|---|---|---|
| Stomach and small intestine | |||||
| Stomach and small intestine/oral gavage/phenol red | Rat | t-DCA DCA |
26–100 mM 5–26 mM |
GE ↓ SIT ↓ GE ↓↓ IT ↓↓ |
(117) |
| Stomach-cecum/ileum infusion/environmental hydrogen analysis | t-CA DCA |
10 mM 10 mM |
SCTT ↑ SCTT ↓ |
(118) | |
| Stomach/nasogastric/paracetamol absorption test | Human (healthy and type 2 diabetes) | CDCA | 1.25 g in 100 mL water | GE ↓ | (119) |
| Small intestine | |||||
| Jejunum and ileum/intraluminal infusion/lactulose and in vivo manometry | Human (healthy) | g-CDCA g-CA |
15–30 mM 15 mM |
SIT ↓ SIT ↓ |
(120, 121) |
| Terminal ileum/intraluminal infusion/ex vivo manometry | Rabbit | Rabbit bile t-DCA |
0.1–10 mM | MA ↓ MA ↓ at 10 mM |
(122) |
| Colon (in vivo) | |||||
| GI and colonic transit/oral/side effect profile | Human (cholesterol gallstone) | UDCA CDCA |
7–8 and 14–15 mg/kg/day (3–12 mo) | BF – BF ↑ |
(123) |
| Proximal colon/ rectal perfusion/in vivo manometry and GI sensation | Human (healthy) | CDCA | 1 mM | Sensory threshold ↓ PS ↑ |
(124) |
| GI and colonic transit/oral capsule/scintigraphy method and bowel function | Human (healthy and IBS-C) | CDCA | 500, 1,000 mg | CT ↑ BF ↑ | (125) (126) |
| GI and colonic transit/rectal perfusion/ GI sensation | Human (healthy) | TCA | 1,500 and 3,500 mg | Sensory threshold ↓ BF ↑ |
(127) |
| Colon (in vitro/ex vivo) | |||||
| Colonic motility/bath addition muscle exposure/contraction assay | Mouse tgr5-wt and ko |
UDCA DCA |
100 µM 1, 10, 100 µM |
WT contraction - WT contraction ↓ KO contraction - |
(128, 129) |
| Colonic motility/ bath addition mucosal exposure/ contraction assay | tgr5-wt and ko | UDCA DCA LCA OA |
100 µM 1, 10, 100 µM 1, 10, 100 µM 100 µM |
WT PR - WT PR ↑ KO - WT PR ↑ KO - WT PR ↑ KO - |
(129) |
| Colonic smooth muscle/ bath addition muscle exposure/ contraction assay | Rat | DCA | 1, 10, 100 µM | Contraction - | (130) |
| Colon | |||||
| Colon and rectum/intraluminal infusion/ex vivo manometry | Rabbit | t-CA | 2–16 mM | MA ↑ | (131) |
| g-CA | 8–24 mM | MA ↑ | (132) | ||
| g-CA | 3–30 mM | MA - | |||
| DCA | MA ↑ |
CA, cholic acid; CDCA, chenodeoxycholic acid; DCA, deoxycholic acid; LCA, lithocholic acid; UDCA, ursodeoxycholic acid; t-, tauro-conjugated; g-, glyco-conjugated bile acid; Rank order of potency for TGR5 is t-LCA > LCA > DCA > CDCA, UDCA and OA are weak TGR5 agonists. Responses: stimulation, ↑; inhibition, ↓; no change, -. BF, bowel function; CT, colonic transit; GE, gastric emptying; IBS-C, irritable bowel syndrome-constipation predominate; MA, motor activity; PR, peristaltic reflex; PS, propagating sequences; SIT, small intestinal transit.
Molecular Mechanisms of Bile Acid-Induced Fluid Transport
BAs induce luminal fluid accumulation by inhibiting Na+ absorption and promoting Cl− secretion in both the small intestine and colon. This has been demonstrated in many animal models using in vitro and in vivo methods to assess fluid and ion transport in the ileum (136–139) and colon (140, 141). Studies using cultured intestinal epithelial cell lines and on isolated human colonic crypts have provided considerable insights into mechanisms by which BAs induce luminal fluid accumulation and diarrhea. In Caco-2 cells, a model of absorptive epithelial cells, conjugated BAs inhibit Cl−/OH− exchange (142). Likewise, when present at high concentrations, CDCA inhibits both Na+/H+ exchange (NHE) activity and Cl−/HCO3− exchange in isolated human colonic crypts (143). As discussed in bile acids as modifiers of ion channel activity, BAs can also modify the function of ENaC, which plays a role in Na+ absorption in the distal colon, particularly when plasma aldosterone levels are increased to minimize renal and intestinal sodium losses. High concentrations of BAs can rapidly induce Cl− secretion from the crypts across both small intestinal and colonic epithelial cells. This effect appears to involve activation of basolateral K+ channels, where K+ leaving the cells provides the electrochemical driving force for Cl− to exit through channels in the apical membrane. This most likely occurs through the cystic fibrosis transmembrane conductance regulator (CFTR) (144).
The actions of BAs on these transport proteins appear to be predominantly mediated by elevations in intracellular Ca2+ (133, 143, 145). BAs have been shown to induce increases in intracellular Ca2+ by both influx across the plasma membrane and release from the endoplasmic reticulum (143, 146, 147). However, the upstream mechanisms leading to increased intracellular Ca2+ are still not well defined. The fact that only dihydroxy BAs are active might point to the involvement of a receptor. However, neither TGR5, which appears to exert antisecretory actions (7, 148), nor the nuclear receptor, FXR, is a likely candidate. As noted previously in this review, BAs have detergent-like properties and by inducing perturbations in the cell membrane, this may lead to activation of downstream signaling pathways, thereby promoting their prosecretory and antiabsorptive actions (149, 150). It appears that BAs can also directly interact with endoplasmic reticulum membranes to increase cytosolic Ca2+ levels (151, 152).
It is clear from studies of isolated epithelial cells that BAs can exert direct actions on intestinal absorption and secretion. However, studies from ex vivo tissues demonstrate that BAs acting on other mucosal cells may also contribute to this dysregulation. As discussed in Actions of Bile Acids on the Mucosa to Promote Colonic Motility, luminal BAs can stimulate enterochromaffin cells (ECC) within the mucosa to release serotonin (5-HT). 5-HT can then activate enteric neural pathways, including those that regulate fluid secretion (153–155). Such neural pathways may provide a mechanism by which changes in intestinal fluid and electrolyte transport can be coordinated with changes in motility and permeability (5, 156–158). BAs can also activate mucosal mast cells to release their mediators (159), most notably histamine, and this may be important in mediating Cl− secretory responses to CDCA (160). It is also important to note that mast cells and enteric nerves exist close to one another in the intestinal mucosa and that interactions between these two cell types are likely to be important in regulating epithelial transport responses to BAs (161, 162).
Roles of Bile Acids in Epithelial Fluid and Electrolyte Transport under Physiological Conditions
In contrast to their acute prosecretory and antiabsorptive effects at high concentrations, BAs have recently been shown to exert antisecretory actions at more physiologically relevant levels (53, 105, 163). Such antisecretory effects are slow in onset (12–24 h) and for DCA occur at concentrations as low as 50 µM, a concentration that is well within the normal physiological range for this BA in the proximal colon (164). Such antisecretory actions of naturally occurring BAs are mimicked by selective FXR agonists and are associated with reduced expression and activity of basolateral Na+-K+-ATPase pumps and apical CFTR Cl− channels (53). We have proposed that under such physiological conditions, BAs may function to inhibit colonic fluid secretion, thereby promoting the normal absorptive function of the colon (summarized in Fig. 1). Thus, by virtue of their opposing actions at low versus high concentrations, dihydroxy BAs may have the capacity to dynamically control the fluidity of the luminal contents. In this context, bacterially mediated deconjugation of luminal BAs would be essential to permit their entry into the cell where FXR activation can occur to induce antisecretory actions. Thus, diet or disease-associated changes in the colonic microbiota that result in altered expression of bile salt hydrolase activity could have important implications for fluid and electrolyte transport (165–169).
Figure 1.

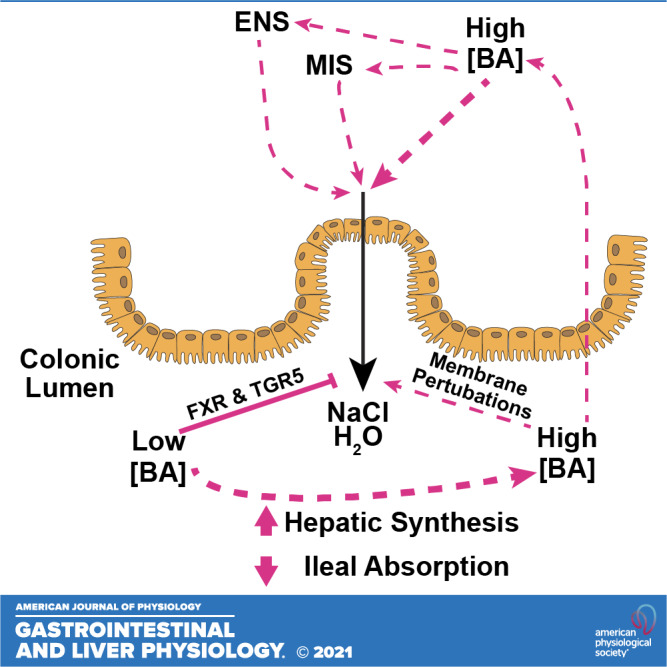
Bile acids (BAs) in regulation of colonic epithelial Cl− secretion. BAs are normally present in the colon at relatively low concentrations where they act via BA receptors, such as Farnesoid X receptor (FXR) and Takeda G protein-coupled receptor 5 (TGR5), to inhibit salt and water secretion into the lumen (left). We propose that such actions serve to promote normal colonic absorptive function. In some pathological conditions, increased delivery of BAs to the colon may result from either increased synthesis of primary BAs by hepatocytes or reduced ileal absorption. The resulting high levels of luminal BAs induce perturbations in the epithelial cell membranes to promote Cl− secretion and inhibit Na+ absorption, thereby promoting luminal fluid accumulation (right). Such direct effects of BAs on the epithelium are amplified by indirect actions involving the release of neurotransmitters and mediators from the enteric nervous and mucosal immune systems (ENS and MIS, respectively). It is thought that, under such circumstances, increased colonic fluid secretion is a protective mechanism that serves to dilute the abnormally high levels of luminal BAs.
Studies have also investigated the potential role for TGR5 in regulating colonic fluid and electrolyte transport. These revealed that TGR5 also exerts antisecretory effects, although its role appears to be distinct to that of FXR. Treatment of muscle-stripped, nerve-free, mucosal preparations of rat colon with the TGR5-selective agonist, INT-777, rapidly and transiently reduced basal Cl− secretion with a concomitant inhibition in the capacity of the tissue to evoke secretory responses to Ca2+-dependent secretagogues (148). Furthermore, the effects of INT-777 were mimicked by LCA, an endogenous ligand for TGR5. Subsequent studies revealed that TGR5 activation may also dampen colonic Cl− secretion by inhibiting submucosal neurons. Such actions are proposed to represent a regulatory mechanism that may counterbalance BA-induced fluid secretion (7). A schematic representation of the roles of BAs in regulating colonic intestinal fluid and electrolyte transport is depicted in Fig. 1.
BILE ACIDS AND INTESTINAL MOTILITY
There is strong, consistent evidence that oral delivery or rectal administration of BAs promotes colonic motility. Although early experimental evidence from in vivo studies in rabbit demonstrated that luminal infusion of secondary BAs promotes colonic motility (131, 132), the most compelling evidence comes from clinical studies (124–126). Rectal perfusion of CDCA to healthy individuals reduces the perceived defecatory urge and enhances the frequency of distension-evoked pressure waves or bowel movements (124, 127). Similarly, oral administration of CDCA significantly accelerates colonic transit, but not gastric emptying or colon filling (125). These effects are not limited to healthy patients since in individuals with constipation, oral administration of BAs increases stool frequency and decreases stool consistency (126, 170). This work highlights the therapeutic potential of targeting BA signaling to treat gastrointestinal motility disorders. A summary of the roles of BAs in GI motility and the relative concentrations required to stimulate these actions is presented in Table 3.
Mechanisms for the Prokinetic Actions of BA and the Case for TGR5
Early literature from clinical studies implicates the role of TGR5 in the prokinetic actions of BAs in the colon. Results from clinical trials testing the efficacy of UDCA and CDCA to dissolve and treat gallstones demonstrated that patients treated with CDCA, but not UDCA, experienced diarrhea as a side effect (123, 171, 172). Furthermore, constipation was relieved by treatment with CDCA, but not UDCA (123). This may be explained by the differing agonist activities of the BAs at TGR5, where UDCA has a lower efficacy and potency for TGR5 agonism than CDCA (9, 173). This aligns with evidence from candidate genotype analysis using PCR-based methods, where TGR5 genotype variations are associated with constipation and diarrhea in patients with IBS (174, 175).
Although selective TGR5 agonists are becoming available, the functional effects of TGR5-selective drugs on colonic motility in humans have not yet been investigated. However, studies in mice and dogs demonstrate that TGR5 is expressed throughout the gastrointestinal tract in enteric neurons, epithelial and enteroendocrine cells, and smooth muscle. TGR5 has been identified as a regulator of colonic motility in these species under normal conditions (5, 128, 176). Indeed, there are likely several mechanisms by which BAs can modulate intestinal motility. Data from in vitro studies suggest that BAs exert prokinetic actions when exposure is limited to the mucosa (5), but exposure to the muscularis externa inhibits intestinal motility (128). In the latter case, BAs may have a direct action on the smooth muscle cells to impair contractile activity (130, 177), or may directly activate inhibitory enteric motor neurons that express TGR5 (128). Such inhibitory actions of BAs on the muscularis externa do not contradict the findings from clinical studies. The absence of transporters, such as ASBT, that permit the absorption of conjugated BAs in the colon limits their access to this tissue layer (178), and most clinical studies involve BA exposure to the gut lumen. Therefore, it is likely that the prokinetic actions of BAs are due to effects initiated in the mucosa. Direct effects of BAs on the muscularis externa may be more relevant in disease conditions, such as inflammation, where loss of barrier function and increased paracellular permeability allows both conjugated and unconjugated BAs to passively pass through the mucosa (179, 180).
Actions of Bile Acids on the Mucosa to Promote Colonic Motility
BAs promote the release of several bioactive mediators from the intestinal mucosa. For example, isolated human ECC release 5-HT following exposure to sodium deoxycholate (57), while mucosal application of BAs to rat jejunum (153) and mouse colon promotes 5-HT release possibly via a TGR5-dependent mechanism (5). This BA-mediated 5-HT release activates neural pathways to promote peristaltic contractions (5).
In addition to 5-HT, it is well documented that BAs also stimulate intestinal L cells to release the peptide hormones glucagon-like peptide-1 (GLP-1) and peptide YY (PYY) (119, 181–185). This involves TGR5-dependent and TGR5-independent mechanisms (8, 9, 186, 187). The release of GLP-1 and PYY is important for regulating appetite and metabolism. One mechanism of action of GLP-1 in metabolism is to promote insulin release, and hence GLP-1 receptor agonists are used clinically to treat diabetes mellitus. GLP-1 and PYY promote digestion and nutrient absorption by delaying gastric emptying and reducing intestinal motility. Infusion of BAs to the lumen of the stomach and small intestine in vivo reduces gastric emptying and intestinal transit in the rat, rabbit, and human (117, 118, 120, 121). The association between BAs, the release of neuropeptides, and delayed small intestinal motility is supported by studies in the rabbit ileum (122) and human stomach (119), where luminal perfusion of BAs increases the plasma concentrations of PYY or GLP-1, respectively, and this is associated with reduced GI motility in these regions.
BAs mediate opposing effects on motility of the small versus large intestines and it is unclear whether neuropeptide release alone can account for these differences. Rectal administration of tauro-CA in humans dose-dependently increases plasma GLP-1 and PYY levels (127, 182) and this is associated with increased defecation frequency (127). GLP-1 or PYY may directly regulate the BA-mediated changes in motility and this could explain the differential actions in the upper gut and colon where they are present at differing concentrations. In vitro studies show that application of GLP-1 inhibits the responses of intestinal muscle strips to electrical stimulation or exogenous application of ACh without influencing basal tone. This inhibition is likely due to an action on enteric inhibitory neurons (188–190), but also involves actions on the smooth muscle cells themselves (189). Myenteric and submucosal neurons functionally express GLP-1 receptors. In studies on wholemount preparations of rat colon, application of the highly selective agonist, exendin-4, stimulated increases in intracellular calcium in a proportion of neurons from both regions (191). In vivo motility studies indicate that regulation from the central nervous system may also be involved (191, 192). There is conflicting data as to whether PYY stimulates (193, 194) or inhibits GI smooth muscle contractility (195, 196) and these effects may be region-specific (197, 198). PYY is a 36-amino acid polypeptide that is endogenously expressed as either full-length PYY1-36 or as the truncated PYY3-36 form. These peptides have different affinities for the Y receptors that PYY targets (199). Therefore, the presence and activity of PYY variants may account for differences in the contractile responses of different regions to PYY. The cell surface enzyme, dipeptidyl peptidase IV (DPP-IV), both inactivates GLP-1 and converts PYY from its long to its truncated form. Thus, the activity of DPP-IV may play a significant role in shaping the contractile responses to PYY and GLP-1 following BA activation. GLP-1 containing L-cells are evenly distributed across the stomach, small intestine, and colon. In contrast, PYY containing L-cells typically populate the distal small intestine and colon only (195, 200, 201). Whether TGR5 is equivalently expressed by all L-cells regardless of if they contain PYY or GLP-1 remains to be determined, and how these profiles may also contribute to the generation of gut motility patterns is also unknown.
Differing results using taurine-conjugated versus unconjugated isoforms and the use of individual BAs versus mixtures of BAs provide better insight on how they influence motility (see Table 3). In a study by Brown et al. (118), perfusion of the ileum with taurocholic acid (tauro-CA) increased stomach to caecum transit time. In contrast, this transit time decreased with perfusion of DCA. Ullmer et al. found that taurine-conjugated and unconjugated BAs have an equal binding affinity for human-TGR5 in cell lines. However, their effectiveness to increase plasma PYY levels in animals depended on systemic versus luminal application of the agonists (10). The taurine-conjugated TGR5 agonist tauro-RO5527239 did not stimulate secretion of PYY from L-cells, whereas the unconjugated form did. Conjugated forms of BAs or TGR5 agonists have limited ability to access the receptor that is located on the basal side of epithelial cells (186). We speculate that this, in turn, would limit the release of factors, such as PYY, by these cells and thus potentially limit their influence on intestinal motility. Given the importance of the colonic microbiome in determining BA deconjugation, it is clear that changes in bacterial populations that express bile salt hydrolases are likely to significantly impact how BAs modulate fecal transit through the colon. (166–169). BA modulation by the colonic microbiome may therefore be influenced by factors such as diet modification or the onset of disease.
Bile Acids and Intestinal Motility Disorders
Enhanced delivery of BAs to the colon has long been associated with diarrhea (114, 202), with ∼25%–33% of patients with chronic functional diarrhea having increased BA delivery to the colon (203). In some rare cases, BA-malabsorption (BAM) can be due to loss-of-function mutations in intestinal bile acid transporters, such as ASBT and organic solute transporter β (Ostβ) (5, 204–206). However, BAM is most commonly due to structural damage brought about by ileal inflammation from conditions such as Crohn’s disease, or following surgical or radiation procedures (207–209). Idiopathic BAM is characterized by increased delivery of BAs in the stool in the absence of structural pathology. Alterations in BA synthesis (174, 210–212) or the enterohepatic circulation (213) are attributed as the causes of BAM and the resultant diarrhea in these conditions. Moreover, studies suggest that 10%–26% of patients with diarrhea predominant IBS (IBS-D) may have BAM as the underlying cause (214). Conversely, recent evidence suggests that some instances of constipation may result from reduced BA delivery to the colon. In a retrospective study, Vijayvargiya et al. (215) reported that 15% of patients with IBS-C had decreased fecal BA levels compared with healthy control patients (45 patients, 108 controls). This evidence for the important roles of BAs in a range of GI motility conditions has prompted research into the therapeutic potential of altering BA delivery to the colon or influencing BA receptor signaling.
More recent clinical studies have shifted away from the direct administration of BAs to patients, to investigating the therapeutic potential of modulating the delivery of endogenous BAs. This approach has produced results consistent with the effects of exogenous administration of BAs. One such strategy is to inhibit the reuptake of BAs in the terminal ileum, using drugs such as IBAT inhibitor, Elobixibat (A3309) (216–218). In vivo studies in constipated dogs (218) and clinical trials conducted in Europe, North America, and Japan have all demonstrated Elobixibat to effectively increase the number of spontaneous bowel movements and to improve stool consistency in patients with chronic constipation (219–222). Elobixibat is now an approved drug for the treatment of constipation in Japan (223). There is limited knowledge as to whether administration of ASBT inhibitors can promote GI motility under control conditions (i.e., nonconstipated patients). However, in vivo experiments in dogs demonstrated that Elobixibat induced giant migrating contractions and increased defecation frequency and fecal wet weight within 10 h of administration (224).
Several other IBAT inhibitors, including volixibat, maralixibat, odevixibat (A4250), and GSK2330672 are in various stages of clinical trials to treat conditions such as nonalcoholic steatohepatitis (225), type 2 diabetes mellitus (226), and cholestatic pruritus in primary biliary cholangitis (227–231). In all of these studies, diarrhea and abdominal pain are cited as treatment-related adverse events, with an incidence ranging from 10% to 80%. Although it is unclear whether a patient subtype is more or less likely to develop these symptoms, the dosage of the drugs can be correlated with the number of reported incidences. Recently published results of a phase 2 clinical trial for GSK2330672 to treat cholestatic pruritus reported diarrhea as an adverse event in 38%, 65%, and 66% of patients receiving 20, 90, and 180 mg of the drug, respectively. This was compared with 11% of patients receiving the placebo (229). This highlights the importance of establishing efficacy, safety, and tolerability of these drugs to treat these conditions. As noted elsewhere, BAs will cause symptoms such as diarrhea when delivered at high, pathophysiological concentrations; therefore, it would be interesting to know how the levels of fecal BAs correlated with symptom severity. In healthy volunteers, volixibat increased fecal BAs by 1.6–3.2 times compared with those that had received placebo. It is worth noting that there was no clear correlation between dosage of volixibat (0.5–5 mg) and excreted BAs. How these levels correlated with symptom severity was not reported. However, these results highlight that there may be variability in the ways in which patients respond to these drugs.
Another approach to modulating endogenous levels of BAs involves the use of BA sequestrants that have opposing effects to ASBT inhibitors and reduce free BA levels in the colon. Such drugs, which include cholestyramine and colesevelam, have been trialed to treat a range of diarrhea-associated conditions including idiopathic BA-diarrhea and BA-associated diarrhea in Crohn’s disease and IBS-D (57, 125, 217, 232–234). As FXR activation leads to the inhibition of BA production in hepatocytes, FXR agonists have also been trialed to treat BA-induced diarrhea (52, 86, 235). Two small clinical trials conducted in the UK and the USA have demonstrated the benefits of the FXR agonists obeticholic acid and tropifexor. Daily tropifexor administration had a small but significant effect on slowing ascending colonic motility in patients with primary BA-diarrhea in a recent safety profiling study [60 µg daily, n = 8 (235)]. Obeticholic acid given to patients with primary BA-diarrhea improved stool frequency and formation during a 2-wk treatment period [25 mg, daily, n = 10 (52)]. However, patients with chronic idiopathic diarrhea did not have improved symptoms (n = 8) indicating that neither FXR activation nor limiting physiological levels of BA production have roles in controlling colonic motility (52). This differs to patients with BA-diarrhea, where removing excess BAs reduces the symptoms of the disease. It is important to note that GI motility and secretion are interlinked, where increasing secretion will stimulate motility. FXR agonists limit intestinal fluid secretion in mouse models of secretory diarrhea (53). However, these studies did not specifically study direct GI motor indices.
When considering the potential for targeting BAs for therapeutic purposes, it is important to consider how the gut microbiome interacts with these molecules and their receptors to modulate intestinal motility (236). There is a growing appreciation for the roles of intestinal bacteria in a range of GI disorders. BAs increase intestinal permeability, providing greater opportunity for microbiota to access the gut wall. Although the BAs can modulate motility directly, the inflammation promoted by the infiltrating bacteria can also alter intestinal motility (237). This pairs with the proposed role of TGR5 in regulating intestinal inflammation, which may involve TGR5 expressing-circulating macrophages (179, 180). Recent studies in mice have highlighted that with age-related changes to the microbiome there is an increase in fecal deconjugated BAs. TGR5 activation by conjugated BAs in young mice stimulated an anti-inflammatory phenotype in muscularis macrophages (mMac) protecting normal motility. With aging, the shift of fecal BA content to deconjugated BAs led to reduced TGR5 activation. This resulted in a phenotypic switch of mMac to a more proinflammatory state, stimulating low-grade inflammation in these tissues and thus delayed GI transit (238). The microbiome-BA balance and its role in intestinal motility seem an obvious consideration when trying to understand complex treatment strategies such as fecal transplants (236). This could have important implications for our understanding of the pathology of a range of GI motility disorders but may also provide novel therapeutic strategies for these conditions.
CONCLUSION
The BA receptor axis is a viable therapeutic target, as evidenced by the clinical use of BA sequestrants and more recently IBAT inhibitors. With the discovery of specific BA receptors, BA-modulated ion channels, and BA transporters, our understanding of the roles of BAs in physiological processes and disease has evolved. BAs have physiological actions as signaling molecules through binding and activation of both cell surface and nuclear receptors and through modifying ion channel function. They also drive pathophysiological changes in disease and may have mechanistic roles in GI disorders including IBS. As outlined in this review, there remains the need for greater understanding of the functional importance of BA receptors, particularly those which have only recently been defined.
GRANTS
S. E. Carbone is supported by an Australian Research Council DECRA Fellow Grant DE200100825. Research in the author’s laboratories is supported by National Health and Medical Research Council Australia 1083480 (to D. P. Poole), National Institutes of Health Grants NS102722, DE026806, DE029951, DK118971 (to N. W. Bunnett), Department of Defense Grant W81XWH1810431 (to N. W. Bunnett), the ELAN program of the Interdisciplinary Center for Clinical Research, Erlangen (IZKF) Grant 17-08-01-1-Ilyaskin/P015 (to A.V. Ilyaskin), and by Science Foundation Ireland Grant No. 16/IA/4445 (to S. J. Keely).
DISCLOSURES
N. W. Bunnett is a founding scientist of Endosome Therapeutics Inc. Research in the laboratories of N. W. Bunnett, D. P. Poole, and S. E. Carbone is funded in part by Takeda Pharmaceuticals. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
S.E.C. analyzed data; D.P.P. and S.E.C. prepared figures; S.J.K., A.U., A.V.I., C.K., N.W.B., D.P.P., and S.E.C. drafted manuscript; S.J.K., A.U., A.V.I., C.K., N.W.B., D.P.P., and S.E.C. edited and revised manuscript; S.J.K., A.U., A.V.I., C.K., N.W.B., D.P.P., and S.E.C. approved final version of manuscript.
REFERENCES
- 1.de Aguiar Vallim TQ, Tarling EJ, Edwards PA. Pleiotropic roles of bile acids in metabolism. Cell Metab 17: 657–669, 2013. doi: 10.1016/j.cmet.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peleman C, Camilleri M, Busciglio I, Burton D, Donato L, Zinsmeister AR. Colonic transit and bile acid synthesis or excretion in patients with irritable bowel syndrome-diarrhea without bile acid malabsorption. Clin Gastroenterol Hepatol 15: 720–727.e1, 2017. doi: 10.1016/j.cgh.2016.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mekhjian HS, Phillips SF, Hofmann AF. Colonic absorption of unconjugated bile acids: perfusion studies in man. Dig Dis Sci 24: 545–550, 1979. doi: 10.1007/BF01489324. [DOI] [PubMed] [Google Scholar]
- 4.Mikov M, Fawcett JP, Kuhajda K, Kevresan S. Pharmacology of bile acids and their derivatives: absorption promoters and therapeutic agents. Eur J Drug Metab Pharmacokinet 31: 237–251, 2006. doi: 10.1007/BF03190714. [DOI] [PubMed] [Google Scholar]
- 5.Alemi F, Poole DP, Chiu J, Schoonjans K, Cattaruzza F, Grider JR, Bunnett NW, Corvera CU. The receptor TGR5 mediates the prokinetic actions of intestinal bile acids and is required for normal defecation in mice. Gastroenterology 144: 145–154, 2013. doi: 10.1053/j.gastro.2012.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castro J, Harrington AM, Lieu T, Garcia-Caraballo S, Maddern J, Schober G, O'Donnell T, Grundy L, Lumsden AL, Miller P, Ghetti A, Steinhoff MS, Poole DP, Dong X, Chang L, Bunnett NW, Brierley SM. Activation of pruritogenic TGR5, MrgprA3, and MrgprC11 on colon-innervating afferents induces visceral hypersensitivity. JCI insight 4: 332, 2019. doi: 10.1172/jci.insight.131712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duboc H, Tolstanova G, Yuan PQ, Wu V, Kaji I, Biraud M, Akiba Y, Kaunitz J, Million M, Tache Y, Larauche M. Reduction of epithelial secretion in male rat distal colonic mucosa by bile acid receptor TGR5 agonist, INT-777: role of submucosal neurons. Neurogastroenterol Motil 28: 1663–1676, 2016. doi: 10.1111/nmo.12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Katsuma S, Hirasawa A, Tsujimoto G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem Biophys Res Commun 329: 386–390, 2005. doi: 10.1016/j.bbrc.2005.01.139. [DOI] [PubMed] [Google Scholar]
- 9.Kuhre RE, Wewer Albrechtsen NJ, Larsen O, Jepsen SL, Balk-Møller E, Andersen DB, Deacon CF, Schoonjans K, Reimann F, Gribble FM, Albrechtsen R, Hartmann B, Rosenkilde MM, Holst JJ. Bile acids are important direct and indirect regulators of the secretion of appetite- and metabolism-regulating hormones from the gut and pancreas. Mol Metab 11: 84–95, 2018. doi: 10.1016/j.molmet.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ullmer C, Alvarez Sanchez R, Sprecher U, Raab S, Mattei P, Dehmlow H, Sewing S, Iglesias A, Beauchamp J, Conde-Knape K. Systemic bile acid sensing by G protein-coupled bile acid receptor 1 (GPBAR1) promotes PYY and GLP-1 release. Br J Pharmacol 169: 671–684, 2013. doi: 10.1111/bph.12158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meixiong J, Vasavda C, Snyder SH, Dong X. MRGPRX4 is a G protein-coupled receptor activated by bile acids that may contribute to cholestatic pruritus. Proc Natl Acad Sci USA 116: 10525–10530, 2019. doi: 10.1073/pnas.1903316116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ibrahim E, Diakonov I, Arunthavarajah D, Swift T, Goodwin M, McIlvride S, Nikolova V, Williamson C, Gorelik J. Bile acids and their respective conjugates elicit different responses in neonatal cardiomyocytes: role of Gi protein, muscarinic receptors and TGR5. Sci Rep 8: 7110–7112, 2018. doi: 10.1038/s41598-018-25569-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sheikh Abdul Kadir SH, Miragoli M, Abu-Hayyeh S, Moshkov AV, Xie Q, Keitel V, Nikolaev VO, Williamson C, Gorelik J. Bile acid-induced arrhythmia is mediated by muscarinic M2 receptors in neonatal rat cardiomyocytes. PLoS One 5: e9689, 2010. doi: 10.1371/journal.pone.0009689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Urso A, D'Ovidio F, Xu D, Emala CW, Bunnett NW, Perez-Zoghbi JF. Bile acids inhibit cholinergic constriction in proximal and peripheral airways from humans and rodents. Am J Physiol Lung Cell Mol Physiol 318: L264–L275, 2020. doi: 10.1152/ajplung.00242.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Studer E, Zhou X, Zhao R, Wang Y, Takabe K, Nagahashi M, Pandak WM, Dent P, Spiegel S, Shi R, Xu W, Liu X, Bohdan P, Zhang L, Zhou H, Hylemon PB. Conjugated bile acids activate the sphingosine-1-phosphate receptor 2 in primary rodent hepatocytes. Hepatology 55: 267–276, 2012. doi: 10.1002/hep.24681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y, Aoki H, Yang J, Peng K, Liu R, Li X, Qiang X, Sun L, Gurley EC, Lai G, Zhang L, Liang G, Nagahashi M, Takabe K, Pandak WM, Hylemon PB, Zhou H. The role of sphingosine 1-phosphate receptor 2 in bile-acid-induced cholangiocyte proliferation and cholestasis-induced liver injury in mice. Hepatology 65: 2005–2018, 2017. doi: 10.1002/hep.29076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen X, Yang D, Shen W, Dong HF, Wang JM, Oppenheim JJ, Howard MZ. Characterization of chenodeoxycholic acid as an endogenous antagonist of the G-coupled formyl peptide receptors. Inflamm Res 49: 744–755, 2000. doi: 10.1007/s000110050656. [DOI] [PubMed] [Google Scholar]
- 18.Chen X, Mellon RD, Yang L, Dong HF, Oppenheim JJ, Howard MZ. Regulatory effects of deoxycholic acid, a component of the anti-inflammatory traditional Chinese medicine Niuhuang, on human leukocyte response to chemoattractants. Biochem Pharmacol 63: 533–541, 2002. doi: 10.1016/S0006-2952(01)00917-0. [DOI] [PubMed] [Google Scholar]
- 19.Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ, Shan B. Identification of a nuclear receptor for bile acids. Science 284: 1362–1365, 1999. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 20.Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, Stimmel JB, Willson TM, Zavacki AM, Moore DD, Lehmann JM. Bile acids: natural ligands for an orphan nuclear receptor. Science 284: 1365–1368, 1999. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- 21.Zhang JH, Nolan JD, Kennie SL, Johnston IM, Dew T, Dixon PH, Williamson C, Walters JRF. Potent stimulation of fibroblast growth factor 19 expression in the human ileum by bile acids. Am J Physiol Gastrointest Liver Physiol 304: G940–G948, 2013. doi: 10.1152/ajpgi.00398.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, Luo G, Jones SA, Goodwin B, Richardson JA, Gerard RD, Repa JJ, Mangelsdorf DJ, Kliewer SA. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2: 217–225, 2005. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 23.Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, MacKenzie KI, LaTour A, Liu Y, Klaassen CD, Brown KK, Reinhard J, Willson TM, Koller BH, Kliewer SA. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci USA 98: 3369–3374, 2001. doi: 10.1073/pnas.051551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xie W, Radominska-Pandya A, Shi Y, Simon CM, Nelson MC, Ong ES, Waxman DJ, Evans RM. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc Natl Acad Sci USA 98: 3375–3380, 2001. doi: 10.1073/pnas.051014398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM, Haussler MR, Mangelsdorf DJ. Vitamin D receptor as an intestinal bile acid sensor. Science 296: 1313–1316, 2002. doi: 10.1126/science.1070477. [DOI] [PubMed] [Google Scholar]
- 26.Ishizawa M, Akagi D, Makishima M. Lithocholic acid is a vitamin D receptor ligand that acts preferentially in the ileum. Int J Mol Sci 19: 1975, 2018. doi: 10.3390/ijms19071975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoneno K, Hisamatsu T, Shimamura K, Kamada N, Ichikawa R, Kitazume MT, Mori M, Uo M, Namikawa Y, Matsuoka K, Sato T, Koganei K, Sugita A, Kanai T, Hibi T. TGR5 signalling inhibits the production of pro-inflammatory cytokines by in vitro differentiated inflammatory and intestinal macrophages in Crohn’s disease. Immunology 139: 19–29, 2013. doi: 10.1111/imm.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jia W, Xie G, Jia W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat Rev Gastroenterol Hepatol 15: 111–128, 2018. doi: 10.1038/nrgastro.2017.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dong X, Han S, Zylka MJ, Simon MI, Anderson DJ. A diverse family of GPCRs expressed in specific subsets of nociceptive sensory neurons. Cell 106: 619–632, 2001. doi: 10.1016/s0092-8674(01)00483-4. [DOI] [PubMed] [Google Scholar]
- 30.Meixiong J, Vasavda C, Green D, Zheng Q, Qi L, Kwatra SG, Hamilton JP, Snyder SH, Dong X. Identification of a bilirubin receptor that may mediate a component of cholestatic itch. eLife 8: e44116, 2019. doi: 10.7554/eLife.44116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gulamhusein AF, Hirschfield GM. Primary biliary cholangitis: pathogenesis and therapeutic opportunities. Nat Rev Gastroenterol Hepatol 17: 93–110, 2020. doi: 10.1038/s41575-019-0226-7. [DOI] [PubMed] [Google Scholar]
- 32.Van Remoortel S, Ceuleers H, Arora R, Van Nassauw L, De Man JG, Buckinx R, De Winter BY, Timmermans J-P. Mas-related G protein-coupled receptor C11 (Mrgprc11) induces visceral hypersensitivity in the mouse colon: a novel target in gut nociception? Neurogastroenterol Motil 31: e13623, 2019. doi: 10.1111/nmo.13623. [DOI] [PubMed] [Google Scholar]
- 33.Avula LR, Buckinx R, Favoreel H, Cox E, Adriaensen D, Van Nassauw L, Timmermans JP. Expression and distribution patterns of Mas-related gene receptor subtypes A-H in the mouse intestine: inflammation-induced changes. Histochem Cell Biol 139: 639–658, 2013. doi: 10.1007/s00418-013-1086-9. [DOI] [PubMed] [Google Scholar]
- 34.Avula LR, Buckinx R, Alpaerts K, Costagliola A, Adriaensen D, Van Nassauw L, Timmermans JP. The effect of inflammation on the expression and distribution of the MAS-related gene receptors MrgE and MrgF in the murine ileum. Histochem Cell Biol 136: 569–585, 2011. doi: 10.1007/s00418-011-0862-7. [DOI] [PubMed] [Google Scholar]
- 35.Inclan-Rico JM, Kim BS, Abdus-Saboor I. Beyond somatosensation: Mrgprs in mucosal tissues. Neurosci Lett 748: 135689, 2021. doi: 10.1016/j.neulet.2021.135689. [DOI] [PubMed] [Google Scholar]
- 36.Luo J, Feng J, Liu S, Walters ET, Hu H. Molecular and cellular mechanisms that initiate pain and itch. Cell Mol Life Sci 72: 3201–3223, 2015. [Erratum in Cell Mol Life Sci 72: 3587–3588, 2015]. doi: 10.1007/s00018-015-1904-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng K, Chen Y, Zimniak P, Raufman JP, Xiao Y, Frucht H. Functional interaction of lithocholic acid conjugates with M3 muscarinic receptors on a human colon cancer cell line. Biochim Biophys Acta 1588: 48–55, 2002. doi: 10.1016/s0925-4439(02)00115-1. [DOI] [PubMed] [Google Scholar]
- 38.Raufman J-P, Chen Y, Cheng K, Compadre C, Compadre L, Zimniak P. Selective interaction of bile acids with muscarinic receptors: a case of molecular mimicry. Eur J Pharmacol 457: 77–84, 2002. doi: 10.1016/s0014-2999(02)02690-0. [DOI] [PubMed] [Google Scholar]
- 39.Nagahashi M, Yuza K, Hirose Y, Nakajima M, Ramanathan R, Hait NC, Hylemon PB, Zhou H, Takabe K, Wakai T. The roles of bile acids and sphingosine-1-phosphate signaling in the hepatobiliary diseases. J Lipid Res 57: 1636–1643, 2016. doi: 10.1194/jlr.R069286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Keitel V, Stindt J, Häussinger D. Bile acid-activated receptors: GPBAR1 (TGR5) and other G protein-coupled receptors. In: Bile Acids and Their Receptors, edited by Fiorucci S, Distrutti E.. Cham: Springer International Publishing, 2019, p. 19–49. [DOI] [PubMed] [Google Scholar]
- 41.Ferrari C, Macchiarulo A, Costantino G, Pellicciari R. Pharmacophore model for bile acids recognition by the FPR receptor. J Comput Aided Mol Des 20: 295–303, 2006. doi: 10.1007/s10822-006-9055-1. [DOI] [PubMed] [Google Scholar]
- 42.Chandrasekharan B, Saeedi BJ, Alam A, Houser M, Srinivasan S, Tansey M, Jones R, Nusrat A, Neish AS. Interactions between commensal bacteria and enteric neurons, via fpr1 induction of ROS, increase gastrointestinal motility in mice. Gastroenterology 157: 179–194, 2019. doi: 10.1053/j.gastro.2019.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anton PA, Targan SR, Shanahan F. Increased neutrophil receptors for and response to the proinflammatory bacterial peptide formyl-methionyl-leucyl-phenylalanine in Crohn’s disease. Gastroenterology 97: 20–28, 1989. doi: 10.1016/0016-5085(89)91410-8. [DOI] [PubMed] [Google Scholar]
- 44.Francis GA, Fayard E, Picard F, Auwerx J. Nuclear receptors and the control of metabolism. Annu Rev Physiol 65: 261–311, 2003. doi: 10.1146/annurev.physiol.65.092101.142528. [DOI] [PubMed] [Google Scholar]
- 45.Li T, Chiang JY. Nuclear receptors in bile acid metabolism. Drug Metab Rev 45: 145–155, 2013. doi: 10.3109/03602532.2012.740048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li T, Chiang JYL. Bile acid signaling in metabolic disease and drug therapy. Pharmacol Rev 66: 948–983, 2014. doi: 10.1124/pr.113.008201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schaap FG, Trauner M, Jansen PL. Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol 11: 55–67, 2014. doi: 10.1038/nrgastro.2013.151. [DOI] [PubMed] [Google Scholar]
- 48.Forman BM, Goode E, Chen J, Oro AE, Bradley DJ, Perlmann T, Noonan DJ, Burka LT, McMorris T, Lamph WW, Evans RM, Weinberger C. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 81: 687–693, 1995. doi: 10.1016/0092-8674(95)90530-8. [DOI] [PubMed] [Google Scholar]
- 49.Wang H, Chen J, Hollister K, Sowers LC, Forman MB. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell 3: 543–553, 1999. doi: 10.1016/S1097-2765(00)80348-2. [DOI] [PubMed] [Google Scholar]
- 50.Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 102: 731–744, 2000. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 51.Keely SJ, Walters JRF. The Farnesoid X receptor: good for bad. Cell Mol Gastroenterol Hepatol 2: 725–732, 2016. doi: 10.1016/j.jcmgh.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Walters JRF, Johnston IM, Nolan JD, Vassie C, Pruzanski ME, Shapiro DA. The response of patients with bile acid diarrhoea to the farnesoid X receptor agonist obeticholic acid. Aliment Pharmacol Ther 41: 54–64, 2015. doi: 10.1111/apt.12999. [DOI] [PubMed] [Google Scholar]
- 53.Mroz MS, Keating N, Ward JB, Sarker R, Amu S, Aviello G, Donowitz M, Fallon PG, Keely SJ. Farnesoid X receptor agonists attenuate colonic epithelial secretory function and prevent experimental diarrhoea in vivo. Gut 63: 808–817, 2014. doi: 10.1136/gutjnl-2013-305088. [DOI] [PubMed] [Google Scholar]
- 54.Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, Chalasani N, Dasarathy S, Diehl AM, Hameed B, Kowdley KV, McCullough A, Terrault N, Clark JM, Tonascia J, Brunt EM, Kleiner DE, Doo E; NASH Clinical Research Network. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet 385: 956–965, 2015. [Erratum in Lancet 385: 946, 2015].doi: 10.1016/S0140-6736(14)61933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chávez-Talavera O, Tailleux A, Lefebvre P, Staels B. Bile acid control of metabolism and inflammation in obesity, type 2 diabetes, dyslipidemia, and nonalcoholic fatty liver disease. Gastroenterology 152: 1679–1694e.3, 2017. doi: 10.1053/j.gastro.2017.01.055. [DOI] [PubMed] [Google Scholar]
- 56.Cheng J, Ma X, Gonzalez FJ. Pregnane X receptor- and CYP3A4-humanized mouse models and their applications. Br J Pharmacol 163: 461–468, 2011. doi: 10.1111/j.1476-5381.2010.01129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kidd M, Modlin IM, Gustafsson BI, Drozdov I, Hauso O, Pfragner R. Luminal regulation of normal and neoplastic human EC cell serotonin release is mediated by bile salts, amines, tastants, and olfactants. Am J Physiol Gastrointest Liver Physiol 295: G260–G272, 2008. doi: 10.1152/ajpgi.00056.2008. [DOI] [PubMed] [Google Scholar]
- 58.Fiorucci S, Carino A, Baldoni M, Santucci L, Costanzi E, Graziosi L, Distrutti E, Biagioli M. Bile acid signaling in inflammatory bowel diseases. Dig Dis Sci 66: 674–693, 2021. doi: 10.1007/s10620-020-06715-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wilson A, Almousa A, Teft WA, Kim RB. Attenuation of bile acid-mediated FXR and PXR activation in patients with Crohn’s disease. Sci Rep 10: 1866, 2020. doi: 10.1038/s41598-020-58644-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Iwamoto J, Saito Y, Honda A, Miyazaki T, Ikegami T, Matsuzaki Y. Bile acid malabsorption deactivates pregnane X receptor in patients with Crohn’s disease. Inflamm Bowel Dis 19: 1278–1284, 2013. doi: 10.1097/MIB.0b013e318281f423. [DOI] [PubMed] [Google Scholar]
- 61.Shah YM, Ma X, Morimura K, Kim I, Gonzalez FJ. Pregnane X receptor activation ameliorates DSS-induced inflammatory bowel disease via inhibition of NF-κB target gene expression. Am J Physiol Gastrointest Liver Physiol 292: G1114–G1122, 2007. doi: 10.1152/ajpgi.00528.2006. [DOI] [PubMed] [Google Scholar]
- 62.Owen RW, Dodo M, Thompson MH, Hill MJ. Fecal steroids and colorectal cancer. Nutr Cancer 9: 73–80, 1987. doi: 10.1080/01635588709513914. [DOI] [PubMed] [Google Scholar]
- 63.Woolbright BL, Li F, Xie Y, Farhood A, Fickert P, Trauner M, Jaeschke H. Lithocholic acid feeding results in direct hepato-toxicity independent of neutrophil function in mice. Toxicol Lett 228: 56–66, 2014. doi: 10.1016/j.toxlet.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yao B, He J, Yin X, Shi Y, Wan J, Tian Z. The protective effect of lithocholic acid on the intestinal epithelial barrier is mediated by the vitamin D receptor via a SIRT1/Nrf2 and NF-κB dependent mechanism in Caco-2 cells. Toxicol Lett 316: 109–118, 2019. doi: 10.1016/j.toxlet.2019.08.024. [DOI] [PubMed] [Google Scholar]
- 65.Kim SD, Kwon S, Lee SK, Kook M, Lee HY, Song K-D, Lee H-K, Baek S-H, Park CB, Bae Y-S. The immune-stimulating peptide WKYMVm has therapeutic effects against ulcerative colitis. Exp Mol Med 45: e40, 2013. doi: 10.1038/emm.2013.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu W, Chen Y, Golan MA, Annunziata ML, Du J, Dougherty U, Kong J, Musch M, Huang Y, Pekow J, Zheng C, Bissonnette M, Hanauer SB, Li YC. Intestinal epithelial vitamin D receptor signaling inhibits experimental colitis. J Clin Invest 123: 3983–3996, 2013. doi: 10.1172/JCI65842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schmidt DR, Holmstrom SR, Fon Tacer K, Bookout AL, Kliewer SA, Mangelsdorf DJ. Regulation of bile acid synthesis by fat-soluble vitamins A and D. J Biol Chem 285: 14486–14494, 2010. doi: 10.1074/jbc.M110.116004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cheng J, Fang Z-Z, Kim J-H, Krausz KW, Tanaka N, Chiang JL, Gonzalez FJ. Intestinal CYP3A4 protects against lithocholic acid-induced hepatotoxicity in intestine-specific VDR-deficient mice. J Lipid Res 55: 455–465, 2014. doi: 10.1194/jlr.M044420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wei P, Zhang J, Egan-Hafley M, Liang S, Moore DD. The nuclear receptor CAR mediates specific xenobiotic induction of drug metabolism. Nature 407: 920–923, 2000. doi: 10.1038/35038112. [DOI] [PubMed] [Google Scholar]
- 70.Miao J, Fang S, Bae Y, Kemper JK. Functional inhibitory cross-talk between constitutive androstane receptor and hepatic nuclear factor-4 in hepatic lipid/glucose metabolism is mediated by competition for binding to the DR1 motif and to the common coactivators, GRIP-1 and PGC-1α. J Biol Chem 281: 14537–14546, 2006. doi: 10.1074/jbc.M510713200. [DOI] [PubMed] [Google Scholar]
- 71.Zhang J, Huang W, Qatanani M, Evans RM, Moore DD. The constitutive androstane receptor and pregnane X receptor function coordinately to prevent bile acid-induced hepatotoxicity. J Biol Chem 279: 49517–49522, 2004. doi: 10.1074/jbc.M409041200. [DOI] [PubMed] [Google Scholar]
- 72.Hudson GM, Flannigan KL, Erickson SL, Vicentini FA, Zamponi A, Hirota CL, Alston L, Altier C, Ghosh S, Rioux KP, Mani S, Chang TK, Hirota SA. Constitutive androstane receptor regulates the intestinal mucosal response to injury. Br J Pharmacol 174: 1857–1871, 2017. doi: 10.1111/bph.13787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Guo GL, Lambert G, Negishi M, Ward JM, Brewer HB Jr, Kliewer SA, Gonzalez FJ, Sinal CJ. Complementary roles of farnesoid X receptor, pregnane X receptor, and constitutive androstane receptor in protection against bile acid toxicity. J Biol Chem 278: 45062–45071, 2003. doi: 10.1074/jbc.M307145200. [DOI] [PubMed] [Google Scholar]
- 74.Stedman CAM, Liddle C, Coulter SA, Sonoda J, Alvarez JGA, Moore DD, Evans RM, Downes M. Nuclear receptors constitutive androstane receptor and pregnane X receptor ameliorate cholestatic liver injury. Proc Natl Acad Sci USA 102: 2063–2068, 2005. doi: 10.1073/pnas.0409794102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Toda T, Saito N, Ikarashi N, Ito K, Yamamoto M, Ishige A, Watanabe K, Sugiyama K. Intestinal flora induces the expression of Cyp3a in the mouse liver. Xenobiotica 39: 323–334, 2009. doi: 10.1080/00498250802651984. [DOI] [PubMed] [Google Scholar]
- 76.Dempsey JL, Wang D, Siginir G, Fei Q, Raftery D, Gu H, Yue Cui J. Pharmacological activation of PXR and CAR downregulates distinct bile acid-metabolizing intestinal bacteria and alters bile acid homeostasis. Toxicol Sci 168: 40–60, 2019. doi: 10.1093/toxsci/kfy271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wiemuth D, Sahin H, Falkenburger BH, Lefevre CMT, Wasmuth HE, Gründer S. BASIC-a bile acid-sensitive ion channel highly expressed in bile ducts. FASEB J 26: 4122–4130, 2012. doi: 10.1096/fj.12-207043. [DOI] [PubMed] [Google Scholar]
- 78.Wiemuth D, Sahin H, Lefevre CM, Wasmuth HE, Gründer S. Strong activation of bile acid-sensitive ion channel (BASIC) by ursodeoxycholic acid. Channels (Austin) 7: 38–42, 2013. doi: 10.4161/chan.22406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lenzig P, Wirtz M, Wiemuth D. Comparative electrophysiological analysis of the bile acid-sensitive ion channel (BASIC) from different species suggests similar physiological functions. Pflugers Arch 471: 329–336, 2019. doi: 10.1007/s00424-018-2223-z. [DOI] [PubMed] [Google Scholar]
- 80.Wiegreffe S, Lohrer D, Wirtz M, Wiemuth D. The bile acid-sensitive ion channel (BASIC) mediates bile acid-dependent currents in bile duct epithelial cells. Pflugers Arch 473: 1841–1850, 2021. doi: 10.1007/s00424-021-02622-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ilyaskin AV, Kirsch SA, Böckmann RA, Sticht H, Korbmacher C, Haerteis S, Diakov A. The degenerin region of the human bile acid-sensitive ion channel (BASIC) is involved in channel inhibition by calcium and activation by bile acids. Pflugers Arch 470: 1087–1102, 2018. doi: 10.1007/s00424-018-2142-z. [DOI] [PubMed] [Google Scholar]
- 82.Lefevre CMT, Diakov A, Haerteis S, Korbmacher C, Gründer S, Wiemuth D. Pharmacological and electrophysiological characterization of the human bile acid-sensitive ion channel (hBASIC). Pflugers Arch 466: 253–263, 2014. doi: 10.1007/s00424-013-1310-4. [DOI] [PubMed] [Google Scholar]
- 83.Wiemuth D, Lefèvre CM, Heidtmann H, Gründer S. Bile acids increase the activity of the epithelial Na+ channel. Pflugers Arch 466: 1725–1733, 2014. doi: 10.1007/s00424-013-1403-0. [DOI] [PubMed] [Google Scholar]
- 84.Ilyaskin AV, Diakov A, Korbmacher C, Haerteis S. Activation of the human epithelial sodium channel (ENaC) by bile acids involves the degenerin site. J Biol Chem 291: 19835–19847, 2016. doi: 10.1074/jbc.M116.726471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang X-P, Im SJ, Balchak DM, Montalbetti N, Carattino MD, Ray EC, Kashlan OB. Murine epithelial sodium (Na+) channel regulation by biliary factors. J Biol Chem 294: 10182–10193, 2019. doi: 10.1074/jbc.RA119.007394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mroz MS, Harvey BJ. Ursodeoxycholic acid inhibits ENaC and Na/K pump activity to restore airway surface liquid height in cystic fibrosis bronchial epithelial cells. Steroids 151: 108461, 2019. doi: 10.1016/j.steroids.2019.108461. [DOI] [PubMed] [Google Scholar]
- 87.Ilyaskin AV, Diakov A, Korbmacher C, Haerteis S. Bile acids potentiate proton-activated currents in Xenopus laevis oocytes expressing human acid-sensing ion channel (ASIC1a). Physiol Rep 5: e13132, 2017. doi: 10.14814/phy2.13132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dopico AM, Bukiya AN. Regulation of Ca2+-sensitive K+ channels by cholesterol and bile acids via distinct channel subunits and sites. Curr Top Membr 80: 53–93, 2017. doi: 10.1016/bs.ctm.2017.07.001. [DOI] [PubMed] [Google Scholar]
- 89.Schubring SR, Fleischer W, Lin JS, Haas HL, Sergeeva OA. The bile steroid chenodeoxycholate is a potent antagonist at NMDA and GABA(A) receptors. Neurosci Lett 506: 322–326, 2012. doi: 10.1016/j.neulet.2011.11.036. [DOI] [PubMed] [Google Scholar]
- 90.Yanovsky Y, Schubring SR, Yao QL, Zhao Y, Li S, May A, Haas HL, Lin JS, Sergeeva OA. Waking action of ursodeoxycholic acid (UDCA) involves histamine and GABA(A) receptor block. Plos One 7: e42512, 2012. doi: 10.1371/journal.pone.0042512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Koch A, Bonus M, Gohlke H, Klöcker N. Isoform-specific inhibition of N-methyl-d-aspartate receptors by bile salts. Sci Rep 9: 10068, 2019. doi: 10.1038/s41598-019-46496-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ilyaskin AV, Sure F, Nesterov V, Haerteis S, Korbmacher C. Bile acids inhibit human purinergic receptor P2X4 in a heterologous expression system. J Gen Physiol 151: 820–833, 2019. doi: 10.1085/jgp.201812291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schmidt A, Joussen S, Hausmann R, Gründer S, Wiemuth D. Bile acids are potent inhibitors of rat P2X2 receptors. Purinergic Signal 15: 213–221, 2019. doi: 10.1007/s11302-019-09657-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sakai H, Lingueglia E, Champigny G, Mattei MG, Lazdunski M. Cloning and functional expression of a novel degenerin-like Na+ channel gene in mammals. J Physiol 519: 323–333, 1999. doi: 10.1111/j.1469-7793.1999.0323m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schaefer L, Sakai H, Mattei MG, Lazdunski M, Lingueglia E. Molecular cloning, functional expression and chromosomal localization of an amiloride-sensitive Na+ channel from human small intestine. Febs Lett 471: 205–210, 2000. doi: 10.1016/S0014-5793(00)01403-4. [DOI] [PubMed] [Google Scholar]
- 96.Li Q, Kresge C, Bugde A, Lamphere M, Park JY, Feranchak AP. Regulation of mechanosensitive biliary epithelial transport by the epithelial Na+ channel. Hepatology 63: 538–549, 2016. doi: 10.1002/hep.28301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Garty H, Palmer LG. Epithelial sodium channels: function, structure, and regulation. Physiol Rev 77: 359–396, 1997. doi: 10.1152/physrev.1997.77.2.359. [DOI] [PubMed] [Google Scholar]
- 98.Schild L. The epithelial sodium channel and the control of sodium balance. Biochim Biophys Acta 1802: 1159–1165, 2010. doi: 10.1016/j.bbadis.2010.06.014. [DOI] [PubMed] [Google Scholar]
- 99.Rossier BC. Epithelial sodium channel (ENaC) and the control of blood pressure. Curr Opin Pharmacol 15: 33–46, 2014. doi: 10.1016/j.coph.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 100.Rotin D, Staub O. Function and regulation of the epithelial Na+ channel ENaC. Compr Physiol 11: 2017–2045, 2021. doi: 10.1002/cphy.c200012. [DOI] [PubMed] [Google Scholar]
- 101.Loffing J, Korbmacher C. Regulated sodium transport in the renal connecting tubule (CNT) via the epithelial sodium channel (ENaC). Pflugers Arch 458: 111–135, 2009. doi: 10.1007/s00424-009-0656-0. [DOI] [PubMed] [Google Scholar]
- 102.Rossier BC, Stutts MJ. Activation of the epithelial sodium channel (ENaC) by serine proteases. Annu Rev Physiol 71: 361–379, 2009. doi: 10.1146/annurev.physiol.010908.163108. [DOI] [PubMed] [Google Scholar]
- 103.Kleyman TR, Kashlan OB, Hughey RP. Epithelial Na+ channel regulation by extracellular and intracellular factors. Annu Rev Physiol 80: 263–281, 2018. doi: 10.1146/annurev-physiol-021317-121143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kleyman TR, Eaton DC. Regulating ENaC’s gate. Am J Physiol Cell Physiol 318: C150–C162, 2020. doi: 10.1152/ajpcell.00418.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bertog M, Reeh L, Bunnett NW, Korbmacher C. Lithocholic acid has a sustained stimulatory effect on the epithelial sodium channel (ENaC) in mouse late distal colon. Acta Physiol 219: 164, 2017. doi: 10.1111/apha.12870. [DOI] [Google Scholar]
- 106.Holzer P. Acid-sensing ion channels in gastrointestinal function. Neuropharmacology 94: 72–79, 2015. doi: 10.1016/j.neuropharm.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gonzales EB, Kawate T, Gouaux E. Pore architecture and ion sites in acid-sensing ion channels and P2X receptors. Nature 460: 599–605, 2009. doi: 10.1038/nature08218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Doctor RB, Matzakos T, McWilliams R, Johnson S, Feranchak AP, Fitz JG. Purinergic regulation of cholangiocyte secretion: identification of a novel role for P2X receptors. Am J Physiol Gastrointest Liver Physiol 288: G779–G786, 2005. doi: 10.1152/ajpgi.00325.2004. [DOI] [PubMed] [Google Scholar]
- 109.Besnard A, Gautherot J, Julien B, Tebbi A, Garcin I, Doignon I, Péan N, Gonzales E, Cassio D, Grosse B, Liu BK, Safya H, Cauchois F, Humbert L, Rainteau D, Tordjmann T. The P2X4 purinergic receptor impacts liver regeneration after partial hepatectomy in mice through the regulation of biliary homeostasis. Hepatology 64: 941–953, 2016. doi: 10.1002/hep.28675. [DOI] [PubMed] [Google Scholar]
- 110.Ruan HZ, Burnstock G. P2X2 and P2X3 receptor expression in the gallbladder of the guinea pig. Auton Neurosci 111: 89–96, 2004. doi: 10.1016/j.autneu.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 111.Castelucci P, Robbins HL, Poole DP, Furness JB. The distribution of purine P2X(2) receptors in the guinea-pig enteric nervous system. Histochem Cell Biol 117: 415–422, 2002. doi: 10.1007/s00418-002-0404-4. [DOI] [PubMed] [Google Scholar]
- 112.Barrett KE, Keely SJ. Electrolyte secretion and absorption in the small intestine and colon. In: Textbook of Gastroenterology, edited by Podolsky D, Camilleri M, Fitz J, Kalloo A, Shanahan F, Wang T.. Hoboken, NJ: Wiley-Blackwell, 2015. [Google Scholar]
- 113.Forth W, Rummel W, Glasner H. [On the absorption inhibiting effect of bile acids]. Naunyn Schmiedebergs Arch Pharmakol Exp Pathol 254: 364–380, 1966. [PubMed] [Google Scholar]
- 114.Hofmann AF. The syndrome of ileal disease and the broken enterohepatic circulation: cholerheic enteropathy. Gastroenterology 52: 752–757, 1967. [PubMed] [Google Scholar]
- 115.Mekjian HS, Phillips SF, Hofmann AF. Colonic secretion of water and electrolytes induced by bile acids: perfusion studies in man. J Clin Invest 50: 1569–1577, 1971. doi: 10.1172/JCI106644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wingate DL, Krag E, Mekhjian HS, Phillips SF. Relationships between ion and water movement in the human jejunum, ileum and colon during perfusion with bile acids. Clin Sci Mol Med 45: 593–606, 1973. doi: 10.1042/cs0450593. [DOI] [PubMed] [Google Scholar]
- 117.Feldman S, Gibaldi M. Effect of bile salts on gastric emptying and intestinal transit in the rat. Gastroenterology 54: 918–921, 1968. [PubMed] [Google Scholar]
- 118.Brown NJ, Read NW, Richardson A, Rumsey RD, Bogentoft C. Characteristics of lipid substances activating the ileal brake in the rat. Gut 31: 1126–1129, 1990. doi: 10.1136/gut.31.10.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hansen M, Scheltema MJ, Sonne DP, Hansen JS, Sperling M, Rehfeld JF, Holst JJ, Vilsbøll T, Knop FK. Effect of chenodeoxycholic acid and the bile acid sequestrant colesevelam on glucagon-like peptide-1 secretion. Diabetes Obes Metab 18: 571–580, 2016. doi: 10.1111/dom.12648. [DOI] [PubMed] [Google Scholar]
- 120.Penagini R, Misiewicz JJ, Frost PG. Effect of jejunal infusion of bile acids on small bowel transit and fasting jejunal motility in man. Gut 29: 789–794, 1988. doi: 10.1136/gut.29.6.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Penagini R, Spiller RC, Misiewicz JJ, Frost PG. Effect of ileal infusion of glycochenodeoxycholic acid on segmental transit, motility, and flow in the human jejunum and ileum. Gut 30: 609–617, 1989. doi: 10.1136/gut.30.5.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Armstrong DN, Krenz HK, Modlin IM, Ballantyne GH. Bile salt inhibition of motility in the isolated perfused rabbit terminal ileum. Gut 34: 483–488, 1993. doi: 10.1136/gut.34.4.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Roda E, Bazzoli F, Labate AM, Mazzella G, Roda A, Sama C, Festi D, Aldini R, Taroni F, Barbara L. Ursodeoxycholic acid vs. chenodeoxycholic acid as cholesterol gallstone-dissolving agents: a comparative randomized study. Hepatology 2: 804–810, 1982. doi: 10.1002/hep.1840020611. [DOI] [PubMed] [Google Scholar]
- 124.Bampton PA, Dinning PG, Kennedy ML, Lubowski DZ, Cook IJ. The proximal colonic motor response to rectal mechanical and chemical stimulation. Am J Physiol Gastrointest Liver Physiol 282: G443–G449, 2002. doi: 10.1152/ajpgi.00194.2001. [DOI] [PubMed] [Google Scholar]
- 125.Odunsi-Shiyanbade ST, Camilleri M, McKinzie S, Burton D, Carlson P, Busciglio IA, Lamsam J, Singh R, Zinsmeister AR. Effects of chenodeoxycholate and a bile acid sequestrant, colesevelam, on intestinal transit and bowel function. Clin Gastroenterol Hepatol 8: 159–165, 2010. doi: 10.1016/j.cgh.2009.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rao AS, Wong BS, Camilleri M, Odunsi-Shiyanbade ST, McKinzie S, Ryks M, Burton D, Carlson P, Lamsam J, Singh R, Zinsmeister AR. Chenodeoxycholate in females with irritable bowel syndrome-constipation: a pharmacodynamic and pharmacogenetic analysis. Gastroenterology 139: 1549–1558, 2010. doi: 10.1053/j.gastro.2010.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wu T, Bound MJ, Standfield SD, Gedulin B, Jones KL, Horowitz M, Rayner CK. Effects of rectal administration of taurocholic acid on glucagon-like peptide-1 and peptide YY secretion in healthy humans. Diabetes Obes Metab 15: 474–477, 2013. doi: 10.1111/dom.12043. [DOI] [PubMed] [Google Scholar]
- 128.Poole DP, Godfrey C, Cattaruzza F, Cottrell GS, Kirkland JG, Pelayo JC, Bunnett NW, Corvera CU. Expression and function of the bile acid receptor GpBAR1 (TGR5) in the murine enteric nervous system. Neurogastroenterol Motil 22: 814–e228, 2010. doi: 10.1111/j.1365-2982.2010.01487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Alemi F, Kwon E, Poole DP, Lieu T, Lyo V, Cattaruzza F, Cevikbas F, Steinhoff M, Nassini R, Materazzi S, Guerrero-Alba R, Valdez-Morales E, Cottrell GS, Schoonjans K, Geppetti P, Vanner SJ, Bunnett NW, Corvera CU. The TGR5 receptor mediates bile acid-induced itch and analgesia. J Clin Invest 123: 1513–1530, 2013. doi: 10.1172/JCI64551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hu LD, Yu BP, Yang B. Deoxycholic acid inhibits smooth muscle contraction via protein kinase C-dependent modulation of L-type Ca2+ channels in rat proximal colon. Mol Med Rep 6: 833–837, 2012. doi: 10.3892/mmr.2012.984. [DOI] [PubMed] [Google Scholar]
- 131.Kirwan WO, Smith AN, Mitchell WD, Falconer JD, Eastwood MA. Bile acids and colonic motility in the rabbit and the human. Gut 16: 894–902, 1975. doi: 10.1136/gut.16.11.894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Falconer JD, Smith AN, Eastwood MA. The effects of bile acids on colonic motility in the rabbit. Q J Exp Physiol Cogn Med Sci 65: 135–144, 1980. doi: 10.1113/expphysiol.1980.sp002497. [DOI] [PubMed] [Google Scholar]
- 133.Dharmsathaphorn K, Huott PA, Vongkovit P, Beuerlein G, Pandol SJ, Ammon HV. Cl- secretion induced by bile salts. A study of the mechanism of action based on a cultured colonic epithelial cell line. J Clin Invest 84: 945–953, 1989. doi: 10.1172/JCI114257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Keely SJ, Scharl MM, Bertelsen LS, Hagey LR, Barrett KE, Hofmann AF. Bile acid-induced secretion in polarized monolayers of T84 colonic epithelial cells: structure-activity relationships. Am J Physiol Gastrointest Liver Physiol 292: G290–G297, 2007. doi: 10.1152/ajpgi.00076.2006. [DOI] [PubMed] [Google Scholar]
- 135.Kelly OB, Mroz MS, Ward JB, Colliva C, Scharl M, Pellicciari R, Gilmer JF, Fallon PG, Hofmann AF, Roda A, Murray FE, Keely SJ. Ursodeoxycholic acid attenuates colonic epithelial secretory function. J Physiol 591: 2307–2318, 2013. doi: 10.1113/jphysiol.2013.252544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Teem MV, Phillips SF. Perfusion of the hamster jejunum with conjugated and unconjugated bile acids: inhibition of water absorption and effects on morphology. Gastroenterology 62: 261–267, 1972. [PubMed] [Google Scholar]
- 137.Gracey M, Papadimitriou J, Burke V, Thomas J, Bower G. Effects on small-intestinal function and structure induced by feeding a deconjugated bile salt. Gut 14: 519–528, 1973. doi: 10.1136/gut.14.7.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Volpe BT, Binder HJ. Bile salt alteration of ion transport across jejunal mucosa. Biochim Biophys Acta 394: 597–604, 1975. doi: 10.1016/0005-2736(75)90145-5. [DOI] [PubMed] [Google Scholar]
- 139.Freel RW, Hatch M, Earnest DL, Goldner AM. Dihydroxy bile salt-induced alterations in NaCl transport across the rabbit colon. Am J Physiol Gastrointest Liver Physiol 245: G808–G815, 1983. doi: 10.1152/ajpgi.1983.245.6.G808. [DOI] [PubMed] [Google Scholar]
- 140.Binder HJ, Rawlins CL. Effect of conjugated dihydroxy bile salts on electrolyte transport in rat colon. J Clin Invest 52: 1460–1466, 1973. doi: 10.1172/JCI107320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mauricio AC, Slawik M, Heitzmann D, von Hahn T, Warth R, Bleich M, Greger R. Deoxycholic acid (DOC) affects the transport properties of distal colon. Pflugers Arch 439: 532–540, 2000. doi: 10.1007/s004249900226. [DOI] [PubMed] [Google Scholar]
- 142.Alrefai WA, Saksena S, Tyagi S, Gill RK, Ramaswamy K, Dudeja PK. Taurodeoxycholate modulates apical Cl−/OH− exchange activity in Caco2 cells. Dig Dis Sci 52: 1270–1278, 2007. doi: 10.1007/s10620-006-9090-8. [DOI] [PubMed] [Google Scholar]
- 143.Pallagi-Kunstár É, Farkas K, Maléth J, Rakonczay Z Jr, Nagy F, Molnár T, Szepes Z, Venglovecz V, Lonovics J, Rázga Z, Wittmann T, Hegyi P. Bile acids inhibit Na+/H+ exchanger and Cl−/HCO3− exchanger activities via cellular energy breakdown and Ca2+ overload in human colonic crypts. Pflugers Arch 467: 1277–1290, 2015. doi: 10.1007/s00424-014-1560-9. [DOI] [PubMed] [Google Scholar]
- 144.Moschetta A, Portincasa P, Debellis L, Petruzzelli M, Montelli R, Calamita G, Gustavsson P, Palasciano G. Basolateral Ca2+-dependent K+-channels play a key role in Cl− secretion induced by taurodeoxycholate from colon mucosa. Biol Cell 95: 115–122, 2003. doi: 10.1016/s0248-4900(03)00011-x. [DOI] [PubMed] [Google Scholar]
- 145.Devor DC, Sekar MC, Frizzell RA, Duffey ME. Taurodeoxycholate activates potassium and chloride conductances via an IP3-mediated release of calcium from intracellular stores in a colonic cell line (T84). J Clin Invest 92: 2173–2181, 1993. doi: 10.1172/JCI116819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gerbino A, Ranieri M, Lupo S, Caroppo R, Debellis L, Maiellaro I, Caratozzolo MF, Lopez F, Colella M. Ca2+-dependent K+ efflux regulates deoxycholate-induced apoptosis of BHK-21 and Caco-2 cells. Gastroenterology 137: 955–964, 2009. doi: 10.1053/j.gastro.2009.03.038. [DOI] [PubMed] [Google Scholar]
- 147.Chien JM, Chou CT, Liang WZ, Cheng JS, Chang HT, Tseng HW, Kuo SY, Kuo CC, Chen FA, Shieh P, Ho CM, Lin JR, Kuo DH, Jan CR. Effect of deoxycholic acid on Ca2+ movement, cell viability and apoptosis in human gastric cancer cells. Toxicol Mech Methods 25: 113–119, 2015. doi: 10.3109/15376516.2014.990597. [DOI] [PubMed] [Google Scholar]
- 148.Ward JB, Mroz MS, Keely SJ. The bile acid receptor, TGR5, regulates basal and cholinergic-induced secretory responses in rat colon. Neurogastroenterol Motil 25: 708–711, 2013. doi: 10.1111/nmo.12148. [DOI] [PubMed] [Google Scholar]
- 149.Akare S, Martinez JD. Bile acid induces hydrophobicity-dependent membrane alterations. Biochim Biophys Acta 1735: 59–67, 2005. doi: 10.1016/j.bbalip.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 150.Jean-Louis S, Akare S, Ali MA, Mash EA Jr, Meuillet E, Martinez JD. Deoxycholic acid induces intracellular signaling through membrane perturbations. J Biol Chem 281: 14948–14960, 2006. doi: 10.1074/jbc.M506710200. [DOI] [PubMed] [Google Scholar]
- 151.Combettes L, Berthon B, Claret M, Erlinger S. Selective permeabilization of the endoplasmic reticulum by monohydroxylated bile acids in liver. Hepatology 9: 663–665, 1989. doi: 10.1002/hep.1840090431. [DOI] [PubMed] [Google Scholar]
- 152.Combettes L, Dumont M, Berthon B, Erlinger S, Claret M. Release of calcium from the endoplasmic reticulum by bile acids in rat liver cells. J Biol Chem 263: 2299–2303, 1988. [PubMed] [Google Scholar]
- 153.Peregrin AT, Ahlman H, Jodal M, Lundgren O. Involvement of serotonin and calcium channels in the intestinal fluid secretion evoked by bile salt and cholera toxin. Br J Pharmacol 127: 887–894, 1999. doi: 10.1038/sj.bjp.0702615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tantisira MH, Jodal M, Lundgren O. Intraluminal bile salt increases rate of firing in afferent fibers from the small intestine of the rat. Experientia 43: 168–169, 1987. doi: 10.1007/BF01942837. [DOI] [PubMed] [Google Scholar]
- 155.Karlström L, Cassuto J, Jodal M, Lundgren O. The importance of the enteric nervous system for the bile-salt-induced secretion in the small intestine of the rat. Scand J Gastroenterol 18: 117–123, 1983. doi: 10.3109/00365528309181570. [DOI] [PubMed] [Google Scholar]
- 156.Karlström L. Evidence of involvement of the enteric nervous system in the effects of sodium deoxycholate on small-intestinal transepithelial fluid transport and motility. Scand J Gastroenterol 21: 321–330, 1986. doi: 10.3109/00365528609003082. [DOI] [PubMed] [Google Scholar]
- 157.Forsgård RA, Korpela R, Stenman LK, Osterlund P, Holma R. Deoxycholic acid induced changes in electrophysiological parameters and macromolecular permeability in murine small intestine with and without functional enteric nervous system plexuses. Neurogastroenterol Motil 26: 1179–1187, 2014. doi: 10.1111/nmo.12383. [DOI] [PubMed] [Google Scholar]
- 158.Sun Y, Fihn BM, Sjövall H, Jodal M. Enteric neurones modulate the colonic permeability response to luminal bile acids in rat colon in vivo. Gut 53: 362–367, 2004. doi: 10.1136/gut.2003.015867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Quist RG, Ton-Nu HT, Lillienau J, Hofmann AF, Barrett KE. Activation of mast cells by bile acids. Gastroenterology 101: 446–456, 1991. doi: 10.1016/0016-5085(91)90024-f. [DOI] [PubMed] [Google Scholar]
- 160.Gelbmann CM, Schteingart CD, Thompson SM, Hofmann AF, Barrett KE. Mast cells and histamine contribute to bile acid-stimulated secretion in the mouse colon. J Clin Invest 95: 2831–2839, 1995. doi: 10.1172/JCI117988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Arizono N, Matsuda S, Hattori T, Kojima Y, Maeda T, Galli SJ. Anatomical variation in mast cell nerve associations in the rat small intestine, heart, lung, and skin. Similarities of distances between neural processes and mast cells, eosinophils, or plasma cells in the jejunal lamina propria. Lab Invest 62: 626–634, 1990. [PubMed] [Google Scholar]
- 162.Stead RH, Dixon MF, Bramwell NH, Riddell RH, Bienenstock J. Mast cells are closely apposed to nerves in the human gastrointestinal mucosa. Gastroenterology 97: 575–585, 1989. doi: 10.1016/0016-5085(89)90627-6. [DOI] [PubMed] [Google Scholar]
- 163.Keating N, Mroz MS, Scharl MM, Marsh C, Ferguson G, Hofmann AF, Keely SJ. Physiological concentrations of bile acids down-regulate agonist induced secretion in colonic epithelial cells. J Cell Mol Med 13: 2293–2303, 2009. doi: 10.1111/j.1582-4934.2009.00838.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hamilton JP, Xie G, Raufman JP, Hogan S, Griffin TL, Packard CA, Chatfield DA, Hagey LR, Steinbach JH, Hofmann AF. Human cecal bile acids: concentration and spectrum. Am J Physiol Gastrointest Liver Physiol 293: G256–G263, 2007. doi: 10.1152/ajpgi.00027.2007. [DOI] [PubMed] [Google Scholar]
- 165.Wei W, Wang HF, Zhang Y, Zhang YL, Niu BY, Yao SK. Altered metabolism of bile acids correlates with clinical parameters and the gut microbiota in patients with diarrhea-predominant irritable bowel syndrome. World J Gastroenterol 26: 7153–7172, 2020. doi: 10.3748/wjg.v26.i45.7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zhan K, Zheng H, Li J, Wu H, Qin S, Luo L, Huang S. Gut microbiota-bile acid crosstalk in diarrhea-irritable bowel syndrome. Biomed Res Int 2020: 3828249, 2020. doi: 10.1155/2020/3828249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Sagar NM, Duboc H, Kay GL, Alam MT, Wicaksono AN, Covington JA, Quince C, Kokkorou M, Svolos V, Palmieri LJ, Gerasimidis K, Walters JRF, Arasaradnam RP. The pathophysiology of bile acid diarrhoea: differences in the colonic microbiome, metabolome and bile acids. Sci Rep 10: 20436, 2020. doi: 10.1038/s41598-020-77374-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Camilleri M, Vijayvargiya P. The role of bile acids in chronic diarrhea. Am J Gastroenterol 115: 1596–1603, 2020. doi: 10.14309/ajg.0000000000000696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Jeffery IB, Das A, O'Herlihy E, Coughlan S, Cisek K, Moore M, Bradley F, Carty T, Pradhan M, Dwibedi C, Shanahan F, O'Toole PW. Differences in fecal microbiomes and metabolomes of people with vs without irritable bowel syndrome and bile acid malabsorption. Gastroenterology 158: 1016–1028.e8, 2020. doi: 10.1053/j.gastro.2019.11.301. [DOI] [PubMed] [Google Scholar]
- 170.Bazzoli F, Malavolti M, Petronelli A, Barbara L, Roda E. Treatment of constipation with chenodeoxycholic acid. J Int Med Res 11: 120–123, 1983. doi: 10.1177/030006058301100211. [DOI] [PubMed] [Google Scholar]
- 171.Fromm H, Sarva RP, Ravitch MM, McJunkin B, Farivar S, Amin P. Effects of jejunoileal bypass on the enterohepatic circulation of bile acids, bacterial flora in the upper small intestine, and absorption of vitamin B12. Metabolism 32: 1133–1141, 1983. doi: 10.1016/0026-0495(83)90060-4. [DOI] [PubMed] [Google Scholar]
- 172.Sackmann M, Pauletzki J, Aydemir U, Holl J, Sauerbruch T, Hasford J, Paumgartner G. Efficacy and safety of ursodeoxycholic acid for dissolution of gallstone fragments: comparison with the combination of ursodeoxycholic acid and chenodeoxycholic acid. Hepatology 14: 1136–1141, 1991. [PubMed] [Google Scholar]
- 173.Iguchi Y, Nishimaki-Mogami T, Yamaguchi M, Teraoka F, Kaneko T, Une M. Effects of chemical modification of ursodeoxycholic acid on TGR5 activation. Biol Pharm Bull 34: 1–7, 2011. doi: 10.1248/bpb.34.1. [DOI] [PubMed] [Google Scholar]
- 174.Camilleri M, Klee EW, Shin A, Carlson P, Li Y, Grover M, Zinsmeister AR. Irritable bowel syndrome-diarrhea: characterization of genotype by exome sequencing, and phenotypes of bile acid synthesis and colonic transit. Am J Physiol Gastrointest Liver Physiol 306: G13–G26, 2014. doi: 10.1152/ajpgi.00294.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Camilleri M, Vazquez-Roque MI, Carlson P, Burton D, Wong BS, Zinsmeister AR. Association of bile acid receptor TGR5 variation and transit in health and lower functional gastrointestinal disorders. Neurogastroenterol Motil 23: 995–e458, 2011. doi: 10.1111/j.1365-2982.2011.01772.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Giaretta PR, Suchodolski JS, Blick AK, Steiner JM, Lidbury JA, Rech RR. Distribution of bile acid receptor TGR5 in the gastrointestinal tract of dogs. Histol Histopathol 34: 69–79, 2019. doi: 10.14670/HH-18-025. [DOI] [PubMed] [Google Scholar]
- 177.Dopico AM, Walsh JV Jr, Singer JJ. Natural bile acids and synthetic analogues modulate large conductance Ca2+-activated K+ (BKCa) channel activity in smooth muscle cells. J Gen Physiol 119: 251–273, 2002. doi: 10.1085/jgp.20028537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Meier Y, Eloranta JJ, Darimont J, Ismair MG, Hiller C, Fried M, Kullak-Ublick GA, Vavricka SR. Regional distribution of solute carrier mRNA expression along the human intestinal tract. Drug Metab Dispos 35: 590–594, 2007. doi: 10.1124/dmd.106.013342. [DOI] [PubMed] [Google Scholar]
- 179.Biagioli M, Carino A, Cipriani S, Francisci D, Marchiano S, Scarpelli P, Sorcini D, Zampella A, Fiorucci S. The bile acid receptor GPBAR1 regulates the M1/M2 phenotype of intestinal macrophages and activation of GPBAR1 rescues mice from murine colitis. J Immunol 199: 718–733, 2017. doi: 10.4049/jimmunol.1700183. [DOI] [PubMed] [Google Scholar]
- 180.Cipriani S, Mencarelli A, Chini MG, Distrutti E, Renga B, Bifulco G, Baldelli F, Donini A, Fiorucci S. The bile acid receptor GPBAR-1 (TGR5) modulates integrity of intestinal barrier and immune response to experimental colitis. PLoS One 6: e25637, 2011. [Erratum in PLoS One 8, 2013. doi: 10.1371/journal.pone.0025637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Adrian TE, Ballantyne GH, Longo WE, Bilchik AJ, Graham S, Basson MD, Tierney RP, Modlin IM. Deoxycholate is an important releaser of peptide YY and enteroglucagon from the human colon. Gut 34: 1219–1224, 1993. doi: 10.1136/gut.34.9.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Adrian TE, Gariballa S, Parekh KA, Thomas SA, Saadi H, Al Kaabi J, Nagelkerke N, Gedulin B, Young AA. Rectal taurocholate increases L cell and insulin secretion, and decreases blood glucose and food intake in obese type 2 diabetic volunteers. Diabetologia 55: 2343–2347, 2012. doi: 10.1007/s00125-012-2593-2. [DOI] [PubMed] [Google Scholar]
- 183.Dumoulin V, Moro F, Barcelo A, Dakka T, Cuber JC. Peptide YY, glucagon-like peptide-1, and neurotensin responses to luminal factors in the isolated vascularly perfused rat ileum. Endocrinology 139: 3780–3786, 1998. doi: 10.1210/endo.139.9.6202. [DOI] [PubMed] [Google Scholar]
- 184.Meyer-Gerspach AC, Steinert RE, Keller S, Malarski A, Schulte FH, Beglinger C. Effects of chenodeoxycholic acid on the secretion of gut peptides and fibroblast growth factors in healthy humans. J Clin Endocrinol Metab 98: 3351–3358, 2013. doi: 10.1210/jc.2012-4109. [DOI] [PubMed] [Google Scholar]
- 185.Plaisancié P, Dumoulin V, Chayvialle JA, Cuber JC. Luminal peptide YY-releasing factors in the isolated vascularly perfused rat colon. J Endocrinol 151: 421–429, 1996. doi: 10.1677/joe.0.1510421. [DOI] [PubMed] [Google Scholar]
- 186.Brighton CA, Rievaj J, Kuhre RE, Glass LL, Schoonjans K, Holst JJ, Gribble FM, Reimann F. Bile acids trigger GLP-1 release predominantly by accessing basolaterally located G protein-coupled bile acid receptors. Endocrinology 156: 3961–3970, 2015. doi: 10.1210/en.2015-1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Harach T, Pols TW, Nomura M, Maida A, Watanabe M, Auwerx J, Schoonjans K. TGR5 potentiates GLP-1 secretion in response to anionic exchange resins. Sci Rep 2: 430, 2012. doi: 10.1038/srep00430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Amato A, Baldassano S, Liotta R, Serio R, Mulè F. Exogenous glucagon-like peptide 1 reduces contractions in human colon circular muscle. J Endocrinol 221: 29–37, 2014. doi: 10.1530/JOE-13-0525. [DOI] [PubMed] [Google Scholar]
- 189.May AT, Crowe MS, Blakeney BA, Mahavadi S, Wang H, Grider JR, Murthy KS. Identification of expression and function of the glucagon-like peptide-1 receptor in colonic smooth muscle. Peptides 112: 48–55, 2019. doi: 10.1016/j.peptides.2018.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Halim MA, Degerblad M, Sundbom M, Karlbom U, Holst JJ, Webb DL, Hellström PM. Glucagon-like peptide-1 inhibits prandial gastrointestinal motility through myenteric neuronal mechanisms in humans. J Clin Endocrinol Metab 103: 575–585, 2018. doi: 10.1210/jc.2017-02006. [DOI] [PubMed] [Google Scholar]
- 191.O’Brien R, O’Malley D. The Glucagon-like peptide-1 receptor agonist, exendin-4, ameliorated gastrointestinal dysfunction in the Wistar Kyoto rat model of Irritable Bowel Syndrome. Neurogastroenterol Motil 32: e13738, 2020. doi: 10.1111/nmo.13738. [DOI] [PubMed] [Google Scholar]
- 192.Tolessa T, Gutniak M, Holst JJ, Efendic S, Hellstrom PM. Glucagon-like peptide-1 retards gastric emptying and small bowel transit in the rat: effect mediated through central or enteric nervous mechanisms. Dig Dis Sci 43: 2284–2290, 1998. doi: 10.1023/a:1026678925120. [DOI] [PubMed] [Google Scholar]
- 193.Ferrier L, Segain JP, Pacaud P, Cherbut C, Loirand G, Galmiche JP, Blottière HM. Pathways and receptors involved in peptide YY induced contraction of rat proximal colonic muscle in vitro. Gut 46: 370–375, 2000. doi: 10.1136/gut.46.3.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Fox-Threlkeld JA, Daniel EE, Christinck F, Woskowska Z, Cipris S, McDonald TJ. Peptide YY stimulates circular muscle contractions of the isolated perfused canine ileum by inhibiting nitric oxide release and enhancing acetylcholine release. Peptides 14: 1171–1178, 1993. doi: 10.1016/0196-9781(93)90172-d. [DOI] [PubMed] [Google Scholar]
- 195.Lundberg JM, Tatemoto K, Terenius L, Hellström PM, Mutt V, Hökfelt T, Hamberger B. Localization of peptide YY (PYY) in gastrointestinal endocrine cells and effects on intestinal blood flow and motility. Proc Natl Acad Sci USA 79: 4471–4475, 1982. doi: 10.1073/pnas.79.14.4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Tough IR, Forbes S, Tolhurst R, Ellis M, Herzog H, Bornstein JC, Cox HM. Endogenous peptide YY and neuropeptide Y inhibit colonic ion transport, contractility and transit differentially via Y1 and Y2 receptors. Br J Pharmacol 164: 471–484, 2011. doi: 10.1111/j.1476-5381.2011.01401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Ferrier L, Segain JP, Bonnet C, Cherbut C, Lehur PA, Jarry A, Galmiche JP, Blottiere HM. Functional mapping of NPY/PYY receptors in rat and human gastro-intestinal tract. Peptides 23: 1765–1771, 2002. doi: 10.1016/s0196-9781(02)00133-x. [DOI] [PubMed] [Google Scholar]
- 198.Krantis A, Potvin W, Harding RK. Peptide YY (PYY) stimulates intrinsic enteric motor neurones in the rat small intestine. Naunyn Schmiedebergs Arch Pharmacol 338: 287–292, 1988. doi: 10.1007/BF00173402. [DOI] [PubMed] [Google Scholar]
- 199.Larhammar D. Structural diversity of receptors for neuropeptide Y, peptide YY and pancreatic polypeptide. Regul Pept 65: 165–174, 1996. doi: 10.1016/0167-0115(96)00110-3. [DOI] [PubMed] [Google Scholar]
- 200.Cho HJ, Kosari S, Hunne B, Callaghan B, Rivera LR, Bravo DM, Furness JB. Differences in hormone localisation patterns of K and L type enteroendocrine cells in the mouse and pig small intestine and colon. Cell Tissue Res 359: 693–698, 2015. doi: 10.1007/s00441-014-2033-3. [DOI] [PubMed] [Google Scholar]
- 201.Svendsen B, Pedersen J, Albrechtsen NJ, Hartmann B, Toräng S, Rehfeld JF, Poulsen SS, Holst JJ. An analysis of cosecretion and coexpression of gut hormones from male rat proximal and distal small intestine. Endocrinology 156: 847–857, 2015. doi: 10.1210/en.2014-1710. [DOI] [PubMed] [Google Scholar]
- 202.Austad WI, Lack L, Tyor MP. Importance of bile acids and of an intact distal small intestine for fat absorption. Gastroenterology 52: 638–646, 1967. [PubMed] [Google Scholar]
- 203.Valentin N, Camilleri M, Altayar O, Vijayvargiya P, Acosta A, Nelson AD, Murad MH. Biomarkers for bile acid diarrhoea in functional bowel disorder with diarrhoea: a systematic review and meta-analysis. Gut 65: 1951–1959, 2016. doi: 10.1136/gutjnl-2015-309889. [DOI] [PubMed] [Google Scholar]
- 204.Oelkers P, Kirby LC, Heubi JE, Dawson PA. Primary bile acid malabsorption caused by mutations in the ileal sodium-dependent bile acid transporter gene (SLC10A2). J Clin Invest 99: 1880–1887, 1997. doi: 10.1172/JCI119355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Ho RH, Leake BF, Urquhart BL, Gregor JC, Dawson PA, Kim RB. Functional characterization of genetic variants in the apical sodium-dependent bile acid transporter (ASBT; SLC10A2). J Gastroenterol Hepatol 26: 1740–1748, 2011. doi: 10.1111/j.1440-1746.2011.06805.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Sultan M, Rao A, Elpeleg O, Vaz FM, Abu-Libdeh B, Karpen SJ, Dawson PA. Organic solute transporter-β (SLC51B) deficiency in two brothers with congenital diarrhea and features of cholestasis. Hepatology 68: 590–598, 2018. doi: 10.1002/hep.29516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Arlow FL, Dekovich AA, Priest RJ, Beher WT. Bile acids in radiation-induced diarrhea. South Med J 80: 1259–1261, 1987. doi: 10.1097/00007611-198710000-00015. [DOI] [PubMed] [Google Scholar]
- 208.Shin A, Camilleri M, Vijayvargiya P, Busciglio I, Burton D, Ryks M, Rhoten D, Lueke A, Saenger A, Girtman A, Zinsmeister AR. Bowel functions, fecal unconjugated primary and secondary bile acids, and colonic transit in patients with irritable bowel syndrome. Clin Gastroenterol Hepatol 11: 1270–1275.e1, 2013. doi: 10.1016/j.cgh.2013.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Aldini R, Roda A, Festi D, Sama C, Mazzella G, Bazzoli F, Morselli AM, Roda E, Barbara L. Bile acid malabsorption and bile acid diarrhea in intestinal resection. Dig Dis Sci 27: 495–502, 1982. doi: 10.1007/BF01296727. [DOI] [PubMed] [Google Scholar]
- 210.Camilleri M, Busciglio I, Acosta A, Shin A, Carlson P, Burton D, Ryks M, Rhoten D, Lamsam J, Lueke A, Donato LJ, Zinsmeister AR. Effect of increased bile acid synthesis or fecal excretion in irritable bowel syndrome-diarrhea. Am J Gastroenterol 109: 1621–1630, 2014. doi: 10.1038/ajg.2014.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Lee JM, Ong JR, Vergnes L, de Aguiar Vallim TQ, Nolan J, Cantor RM, Walters JRF, Reue K. Diet1, bile acid diarrhea, and FGF15/19: mouse model and human genetic variants. J Lipid Res 59: 429–438, 2018. doi: 10.1194/jlr.M078279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Walters JR, Tasleem AM, Omer OS, Brydon WG, Dew T, Le Roux CW. A new mechanism for bile acid diarrhea: defective feedback inhibition of bile acid biosynthesis. Clin Gastroenterol Hepatol 7: 1189–1194, 2009. doi: 10.1016/j.cgh.2009.04.024. [DOI] [PubMed] [Google Scholar]
- 213.Montagnani M, Love MW, Rössel P, Dawson PA, Qvist P. Absence of dysfunctional ileal sodium-bile acid cotransporter gene mutations in patients with adult-onset idiopathic bile acid malabsorption. Scand J Gastroenterol 36: 1077–1080, 2001. doi: 10.1080/003655201750422693. [DOI] [PubMed] [Google Scholar]
- 214.Wedlake L, Thomas K, Lalji A, Anagnostopoulos C, Andreyev HJ. Effectiveness and tolerability of colesevelam hydrochloride for bile-acid malabsorption in patients with cancer: a retrospective chart review and patient questionnaire. Clin Ther 31: 2549–2558, 2009. doi: 10.1016/j.clinthera.2009.11.027. [DOI] [PubMed] [Google Scholar]
- 215.Vijayvargiya P, Busciglio I, Burton D, Donato L, Lueke A, Camilleri M. Bile acid deficiency in a subgroup of patients with irritable bowel syndrome with constipation based on biomarkers in serum and fecal samples. Clin Gastroenterol Hepatol 16: 522–527, 2018. doi: 10.1016/j.cgh.2017.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Chedid V, Vijayvargiya P, Camilleri M. Elobixibat for the treatment of constipation. Expert Rev Gastroenterol Hepatol 12: 951–960, 2018. doi: 10.1080/17474124.2018.1522248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Simrén M, Tack J. New treatments and therapeutic targets for IBS and other functional bowel disorders. Nat Rev Gastroenterol Hepatol 15: 589–605, 2018. doi: 10.1038/s41575-018-0034-5. [DOI] [PubMed] [Google Scholar]
- 218.Gillberg P-G, Dahlström M, Starke I, Östlund-Lindqvist A-M. The IBAT inhibition by A3309 a potential mechanism for the treatment of constipation. Gastroenterology 138: S224, 2010. doi: 10.1016/S0016-5085(10)61017-7. [DOI] [Google Scholar]
- 219.Chey WD, Camilleri M, Chang L, Rikner L, Graffner H. A randomized placebo-controlled phase IIb trial of a3309, a bile acid transporter inhibitor, for chronic idiopathic constipation. Am J Gastroenterol 106: 1803–1812, 2011. doi: 10.1038/ajg.2011.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Nakajima A, Seki M, Taniguchi S. Determining an optimal clinical dose of elobixibat, a novel inhibitor of the ileal bile acid transporter, in Japanese patients with chronic constipation: a phase II, multicenter, double-blind, placebo-controlled randomized clinical trial. J Gastroenterol 53: 525–534, 2018. doi: 10.1007/s00535-017-1383-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Nakajima A, Seki M, Taniguchi S, Ohta A, Gillberg PG, Mattsson JP, Camilleri M. Safety and efficacy of elobixibat for chronic constipation: results from a randomised, double-blind, placebo-controlled, phase 3 trial and an open-label, single-arm, phase 3 trial. Lancet Gastroenterol Hepatol 3: 537–547, 2018. doi: 10.1016/S2468-1253(18)30123-7. [DOI] [PubMed] [Google Scholar]
- 222.Simrén M, Bajor A, Gillberg P-G, Rudling M, Abrahamsson H. Randomized clinical trial: the ileal bile acid transporter inhibitor A3309 vs. placebo in patients with chronic idiopathic constipation—a double-blind study. Aliment Pharmacol Ther 34: 41–50, 2011. doi: 10.1111/j.1365-2036.2011.04675.x. [DOI] [PubMed] [Google Scholar]
- 223.Nakajima A, Taniguchi S, Kurosu S, Gillberg PG, Mattsson JP, Camilleri M. Efficacy, long-term safety, and impact on quality of life of elobixibat in more severe constipation: post hoc analyses of two phase 3 trials in Japan. Neurogastroenterol Motil 31: e13571, 2019. doi: 10.1111/nmo.13571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Taniguchi S, Yano T, Imaizumi M, Manabe N. Elobixibat, an ileal bile acid transporter inhibitor, induces giant migrating contractions during natural defecation in conscious dogs. Neurogastroenterol Motil 30: e13448, 2018. doi: 10.1111/nmo.13448. [DOI] [PubMed] [Google Scholar]
- 225.Newsome PN, Palmer M, Freilich B, Sheikh MY, Sheikh A, Sarles H, Herring R, Mantry P, Kayali Z, Hassanein T, Lee H-M, Aithal GP; Volixibat in Adults study group . Volixibat in adults with non-alcoholic steatohepatitis: 24-week interim analysis from a randomized, phase II study. J Hepatol 73: 231–240, 2020. doi: 10.1016/j.jhep.2020.03.024. [DOI] [PubMed] [Google Scholar]
- 226.Tiessen RG, Kennedy CA, Keller BT, Levin N, Acevedo L, Gedulin B, van Vliet AA, Dorenbaum A, Palmer M. Safety, tolerability and pharmacodynamics of apical sodium-dependent bile acid transporter inhibition with volixibat in healthy adults and patients with type 2 diabetes mellitus: a randomised placebo-controlled trial. BMC Gastroenterol 18: 3, 2018. doi: 10.1186/s12876-017-0736-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Mayo MJ, Pockros PJ, Jones D, Bowlus CL, Levy C, Patanwala I, Bacon B, Luketic V, Vuppalanchi R, Medendorp S, Dorenbaum A, Kennedy C, Novak P, Gu J, Apostol G, Hirschfield GM. A randomized, controlled, phase 2 study of maralixibat in the treatment of itching associated with primary biliary cholangitis. Hepatol Commun 3: 365–381, 2019. doi: 10.1002/hep4.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Shneider BL, Spino C, Kamath BM, Magee JC, Bass LM, Setchell KD, Miethke A, Molleston JP, Mack CL, Squires RH, Murray KF, Loomes KM, Rosenthal P, Karpen SJ, Leung DH, Guthery SL, Thomas D, Sherker AH, Sokol RJ; Childhood Liver Disease Research Network. Placebo-controlled randomized trial of an intestinal bile salt transport inhibitor for pruritus in alagille syndrome. Hepatol Commun 2: 1184–1198, 2018. doi: 10.1002/hep4.1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.United States National Library of Medicine. Dose Response Study of GSK2330672 for the Treatment of Pruritus in Participants With Primary Biliary Cholangitis. 4th May 2021. https://clinicaltrials.gov/show/NCT02966834.
- 230.Al-Dury S, Wahlström A, Wahlin S, Langedijk J, Elferink RO, Ståhlman M, Marschall HU. Pilot study with IBAT inhibitor A4250 for the treatment of cholestatic pruritus in primary biliary cholangitis. Sci Rep 8: 6658, 2018. doi: 10.1038/s41598-018-25214-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Hegade VS, Kendrick SFW, Dobbins RL, Miller SR, Thompson D, Richards D, Storey J, Dukes GE, Corrigan M,, Elferink RPJO, Beuers U, Hirschfield GM, Jones DE. Effect of ileal bile acid transporter inhibitor GSK2330672 on pruritus in primary biliary cholangitis: a double-blind, randomised, placebo-controlled, crossover, phase 2a study. Lancet 389: 1114–1123, 2017. doi: 10.1016/S0140-6736(17)30319-7. [DOI] [PubMed] [Google Scholar]
- 232.Beigel F, Teich N, Howaldt S, Lammert F, Maul J, Breiteneicher S, Rust C, Göke B, Brand S, Ochsenkühn T. Colesevelam for the treatment of bile acid malabsorption-associated diarrhea in patients with Crohn's disease: a randomized, double-blind, placebo-controlled study. J Crohns Colitis 8: 1471–1479, 2014. doi: 10.1016/j.crohns.2014.05.009. [DOI] [PubMed] [Google Scholar]
- 233.Money ME, Hofmann AF, Hagey LR, Walkowiak J, Talley NJ. Treatment of irritable bowel syndrome-diarrhea with pancrealipase or colesevelam and association with steatorrhea. Pancreas 38: 232–233, 2009. doi: 10.1097/MPA.0b013e31817c1b36. [DOI] [PubMed] [Google Scholar]
- 234.Camilleri M, Acosta A, Busciglio I, Boldingh A, Dyer RB, Zinsmeister AR, Lueke A, Gray A, Donato LJ. Effect of colesevelam on faecal bile acids and bowel functions in diarrhoea-predominant irritable bowel syndrome. Aliment Pharmacol Ther 41: 438–448, 2015. doi: 10.1111/apt.13065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Camilleri M, Nord SL, Burton D, Oduyebo I, Zhang Y, Chen J, Im K, Bhad P, Badman MK, Sanders DS, Walters JRF. Randomised clinical trial: significant biochemical and colonic transit effects of the farnesoid X receptor agonist tropifexor in patients with primary bile acid diarrhoea. Aliment Pharmacol Ther 52: 808–820, 2020. doi: 10.1111/apt.15967. [DOI] [PubMed] [Google Scholar]
- 236.Ge X, Zhao W, Ding C, Tian H, Xu L, Wang H, Ni L, Jiang J, Gong J, Zhu W, Zhu M, Li N. Potential role of fecal microbiota from patients with slow transit constipation in the regulation of gastrointestinal motility. Sci Rep 7: 441, 2017. doi: 10.1038/s41598-017-00612-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Munch A, Strom M, Soderholm JD. Dihydroxy bile acids increase mucosal permeability and bacterial uptake in human colon biopsies. Scand J Gastroenterol 42: 1167–1174, 2007. doi: 10.1080/00365520701320463. [DOI] [PubMed] [Google Scholar]
- 238.Becker L, Spear ET, Sinha SR, Haileselassie Y, Habtezion A. Age-related changes in gut microbiota alter phenotype of muscularis macrophages and disrupt gastrointestinal motility. Cell Mol Gastroenterol Hepatol 7: 243–245.e2, 2019. doi: 10.1016/j.jcmgh.2018.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]