

Abstract
Heart failure (HF) is a clinical condition distinguished by structural and functional defects in the myocardium, which genetic and environmental factors can induce. HF is caused by various genetic factors that are both heterogeneous and complex. The incidence of HF varies depending on the definition and area, but it is calculated to be between 1 and 2% in developed countries. There are several factors associated with the progression of HF, ranging from coronary artery disease to hypertension, of which observed the most common genetic cause to be cardiomyopathy. The main objective of this study is to investigate heart failure and its association with cardiomyopathy with their genetic variants. The selected novel genes that have been linked to human inherited cardiomyopathy play a critical role in the pathogenesis and progression of HF. Research sources collected from the human gene mutation and several databases revealed that numerous genes are linked to cardiomyopathy and thus explained the hereditary influence of such a condition. Our findings support the understanding of the genetics aspect of HF and will provide more accurate evidence of the role of changing disease accuracy. Furthermore, a better knowledge of the molecular pathophysiology of genetically caused HF could contribute to the emergence of personalized therapeutics in future.
Keywords: Heart failure, Cardiomyopathy, Genetic association, Hypertrophy, Ventricular ejection
Background
HF is a kind of clinical condition distinguished by functional and structural defects in the myocardium that hinder ventricular filling or blood ejection [1]. Due to its rising prevalence and higher mortality rate, HF is considered a significant cardiovascular disease. It is linked to a wide variety of consequences during the disease, including hospitalization, lethal arrhythmia, and mortality [2]. HF is a clinical disease characterized by common patient complaints and physical test outcomes due to ventricular failure. Since the term covers a variety of manifestations, treating it can be complicated. HF can be caused by several illnesses, including heart disease, genetic disorders, and systemic diseases [3]. The deficit site determines whether the heart failure is primarily left ventricular, right ventricular, or biventricular. HF is graded as acute or chronic depending on when it manifests [4]. The most commonly used nomenclature to characterize HF is the left ventricular ejection fraction (LVEF). The person in which Heart failure with normal LVEF levels (50%) are seen is classified as HF with preserved ejection fraction (HFpEF). Those with reduced LVEF (40%) is termed as HF with reduced ejection fraction (HFrEF), and patients who have an LVEF of 40% to 49% are classified as having HF with a mildly reduced ejection fraction (HFmrEF), and it is now classified as a distinct disease, but its epidemiology, pathophysiology, treatment, and prognosis are unknown [5]. Recognizing and handling the potential risks and subclinical precursors to heart failure is a significant concern at the moment. Growing evidence indicates that genetic predisposition influences the probability and progression of heart failure. Moreover, the mutations that arise in various genes associated with the heart can lead to this condition [6]. Therefore, even other aspects are involved in the progression of heart failure; genetic factors play a crucial role in it. The purpose of this review is to discuss the clear view on the prevalence, risk factors, types of cardiomyopathies, current insight on the genetic basis of heart attack with associated cardiomyopathy, significant genes associated with cardiomyopathy, and investigate the early diagnosis and treatment of heart failure by this molecular analysis.
Methodology
The following information regarding the prevalence, risk factors, significant genes listed in this research are found from the literature papers published throughout the past thirty years from the Web of Science, PubMed, and several other databases. The research studies were selected based on the following key terms: heart failure or HF, cardiomyopathy, dilated cardiomyopathy or DCM, restricted cardiomyopathy or RCM, hypertrophic cardiomyopathy (HCM), arrhythmogenic right ventricular cardiomyopathy or ARVCM, the incidence of heart failure or cardiomyopathy, genetic association, mutation, polymorphism, SNP, allele. The genes selected have both intronic and exonic mutations have found that gene expression in diverse places leads to many nucleotide polymorphisms (SNP). The case–control, cohort, systematic review, and meta-analysis study design for assessing heart failure, cardiomyopathy, and links between genetic polymorphism have been included. Investigations have been undertaken on cell lines, case reports and up-to-date assessment reports have also been included in this study. This review has not included research articles other than the English language.
Prevalence
The incidence of HF varies according to their definition and area, but it is calculated to be between 1–2% in developed countries. The incidence rate rises with age, reaching over 10% for people over 70 [7]. HF affects 5.7 million people in the USA today, but reports indicate that by 2030, over 8 million individuals will have the disease, representing a 46% rise in incidence [8]. In research conducted by Epidemiology of Heart Failure and Learning—EPICA in the late 1990s in Portugal, it was found that about 1.36% of people between 25–49 years of age were affected, followed by 2.93% in the 50–59 year old individuals, 7.63% in the 60–69 year old individuals, 12.67% in the 70–79-year-old individuals and 16.14% in patients > 80 years [9]. A further study in Spain examined that HF incidence rates consistently increased from 895 cases/100,000 individuals each year in 2000 to 2126 cases/100,000 individuals in 2007, with men having elevated incidence than women. HFpEF was much more familiar than HFrEF, with the former having increased rates in females and the latter elevated rates in males [10]. Overall, the incidence of HF raised with age, especially in affected people over 64 years old and people with HFpEF. In Germany, the HF incidence was 1.6% in females and 1.8% in males in 2006, rising dramatically with age [11]. In 2010, evaluated a rough incidence of HF in Sweden to be 1.8%, with equal rates in males and females. However, after adjusting for demographic composition, the incidence rate was 2.2%, with a weak downward trend in females and not in males between 2006 and 2010 [12]. According to the latest report, 1.44% of the Italian people have HF, with rates that as people get older [13]. HF is a significant public health concern in Asia, where severity appears to be much higher than in Western countries, ranging from 1.3 to 6.7% [14]. HF affects 4.2 million Chinese people, with a 1.3% incidence rate [15, 16]. About 1 million Japanese people have this disease, responsible for 1% of the community [17–19]. About 1.3 to 4.6 million people, contributing to a prevalence of 0.12–0.44%, have been affected in India, but this estimate may be underestimated [20]. HF affects 9 million individuals in Southeast Asia, with Malaysia reporting 6.7% and Singapore having a prevalence of 4.5% [21, 22]. The incidence of HF in South America is 1% and 1 to 2% in Australia, which is comparable to Western countries [23, 24]. While etiologies and disease manifestations have been examined in Sub-Saharan Africa [25], no population studies on susceptibility or incidence have been performed [26]. The PREVEND research, which involved every 28–75 year old residents of Groningen (85,421 subjects) in the Netherlands in 1997–1998 and monitored them till the later part of 2009, found an overall HF incidence of 4.4%, with 34% of new-onset cases identified as HFpEF and 66% as HFrEF [27]. Bragazzi and co-workers published an epidemiology study of heart failure in 2021 and reported that from 1990 to 2017, there was substantial regional and socio-demographic diversity in the prevalence and patterns of HF burden. Among all types of HF, IHD contributed for the most significant percentage (26.5%) of age-standardized incidence rates of HF in 2017, followed by HHD (26.2%) and COPD (23.4%) [28]. The prevalence of the topmost affected countries is given in Fig. 1.
Fig. 1.
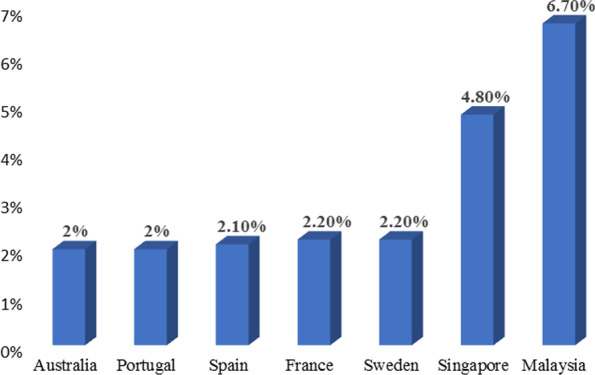
Percentage of people affected by cardiomyopathy-globally
Risk factors
The most significant threats observed for the past couple of decades include coronary artery disease (CAD), myocardial infarction (MI), hypertension, diabetes mellitus, age, sex, and obesity [29]. Along with this, several other risk factors are also involved in HF, which are mentioned in Fig. 2. Regardless of numerous studies which have conveyed that decrement of risk factors is effective, only 25%, 4% and 59% of the population with hypertension, hypercholesterolemia, and diabetes got preventive medication in the Lifelines Study Cohort, which included about 150,000 individuals in the regions of Netherlands between 2008 and 2012, respectively [30]. The 2021 European Society of Cardiology (ESC) regulations on the management and therapy of HF recommend the need for drugs like statins with an angiotensin-converting enzyme (ACE) and Sodium-glucose co-transporter-2 (SGLT2) inhibitors to prohibit HF [31]. Furthermore, to make case detection easier, the use of NT-pro BNP in the prognosis of HF was recommended. Healthy patients with elevated NT-pro BNP could be targeted to reduce the risk of new-onset HF, according to two recent trials, the PONTIAC and STOP-HF [32, 33].
Fig. 2.

Various risk factors associated with heart failure
Etiology of heart failure
HF can be triggered by a mixture of cardiac problems, genetic abnormalities, and systemic disorders (Table 1). Affected people may have a mix of aetiologies that are not mutually exclusive, and HF aetiologies vary dramatically between high-income and low-income countries [34, 35]. As per the Global Burden of Disease Study, about 17 primary causes of HF [36]. COPD, Ischaemic heart disease, Hypertensive, and Rheumatic heart disease account for more than half of all instances of cardiac failure. Even though this study attempts to evaluate the inconvenience of right-sided HF from COPD, studies exploring the incidence are sparse, necessitating further research [37]. Hypertensive heart disease, rheumatic heart disease, cardiomyopathy, and myocarditis2, high-income areas are disproportionately impacted by ischemic heart disease and chronic obstructive pulmonary disease [38]. Therefore, the evaluation and control of HF risk require policies to be adjusted to the threats and underlying causes of distinct communities all over the globe [39–42].
Table 1.
| S. no. | HF—etiologies | Corresponding complications |
|---|---|---|
| 1 | CAD | Myocardial infarction and Ischaemia |
| 2 | Cardiomyopathy | Dilated, Hypertrophic, Restrictive and Obliterative |
| 3 | Valvar and Congenital heart disease | Mitral valve disease, Aortic valve disease, Atrial septal defect and Ventricular septal defect |
| 4 | Arrhythmias | Tachycardia, Bradycardia and Loss of Atrial support (Atrial fibrillation) |
| 5 | Alcohol and Drugs | Alcohol and Cardiac depressant drugs |
| 6 | High output failure | Anemia, thyrotoxicosis, arteriovenous fistulae, Paget's disease |
| 7 | Pericardial disease | Constrictive pericarditis, Pericardial effusion |
| 8 | Primary right HF | Pulmonary hypertension, Tricuspid incompetence |
| 9 | Hypertension | HFrEF and HFpEF |
Main text
Genetic susceptibility for heart failure
HF predisposition is frequently heritable due to genetic variants. According to the Framingham Offspring Study, parental HF was connected to asymptomatic left ventricular dysfunction and increased the likelihood of overt heart failure in the progeny [43]. This study highlighted the significance of familial (genetic) factors as predictors of HF. The emphasis on hereditary variables as independent predictors for HF was also demonstrated in a large Swedish community-based study. People who had more than one sibling with heart failure were at an even higher risk of HF [44].
Furthermore, this circumstance was linked to the start of heart failure at an early age, and a genetic origin is usually observed in children with HF [45]. The incidence of HF caused by (monogenic) cardiomyopathies in unselected adult heart failure communities is likely lower than in pediatric groups. HF is caused by diverse genetic factors, some of which are sophisticated. There are hereditary monogenic HF syndromes with greater penetrance and monogenic causal congenital abnormalities on one end of the disease spectrum [46]. They are usually inherited as an autosomal dominant trait, but they can also be inherited as a recessive, X-linked trait, or mitochondrial. On the other hand, HF vulnerability may be affected by high frequent but low penetrant genetic variations (Fig. 3). In this situation, the combined effect of common variations communicates with external factors to predict susceptibility to HF, and it should be viewed as a complex disease [2]. So the more frequent familial cause of HF, i.e., Cardiomyopathy and its associated genes are studied from here.
Fig. 3.

Genetic risk factors of heart failure
Familial cardiomyopathy and its types
Cardiomyopathies are the class of cardiac muscle disorder mainly associated with the electrical or muscle dysfunction of the heart. Usually, they result in abnormal myocardial structure, function, and loading conditions [47]. AHA states it as a diverse community of diseases of myocardium, generally with improper ventricular hypertrophy or dilation [40]. It can be limited to the heart or part of a more significant systemic disorder, resulting in cardiac mortality or progressive heart failure-related impairment [48]. Restrictive cardiomyopathy (RCM), hypertrophic cardiomyopathy (HCM), Dilated cardiomyopathy (DCM), arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D), right ventricular (RV) are the four different types of cardiomyopathies that have been historically categorized based on morphological and physiological [49]. The above four categories are genetic (familial) and non-genetic (non-familial) types. The World Heart Federation determined in 2013 that "significant advances in our interpretation of the genetic basis of cardiomyopathy necessitated the development of a standardized, generally recognized classification/nosology system that incorporates phenotype description as well as genetic makeup [50]. As a result, the MOGE(S) nosology strategy was developed, which classifies cardiomyopathies not only by morphofunctional phenotype (M) but also by organ involvement (O), genetic inheritance pattern (G), and aetiological annotation (E) such as genetic disorder or underlying disease/substrate, as well as disease functional status (S). This categorization approach provides greater flexibility in defining genetic and phenotypic disorders that overlap [51].
Dilated cardiomyopathy
DCM is a heart condition marked by systolic failure and enlargement of a minimum of one ventricle. The ventricle's contractile capacity diminishes, and the ventricular wall also gets shrunk. Thromboembolic events, arrhythmias, like stroke, and the symptoms of heart failure, such as edema, exhaustion, orthopnea, and dyspnea, are the most common clinical manifestations [52]. DCM can be induced by genetic or acquired factors like myocardial infarction, medications, toxins, inflammatory diseases, chest irradiation, valve disease, and severe long-term hypertension. Although DCM is mostly an adult-onset disease, the average age has proved to be widely varied. The prevalence rate is 1 in every 2700 people [53]. It is possible to categorize it as acquired, syndromic, or non-syndromic. When both left ventricular hypertrophy and systolic dysfunction are confirmed, then the condition is characterized. DCM characterization also requires the patient's medical history, physical examination, and echocardiography (ECG) [54]. The genetic test could be employed for validation, differential diagnosis, recurrence risk assessment, and prenatal diagnosis in individuals with confirmed variants. Acquired DCM, syndromic variants, and other cardiomyopathies which could manifest with the action of the left ventricle should all be monitored in the differential diagnosis [55]. Emery-Dreifuss muscular dystrophy, Barth syndrome, Duchenne and Becker muscular dystrophy, Carvajal syndrome, Laing distal myopathy, and mitochondrial DCM are syndromic forms of HFE-associated hereditary hemochromatosis [56]. DCM is a genetically diverse condition with multiple inheritance patterns. Missense, nonsense, splicing, and minor indels are all examples of pathogenic variations. MYH7, SCN5A, BAG3, FKT, DES, RAF1, TAZ EYA4, DND, SGCD, TNNI3, MYBPC3, PSEN1, NEXN, PRDM16, and LMNA have also been shown to have substantial deletions/duplications. The mutation range for the most often mutant DCM related genes are TNNT2 3%, LMNA 6%, MYH7 4–5%, MYBPC3 2–4%, MYH6 3–4%, BAG3 2–3%, TTN 18–25% [57].
Hypertrophic cardiomyopathy
A rise in the number of cardiac muscle cells characterizes HCM. Mutations in genes encoding sarcomeric proteins are frequently responsible, resulting in myocyte disarray, a characteristic of HCM [58]. Clinical symptoms vary from person to person, even within a family, ranging from asymptomatic left ventricular hypertrophy to progressive heart failure or sudden cardiac death. Common symptoms include dyspnoea, chest discomfort, palpitations, orthostasis, presyncope, and syncope. HCM usually manifests in adolescence or early adulthood; however, it can manifest during any point of life, including old age, infancy, or childhood [59]. With a prevalence of 1:500 in the community, HCM is a reasonably prevalent genetic cardiac condition. A clinical diagnosis is made depending on the patient's medical history, physical examination, and ECG to determine hypertrophy. In families with a known mutation, the genetic test is beneficial for establishing a diagnosis, differential diagnosis, recurrence risk assessment, and prenatal diagnosis [60]. Acquired left ventricular hypertrophy, cardiac amyloidosis, Danon disease, Fabry disease, glycogen storage disease type II, Noonan syndrome, and Friedreich ataxia should all be monitored in the differential diagnosis [61]. Autosomal dominance is the common inheritance in HCM. Splicing, nonsense, missense, and minor indels are all examples of pathogenic variations. The NEXN, TNNI3, MYBPC3, CAV3, and MYH7 genes have been shown to have significant deletions/duplications. The most frequent mutant genes have a mutation detection rate of 56% (TPM1 1–3%; MYH7 20–30%; TNNT2 3–5%; MYBPC3 20–30%; TNNI3 3–5%) [62].
Restrictive cardiomyopathy
RCM is a rare genetic, primarily genetic, as the genetic disorder can explain only 75% of idiopathic RCM [63]. It is characterized by diastolic dysfunction and constrained ventricle filling due to heart muscle stiffness that causes unusual ventricle relaxation, even though thicknesses and systolic activity are generally average until late in the condition [64]. It can happen at whatever stage in life, from the outset to adulthood. Difficulty in weight gaining and flourishing, exhaustion, and fainting could be the initial indicators in children. Edema, ascites, hepatomegaly, and lung congestion may arise as the condition progresses. Some youngsters are completely asymptomatic, with abrupt death being the first symptom. Dyspnea, tiredness, and a diminished ability to exercise are the early symptoms of RCM in adults. Adults with RCM frequently encounter arrhythmia and palpitations [65]. RCM is uncommon, accounting for fewer than 5% in US and Europe, 20% in Uganda, and 8.6% in Mozambique [66]. The incidence of this disease was also studied in middle countries like Egypt, Ethiopia, Congo, Kenya, Sudan, Zimbabwe, South Africa, Ghana, Zambia, Senegal, and Tanzania, but inconsistent results have been found [67, 68].
Medical and physical examination, ECG, family history, Holter monitoring, stress test, cardiac MRI, chest X-ray, myocardial biopsy, cardiac catheterization and coronary angiography are all used to run a medical assessment [69]. Genetic testing plays a prime role in diagnostic analysis, thereby exhibiting differential diagnosis, systematic risk evaluations, and prenatal diagnosis among families. Constrictive pericarditis, idiopathic forms such as Loeffler eosinophilic endomyocardial ailment, secondary structures like infiltrative illness (sarcoidosis, Friedreich ataxia Fabry sickness, amyloidosis, hemochromatosis, and Danon illness), and therapy instigated RCM should all be considered in the differential diagnosis [70]. The inheritance pattern of RCM is of the autosomal dominant type. Missense, nonsense, splicing, and minor indels are all examples of pathogenic variations. In the TNNI3, MYBPC3, and MYH7 genes, massive deletions/duplications have been identified [71].
Arrhythmogenic right ventricular cardiomyopathy
ARVC is heritable cardiomyopathy characterized by fibrosis and fatty infiltration of the RV myocardium and signs of ventricular tachycardia and ventricular fibrillation. It was recently discovered that the condition is not controlled solely within the right ventricle, as the name implies, because up to 75% of people damage their left ventricle [72]. ARVC is predicted to have an incidence of 1:1000–1250 in the typical community [73], although it seems to be far more similar in regions with extensive family screening [74]. As reported in a community investigation, males were 3.3% substantially more prone than females to be linked with arrhythmia episodes with an even distribution of males and females [75]. The condition manifests itself in various ways, and penetrance is imperfect and age-related [76]. This condition contributes to 20% of SCD instances, and the incidence of this cardiomyopathy is markedly higher in athletes who die abruptly. ARVC is a hereditary disorder in which 30–50% of individuals have an autosomal-dominant inheritance of genetic variations that express desmosomal proteins [77]. Cardiac sarcoidosis, Idiopathic right ventricular outflow tract tachycardia, and CHD resulting in the overload of right ventricle volume should all be considered in the differential diagnosis [78]. Variants induce ARVC in genes that code for desmosomal proteins in almost 50% of all cases. Plakophilin 2 (PKP2) gene mutations are prevalent [79]. Desmocollin (DSC), desmoplakin (DSP), desmoglein 2 (DSG2), and plakoglobin (JUP) genes all have similar mutations (DSC2) [80, 81].
Other cardiomyopathies
Metabolic or mitochondrial disorders induce other types of hereditary cardiomyopathy that trigger heart failure. These disorders are classified depending on the genetic variations in proteins associated with fat or glucose metabolism and mitochondrial biogenesis. Most of them are interconnected with unspecified left ventricular hypertrophy that mimics HCM, DCM, or RCM phenotypes [49]. However, the genetic etiology of such disorders might not be discovered in the genes that cause cardiomyopathies. They are so-called phenocopies since they have entirely different pathologies, extracardiac symptoms, and treatments. Fabry disease, a class of lysosomal storage disease, which is figured by a variant in the GLA gene, which codes for galactosidase A, is an example of such photocopy [82].
Significant genes associated with cardiomyopathies
About 100 different types of genetic mutations can cause various types of cardiomyopathies that have been discovered in recent decades [83]. Most of these genes are linked to HCM and DCM, with ARVC and RCM exceptions. Figure 4 shows a list of frequent genes whose mutations can cause cardiomyopathies. These genes play a crucial role in the progression of cardiomyopathies associated with HF. The specified genes have a transformation in regions of both introns and exons, and the expression of genes in different locations results in distinct SNP (Table 2).
Fig. 4.

Role of selected genes in cardiomyopathy
Table 2.
Significant genes associated with Cardiomyopathies
| Gene | Protein name | OMIM ID | Chromosome location | Exon count | Amino acids | Inheritance type | Cardiomyopathy form | Functions | References |
|---|---|---|---|---|---|---|---|---|---|
| MYH7 | Beta myosin heavy chain | 160,760 | 14q12 | 40 | 1935 | AD | HCM/DCM/RCM | Beta heavy chain subunit of cardiac myosin | [49, 81–84] |
| TNNT2 | Troponin T | 191,045 | 1q32.1 | 17 | 297 | AD | HCM/DCM/RCM | Ca2 + -dependent regulator of muscle contraction | [90–97] |
| MYBPC3 | Cardiac Myosin binding protein C | 160,710 | 11p11.2 | 35 | 1274 | AD | HCM/DCM/RCM | A cardiac isoform of myosin-binding protein C was found in the cross-bridge-bearing zone (C area) of A bands | [103–106] |
| TNNI3 | Troponin I | 191,044 | 19q13.42 | 8 | 210 | AD | HCM/DCM/RCM | This cardiac mediator mediates striated muscle relaxation | [110–112] |
| TPM1 | Alpha-Tropomyosin | 191,010 | 15q22.2 | 15 | 284 | AD | HCM/DCM/RCM | Ca2 + -dependent striated muscle contraction regulator | [95, 117–122] |
| LMNA | Lamin A/C | 150,330 | 1q22 | 17 | 572 | AD | DCM/ARVC | Cardiac homeostasis is maintained | [128–132] |
| PKP2 | Plakophilin 2 | 602,861 | 12p11.21 | 14 | 881 | AR | ARVC | Plays a role in junctional plaques | [137–144] |
| DSC2 | Desmocollin | 125,645 | 18q12.1 | 18 | 901 | AD | ARVC | Major components of desmosomes | [146, 148–153] |
| DSG2 | Desmoglein 2 | 125,671 | 18q12.1 | 16 | 1118 | AD | ARVC/DCM | Ca2 + -binding transmembrane glycoprotein components of desmosomes between myocardial cells | [157–161] |
| DSP | Desmoplakin | 125,647 | 6p24.3 | 24 | 2871 | AD/AR | ARVC/DCM | It is an essential component of functional desmosomes | [167–169] |
| JUP | Plakoglobin | 173,325 | 17q21.2 | 19 | 745 | AD | ARVC | The common component of desmosomes and intermediate junctions | [146, 178–180] |
| TTN | Titin | 188,840 | 2q31.2 | 365 | 34,350 | AD | ARVC/HCM/DCM | Essential for striated muscle assembly and functioning; connects microfilaments | [186–189] |
Myosin heavy chain 7 (MYH7)
MYH7 gene is located on chromosome 14q12 and has 40 exons, with a total exon length of 5808 bp [84]. Furthermore, it has 3–21 exons in the head, 21–25 exons in the neck (also called the head-rod joints), and 25–40 exons in the valve system [85]. Aggregation occurs when this gene is mutated, and it could be detected in the head and head-rod joints [86]. Gly425Arg, Thr441Met, and Arg453Ser are the head mutations that have elevated clinically extraneous rates. Mutations in this location have been observed to increase the activity of myosin S1's adenosine triphosphate enzyme, which may limit changes in myosin conformation or modify its connection with actin and other components. As a result, HCM may develop [87–89]. Research suggests that exon 14 in the MYH7 gene displays the missense mutation Thr446Pro and Phe468Leu, which tends to cause HCM [90]. MYH7 missense variants are also detected in DCM. MYH7 missense mutations linked to DCM are already mimicked in mice, but only in the context of Myh6. On the other hand, these mutants have a higher tension cost, requiring larger ATP for a given degree of shortening, lower actin sliding velocities, and extensive dilatation [91]. In contrast to hypercontractile DCM mutations, HCM mutations in articulated myosin create a hypocontractile condition, eventually leading to HF [92].
Troponin T (TNNT2)
TNNT2 encodes a thin filament contractile protein that binds troponin complex and tropomyosin [93], which contains about 17 exons and covers 25 kb on chromosome 1q32 [94]. HCM, RCM, and DCM can all be caused by mutations in the TNNT2 gene [95–97]. TNNT2 mutations are thought to be responsible for about 15% of all instances of familial HCM [93, 98, 99]. Recent studies show that TNNT2 mutations have been linked to DCM, and the overall prevalence of TNNT2 variants in DCM is estimated to be 3–6% [100, 101]. The troponin complex significantly impacts muscle contraction since it acts as both a Ca2+ sensor regulator and an intercellular free Ca2+ concentration. Although TNNT2 mutations are frequently associated with HCM, TNNT2 genes also can provoke DCM [102]. Mutations in the TNNT2 gene may impair complex stability and the interrelationship between tropomyosin and troponin T, affecting actin interaction [103]. TNNT2 gene variants are demonstrated to impair the susceptibility of the complex to Ca2+, thereby lowering the contractile force of the myocardium. Genetic variants in sarcomere proteins linked to HCM can enhance Ca2+ sensitivity, increasing cardiac contractility [104]. Thus, based on the net impact of the mutation on cardiac myocardial contractile strength, mutations in various proteins may be linked with a shared cardiac disease pattern (DCM or HCM) [105].
Cardiac myosin binding protein 3 (MYBPC3)
MYBPC3 is a gene that codes for cardiac myosin binding protein-C (cMyBP-C), a crucial protein required for cardiac function6 and a regulator of ventricular contractility concerning adrenergic stimulation [106, 107]. About 40–50% of all HCM cases occur due to the MYBPC3 gene, the most mutated gene in HCM. A considerable proportion of MYBPC3-related HCM mutations are heterozygous, and people often develop the disease later in life with a benign course. According to studies, 70% of MYBPC3 mutations are truncating mutations, which generate a more severe HCM phenotype than missense and in-frame deletions [108]. Homozygous or compound heterozygous truncating pathogenic MYBPC3 variations potentially cause neonatal cardiomyopathy, which leads to HF and mortality within the first year of life, unlike heterozygous pathogenic mutations [109]. In addition, when compared to individuals with a single mutation, HCM patients with several pathogenic genetic variations have a worse prognosis concerning earlier disease onset, higher left ventricular hypertrophy, and greater frequency of HF and sudden cardiac death [110–112]. A non-polar residue (Pro) is presumed to be replaced with a positively charged residue (His) in this MYBPC3 variation. It is found in the protein's C7 domain and may disrupt protein integration in the sarcomere's A-band. According to the 'poison peptide' concept, mutated sarcomeric proteins integrate into myofibrils and operate as dominant-negative proteins [113]. As a result, the current instance backs up prior claims that rare homozygous mutations can worsen HCM clinical severity [114].
Troponin I (TNNI3)
The most intact structure of TNNI3 (amino acids 184–210 in human cardiac TnI) is the C-terminal, which links with tropomyosin in a calcium-regulated way, implying a vital role. This area is very flexible, with no fixed secondary structure, indicating a dynamic system in troponin that functions [115]. When Ca2+ is activated, the terminal segment of cTnI (amino acids 190–210) has been demonstrated to have a role in tropomyosin stability in the actin filament [116]. In vivo evidence suggests that variations in this area produce myofibril Ca2+ hypersensitivity and subsequent improper relaxation, the primary cause of RCM [117]. Several variations in the end segment of the protein's C-terminus have been linked to HCM, three of which (p.Asp190Gly, p.Arg192His, and p.Arg204His) were also linked to RCM [118]. A vast family's genetic analysis indicated that carriers of the same variation (p.Asp190Gly) showed significant phenotypic variability, with the majority of them meeting HCM clinical criteria though some were diagnosed with RCM [95]. Furthermore, mutations impacting the same amino acid position have been linked toward the same (i.e., p.Arg192His and p.Arg192Cys are both related to RCM) or distinct phenotypes [119]. (i.e., p.Leu144Gln is linked to RCM while p.Leu144Pro is linked to HCM) [120].
Tropomyosin alpha-1 (TPM1)
TPM1 protein is a sarcomere constituent that stabilizes thin filaments and facilitates actin-myosin interrelationship during muscular contraction [121]. Moreover, in response to Ca2+ signaling, its action is linked to the troponin complex. Besides this, its function is also correlated with the troponin complex in response to Ca2+ signaling [122]. A gene encodes TPM1 protein on 15q22.2, and the two isoforms of the TPM1 gene is TPM1α and TPM1, differ only by the presence of exon 2b or 2a is present. Both are expressed evenly in the fetal and adult heart [123–125]. The missense mutations of TPM1 gene Glu54Lys and Glu40Lys lead to autosomal dominant inheritance of DCM, as portrayed by the genetic research of DCM patients [126]. In recent times, it has been discovered that several TPM1 gene variants are linked to DCM [127]. Most current research into the link between TPM1 gene mutation and disease focuses on the gene's coding region. The T230 N mutation in the TPM1 gene, for example, lowers the flexibility of the protein's C-terminus by changing its helical structure, lowering the flexibility of the TPM1 overlap, and affecting its ability to regulate contraction [128].
Lamin A/C (LMNA/C)
The Lamin A/C gene (LMNA/C) is positioned on the human chromosome 1q21.2 [129] and encodes for the subcellular proteins lamin A and C. Lamin A is translated as a precursor, prelamin A, and needs significant C-terminal processing to develop. Meanwhile, Lamin C has already been translated as a mature protein [130]. The LMNA gene is frequently mutated in DCM, accounting for about 6–8% of DCM instances in humans [131, 132]. Heterozygous mutation in LMNA can provoke DCM with AV conduction disruption in an autosomal dominant manner [133]. Recent research has discovered that in a multigenerational family, a unique splice-site mutation in the Lamin A/C gene, LMNA c.357-2A>G (p.N120Lfs*5), causes DCM, HF, and sudden death [134]. LMNA (lamin A/C) mutation sites related to DCM have been identified, including LMNA-D300N, LMNA-H222P, and LMNA-N195K [135–137]. Furthermore, a growing body of evidence suggests that LMNA mutations cause DCM by disrupting various cellular pathways, including the mitogen-activated protein kinases (MAPK) pathway, the AKT/mTOR network, and the WNT route [138, 139]. The MAPK pathway and the mammalian target of the rapamycin complex 1 (mTORC1) pathway have previously been demonstrated to have aberrant activity in LMNA mutation induced-DCM [140–143].
Plakophilin 2 (PKP2)
PKP2 is a protein found in the intercalated discs of cardiac desmosomes, intercellular mechanical junctions [144]. Furthermore, these protein complexes offer mechanical strength and force transfer among cells by linking intermediate filaments over cell membranes [145]. The PKP2 c.2146-1G>C mutation found in the four Sweden families included in this investigation has previously been identified in people who have been diagnosed with ARVC [146–148]. These PKP2 c.2146-1G>C variants identified in a study of patients showed a splice site mutation anticipated to trigger a cryptic splice acceptor site in intron 12 or another cryptic splice acceptor site in PKP2 exon 13 [149]. Gerull and his teammates reported earlier that pathogenic variants in PKP2, encoding the desmosomal protein plakophilin 2, are connected with ARVC [146]. A cardiomyocyte-specific, tamoxifen-activated PKP2-cKO mouse model was recently published, allowing control of the onset of PKP2 loss of expression. The findings showed that the absence of PKP2 in adult ventricular myocytes was sufficient to cause ARVC [150].
Desmocollin 2 (DSC2)
DSC2 belongs to the desmocollin protein subfamily on human chromosome 18q12 in a cluster with other desmocollin family members [151]. Desmosomal cadherins, or DSC2, are single-pass transmembrane glycoproteins that coordinate Ca2+-dependent cell–cell adhesion by communicating laterally and transcellular with one another and recruiting cytoplasmic plaque proteins that aid intermediate filament binding [152]. Dominant mutations in the DSC2 gene induce ARVC, a progressive heart muscle condition characterized by ventricular tachyarrhythmias, heart failure, and risk of unexpected juvenile death [153]. In ACM patients, numerous missense [154], non-sense [155], and splice site variations [156] of DSC2 have been identified. As per investigations, two separate DSC2 mutations and one DSC2 variant were involved in ARVC progression in about 7 out of 112 unrelated ARVC/D index cases, and p.A897KfsX4 was previously referred to represent a causative mutation [157, 158]. The frameshift variation was detected in a recent analysis, executed with five different patients, four carriers for one or two mutations in recognized ARVC/D genes [159].
Desmoglein 2 (DSG2)
DSG2 is a cadherin-family cell adhesion protein essential for cardiomyocyte cohesion and function [160]. Its position is to control cell–cell interaction with neighboring cells. Desmosomal cadherins' altered expression and activity are linked with various tumorigenesis [161]. Variations in the DSG2 gene have been linked to severe cardiac muscle illnesses, including ARVC, marked by a gradual loss of cardiomyocytes and the replacement of fibrofatty tissue in the right ventricle [162]. A recent analysis detected DCM in a family in 33% of Thr335Ala carriers [163, 164]. Several other homozygous DSG2 variations, such as p.(Val55Met), have also been found to be associated with DCM instances [165]. As per recent research, mutant DSG2 proteins integrated into desmosomes have proved highly adverse effects in ARVC [166]. Furthermore, DSG2 mutations have a significant degree of penetrance and cause varying degrees of condition severity [147, 167]. Furthermore, individuals with numerous desmosomal variants have a much more severe clinical outcome, with more ventricular arrhythmias and cardiac failure than those with a single mutation [168–170].
Desmoplakin (DSP)
DSP is indeed a cytoplasmic plaque protein with no transmembrane domains produced by alternative splicing of the same mRNA into two isoforms, DSPI and DSPII [171, 172]. These isoforms' globular head and tail domains are identical at the C and N termini, but the rod domain that connects them differs in length [173]. About 2–12% of all ARVC cases are caused due to DSP mutations [169, 174]. Recent investigations claim that Desmoplakin mutation Gly2375Arg has been documented in a syndrome expressing with ARVC, and other dominant mutations in desmoplakin and plakophilin‐2 have also been correlated with non‐syndromic ARVC [146, 175–178]. Thus, specific missense desmoplakin mutations may cause ARVC, characterized by severe left ventricular involvement, fibrosis, and abrupt mortality. Further, research revealed that the primary impact of DSP R451G is significantly contributed to protein cleavage by Calpin, contributing to desmoplakin deficiency [179]. Modern-day studies have also demonstrated that mutations that affect ion channel function could also be a cause or modulator of ARVC [180, 181].
Plakoglobin (JUP)
JUP codes for junction plakoglobin protein, situated on chromosome 17q21.2. It comprises 14 exons, spreading over roughly 42 kilobases [182]. Human JUP can be partitioned into three locales: the C-terminal, 12 ARM repeats domain, and N-terminal [183]. JUP gene is associated with diffuse palmoplantar keratoderma, skin fragility, ARVC [184]. Until now, 41 changes of JUP have been discovered in patients with ARVC and cutaneous ailment issues. The p.R577C mutation is situated in the 10th ARM repeat domain of the JUP gene [185]. However, previous reports have exhibited that this conserved domain has a pivotal part in desmosomes activity. In addition, one mutation (p.Q539X) was accounted to cause epidermolysis bullosa, and another modification (p.V603L) was recognized in ARVC patients in the 10th ARM region [186]. In a blend of Sanger sequencing and bioinformatics investigation, a de novo mutation (c.1729C>T/p.R577C) of JUP was found in suspected ARVC patients from southern China. The specified modification means that arginine is substituted by cysteine in the location codon 577 of JUP [187]. With the application of bioinformatical knowledge, the cross-species alignment analysis of JUP amino acid sequences uncovered that this mutated site was a profoundly conserved domain. Western Blot method revealed that JUP directly affects Connexin 43 and DSG2 expression, related to desmosome junction stability. Cardiovascular-related interpretations presented age-associated penetrance of ARVC. Hence, phenotypically young ordinary patients with biallelic JUP changes ought to be checked for improvement of ARVC [188].
Titin
Titin is the most significant known human protein, and it frames the third myofilament structure traversing the sarcomere from the Z-circle to the M-band. TTN gene consisting of 364 exons codes the Titin protein [189]. The titin I-band acts as a molecular spring, creating a vital force for sarcomeric integrity maintenance. Pathogenic TTN variations lead to a broad scope of skeletal muscle and cardiovascular problems [190]. Titin-related myopathies are a heterogeneous gathering of acquired muscle problems that change as inheritance mode such as dominant and recessive, age at the beginning of ailments, the pattern of muscle activity, prognosis, and disease progression [191]. TTN variations cause muscle-related ailments like Myopathic congenital arthrogryposis, distal myopathies, and other forms of myopathies. Although dilated cardiomyopathy is associated with many genes, truncating mutations observed in the TTN gene are often found. Truncating variations in the TTN gene termed TTNtv is the most popular reason for heritable dilated cardiomyopathy (HCM). TTNtv expression causes incessant arrhythmia, and harmful ventricular arrhythmias are usually connected with severe left ventricular systolic dysfunction (LVSD) [192]. Phosphorylation levels of TnI and MyBP-C found in the left ventricles are fundamental for the length-subordinate changes in Ca2+ for healthy cardiac muscle activity. However, they are decreased in HCM patients with TTN-truncating variants [193]. The pervasiveness of TTN mutations among HCM affected subjects was analyzed in a meta-analysis by Fang HJ et al., which showed that the prevalence of familial dilated cardiomyopathy was 0.23 (95% CI 0.20–0.26), 0.17 (95% CI 0.14–0.19), and sporadic dilated cardiomyopathy was 0.16 (95% CI 0.12–0.21), respectively [194]. TTN was also listed in massively parallel sequencing in an attempt to spot rare variants of genes causing distal myopathy, cardiac muscle, and skeletal muscle diseases [195].
Treatment of heart failure by gene testing
It is possible to enhance therapeutic care by determining the specific genetic etiology of HF in patients. The discovery of a harmful variant enhances therapeutic precision and reduces ambiguity linked with phenotypic variance. Genetic information may also influence the application of developing medicines that target the physical and biological implications caused by polymorphisms [196]. In addition, molecular identification offers value testing of first-degree relatives and reduces medical expenses for families lacking harmful variants, bringing in considerable cost of medical care reductions [197].
Cardiomyopathy genetic arrays tend to change but now contain thorough assessments of all genes involved in HCM, DCM, ARVC, or LVNC. Marketed accessible genetic screening for cardiac diseases may be obtained at the GeneTests webpage [198]. This site provides accessible genotype arrays and executing laboratories with details and pricing. Further genetic screening guidance is available in the National Institutes of Health Genetic Testing Registry [199]. Genetic screening prices vary significantly across corporate and university facilities, equipment employed, and quantities of alleles tested. In the USA, patients are first examined utilizing broad polygenic cardiac arrays analyzed by next-generation techniques. If negative, pretty much the entire genome sequencing studies are explored. With the continuous decline in analytical and mechanical expenses, such comprehensive ranging technologies could emerge as the favored technique for genetic analysis of cardiac diseases. The detection of a definite variation permits focused investigation of first-degree families at considerably lower expenses.
Conclusions
Heart failure is a condition marked by differences in the morphology and function of the myocardium, which both genetic and environmental factors can induce. Cardiomyopathy is the most frequent genetic cause of HF and comes in various forms, each genetic variation. Hence, it is critical to determine heart failure's genetic drivers to prevent and treat it. Several disease-associated genes can lead to heart failure, and it can indeed be caused by a complicated mix of genetic and environmental conditions. Our findings reveal that selected genetic variants are more frequently associated with various cardiomyopathy and are determined to be the primary cause of familial cardiomyopathy. The innovative genetic testing is scientifically and therapeutically beneficial for heart failure, most likely caused by a combination of hereditary and environmental variables. A comprehensive genetic analysis of HF can increase understanding of molecular etiology, influence treatment, and improve prognosis. Furthermore, extensive genetic testing allows for the early identification of other family members at risk for heart failure. A more extensive research study is needed to determine how genetic pathways play a significant role in developing heart disease and risk stratification.
Acknowledgments
The authors thank the Chettinad Academy of Research Education (CARE) for the constant support and encouragement.
Abbreviations
- HF
Heart Failure
- LVEF
Left ventricular ejection fraction
- HFpEF
HF with preserved ejection fraction
- HFrEF
HF with reduced ejection fraction
- DCM
Dilated cardiomyopathy
- RCM
Restricted cardiomyopathy
- HCM
Hypertrophic cardiomyopathy
- SNP
Single nucleotide polymorphism
- CAD
Coronary artery disease CAD
- MI
Myocardial infarction
- ACE
Angiotensin-converting enzyme
- SGLT2
Sodium-glucose co-transporter-2
- COPD
Chronic obstructive pulmonary disease
- ARVC/D
Arrhythmogenic right ventricular cardiomyopathy/dysplasia
- RV
Right ventricular
- ECG
Echocardiography
- MYH7
Myosin Heavy Chain 7
- TNNT2
Troponin T
- MYBPC3
Cardiac Myosin Binding Protein 3
- TNNI3
Troponin I
- TPM1
Tropomyosin alpha-1
- LMNA/C
Lamin A/C
- DSC2
Plakophilin 2
- DSC2
Desmocollin 2
- DSG2
Desmoglein 2
- DSP
Desmoplakin
- JUP
Plakoglobin
- TTN
Titin
- LVSD
Left ventricular systolic dysfunction
Authors' contributions
VK and VM written the contents, edited the figures and tables. RV designed the study, corrected, revised, and approved the manuscript for submission. All authors read and approved the final manuscript.
Funding
Not applicable.
Availability of data and materials
Not applicable.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Dassanayaka S, Jones SP. Recent developments in heart failure. Circ Res. 2015;117(7):e58–e63. doi: 10.1161/CIRCRESAHA.115.305765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Skrzynia C, Berg JS, Willis MS, Jensen BC. Genetics and heart failure: a concise guide for the clinician. Curr Cardiol Rev. 2015;11(1):10–17. doi: 10.2174/1573403X09666131117170446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ziaeian B, Fonarow GC. Epidemiology and etiology of heart failure. Nat Rev Cardiol. 2016;13(6):368–378. doi: 10.1038/nrcardio.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ohtani T, Mohammed SF, Yamamoto K, Dunlay SM, Weston SA, Sakata Y, et al. Diastolic stiffness as assessed by diastolic wall strain is associated with adverse remodelling and poor outcomes in heart failure with preserved ejection fraction. Eur Heart J. 2012;33(14):1742–1749. doi: 10.1093/eurheartj/ehs135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016;37(27):2129–2200. doi: 10.1093/eurheartj/ehw128. [DOI] [PubMed] [Google Scholar]
- 6.Czepluch FS, Wollnik B, Hasenfuß G. Genetic determinants of heart failure: facts and numbers. ESC heart failure. 2018;5(3):211–217. doi: 10.1002/ehf2.12267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mosterd A, Hoes AW. Clinical epidemiology of heart failure. Heart. 2007;93(9):1137–1146. doi: 10.1136/hrt.2003.025270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart disease and stroke statistics—2016 update: a report from the American Heart Association. Circulation. 2016;133(4):e38–e360. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 9.Ceia F, Fonseca C, Mota T, Morais H, Matias F, de Sousa A, et al. Prevalence of chronic heart failure in Southwestern Europe: the EPICA study. Eur J Heart Fail. 2002;4(4):531–539. doi: 10.1016/s1388-9842(02)00034-x. [DOI] [PubMed] [Google Scholar]
- 10.Gomez-Soto FM, Andrey JL, Garcia-Egido AA, Escobar MA, Romero SP, Garcia-Arjona R, et al. Incidence and mortality of heart failure: a community-based study. Int J Cardiol. 2011;151(1):40–45. doi: 10.1016/j.ijcard.2010.04.055. [DOI] [PubMed] [Google Scholar]
- 11.Ohlmeier C, Mikolajczyk R, Frick J, Prütz F, Haverkamp W, Garbe E. Incidence, prevalence and 1-year all-cause mortality of heart failure in Germany: a study based on electronic healthcare data of more than six million persons. Clin Res Cardiol. 2015;104(8):688–696. doi: 10.1007/s00392-015-0841-4. [DOI] [PubMed] [Google Scholar]
- 12.Zarrinkoub R, Wettermark B, Wändell P, Mejhert M, Szulkin R, Ljunggren G, Kahan T. The epidemiology of heart failure, based on data for 2.1 million inhabitants in Sweden. Eur J Heart Fail. 2013;15(9):995–1002. doi: 10.1093/eurjhf/hft064. [DOI] [PubMed] [Google Scholar]
- 13.Buja A, Solinas G, Visca M, Federico B, Gini R, Baldo V, et al. Prevalence of heart failure and adherence to process indicators: which socio-demographic determinants are involved? Int J Environ Res Public Health. 2016;13(2):238. doi: 10.3390/ijerph13020238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sakata Y, Shimokawa H. Epidemiology of heart failure in Asia. Circ J. 2013;77:2209–2217. doi: 10.1253/circj.cj-13-0971. [DOI] [PubMed] [Google Scholar]
- 15.Hu SS, Kong LZ, Gao RL, Zhu ML, Wen WANG, Wang YJ, et al. Outline of the report on cardiovascular disease in China, 2010. Biomed Environ Sci. 2012;25(3):251–256. doi: 10.3967/0895-3988.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 16.Yang YN, Ma YT, Liu F, Huang D, Li XM, Huang Y, et al. Incidence and distributing feature of chronic heart failure in adult population of Xinjiang. Zhonghua Xin Xue Guan Bing Za Zhi. 2010;38(5):460–464. [PubMed] [Google Scholar]
- 17.Okamoto H, Kitabatake A. The epidemiology of heart failure in Japan. Nihon rinsho. Jpn J Clin Med. 2003;61(5):709–714. [PubMed] [Google Scholar]
- 18.Okura Y, Ramadan MM, Ohno Y, Mitsuma W, Tanaka K, Ito M, et al. Impending epidemic future projection of heart failure in Japan to the year 2055. Circ J. 2008;72(3):489–491. doi: 10.1253/circj.72.489. [DOI] [PubMed] [Google Scholar]
- 19.Konishi M, Ishida J, Springer J, von Haehling S, Akashi YJ, Shimokawa H, Anker SD. Heart failure epidemiology and novel treatments in Japan: facts and numbers. ESC Heart Fail. 2016;3:145–151. doi: 10.1002/ehf2.12103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huffman MD, Prabhakaran D. Heart failure: epidemiology and prevention in India. Natl Med J India. 2010;23(5):283. [PMC free article] [PubMed] [Google Scholar]
- 21.Ponikowski P, Anker SD, AlHabib KF, Cowie MR, Force TL, Hu S, et al. Heart failure: preventing disease and death worldwide. ESC Heart Fail. 2014;1(1):4–25. doi: 10.1002/ehf2.12005. [DOI] [PubMed] [Google Scholar]
- 22.Lam CS. Heart failure in Southeast Asia: facts and numbers. ESC Heart Fail. 2015;2:46–49. doi: 10.1002/ehf2.12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ciapponi A, Alcaraz A, Calderon M, Matta MG, Chaparro M, Soto N, Bardach A. Burden of heart failure in Latin America: a systematic review and meta-analysis. Revista Española de Cardiología (English Edition) 2016;69(11):1051–1060. doi: 10.1016/j.rec.2016.04.054. [DOI] [PubMed] [Google Scholar]
- 24.Sahle BW, Owen AJ, Mutowo MP, Krum H, Reid CM. Prevalence of heart failure in Australia: a systematic review. BMC Cardiovasc Disord. 2016;16(1):1–6. doi: 10.1186/s12872-016-0208-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Makubi A, Hage C, Lwakatare J, Kisenge P, Makani J, Rydén L, Lund LH. Contemporary aetiology, clinical characteristics and prognosis of adults with heart failure observed in a tertiary hospital in Tanzania: the prospective Tanzania Heart Failure (TaHeF) study. Heart. 2014;100(16):1235–1241. doi: 10.1136/heartjnl-2014-305599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ntusi NB, Mayosi BM. Epidemiology of heart failure in sub-Saharan Africa. Expert Rev Cardiovasc Ther. 2009;7(2):169–180. doi: 10.1586/14779072.7.2.169. [DOI] [PubMed] [Google Scholar]
- 27.Brouwers FP, de Boer RA, van der Harst P, Voors AA, Gansevoort RT, Bakker SJ, et al. Incidence and epidemiology of new-onset heart failure with preserved vs. reduced ejection fraction in a community-based cohort: 11-year follow-up of PREVEND. Eur Heart J. 2013;34(19):1424–1431. doi: 10.1093/eurheartj/eht066. [DOI] [PubMed] [Google Scholar]
- 28.Bragazzi NL, Zhong W, Shu J, Abu Much A, Lotan D, Grupper A, et al. Burden of heart failure and underlying causes in 195 countries and territories from 1990 to 2017. Eur J Prev Cardiol. 2021;28:1682–1690. doi: 10.1093/eurjpc/zwaa147. [DOI] [PubMed] [Google Scholar]
- 29.Komanduri S, Jadhao Y, Guduru SS, Cheriyath P, Wert Y. Prevalence and risk factors of heart failure in the USA: NHANES 2013–2014 epidemiological follow-up study. J Community Hosp Intern Med Perspect. 2017;7(1):15–20. doi: 10.1080/20009666.2016.1264696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van der Ende MY, Hartman MH, Hagemeijer Y, Meems LM, de Vries HS, Stolk RP, et al. The LifeLines Cohort Study: prevalence and treatment of cardiovascular disease and risk factors. Int J Cardiol. 2017;228:495–500. doi: 10.1016/j.ijcard.2016.11.061. [DOI] [PubMed] [Google Scholar]
- 31.McDonagh TA, Metra M, Adamo M, Gardner RS, Baumbach A, Böhm M, Burri H, et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) With the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2021;42(36):3599–3726. [Google Scholar]
- 32.Ledwidge M, Gallagher J, Conlon C, Tallon E, O’Connell E, Dawkins I, et al. Natriuretic peptide-based screening and collaborative care for heart failure: the STOP-HF randomized trial. JAMA. 2013;310(1):66–74. doi: 10.1001/jama.2013.7588. [DOI] [PubMed] [Google Scholar]
- 33.Huelsmann M, Neuhold S, Resl M, Strunk G, Brath H, Francesconi C, et al. PONTIAC (NT-proBNP Selected PreventiOn of cardiac events in a populaTion of dIabetic patients without A history of Cardiac disease) A Prospective Randomized Controlled Trial. J Am Coll Cardiol. 2013;62(15):1365–1372. doi: 10.1016/j.jacc.2013.05.069. [DOI] [PubMed] [Google Scholar]
- 34.Yusuf S, Rangarajan S, Teo K, Islam S, Li W, Liu L, et al. Cardiovascular risk and events in 17 low-, middle-, and high-income countries. N Engl J Med. 2014;371(9):818–827. doi: 10.1056/NEJMoa1311890. [DOI] [PubMed] [Google Scholar]
- 35.Baldasseroni S, Opasich C, Gorini M, Lucci D, Marchionni N, Marini M, et al. Left bundle-branch block is associated with increased 1-year sudden and total mortality rate in 5517 outpatients with congestive heart failure: a report from the Italian network on congestive heart failure. Am Heart J. 2002;143(3):398–405. doi: 10.1067/mhj.2002.121264. [DOI] [PubMed] [Google Scholar]
- 36.Hawkins NM, Petrie MC, Jhund PS, Chalmers GW, Dunn FG, McMurray JJ. Heart failure and chronic obstructive pulmonary disease: diagnostic pitfalls and epidemiology. Eur J Heart Fail. 2009;11(2):130–139. doi: 10.1093/eurjhf/hfn013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ziaeian B, Fonarow GC. Epidemiology and aetiology of heart failure. Nat Rev Cardiol. 2016;13(6):368–378. doi: 10.1038/nrcardio.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vos T, Flaxman AD, Naghavi M, Lozano R, Michaud C, Ezzati M, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. The lancet. 2012;380(9859):2163–2196. doi: 10.1016/S0140-6736(12)61729-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao D, Liu J, Xie W, Qi Y. Cardiovascular risk assessment: a global perspective. Nat Rev Cardiol. 2015;12(5):301. doi: 10.1038/nrcardio.2015.28. [DOI] [PubMed] [Google Scholar]
- 40.Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association scientific statement from the council on clinical cardiology, heart failure and transplantation committee; quality of care and outcomes research and functional genomics and translational biology interdisciplinary working groups; and council on epidemiology and prevention. Circulation. 2006;113(14):1807–1816. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- 41.Maron BJ, Thiene G. Hurst’s hear. McGraw Hill Medical; 2011. [Google Scholar]
- 42.Lip GY, Gibbs CR, Beevers DG. ABC of heart failure: aetiology. BMJ (Clin Res Ed) 2000;320(7227):104–107. doi: 10.1136/bmj.320.7227.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee DS, Pencina MJ, Benjamin EJ, Wang TJ, Levy D, O'Donnell CJ, et al. Association of parental heart failure with risk of heart failure in offspring. N Engl J Med. 2006;355(2):138–147. doi: 10.1056/NEJMoa052948. [DOI] [PubMed] [Google Scholar]
- 44.Lindgren MP, Smith JG, Li X, Sundquist J, Sundquist K, Zöller B. Sibling risk of hospitalization for heart failure—a nationwide study. Int J Cardiol. 2016;223:379–384. doi: 10.1016/j.ijcard.2016.08.067. [DOI] [PubMed] [Google Scholar]
- 45.Towbin JA, Lowe AM, Colan SD, Sleeper LA, Orav EJ, Clunie S, et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA. 2006;296(15):1867–1876. doi: 10.1001/jama.296.15.1867. [DOI] [PubMed] [Google Scholar]
- 46.Monserrat L, Hermida M, Bouzas B, Mosquera I, Mahon N, Peteiro J, et al. Familial dilated cardiomyopathy in patients transplanted for idiopathic dilated cardiomyopathy. Rev Esp Cardiol. 2002;55(7):725–732. doi: 10.1016/s0300-8932(02)76691-8. [DOI] [PubMed] [Google Scholar]
- 47.Jacoby D, McKenna WJ. Genetics of inherited cardiomyopathy. Eur Heart J. 2012;33(3):296–304. doi: 10.1093/eurheartj/ehr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wexler RK, Elton T, Pleister A, Feldman D. Cardiomyopathy: an overview. Am Fam Phys. 2009;79(9):778–784. [PMC free article] [PubMed] [Google Scholar]
- 49.Sisakian H. Cardiomyopathies: evolution of pathogenesis concepts and potential for new therapies. World J Cardiol. 2014;6(6):478–494. doi: 10.4330/wjc.v6.i6.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Davies MJ. The cardiomyopathies: an overview. Heart (Br Cardiac Soc) 2000;83(4):469–474. doi: 10.1136/heart.83.4.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arbustini E, Narula N, Tavazzi L, Serio A, Grasso M, Favalli V, et al. The MOGE (S) classification of cardiomyopathy for clinicians. J Am Coll Cardiol. 2014;64(3):304–318. doi: 10.1016/j.jacc.2014.05.027. [DOI] [PubMed] [Google Scholar]
- 52.Weintraub RG, Semsarian C, Macdonald P. Dilated cardiomyopathy. Lancet. 2017;390(10092):400–414. doi: 10.1016/S0140-6736(16)31713-5. [DOI] [PubMed] [Google Scholar]
- 53.Codd MB, Sugrue DD, Gersh BJ, Melton LJ., III Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in Olmsted County, Minnesota, 1975–1984. Circulation. 1989;80(3):564–572. doi: 10.1161/01.cir.80.3.564. [DOI] [PubMed] [Google Scholar]
- 54.Hershberger RE, Givertz MM, Ho CY, Judge DP, Kantor PF, McBride KL, et al. Genetic evaluation of cardiomyopathy: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG) Genet Med. 2018;20(9):899–909. doi: 10.1038/s41436-018-0039-z. [DOI] [PubMed] [Google Scholar]
- 55.Sen-Chowdhry S, Syrris P, Prasad SK, Hughes SE, Merrifield R, Ward D, et al. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol. 2008;52(25):2175–2187. doi: 10.1016/j.jacc.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 56.Hershberger RE, Cowan J, Morales A, Siegfried JD. Progress with genetic cardiomyopathies: screening, counseling, and testing in dilated, hypertrophic, and arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Heart Fail. 2009;2(3):253–261. doi: 10.1161/CIRCHEARTFAILURE.108.817346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Posafalvi A, Herkert JC, Sinke RJ, Van Den Berg MP, Mogensen J, Jongbloed JD, Van Tintelen JP. Clinical utility gene card for: dilated cardiomyopathy (CMD) Eur J Hum Genet. 2013;21(10):1185–1185. doi: 10.1038/ejhg.2012.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Callis TE, Jensen BC, Weck KE, Willis MS. Evolving molecular diagnostics for familial cardiomyopathies: at the heart of it all. Expert Rev Mol Diagn. 2010;10(3):329–351. doi: 10.1586/erm.10.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Niimura H, Patton KK, McKenna WJ, Soults J, Maron BJ, Seidman JG, Seidman CE. Sarcomere protein gene mutations in hypertrophic cardiomyopathy of the elderly. Circulation. 2002;105(4):446–451. doi: 10.1161/hc0402.102990. [DOI] [PubMed] [Google Scholar]
- 60.Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287(10):1308–1320. doi: 10.1001/jama.287.10.1308. [DOI] [PubMed] [Google Scholar]
- 61.Cirino AL, Ho C (2016) Hypertrophic cardiomyopathy overview. 2008 Aug 5 [updated 2014 Jan 16]. GeneReviews® [Internet]. University of Washington
- 62.Pinto YM, Wilde AA, Van Rijsingen IA, Christiaans I, Deprez RHL, Elliott PM. Clinical utility gene card for: hypertrophic cardiomyopathy (type 1–14) Eur J Hum Genet. 2011;19(8):3–4. doi: 10.1038/ejhg.2010.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gallego-Delgado M, Delgado JF, Brossa-Loidi V, Palomo J, Marzoa-Rivas R, Perez-Villa F, et al. Idiopathic restrictive cardiomyopathy is primarily a genetic disease. J Am Coll Cardiol. 2016;67(25):3021–3023. doi: 10.1016/j.jacc.2016.04.024. [DOI] [PubMed] [Google Scholar]
- 64.Muchtar E, Blauwet LA, Gertz MA. Restrictive cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res. 2017;121(7):819–837. doi: 10.1161/CIRCRESAHA.117.310982. [DOI] [PubMed] [Google Scholar]
- 65.Pereira NL, Grogan M, Dec GW. Spectrum of restrictive and infiltrative cardiomyopathies: part 2 of a 2-part series. J Am Coll Cardiol. 2018;71(10):1149–1166. doi: 10.1016/j.jacc.2018.01.017. [DOI] [PubMed] [Google Scholar]
- 66.Sliwa K, Damasceno A, Mayosi BM. Epidemiology and etiology of cardiomyopathy in Africa. Circulation. 2005;112(23):3577–3583. doi: 10.1161/CIRCULATIONAHA.105.542894. [DOI] [PubMed] [Google Scholar]
- 67.Mocumbi AO, Falase AO. Recent advances in the epidemiology, diagnosis and treatment of endomyocardial fibrosis in Africa. Heart. 2013;99(20):1481–1487. doi: 10.1136/heartjnl-2012-303193. [DOI] [PubMed] [Google Scholar]
- 68.Elasfar AA, Alhabeeb W, Elasfar S. Heart failure in the middle east arab countries: current and future perspectives. J Saudi Heart Assoc. 2020;32(2):236. doi: 10.37616/2212-5043.1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xu Q, Dewey S, Nguyen S, Gomes AV. Malignant and benign mutations in familial cardiomyopathies: insights into mutations linked to complex cardiovascular phenotypes. J Mol Cell Cardiol. 2010;48(5):899–909. doi: 10.1016/j.yjmcc.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 70.Precone V, Krasi G, Guerri G, Madureri A, Piazzani M, Michelini S, Barati S, Maniscalchi T, Bressan S, Bertelli M. Cardiomyopathies. Acta Bio-Med Atenei Parm. 2019;90(10-S):32–43. doi: 10.23750/abm.v90i10-S.8755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Towbin JA. Inherited cardiomyopathies. Circ J. 2014;78:2347–2356. doi: 10.1253/circj.cj-14-0893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Falase AO, Ogah OS. Cardiomyopathies and myocardial disorders in Africa: present status and the way forward. Cardiovasc J Afr. 2012;23(10):552. doi: 10.5830/CVJA-2012-046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Peters S. Advances in the diagnostic management of arrhythmogenic right ventricular dysplasia—cardiomyopathy. Int J Cardiol. 2006;113(1):4–11. doi: 10.1016/j.ijcard.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 74.Peters S, Trümmel M, Meyners W. Prevalence of right ventricular dysplasia-cardiomyopathy in a non-referral hospital. Int J Cardiol. 2004;97(3):499–501. doi: 10.1016/j.ijcard.2003.10.037. [DOI] [PubMed] [Google Scholar]
- 75.Protonotarios A, Anastasakis A, Panagiotakos DB, Antoniades L, Syrris P, Vouliotis A, et al. Arrhythmic risk assessment in genotyped families with arrhythmogenic right ventricular cardiomyopathy. Europace. 2016;18(4):610–616. doi: 10.1093/europace/euv061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Poloni G, De Bortoli M, Calore M, Rampazzo A, Lorenzon A. Arrhythmogenic right ventricular cardiomyopathy: molecular genetics into clinical practice in the era of next-generation sequencing. J Cardiovasc Med. 2016;17(6):399–407. doi: 10.2459/JCM.0000000000000385. [DOI] [PubMed] [Google Scholar]
- 77.McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom-Lundqvist C, Fontaine G, Camerini F. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the working group myocardial and pericardial disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J. 1994;71(3):215. doi: 10.1136/hrt.71.3.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Corrado D, Link MS, Calkins H. Arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2017;376(1):61–72. doi: 10.1056/NEJMra1509267. [DOI] [PubMed] [Google Scholar]
- 79.Van Der Smagt JJ, Van Der Zwaag PA, Van Tintelen JP, Cox MG, Wilde AA, Van Langen IM, et al. Clinical and genetic characterization of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy caused by a plakophilin-2 splice mutation. Cardiology. 2012;123(3):181–189. doi: 10.1159/000342717. [DOI] [PubMed] [Google Scholar]
- 80.Schulze-Bahr E, Klaassen S, Abdul-Khaliq H, Schunkert H. Molecular diagnostics of cardiovascular diseases. Expert consensus statement by the German Cardiac Society (DGK) and the German Society of Pediatric Cardiology (DGPK) Cardiology. 2015;9:213–243. [Google Scholar]
- 81.Pinamonti B, Brun F, Mestroni L, Sinagra G. Arrhythmogenic right ventricular cardiomyopathy: from genetics to diagnostic and therapeutic challenges. World J Cardiol. 2014;6(12):1234. doi: 10.4330/wjc.v6.i12.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Van Rijsingen IA, Arbustini E, Elliott PM, Mogensen J, Hermans-van Ast JF, Van Der Kooi AJ, et al. Risk factors for malignant ventricular arrhythmias in lamin A/C mutation carriers: a European cohort study. J Am Coll Cardiol. 2012;59(5):493–500. doi: 10.1016/j.jacc.2011.08.078. [DOI] [PubMed] [Google Scholar]
- 83.McNally EM, Barefield DY, Puckelwartz MJ. The genetic landscape of cardiomyopathy and its role in heart failure. Cell Metab. 2015;21(2):174–182. doi: 10.1016/j.cmet.2015.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Epstein ND, Cohn GM, Cyran F, Fananapazir L. Differences in clinical expression of hypertrophic cardiomyopathy associated with two distinct mutations in the beta-myosin heavy chain gene. A 908Leu––Val mutation and a 403Arg––Gln mutation. Circulation. 1992;86(2):345–352. doi: 10.1161/01.cir.86.2.345. [DOI] [PubMed] [Google Scholar]
- 85.Oldfors A. Hereditary myosin myopathies. Neuromuscul Disord. 2007;17(5):355–367. doi: 10.1016/j.nmd.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 86.Walsh R, Rutland C, Thomas R, Loughna S. Cardiomyopathy: a systematic review of disease-causing mutations in myosin heavy chain 7 and their phenotypic manifestations. Cardiology. 2010;115(1):49–60. doi: 10.1159/000252808. [DOI] [PubMed] [Google Scholar]
- 87.Perrot A, Schmidt-Traub H, Hoffmann B, Prager M, Bit-Avragim N, Rudenko RI, et al. Prevalence of cardiac beta-myosin heavy chain gene mutations in patients with hypertrophic cardiomyopathy. J Mol Med. 2005;83(6):468–477. doi: 10.1007/s00109-005-0635-7. [DOI] [PubMed] [Google Scholar]
- 88.Stockler S, Corvera S, Lambright D, Fogarty K, Nosova E, Leonard D, et al. Single point mutation in Rabenosyn-5 in a female with intractable seizures and evidence of defective endocytotic trafficking. Orphanet J Rare Dis. 2014;9(1):1–11. doi: 10.1186/s13023-014-0141-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fan XP, Yang ZW, Feng XL, Yang FH, Xiao B, Liang Y. Mutation analysis of beta myosin heavy chain gene in hypertrophic cardiomyopathy families. Zhonghua yi xue yi chuan xue za zhi= Zhonghua yixue yichuanxue zazhi= Chin J Med Genet. 2011;28(4):387–392. doi: 10.3760/cma.j.issn.1003-9406.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 90.Liu HT, Ji FF, Wei L, Zuo AJ, Gao YX, Qi L, et al. Screening of MYH7 gene mutation sites in hypertrophic cardiomyopathy and its significance. Chin Med J. 2019;132(23):2835. doi: 10.1097/CM9.0000000000000428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schmitt JP, Debold EP, Ahmad F, Armstrong A, Frederico A, Conner DA, et al. Cardiac myosin missense mutations cause dilated cardiomyopathy in mouse models and depress molecular motor function. Proc Natl Acad Sci. 2006;103(39):14525–14530. doi: 10.1073/pnas.0606383103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bloemink M, Deacon J, Langer S, Vera C, Combs A, Leinwand L, Geeves MA. The hypertrophic cardiomyopathy myosin mutation R453C alters ATP binding and hydrolysis of human cardiac β-myosin. J Biol Chem. 2014;289(8):5158–5167. doi: 10.1074/jbc.M113.511204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Garcıa-Castro M, Reguero JR, Batalla A, Dıaz-Molina B, Gonzalez P, Alvarez V, et al. Hypertrophic cardiomyopathy: low frequency of mutations in the β-myosin heavy chain (MYH7) and cardiac troponin t (TNNT2) genes among Spanish patients. Clin Chem. 2003;49(8):1279–1285. doi: 10.1373/49.8.1279. [DOI] [PubMed] [Google Scholar]
- 94.Gomes AV, Barnes JA, Harada K, Potter JD. Role of troponin T in disease. Mol Cell Biochem. 2004;263(1):115–129. doi: 10.1023/B:MCBI.0000041853.20588.a0. [DOI] [PubMed] [Google Scholar]
- 95.Mogensen J, Kubo T, Duque M, Uribe W, Shaw A, Murphy R, et al. Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. J Clin Investig. 2003;111(2):209–216. doi: 10.1172/JCI16336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Thierfelder L, Watkins H, MacRae C, Lamas R, McKenna W, Vosberg HP, et al. α-Tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell. 1994;77(5):701–712. doi: 10.1016/0092-8674(94)90054-x. [DOI] [PubMed] [Google Scholar]
- 97.Townsend PJ, Farza H, Macgeoch C, Spurr NK, Wade R, Gahlmann R, et al. Human cardiac troponin T: identification of fetal isoforms and assignment of the TNNT2 locus to chromosome 1q. Genomics. 1994;21(2):311–316. doi: 10.1006/geno.1994.1271. [DOI] [PubMed] [Google Scholar]
- 98.Palm T, Graboski S, Hitchcock-DeGregori SE, Greenfield NJ. Disease-causing mutations in cardiac troponin T: identification of a critical tropomyosin-binding region. Biophys J . 2001;81(5):2827–2837. doi: 10.1016/S0006-3495(01)75924-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sehnert AJ, Huq A, Weinstein BM, Walker C, Fishman M, Stainier DY. Cardiac troponin T is essential in sarcomere assembly and cardiac contractility. Nat Genet. 2002;31(1):106–110. doi: 10.1038/ng875. [DOI] [PubMed] [Google Scholar]
- 100.Chang AN, Parvatiyar MS, Potter JD. Troponin and cardiomyopathy. Biochem Biophys Res Commun. 2008;369(1):74–81. doi: 10.1016/j.bbrc.2007.12.081. [DOI] [PubMed] [Google Scholar]
- 101.Hershberger RE, Pinto JR, Parks SB, Kushner JD, Li D, Ludwigsen S, et al. Clinical and functional characterization of TNNT2 mutations identified in patients with dilated cardiomyopathy. Circ Cardiovasc Genet. 2009;2(4):306–313. doi: 10.1161/CIRCGENETICS.108.846733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li X, Wang H, Luo R, Gu H, Zhang C, Zhang Y, et al. TNNT2 gene polymorphisms are associated with susceptibility to idiopathic dilated cardiomyopathy in the Han Chinese population. BioMed Res Int. 2013;2013(2013):201372. doi: 10.1155/2013/201372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mirza M, Marston S, Willott R, Ashley C, Mogensen J, McKenna W, et al. Dilated cardiomyopathy mutations in three thin filament regulatory proteins result in a common functional phenotype. J Biol Chem. 2005;280(31):28498–28506. doi: 10.1074/jbc.M412281200. [DOI] [PubMed] [Google Scholar]
- 104.Villard E, Perret C, Gary F, Proust C, Dilanian G, Hengstenberg C, et al. A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Eur Heart J. 2011;32(9):1065–1076. doi: 10.1093/eurheartj/ehr105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kimura A. Contribution of genetic factors to the pathogenesis of dilated cardiomyopathy-the cause of dilated cardiomyopathy: acquired or genetic? (Genetic-side) Circ J. 2011;75:1756–1765. doi: 10.1253/circj.cj-11-0368. [DOI] [PubMed] [Google Scholar]
- 106.Sadayappan S, Osinska H, Klevitsky R, Lorenz JN, Sargent M, Molkentin JD, et al. Cardiac myosin-binding protein C phosphorylation is cardioprotective. Proc Natl Acad Sci. 2006;103(45):16918–16923. doi: 10.1073/pnas.0607069103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gruen M, Gautel M. Mutations in β-myosin S2 that cause familial hypertrophic cardiomyopathy (FHC) abolish the interaction with the regulatory domain of myosin-binding protein-C. J Mol Biol. 1999;286(3):933–949. doi: 10.1006/jmbi.1998.2522. [DOI] [PubMed] [Google Scholar]
- 108.Carrier L, Mearini G, Stathopoulou K, Cuello F. Cardiac myosin-binding protein C (MYBPC3) in cardiac pathophysiology. Gene. 2015;573(2):188–197. doi: 10.1016/j.gene.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dorsch LM, Schuldt M, Knežević D, Wiersma M, Kuster DW, van der Velden J, Brundel BJ. Untying the knot: protein quality control in inherited cardiomyopathies. Pflügers Archiv-Eur J Physiol. 2019;471(5):795–806. doi: 10.1007/s00424-018-2194-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fourey D, Care M, Siminovitch KA, Weissler-Snir A, Hindieh W, Chan RH, et al. Prevalence and clinical implication of double mutations in hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2017;10(2):e001685. doi: 10.1161/CIRCGENETICS.116.001685. [DOI] [PubMed] [Google Scholar]
- 111.Mohiddin SA, Begley DA, McLam E, Cardoso JP, Winkler JB, Sellers JR, Fananapazir L. Utility of genetic screening in hypertrophic cardiomyopathy: prevalence and significance of novel and double (homozygous and heterozygous) β-myosin mutations. Genet Test. 2003;7(1):21–27. doi: 10.1089/109065703321560895. [DOI] [PubMed] [Google Scholar]
- 112.Maron BJ, Maron MS, Semsarian C. Double or compound sarcomere mutations in hypertrophic cardiomyopathy: a potential link to sudden death in the absence of conventional risk factors. Heart Rhythm. 2012;9(1):57–63. doi: 10.1016/j.hrthm.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 113.Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, Seidman JG. A molecular basis for familial hypertrophic cardiomyopathy: a β cardiac myosin heavy chain gene missense mutation. Cell. 1990;62(5):999–1006. doi: 10.1016/0092-8674(90)90274-i. [DOI] [PubMed] [Google Scholar]
- 114.Kissopoulou A, Trinks C, Green A, Karlsson JE, Jonasson J, Gunnarsson C. Homozygous missense MYBPC3 Pro873His mutation associated with increased risk for heart failure development in hypertrophic cardiomyopathy. ESC Heart Fail. 2018;5(4):716–723. doi: 10.1002/ehf2.12288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Julien O, Mercier P, Allen CN, Fisette O, Ramos CH, Lagüe P, et al. Is there nascent structure in the intrinsically disordered region of troponin I? Proteins Struct Funct Bioinform. 2011;79(4):1240–1250. doi: 10.1002/prot.22959. [DOI] [PubMed] [Google Scholar]
- 116.Galinska A, Hatch V, Craig R, Murphy AM, Van Eyk JE, Wang CLA, et al. Novelty and significance. Circ Res. 2010;106(4):705–711. doi: 10.1161/CIRCRESAHA.109.210047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Li Y, Charles PYJ, Nan C, Pinto JR, Wang Y, Liang J, et al. Correcting diastolic dysfunction by Ca2+ desensitizing troponin in a transgenic mouse model of restrictive cardiomyopathy. J Mol Cell Cardiol. 2010;49(3):402–411. doi: 10.1016/j.yjmcc.2010.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mogensen J, Hey T, Lambrecht S. A systematic review of phenotypic features associated with cardiac troponin I mutations in hereditary cardiomyopathies. Can J Cardiol. 2015;31(11):1377–1385. doi: 10.1016/j.cjca.2015.06.015. [DOI] [PubMed] [Google Scholar]
- 119.van den Wijngaard A, Volders P, Van Tintelen JP, Jongbloed JD, Van Den Berg MP, Deprez RL, et al. Recurrent and founder mutations in the Netherlands: cardiac Troponin I (TNNI3) gene mutations as a cause of severe forms of hypertrophic and restrictive cardiomyopathy. Neth Hear J. 2011;19(7):344–351. doi: 10.1007/s12471-011-0135-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lippi G, Targher G, Franchini M, Plebani M. Genetic and biochemical heterogeneity of cardiac troponins: clinical and laboratory implications. Clin Chem Lab Med. 2009;47(10):1183–1194. doi: 10.1515/CCLM.2009.322. [DOI] [PubMed] [Google Scholar]
- 121.England J, Granados-Riveron J, Polo-Parada L, Kuriakose D, Moore C, Brook JD, et al. Tropomyosin 1: multiple roles in the developing heart and in the formation of congenital heart defects. J Mol Cell Cardiol. 2017;106:1–13. doi: 10.1016/j.yjmcc.2017.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jagatheesan G, Rajan S, Ahmed RP, Petrashevskaya N, Boivin G, Arteaga GM, et al. Striated muscle tropomyosin isoforms differentially regulate cardiac performance and myofilament calcium sensitivity. J Muscle Res Cell Motil. 2010;31(3):227–239. doi: 10.1007/s10974-010-9228-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dube S, Panebianco L, Matoq AA, Chionuma HN, Denz CR, Poiesz BJ, Dube DK. Expression of TPM1, a novel sarcomeric isoform of the TPM1 gene, in mouse heart and skeletal muscle. Mol Biol Int. 2014;2014:1–9. doi: 10.1155/2014/896068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang J, Sanger JM, Kang S, Thurston H, Abbott LZ, Dube DK, Sanger JW. Ectopic expression and dynamics of TPM1α and TPM1κ in myofibrils of avian myotubes. Cell Motil Cytoskelet. 2007;64(10):767–776. doi: 10.1002/cm.20221. [DOI] [PubMed] [Google Scholar]
- 125.Lohmeier-Vogel EM, Heeley DH. Biochemical comparison of Tpm1. 1 (α) and Tpm2. 2 (β) tropomyosins from rabbit skeletal muscle. Biochemistry. 2016;55(9):1418–1427. doi: 10.1021/acs.biochem.5b01140. [DOI] [PubMed] [Google Scholar]
- 126.Olson TM, Kishimoto NY, Whitby FG, Michels VV. Mutations that alter the surface charge of alpha-tropomyosin are associated with dilated cardiomyopathy. J Mol Cell Cardiol. 2001;33(4):723–732. doi: 10.1006/jmcc.2000.1339. [DOI] [PubMed] [Google Scholar]
- 127.Redwood C, Robinson P. Alpha-tropomyosin mutations in inherited cardiomyopathies. J Muscle Res Cell Motil. 2013;34(3–4):285–294. doi: 10.1007/s10974-013-9358-5. [DOI] [PubMed] [Google Scholar]
- 128.Yao Q, Zhang W, Zhang T. Association of single nucleotide polymorphisms in the 3′ UTR region of TPM1 gene with dilated cardiomyopathy: a case-control study. Medicine. 2019;98(44):e17710. doi: 10.1097/MD.0000000000017710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lin F, Worman HJ. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J Biol Chem. 1993;268(22):16321–16326. [PubMed] [Google Scholar]
- 130.Harborth J, Elbashir SM, Bechert K, Tuschl T, Weber K. Identification of essential genes in cultured mammalian cells using small interfering RNAs. J Cell Sci. 2001;114(24):4557–4565. doi: 10.1242/jcs.114.24.4557. [DOI] [PubMed] [Google Scholar]
- 131.Taylor MR, Fain PR, Sinagra G, Robinson ML, Robertson AD, Carniel E, et al. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J Am Coll Cardiol. 2003;41(5):771–780. doi: 10.1016/s0735-1097(02)02954-6. [DOI] [PubMed] [Google Scholar]
- 132.Parks SB, Kushner JD, Nauman D, Burgess D, Ludwigsen S, Peterson A, et al. Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Am Heart J. 2008;156(1):161–169. doi: 10.1016/j.ahj.2008.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction system disease. N Engl J Med. 1999;341(23):1715–1724. doi: 10.1056/NEJM199912023412302. [DOI] [PubMed] [Google Scholar]
- 134.Perovanovic J, Dell’Orso S, Gnochi VF, Jaiswal JK, Sartorelli V, Vigouroux C, et al. Laminopathies disrupt epigenomic developmental programs and cell fate. Sci Transl Med. 2016;8(335):335ra58. doi: 10.1126/scitranslmed.aad4991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Markandeya YS, Tsubouchi T, Hacker TA, Wolff MR, Belardinelli L, Balijepalli RC. Inhibition of late sodium current attenuates ionic arrhythmia mechanism in ventricular myocytes expressing LaminA-N195K mutation. Heart Rhythm. 2016;13(11):2228–2236. doi: 10.1016/j.hrthm.2016.08.007. [DOI] [PubMed] [Google Scholar]
- 136.Gerbino A, Procino G, Svelto M, Carmosino M. Role of lamin A/C gene mutations in the signaling defects leading to cardiomyopathies. Front Physiol. 2018;9:1356. doi: 10.3389/fphys.2018.01356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chen SN, Lombardi R, Karmouch J, Tsai JY, Czernuszewicz G, Taylor MR, et al. DNA damage response/TP53 pathway is activated and contributes to the pathogenesis of dilated cardiomyopathy associated with LMNA (Lamin A/C) mutations. Circ Res. 2019;124(6):856–873. doi: 10.1161/CIRCRESAHA.118.314238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Dauer WT, Worman HJ. The nuclear envelope as a signaling node in development and disease. Dev Cell. 2009;17(5):626–638. doi: 10.1016/j.devcel.2009.10.016. [DOI] [PubMed] [Google Scholar]
- 139.Cheedipudi SM, Matkovich SJ, Coarfa C, Hu X, Robertson MJ, Sweet M, et al. Genomic reorganization of lamin-associated domains in cardiac myocytes is associated with differential gene expression and DNA methylation in human dilated cardiomyopathy. Circ Res. 2019;124(8):1198–1213. doi: 10.1161/CIRCRESAHA.118.314177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Muchir A, Pavlidis P, Decostre V, Herron AJ, Arimura T, Bonne G, Worman HJ. Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J Clin Investig. 2007;117(5):1282–1293. doi: 10.1172/JCI29042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wu W, Muchir A, Shan J, Bonne G, Worman HJ. Mitogen-activated protein kinase inhibitors improve heart function and prevent fibrosis in cardiomyopathy caused by mutation in lamin A/C gene. Circulation. 2011;123(1):53–61. doi: 10.1161/CIRCULATIONAHA.110.970673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Choi JC, Wu W, Muchir A, Iwata S, Homma S, Worman HJ. Dual specificity phosphatase 4 mediates cardiomyopathy caused by lamin A/C (LMNA) gene mutation. J Biol Chem. 2012;287(48):40513–40524. doi: 10.1074/jbc.M112.404541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ramos FJ, Chen SC, Garelick MG, Dai DF, Liao CY, Schreiber KH, et al. Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci Transl Med. 2012;4(144):144ra103. doi: 10.1126/scitranslmed.3003802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mertens C, Kuhn C, Franke WW. Plakophilins 2a and 2b: constitutive proteins of dual location in the karyoplasm and the desmosomal plaque. J Cell Biol. 1996;135(4):1009–1025. doi: 10.1083/jcb.135.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bass-Zubek AE, Hobbs RP, Amargo EV, Garcia NJ, Hsieh SN, Chen X, et al. Plakophilin 2: a critical scaffold for PKCα that regulates intercellular junction assembly. J Cell Biol. 2008;181(4):605–613. doi: 10.1083/jcb.200712133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA, et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet. 2004;36(11):1162–1164. doi: 10.1038/ng1461. [DOI] [PubMed] [Google Scholar]
- 147.Syrris P, Ward D, Asimaki A, Evans A, Sen-Chowdhry S, Hughes SE, McKenna WJ. Desmoglein-2 mutations in arrhythmogenic right ventricular cardiomyopathy: a genotype-phenotype characterization of familial disease. Eur Heart J. 2007;28(5):581–588. doi: 10.1093/eurheartj/ehl380. [DOI] [PubMed] [Google Scholar]
- 148.Watkins DA, Hendricks N, Shaboodien G, Mbele M, Parker M, Vezi BZ, et al. Clinical features, survival experience, and profile of plakophylin-2 gene mutations in participants of the arrhythmogenic right ventricular cardiomyopathy registry of South Africa. Heart Rhythm. 2009;6(11):S10–S17. doi: 10.1016/j.hrthm.2009.08.018. [DOI] [PubMed] [Google Scholar]
- 149.Svensson A, Åström-Aneq M, Widlund KF, Fluur C, Green A, Rehnberg M, Gunnarsson C. Arrhythmogenic Right Ventricular Cardiomyopathy—4 Swedish families with an associated PKP2 c.2146–1G>C variant. Am J Cardiovasc Dis. 2016;6(2):55–65. [PMC free article] [PubMed] [Google Scholar]
- 150.Cerrone M, Montnach J, Lin X, Zhao YT, Zhang M, Agullo-Pascual E, et al. Plakophilin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm. Nat Commun. 2017;8(1):1–16. doi: 10.1038/s41467-017-00127-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Flemming S, Luissint AC, Kusters DH, Raya-Sandino A, Fan S, Zhou DW, et al. Desmocollin-2 promotes intestinal mucosal repair by controlling integrin-dependent cell adhesion and migration. Mol Biol Cell. 2020;31(6):407–418. doi: 10.1091/mbc.E19-12-0692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Evander M, Frazer IH, Payne E, Qi YM, Hengst K, McMillan NA. Identification of the alpha6 integrin as a candidate receptor for papillomaviruses. J Virol. 1997;71(3):2449. doi: 10.1128/jvi.71.3.2449-2456.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lorenzon A, Pilichou K, Rigato I, Vazza G, De Bortoli M, Calore M, et al. Homozygous desmocollin-2 mutations and arrhythmogenic cardiomyopathy. Am J Cardiol. 2015;116(8):1245–1251. doi: 10.1016/j.amjcard.2015.07.037. [DOI] [PubMed] [Google Scholar]
- 154.Liu JS, Fan LL, Li JJ, Xiang R. Whole-exome sequencing identifies a novel mutation of desmocollin 2 in a Chinese family with arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2017;119(9):1485–1489. doi: 10.1016/j.amjcard.2017.01.011. [DOI] [PubMed] [Google Scholar]
- 155.Gehmlich K, Syrris P, Peskett E, Evans A, Ehler E, Asimaki A, et al. Mechanistic insights into arrhythmogenic right ventricular cardiomyopathy caused by desmocollin-2 mutations. Cardiovasc Res. 2011;90(1):77–87. doi: 10.1093/cvr/cvq353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Heuser A, Plovie ER, Ellinor PT, Grossmann KS, Shin JT, Wichter T, et al. Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2006;79(6):1081–1088. doi: 10.1086/509044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Beffagna G, De Bortoli M, Nava A, Salamon M, Lorenzon A, Zaccolo M, et al. Missense mutations in desmocollin-2 N-terminus, associated with arrhythmogenic right ventricular cardiomyopathy, affect intracellular localization of desmocollin-2 in vitro. BMC Med Genet. 2007;8(1):1–10. doi: 10.1186/1471-2350-8-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Syrris P, Ward D, Evans A, Asimaki A, Gandjbakhch E, Sen-Chowdhry S, McKenna WJ. Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. Am J Hum Genet. 2006;79(5):978–984. doi: 10.1086/509122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.De Bortoli M, Beffagna G, Bauce B, Lorenzon A, Smaniotto G, Rigato I, Calore M, Li Mura IE, Basso C, Thiene G, Lanfranchi G, Danieli GA, Nava A, Rampazzo A. The p.A897KfsX4 frameshift variation in desmocollin-2 is not a causative mutation in arrhythmogenic right ventricular cardiomyopathy. Eur J Hum Genet. 2010;18(7):776–782. doi: 10.1038/ejhg.2010.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hermida A, Fressart V, Hidden-Lucet F, Donal E, Probst V, Deharo JC, et al. High risk of heart failure associated with desmoglein-2 mutations compared to plakophilin-2 mutations in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Eur J Heart Fail. 2019;21(6):792–800. doi: 10.1002/ejhf.1423. [DOI] [PubMed] [Google Scholar]
- 161.Zhou G, Yang L, Gray A, Srivastava AK, Li C, Zhang G, Cui T. The role of desmosomes in carcinogenesis. Onco Targets Ther. 2017;10:4059. doi: 10.2147/OTT.S136367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Maron BJ. The 2006 American Heart Association classification of cardiomyopathies is the gold standard. Circ Heart Fail. 2008;1(1):72–76. doi: 10.1161/CIRCHEARTFAILURE.108.770826. [DOI] [PubMed] [Google Scholar]
- 163.Garcia-Pavia P, Syrris P, Salas C, Evans A, Mirelis JG, Cobo-Marcos M, et al. Desmosomal protein gene mutations in patients with idiopathic dilated cardiomyopathy undergoing cardiac transplantation: a clinicopathological study. Heart. 2011;97(21):1744–1752. doi: 10.1136/hrt.2011.227967. [DOI] [PubMed] [Google Scholar]
- 164.Pugh TJ, Kelly MA, Gowrisankar S, Hynes E, Seidman MA, Baxter SM, et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med. 2014;16(8):601–608. doi: 10.1038/gim.2013.204. [DOI] [PubMed] [Google Scholar]
- 165.Posch MG, Posch MJ, Geier C, Erdmann B, Mueller W, Richter A, et al. A missense variant in desmoglein-2 predisposes to dilated cardiomyopathy. Mol Genet Metab. 2008;95(1–2):74–80. doi: 10.1016/j.ymgme.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 166.Dieding M, Debus JD, Kerkhoff R, Gaertner-Rommel A, Walhorn V, Milting H, Anselmetti D. Arrhythmogenic cardiomyopathy related DSG2 mutations affect desmosomal cadherin binding kinetics. Sci Rep. 2017;7(1):1–9. doi: 10.1038/s41598-017-13737-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ohno S, Nagaoka I, Fukuyama M, Kimura H, Itoh H, Makiyama T, et al. Age-dependent clinical and genetic characteristics in Japanese patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circ J. 2013;77(6):1534–1542. doi: 10.1253/circj.cj-12-1446. [DOI] [PubMed] [Google Scholar]
- 168.Bauce B, Nava A, Beffagna G, Basso C, Lorenzon A, Smaniotto G, et al. Multiple mutations in desmosomal proteins encoding genes in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart Rhythm. 2010;7(1):22–29. doi: 10.1016/j.hrthm.2009.09.070. [DOI] [PubMed] [Google Scholar]
- 169.Bhonsale A, Groeneweg JA, James CA, Dooijes D, Tichnell C, Jongbloed JD, et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur Heart J. 2015;36(14):847–855. doi: 10.1093/eurheartj/ehu509. [DOI] [PubMed] [Google Scholar]
- 170.Sen-Chowdhry S, Syrris P, Ward D, Asimaki A, Sevdalis E, McKenna WJ. Clinical perspective. Circulation. 2007;115(13):1710–1720. doi: 10.1161/CIRCULATIONAHA.106.660241. [DOI] [PubMed] [Google Scholar]
- 171.Green KJ, Parry DA, Steinert PM, Virata ML, Wagner RM, Angst BD, Nilles LA. Structure of the human desmoplakins. Implications for function in the desmosomal plaque. J Biol Chem. 1990;265(5):2603–2612. [PubMed] [Google Scholar]
- 172.Green KJ, Stappenbeck TS, Parry DA, Virata MLA. Structure of desmoplakin and its association with intermediate filaments. J Dermatol. 1992;19(11):765–769. doi: 10.1111/j.1346-8138.1992.tb03777.x. [DOI] [PubMed] [Google Scholar]
- 173.O'Keefe EJ, Erickson HP, Bennett V. Desmoplakin I and desmoplakin II: purification and characterization. J Biol Chem. 1989;264(14):8310–8318. [PubMed] [Google Scholar]
- 174.Keplinger JD, Landstrom AP, Salisbury BA, Callis TE, Pollevick GD, Tester DJ, et al. Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia—associated mutations from background genetic noise. J Am Coll Cardiol. 2011;57(23):2317–2327. doi: 10.1016/j.jacc.2010.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Alcalai R, Metzger S, Rosenheck S, Meiner V, Chajek-Shaul T. A recessive mutation in desmoplakin causes arrhythmogenic right ventricular dysplasia, skin disorder, and woolly hair. J Am Coll Cardiol. 2003;42(2):319–327. doi: 10.1016/s0735-1097(03)00628-4. [DOI] [PubMed] [Google Scholar]
- 176.Armstrong DKB, McKenna KE, Purkis PE, Green KJ, Eady RA, Leigh IM, Hughes AE. Haploinsufficiency of desmoplakin causes a striate subtype of palmoplantar keratoderma. Hum Mol Genet. 1999;8(1):143–148. doi: 10.1093/hmg/8.1.143. [DOI] [PubMed] [Google Scholar]
- 177.Whittock NV, Ashton GH, Dopping-Hepenstal PJ, Gratian MJ, Keane FM, Eady RA, McGrath JA. Striate palmoplantar keratoderma resulting from desmoplakin haploinsufficiency. J Investig Dermatol. 1999;113(6):940–946. doi: 10.1046/j.1523-1747.1999.00783.x. [DOI] [PubMed] [Google Scholar]
- 178.Rampazzo A, Nava A, Malacrida S, Beffagna G, Bauce B, Rossi V, et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2002;71(5):1200–1206. doi: 10.1086/344208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Ng R, Manring H, Papoutsidakis N, Albertelli T, Tsai N, See CJ, Li X, Park J, Stevens TL, Bobbili PJ, Riaz M, Ren Y, Stoddard CE, Janssen PM, Bunch TJ, Hall SP, Lo YC, Jacoby DL, Qyang Y, Wright N, et al. Patient mutations linked to arrhythmogenic cardiomyopathy enhance calpain-mediated desmoplakin degradation. JCI Insight. 2019;5(14):e128643. doi: 10.1172/jci.insight.128643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Te Riele AS, Agullo-Pascual E, James CA, Leo-Macias A, Cerrone M, Zhang M, et al. Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non-canonical mechanisms for disease pathogenesis. Cardiovasc Res. 2017;113(1):102–111. doi: 10.1093/cvr/cvw234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Xiong Q, Cao Q, Zhou Q, Xie J, Shen Y, Wan R, et al. Arrhythmogenic cardiomyopathy in a patient with a rare loss-of-function KCNQ 1 mutation. J Am Heart Assoc. 2015;4(1):e001526. doi: 10.1161/JAHA.114.001526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Asimaki A, Syrris P, Wichter T, Matthias P, Saffitz JE, McKenna WJ. A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2007;81(5):964–973. doi: 10.1086/521633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Asimaki A, Protonotarios A, James CA, Chelko SP, Tichnell C, Murray B, et al. Characterizing the molecular pathology of arrhythmogenic cardiomyopathy inpatient buccal mucosa cells. Circ Arrhythm Electrophysiol. 2016;9(2):e003688. doi: 10.1161/CIRCEP.115.003688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Cabral RM, Liu L, Hogan C, Dopping-Hepenstal PJ, Winik BC, Asia RA, et al. Homozygous mutations in the 5′ region of the JUP gene result in cutaneous disease but normal heart development in children. J Investig Dermatol. 2010;130(6):1543–1550. doi: 10.1038/jid.2010.7. [DOI] [PubMed] [Google Scholar]
- 185.Pigors M, Kiritsi D, Krümpelmann S, Wagner N, He Y, Podda M, et al. Lack of plakoglobin leads to lethal congenital epidermolysis bullosa: a novel clinico-genetic entity. Hum Mol Genet. 2011;20(9):1811–1819. doi: 10.1093/hmg/ddr064. [DOI] [PubMed] [Google Scholar]
- 186.Christensen AH, Benn M, Bundgaard H, Tybjærg-Hansen A, Haunso S, Svendsen JH. Wide spectrum of desmosomal mutations in Danish patients with arrhythmogenic right ventricular cardiomyopathy. J Med Genet. 2010;47(11):736–744. doi: 10.1136/jmg.2010.077891. [DOI] [PubMed] [Google Scholar]
- 187.Liu L, Chen C, Li Y, Yu R. Whole-exome sequencing identified a de novo mutation of junction plakoglobin (p. R577C) in a Chinese patient with arrhythmogenic right ventricular cardiomyopathy. BioMed Res Int. 2019;2019:9103860. doi: 10.1155/2019/9103860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Vahidnezhad H, Youssefian L, Faghankhani M, Mozafari N, Saeidian AH, Niaziorimi F, et al. Arrhythmogenic right ventricular cardiomyopathy in patients with biallelic JUP-associated skin fragility. Sci Rep. 2020;10(1):1–10. doi: 10.1038/s41598-020-78344-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Bang ML, Centner T, Fornoff F, Geach AJ, Gotthardt M, McNabb M, et al. The complete gene sequence of titin, expression of an unusual≈ 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ Res. 2001;89(11):1065–1072. doi: 10.1161/hh2301.100981. [DOI] [PubMed] [Google Scholar]
- 190.Savarese M, Sarparanta J, Vihola A, Udd B, Hackman P. Increasing role of titin mutations in neuromuscular disorders. J Neuromuscul Dis. 2016;3(3):293–308. doi: 10.3233/JND-160158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Savarese M, Vihola A, Oates EC, Barresi R, Fiorillo C, Tasca G, et al. Genotype–phenotype correlations in recessive titinopathies. Genet Med. 2020;22(12):2029–2040. doi: 10.1038/s41436-020-0914-2. [DOI] [PubMed] [Google Scholar]
- 192.Akhtar MM, Lorenzini M, Cicerchia M, Ochoa JP, Hey TM, Sabater Molina M, et al. Clinical phenotypes and prognosis of dilated cardiomyopathy caused by truncating variants in the TTN gene. Circ Heart Fail. 2020;13(10):e006832. doi: 10.1161/CIRCHEARTFAILURE.119.006832. [DOI] [PubMed] [Google Scholar]
- 193.Vikhorev PG, Vikhoreva NN, Yeung W, Li A, Lal S, Dos Remedios CG, et al. Titin-truncating mutations associated with dilated cardiomyopathy alter length-dependent activation and its modulation via phosphorylation. Cardiovasc Res. 2020;118:241–253. doi: 10.1093/cvr/cvaa316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Fang HJ, Liu BP. Prevalence of TTN mutations in patients with dilated cardiomyopathy. Herz. 2019;45:1–8. doi: 10.1007/s00059-019-4825-4. [DOI] [PubMed] [Google Scholar]
- 195.Savarese M, Sarparanta J, Vihola A, Jonson PH, Johari M, Rusanen S, et al. Panorama of the distal myopathies. Acta Myol. 2020;39(4):245. doi: 10.36185/2532-1900-028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Green EM, Wakimoto H, Anderson RL, Evanchik MJ, Gorham JM, Harrison BC, et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science. 2016;351(6273):617–621. doi: 10.1126/science.aad3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Alfares AA, Kelly MA, Mcdermott G, Funke BH, Lebo MS, Baxter SB, et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med. 2015;17(11):880–888. doi: 10.1038/gim.2014.205. [DOI] [PubMed] [Google Scholar]
- 198.GeneTests (2016) https://www.genetests.org. Accessed 17 Oct 2016
- 199.GTR: Genetic Testing Registry. National Institutes of Health. https://www.ncbi.nlm.nih.gov/gtr/. Accessed 1 Aug 2016
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.