

Abstract
Platelets are anucleate blood cells that are shed from megakaryocytes (MKs) into the bloodstream to maintain hemostasis and promote wound healing after vascular injury. To carry out their functions, platelets become activated and release bioactive substances from their secretory granules. As alpha granules (αGs) in resting platelets store proteins and release them only after activation, the packaging of proteins into αGs is an attractive strategy to deliver therapeutic proteins. Here, we propose an adjustable model for targeting transgenic proteins to platelet αGs using third-generation self-inactivating lentiviral vectors. The vectors express from the murine platelet factor 4 promoter (mPf4P), restricting transgene expression to the MK lineage. For the delivery and retention of expressed proteins in αGs, proteins are fused to short peptide sorting signals derived from the human cytokine RANTES or from the transmembrane protein P-selectin. We demonstrate effective targeting of GFP to αGs of murine and human in vitro-differentiated MKs and murine platelets in vivo. Furthermore, interferon-α (IFNα), as a potentially therapeutic cytokine, was successfully delivered to and stored in murine platelets in vivo, was released after activation, and inhibited virus replication in vitro. Our vectors create possibilities for numerous applications in cell therapy utilizing platelets as carriers of therapeutic proteins.
Keywords: platelet-directed gene therapy, platelet alpha granules, sorting signals, lentiviral vectors, bone marrow transplantation model, alpha granular targeting
Graphical Abstract
We propose an adaptable platform for targeting proteins to platelet alpha granules based on lentiviral vectors to utilize platelets as vehicles to store, hide, and specifically deliver therapeutic proteins.
Introduction
Platelets are small anucleate blood cells that are released from megakaryocytes (MKs) in the bone marrow (BM). They circulate in a resting state in the blood, carrying numerous bioactive substances in their granules, which they release after activation. Platelet activation is triggered by the environment, such as by vessel injuries or pathogens, and mediated via specific platelet surface receptors, including the glycoprotein (GP) IIb/IIIa and GPIb/IX/V complexes or the thrombin protease-activated receptor (PAR). This unique physiology makes platelets attractive cell therapy targets. Researchers have studied the use of platelets and of platelet membrane-coated nanoparticles as drug delivery tools (e.g., to target tumors or cardiovascular disease).1, 2, 3, 4 Platelet-directed gene therapy has been explored for the delivery of coagulation factors to treat hemophilia.5 In this case, gene transfer is done in the patient’s autologous hematopoietic stem and progenitor cells (HSPCs), which are then transplanted back into the patient in order to replace the blood system with gene-modified cells. With this strategy, a long-lasting therapeutic effect is guaranteed. To restrict transgene expression to platelets, lentiviral vectors with MK-specific promoters are generally used. In this context, a clinical trial in patients with preexisting inhibitors has been opened (NCT03818763). Our focus in this study was to load transgenes into platelet alpha granules (αGs) so that they will be secreted or presented after activation but will be hidden from circulation until needed, thereby reducing immunogenicity or toxicity. This strategy will allow the delivery of transgenic bioactive substances with high local doses that may not be well tolerated after systemic administration.
αGs are the most abundant of the three types of platelet granules. They store more than 300 different proteins, such as growth factors (e.g., vascular endothelial growth factor [VEGF]), cytokines (e.g., platelet factor 4, tumor necrosis factor alpha [TNF-α]), proangiogenic and antiangiogenic factors (e.g., platelet-derived growth factor, thrombospondin), and von Willebrand factor (vWF). The majority of αG proteins are produced and packaged in MKs, but cargo can also be acquired via selective endocytosis from the plasma.6 Also, αGs have specific proteins on their membranes, including GPIIb/IIIa (CD41) and P-selectin (CD62P). Whereas CD41 is also expressed on the surface of resting platelets, P-selectin is specifically translocated to the platelet surface after activation. P-selectin is incorporated into the granule membrane at the trans-Golgi network,7 with the signals responsible for sorting located in the transmembrane domain and cytoplasmic tail.8,9 However, the intracellular part was shown to be necessary only for sorting to the endothelial cell-specific granules (Weibel-Palade bodies), not αGs in platelets.9
Soluble proteins are sorted into αGs by various mechanisms. A four amino acid (aa) sequence has been identified in the platelet factor 4 (Pf4) cytokine, and subsequently also in the cytokines Nap2 and RANTES, as responsible for αG sorting.10,11 In contrast, large proteins, such as vWF or multimerin, probably localize to granules by a mechanism of self-assembly.12,13 Other proteins, such as fibrinogen, are endocytosed at the plasma membrane of MKs or platelets by the αIIbβ3 integrin receptor.14 The respective sorting signals are thought to be recognized by cytosolic proteins that act as adapters for the recruitment of clathrin, although the precise molecular underpinnings of granular sorting are poorly understood.
As protein sorting primarily takes place in MKs, we postulated that the expression of transgenic proteins equipped with suitable sorting signals in MKs may allow their specific loading into the αGs. The granules will then be transferred to platelets with the therapeutic cargo. A strategy for loading proteins to αGs has been explored by fusing FVIII to the D2 domain of the vWF propeptide.15 This domain was shown to be responsible for the sorting and retention of vWF in αGs.16,17 Effective sorting of D2.FVIII to platelet αGs was demonstrated, but release was not improved. In addition, the D2 domain spans >400 aa, and with this, it may not be suitable to target small proteins, while also taking up considerable coding capacity in gene transfer vectors.
We have previously shown that by using the promoter of Pf4, we could target gene expression into the MK lineage after lentiviral gene transfer to hematopoietic stem cells.18 In building on these vectors, we have generated lentiviral vectors utilizing the sorting signals of P-selectin and RANTES for targeting proteins to αGs in MKs and platelets. If the protein is fused to the RANTES sorting domain, its release into the blood after activation is expected; when fused to the P-selectin intracellular part and transmembrane domain, it will be presented on the surface of activated platelets. Our platform has great potential for gene and cell therapy, allowing for the adjustment of the platelet protein cargo as a viable therapeutic tool.
Results
Lentiviral vectors for alpha granular targeting
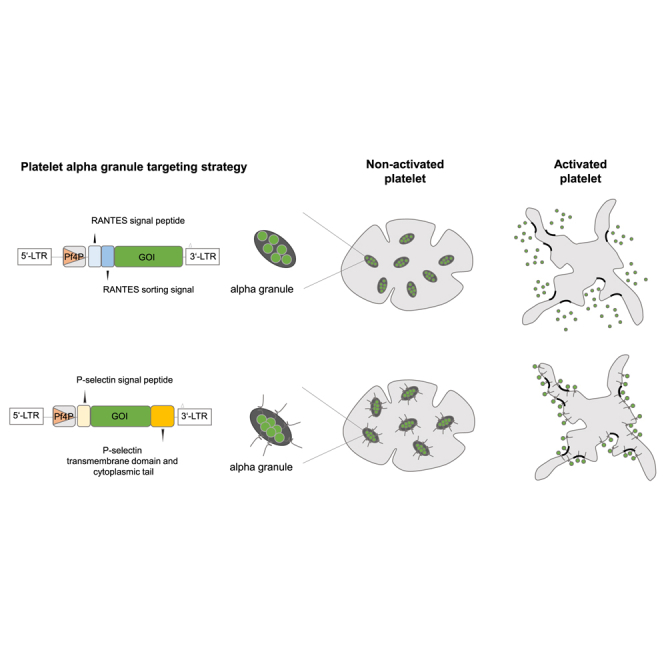
To deliver therapeutic proteins to the αGs in MKs, we designed lentiviral vectors for expression in MKs coding for transgenic proteins with specific αG-sorting signals. We chose two strategies. In the first, we used the sorting signal of 10 aa described in the human chemokine RANTES.10 This sequence was fused downstream of the signal peptide of RANTES to the N terminus of destabilized (ds)GFP (RANTES sorting domain [RSD] vector, Figure 1). In our second strategy, dsGFP was fused at its C terminus to the transmembrane and cytoplasmic tail (TDCT) of human P-selectin. For transport to the endoplasmic reticulum, the signal peptide of P-selectin was fused to the N terminus of dsGFP (TDCT vector, Figure 1). All lentiviral vectors were expressed from the murine platelet factor 4 promoter (Pf4P), which has strong activity in MKs.18 As the control, a lentiviral vector expressing dsGFP without any modification was used.
Figure 1.

Lentiviral vector designs for alpha granular targeting
Third-generation self-inactivating (SIN) lentiviral vectors driving the expression of dsGFP from the internal Pf4 promoter. Protein-sorting sequences of RANTES and its signal peptide were fused 5′ of dsGFP (RSD vector). P-selectin’s TDCT was fused 3′ of dsGFP, while its signal peptide was inserted 5′ of dsGFP (TDCT vector). ΔU3, 3′ unique region with deletion; SP, signal peptide; SoSi, sorting signal; dsGFP, destabilized green fluorescent protein; wPRE, woodchuck hepatitis virus posttranscriptional regulatory element; Pf4P, platelet factor 4 promoter; TDCT, transmembrane domain, cytoplasmic tail.
Expression from lentiviral vectors with alpha granular targeting domains
The RSD, TDCT, and control vectors were produced with similar high titers (1 × 108 to 1 × 109 infectious particles/mL after concentration) (Figure 2A). Vector performance was evaluated by transducing the megakaryoblastic leukemia cell line MEG-01 with the RSD and TDCT targeting vectors, as well as the control vector at defined multiplicities of infection (MOIs). We observed similar transduction efficiencies of 23.3%, 25.4%, and 22.6% for the RSD, TDCT, and control vectors, respectively, with the MOI of 0.25 (Figure 2B). To further characterize the vectors, MEG-01 cells were transduced with increasing MOIs (0.075, 0.13, 0.25, 0.6) and evaluated for transduction efficiency, mean fluorescence intensity (MFI), vector copy number (VCN), and relative GFP mRNA expression (Figures 2C–2F). There was a dose-dependent increase in transduction efficacy ranging from 6% to 52% GFP for all three vectors (Figure 2C). However, the intensity of GFP expression (MFI of GFP+ cells) from the TDCT vector was only about half of that from the RSD and the control vector (Figure 2D). We then evaluated the efficiency of vector integration in MEG-01 cells. The mean VCN increased with higher vector doses, but there was no linear correlation, probably due to the overall low values (Figure 2E). When we investigated transcription from the integrated vectors by quantifying GFP mRNA, we observed a similar dose-dependent increase in the relative GFP expression for all three vectors; however, the expression from the TDCT vector was the lowest. This was in agreement with the lower MFI of GFP expression from this vector (Figure 2F). Since the TDCT vector expresses a membrane-anchored protein, we wanted to specifically highlight this capability. Therefore, we transduced MEG-01 cells with increasing vector doses and confirmed the presence of cell-surface GFP by staining live cells with an anti-GFP antibody (Figures 2G and 2H). P-selectin expression in MEG-01 cells was also confirmed by flow cytometry (Figure S1). Taken together, our analysis confirmed good expression from the vectors with reliable transduction efficiencies, similar to our well-studied control vector. Only the TDCT vector showed overall lower GFP intensities, which could be explained by lower levels of transcription from the vector.
Figure 2.

Vector characterization in MEG-01 cells
(A) Vector titers of all three vectors. (B) Representative flow cytometry plots. (C) Transduction efficiency. (D) MFI of GFP+ cells normalized to mock MFI. (E) VCN and (F) GFP mRNA expression in cells transduced with MOIs 0.075, 0.13, 0.25, and 0.6 of the RSD, TDCT, or control vector. Data represent mean ± SD, n = 3–5 independent cultures; in (A) one-way ANOVA, ns, no significant difference; (C–F) two-way ANOVA, value from one vector at one MOI was compared with the other two vectors at the same MOIs; significance is relative to RSD and control; ∗p < 0.05, ∗∗p < 0.01. (G) Flow cytometric histograms showing surface GFP expression in MEG-01 cells transduced with TDCT (MOI 0.25 and 1) in comparison with RSD and untransduced cells. GFP was detected by antibody staining (clone 1A12-6-18, BD Bioscience). (H) Summarized quantification of the MFI of cell surface GFP on MEG-01 cells. n = 3 independent transductions, mean ± SD, one-way ANOVA, ∗∗∗∗p < 0.0001. MOI, multiplicity of infection; MFI, mean fluorescence intensity; VCN, vector copy number.
Colocalization of proteins in the αGs of murine and human megakaryocytes
To address the level of αG targeting, we evaluated the colocalization of the transgenic reporter protein dsGFP encoded by the RSD and TDCT vectors with the αG marker proteins vWF and P-selectin in murine and human MKs derived from transduced HSPCs. The colocalization analysis was performed on confocal images of MK immunostained with anti-vWF or P-selectin antibodies. Both murine and human MKs demonstrated the characteristic polyploid phenotype of mature MKs (Figures 3A and 3B), which was further confirmed by flow cytometry showing up to 70% CD41+/CD42b+ surface-marker expression in human and up to 40% in murine MKs and >4N ploidy (Figures S2 and S3). The differentiation potential of HSPCs was not affected by vector transduction, as demonstrated in human and mouse MKs by their cell-surface-marker expression and polyploidization (Figures S2 and S3).
Figure 3.

Colocalization analysis of αG targeting in murine and human primary megakaryocytes (MKs)
Immunofluorescence images of MKs differentiated from transduced (A) murine lineage-marker-negative BM cells or (B) human CD34+ HSPCs. MKs transduced with RSD, TDCT, or control vector (green) were stained with antibodies against αG proteins (red) and DAPI (blue). vWF+ αGs were visualized with 1° rabbit antibodies (Abs) and 2° goat-anti-rabbit Alexa Fluor 647 Abs, while P-selectin+ αGs were visualized with 1° human/mouse Abs and 2° goat-anti-mouse Cy3 Abs. Images were acquired with a Zeiss confocal microscope and a 100× oil immersion objective for murine MKs or a Leica confocal microscope with a 63× oil immersion objective for human MKs. The merged images show colocalization of vector and αG protein; scale bar, 10 μm. Quantification of colocalization of GFP in the three vectors with vWF or P-selectin in (C) murine MKs (n = 11–28) and (D) human MKs (n = 8–28). MKs were analyzed from two (mouse) and three (human) independent transductions and differentiations. Line at median on box-whisker plots, Mann-Whitney test, ∗∗∗∗p < 0.0001.
The confocal images demonstrated a broad punctate distribution of vWF and P-selectin throughout the cytoplasm of murine and human MKs; however, there was a clear difference in the distribution pattern of the transgenic GFP (Figures 3A and 3B). While cells transduced with RSD or TDCT targeting vectors showed GFP distributed with a dotted appearance in the cytoplasm only, the non-targeting control vector resulted in an even GFP expression in the cytoplasm, as well as high GFP levels in the nucleus (Figures 3A and 3B). No GFP fluorescence was observed in the mock controls (Figure S4). To quantify the colocalization between the GFP encoded by the vectors and the αG proteins, weighted colocalization coefficients from 0 (no colocalization) to 1 (complete colocalization) were used. This allowed us to record the pixel/intensity signal from the GFP (green) that also had an overlap with the pixel/intensity signal from the αG proteins (red), as shown in the merged images in Figures 3A and 3B. There was a significantly higher colocalization of transgenic GFP in vWF- and P-selectin-positive αGs achieved with the targeting RSD and TDCT vectors compared with the non-targeting control vector in murine and human MKs (Figures 3C and 3D). Thus, approximately 70%, 80%, and 20% GFP colocalization to vWF+ αGs was detected in murine MKs following RSD, TDCT, and control vector transduction, respectively (Figure 3C). Moreover, colocalization of transgenic GFP to P-selectin+ αGs was very similar. Differences between both targeting vectors and the control vector were highly significant (∗∗∗∗p < 0.0001), regardless of whether αG staining was performed with vWF or P-selectin (Figure 3C). Human MKs showed approximately 70% colocalization of transgenic GFP with vWF+ αGs and 68% and 62% with P-selectin after RSD and TDCT transduction, but only 32%–35% for the control vector (Figure 3D). Again, differences between RSD and TDCT compared with the control construct in human MKs were highly significant (∗∗∗∗p < 0.0001) (Figure 3D). As a further control, we mutated the four core amino acids in the RSD (TRKN) to alanine (AAAA) and demonstrated that this mutation reduced colocalization to vWF-positive granules in MEG-01 cells to 27% (Figure S5). Taken together, these data show highly effective αG targeting of the transgenic GFP by our RSD and TDCT vectors in murine as well as human MK. Moreover, the use of two different αG proteins (vWF and P-selectin) in the analysis increased the validity of our results because it is known that the loading of αG with these proteins is not identical (Figure S6).19,20
Secretion of proteins from the αGs of murine platelets
After demonstrating the effective sorting of our αG-targeted fusion proteins to the αGs of murine and human MKs, we next wanted to study whether the cargo would also be transferred to platelets and whether it would be released after platelet activation. This question was addressed in a murine gene transfer/bone marrow transplantation (BMT) model using mTmG marker mice as donors. Platelets of donor mTmG mice are marked by membrane-bound dTomato and can therefore be distinguished from recipient platelets in C57BL/6 hosts (Figure S7). HSPCs from the mTmG marker mice were in vitro transduced with the RSD, TDCT, or control vector before transplantation. Successful engraftment in all mice was confirmed by good chimerism in leukocytes and platelets (Figure S8), as well as the recovery of peripheral blood cell counts (Figure S9). The GFP expression in donor platelets was contained over 16 weeks posttransplant (Figure S10). The VCN in TDCT mice was higher than in RSD mice. Accordingly, the control mice were transplanted with BM cells transduced with higher or lower VCN (Figure S11). The high-VCN groups showed lower red blood cell (RBC) counts at 6 weeks posttransplantation, perhaps due to the high transduction rate.
In the supernatants of activated (A) and non-activated (NA) platelets taken from the mice, GFP protein content was measured by ELISA (Figure 4A). Upon activation, RSD platelets released up to 40,000 pg/mL GFP, compared with ∼5,000 pg/mL GFP without activation (∼8-fold increase). Consistent with the role of P-selectin as a membrane anchor protein, TDCT platelets showed no effective release of GFP into the supernatant in the NA (500 pg/mL) or A (∼2,000 pg/mL) state. On the other hand, control platelets released up to 30,000 pg/mL GFP and 25,000 pg/mL GFP in the NA and A states, respectively (Figure 4A), indicating that there was neither effective retention in the NA state nor increased secretion after activation.
Figure 4.

GFP release from murine platelets activated with thrombin
(A) Bar chart quantifying GFP in the supernatant of activated platelets by ELISA (n = 4–8 individually transplanted mice, Student’s t test, ∗∗∗∗p < 0.0001). Open circles: high-VCN mice; closed circles: low-VCN mice of the control group. (B) MFI of GFP in activated (A) and non-activated (NA) platelets (n = 11–15, Student’s t test, paired, ∗p = 0.02, ∗∗∗∗p < 0.001). (C) Representative flow cytometry plots demonstrating GFP release after platelet activation for all three vectors (control showing a high and a low copy number example) and their corresponding overlay histogram. Activation is shown by the upregulation of P-selectin in A versus NA platelets. (D) Ratio of the MFI of GFP in platelets after activation compared with the MFI before activation with thrombin (ratio A/NA, n = 11–15, Student’s t test, ∗∗∗∗p < 0.0001). The platelets for analysis shown in (B) and (D) were taken several times from five (RSD), four (TDCT), or eight (control) mice by blood sampling at different times. (E) Quantification of surface GFP expression (MFI) from NA and A platelets from donor cells (TDCT) and recipient (non-transduced) (n = 4, individually transplanted mice, one-way ANOVA with Dunett's correction; ns, not significant; ∗∗p < 0.01).
Since the secretion of GFP will reduce the amount of GFP in the platelets, we correlated the MFI of GFP expression with platelet activation, assuming that the release of GFP should lower the GFP MFI in platelets (Figure 4B) We observed a clear reduction of GFP levels in RSD platelets after activation, with a ratio of ∼0.4 for the MFIs in NA to A platelets (Figures 4C and 4D). This was much less the case for TDCT platelets, and no reduction in GFP levels upon activation was observed in platelets after control vector transduction, indicating that RSD-transduced platelets lost their GFP cargo after activation, while this was not the case for the control vector platelets and was greatly reduced for the TDCT platelets. GFP presentation could be detected on the surface of activated TDCT platelets (Figures 4E and S12). Taken together, our data show that functional platelets that were differentiated from vector-transduced HSPCs carried the GFP cargo. After activation, RSD platelets efficiently released the GFP, while TDCT platelets retained their GFP for presentation on the platelet surface.
Targeting IFNα as a therapeutic cytokine to αGs of human MKs and murine platelets
Since we proved the effective targeting of GFP to the αGs of murine and human MK, we next expanded our platform to deliver IFNα as a proof of concept for therapeutic translation. Therefore, we generated two additional lentiviral vectors encoding the human IFNα1 and coexpressing EGFP by an internal ribosome entry site (IRES). The targeting vector incorporated the human cytokine RANTES signal peptide and sorting signal fused to the N terminus of IFNα1 (lacking its own signal peptide), so that the IFNα could be secreted from activated platelet αGs (RSD.IFNα vector). As the control, a second vector encoded the complete coding sequence of IFNα1 with its signal peptide and without any additional sorting signals (IFNα vector) (Figure 5A).
Figure 5.

Targeting of IFNα to the alpha granules
(A) Design of third-generation SIN lentiviral vectors expressed from Pf4P. For the targeting of IFNα to αGs, the sorting domain and signal peptide of RANTES were fused to the N terminus of the IFNα1 coding sequence without the IFNα signal peptide, plus an IRES-EGFP sequence (RSD.IFNα vector). As the control, only the full coding sequence of IFNα with its signal peptide was fused to the IRES.eGFP (IFNα vector). (B) Confocal images of RSD.IFNα- and IFNα-transduced CB-derived MKs showing transgenes (green), P-selectin (red), and DAPI (blue). IFNα was visualized with 1° human IFNα-1 Abs and 2° goat-anti-rabbit Alexa Fluor 647 Abs, P-selectin+ αGs were visualized with 1° human/mouse Abs and 2° goat-anti-mouse Cy3 Abs, and GFP was detected in the 488 channel without Ab staining. Images were acquired with a Leica Dmi8 Inverted-3 confocal microscope with a 63× oil immersion objective and 4× digital zoom; scale bar, 10 μm. (C) Box-whisker plots showing colocalization of the GFP and IFNα transgenes with P-selectin+ αGs in CB-derived MKs. Line at median on boxplots, n = 8 MKs from two independent transductions; ns, not significant; ∗∗p < 0.01, Mann-Whitney test. (D) ELISA results showing secretion of GFP and IFNα protein from NA and A murine platelets. Data represent meadian and range, n = 5 individually transplanted mice, ∗∗p < 0.01, Mann-Whitney test. (E) MFI of P-selectin expression in A and NA GFP+ and GFP− platelets. Native platelets shown for comparison. (F) Platelet size measured by the FSC-A of IFNα-positive and native platelets. (E and F) n = 5, platelets from five independent RSD.IFNα and mock mice, Student's t test with Welch's correction, ns, not significant.
We first evaluated αG targeting efficiency. Therefore, we transduced cord blood (CB)-derived HSPCs with virus supernatants of RSD.IFNα and the IFNα control at MOI 40. We differentiated the cells toward MKs and performed colocalization analysis on immunostained, vector-transduced, and mock cells. αGs were visualized by immunostaining against P-selectin (red) and IFNα (Alexa Fluor 647, shown here in green), while GFP was detected without antibody staining (green) (Figure 5B). No IFNα or GFP fluorescent signals were observed in the mock controls (Figure S13). The colocalization analysis showed that there was a significant colocalization of IFNα with P-selectin-positive αG (∗∗p < 0.01) in RSD.IFNα MK compared with the control without the RSD domain (Figure 5C). No preferential colocalization of GFP expressed via the IRES to αGs was detected, although the colocalization of EGFP was higher than in our previous experiments, probably because EGFP was expressed, in contrast to the destabilized version in the previous vectors. Most important, IFNα expression did not impair MK differentiation or proliferation (Figures S14A–S14E). IFNα expression also was confirmed by mRNA detection (Figure S14F). Taken together, these data showed that we could target IFNα to MK αG using the RSD vector, and none of the repressive effects from exogenous IFNα administration on megakaryopoiesis were observed in this setting.21
To further investigate αG targeting in vivo, we employed our murine BMT model as previously described. In this context, HSPCs from mTmG marker mice were transduced with the RSD.IFNα vector supernatant at MOI 2.5–5 and then transplanted into C57BL/6 recipients (Figures S15A and S15B). At this stage, we did not include the IFNα control group because major side effects during hematopoietic recovery after BM transplantation due to IFNα expression were expected. We next activated platelets isolated from transplanted mice with thrombin to investigate if we could release IFNα from the αGs. Similar to what was observed for dsGFP, there was neither effective release nor retention of the EGFP transgene in A and NA platelets, respectively (Figure 5D). However, there was a highly significant amount of IFNα secreted from A platelets (up to 20,000 pg/mL) compared with NA platelets (∼650 pg/mL) (∗∗p < 0.01). These results confirmed that even though the RSD.IFNα encoded both IFNα and EGFP as transgenes, only the IFNα was specifically targeted to platelet αGs owing to the RSD fusion.
Although the PF4 promoter has high specificity for expression in MK, there was off-target expression in other blood cell lineages of between 5% and 13% (Figure S15C, also reported in Latorre-Rey et al.18). These cells would release IFNα, and accordingly we detected human IFNα in the blood (11.2 ± 3.3 ng/mL, Figure S15D) and the activation of IFNα-inducible genes in the BM (Figure S15E). This did not induce disease symptoms in mice, but white blood cell counts recovered delayed after transplantation and the B cell contribution to lymphocytes was slightly reduced (Figure S9). Effects on B cell development after long-term exposure to IFNα have been reported.22,23 We did not observe alterations of platelet function, size, or numbers or neutropenia in these mice (Figures 5E and 5F; Figure S9C).
IFNα released from modified platelets inhibits measles virus replication
After proving the specific targeting of IFNα to the αGs and release after stimulation, we tested the efficacy of the IFNα released from platelets to suppress measles virus replication in vitro (vaccine strain MeVvac2(GFP)). Virus replication was quantified by the GFP-positive area. Platelets isolated from transplanted mice that expressed from the RSD.IFNα vector and activated with thrombin exhibited a dose-dependent inhibitory effect against MeVVac2(GFP) replication (Figures 6A–6C; Figure S16). Inhibition by RSD.IFNα-activated platelets at the highest amount was comparable to 10 U/mL recombinant IFNα at 72 h postinfection. There was no (5 × 105 platelets) or only minor (5 × 106 platelets) inhibition of virus replication in wells that were treated with NA RSD.IFNα platelets, indicating that there was no release from platelets without activation (Figures S16 and S17). RSD.eGFP platelets, regardless of activation, did not suppress MeVvac2(GFP) replication and allowed replication similar to the MeVvac2(GFP) control (Figures 6A–6C).
Figure 6.

Inhibition of virus replication by IFNα released from engineered platelets
(A) Left: fluorescence images of Vero-E6 cells infected with MeVvac2(GFP) at MOI 0.01 at 72 h after infection (left top image) and Vero-E6 cells infected and treated with decreasing doses of IFNα (100–0.01 U). Right: Vero-E6 cells infected with MeVvac2(GFP) at MOI 0.01 and treated with 5 × 106, 5 × 105, and 5 × 104 A or NA platelets from RSD.IFNα or RSD.GFP mice. Images were taken 72 h post infection. Magnification 40×; scale bar, 500 μm. (B) Quantification of the fluorescence area, 72 h post infection (platelets of n = 3 independent mice, mean ± SD, three images per well). One-way ANOVA; multiple comparisons between A or NA platelets RSD.IFNα with RSD.eGFP within each platelet dilution ∗∗p > 0.01, ∗∗∗∗p > 0.0001. (C) Time course of virus replication measured by the fluorescence area 48, 72, and 144 h post infection (platelets of n = 3–5 independent mice, mean ± SD, three images per well). Two-way ANOVA; multiple comparisons of RSD.IFNα with MeVVac2(GFP) control. ∗∗∗∗p < 0.0001.
Discussion
In our present work, lentiviral vector-mediated gene transfer was used to effectively target GFP and IFNα to αGs in MKs and platelets. In addressing some of the limitations of using larger αG targeting domains such as the 400 aa vWF D2 by Du et al.,15 we opted for the smaller and potentially more efficient sorting signal of RANTES (10 aa, RSD) and P-selectin transmembrane domain and cytoplasmic tail (57 aa, TDCT). Although both sequences have a human origin, they exhibit a high homology to the murine sequence (Figure S18). The RSD and TDCT vectors were very well equipped to load the transgenic proteins into αGs in murine and human MKs. The colocalization of the RSD.GFP with the αG markers vWF and P-selectin was around 70% and recapitulates the observations by El Golli et al. in transfected AtT20 cells, demonstrating that the targeting was also efficient in MKs in the context of lentiviral transduction.10 In platelets, the RSD fusion proteins were well retained in resting platelets and could be effectively released after activation. By comparison, the TDCT fusion retained the GFP signal on the platelets, and no release after activation could be measured. Cell-surface presentation of the TDCT-GFP fusion protein was further confirmed in the P-selectin-positive MEG-01 cells and activated platelets. However, the overall intensity of expression from the TDCT vector was lower compared with the RSD vector, although vector titers and transduction efficiencies were the same. One might argue that the differences in vector performance may be attributed to the nature of the protein fusion to dsGFP. The TDCT vector was designed with a polyglycine linker between dsGFP’s C-terminal and TDCT’s N-terminal domains in order to optimally circumvent steric effects, which have been described as having an impact on efficient protein folding, processing, or stability.24, 25, 26 However, the reduced GFP intensity was accompanied by lower mRNA levels by the same VCN, arguing for reduced transcription from the genome copies and against issues with translation of TDCT. Notwithstanding, the TDCT vector can be improved in further studies. It is also not clear if the discrepancies observed in the functional characteristics would persist if other proteins were to be fused to the TDCT. The presentation of proteins on the platelets surface may be of special interest, e.g., in the context of coagulation factors that need the procoagulative platelet surface.
We were able to validate our proof of concept by targeting IFNα as a therapeutic cytokine showing effective packaging and release from platelet αGs. IFNα release was also effective at inhibiting measles virus replication in vitro in a controlled manner, as there was only minor inhibition when platelets were not activated. The rationale of targeting IFNα to αGs was due to its established antiviral and antitumor effects, which have already been applied in clinical settings.27, 28, 29 Therefore, our successful strategy of packaging this potent cytokine in the αGs of the MK lineage serves as a solid platform to build upon with more advanced studies. Although we used the BMT model in our study, this was mainly done to generate fully functional platelets, which we then studied ex vivo. One may envision the use of in vitro-generated and modified platelets for direct transfusion. In this case, the therapeutic effect would be very short term. On the other hand, IFNα expression in tumor-resident macrophages has been shown to be effective as antitumor therapy after transplantation of lentiviral-transduced hematopoietic stem cells (HSCs).30,31 As platelets are attracted by tumor vessels,32,33 a platelet-mediated IFNα delivery may be similarly applicable. However, to allow the safe expression of IFNα in platelets after transplantation of transduced HSCs, further modifications to the vector must be made to reduce the background expression in other blood cell lineages and, with this, limit systemic IFNα levels.
In summary, our study presents an adaptable platform for targeting proteins to platelet αGs based on lentiviral vectors to utilize platelets as vehicles to store, hide, and specifically deliver therapeutic proteins. Our novel vector platform should be applicable to a broader range of applications.
Materials and methods
Lentiviral vectors
The coding sequence of the fusion protein consisting of dsGFP fused with the signal peptide and sorting signal of human RANTES was amplified by PCR and inserted into the lentiviral vector expressing from the internal Pf4P (RSD vector). The RANTES signal peptide contains the first 23 aa, and the sorting signal comprises aa 62–72 of the RANTES protein. The human P-selectin transmembrane (aa 772–795) and cytoplasmic tail (aa 796–830) were fused to dsGFP by a polyglycine linker by ligating two PCR products. At the N terminus, the P-selectin signal peptide (41 aa) was fused to dsGFP. The whole coding sequence was then inserted downstream of the Pf4P in the lentiviral vectors (TDCT vector). To generate the dsGFP-expressing control vector, the EGFP of the vector RRL.PPT.Pf4P.eGFP.wPre, published by Latorre-Rey et al.,18 was replaced by dsGFP.
Lentiviral vectors encoding IFNα were generated. In the first vector, the human cDNA of IFNα1 without its signal peptide was PCR amplified and inserted into the lentiviral vector to generate a fusion protein with the RANTES signal peptide and sorting signal. The IFNα sequence was followed by an IRES-EGFP (RSD.IFNα vector). In the second vector, the full coding sequence of IFNα1 with its signal peptide was PCR amplified and inserted into the lentiviral vector upstream of an IRES-EGFP sequence. This second vector was void of the RANTES sorting signal, thereby functioning as a control (IFNα vector).
Generation of lentiviral vector supernatants
We seeded 5 × 106 293T cells in 10 cm plates in DMEM, 10% fetal calf serum (FCS), 2 mM L-glutamine, 1% penicillin/streptomycin, and 20 mM HEPES and transfected them by calcium-phosphate transfection with 10 μg of vector plasmid, 10 μg of gag/pol plasmid (pCDA3.GP.4XC plasmid), 1.5 μg of plasmid encoding the VSV-G envelope (pMD.G plasmid), and 5 μg of Rev-encoding plasmid in DMEM supplemented with 25 μM chloroquine. Supernatants were collected after 36 and 48 h, concentrated by high-speed centrifugation at 80,000g × 2 h, resuspended in StemSpan medium (STEMCELL Technologies, Köln, Germany), and stored at −80°C. Vector titers were determined by transduction of murine SC-1 cells in serial dilutions followed by flow cytometry or VCN detection by qPCR.
Isolation and transduction of murine lineage marker-negative (lin−) BM cells
BM was flushed from femora, tibiae, and hip bones of C57BL/6 mice or mTmG marker mice (B6.129-Gt(ROSA)26Sor<tm4(ACTB-tdTomato,-EGFP)Luo>/J). Mononuclear cells were purified by gradient centrifugation (Histopaque 1083, Sigma-Aldrich, Munich, Germany) at 800g, 20 min, room temperature (RT). Lin− cells were selected by magnetic cell sorting using the following lineage-specific antibodies: anti-CD5, CD45R (B220), CD11b, Gr-1 (Ly-6G/C), 7-4, and Ter-119 (Miltenyi Biotech, Bergisch Gladbach, Germany). Briefly, cells were incubated with the biotin-labeled antibody cocktail for 20 min at 4°C, followed by the secondary antibody (anti-biotin microbeads) for 20 min at 4°C. Antibody-labeled cells were run over a separation column and the lin− population was collected from the flow-through. Purified cells were cultivated in a serum-free medium (StemSpan SFEM, STEMCELL Technologies, Köln, Germany), 10 ng/mL mSCF, 20 ng/mL mTHPO, 20 ng/mL mIGF2, 10 ng/mL hFGF-1, 2 mM glutamine, and 1% penicillin/streptomycin. After prestimulation for 24 h, lin− cells were transduced twice on two consecutive days with lentiviral vectors on RetroNectin-coated wells (Takara-Clontech, 10 μg/cm2, 24 or 12 well plates, depending on cell number), with a defined MOI of 2.5–5. For the differentiation to MKs, cells were cultured in a serum-free medium containing 50 ng/mL THPO for up to 3 weeks. Surface-marker expression was evaluated via flow cytometry by staining harvested murine MKs with anti-CD41-PerCP Cy5.5 (MWReg30, BioLegend, San Diego, CA) and anti-CD42d-APC (1C2, eBioscience, Thermo Fisher Scientific, Waltham, MA, USA) antibodies. DNA content/polyploidy analysis was performed by staining ethanol-fixed cells with propidium iodide (PI) staining solution (50 μg/mL PI, 0.1 mg/mL RNase A, 0.05% Trition-X-100, PBS) and then performing flow cytometry. The Cytoflex flow cytometer was used for analysis (Beckman Coulter, Krefeld, Germany).
Isolation and transduction of primary human CD34+ cells
Human umbilical CB or G-CSF-mobilized peripheral blood (PB) and BM samples were collected after written donor consent at the Hannover Medical School and German Red Cross, respectively. Mononuclear cells were purified by gradient centrifugation (Biocoll Separating Solution, Biochrom, Berlin, Germany) at 400g, 40 min, at RT. CD34+ HSPCs were positively selected by magnetic cell sorting using the human CD34 MicroBead Kit (Miltenyi Biotech, Bergisch Gladbach, Germany). Briefly, cells were incubated with the CD34 microbeads and FcR blocking reagent for 30 min at 4°C. The cells were run over a separation column and the flow-through was discarded, and magnetically labeled CD34+ cells were eluted into a new collection tube after removal of the separation column from the magnetic rack. CD34+ cells were cultivated in StemSpan SFEM (STEMCELL Technologies), 100 ng/mL hFLT-3 ligand, 100 ng/mL hSCF, 50 ng/mL hTHPO, 2 mM L-glutamine, and 1% penicillin/streptomycin. After 24 h expansion, CD34+ cells were subjected to a single round of transduction with lentiviral vectors on RetroNectin-coated wells (Takara-Clontech, Kusatsu, Japan; 10 μg/cm2, 48 well plates, 1 × 105 cells/well) with defined MOI of 5–40.
Differentiation and characterization of primary human megakaryocytes
Lentiviral- and mock-transduced CD34+ cells were seeded in 24 well suspension plates at a cell density of 5 × 105 cells/well for differentiation in StemSpan SFEMII medium (STEMCELL Technologies), 50 ng/mL hTHPO, 25 ng/mL hSCF, and 7.5 ng/mL hIL-6. From day 6, the SFEMII medium was supplemented with only 50 ng/mL hTHPO and 25 ng/mL hSCF until the end of the differentiation period of 14 days. Surface-marker expression was evaluated via flow cytometry by staining harvested MKs with anti-CD41-APC-Cy7 (HIP8; BioLegend, San Diego, CA, USA) and anti-CD42b-PerCP (HIP1; BioLegend, San Diego, CA, USA) antibodies. In addition, DNA content analysis was performed by staining ethanol-fixed cells with PI staining solution (50 μg/mL PI, 0.1 mg/mL RNase A, 0.05% Trition-X-100, PBS) and then performing flow cytometry. The Cytoflex flow cytometer was used for analysis (Beckman Coulter, Krefeld, Germany).
Immunofluorescence and confocal analysis
Human and murine culture-derived MKs were spun onto coverslips at 400 rpm for 10 min using the CytoSpin4 cytocentrifuge (Thermo Fisher Scientific). MKs were fixed in 4% paraformaldehyde (PFA) for 15 min at RT and permeabilized with 0.5% Triton X-100 in PBS for 10 min at RT. After rehydration with PBS, blocking was done in 10% bovine serum albumin (BSA) (Carl Roth, Karlsruhe, Germany) in PBS for 1 h at RT. Incubation with the primary antibodies anti-vWF (1:100) (Abcam, Catalog No. ab9378), murine anti-human/mouse CD62P-PE (1:100) (eBioscience, Catalog No. 12-0626-82), and rabbit anti-IFN-α (1:50) (Novus Biologicals, Catalog No. 31101-1) was carried out for 1 h at RT. After subsequent washing steps, incubation with the secondary antibodies goat anti-rabbit IgG Alexa Fluor 647 (1:2,000) (Invitrogen, Catalog No. A21245) and goat anti-mouse IgG Cy3 (Invitrogen, Catalog No. A10521) and nuclear counterstain 4′,6-diamidino-2-phenylindole (DAPI) (1:5,000) (Sigma-Aldrich, Seelze, Germany) was performed for 1 h at RT. Washed coverslips were then mounted onto glass slides in Dako Faramount aqueous mounting medium (Dako, Carpinteria, CA, USA), allowed to dry overnight at RT in the dark, and then transferred to 4°C for storage. Cells were acquired with the LSM510 Axiovert200M confocal microscope (Zeiss, Oberkochen, Germany) or the DMi8 Inverted-3 confocal microscope (Leica, Wetzlar, Germany) using appropriate filter sets. MEG-01 cells transduced with the targeting vectors (MOI 1) were fixed with 4% PFA for 15 min and permeabilized with 0.5% Triton X-100 in PBS for 30 min. Cells were incubated with primary antibodies anti-vWF (1:100) in PBS with 2% FCS overnight at 4°C. Cells were washed and incubated with secondary antibodies goat anti-rabbit IgG Alexa Fluor 647 (1:1,000) and DAPI (1:5,000) for 1 h at RT. About 5 × 105 cells/well were transferred into the CellCarrier-96 plate (PerkinElmer, Waltham, MA, USA) and confocal images were acquired with the Operetta high-content imaging system (PerkinElmer, Waltham, MA, USA).
Colocalization analysis of murine and human megakaryocytes
αG targeting efficiency of the vectors was determined by evaluating the colocalization of transgenes with the αG marker proteins vWF and P-selectin. The Zen Blue software (Zeiss) and the JaCoPx plug-in34 of the FIJI image analysis software35 was used. Murine MKs were acquired with the LSM 510, Axiovert200M confocal microscope (Zeiss) using a 100× oil immersion objective in a 512 × 512 format with an image size of 127.28 × 127.28 μm. Colocalization of the samples with the Zen Blue software was determined by using single-stained controls to set the crosshairs for intensity thresholds so that a pixel-by-pixel comparison could be made for the selected channels. Applying the weighted colocalization coefficient (WCC) took into account the intensity value of the summed pixels, where the value of each pixel is equal to its intensity value. Human MKs were acquired with the DMi8 Inverted-3 confocal microscope (Leica), using a 63× oil immersion objective in a 1,024 × 1,024 format. Image sizes were 92.26 × 92.26 μm or 46.13 × 46.13 μm when 4× digital zoom was applied. Since the LAS X (Leica) image processing software does not include a built-in weighted colocalization tool, FIJI was used for this purpose. Confocal images of transduced MEG-01 cells were acquired with the Operetta high-content imaging system using a 40× high numerical aperture objective in a 1,360 × 1,024 format with an image size of 88.0 × 66.0 μm. The rank weighted colocalization (RWC) algorithm of JaCoPx was applied to cell images using default thresholds in order to obtain a pixel and intensity correlation value, which has been described as being superior to traditional methods of colocalization.36 For both RWC and WCC analyses, values ranging from 0 (no colocalization) to 1 (complete colocalization) were obtained corresponding to the colocalization of the transgenes encoded by targeting and non-targeting vectors with the individual αG marker proteins. Although murine and human cells were analyzed differently, the bases of pixel and intensity cooccurrence and correlation were comparable, and the colocalization coefficients are derivatives from Pearson’s correlation coefficient.37 Therefore, colocalization results in the present work are displayed as “colocalization” for uniformity.
Bone marrow transplantation
Animal experiments were carried out according to the German animal protection laws and were approved by the local animal welfare committee (Regierungspräsidium Darmstadt). C57BL/6 mice (obtained from Janvier or Charles River) were preconditioned with 7 Gy, 137Cs-g rays, 0.036 Gy/s irradiation and transplanted with a minimum of 5 × 105 cells 1 day after their transduction. Donor cells were derived from mTmG marker mice. Cell infusion was performed via tail vein injection in 100 μL PBS. All mice were kept in the specific-pathogen-free animal facilities of the Paul-Ehrlich-Institute, Langen, Germany. To circumvent infection-associated complications after irradiation, antibiotics were supplied in the first 3 weeks after transplantation (Ciproflaxin 0.1 mg/mL in drinking water). After 6 weeks, and then every 3 weeks, blood samples of transplanted mice were collected via retro-orbital bleeding. Blood cell counts were determined using a VetABC hemocytometer (Scil Animal Care Company, Viernheim, Germany). Chimerism was determined by detecting dTomato from donor mice on leukocytes (after erythrocyte lysis) or in platelets. Platelets were stained with CD41-APC (clone eBioMWReg30, Thermo Fisher Scientific). Flow cytometry was carried out using the Cytoflex (Beckman Coulter).
Platelet activation and ELISA
One hundred microliters of mouse blood was collected in 300 μL heparin in PBS (25 U/mL). Blood cells were then washed twice with Tyrode’s buffer (20× Tyrode’s buffer: 2.73 M NaCl, 53.6 mM KCl, 238 mM NaHCO3, 8 mM Na2HPO4, 0.5 M HEPES) plus 0.36% BSA, 0.1% glucose, and 1 mM MgCl2 and separated by centrifugation with 800g, 5 min at RT. Cells were then resuspended in 750 μL Tyrode’s buffer containing 2 mM CaCl2. Fifty microliters from the sample was activated with thrombin (0.1 U, 6 min at 37°C, 6 min at RT) and stained with antibodies detecting CD41-PerCP-Cy5.5 (clone MWReg30; BioLegend) and P-selectin-APC (clone Psel.KO2.3; Thermo Fisher Scientific). Before cytometric analysis, 1–2 mL fluorescence-activated cell sorting (FACS) buffer (5 mM EDTA, 2% FCS, PBS) was added. If GFP or IFNα was measured in the platelet supernatant by ELISA, the remaining blood sample was split into two tubes, of which only one was activated with thrombin. Leukocytes were separated by centrifugation at 250g, 5 min at RT, and platelet-containing supernatants were collected. A and NA platelet samples were centrifuged at 800g, 5 min at RT, and supernatants (150 μL each) and pellets were collected. GFP or IFNα proteins in the platelet supernatants were measured using the GFP ELISA kit (Cell Biolabs, San Diego, CA, USA) or human IFN-α ELISA PRO kit (Mabtech, Nacka Strand, Sweden), respectively, following the manufacturers’ instructions.
Virus replication assay
African green monkey kidney Vero-E6 cells (8 × 104) were seeded in 48 well plates and grown for 24 h in DMEM (BioWest) supplemented with 5% FCS and 2 mM L-glutamine. The medium was replaced, and the 90% confluent cells were infected with a vaccine measles virus (MeV) strain (Vac2) expressing enhanced GFP from an additional transcription unit downstream of H (MeVVac2(GFP),38 kindly provided by C.K. Pfaller, Paul-Ehrlich-Institute) at an MOI of 0.01 in 250 μL Opti-MEM (Thermo Fisher Scientific) without additives for 2 h at 32°C. The medium was then replaced with DMEM containing 10% FCS, 2 mM L-glutamine, and 1% penicillin/streptomycin. Platelets were isolated into Tyrode’s buffer as described above. Then, 5 × 106, 5 × 105, or 5 × 104 platelets were added to the infected Vero cells and activated with thrombin (5 U/mL) (A) or not activated (NA). As a control, recombinant human IFNα (PBL Assay Science, Piscataway, NJ, USA) was used at concentrations of 100–0.01 U per well. Cells were incubated at 32°C in a humidified atmosphere containing 5% CO2 for 6 days. MeVVac2(GFP) replication was measured by GFP-fluorescence microscopy (Nikon ECLIPSE Ti-S) at 48, 72, and 144 h post infection. Three pictures per well and per condition were taken (magnification 4×, exposure 100 ms, camera Nikon DIGITAL SIGHT DS-QilMc-U3, and software Nikon Nis-Elements 4.20.00 LO) and the area of GFP fluorescence was analyzed with ImageJ with set thresholds from 40 to 255 to exclude the background signal from the GFP-positive platelets.
Detection of the mean vector copy number by PCR
TaqMan real-time PCR was performed with the TaqMan Fast Advanced Master Mix (Thermo Fisher Scientific). VCN was determined on genomic DNA from blood cells or culture-derived cells isolated using the NucleoSpin Tissue Kit (Macherey-Nagel, Düren, Germany), using specific primers and probes complementary to the wPre element or PTBP2 as a housekeeping gene. VCN was calculated by normalization to the housekeeping gene and quantification by a plasmid standard. PCRs were run on the StepOnePlus Real-Time PCR System (Applied Biosystems, Waltham, MA, USA).
Gene-expression PCR
RNA was isolated using the Direct-zol RNA Microprep Kit (Zymo Research, Irvine, CA, USA) and reverse transcribed using RevertAid H Minus reverse transcriptase (Thermo Fisher Scientific) following the manufacturers’ protocols. Expression levels of dsGFP were analyzed relative to β-actin (Primer Probe Set, Thermo Fisher Scientific) as a housekeeping gene. Primers and probe for dsGFP were used in concentrations of 900 and 250 nM, respectively.
Acknowledgments
We thank Christian Pfaller, Division of Veterinary Medicine, Paul-Ehrlich-Institute, for providing the MeVvac2(GFP) virus and Nina Hein-Fuchs, Research Group Host-Pathogen Interactions, Paul-Ehrlich-Institute, for assistance with the Operetta system for confocal images. This work was supported by the German Research Foundation (DFG; individual grants Mo860/5-1, Mo886/8-1, and REBIRTH Cluster of Excellence [EXC62]) and the REBIRTH Research Center for Translational Regenerative Medicine (ZN3440, State of Lower Saxony, Ministry of Science and Culture [Nieders. Vorab]).
Author contributions
V.W. and L.L. generated vectors. V.W., L.L., and M.R. performed colocalization experiments, V.W and L.L. assisted with the BM transplantation experiments, and analyzed data. M.R. and F.S. performed BM transplantation and virus replication assays and analyzed data. T.M. and U.M. designed the project and analyzed data. V.W., M..R., T.M., and U.M. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.12.038.
Contributor Information
Thomas Moritz, Email: moritz.thomas@mh-hannover.de.
Ute Modlich, Email: ute.modlich@pei.de.
Supplemental information
References
- 1.Hu C.-M.J., Fang R.H., Wang K.-C., Luk B.T., Thamphiwatana S., Dehaini D., Nguyen P., Angsantikul P., Wen C.H., Kroll A.V., et al. Nanoparticle biointerfacing by platelet membrane cloaking. Nature. 2015;526:118–121. doi: 10.1038/nature15373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu P., Zuo H., Chen B., Wang R., Ahmed A., Hu Y., Ouyang J. Doxorubicin-loaded platelets as a smart drug delivery system: an improved therapy for lymphoma. Sci. Rep. 2017;7:42632. doi: 10.1038/srep42632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lyde R., Sabatino D., Sullivan S.K., Poncz M. Platelet-delivered therapeutics. J. Thromb. Haemost. 2015;13:S143–S150. doi: 10.1111/jth.12938. [DOI] [PubMed] [Google Scholar]
- 4.Hyslop S.R., Josefsson E.C. Undercover agents: targeting tumours with modified platelets. Trends Cancer. 2017;3:235–246. doi: 10.1016/j.trecan.2017.01.006. [DOI] [PubMed] [Google Scholar]
- 5.Cai Y., Shi Q. Platelet-targeted FVIII gene therapy restores hemostasis and induces immune tolerance for hemophilia A. Front. Immunol. 2020;11:964. doi: 10.3389/fimmu.2020.00964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harrison P., Savidge G.F., Cramer E.M. The origin and physiological relevance of alpha-granule adhesive proteins. Br. J. Haematol. 1990;74:125–130. doi: 10.1111/j.1365-2141.1990.tb02554.x. [DOI] [PubMed] [Google Scholar]
- 7.Harrison-Lavoie K.J., Michaux G., Hewlett L., Kaur J., Hannah M.J., Lui-Roberts W.W.Y., Norman K.E., Cutler D.F. P-selectin and CD63 use different mechanisms for delivery to Weibel-Palade bodies. Traffic. 2006;7:647–662. doi: 10.1111/j.1600-0854.2006.00415.x. [DOI] [PubMed] [Google Scholar]
- 8.Disdier M., Morrissey J.H., Fugate R.D., Bainton D.F., McEver R.P. Cytoplasmic domain of P-selectin (CD62) contains the signal for sorting into the regulated secretory pathway. Mol. Biol. Cell. 1992;3:309–321. doi: 10.1091/mbc.3.3.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hartwell D.W., Mayadas T.N., Berger G., Frenette P.S., Rayburn H., Hynes R.O., Wagner D.D. Role of P-selectin cytoplasmic domain in granular targeting in vivo and in early inflammatory responses. J. Cell Biol. 1998;143:1129–1141. doi: 10.1083/jcb.143.4.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.El Golli N., Issertial O., Rosa J.-P., Briquet-Laugier V. Evidence for a granule targeting sequence within platelet factor 4. J. Biol. Chem. 2005;280:30329–30335. doi: 10.1074/jbc.M503847200. [DOI] [PubMed] [Google Scholar]
- 11.Briquet-Laugier V., Lavenu-Bombled C., Schmitt A., Leboeuf M., Uzan G., Dubart-Kupperschmitt A., Rosa J.-P. Probing platelet factor 4 alpha-granule targeting. J. Thromb. Haemost. 2004;2:2231–2240. doi: 10.1111/j.1538-7836.2004.01037.x. [DOI] [PubMed] [Google Scholar]
- 12.Blagoveshchenskaya A.D., Hannah M.J., Allen S., Cutler D.F. Selective and signal-dependent recruitment of membrane proteins to secretory granules formed by heterologously expressed von Willebrand factor. Mol. Biol. Cell. 2002;13:1582–1593. doi: 10.1091/mbc.01-09-0462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayward C.P., Song Z., Zheng S., Fung R., Pai M., Massé J.M., Cramer E.M. Multimerin processing by cells with and without pathways for regulated protein secretion. Blood. 1999;94:1337–1347. [PubMed] [Google Scholar]
- 14.Handagama P., Scarborough R.M., Shuman M.A., Bainton D.F. Endocytosis of fibrinogen into megakaryocyte and platelet alpha-granules is mediated by alpha IIb beta 3 (glycoprotein IIb-IIIa) Blood. 1993;82:135–138. [PubMed] [Google Scholar]
- 15.Du L.M., Nurden P., Nurden A.T., Nichols T.C., Bellinger D.A., Jensen E.S., Haberichter S.L., Merricks E., Raymer R.A., Fang J., et al. Platelet-targeted gene therapy with human factor VIII establishes haemostasis in dogs with haemophilia A. Nat. Commun. 2013;4:2773. doi: 10.1038/ncomms3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haberichter S.L., Jozwiak M.A., Rosenberg J.B., Christopherson P.A., Montgomery R.R. The von Willebrand factor propeptide (VWFpp) traffics an unrelated protein to storage. Arterioscler. Thromb. Vasc. Biol. 2002;22:921–926. doi: 10.1161/01.atv.0000017063.36768.87. [DOI] [PubMed] [Google Scholar]
- 17.Haberichter S.L., Jacobi P., Montgomery R.R. Critical independent regions in the VWF propeptide and mature VWF that enable normal VWF storage. Blood. 2003;101:1384–1391. doi: 10.1182/blood-2002-07-2281. [DOI] [PubMed] [Google Scholar]
- 18.Latorre-Rey L.J., Wintterle S., Dütting S., Kohlscheen S., Abel T., Schenk F., Wingert S., Rieger M.A., Nieswandt B., Heinz N., et al. Targeting expression to megakaryocytes and platelets by lineage-specific lentiviral vectors. J. Thromb. Haemost. 2017;15:341–355. doi: 10.1111/jth.13582. [DOI] [PubMed] [Google Scholar]
- 19.Battinelli E.M., Thon J.N., Okazaki R., Peters C.G., Vijey P., Wilkie A.R., Noetzli L.J., Flaumenhaft R., Italiano J.E. Megakaryocytes package contents into separate α-granules that are differentially distributed in platelets. Blood Adv. 2019;3:3092–3098. doi: 10.1182/bloodadvances.2018020834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Italiano J.E., Battinelli E.M. Selective sorting of alpha-granule proteins. J. Thromb. Haemost. 2009;7:173–176. doi: 10.1111/j.1538-7836.2009.03387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Q., Miyakawa Y., Fox N., Kaushansky K. Interferon-alpha directly represses megakaryopoiesis by inhibiting thrombopoietin-induced signaling through induction of SOCS-1. Blood. 2000;96:2093–2099. [PubMed] [Google Scholar]
- 22.Di Scala M., Gil-Fariña I., Vanrell L., Sánchez-Bayona R., Alignani D., Olagüe C., Vales A., Berraondo P., Prieto J., González-Aseguinolaza G. Chronic exposure to IFNα drives medullar lymphopoiesis towards T-cell differentiation in mice. Haematologica. 2015;100:1014–1022. doi: 10.3324/haematol.2014.115410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fallet B., Narr K., Ertuna Y.I., Remy M., Sommerstein R., Cornille K., Kreutzfeldt M., Page N., Zimmer G., Geier F., et al. Interferon-driven deletion of antiviral B cells at the onset of chronic infection. Sci. Immunol. 2016;1:eaah6817. doi: 10.1126/sciimmunol.aah6817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang L., Pike D., Sleat D.E., Nanda V., Lobel P. Potential pitfalls and solutions for use of fluorescent fusion proteins to study the lysosome. PLoS One. 2014;9:e88893. doi: 10.1371/journal.pone.0088893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krishna M.M.G., Englander S.W. The N-terminal to C-terminal motif in protein folding and function. Proc. Natl. Acad. Sci. U S A. 2005;102:1053–1058. doi: 10.1073/pnas.0409114102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomas C.L., Maule A.J. Limitations on the use of fused green fluorescent protein to investigate structure-function relationships for the cauliflower mosaic virus movement protein. J. Gen. Virol. 2000;81:1851–1855. doi: 10.1099/0022-1317-81-7-1851. [DOI] [PubMed] [Google Scholar]
- 27.Li S.-F., Gong M.-J., Zhao F.-R., Shao J.-J., Xie Y.-L., Zhang Y.-G., Chang H.-Y. Type I interferons: distinct biological activities and current applications for viral infection. Cell Physiol. Biochem. 2018;51:2377–2396. doi: 10.1159/000495897. [DOI] [PubMed] [Google Scholar]
- 28.Saleki K., Yaribash S., Banazadeh M., Hajihosseinlou E., Gouravani M., Saghazadeh A., Rezaei N. Interferon therapy in patients with SARS, MERS, and COVID-19: a systematic review and meta-analysis of clinical studies. Eur. J. Pharmacol. 2021;906:174248. doi: 10.1016/j.ejphar.2021.174248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zitvogel L., Galluzzi L., Kepp O., Smyth M.J., Kroemer G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015;15:405–414. doi: 10.1038/nri3845. [DOI] [PubMed] [Google Scholar]
- 30.Escobar G., Barbarossa L., Barbiera G., Norelli M., Genua M., Ranghetti A., Plati T., Camisa B., Brombin C., Cittaro D., et al. Interferon gene therapy reprograms the leukemia microenvironment inducing protective immunity to multiple tumor antigens. Nat. Commun. 2018;9:2896. doi: 10.1038/s41467-018-05315-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Catarinella M., Monestiroli A., Escobar G., Fiocchi A., Tran N.L., Aiolfi R., Marra P., Esposito A., Cipriani F., Aldrighetti L., et al. IFNα gene/cell therapy curbs colorectal cancer colonization of the liver by acting on the hepatic microenvironment. EMBO Mol. Med. 2016;8:155–170. doi: 10.15252/emmm.201505395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gaertner F., Massberg S. Patrolling the vascular borders: platelets in immunity to infection and cancer. Nat. Rev. Immunol. 2019;19:747–760. doi: 10.1038/s41577-019-0202-z. [DOI] [PubMed] [Google Scholar]
- 33.Yan M., Jurasz P. The role of platelets in the tumor microenvironment: from solid tumors to leukemia. Biochim. Biophys. Acta. 2016;1863:392–400. doi: 10.1016/j.bbamcr.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 34.Singan V.R., Simpson J.C. Implementation of the Rank-Weighted Co-localization (RWC) algorithm in multiple image analysis platforms for quantitative analysis of microscopy images. Source Code Biol. Med. 2016;11:2. doi: 10.1186/s13029-016-0048-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singan V.R., Jones T.R., Curran K.M., Simpson J.C. Dual channel rank-based intensity weighting for quantitative co-localization of microscopy images. BMC Bioinformatics. 2011;12:407. doi: 10.1186/1471-2105-12-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dunn K.W., Kamocka M.M., McDonald J.H. A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 2011;300:C723–C742. doi: 10.1152/ajpcell.00462.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Del Valle J.R., Devaux P., Hodge G., Wegner N.J., McChesney M.B., Cattaneo R. A vectored measles virus induces hepatitis B surface antigen antibodies while protecting macaques against measles virus challenge. J. Virol. 2007;81:10597–10605. doi: 10.1128/JVI.00923-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.