

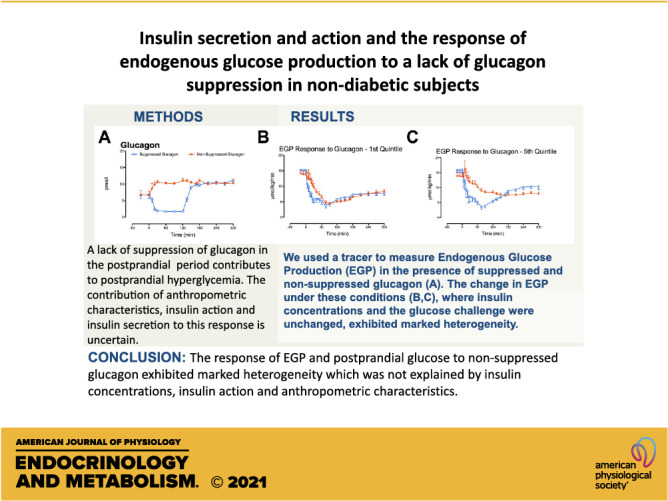
Keywords: endogenous glucose production, glucagon, hepatic insulin action, insulin action, minimal model
Abstract
Type 2 diabetes is a disease characterized by impaired insulin secretion and defective glucagon suppression in the postprandial period. We examined the effect of impaired glucagon suppression on glucose concentrations and endogenous glucose production (EGP) at different degrees of insulin secretory impairment. The contribution of anthropometric characteristics, peripheral, and hepatic insulin action to this variability was also examined. To do so, we studied 54 nondiabetic subjects on two occasions in which endogenous hormone secretion was inhibited by somatostatin, with glucagon infused at a rate of 0.65 ng/kg/min, at 0 min to prevent a fall in glucagon (nonsuppressed day) or at 120 min to create a transient fall in glucagon (suppressed day). Subjects received glucose (labeled with [3-3H]-glucose) infused to mimic the systemic appearance of 50-g oral glucose. Insulin was infused to mimic a prandial insulin response in 18 subjects, another 18 received 80% of the dose, and the remaining 18 received 60%. EGP was measured using the tracer-dilution technique. Decreased prandial insulin resulted in greater % increase in peak glucose but not in integrated glucose concentrations attributable to nonsuppressed glucagon. The % change in integrated EGP was unaffected by insulin dose. Multivariate regression analysis, adjusted for age, sex, weight, and insulin dose, did not show a relationship between the EGP response to impaired suppression of glucagon and insulin action as measured at the time of screening by oral glucose tolerance. A similar analysis for hepatic insulin action also did not show a relationship with the EGP response. These data indicate that the effect of impaired glucagon suppression on EGP is independent of anthropometric characteristics and insulin action.
NEW & NOTEWORTHY In prediabetes, anthropometric characteristics as well as insulin action do not alter the hepatic response to glucagon. The postprandial suppression or lack of suppression of glucagon secretion is an important factor governing postprandial glucose tolerance independent of insulin secretion.
INTRODUCTION
Defects in both insulin and glucagon secretion contribute to the metabolic abnormalities observed in type 2 diabetes (1). In lean, nondiabetic subjects, Shah et al. (2) demonstrated that a lack of glucagon suppression caused hyperglycemia that was especially marked when the normal rise in postchallenge insulin concentrations was delayed and decreased. Similar results were observed in subjects with type 2 diabetes who received an oral challenge (3). These data implied that abnormal postprandial glucagon suppression was only relevant to glucose homeostasis in the presence of significant β-cell dysfunction. Although insulin may play a role in the regulation (and restraint) of α-cell secretion (4), there is increasing evidence that defects in α-cell function seem to occur independently of changes in insulin secretion in the pathogenesis of prediabetes (5–7).
The demonstration that a diabetes-associated genetic variant (in the TCF7L2 locus) is associated with α-cell dysfunction implies that defects in glucagon secretion play a role in predisposition to diabetes (8). Elevated fasting glucagon concentrations also predict a longitudinal decline in β-cell function (9). Impaired glucagon suppression increases postprandial glucose concentrations by impairing the suppression of endogenous glucose production (EGP) by insulin and glucose (2). Prior work had demonstrated that defects in insulin secretion interact with impaired insulin action to alter postprandial suppression of EGP (10). The effect of the interaction of these factors with impaired glucagon suppression on postprandial glucose homeostasis is unknown.
This is a significant gap in our ability to quantify the effect of islet function on glucose tolerance. Currently, the oral minimal model is used to quantify the β-cell response to an oral challenge expressing insulin secretion as a function of the prevailing insulin action (11). A measure of β-cell function calculated from the minimal model—the Disposition Index—can correctly predict risk of progression to type 2 diabetes (12). Using this methodology, insulin action is calculated from the relationship of postchallenge insulin and glucose concentrations (11). However, in this scenario, glucagon could raise glucose concentrations independently of the ability of insulin to suppress EGP and confound the estimation of insulin action. Quantification of glucagon’s effects on postprandial glucose metabolism might enhance the ability of the minimal model to predict the risk of progression to type 2 diabetes.
Therefore, to quantify how impaired glucagon suppression interacts with insulin secretion and action to alter EGP we studied subjects on two occasions in the presence of suppressed and nonsuppressed glucagon (2). The cohort was divided into three subgroups receiving 60%, 80%, and 100% (the 0.6, 0.8, and 1.0 dose groups, respectively) of a “prandial” insulin infusion. Under these experimental conditions, the hepatic response to a lack of postprandial glucagon suppression was not affected by prevailing whole body or hepatic insulin action at the time of the experiment. Our data indicate that the effect of impaired glucagon suppression on EGP is independent of anthropometric characteristics and insulin action. A method of quantifying glucagon action will be required to examine the relative contributions of glucagon and insulin to glucose tolerance in a given individual.
METHODS
Screening
We recruited subjects using methods established previously to study the effect of diabetes-associated variation in TCF7L2 (rs7903146) on glucose metabolism (8). Subjects were randomly selected from the biobank cohort if they had no history of diabetes and they resided within a 100-mile radius of Mayo Clinic in Rochester, MN. Subjects homozygous for the diabetes-associated allele (TT) were matched for age, sex, fasting glucose concentrations, and body weight with subjects homozygous for the disease-protective allele (CC) and invited to come for a screening visit. They were then invited to come the Clinical Research and Translation Unit (CRTU) for a screening visit. After written, informed consent was obtained, participants underwent a 2-h, 75-g oral glucose tolerance test (OGTT) to characterize their glucose tolerance status as previously described (13). All participants were not taking medications that could affect glucose metabolism and had no history of chronic illness or upper gastrointestinal surgery. Subjects were in good health, at a stable weight, and did not engage in regular vigorous exercise. All subjects were instructed to follow a weight-maintenance diet containing 55% carbohydrate, 30% fat, and 15% protein for at least 3 days before the study. Body composition was measured using dual-energy X-ray absorptiometry (Lunar, Madison, WI) at screening.
Experimental Design
Subjects underwent randomization to one of three groups; one group of subjects received an insulin profile mimicking the insulin secretory response in subjects with normal glucose tolerance to a glucose challenge (Insulin 1.0). Another group of subjects received an insulin profile providing 0.8 of the insulin infused in the first group (Insulin 0.8), and the final group received 0.6 of the insulin infused in Group 1 (Insulin 0.6). All were studied on two occasions in random order, at least 2 wk apart.
On the nonsuppressed glucagon study day (NSG), subjects were admitted to the CRTU at 1700 on the day before study. They then consumed a standard 10 kcal/kg meal (55% carbohydrate, 30% fat, 15% protein) and fasted overnight. The following (0530), a forearm vein was cannulated to allow infusions to be performed. In addition, a cannula was inserted retrogradely into a vein of the contralateral dorsum of the hand, which was then placed in a heated Plexiglas box maintained at 55°C to allow sampling of arterialized venous blood. At ∼0600 (−180 min), a primed (10 µCi prime, 0.1 µCi/min continuous) infusion containing trace amounts of glucose labeled with [3-3H] glucose was started and continued till 0900 (0 min). At 0900 (0 min), the infusion was decreased in a way that mimics the anticipated fall in EGP. In addition, a “prandial” glucose infusion also labeled with [3-3H] glucose was started so as to produce glucose concentrations similar to those observed after oral ingestion of 75-g glucose (14). This minimizes variation in specific activity ensuring accurate measure of glucose turnover (15).
Simultaneously, an infusion of somatostatin (60 ng/kg/min) was started at time 0 to inhibit endogenous islet secretion and therefore ensure identical portal insulin concentrations on the two study days (16). Insulin was infused using a variable insulin infusion that mimics postprandial insulin secretion rates depending on the subject groups described above. Beginning at time 0, on the NSG study day, glucagon was infused at 0.65 ng/kg/min and maintained till the end of the study to maintain glucagon concentrations constant mimicking the lack of postprandial suppression as previously described (3).
The study visit for the suppressed glucagon (SG) study day was similar. However, no glucagon was infused for the first 120 min then infused at 0.65 ng/kg/min from 120 to 300 min to mimic normal postprandial glucagon suppression (8). Data on glucagon appearance during the SG day have been used to develop a population-based model of glucagon kinetics (17).
Analytical Techniques
All blood was immediately placed on ice after collection, centrifuged at 4°C, separated, and stored at −80°C until assay. Plasma glucose concentrations were measured using a Yellow Springs glucose analyzer. Plasma insulin concentrations were measured using a chemiluminescence assay (Access Assay, Beckman, Chaska, MN). Plasma C-peptide was measured using a 2-site immunenzymatic sandwich assay (Roche Diagnostics, Indianapolis, IN). Glucagon was measured using a two-site ELISA (Mercodia, Winston Salem, NC) in accordance with the manufacturer’s instructions. [3-3H] glucose-specific activity was measured by liquid scintillation counting following deproteinization (18).
Calculations
The oral minimal model was used to calculate β-cell responsivity, insulin action, and disposition index from the screening OGTT data (11). Total insulin sensitivity (Si) and hepatic insulin sensitivity (SiL) were also estimated from the experimental data as previously reported (19, 20). Two-segment smoothing using the method of Bradley et al. was used to decrease variation in specific activity (21). Glucose appearance and disappearance were calculated using the nonsteady-state equations of Steele et al., using the tracer infusion rate for each interval. The volume of distribution of glucose was assumed to equal 200 mL/kg, with a pool correction factor of 0.65 (22). EGP was calculated by subtracting the glucose infusion rate from the tracer-determined rate of glucose appearance (15). All rates of turnover are expressed per kg/lean body mass.
Statistical Analysis
All continuous data are summarized as means ± SE. Area under the curve (AUC) and area above basal (AAB) were calculated using the trapezoidal rule. For the purposes of these analyses, we focused on differences occurring during the first 120 min of either study day, where the differences in glucagon concentrations are most pronounced. A paired, two-way student t test (parametric) or a Wilcoxon matched-pairs signed rank test (nonparametric) was used to examine changes between study days. Differences between the three insulin dose groups were assessed using repeated-measures ANOVA. In selected instances, we used (symmetric) percent differences calculated as 100 × Loge (SG study day value/NSG study day value) (23). Multivariate analysis adjusting for the effects of age, sex, and weight was performed in JMP Pro 14 (SAS Institute Inc., Cary, NC) and in Prism 5 (GraphPad Software, San Diego, CA). Residuals for the conditional logistic regression of a particular parameter with the covariates were used to confirm or refute the contribution of that parameter to variation in the tested index. A P value <0.05 was considered statistically significant.
RESULTS
Subject Characteristics by Insulin Group
Subjects were assigned to an insulin dose group, ensuring that each insulin group was balanced for age, weight, and sex distribution (Table 1). There was a small, but significant, difference in fasting glucose at the time of screening between the 0.8 and 1.0 insulin dose groups. However, β-cell responsivity and insulin action at the time of screening did not differ among groups.
Table 1.
Participant characteristics at the time of screening when characterized by insulin group
| 0.6 | 0.8 | 1.0 | P Value | |
|---|---|---|---|---|
| n | 18 | 18 | 18 | |
| Age, yr | 55 ± 3 | 52 ± 3 | 55 ± 3 | 0.73 |
| Sex, M/F | 3/15 | 6/12 | 8/10 | 0.16* |
| rs7903146 genotype, CC/TT | 8/10 | 9/9 | 8/10 | 0.95* |
| Total body mass, kg | 81 ± 3 | 77 ± 4 | 85 ± 4 | 0.30 |
| BMI, kg/m2 | 28 ± 1 | 27 ± 1 | 29 ± 1 | 0.36 |
| LBM, kg | 45 ± 2 | 45 ± 2 | 51 ± 3 | 0.19 |
| Fasting glucose, mmol/L | 4.9 ± 0.1 | 4.8 ± 0.1 | 5.2 ± 0.1 | 0.04° |
| Peak glucose, mmol/L | 9.6 ± 0.4 | 10.1 ± 0.3 | 9.4 ± 0.3 | 0.41 |
| 120-min glucose, pmol/L | 7.0 ± 0.4 | 7.4 ± 0.3 | 7.3 ± 0.3 | 0.72 |
| Si, 10−4 dL/kg/min/μU/mL | 15 ± 3 | 16 ± 3 | 17 ± 3 | 0.87 |
| Φ, 10−9 min−1 | 65 ± 7 | 52 ± 4 | 66 ± 7 | 0.24 |
| DI, 10−14 dL/kg/min/pmol | 1,631 ± 354 | 1,308 ± 258 | 1,385 ± 220 | 0.80 |
P values represent results of a one-way analysis of variance (ANOVA) test except for * which represent results of a chi-squared test. °Post hoc Tukey’s test suggests a significant difference for the fasting glucose values in the 0.8 group vs. the 1.0 group. BMI, body mass index; CC, genotype at rs7903146 - 2 C-alleles; DI, disposition index; F, female; LBM, lean body mass; M, male; TT, genotype at rs7903146 - 2 T-alleles.
Glucagon, Insulin, and C-Peptide Concentrations during the Suppressed and Nonsuppressed Glucagon Study Days
Fasting glucagon concentrations did not differ significantly between study days in all insulin dose groups (Fig. 1, A and B). During the SG day, between 0 and 120 min, glucagon concentrations fell to ∼2pmol/L. After glucagon infusion commenced at 120 min, concentrations rose to ∼10 pmol/L (Fig. 1A). In contrast, on the NSG day (Fig. 1B), glucagon infusion commenced at 0 min and raised peripheral concentrations to values that did not differ from those observed in the later part of the SG day. Glucagon concentrations over time did not differ between insulin dose groups during both the SG and NSG days.
Figure 1.

Glucagon, insulin, and C-peptide concentrations during the nonsuppressed (A, C, and E) and the suppressed (B, D, and F) study days, respectively, for subjects receiving the 0.6 (○), the 0.8 (▲), and the 1.0 insulin doses (•).
Fasting insulin concentrations did not differ significantly between study days in all insulin dose groups (Fig. 1, C and D). However, by design, peak and integrated insulin concentrations increased progressively in the 0.8 and 1.0 groups as compared with the values observed in the 0.6 group (Table 2). Insulin concentrations after the start of the study did not differ between the SG and NSG days.
Table 2.
Glucose, insulin, endogenous glucose production, and glucose disappearance by insulin group during the suppressed and nonsuppressed glucagon study days
| 0.6 | 0.8 | 1.0 | °P value | |
|---|---|---|---|---|
| Suppressed Glucagon Study Day | ||||
| Peak glucose, mmol/L | 9.3 ± 0.4 | 9.2 ± 0.3 | 9.9 ± 0.4 | 0.37 |
| AAB glucose, mmol/2 h | 378 ± 27 | 341 ± 28 | 380 ± 35 | 0.49 |
| Peak insulin, pmol/L | 533 ± 33 | 766 ± 43 | 921 ± 87 | <0.01 |
| AAB insulin, nmol/2 h | 11.0 ± 2.1 | 22.6 ± 2.4 | 26.4 ± 2.7 | <0.01 |
| Nadir EGP, µmol/kg/min | 4.7 ± 0.6 | 4.5 ± 0.5 | 4.2 ± 0.5 | 0.83 |
| AUC EGP, mmol/kg/2 h | 0.91 ± 0.12 | 0.98 ± 0.09 | 1.02 ± 0.10 | 0.02 |
| Peak Rd, µmol/kg/min | 74 ± 3 | 77 ± 3 | 74 ± 3 | 0.64 |
| AAB Rd, mmol/kg/2 h | 2.5 ± 0.1 | 2.5 ± 0.1 | 2.5 ± 0.1 | 0.95 |
| Nonsuppressed Glucagon Study Day | ||||
| Peak glucose, mmol/L | 12.2 ± 0.4 | 11.4 ± 0.4 | 11.4 ± 0.4 | 0.30 |
| AAB glucose, mmol/2 h | 550 ± 22 | 496 ± 29 | 507 ± 29 | 0.42 |
| Peak insulin, pmol/L | 533 ± 33 | 716 ± 46 | 851 ± 32 | < 0.01 |
| AAB insulin, nmol/2 h | 12.8 ± 1.8 | 19.8 ± 3.7 | 27.1 ± 3.1 | < 0.01 |
| Nadir EGP, µmol/kg/min | 6.7 ± 0.5 | 6.6 ± 0.7 | 6.0 ± 0.6 | 0.35 |
| AUC EGP, mmol/kg/2 h | 1.28 ± 0.11 | 1.21 ± 0.11 | 1.22 ± 0.11 | < 0.01 |
| Peak Rd, µmol/kg/min | 77 ± 3 | 77 ± 3 | 72 ± 3 | 0.32 |
| AAB Rd, mmol/kg/2 h | 2.9 ± 0.2 | 2.8 ± 0.2 | 2.7 ± 0.2 | 0.81 |
| Within-Group Differences between the Two Study Days | ||||
| *P value for peak glucose | <1.0 × 10−3 | <1.0 × 10−3 | <1.0 × 10−3 | |
| *P value for AAB glucose | <1.0 × 10−3 | <1.0 × 10−3 | <1.0 × 10−3 | |
| *P value for peak insulin | 0.90 | 0.40 | 0.39 | |
| *P value for AAB insulin | 0.28 | 0.24 | 0.79 | |
| *P value for nadir EGP | 0.01 | 0.02 | 0.02 | |
| *P value for AUC EGP | 0.02 | 0.05 | 0.04 | |
| *P value for peak Rd | 0.37 | 0.97 | 0.37 | |
| *P value for AAB Rd | 0.06 | 0.11 | 0.08 | |
°P values represent results of a one‐way analysis of variance (ANOVA) test between insulin groups. *P values represent results of a two‐way, paired Student’s t test for within‐group changes between the two study days. AAB, area above basal; AUC, area under the curve; EGP, endogenous glucose production; Rd, rates of glucose disappearance.
Fasting C-peptide concentrations did not differ significantly between study days in all insulin dose groups (Fig. 1, E and F). Somatostatin suppressed endogenous insulin secretion equally in all groups for the first 120 min of the study. There was some breakthrough secretion during the NSG day (Fig. 1F), but these differences were only significant at 180 min.
Glucose, Endogenous Glucose Production, and Glucose Disappearance during the Suppressed and Nonsuppressed Glucagon Study Days
Fasting glucose concentrations did not differ significantly between study days in all insulin dose groups (Fig. 2, A and B). Although peak glucose concentrations during the first 120 min did not differ between groups, lack of glucagon suppression during the NSG study day significantly increased peak and integrated glucose concentrations compared with the SG day (Table 2).
Figure 2.

Glucose concentrations and rates of endogenous glucose production and glucose disappearance during the nonsuppressed (A, C, and E) and the suppressed (B, D, and F) study days, respectively, for subjects receiving the 0.6 (○), the 0.8 (▲), and the 1.0 insulin doses (•).
Fasting EGP did not differ significantly between study days in all insulin dose groups (Fig. 2, C and D). Rising glucose and insulin concentrations during the SG study day suppressed EGP to a nadir that did not differ between groups. A lack of glucagon suppression during the NSG study day significantly increased nadir EGP compared with the SG day (Table 2).
Fasting, peak, and integrated rates of glucose disappearance (Rd) did not differ significantly between study days in all dose groups (Fig. 2, E and F and Table 2).
Nadir and Integrated Endogenous Glucose Production during the Suppressed and Nonsuppressed Glucagon Study Days
As expected, individual values of fasting EGP (Fig. 3A) did not differ significantly between study days in all insulin dose groups. Nadir EGP (Fig. 3B) also did not differ between insulin dose groups during both study days. However, nadir EGP during the NSG day was higher than that observed during the SG day (Table 2).
Figure 3.
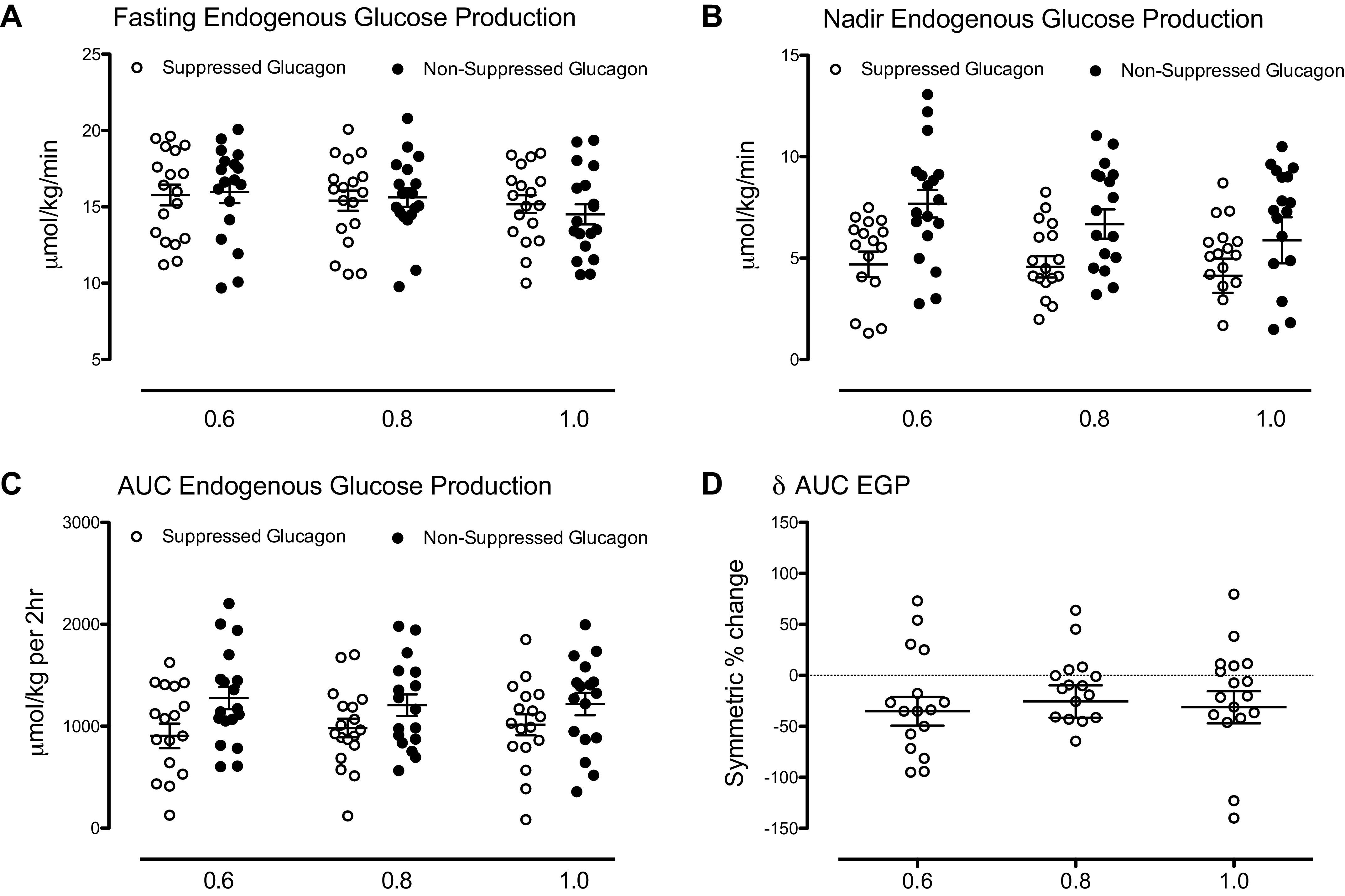
Fasting (A), nadir (B), area under the curve (AUC; C), and symmetric % change of area under the curve (D) of rates of endogenous glucose production (EGP) in each of the three insulin dose groups. Data in A−C represent values observed during the suppressed (○) and the nonsuppressed (•) glucagon study day.
Integrated EGP (AUC, Fig. 3C) during the first 120 min of the study did not differ between insulin dose groups during the SG day. Similarly, AUC EGP during the NSG day, though higher than during the SG day, did not differ between groups (Table 2). The symmetric percent change for AUC EGP (Fig. 3D) did not differ between insulin dose groups.
Relationship of Nadir and Integrated Endogenous Glucose Production during the Nonsuppressed Glucagon Study with Insulin Action
When adjusted for age, sex, and weight, nadir EGP during the NSG study day did not correlate with insulin action measured during the screening OGTT (Fig. 4A). Similarly, no relationship with hepatic insulin action (SiL) was observed (Fig. 4B). Integrated values (AUC) for EGP observed during the NSG study day and adjusted for age, sex, and weight also did not correlate with insulin action measured during the screening OGTT (Fig. 4C). No relationship with hepatic insulin action (SiL) was observed (Fig. 4D).
Figure 4.
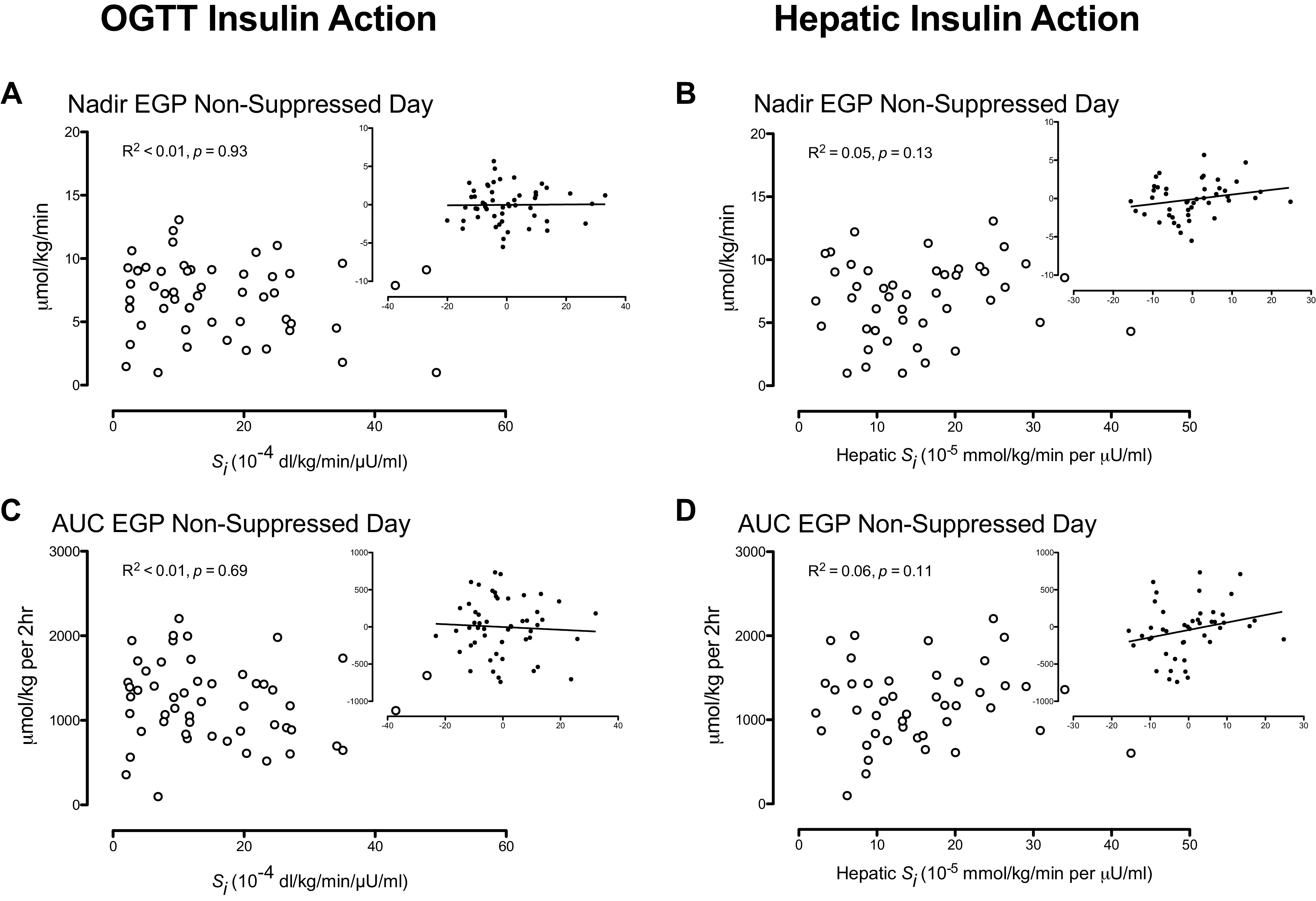
Insulin action measured during an oral glucose tolerance test (OGTT) and hepatic insulin action measured from the endogenous glucose production (EGP) response to insulin during the suppressed glucagon study day did not correlate with nadir (A and B) and area under the curve (AUC; C and D) EGP. R2 and P values represent results from a multivariate analysis adjusting for the effects of age, sex, and weight. The inset panels represent residuals after adjusting for these covariates.
DISCUSSION
Type 2 diabetes is a bihormonal disease characterized by impaired insulin secretion as well as impaired glucagon suppression in response to meal ingestion (1). Shah et al. had previously studied healthy, young, and lean individuals in a fashion similar to that described in this experiment (2). The investigators concluded that the effects of impaired glucagon suppression, while evident even in the presence of a normal “prandial” insulin profile, were most marked in the presence of a “diabetic” insulin profile with a significantly decreased and delayed insulin peak. This implied that if insulin secretion is relatively intact, abnormal glucagon suppression has relatively minor effects on glucose tolerance. However, in an older, overweight population, we demonstrate that a lack of glucagon suppression has marked effects on peak and integrated postprandial glucose concentrations explained by impaired suppression of EGP. This is also notable because the insulin profile utilized preserved the timing of peak insulin secretion regardless of the insulin dose that subjects received (10).
Intriguingly, despite a 20% and 40% reduction in insulin exposure, there were no significant between-group differences in peak and integrated glucose excursions during the first 2 h of the glucose challenge. A similar pattern was observed for nadir and integrated EGP in each insulin dose group where the change in integrated EGP attributable to nonsuppressed glucagon did not differ between groups. One implication of these findings is that the timing of the postprandial insulin peak—usually delayed in type 2 diabetes—or a delay in the onset of insulin action may be more important than absolute insulin concentrations in suppressing EGP regardless of whether glucagon is appropriately suppressed in the postprandial period (10). Another corollary of these results is that in the absence of endogenous insulin secretion, postprandial suppression of glucagon may be more important for postprandial glycemic control that modest increases in insulin dosing.
It is also notable that significant heterogeneity in EGP was observed in either study day. This heterogeneity was not explained by age, weight, and sex (data not shown). We also examined whether differences in insulin action measured at the time of screening could contribute to the response to nonsuppressed glucagon (11). Insulin action was first calculated from the screening OGTT data and exhibited no relationship with the EGP response to glucagon. Subsequently, hepatic insulin action calculated from the relationship between EGP and insulin concentrations (19, 20) observed on the SG study day also did not correlate with the EGP response to nonsuppressed glucagon. These findings imply that factors beyond responsiveness to insulin, absolute insulin concentrations, and anthropometric characteristics contribute to individual heterogeneity in the hepatic response to glucagon. Indeed there is some evidence to suggest that circulating amino acid concentrations and the ability of glucagon to stimulate hepatic amino acid metabolism might govern postprandial glucagon responses (24, 25).
A contribution of diabetes-associated genetic variation to α-cell dysfunction implies that defects in glucagon secretion contribute to the early pathogenesis of type 2 diabetes (8). Currently, β-cell function as measured by the minimal model is useful in predicting progression to diabetes (26). However, the measurement of insulin action ignores any effects of glucagon on glucose (via changes in EGP) independently of insulin secretion and action (11). It is likely that incorporation of indices of glucagon secretion and action will improve the ability of modeling to assess islet function and future risk of diabetes. Data from this experiment have already been utilized to enable deconvolution of peripheral rates of glucagon appearance from glucagon concentrations (17). Future work will focus on quantifying the effect of changes in glucagon concentrations on EGP to better characterize glucagon action in individuals.
This study suffers from some limitations. The first is that glucose, insulin, and glucagon are delivered intravenously into the peripheral circulation. Although we can estimate portal concentrations of insulin from peripheral concentrations, there is some uncertainty regarding the magnitude of hepatic extraction of glucagon in humans (27). Certainly, at the start of the study on the NSG study day, peripheral concentrations of glucagon rose from fasting concentrations. Although these differences may be accounted for by (a lack of) hepatic extraction of glucagon during infusion, implying that portal concentrations of glucagon are unchanged before and after time 0, at present, this is unclear. Although insulin concentrations during the experiment were likely lower than the portal concentrations observed during an oral challenge, due to hepatic extraction (13, 28), even at these concentrations, further decreases in insulin (0.6 vs. 0.8 vs. 1.0) did not alter the EGP response to nonsuppressed glucagon.
We note evidence of endogenous insulin secretion in some subjects at the time of peak hyperglycemia during the NSG day (∼180 min). This would not explain the heterogeneity of the EGP response observed during either study day during the first 120 min of the experiment. Although gut incretin hormones secreted in response to meal ingestion may restrain α-cell function and stimulate insulin secretion (29), our experimental design allowed matching of insulin concentrations between study days and sought to quantify the effect of α-cell dysfunction—as observed in type 2 diabetes (1, 30)—on EGP and postprandial glucose metabolism .
Another consideration is that this cohort was recruited by genotype at rs7903146 in the TCF7L2 locus. Previously, we utilized a euglycemic clamp to show that genotype at rs7903146 did not alter hepatic glucose metabolism (16). Indeed, a study of 550 individuals using a euglycemic, hyperinsulinemic clamp also demonstrated that rs7903146 genotype was not associated with differences in insulin action (31). These data demonstrate that when insulin and glucagon concentrations are matched, genotype at rs7903146 does not directly influence glucose metabolism. In this experiment, in keeping with prior observations (16, 31), there was no effect of genotype on the response of EGP or Rd to a lack of glucagon suppression (data not shown).
We conclude by noting that a lack of suppression of glucagon produces marked changes in glucose tolerance in overweight, middle-aged individuals even when the normal pattern of postprandial insulin secretion is replicated. The changes in EGP are relatively unaffected even by a 40% decrease in insulin concentrations provided that peak insulin concentrations are achieved early in the postprandial period. Finally, heterogeneity in the response to glucagon seems to be relatively independent of anthropometric characteristics and insulin signaling. This implies that further work is necessary to identify and quantify the factors that modulate the hepatic response to glucagon in humans.
GRANTS
Dr. Vella is supported by National Institutes of Health Grants DK78646, DK116231, and DK126206.
DISCLOSURES
Dr. Vella is the recipient of an investigator-initiated grant from Novo Nordisk and has consulted for vTv Therapeutics and Zeeland Pharmaceuticals. None of the other authors has any conflict of interests, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
C.C. and A.V. conceived and designed research; J.D.A., A.M.E., and A.V. performed experiments; J.D.A., A.M.E., M.C.L., D.S.W., K.R.B., C.D.M., and A.V. analyzed data; J.D.A., M.C.L., K.R.B., C.C., C.D.M., and A.V. interpreted results of experiments; M.C.L., D.S.W., and A.V. prepared figures; A.M.E. and M.C.L. drafted manuscript; K.R.B., C.C., C.D.M., and A.V. edited and revised manuscript; J.D.A., A.M.E., M.C.L., D.S.W., K.R.B., C.C., C.D.M., and A.V. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors acknowledge the support of the Mayo Clinic General Clinical Research Center (DK TR000135).
REFERENCES
- 1.Unger RH, Orci L. The essential role of glucagon in the pathogenesis of diabetes mellitus. Lancet 1: 14–16, 1975. doi: 10.1016/S0140-6736(75)92375-2. [DOI] [PubMed] [Google Scholar]
- 2.Shah P, Basu A, Basu R, Rizza R. Impact of lack of suppression of glucagon on glucose tolerance in humans. Am J Physiol Endocrinol Physiol 277: E283–E290, 1999. doi: 10.1152/ajpendo.1999.277.2.E283. [DOI] [PubMed] [Google Scholar]
- 3.Shah P, Vella A, Basu A, Basu R, Schwenk WF, Rizza RA. Lack of suppression of glucagon contributes to postprandial hyperglycemia in subjects with type 2 diabetes mellitus. J Clin Endocrinol Metab 85: 4053–4059, 2000. doi: 10.1210/jc.85.11.4053. [DOI] [PubMed] [Google Scholar]
- 4.Meier JJ, Kjems LL, Veldhuis JD, Lefebvre P, Butler PC. Postprandial suppression of glucagon secretion depends on intact pulsatile insulin secretion: further evidence for the intraislet insulin hypothesis. Diabetes 55: 1051–1056, 2006. doi: 10.2337/diabetes.55.04.06.db05-1449. [DOI] [PubMed] [Google Scholar]
- 5.Sharma A, Varghese RT, Shah M, Man CD, Cobelli C, Rizza RA, Bailey KR, Vella A. Impaired insulin action is associated with increased glucagon concentrations in non-diabetic humans. J Clin Endocrinol Metab 103: 314–319, 2018. doi: 10.1210/jc.2017-01197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Færch K, Vistisen D, Pacini G, Torekov SS, Johansen NB, Witte DR, Jonsson A, Pedersen O, Hansen T, Lauritzen T, Jørgensen ME, Ahrén B, Holst JJ. Insulin resistance is accompanied by increased fasting glucagon and delayed glucagon suppression in individuals with normal and impaired glucose regulation. Diabetes 65: 3473–3481, 2016. doi: 10.2337/db16-0240. [DOI] [PubMed] [Google Scholar]
- 7.Ferrannini E, Muscelli E, Natali A, Gabriel R, Mitrakou A, Flyvbjerg A, Golay A, Hojlund K; Relationship between Insulin Sensitivity and Cardiovascular Disease Risk (RISC) Project Investigators. Association of fasting glucagon and proinsulin concentrations with insulin resistance. Diabetologia 50: 2342–2347, 2007. doi: 10.1007/s00125-007-0806-x. [DOI] [PubMed] [Google Scholar]
- 8.Shah M, Varghese RT, Miles JM, Piccinini F, Dalla Man C, Cobelli C, Bailey KR, Rizza RA, Vella A. TCF7L2 genotype and α-cell function in humans without diabetes. Diabetes 65: 371–380, 2016. doi: 10.2337/db15-1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adams JD, Dalla Man C, Laurenti MC, Andrade MDH, Cobelli C, Rizza RA, Bailey KR, Vella A. Fasting glucagon concentrations are associated with longitudinal decline of β-cell function in non-diabetic humans. Metabolism 105: 154175, 2020. doi: 10.1016/j.metabol.2020.154175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Basu A, Alzaid A, Dinneen S, Caumo A, Cobelli C, Rizza RA. Effects of a change in the pattern of insulin delivery on carbohydrate tolerance in diabetic and nondiabetic humans in the presence of differing degrees of insulin resistance. J Clin Invest 97: 2351–2361, 1996. doi: 10.1172/JCI118678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cobelli C, Dalla Man C, Toffolo G, Basu R, Vella A, Rizza R. The oral minimal model method. Diabetes 63: 1203–1213, 2014. doi: 10.2337/db13-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watanabe RM, Valle T, Hauser ER, Ghosh S, Eriksson J, Kohtamäki K, Ehnholm C, Tuomilehto J, Collins FS, Bergman RN, Boehnke M. Familiarity of quantitative metabolic traits in Finnish families with non-insulin-dependent diabetes mellitus. Finland-United States Investigation of NIDDM Genetics (FUSION) Study investigators. Hum Hered 49: 159–168, 1999. doi: 10.1159/000022865. [DOI] [PubMed] [Google Scholar]
- 13.Sathananthan A, Man CD, Zinsmeister AR, Camilleri M, Rodeheffer RJ, Toffolo G, Cobelli C, Rizza RA, Vella A. A concerted decline in insulin secretion and action occurs across the spectrum of fasting and postchallenge glucose concentrations. Clin Endocrinol (Oxf) 76: 212–219, 2012. doi: 10.1111/j.1365-2265.2011.04159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vella A, Shah P, Basu R, Basu A, Holst JJ, Rizza RA. Effect of glucagon-like peptide 1(7-36) amide on glucose effectiveness and insulin action in people with type 2 diabetes. Diabetes 49: 611–617, 2000. doi: 10.2337/diabetes.49.4.611. [DOI] [PubMed] [Google Scholar]
- 15.Vella A, Rizza RA. Application of isotopic techniques using constant specific activity or enrichment to the study of carbohydrate metabolism. Diabetes 58: 2168–2174, 2009. doi: 10.2337/db09-0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Varghese RT, Viegas I, Barosa C, Marques C, Shah M, Rizza RA, Jones JG, Vella A. Diabetes-associated variation in TCF7L2 is not associated with hepatic or extrahepatic insulin resistance. Diabetes 65: 887–892, 2016. doi: 10.2337/db15-1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laurenti MC, Vella A, Adams JD, Schembri Wismayer DJ, Egan AM, Dalla Man C. Assessment of individual and standardized glucagon kinetics in healthy humans. Am J Physiol Endocrinol Metab 320: E71–E77, 2021. doi: 10.1152/ajpendo.00488.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Basu R, Di Camillo B, Toffolo G, Basu A, Shah P, Vella A, Rizza R, Cobelli C. Use of a novel triple-tracer approach to assess postprandial glucose metabolism. Am J Physiol Endocrinol Metab 284: E55–E69, 2003. doi: 10.1152/ajpendo.00190.2001. [DOI] [PubMed] [Google Scholar]
- 19.Caumo A, Bergman RN, Cobelli C. Insulin sensitivity from meal tolerance tests in normal subjects: a minimal model index. J Clin Endocrinol Metab 85: 4396–4402, 2000. doi: 10.1210/jcem.85.11.6982. [DOI] [PubMed] [Google Scholar]
- 20.Dalla Man C, Piccinini F, Basu R, Basu A, Rizza RA, Cobelli C. Modeling hepatic insulin sensitivity during a meal: validation against the euglycemic hyperinsulinemic clamp. Am J Physiol Endocrinol Metab 304: E819–E825, 2013. doi: 10.1152/ajpendo.00482.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bradley DC, Steil GM, Bergman RN. Quantitation of measurement error with optimal segments: basis for adaptive time course smoothing. Am J Physiol Endocrinol Physiol 264: E902–E911, 1993. doi: 10.1152/ajpendo.1993.264.6.E902. [DOI] [PubMed] [Google Scholar]
- 22.Steele R, Wall JS, De Bodo RC, Altszuler N. Measurement of size and turnover rate of body glucose pool by the isotope dilution method. Am J Physiol 187: 15–24, 1956. doi: 10.1152/ajplegacy.1956.187.1.15. [DOI] [PubMed] [Google Scholar]
- 23.Cole TJ. Sympercents: symmetric percentage differences on the 100 log(e) scale simplify the presentation of log transformed data. Stat Med 19: 3109–3125, 2000. doi:. [DOI] [PubMed] [Google Scholar]
- 24.Galsgaard KD, Winther-Sørensen M, Pedersen J, Kjeldsen SAS, Rosenkilde MM, Wewer Albrechtsen NJ, Holst JJ. Glucose and amino acid metabolism in mice depend mutually on glucagon and insulin receptor signaling. Am J Physiol Endocrinol Metab 316: E660–E673, 2019. doi: 10.1152/ajpendo.00410.2018. [DOI] [PubMed] [Google Scholar]
- 25.Suppli MP, Bagger JI, Lund A, Demant M, van Hall G, Strandberg C, Kønig MJ, Rigbolt K, Langhoff JL, Wewer Albrechtsen NJ, Holst JJ, Vilsbøll T, Knop FK. Glucagon resistance at the level of amino acid turnover in obese subjects with hepatic steatosis. Diabetes 69: 1090–1099, 2020. doi: 10.2337/db19-0715. [DOI] [PubMed] [Google Scholar]
- 26.Xiang AH, Watanabe RM, Buchanan TA. HOMA and Matsuda indices of insulin sensitivity: poor correlation with minimal model-based estimates of insulin sensitivity in longitudinal settings. Diabetologia 57: 334–338, 2014. doi: 10.1007/s00125-013-3121-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ishida T, Chap Z, Chou J, Lewis R, Hartley C, Entman M, Field JB. Differential effects of oral, peripheral intravenous, and intraportal glucose on hepatic glucose uptake and insulin and glucagon extraction in conscious dogs. J Clin Invest 72: 590–601, 1983. doi: 10.1172/JCI111007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meier JJ, Veldhuis JD, Butler PC. Pulsatile insulin secretion dictates systemic insulin delivery by regulating hepatic insulin extraction in humans. Diabetes 54: 1649–1656, 2005. doi: 10.2337/diabetes.54.6.1649. [DOI] [PubMed] [Google Scholar]
- 29.Holst JJ, Christensen M, Lund A, de Heer J, Svendsen B, Kielgast U, Knop FK. Regulation of glucagon secretion by incretins. Diabetes Obes Metab 13, Suppl 1: 89–94, 2011. doi: 10.1111/j.1463-1326.2011.01452.x. [DOI] [PubMed] [Google Scholar]
- 30.Butler PC, Rizza RA. Contribution to postprandial hyperglycemia and effect on initial splanchnic glucose clearance of hepatic glucose cycling in glucose-intolerant or NIDDM patients. Diabetes 40: 73–81, 1991. doi: 10.2337/diab.40.1.73. [DOI] [PubMed] [Google Scholar]
- 31.Rasmussen-Torvik LJ, Pankow JS, Jacobs DR Jr, Sinaiko AR. Preliminary report: no association between TCF7L2 rs7903146 and euglycemic-clamp-derived insulin sensitivity in a mixed-age cohort. Metabolism 58: 1369–1371, 2009. doi: 10.1016/j.metabol.2009.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]