

Keywords: apoptosis, interleukin-1, kidney, podocyte, proteinuria
Abstract
Interleukin (IL)-1 receptor type 1 (IL-1R1) activation triggers a proinflammatory signaling cascade that can exacerbate kidney injury. However, the functions of podocyte IL-1R1 in glomerular disease remain unclear. To study the role of IL-1R1 signaling in podocytes, we selectively ablated podocyte IL-1R1 in mice (PKO mice). We then subjected PKO mice and wild-type controls to two glomerular injury models: nephrotoxic serum (NTS)- and adriamycin-induced nephropathy. Surprisingly, we found that IL-1R1 activation in podocytes limited albuminuria and podocyte injury during NTS- and adriamycin-induced nephropathy. Moreover, deletion of IL-1R1 in podocytes drove podocyte apoptosis and glomerular injury through diminishing Akt activation. Activation of Akt signaling abrogated the differences in albuminuria and podocyte injury between wild-type and PKO mice during NTS. Thus, IL-1R1 signaling in podocytes limits susceptibility to glomerular injury via an Akt-dependent signaling pathway. These data identify an unexpected protective role for IL-1R1 signaling in podocytes in the pathogenesis of glomerular disease.
NEW & NOTEWORTHY The present study establishes that activation of the receptor for interleukin-1 limits susceptibility to damage to the kidney glomerulus in preclinical mouse models by stimulating Akt signaling cascades inside the podocyte.
INTRODUCTION
The interleukin (IL)-1 cytokine isoforms IL-1α and IL-1β are well known for their primary roles in initiating innate inflammatory cascades in response to danger signals. These cytokines exclusively bind to a common receptor, IL-1 receptor type 1 (IL-1R1), to activate downstream signaling pathways, which have been implicated in acute kidney injury, glomerulopathies, and kidney fibrosis (1–5). During kidney injury, infiltrating myeloid cells including monocytes/macrophages and neutrophils express and secrete IL-1 upon NLR family pyrin domain containing 3 (NLRP3)-apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (ASC)-caspase-1 axis activation, but renal parenchymal cells can also contribute to IL-1 production in kidney disease (5, 6). Moreover, during injury, kidney epithelial cells via their expression of IL-1R1 can act as key targets of IL-1 produced by immune cells (5, 7, 8). These observations suggest that the functions of IL-1 reach beyond the innate immune response to regulate cellular functions in diverse tissue beds. For example, IL-1 signals can promote kidney stromal cell proliferation and fibrogenic differentiation (9). IL-1β mediates angiogenesis and contributes to recovery after stroke via a VEGF signaling pathway (10). Within bone, IL-1 regulates osteoclast precursor fusion, differentiation, survival, and activation (11).
The pathogenesis of glomerular disease incorporates complex inflammatory responses, characterized by renal accumulation of a range of immune cell populations, including monocytes/macrophages, neutrophils, and T lymphocytes (12). As in other tissues, global IL-1 signaling is proinflammatory and worsens glomerular disease. Accordingly, genetic inactivation or pharmacological inhibition of IL-1α, IL-1β, or IL-1R1 can limit the severity of glomerular disease and preserve kidney function (13–15). However, prior studies have not parsed tissue-specific functions of IL-R1 during glomerular disease. Moreover, human studies have suggested that IL-1 signals may not be uniformly injurious. First, in human idiopathic membranous glomerulonephritis and minimal change disease, upregulation of both isoforms of IL-1 and IL-1R1 in podocytes are associated with generally well-preserved glomerular structure, indicating that intrinsic glomerular production of IL-1 may help to protect glomeruli (16). In patients with chronic kidney disease, inhibition of IL-1β by canakinumab reduced major cardiovascular event rates but did not suppress the urine albumin-to-creatinine ratio or preserve renal function. In the high-dose treatment group, estimated glomerular filtration rate fell compared with placebo over 48 mo (17). Thus, IL-1 signaling on different cell lineages may have divergent biological functions with a discrepant impact on the development and progression of glomerular diseases.
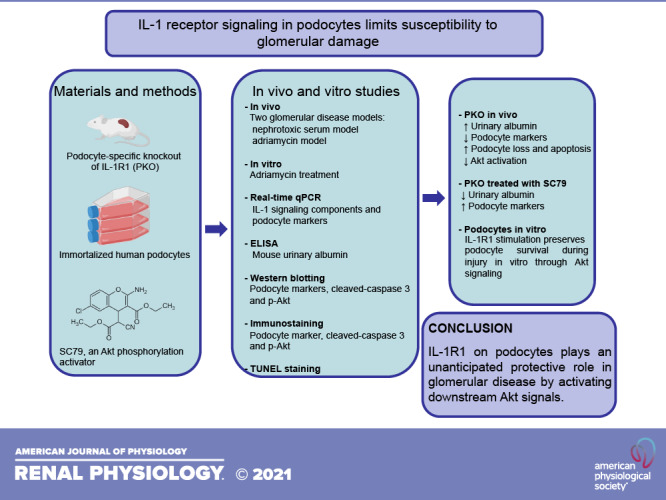
Podocytes are a primary target in a wide range of glomerular diseases marked by albuminuria and tuft collapse, such that podocyte injury and loss are considered a necessary precursor of glomerulosclerosis (18). However, reports on the role and mechanism of IL-1/IL-1R1 signaling in podocytes are lacking. In the present study, we found the upregulation of IL-1 signaling components in glomeruli after nephrotoxic serum nephritis (NTS) and adriamycin (ADR)-induced glomerular injury. In our hands, NTS induction causes robust glomerular matrix deposition, and ADR causes focal segmental glomerulosclerosis-like glomerular disease. To ascertain the role of IL-1/IL-1R1 signaling in podocytes in these models, we generated conditional knockout mice in which the gene encoding IL-1R1 was selectively disrupted in glomerular podocytes (IL-1R1 PKO). Mice with podocyte-specific deletion of IL-1R1 developed more albuminuria and podocyte injury than their control littermates after NTS and ADR injury. Deletion of IL-1R1 in podocytes also resulted in more profound podocyte loss and apoptosis after injury by limiting Akt activation. In vitro, IL-1R1 activation induced by IL-1β protected cultured podocytes from apoptosis after ADR exposure through an Akt-dependent pathway. Accordingly, the activation of Akt signaling preserved podocyte health and diminished proteinuria, abrogating the differences in these parameters between IL-1R1 PKO and wild-type (WT) controls. Our results suggest that IL-1R1 activation in podocytes protects against glomerular injury and podocyte apoptosis by activating the Akt signaling cascade.
METHODS AND METHODS
Animals
In this study, all mice were housed and bred in the animal facilities at the Durham Veterans Affairs Medical Center according to National Institutes of Health guidelines. IL-1R1 floxed mice, a generous gift from Dr. Randy Blakely (19), were backcrossed more than six generations onto the 129/SvEv background (stock no. 028398, Jackson Laboratory, Bar Harbor, ME) and then intercrossed with 129/SvEv Nphs2-Cre mice (20) to generate Nphs2-Cre+ IL1R1fl/fl (PKO) and WT littermates. To map the distribution pattern of Cre recombinase expression in the kidney, mT/mG mice from the Jackson Laboratory were crossed with the Nphs2-Cre transgenic line. mT/mG mice normally express red fluorescent protein in all tissues. When Cre is present, the mT cassette is deleted, triggering expression of membrane-targeted enhanced green fluorescent protein. Female and male mice aged 8–12 wk were randomly used for the NTS and ADR models, respectively. All animals were born phenotypically normal with the expected Mendelian frequency and maintained in specific pathogen-free conditions. Animals had free access to standard rodent chow (Cat. No. 5001, LabDiet) and water.
Histological Analysis and Immunofluorescence Staining
Mouse kidney samples were fixed in 10% formalin (Sigma-Aldrich) overnight and embedded in paraffin. Paraffin sections were stained by periodic acid-Schiff reagent, and histological changes were assessed by light microscopy. An investigator who was blinded to the experimental conditions scored at least 20 random glomeruli under the microscope. A score of 0 denotes that the percentage of sclerotic glomeruli was <5%, and scores of 1, 2, 3, 4, and 5 denote 6–20, 21%–40%, 41–60%, 61–80%, and >81% of the selected glomeruli, respectively. A score was assigned to each mouse.
Frozen kidney sections were blocked with 1% BSA containing 0.3% Triton X-100 for 30 min and then incubated with IL-1R1 (Cat. No. PA5-97866, Invitrogen, 1:100), Wilms’ tumor-1 (WT1; Cat. No. 89901, Abcam, 1:200), nephrin (Cat. No. 20R-NP002, Fitzgerald, 1:100), phosphorylated (p-)Akt (Ser473) (Cat. No. 4060, Cell Signaling Technology, 1:100), or cleaved caspase-3 (Cat. No. 9664, Cell Signaling Technology, 1:100) primary antibody at 4°C overnight. After the primary antibody was washed off, sections were incubated for 1 h at room temperature with the respective secondary antibodies from Thermo Fisher (1:400). After three PBS washes, slides were mounted and visualized by Zeiss Axio Imager Z2 Upright fluorescence microscopy. To compare fluorescence intensities, all images were taken at the same exposure time. An investigator who was blinded to the experimental conditions quantified positive cells in glomeruli among groups. WT1+ cells were counted in 20 randomly chosen glomerular sections, and average numbers of WT1+ cells per glomerular section were calculated. Cleaved caspase-3-positive dots were counted in 50 randomly chosen glomerular sections, and averages of the cleaved caspase-3 dots per glomerulus for each section were calculated. Negative controls for immunofluorescence staining included antisera from the same species and substitution with PBS.
Animal Models of Podocyte Injury and Proteinuria
Mouse models for podocyte injury and proteinuria were established by intravenous injection of NTS or ADR. For the NTS model, mice received an intraperitoneal injection of sheep IgG (250 µg/mouse) at day 0. Tail injection of sheep NTS (Cat. No. PTX-001S, Probetex, San Antonio, TX) was performed at day 5. At day 9, mice were euthanized and urine and kidney tissues were harvested for further analysis. For the ADR model, mice were administered ADR (18 mg/kg) intravenously by tail vein injection. Urine was collected at days 1 and 3, and all mice were euthanized. Kidneys were collected at day 3 after ADR exposure. Investigators were blinded to the group allocation when assessing the outcome of the NTS or ADR experiments.
Glomerular Isolation
Mouse glomeruli were isolated according to previously published methods (21). Briefly, mice were anesthetized with isoflurane and perfused through the heart with HBSS containing 2.5 mg/mL iron oxide (Sigma) and 0.1% BSA. After perfusion, the kidneys were removed, cut into 1-mm3 pieces, and digested in HBSS containing 1 mg/mL collagenase A and 100 U/mL deoxyribonuclease I. Digested tissue was then passed through a 100-μm cell strainer and collected by centrifugation. The pellet was resuspended in 2 mL HBSS, and glomeruli were collected using a magnet. The purity of glomeruli was verified under microscopy, attaining >95% by this method. The isolated glomeruli were used for subsequent RNA or protein analysis.
Cell Culture and Treatment
Conditionally immortalized human podocytes derived from the urine of healthy volunteers (22) were cultured at 33°C in RPMI-1640 medium supplemented with 10% FBS and insulin-transferrin-selenium (Gibco). To induce differentiation, podocytes were grown under nonpermissive conditions at 37°C. Cells were seeded on six-well culture plates to 80–90% confluence in complete medium and then changed to serum-free medium for 16 h. After serum starvation, cells were treated with ADR in the absence or presence of human recombinant IL-1β (Cat. No. 201-LB, R&D) for various groups as indicated. In some experiments, cells were pretreated with the Akt inhibitor wortmannin (Cat. No. 12-338, Sigma) at given concentrations or vehicle 0.5 h before incubation with IL-1β.
Urinary Albumin Measurement
Levels of urinary albumin and creatinine were measured with kits from Exocell (Cat. Nos. 1011 and 1012, respectively, Ethos Biosciences) according to the instructions. Urinary albumin excretion was expressed as the albumin-to-creatinine ratio. Urinary proteins were also analyzed by SDS-PAGE after normalization to urinary creatinine. After separation by SDS-PAGE, urine proteins were stained with Coomassie blue R-250.
Electron Microscopy
Mouse kidneys were perfusion fixed with 2.5% glutaraldehyde in PBS by left cardiac ventricular injection and postfixed in aqueous 1% OsO4. Specimens were dehydrated through an ethanol series, infiltrated in a 1:1 mixture of propylene oxide-polybed 812 epoxy resin, and then embedded. Ultrathin sections were stained with 2% uranyl acetate followed by 1% lead citrate. Sections were observed and photographed using transmission electron microscopy in the Core facility at Duke University.
Western Blot Analysis
Protein expression was detected by Western blot analysis as previously described. Briefly, the isolated glomeruli were homogenized in RIPA buffer (Sigma-Aldrich). The supernatants were collated after centrifugation at 13,000 g at 4°C for 30 min. Concentrations of protein were quantitated using the DC Protein Assay Kit (Bio-Rad Laboratories). Equal amounts of sample were subjected to electrophoresis through 4–12% bis-Tris gels and transferred to polyvinylidene difluoride membranes. After being blocked with 5% milk in Tris-buffered saline with Tween 20, blots were incubated with anti-nephrin (Cat. No. PA5-91907, Thermo Fisher, 1:2,000), anti-podocin (Cat. No. P0372, Sigma, 1:1,000), anti-WT1 (Cat. No. 89901, Abcam, 1:2,000), anti-p-Akt (Ser473) (Cat. No. 4060, Cell Signaling Technology, 1:1,000), anti-caspase-3 (Cat. No. 9662, Cell Signaling Technology, 1:1,000), anti-Akt (Cat. No. 4691, Cell Signaling Technology, 1:2,000), anti-cleaved caspase-3 (Cat. No. 9664, Cell Signaling Technology, 1:1,000), or anti-GAPDH (Cat. No. 2118, Cell Signaling Technology, 1:2,000) overnight at 4°C. The blots were then washed and incubated for 1 h at room temperature with the respective secondary antibodies from Cell Signaling Technology (1:10,000). Bands were detected using an enhanced chemiluminescence detection system. The detected bands were quantified by densitometry through ImageJ for Windows.
Akt Activator SC79 Treatment Experiments
IL-1R1 PKO and WT mice received the Akt activator SC79 (Cat. No. 14972, Cayman, 0.04 mg/g/day) (23) or vehicle by intraperitoneal injection beginning on the day of NTS injection until the day of mouse euthanasia.
Statistics
All data are expressed as means ± SE. Amounts of target proteins were quantified by normalization of their bands to those of GAPDH or caspase-3 bands on the same blots. For comparisons between two groups with normally distributed data, statistical significance was assessed using a two-tailed unpaired Student’s t test. For comparisons between two groups with non-normally distributed variables, a Wilcoxon test was used. Comparison among groups was performed with a one-way ANOVA test followed by a Student–Newman–Keuls test or Kruskal–Wallis test.
RESULTS
IL-1/IL-1R1 Signaling Components Are Upregulated After Glomerular Injury
IL-1 signaling is activated in glomerulonephritis (5). However, the functions of podocyte IL-1 signaling during glomerular injury have not been established. To investigate the role of IL-1 signaling in proteinuric kidney disease, we first examined the glomerular expression of key components of IL-1 signaling, including IL-1α, IL-1β, and IL-1R1. mRNA expression of IL-1α (P = 0.023), IL-1β (P = 0.004), and IL-1R1 (P = 0.004) was upregulated in isolated glomeruli during NTS (Fig. 1A). Immunofluorescent stains localized IL-1R1 to glomerular podocytes after NTS and ADR injection (Fig. 1B). This upregulation of several components of IL-1/IL-1R1 signaling in the injured glomerulus suggested that the NTS and ADR models are suitable for studying the podocyte-specific contributions of IL-1 signaling to glomerular injury.
Figure 1.

Induction of interleukin (IL)-1 signaling components in glomeruli from mice after glomerular injury. A: glomerular IL-1α, IL-1β, and IL-1 receptor type 1 (IL-1R1) mRNA abundance at day 9 after nephrotoxic serum (NTS)-induced glomerular injury (n = 4). B: representative IL-1R1- and nephrin-stained kidney sections from vehicle (Veh), NTS-, and adriamycin (ADR)-injected animals. The white arrows indicate the colocalization of IL-1R1 and nephrin in glomeruli. C: representative sections of kidneys from podocin (Pod) Cre− mT/mG and Pod Cre+ mT/mG reporter mice. Green fluorescence indicates the presence of podocin Cre expression; red fluorescence marks the absence of podocin Cre expression. Blue fluorescence indicates nuclear DAPI stain. D: genotyping of the mice by PCR analysis of genomic DNA. E and F: body weights (E) and kidney-to-body weight ratios (KW/BW; F) of naïve podocyte-specific knockout (PKO) and wild-type (WT) mice at 1 and 6 mo (n = 3–5). Data are presented as means ± SE. P values in A were analyzed by a two-tailed Student’s t test. *P < 0.05. Scale bars = 40 µm.
Generation of Mice With Podocyte-Specific Ablation of IL-1R1
We generated conditional knockout mice in which the IL-1R1 gene was specifically disrupted in glomerular podocytes using a Cre-Lox gene targeting approach. To observe Cre recombinase efficiency in podocytes, we bred an NPHS2-Cre mouse line (20) with a double fluorescence reporter mouse (mT/mG). We detected robust Cre expression marked by green fluorescent protein within the glomeruli of NPHS2-Cre+ (“Pod-Cre”) mT/mG mice but the absence of Cre expression marked by red fluorescent protein in nonglomerular tissues (Fig. 1C). We, therefore, bred the NPHS2-Cre mouse line with an IL-1R1flox line harboring loxp sites on either side of the coding region for the IL-1R1 gene. For our experiments, we used NPHS2-Cre+IL-1R1flox/flox mice (PKO mice; Fig. 1D, lane 4) and NPHS2-Cre-IL-1R1flox/flox littermates (WT mice; Fig. 1D, lane 3). All mice were viable and fertile. There was no difference in sex, body weight, or kidney-to-body weight ratios between PKO mice and control littermates at 1 and 6 mo after birth (Fig. 1, E and F).
Figure 2.

Interleukin-1 receptor type 1 (IL-1R1) signaling in podocytes limits albuminuria and podocyte injury induced by nephrotoxic serum (NTS) in mice. A: urinary albumin concentration in wild-type (WT) and podocyte-specific knockout (PKO) mice at day 9 after vehicle (Veh) or NTS injection (n = 3–9). B: representative SDS-PAGE showing urine proteins in cohorts of mice. C: representative images of kidney sections from WT and PKO mice at day 9 after Veh or NTS injection. Scale bar = 40 µm. D: kidney glomerulosclerosis score in groups as indicated (n = 3–9). E: representative Western blots for nephrin, podocin, and Wilms’ tumor-1 (WT1) protein in glomeruli isolated from WT and PKO mice at day 9 after NTS. F: semiquantitative determination of nephrin, podocin, and WT1 protein from the blots shown in E (n = 6). G: representative immunostaining for nephrin protein and electron microscopy (EM) showing the glomerular filtration barrier in different groups. The red arrowheads indicate the secondary foot process and slit diaphragm. Scale bars = 40 µm (top) and 1 µm (bottom). Data are presented as means ± SE. P values in A and D were analyzed by a one-way ANOVA test followed by a Student–Newman–Keuls or Kruskal–Wallis test. P values in F were analyzed by a two-tailed Student’s t test. *P < 0.05. PAS, periodic acid-Schiff stain; Ucr, urinary creatinine.
IL-1R1 Signals Limit Albuminuria and Podocyte Marker Loss After Glomerular Injury
To explore the role of IL-1R1 activation in podocyte injury, we challenged PKO and WT mice with NTS or ADR. At day 9 of NTS, WT mice developed robust albuminuria compared with vehicle-treated WT controls (PANOVA = 0.002; Fig. 2A). However, NTS-treated PKO mice developed more severe albuminuria (PANOVA = 0.003; Fig. 2A). Analysis of urine samples by SDS-PAGE and Coomassie staining revealed that albumin was the major constituent of urine proteins in mice after NTS injury (Fig. 2B). Analysis of renal pathology showed that PKO glomeruli exhibited more matrix deposition compared with WT glomeruli after NTS (PANOVA = 0.002; Fig. 2, C and D). We further examined the glomerular protein expression of several podocyte markers after NTS. Protein levels of nephrin (P = 0.002), podocin (P = 0.005), and WT1 (P = 0.002) were significantly downregulated in NTS glomeruli from PKO mice compared with WT mice (Fig. 2, E and F). Immunostaining for the podocyte marker nephrin confirmed the blot findings (Fig. 2G). Electron microscopy revealed foot process effacement and loss of the slit diaphragm in some areas of the WT glomeruli after NTS with more severe disruptions in podocyte architecture in PKO kidneys (Fig. 2G).
To confirm these findings in a separate model of glomerular injury, WT and PKO animals were subjected to ADR nephropathy. ADR-treated IL-1R1 PKO mice exhibited significant increases in urinary albumin excretion (PANOVA = 0.007; Fig. 3A). Here again, SDS-PAGE and Coomassie staining revealed that albumin was the major constituent of urine proteins in mice after ADR injury (Fig. 3B). Glomeruli from PKO animals showed significant decreases in podocin (PANOVA < 0.0001) and nephrin (PANOVA = 0.0004) at the protein level compared with ADR-treated WT controls (Fig. 3, C and D). Immunostaining for nephrin confirmed the blot findings (Fig. 3E). These data suggest that IL-1R1 activation in podocytes ameliorates proteinuria and podocyte injury in both NTS and ADR glomerular disease models.
Figure 3.

Interleukin-1 receptor type 1 (IL-1R1) deletion in podocytes augments albuminuria and podocyte injury induced by adriamycin (ADR) in mice. A: urinary albumin concentration in wild-type (WT) and podocyte-specific knockout (PKO) mice at days 1 and 3 after ADR injection (n = 10). B: representative SDS-PAGE showing the urine proteins at day 3 in groups. C: Western blots for podocin and nephrin in glomeruli isolated from WT and PKO mice 3 days after ADR injection. D: semiquantitative determination of podocin and nephrin protein from the blots shown in C (n = 4–6). E: representative immunostaining for nephrin protein in different groups. P values in A and D were analyzed by one-way ANOVA followed by a Student–Newman–Keuls test. *P < 0.05. Ucr, urinary creatinine; Veh, vehicle. Scale bar = 40 µm.
Ablation of IL-1R1 Promotes Podocyte Loss and Apoptosis After Injury
Injured podocytes can undergo apoptosis leading to podocyte loss with consequent glomerulosclerosis (18, 24, 25). To investigate whether podocyte loss occurred in the NTS and ADR models, we counted the number of WT1-positive podocytes in the glomeruli from our experimental animals. NTS caused a significant decrease in the number of WT1-expressing podocytes in the glomeruli of WT mice that was exacerbated in the PKO cohort (PANOVA = 0.003; Fig. 4, A and B). In view of the decrease in WT1-positive cells in the glomeruli after NTS-induced glomerular injury, we next examined whether podocytes undergo apoptosis after NTS injury. We performed an immunofluorescent study of cleaved caspase-3 to detect apoptotic cells, and quantitative determination showed that the cleaved caspase-3-positive cells in the glomerulus were significantly augmented after NTS compared with vehicle-treated mice and further increased in NTS-treated PKO mice (PANOVA = 0.018; Fig. 4, A and B). These findings were recapitulated after ADR-induced injury (PANOVA = 0.01, Fig. 4D, top; PANOVA = 0.023, Fig. 4D, bottom; Fig. 4, C and D). We, therefore, posited that IL-1R1 activation in podocytes protects them from apoptosis and loss after injury.
Figure 4.

Podocyte-specific interleukin-1 receptor type 1 (IL-1R1) deletion promotes podocyte loss and apoptosis induced by nephrotoxic serum (NTS) or adriamycin (ADR) in mice. A: representative immunostaining for Wilms’ tumor 1 (WT1) and cleaved caspase-3 (CC3) in glomeruli from the different groups after NTS. B: quantitative determination of WT1 and cleaved caspase-3 in glomeruli from A (n = 3–6). C: representative immunostaining for WT1 and cleaved caspase-3 in glomeruli from the different groups after ADR. D: quantitative determination of WT1 and cleaved caspase-3 in glomeruli from C (n = 3–6). P values in B and D were analyzed by one-way ANOVA followed by a Student–Newman–Keuls test. *P < 0.05. Scale bar = 40 µm. Veh, vehicle.
IL-1R1 Deletion Diminishes Akt Phosphorylation in Podocytes During NTS
Signaling targets downstream of IL1-R1 include NF-κB, phosphatidylinositol 3-kinase/Akt, JNK, and MAPK (6, 26). Noting that Akt kinase plays a critical role in maintaining podocyte viability and function (27–30), we investigated Akt kinase activation in podocytes after NTS injection. As shown in Fig. 5, A and B, Akt kinase activation marked by Akt phosphorylation (Ser473) was enhanced in glomeruli after NTS injection compared with vehicle-treated controls (PANOVA = 0.042), whereas in PKO glomeruli, p-Akt (Ser473) abundance was much less compared with WT glomeruli after NTS injection (PANOVA = 0.038). Immunofluorescent staining confirmed the increase in p-Akt (Ser473) in podocytes from WT mice at day 9 after NTS injection with an attenuated signal in PKO mice (Fig. 5C). Such downregulation of Akt phosphorylation in the podocyte may permit enhanced susceptibility to cell death. These results suggest that IL-1R1 activation favors podocyte survival by preserving Akt signaling after NTS-induced injury.
Figure 5.

Ablation of interleukin-1 receptor type 1 (IL-1R1) diminishes Akt phosphorylation in podocytes after nephrotoxic serum (NTS). A: Western blot assay showing Akt phosphorylation (Ser473) in glomeruli isolated from wild-type (WT) and podocyte-specific knockout (PKO) mice in the different groups. B: semiquantitative determination of Akt phosphorylation from the blots shown in A (n = 3–8). C: representative immunostaining for Akt phosphorylation in glomeruli from the different groups. P values in B were analyzed by one-way ANOVA followed by a Student–Newman–Keuls test. *P < 0.05. Scale bar = 40 µm. p-Akt, phosphorylated Akt; Veh, vehicle.
IL-1R1 Stimulation Preserves Podocyte Survival During Injury In Vitro
To further investigate whether IL-1R1 stimulation affords protection to podocytes during injury, we cultured human podocytes in vitro (22). As shown in Fig. 6, A and B, incubation of human podocytes with ADR (1 µg/mL) for 16 h induced podocyte apoptosis, characterized by caspase-3 cleavage and activation (P < 0.05). Treatment of podocytes with IL-1β largely prevented caspase-3 cleavage induced by ADR (P < 0.05). The antiapoptotic activity of IL-1β was also dose dependent. TUNEL confirmed the cytoprotective capacity of IL-1β in podocytes after ADR treatment (PANOVA = 0.006; Fig. 6, C and D). We next investigated whether Akt kinase activation mediated the protective role of IL-1β. As shown in Fig. 7, A and B, IL-1β markedly and rapidly increased levels of p-Akt. Incubation with wortmannin, an Akt inhibitor, blocked Akt phosphorylation (Fig. 7C). As shown in Fig. 7, D and E, blockade of Akt activation by wortmannin abolished the ability of IL-1β to prevent apoptosis induced by ADR (PANOVA = 0.0002), suggesting that IL-1β promotes survival through an Akt-dependent pathway.
Figure 6.

Interleukin-1 receptor type 1 (IL-1R1) stimulation ameliorates adriamycin (ADR)-induced podocyte apoptosis in vitro. A: Western blots for cleaved caspase-3 from cultured podocytes. B: semiquantitative analysis for cleaved caspase 3 from A (n = 3). C: representative images showing TUNEL staining in the different groups. D: quantitative determination of TUNEL-positive cells (n = 3). P values in B and D were analyzed by one-way ANOVA followed by a Student–Newman–Keuls test. *P < 0.05 vs. vehicle (Veh) controls; #P < 0.05 vs. ADR alone. Scale bar = 20 µm.
Figure 7.

Interleukin (IL)-1β attenuates podocyte apoptosis induced by adriamycin (ADR) through an Akt-dependent signaling pathway. A: Western blot showing phosphorylated (p-)Akt (Ser473) abundance after IL-1β treatment. B: graphic presentation showing the dynamic changes for Akt phosphorylation abundance normalized to GAPDH. C: Western blot showing p-Akt abundance after Akt inhibition with wortmannin (Wort; 10 nM). D: Western blot showing cleaved caspase-3 after ADR treatment. E: semiquantitative determination of cleaved caspase-3 from the blots shown in D (n = 4). P values in D were analyzed by one-way ANOVA followed by a Student–Newman–Keuls test. *P < 0.05.
Akt Activation Ameliorates Albuminuria and Podocyte Damage in IL-1R1 PKO Mice After NTS-Induced Glomerular Injury
Finally, to directly test whether the diminished Akt activation associated with IL-1R1 deficiency in podocytes exacerbates albuminuria and podocyte injury, we treated WT and IL-1R1 PKO animals with the Akt activator SC79 during NTS. As before, IL-1R1 PKO mice developed more severe albuminuria compared with vehicle-treated WT mice after NTS (PANOVA = 0.013). SC79 treatment reduced albuminuria in PKO mice compared with vehicle-treated PKO mice after NTS and abrogated the differences in albuminuria (PANOVA = 0.003; Fig. 8A) and expression of podocyte differentiation markers WT1, nephrin, and podocin between WT and PKO cohorts (P < 0.05; Fig. 8, B–D). These data suggest that Akt inactivation accruing from loss of IL-1R1 stimulation on podocytes exacerbates albuminuria and podocyte injury and loss.
Figure 8.

Akt activation abrogates exaggerated albuminuria and loss of podocyte marker expression in podocyte-specific knockout (PKO) animals after nephrotoxic serum (NTS) injury. A: urinary albumin concentration in the different groups after NTS (n = 7–12). B–D: mRNA expression for Wilms’ tumor-1 (WT1; B), nephrin (C), and podocin (D) in kidney tissues from the different groups as indicated (n = 7–12). P values were analyzed by one-way ANOVA followed by a Student–Newman–Keuls test. *P < 0.05. Ucr, urinary creatinine; Veh, vehicle.
DISCUSSION
Podocytes are highly differentiated, postmitotic cells that play a critical role in maintaining the integrity of the glomerular filtration barrier (31, 32). The ability of adult podocytes to proliferate or regenerate is very limited. Podocyte dysfunction and loss are recognized as causative events in the pathogenesis of proteinuria and glomerulosclerosis in a variety of proteinuric kidney diseases, regardless of etiology (33, 34). Identifying the important factors that regulate podocyte survival, differentiation, and injury will help to elucidate underlying mechanisms of glomerular damage and provide potential therapeutic targets for the treatment of proteinuric kidney disease. In the present study, we demonstrated that IL-1R1 signaling can limit podocyte injury and loss under pathological conditions. Accordingly, IL-1R1 ablation in podocytes renders mice more susceptible to NTS- and ADR-induced podocyte injury and apoptosis, resulting in more severe albuminuria. Mechanistically, we found that Akt kinase activation induced by IL-1R1 signaling is important for podocyte survival after injury. Thus, ectopic activation of Akt ameliorates albuminuria and preserves podocyte marker expression in IL-1R1 PKO mice with NTS-induced nephropathy. Our in vivo and in vitro findings establish that IL-1R1 signaling is cytoprotective in preventing podocyte dysfunction and apoptosis in proteinuric kidney disease.
As an activator of innate immunity, IL-1 triggers a broad inflammatory response through its functions in leukocytes. Consistent with these functions, IL-1R1−/− and IL-1β−/− mice exhibit less proteinuria, crescent formation, immune cell infiltration, and kidney injury during crescentic glomerulonephritis (13), and treatment with an IL-1 receptor antagonist ameliorates experimental antiglomerular basement membrane glomerulonephritis (14, 35). IL-1R1 activation on leukocytes drives them to secret several other secondary inflammatory mediators, including IL-6, TNF-α, IL-8, and IL-17 (2, 36, 37), that can exaggerate glomerular injury. Renal accumulation of macrophages and CD4-positive T cells is, therefore, dramatically decreased in IL-1R1−/− and IL-1β−/− mice after injury (13). Nevertheless, the results of our study demonstrate that IL-1R1 signaling in podocytes facilitates their survival. Thus, IL-1 binds to IL-1R1 on macrophages and T cells to recruit them into glomeruli and enhance the inflammatory response but concomitantly activates IL-1R1 on podocytes to constrain injury and apoptosis. Our findings, therefore, elucidate paradoxical actions of podocyte IL-1R1 signals in proteinuric kidney disease that could help facilitate the design of pharmacological interventions to precisely target IL-1 signaling in the glomerulus while limiting off-target side effects. Interestingly, in the CANTOS trial of patients with cardiovascular disease, global IL-1β blockade was associated with a small but significant loss of kidney function at 48 mo, again suggesting a renoprotective effect of IL-1, but we are not aware of human trials examining the longitudinal effects of IL-1 or its blockade on proteinuric kidney disease.
IL-1R1 governs multiple biological functions that depend on the cellular context. Besides its well-known proinflammatory activities, IL-1R1 signaling is involved in cell differentiation, growth, proliferation, survival, and injury. During the evolution of kidney fibrosis, IL-1β induces stromal cell proliferation and fibrogenic differentiation to mediate tubulointerstitial expansion that depends on the transcription factor MYC (9, 38, 39). In renal tubular cells, IL-1β induces cellular dilation and epithelial cell cycle arrest (9) and can also regulate Na+ handling in the nephron (8, 40). In myeloid cells, IL-1R1 signals can promote activation and survival through an autocrine effect (41, 42). In cancer cells, IL-1β induces phosphatidylinositol 3-kinase/Akt activation to promote expression of tumor protein p63, a member of the p53 suppressor gene family involved in chemoresistance (24). In our study, we found that IL-1R1 signaling is crucial for podocyte survival and phenotype preservation after glomerular injury.
Akt kinase promotes cell survival through several downstream signaling pathways (43–46). In turn, Akt signaling sustains podocyte survival in several forms of glomerular disease (27, 29, 30). Targeting this pathway by genetic or pharmacological intervention, therefore, has a dramatic impact on albuminuria, podocyte death, and glomerulosclerosis (28, 29, 47, 48). Furthermore, Akt activation can suppress podocyte cytoskeletal alterations by inhibiting GTP-Rac1 activation, a key promoter of cell migration and lamellipodia formation in podocytes (27). In the present study, Akt phosphorylation was augmented in podocytes after injury, consistent with a protective response that was lost with ablation of IL-1R1.The PKO cohort, therefore, exhibited less Akt phosphorylation with greater podocyte apoptosis and loss. Inversely, PKO mice treated with an Akt activator had less albuminuria and showed podocyte preservation. We speculate that Akt activation could not further activate Akt signaling in the WT podocyte to reduce injury and podocyte apoptosis after injury in the WT cohort.
In summary, we found that IL-1R1 on podocytes plays an unanticipated protective role in glomerular disease by activating downstream Akt signals. Deficiency of IL-1R1 on podocytes, therefore, enhances susceptibility to podocyte apoptosis and loss after glomerular injury. IL-1R1 activation drives downstream Akt phosphorylation in podocytes, protecting them from injury, apoptosis, and loss during glomerular disease. Additional studies are warranted to explore how pathways downstream of IL-1R1 can be activated selectively within the podocyte for patients with glomerulopathy.
GRANTS
This work was supported by National Institutes of Health (NIH) Grants DK118019 and HL128355, United States Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development Grant BX000893, a Veterans Affairs Senior CSI Award, and American Heart Association Grant 18TPA34170047. This work was also supported by National Institutes of Health Grant 1K08DK111940-01 and the American Society of Nephrology/Amos Medical Faculty Development Program/Robert Wood Johnson Foundation (to G.H.). This work was also supported by National Natural Science Foundation of China Grant 82100720 (to J.R.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.R. and S.D.C. conceived and designed research; J.R., X.L., G.H., M.J.R., and R.D.B. performed experiments; J.R. and S.D.C. analyzed data; J.R., J.R.P., and S.D.C. interpreted results of experiments; J.R. prepared figures; J.R. drafted manuscript; J.R., X.L., G.H., J.R.P., M.J.R., R.D.B., and S.D.C. edited and revised manuscript; J.R., X.L., G.H., J.R.P., M.J.R., R.D.B., and S.D.C. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors acknowledge the superlative husbandry work done by Taylor Robinette and Cindy Chen in maintaining the animal colonies used in conducting this study.
REFERENCES
- 1.Jones LK, O'Sullivan KM, Semple T, Kuligowski MP, Fukami K, Ma FY, Nikolic-Paterson DJ, Holdsworth SR, Kitching AR. IL-1RI deficiency ameliorates early experimental renal interstitial fibrosis. Nephrol Dial Transplant 24: 3024–3032, 2009. doi: 10.1093/ndt/gfp214. [DOI] [PubMed] [Google Scholar]
- 2.Privratsky JR, Zhang J, Lu X, Rudemiller N, Wei Q, Yu YR, Gunn MD, Crowley SD. Interleukin 1 receptor (IL-1R1) activation exacerbates toxin-induced acute kidney injury. Am J Physiol Renal Physiol 315: F682–F691, 2018. [Erratum in Am J Physiol Renal Physiol 316: F1090, 2019]. doi: 10.1152/ajprenal.00104.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Navarro-Gonzalez JF, Mora-Fernandez C. The role of inflammatory cytokines in diabetic nephropathy. J Am Soc Nephrol 19: 433–442, 2008. doi: 10.1681/ASN.2007091048. [DOI] [PubMed] [Google Scholar]
- 4.Navarro-González JF, Mora-Fernández C, Muros de Fuentes M, García-Pérez J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat Rev Nephrol 7: 327–340, 2011. doi: 10.1038/nrneph.2011.51. [DOI] [PubMed] [Google Scholar]
- 5.Anders HJ. Of inflammasomes and alarmins: IL-1β and IL-1α in kidney disease. J Am Soc Nephrol 27: 2564–2575, 2016. doi: 10.1681/ASN.2016020177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lichtnekert J, Kulkarni OP, Mulay SR, Rupanagudi KV, Ryu M, Allam R, Vielhauer V, Muruve D, Lindenmeyer MT, Cohen CD, Anders HJ. Anti-GBM glomerulonephritis involves IL-1 but is independent of NLRP3/ASC inflammasome-mediated activation of caspase-1. PLoS One 6: e26778, 2011. doi: 10.1371/journal.pone.0026778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity 39: 1003–1018, 2013. doi: 10.1016/j.immuni.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang J, Rudemiller NP, Patel MB, Karlovich NS, Wu M, McDonough AA, Griffiths R, Sparks MA, Jeffs AD, Crowley SD. Interleukin-1 receptor activation potentiates salt reabsorption in angiotensin ii-induced hypertension via the NKCC2 Co-transporter in the nephron. Cell Metab 23: 360–368, 2016. doi: 10.1016/j.cmet.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lemos DR, McMurdo M, Karaca G, Wilflingseder J, Leaf IA, Gupta N, Miyoshi T, Susa K, Johnson BG, Soliman K, Wang G, Morizane R, Bonventre JV, Duffield JS. Interleukin-1β activates a MYC-dependent metabolic switch in kidney stromal cells necessary for progressive tubulointerstitial fibrosis. J Am Soc Nephrol 29: 1690–1705, 2018. doi: 10.1681/ASN.2017121283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rodriguez-Grande B, Varghese L, Molina-Holgado F, Rajkovic O, Garlanda C, Denes A, Pinteaux E. Pentraxin 3 mediates neurogenesis and angiogenesis after cerebral ischaemia. J Neuroinflammation 12: 15, 2015. doi: 10.1186/s12974-014-0227-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Braun T, Zwerina J. Positive regulators of osteoclastogenesis and bone resorption in rheumatoid arthritis. Arthritis Res Ther 13: 235, 2011. doi: 10.1186/ar3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kitching AR, Hutton HL. The players: cells involved in glomerular disease. Clin J Am Soc Nephrol 11: 1664–1674, 2016. doi: 10.2215/CJN.13791215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Timoshanko JR, Kitching AR, Iwakura Y, Holdsworth SR, Tipping PG. Contributions of IL-1β and IL-1α to crescentic glomerulonephritis in mice. J Am Soc Nephrol 15: 910–918, 2004. doi: 10.1097/01.asn.0000115704.86897.f4. [DOI] [PubMed] [Google Scholar]
- 14.Tang WW, Feng L, Vannice JL, Wilson CB. Interleukin-1 receptor antagonist ameliorates experimental anti-glomerular basement membrane antibody-associated glomerulonephritis. J Clin Invest 93: 273–279, 1994. doi: 10.1172/JCI116956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lan HY, Nikolic-Paterson DJ, Mu W, Vannice JL, Atkins RC. Interleukin-1 receptor antagonist halts the progression of established crescentic glomerulonephritis in the rat. Kidney Int 47: 1303–1309, 1995. doi: 10.1038/ki.1995.185. [DOI] [PubMed] [Google Scholar]
- 16.Niemir ZI, Stein H, Dworacki G, Mundel P, Koehl N, Koch B, Autschbach F, Andrassy K, Ritz E, Waldherr R, Otto HF. Podocytes are the major source of IL-1α and IL-1β in human glomerulonephritides. Kidney Int 52: 393–403, 1997. doi: 10.1038/ki.1997.346. [DOI] [PubMed] [Google Scholar]
- 17.Cherney DZI, Lytvyn Y, McCullough PA. Cardiovascular risk reduction in patients with chronic kidney disease: potential for targeting inflammation with canakinumab. J Am Coll Cardiol 71: 2415–2418, 2018. doi: 10.1016/j.jacc.2018.04.008. [DOI] [PubMed] [Google Scholar]
- 18.Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, Saunders TL, Dysko RC, Kohno K, Holzman LB, Wiggins RC. Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol 16: 2941–2952, 2005. doi: 10.1681/ASN.2005010055. [DOI] [PubMed] [Google Scholar]
- 19.Liu X, Nemeth DP, McKim DB, Zhu L, DiSabato DJ, Berdysz O, Gorantla G, Oliver B, Witcher KG, Wang Y, Negray CE, Vegesna RS, Sheridan JF, Godbout JP, Robson MJ, Blakely RD, Popovich PG, Bilbo SD, Quan N. Cell-type-specific interleukin 1 receptor 1 signaling in the brain regulates distinct neuroimmune activities. Immunity 50: 317–333.e6, 2019. doi: 10.1016/j.immuni.2018.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moeller MJ, Sanden SK, Soofi A, Wiggins RC, Holzman LB. Podocyte-specific expression of cre recombinase in transgenic mice. Genesis 35: 39–42, 2003. doi: 10.1002/gene.10164. [DOI] [PubMed] [Google Scholar]
- 21.Katsuya K, Yaoita E, Yoshida Y, Yamamoto Y, Yamamoto T. An improved method for primary culture of rat podocytes. Kidney Int 69: 2101–2106, 2006. doi: 10.1038/sj.ki.5000398. [DOI] [PubMed] [Google Scholar]
- 22.Sakairi T, Abe Y, Kajiyama H, Bartlett LD, Howard LV, Jat PS, Kopp JB. Conditionally immortalized human podocyte cell lines established from urine. Am J Physiol Renal Physiol 298: F557–F567, 2010. doi: 10.1152/ajprenal.00509.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jo H, Mondal S, Tan D, Nagata E, Takizawa S, Sharma AK, Hou Q, Shanmugasundaram K, Prasad A, Tung JK, Tejeda AO, Man H, Rigby AC, Luo HR. Small molecule-induced cytosolic activation of protein kinase Akt rescues ischemia-elicited neuronal death. Proc Natl Acad Sci USA 109: 10581–10586, 2012. doi: 10.1073/pnas.1202810109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagata M. Podocyte injury and its consequences. Kidney Int 89: 1221–1230, 2016. doi: 10.1016/j.kint.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 25.Shankland SJ. The podocyte's response to injury: role in proteinuria and glomerulosclerosis. Kidney Int 69: 2131–2147, 2006. doi: 10.1038/sj.ki.5000410. [DOI] [PubMed] [Google Scholar]
- 26.Fahey E, Doyle SL. IL-1 family cytokine regulation of vascular permeability and angiogenesis. Front Immunol 10: 1426, 2019. doi: 10.3389/fimmu.2019.01426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Canaud G, Bienaimé F, Viau A, Treins C, Baron W, Nguyen C, Burtin M, Berissi S, Giannakakis K, Muda AO, Zschiedrich S, Huber TB, Friedlander G, Legendre C, Pontoglio M, Pende M, Terzi F. AKT2 is essential to maintain podocyte viability and function during chronic kidney disease. Nat Med 19: 1288–1296, 2013. doi: 10.1038/nm.3313. [DOI] [PubMed] [Google Scholar]
- 28.Welsh GI, Hale LJ, Eremina V, Jeansson M, Maezawa Y, Lennon R, Pons DA, Owen RJ, Satchell SC, Miles MJ, Caunt CJ, McArdle CA, Pavenstädt H, Tavaré JM, Herzenberg AM, Kahn CR, Mathieson PW, Quaggin SE, Saleem MA, Coward RJM. Insulin signaling to the glomerular podocyte is critical for normal kidney function. Cell Metab 12: 329–340, 2010. doi: 10.1016/j.cmet.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tejada T, Catanuto P, Ijaz A, Santos JV, Xia X, Sanchez P, Sanabria N, Lenz O, Elliot SJ, Fornoni A. Failure to phosphorylate AKT in podocytes from mice with early diabetic nephropathy promotes cell death. Kidney Int 73: 1385–1393, 2008. doi: 10.1038/ki.2008.109. [DOI] [PubMed] [Google Scholar]
- 30.Dai C, Saleem MA, Holzman LB, Mathieson P, Liu Y. Hepatocyte growth factor signaling ameliorates podocyte injury and proteinuria. Kidney Int 77: 962–973, 2010. doi: 10.1038/ki.2010.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schell C, Huber TB. The evolving complexity of the podocyte cytoskeleton. J Am Soc Nephrol 28: 3166–3174, 2017. doi: 10.1681/ASN.2017020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fissell WH, Miner JH. What is the glomerular ultrafiltration barrier? J Am Soc Nephrol 29: 2262–2264, 2018. doi: 10.1681/ASN.2018050490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kopp JB, Anders HJ, Susztak K, Podestà MA, Remuzzi G, Hildebrandt F, Romagnani P. Podocytopathies. Nat Rev Dis Primers 6: 68, 2020. doi: 10.1038/s41572-020-0196-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Torban E, Braun F, Wanner N, Takano T, Goodyer PR, Lennon R, Ronco P, Cybulsky AV, Huber TB. From podocyte biology to novel cures for glomerular disease. Kidney Int 96: 850–861, 2019. doi: 10.1016/j.kint.2019.05.015. [DOI] [PubMed] [Google Scholar]
- 35.Lan HY, Nikolic-Paterson DJ, Zarama M, Vannice JL, Atkins RC. Suppression of experimental crescentic glomerulonephritis by the interleukin-1 receptor antagonist. Kidney Int 43: 479–485, 1993. doi: 10.1038/ki.1993.70. [DOI] [PubMed] [Google Scholar]
- 36.Pindjakova J, Hanley SA, Duffy MM, Sutton CE, Weidhofer GA, Miller MN, Nath KA, Mills KH, Ceredig R, Griffin MD. Interleukin-1 accounts for intrarenal Th17 cell activation during ureteral obstruction. Kidney Int 81: 379–390, 2012. doi: 10.1038/ki.2011.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O'Neill LAJ. The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol Rev 226: 10–18, 2008. doi: 10.1111/j.1600-065X.2008.00701.x. [DOI] [PubMed] [Google Scholar]
- 38.Postlethwaite AE, Raghow R, Stricklin GP, Poppleton H, Seyer JM, Kang AH. Modulation of fibroblast functions by interleukin 1: increased steady-state accumulation of type I procollagen messenger RNAs and stimulation of other functions but not chemotaxis by human recombinant interleukin 1 alpha and beta. J Cell Biol 106: 311–318, 1988. doi: 10.1083/jcb.106.2.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lertchirakarn V, Birner R, Messer HH. Effects of interleukin-1β on human pulpal fibroblast proliferation and collagen synthesis. J Endod 24: 409–413, 1998. doi: 10.1016/S0099-2399(98)80022-8. [DOI] [PubMed] [Google Scholar]
- 40.Sakairi Y, Ando Y, Tabei K, Kusano E, Asano Y. Interleukin-1 inhibits sodium and water transport in rabbit cortical collecting duct. Am J Physiol Renal Physiol 266: F674–F680, 1994. doi: 10.1152/ajprenal.1994.266.4.F674. [DOI] [PubMed] [Google Scholar]
- 41.Watson RW, Rotstein OD, Parodo J, Bitar R, Marshall JC, William R, Watson G. The IL-1 beta-converting enzyme (caspase-1) inhibits apoptosis of inflammatory neutrophils through activation of IL-1 beta. J Immunol 161: 957–962, 1998. [Erratum in J Immunol 162: 3103, 1999]. [PubMed] [Google Scholar]
- 42.Kim SO, Ono K, Han J. Apoptosis by pan-caspase inhibitors in lipopolysaccharide-activated macrophages. Am J Physiol Lung Cell Mol Physiol 281: L1095–L1105, 2001. doi: 10.1152/ajplung.2001.281.5.L1095. [DOI] [PubMed] [Google Scholar]
- 43.Lazorchak AS, Liu D, Facchinetti V, Di Lorenzo A, Sessa WC, Schatz DG, Su B. Sin1-mTORC2 suppresses rag and il7r gene expression through Akt2 in B cells. Mol Cell 39: 433–443, 2010. doi: 10.1016/j.molcel.2010.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Z, Havasi A, Gall J, Bonegio R, Li Z, Mao H, Schwartz JH, Borkan SC. GSK3β promotes apoptosis after renal ischemic injury. J Am Soc Nephrol 21: 284–294, 2010. doi: 10.1681/ASN.2009080828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumar A, Harris TE, Keller SR, Choi KM, Magnuson MA, Lawrence JC Jr.. Muscle-specific deletion of rictor impairs insulin-stimulated glucose transport and enhances Basal glycogen synthase activity. Mol Cell Biol 28: 61–70, 2008. doi: 10.1128/MCB.01405-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim EK, Yun SJ, Ha JM, Kim YW, Jin IH, Yun J, Shin HK, Song SH, Kim JH, Lee JS, Kim CD, Bae SS. Selective activation of Akt1 by mammalian target of rapamycin complex 2 regulates cancer cell migration, invasion, and metastasis. Oncogene 30: 2954–2963, 2011. doi: 10.1038/onc.2011.22. [DOI] [PubMed] [Google Scholar]
- 47.Logar CM, Brinkkoetter PT, Krofft RD, Pippin JW, Shankland SJ. Darbepoetin alfa protects podocytes from apoptosis in vitro and in vivo. Kidney Int 72: 489–498, 2007. doi: 10.1038/sj.ki.5002362. [DOI] [PubMed] [Google Scholar]
- 48.Lan A, Du J. Potential role of Akt signaling in chronic kidney disease. Nephrol Dial Transplant 30: 385–394, 2015. doi: 10.1093/ndt/gfu196. [DOI] [PubMed] [Google Scholar]