

Abstract
Tongue squamous cell carcinoma (TSCC) is closely linked to head and neck cancers. Here, we sought to explore the role and mechanism of lncRNAs in the occurrence and progression of TSCC and cisplatin resistance. The results of next-generation transcriptomic sequencing revealed that lncRNA-SNHG26 was differentially expressed and was associated with TSCC cisplatin resistance. The Cancer Genome Atlas dataset and tumor tissue analysis revealed that high SHNG26 expression was associated with the occurrence, progression, and poor prognosis of TSCC. Evidence from cell and animal experiments showed that SNHG26 expression was positively correlated with TSCC proliferation, epithelial-mesenchymal transformation, migration, invasion, and cisplatin resistance. Furthermore, in TSCC cells, SNHG26 was found to bind directly to the PGK1 protein, inhibiting its ubiquitination and activating the Akt/mTOR signaling pathway. These findings suggest that lncRNA-SNHG26 may be a promising target for inhibiting TSCC progression and improving sensitivity to cisplatin chemotherapy in TSCC.
Keywords: ubiquitination.Tongue squamous cell carcinoma, lncRNA-SNHG26, cisplatin, chemotherapy resistance, epithelial-mesenchymal transformation, ubiquitination, PGK1, Akt/mTOR
Graphical abstract
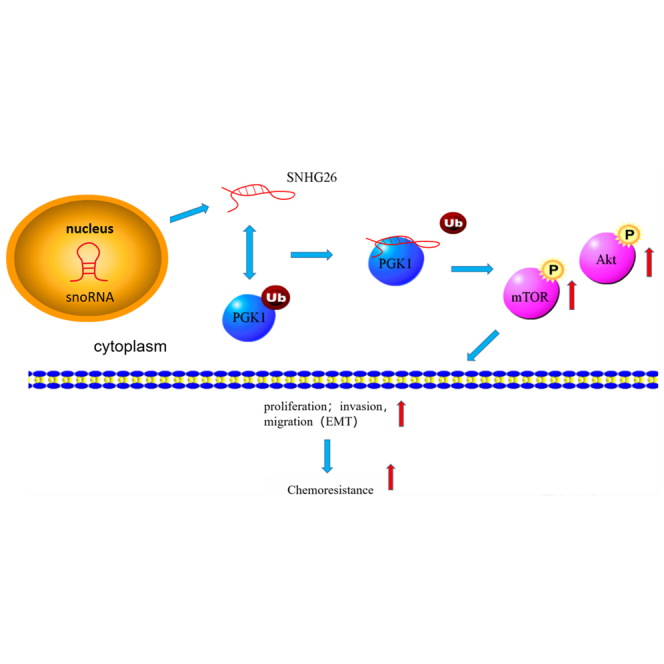
lncRNA-SNHG26 can be used as a prognostic indicator for tongue cancer patients and has potential value in targeted diagnosis and development of targeted therapy.
Introduction
Head and neck malignancies are the sixth leading malignancy cases worldwide.1,2 The vast majority of head and neck malignancies are tongue squamous cell carcinoma (TSCC).3,4 TSCC treatment has evolved from simple surgical treatment in the past to comprehensive sequential treatment approaches, including surgery, radiotherapy, and chemotherapy.5 Previous evidence shows that the 5-year survival rate of advanced TSCC patients is less than 50%.6 Scholars have noted that the poor prognosis of TSCC patients is closely associated with poor chemotherapy efficacy due to chemotherapy resistance.7,8 Therefore, the present investigation explores the molecular mechanisms related to TSCC chemotherapy resistance and provides a basis for improving the efficacy of TSCC chemotherapy.
Long non-coding RNAs (lncRNAs) are a class of transcripts with more than 200 bases and lack protein-coding ability.9 lncRNAs were regarded as background noise generated during transcription.10 According to recent research, lncRNA plays an important role in the occurrence and progression of cancer, as well as the process of chemotherapy resistance.11,12 lncRNAs, through their effects on DNA, mRNA, and proteins, have a potential role in regulating proliferation, invasion, migration, apoptosis, epithelial-mesenchymal transition, and other important biological processes of tumor cells.13, 14, 15, 16, 17, 18, 19 lncRNAs are yet to realize real clinical transformation application; however, in view of the multiple specific expression patterns of lncRNAs in cancer and the various effects in the field of molecular function, they have great application potential in cancer diagnosis, prognosis evaluation, and targeted therapy.20, 21, 22, 23
Herein, TSCC chemo-resistant cell line (CAL27/CDDP) and TSCC cell line (CAL27) were analyzed via next-generation sequencing technology to reveal the differentially expressed lncRNAs. Because the results showed that lncRNA-SNHG26 was significantly upregulated in CAL27/CDDP, it was chosen for further investigation.
Small nucleolar RNA host genes (SNHGs) are a class of lncRNAs derived from small nucleolar RNAs (snoRNAs).24 In recent years, SNHGs have become a research hotspot. For example, evidence shows that SNHGs, including SNHG1, SNHG3, SNHG5, SNHG7, and SNHG15, have an oncogenic role in cancer through the versatility of their interactions at the DNA-RNA-protein level.25 These SNHGs could regulate methylase26 or interact with transcription factors27 in the nucleus, sponge miRNA,28 directly bind to mRNA,29 or bind to a protein30 in the cytoplasm. In particular, for SHNG26, a member of SNHGs class, its function is unknown.
The current study found that SHNG26 expression was significantly higher in TSCC tissues than in paracancer tissues. In addition, the expression level of SHNG26 was correlated with the malignancy degree and poor prognosis in TSCC patients. In vivo and in vitro experiments revealed that SNHG26 promoted proliferation and epithelial-mesenchymal transformation (EMT) of TSCC, as well as increased TSCC drug resistance. RNA pull-down and mass spectrometry analysis revealed that SNHG26 interacts with PGK1 and inhibits its ubiquitination, thereby increasing its stability and activating the Akt/mTOR pathway.
Results
lncRNAs are differentially expressed between CAL27/CDDP and CAL27
To identify the transcripts involved in cisplatin (CDDP) resistance in TSCC cells in vitro, next-generation sequencing was employed to investigate the expression profiles of lncRNAs and mRNAs in CAL27/CDDP and its parental cell CAL27 (Figure 1A). Scatter and volcano plots showed differential expression of lncRNAs between CAL27/CDDP and CAL27 (Figure 1B). A total of 734 upregulated lncRNAs and 842 downregulated lncRNAs (fold change >2.0 or <0.5; p value < 0.05) were identified in CAL27/CDDP (Table S1). The differentially expressed lncRNAs were explored through hierarchical clustering analysis (Figure 1C). Figure 1D shows the top 10 upregulated lncRNAs and top 10 downregulated lncRNAs. Subsequently, five upregulated lncRNAs, five downregulated lncRNAs, three upregulated mRNAs, and three downregulated mRNAs were selected randomly to validate the sequencing results, and their relative expression in CAL27/CDDP and CAL27 was determined (Figure S1). Quantitative real-time PCR (qRT-PCR) analysis confirmed the reliability of sequencing results and aberrant expression of a cluster of lncRNAs in CAL27/CDDP cells.
Figure 1.

lncRNA expression profile in TSCC cells
(A) Drug sensitivity of CAL27 and CAL27/CDDP cells against CDDP. (B) MA plot showing differential expression of lncRNAs between CAL27 and CAL27/CDDP cells. The volcano plot was constructed using fold-change values and p values. The green and red points in the plots represent statistically differentially expressed lncRNAs. (C) Cluster heatmap showing differentially expressed lncRNAs over 2.0-fold change. Red color indicates high expression level, and green color indicates low expression level. (D) The top 10 upregulated lncRNAs and top 10 downregulated lncRNAs. The arrow indicates SNHG26.
SNHG26 was selected from the differentially expressed lncRNAs for subsequent analysis. The Sequencing platform and the UCSC genome database (http://genome.ucsc.edu/) revealed that SNHG26 is located on human chromosome 7: 22,895,705–22,896,694, with a genomic length of 574 bp. The full-length sequence and predicted secondary structure of SNHG26 are presented in Figure S2A. According to the prediction by bioinformatics webservers (Coding Potential Assessment Tool, http://lilab.research.bcm.edu/cpat/), SNHG26 is a noncoding RNA (Figure S2B).
The subcellular localization of lncRNA was found to be closely related to its biological function. Using the lncLocator webserver (www.csbio.sjtu.edu.cn/bioinf/lncLocator), bioinformatics analysis revealed that SNHG26 is found in the cytoplasm (Figure 2A). Furthermore, fluorescence in situ hybridization (FISH) analysis of CAL27 and SCC15 (Figure 2B) and nucleocytoplasmic separation verified that SNHG26 is mainly located in the cytoplasm (Figure 2C).
Figure 2.

Subcellular localization of SNHG26
(A) Prediction analysis using lncLocator showed that SNHG26 was mainly localized in the cytoplasm. (B) FISH analysis showed that SNHG26 was mainly localized in the cytoplasm (×400 magnification). (C) Nucleocytoplasmic separation analysis using qRT-PCR confirmed that SNHG26 was mainly expressed in the cytoplasm.
High SNHG26 expression in TSCC tissues is correlated with the prognosis of TSCC patients
To explore the role of SNHG26 in cancer, cancer patient data were retrieved from The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/). Analysis showed high SNHG26 expression in 15 cancers, including head and neck squamous cell carcinoma (HNSC) (Figure 3A). Further in-depth analysis of HNSC revealed significantly higher SNHG26 expression in cancer tissues compared with that of adjacent normal tissues (Figure 3B). These findings demonstrate that SNHG26 has an upstanding diagnostic potential for HNSC patients (area under the curve = 0.820) (Figure 3C). Kaplan-Meier (KM) curve analysis of HNSC patients was performed based on SNHG26 expression level. The analysis showed that the survival of patients in the high SNHG26 expression group was higher compared with that of patients in the low SNHG26 expression group (Figure 3D). By analyzing the treatment outcome of HNSC patients, we found that patients expressing high SNHG26 levels were highly correlated with poor prognosis compared with those expressing low SNHG26 levels (Figure 3E).
Figure 3.

Bioinformatics analysis of SNHG26 expression using TCGA-HNSC dataset
(A) SNHG26 was highly expressed in 15 cancers including HNSC. (B) Expression level of SNHG26 in HNSC tissues was significantly higher compared with that in adjacent noncancerous tissues. (C) receiver operating characteristic curve analysis showed SNHG26 had a good diagnostic ability for patients with HNSC. (D) Kaplan-Meier analysis showed that the survival of patients in SNHG26 high expression group was higher compared with that of patients in the SNHG26 low expression group (high versus low expression, based on the best cutoff values of SNHG26 expression). (E) Analysis of the treatment outcome of patients with HNSC showed that expression level of SNHG26 in patients with poor prognosis (progressive or stable disease) was higher compared with that in patients with good prognosis (complete or partial remission). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
The transcript levels of SNHG26 in 42 pairs of TSCC tissues and their adjacent noncancerous tissues obtained from First Affiliated Hospital of Nanchang University were assessed by qRT-PCR. The results showed that the transcript levels of SNHG26 were significantly higher in TSCC tissues compared with the levels in adjacent noncancerous tissues (Figure 4A). TSCC patients were divided into high- and low-SNHG26 groups based on the median relative SNHG26 expression in TSCC tissues to explore the correlation of the transcript levels of SNHG26 with the prognosis of TSCC patients. Based on clinical and pathologic characteristics of patients, the analysis showed that the expression level of SNHG26 was significantly related to the T stage and histological grade (Table 1). Further t test analysis revealed that the expression level of SNHG26 in the T3/T4 group was significantly higher than that in the T1/T2 group and that the expression level of SNHG26 in the G2/G3 group was significantly higher than that in the G1 group (Figure 4B). These findings suggest that SNHG26 is highly expressed in TSCC cells and that its level of expression is related to clinical severity and prognosis.
Figure 4.

Expression profile of SNHG26 in TSCC tissues
(A) SNHG26 was significantly upregulated in TSCC tissues compared with the level in adjacent normal tissues. (B) Expression level of SNHG26 in T3/T4 group was significantly higher compared with that of T1/T2 group, and expression level of SNHG26 in G2/G3 group was significantly higher compared with that in G1 group (TNM staging was performed using the eighth edition of the AJCC staging system for oral cancer). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Table 1.
The association of SNHG26 expression in forty-two TSCC patients with clinicopathologic characteristics
| Characteristic | Low expression of SNHG26 | High expression of SNHG26 | p value |
|---|---|---|---|
| n | 21 | 21 | |
| Age, n (%) | 0.758 | ||
| ≤50 | 10 (47.6%) | 11 (52.4%) | |
| >50 | 11 (52.4%) | 10 (47.6%) | |
| Gender, n (%) | 0.217 | ||
| Female | 9 (42.9%) | 13 (61.9%) | |
| Male | 12 (57.1%) | 8 (38.1%) | |
| T stage, n (%) | 0.030∗ | ||
| T1/T2 | 15 (71.4%) | 8 (28.6%) | |
| T3/T4 | 6 (38.1%) | 13 (61.9%) | |
| N stage, n (%) | 0.537 | ||
| N0 | 11 (52.4%) | 9 (42.9%) | |
| N+ | 10 (47.6%) | 12 (57.1%) | |
| Clinical stage, n (%) | 0.533 | ||
| I/II | 10 (47.6%) | 8 (38.1%) | |
| III/IV | 11 (52.4%) | 13 (61.9%) | |
| Histologic grade, n (%) | 0.013∗ | ||
| G1 | 14 (66.7%) | 6 (28.6%) | |
| G2/G3 | 7 (33.3%) | 15 (71.4%) | |
| Lymphovascular invasion, n (%) | 0.346 | ||
| No | 10 (47.6%) | 7 (33.3%) | |
| Yes | 11 (52.4%) | 14 (66.7%) | |
∗p < 0.05 TNM staging was performed using the 8th edition of the AJCC staging system for oral cancer.
SNHG26 promotes the proliferation, migration, and invasion abilities of TSCC cells in vitro
The expression levels of SNHG26 in CAL27/CDDP, CAL27, SCC15, and HGE cells were evaluated by qRT-PCR. The findings showed that SNHG26 expression was significantly upregulated in CAL27/CDDP and downregulated in HGE compared with the expression level in CAL27 and SCC15 cells (Figure 5A). Furthermore, to explore the role of SNHG26 in TSCC, CAL27 and SCC15 cells were transfected with siRNAs against SNHG26, and plasmids containing pcDNA3.1-SNHG26 were transfected into CAL27 and SCC15 cells to upregulate expression of SNHG26. Knockdown and overexpression efficiency was validated by qRT-PCR (Figure 5B). SNHG26 knockdown significantly increased CAL27 and SCC15 cell sensitivity to CDDP, whereas SNHG26 overexpression increased CAL27 and SCC15 cell IC50 values to CDDP (Figure 5C).
Figure 5.

Expression levels of SNHG26 in TSCC cells
(A) SNHG26 was significantly upregulated in CAL27/CDDP and downregulated in HGE compared with the expression in CAL27 and SCC15 cells. (B) Knockdown and overexpression efficiency was validated by qRT-PCR. (C) SNHG26 knockdown significantly increased sensitivity of CAL27 and SCC15 cells to CDDP, whereas SNHG26 overexpression increased IC50 values of CAL27 and SCC15 cells to CDDP. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Findings from 5-ethynyl-2′-deoxyuridine (EdU) and CCK-8 assays indicated that SNHG26 knockdown significantly decreased the proliferation capacity of CAL27 and SCC15 cells (Figure 6A), whereas SNHG26 overexpression promoted their proliferation capacity (Figure 6B). Flow cytometry analysis demonstrated that SNHG26 knockdown inhibited cell cycle progression of CAL27 and SCC15 cells (Figure 6C).
Figure 6.

Relationship between expression level of SNHG26 and proliferation of TSCC cells
(A and B) EdU (×200 magnification) and CCK-8 assays showed that knockdown of SNHG26 significantly reduced proliferation capacity of CAL27 and SCC15 cells, whereas overexpression of SNHG26 enhanced their proliferation capacity. (C) Flow cytometry analysis showed that knockdown of SNHG26 inhibited cell cycle progression of CAL27 and SCC15. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Wound healing assay demonstrated that SNHG26 knockdown significantly decreased migration ability of CAL27 and SCC15 cells (Figure 7A), whereas SNHG26 overexpression promoted the migration ability of CAL27 and SCC15 cells (Figure 7B). Findings from the transwell assay revealed that SNHG26 f significantly reduced the invasion ability of CAL27 and SCC15 cells, whereas SNHG26 overexpression promoted their invasion ability (Figure 7C).
Figure 7.

Relationship between expression of SNHG26 and metastasis of TSCC cells
(A and B) Wound healing assay (×200 magnification) showed that SNHG26 knockdown significantly reduced migration ability of CAL27 and SCC15 cells, whereas overexpression of SNHG26 enhanced their migration ability. (C) Transwell assay (×200 magnification) showed that knockdown of SNHG26 significantly reduced invasion ability of CAL27 and SCC15 cells, whereas overexpression of SNHG26 enhanced their invasion ability. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Western blot (WB) assay results showed that SNHG26 knockdown decreased expression level of Ki-67, PCNA, Snail, N-cadherin, vimintin, MMP-2, and MMP-9 in CAL27 and SCC15 cells, and increased expression level of E-cadherin. Contrarily, SNHG26 overexpression increased the expression level of Ki-67, PCNA, Snail, N-cadherin, vimintin, MMP-2, and MMP-9, and decreased the expression level of E-cadherin in CAL27 and SCC15 cells (Figure 8).
Figure 8.

Effect of SNHG26 on expression of proliferation- and EMT-related proteins
(A) SNHG26 knockdown decreased expression levels of Ki-67, PCNA, Snail, N-cadherin, vimintin, MMP-2, and MMP-9 in CAL27 and SCC15 cells, and increased expression level of E-cadherin. (B) Overexpression of SNHG26 increased expression of Ki-67, PCNA, Snail, N-cadherin, vimintin, MMP-2, and MMP-9, and decreased expression of E-cadherin in CAL27 and SCC15 cells. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
SNHG26 directly interacts with PGK1 and upregulates its protein level by inhibiting PGK1 ubiquitination in TSCC cells in vitro
To investigate the underlying molecular mechanism by which SNHG26 promotes TSCC cell proliferation, migration, and invasion, an RNA pull-down assay was used to identify the protein partners of SNHG26 in CAL27. Silver staining was used to examine RNA-associated proteins that were pulled down by biotin-labeled sense- or antisense-SNHG26 (Figure 9A). Mass spectrometry was then used to analyze the two protein bands. The results revealed that PGK1 was expressed differently in the SNHG26 pull-down group than in the control group (Table S2). A possible interaction between SNHG26 and PGK1 was predicted using the catRAPID tool (http://service.tartaglialab.com/page/catrapid_group) (Figure 9B). PGK1 catalyzes an important step in glycolysis and is essential in the production of glycolytic ATP.31 According to recent research, PGK1 is involved in the occurrence and development of tumors in a variety of ways.32 The immunoblotting analysis further revealed that SNHG26 interacted with PGK1 directly (Figure 9C). The results of the RNA immunoprecipitation (RIP) assay revealed that the antibody to PGK1 captured significantly more SNHG26 than the antibody to IgG in CAL27 and SCC15 cells (Figure 9D). These suggest that SNHG26 interacts with PGK1 specifically in TSCC cells.
Figure 9.

SNHG26 directly interacts with PGK1 in TSCC cells
(A) Silver-stained SDS-PAGE gel of proteins immunoprecipitated from CAL27 cell extract by the sense and antisense RNA of SNHG26. The two lanes were used for mass spectrum determination using liquid chromatography dual mass spectrometry. The frame indicates PGK1. (B) Analysis of the interaction propensities between SNHG26 and PGK1 using catRAPID tool. (C) RNA pull-down assay conducted using biotin-labeled SNHG26 probe and PGK1 expression was determined by western blot assay. (D) Amount of SNHG26 bound to SNRNP70 (a positive control), PGK1, or IgG (a negative control) was detected by qRT-PCR after RIP in TSCC cells. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To investigate the effects of SNHG26 on PGK1, SNHG26 expression levels in TSCC cells were altered, and PGK1 mRNA and protein levels were measured. The results showed that when SNHG26 was silenced or overexpressed in TSCC cells, mRNA levels of PGK1 did not change significantly (Figure 10A). PGK1 protein levels, on the other hand, were reduced in CAL27 and SCC15 cells when SNHG26 was silenced and increased in CAL27 and SCC15 cells when SNHG26 was overexpressed (Figure 10B).
Figure 10.

SNHG26 inhibits PGK1 ubiquitination in TSCC cells
(A) mRNA levels of PGK1 were not significantly different when SNHG26 was silenced or overexpressed in TSCC cells. (B) Protein levels of PGK1 were reduced in CAL27 and SCC15 cells when SNHG26 was silenced and were upregulated in CAL27 and SCC15 cells when SNHG26 was overexpressed. (C) Knockdown of SNHG26 significantly decreased the half-life of PGK1. (D) Overexpression of SNHG26 in CAL27 and SCC15 had similar effect as addition of MG-132. (E) Co-IP assay showed that ubiquitination of PGK1 in CAL27 and SCC15 cells was significantly increased by SNHG26 knockdown. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
These findings suggest that SNHG26 may regulate the synthesis and/or degradation of PGK1 protein. Thus, CAL27 and SCC15 cells were treated with the protein synthesis inhibitor CHX, and the results revealed that knocking down SNHG26 significantly reduced the half-life of PGK1 (Figure 10C). Moreover, the analysis revealed that the overexpression of SNHG26 in CAL27 and SCC15 had a similar effect as the addition of MG-132 (a widely used and efficient proteasome inhibitor), which increased PGK1 expression (Figure 10D). These findings suggest that SNHG26 inhibits proteasome-dependent degradation of PGK1 in TSCC cells.
To confirm that the ubiquitin-proteasome pathway was responsible for SNHG26-mediated PGK1 degradation, a co-immunoprecipitation (co-IP) assay was used to detect PGK1 ubiquitination. The findings showed that ubiquitination of PGK1 in CAL27 and SCC15 cells was significantly increased by SNHG26 knockdown (Figure 10E). These results show that a direct interaction between SNHG26 and PGK1 prevents ubiquitination and degradation of PGK1 in TSCC cells in vitro.
SNHG26-mediated regulation of Akt/mTOR signaling pathway through PGK1 influences proliferation and EMT-related protein expression
Previous research has shown that PGK1 can regulate the expression of the Akt/mTOR pathway33, 34, 35 and that activating the Akt/mTOR pathway can promote cancer cell proliferation, invasion, and migration.36 The STRING (https://www.string-db.org/) and TCGA-HNSC databases were used to investigate the potential regulatory relationship between PGK1 and Akt/mTOR (Figure 11A). Experiments with knockdown and overexpression of PGK1 showed that it can promote Akt and mTOR phosphorylation (Figure 11B).
Figure 11.

SNHG26 regulates the Akt/mTOR signaling pathway via PGK1 to affect proliferation and EMT-related protein expression
(A) STRING and TCGA-HNSC databases showed that PGK1 could interact with AKt/mTOR pathway and has a positive co-expression relationship. (B and C) WB experiment confirmed that PGK1 and SNHG26 could improve the phosphorylation level of Akt/mTOR. (D) The addition of mTOR activator (MHY1485) could reverse the effects of SNHG26 knockdown on proliferation and EMT-related proteins. (E) Overexpression of PGK1 could reverse the effects of SNHG26 knockdown on proliferation and EMT-related proteins.
Further investigation showed that SNHG26 promoted Akt and mTOR phosphorylation (Figure 11C), and that mTOR activators reversed the effects of SNHG26 knockdown on proliferation and EMT-related proteins (Figure 11D). Moreover, it was discovered that PGK1 overexpression reversed the effects of SNHG26 knockdown on Akt/mTOR, proliferation, and EMT-related proteins (Figure 11E). These findings show that SNHG26 activates the Akt/mTOR signaling pathway to influence proliferation and EMT-related protein expression by interacting with PGK1.
Effects of SNHG26 knockdown or overexpression on tumor growth in vivo
To investigate the role of SNHG26 on TSCC cells in vivo, CAL27 with stable transfection with Lv-shRNA-SNHG26, Lv-OV-SNHG26, or Lv-control were injected subcutaneously into nude mice (n = 15). The nude mice were given CDDP (2 mg/kg) intraperitoneally once a week for 4 weeks after tumor cell inoculation. Tumor sizes were measured every week using micrometer calipers. All nude mice were killed after 5 weeks from the tumor cell inoculation and the tumors were removed.
To confirm SNHG26 knockdown or overexpression in the Lv-shRNA-SNHG26 group or Lv-OV-SNHG26 group, qRT-PCR was performed on mouse tumor tissues (Figure 12A). The tumor growth in the Lv-control group was significantly faster than that in the Lv-shRNA-SNHG26 group, but slower than that in the Lv-OV-SNHG26 group (Figure 12B). Furthermore, immunohistochemistry (IHC) staining and WB revealed that compared with the Lv-control group, Ki-67, PGK1, N-cadherin, vimentin, and phosphorylated Akt/mTOR were significantly downregulated in the Lv-shRNA-SNHG26 group, but significantly upregulated in the Lv-OV-SNHG26 group (Figure 12C). These findings showed that SNHG26 knockdown inhibited tumorigenicity activity of TSCC cells in vivo, while overexpression of SNHG26 enhanced their tumorigenicity activity, which is consistent with the in vitro findings.
Figure 12.

Effects of SNHG26 knockdown or overexpression on tumor growth in vivo
(A) Relative expression level of SNHG26 in mouse tumor tissues under different transfection conditions as determined by qRT-PCR. (B) Representative images of xenograft tumors in the three groups and effect of SNHG26 modulation on tumor growth and tumor weight in nude mice are shown. (C) Expression levels of PGK1, p-Akt/Akt, p-mTOR/mTOR, Ki-67, PCNA, Snail, E-cadherin, N-cadherin, and vimentin in tumor samples from nude mice in the three groups as detected by WB analysis. (D) Expression levels of PGK1, Ki-67, E-cadherin, and N-cadherin in tumor samples from nude mice in the three groups as determined by IHC (×400 magnification).
To investigate the relationship between SNHG26 and PGK1, Ki-67, E-cadherin, and N-cadherin further, IHC staining was performed to determine the expression levels of PGK1, Ki-67, E-cadherin, and N-cadherin in the high-SNHG26 (≥ median value) and low-SNHG26 (< median value) human TSCC tissues (n = 42). The findings revealed that the tissues with high SNHG26 expression levels had higher levels of PGK1, Ki-67, and N-cadherin staining, and lower levels of E-cadherin when compared with tissues with low SNHG26 expression (Figure 13).
Figure 13.

Correlation between SNHG26 and PGK1, Ki-67, E-cadherin, and N-cadherin in TSCC tissues (×200 magnification)
Discussion
Several studies have found that lncRNAs play important roles in tumorigenesis and the development of different cancers. According to research evidence, lncRNAs influence TSCC proliferation, metastasis, apoptosis, and chemotherapy resistance.37, 38, 39 In comparison with other tumors, research on lncRNAs in TSCC is limited. The current study aimed to investigate the key lncRNAs associated with drug resistance in TSCC as well as their functional effects on TSCC and the specific molecular mechanism.
SNHGs, a gene family derived from snoRNA, and its members, SNHG1, SNHG5, SNHG7, and SNHG15, have been linked to cancer development.25 SNHG26 has never been studied before. The current study found that lncRNA SNHG26 can promote TSCC proliferation, EMT, and cisplatin resistance.
According to the mechanism, SNHG26 binds to PGK1 in the cytoplasm, inhibits ubiquitination of PGK1, and promotes PGK1 stability. PGK1 is a glycolytic enzyme that plays an important role in metabolism. In addition to regulating glycolytic metabolism, PGK1 is involved in the initiation of autophagy, DNA replication, and repair of mammalian nuclear cells.40
Previous research has shown that PGK1 can promote the growth and progression of a variety of tumors, as well as their tolerance to a variety of chemotherapy drugs.32 Using TCGA-HNSC and GEO (GSE6631,GSE65858) data, we showed that PGK1 is highly correlated with the occurrence, progression, and prognosis of HNSC (Figure 14).
Figure 14.

PGK1 is highly correlated with occurrence, development, and prognosis of HNSC as shown by analysis using TCGA-HNSC and GEO data
(A) PGK1 was upregulated in cancer tissues compared with the adjacent paracancer tissues. (B) KM survival curve showing poor overall survival in PGK1-high expression group of patients with HNSC. (C) Representative images of IHC staining of PGK1 from the Human Protein Atlas database. (D) Expression of PGK1 is associated with diagnosis of HNSC. (E) High expression of PGK1 is associated with poor pathological stage of HNSC.
Previous evidence33,35,41 has linked PGK1 to the inactivation of the Akt/mTOR pathway. The analysis of STRING and TCGA databases showed that there was experimental evidence for the interaction between PGK1 and Akt/mTOR, as well as a positive co-expression relationship (Figure 11A). Our research also confirms this. The activation of Akt/mTOR pathway by PGK1 comes from the glycolysis process mediated by PGK1. However, the specific interaction mechanism between PGK1 and Akt/mTOR is lacking in our and previous studies. This is the defect of this study and the goal of our future research.
The Akt/mTOR signaling pathway is a well-known signaling pathway that promotes cancer growth and metastasis.42,43 Previous studies looked into the role of lncRNA in regulating the Akt/mTOR signaling pathway via the ceRNA mechanism.44, 45, 46 The current study found that SNHG26 can inhibit ubiquitination of PGK1 and regulate the Akt/mTOR pathway via the direct action of lncRNA-protein.
Mounting evidence shows that apoptosis and autophagy are closely linked to tumor chemotherapy resistance.47, 48, 49, 50 Herein, the findings showed that SNHG26 did not affect apoptosis and autophagy of TSCC cells. SNHG26 could indirectly increase cisplatin resistance of tongue cancer by enhancing its proliferative capacity. Recent studies indicate that the emergence of acquired drug resistance of tumor cells is accompanied by EMT-like changes.51, 52, 53, 54 This is due to the fact that cancer EMT can enhance cancer cell metastasis, thereby indirectly weakening the effect of chemotherapy drugs. Furthermore, there may be common signaling molecules that are shared by the two processes. Snail, Twist, ZEB, and other important EMT regulators, for example, have been shown in studies to mediate the development of chemotherapy resistance in different tumors.54, 55, 56 The current study found changes in SNHG26 expression were positively correlated with TSCC proliferation and EMT, which led to the effect of SNHG26 on TSCC cisplatin resistance.
Conclusions
In summary, the findings of the current study show that SNHG26 is an oncogene associated with malignant progression and poor prognosis in TSCC patients and that it promotes TSCC cell growth, metastasis, and cisplatin resistance. Furthermore, the study shows that SNHG26 promotes TSCC proliferation and EMT by inhibiting PGK1 ubiquitination and activating the Akt/mTOR pathway. These findings suggest that SNHG26 can be used as a prognostic indicator for TSCC patients and has potential utility in targeted diagnosis and therapy development.
Materials and methods
Cells and human tissues
Human TSCC cell lines CAL27 and SCC15 were obtained from Shaanxi Provincial Key Laboratory of Craniomaxillofacial Precision Medicine, Hospital of Stomatology, Xi'an Jiaotong University (Xi'an, China). Human TSCC cell line CAL27/CDDP was obtained from Sun Yat-sen Memorial Hospital (Guangzhou, China).57
TSCC cells were maintained in DMEM high-glucose medium supplemented with 10% fetal blood sampling (FBS), 100 units/mL penicillin, and 100 μg/mL streptomycin. Cells were maintained under a humidified atmosphere at 37°C with 5% CO2. Forty-two pairs of TSCC tissues and their corresponding adjacent noncancerous tissues were obtained from TSCC patients who had undergone surgery at the First Affiliated Hospital of Nanchang University (Nanchang, China) between September 2019 and June 2021. Patients were diagnosed with TSCC based on histopathological examination, and no pre-operative treatment was administered. All patients provided written informed consent. The Human Ethics Committee of First Affiliated Hospital of Nanchang University approved this study (No. 2020054).
Next-generation RNA sequencing analysis
RNA sequencing was performed by Oebiotech (Shanghai, China). The mirVana miRNA Isolation Kit was used to extract total RNA. TruSeq Stranded mRNA LTSample Prep Kit (Illumina, USA) was used to create libraries, which were then sequenced on the Illumina sequencing platform (HiSeqTM 2500 or Illumina HiSeq X Ten). Differentially expressed genes were identified using the functions estimateSizeFactors and nbinomTest in the DESeq R package.58 The threshold for significant differential expression was set at p value <0.05 and fold-change >2 or fold-change <0.5.59
RNA extraction and qRT-PCR
Using Trizol reagent, total RNA was extracted from TSCC tissues or cell lines (Invitrogen, USA). RNA was then reverse-transcribed into cDNA using the PrimeScript RT reagent Kit (TaKaRa, China). The qRT-PCR was performed using the SYBR premix DimmerEraser kit (TaKaRa, China) in accordance with the manufacturer's instructions. The u2−ΔΔCt method was used to calculate the fold change in relative mRNA expression. Table S3 contains a list of the primer sequences that were used.
RNA interference and overexpression
Small interfering RNAs (siRNAs) corresponding to the target human SNHG26 and PGK1 sequences were purchased from Ribo (Guangzhou, China). pcDNA3.1-SNHG26 and pcDNA3.1-PGK1 vectors were purchased from Ribo (Guangzhou, China). Transfection was performed using Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer's protocol. Lentiviral vectors containing shRNA-SNHG26 and overexpressing SNHG26 were purchased from Ribo (Guangzhou, China). RNA interference sequences are listed in Table S3.
RNA fluorescence in situ hybridization
The FISH kit and SNHG26 FISH probe were purchased from RiboBio (Guangzhou, China). CAL27 and SCC15 cells were inoculated and fixed with 4% paraformaldehyde for pre-hybridization. Cells were then hybridized overnight in the dark with a 5-mM SNHG26 probe. Cells were counterstained with Hoechst and imaged on the following day.
Western blot
Total protein was extracted from tissue samples and cell lines using cell lysis buffer (Beyotime, China). BCA protein assay kit (Bio-rad, USA) was used to determine protein concentration. Protein samples were heated to 100°C and incubated for 5 min before being electrophoresed on SDS- polyacrylamide gels. The protein was extracted from an SDS gel and transferred to a polyvinylidene fluoride membrane. The membrane was blocked with 5% skimmed milk for 60 min. The membrane was incubated with anti-rabbit polyclonal antibody E-cadherin (1:2,000, Proteintech, China), anti-rabbit polyclonal antibody N-cadherin (1:2,000, Proteintech, China), anti-rabbit polyclonal antibody vimentin (1:2,000, Proteintech, China), anti-rabbit polyclonal antibody MMP-2 (1:1,000, Proteintech, China), anti-rabbit polyclonal antibody MMP-9 (1:2,000, Proteintech, China), anti-rabbit polyclonal antibody Ki-67 (1:1,000, Servicebio, China), anti-rabbit polyclonal antibodies mTOR/p-mTOR (1:1,000, Servicebio, China), anti-rabbit monoclonal antibodies Akt/p-Akt (1:2,000, Cell Signaling Technology, USA), anti-rabbit monoclonal antibody PGK1 (1:2,000, Abcam, USA), antirabbit polyclonal antibody ubiquitin (1:1,000, Proteintech, China), anti-mouse monoclonal antibody PCNA (1:2,000, Cell Signaling Technology, USA), anti-rabbit monoclonal antibody Snail (1:2,000, Proteintech, China), and anti-mouse polyclonal antibody β-actin (1:5,000, Proteintech, China) at 4°C overnight. Following incubation, samples were rinsed three times after incubation. The membrane was then incubated for 1 h at room temperature with goat-anti-mouse second antibody or goat-anti-rabbit second antibody (1:5,000, Proteintech, China), then rinsed several times. The membrane was then imaged with X-ray film and analyzed with an enhanced chemiluminescence performance system. ImageJ software was then used to quantify the images.
Immunohistochemistry assay
Tissue slices were immersed in 0.01 M citrate buffer (pH 6.0; Wellbio, China). The buffer was brought to a boil and cooked for 20 min before being cooled to room temperature. Tissue slices were washed with PBS (pH 7.2–7.6; China) after cooling. The samples were then incubated at 4°C overnight with diluted primary antibody. Following incubation with primary antibody, tissue slices were washed with PBS, and 50–100 μL of anti-rabbit-IgG antibody-HRP polymer (Thermo Fisher Scientific, China) was added. A working solution of 50–100 μL of the chromogenic reagent DAB (Zhongshan Jinqiao Biotechnology, China) was also added. Tissues were counterstained with hematoxylin before being mounted with neutral gum (Sigma, USA). Tissue slices were then examined under a microscope (OLYMPUS, Japan, BX43). Antibodies for IHC staining included Ki-67 (1:200, Servicebio, China), PGK1 (1:200, Proteintech, China), E-cadherin (1:200, Proteintech, China), and N-cadherin (1:200, Proteintech, China). The Ki-67 expression level was graded based on the percentage of staining. The histochemistry score (H-score) was used to determine the expression levels of PGK1, E-cadherin, and N-cadherin. H-score = ΣPi (i + 1), where i denotes the intensity score and Pi denotes the percentage of the cells.
In vitro and in vivo chemosensitivity assay
For in vitro experiments, drug-resistant or normal TSCC cells were seeded into 96-well plates (5 × 103 cells/well) after transfection. Following a 24-h incubation period, a medium containing different concentrations of CDDP was added to each well. Cell Counting Kit-8 (CCK-8, Dojindo, Japan) was added after 24 h of treatment, and the absorbance (450 nm) was measured according to the manufacturer's protocol. The dose-response curves were then used to calculate the 50% inhibition of growth (IC50) value for CDDP.
For in vivo experiments, CAL27 cells (1.0 × 106 cells in 100 μL) that were stably transfected with Lv-shRNA-SNHG26, Lv-OV-SNHG26, or Lv-control were injected subcutaneously into 4-week-old female nude mice. CDDP (2 mg/kg) was injected intraperitoneally once a week for 4 weeks after tumor cell inoculation. Every week, tumor sizes were measured, and the tumor volumes were calculated as 0.5 × length × width2 every week. Mice were sacrificed 5 weeks after implantation, and tumors were excised, weighed, and photographed. IHC staining and WB were used to identify all tumor grafts. The total or nuclear proteins of tumor grafts were extracted using a total and nuclear protein extraction kit (Beyotime, Shanghai, China). All animal experiments were performed in the animal laboratory center of First Affiliated Hospital of Nanchang University. The Animal Care and Use Committee of First Affiliated Hospital of Nanchang University approved the study protocol.
RNA pull-down and mass spectrometry analysis
A positive and reverse SNHG26 gene in vitro transcription template with the T7 promoter sequence was created and used as a probe for RNA pull-down. Biotin-labeled RNAs were transcribed in vitro with Biotin RNA Labeling Mix (Roche, USA) and T7 RNA polymerase (Promega, USA). RNA samples were purified using the RNeasy Mini Kit (Qiagen, Germany) and treated with RNase-free DNase (Roche, USA). Electrophoresis was used to separate the retrieved protein, which was then silver-stained. Following that, bands appearing in the experimental group were removed and sent for mass spectrometry analysis. Finally, the standard WB was used to validate the retrieved protein.
RNA immunoprecipitation
The RIP assay was performed according to the manufacturer's instructions using EZ-Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore, USA). Lysates of cells were prepared in RIP lysis buffer and then incubated with RIP buffer containing magnetic beads conjugated to the human anti-PGK1 antibody. The negative control was mouse IgG (Beyotime, China), and the positive control was anti-SNRNP70 (Millipore, USA). The co-precipitated RNAs were isolated with Trizol reagent (Invitrogen, USA) and quantified using qRT-PCR. Primer sequences used for the qRT-PCR are listed in Table S3.
Co-immunoprecipitation assay
The Pierce Co-IP kit (Thermo, USA) was used for the co-IP assay, according to the manufacturer's instructions. The cell lysates were prepared in RIPA buffer and primary antibodies or IgG were added to the lysates for overnight incubation at 4°C. Protein A/G agarose beads were added and incubated for 3 h at 4°C. Beads were collected after centrifugation and washed five times with RIPA buffer. As previously described, WB was performed using the indicated antibodies.
Cell proliferation and cell cycle analysis
Cell proliferation rate was determined using CCK-8 and EdU assay. The cell cycle was assessed using a flow cytometer. Cells were seeded into 96-well plates (1 × 103 cells/well) for the CCK-8 assay, and the absorbance (450 nm) was measured every 24 h for 72 h after treatment with CCK-8 reagent. EdU assay was performed using the EdU Kit (BeyoClickTM, China). The cells were co-cultured with EdU working solution for 2 h. After the cells were fixed, they were incubated in a dark environment at room temperature for 30 min with click reaction solution before being treated with Hoechst solution for 10 min. Using a fluorescence microscope, we captured images and counted cells by ImageJ software. Cell cycle analysis was performed using the Cell Cycle Analysis kit (Beyotim, China). The flow cytometry and FlowJo software 7.6 (Tree Star, USA) were used to evaluate and calculate the percentage of cells at different stages.
Wound healing assay
The rate of cell migration was determined using a wound healing assay. Transfected cells were plated in 6-well plates (1 × 105 cells/well), and incubated at 37°C in DMEM high-glucose medium without FBS until confluence reached 80%. Cells were scratched across the surface of the well using a 100-μL pipette. Cell migration to the scratches was observed after incubation at 37°C for 12, 24, 36, and 48 h.
Transwell invasion assay
Transwell invasion assay was performed in 24-well plates using 8-μm chamber inserts with Matrigel. In the upper chamber, the FBS-free medium was seeded at a density of 3 × 104 cells. Medium containing 20% FBS was added to the lower chamber. After 36 h, cells below the membrane were fixed and stained for 10 min with 0.1% crystal violet.
Statistical analysis
Data were presented as mean ± standard deviation (SD). The paired-samples t test was used to compare the expression of SNHG26 in TSCC tissues and adjacent noncancerous tissues. The independent samples t test was used to compare two groups. To compare differences between groups, a one-way ANOVA was used, followed by a Bonferroni post hoc test. Chi-square, Fisher exact test, or Pearson's Chi-square test were used to analyze categorical variables. For survival analysis, the KM method was used and the log rank test was used to determine the significance. Statistical significance was defined as p values less than 0.05.
Ethics approval and consent to participate
All experimental protocols were approved by the Ethics Committee of First Affiliated Hospital of Nanchang University (No. 2020054) and conducted in accordance with the principles of the World Medical Association Declaration of Helsinki. Written informed consent was obtained from all participants or their legal guardians prior to sample collection. All animal experiments were approved by the Medical Ethics Committee of First Affiliated Hospital of Nanchang University (No. 2020054) and conducted according to the Guidelines for the Care and Use of Animals for Scientific Research.
Consent for publication
All authors have read and agreed to publish this manuscript.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
The present study was supported by the National Natural Science Foundation of China (Grant No. 81860477) and Jiangxi Provincial Key R&D Plan (Grant No. 20212BBG71005).
Author contributions
Q.K.J. and J.X.Q. conceived and designed the experiments. Q.K.J., Z.H.W., and Q.Q. performed most of the experiments and data analysis. Q.K.J. and Z.H.W. drafted the manuscript. J.L.L. helped with the in vivo experiments. Y.Q.X. collected the clinical samples and information. All authors read and approved the final manuscript.
Declaration of interests
There is no conflict of interest to disclose.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2021.12.021.
Supplemental information
References
- 1.Siegel R.L., Miller K.D., Fuchs H.E., Jemal A. Cancer statistics, 2021. CA Cancer J. Clin. 2021;71:7–33. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 2.Gatta G., Botta L., Sánchez M.J., Anderson L.A., Pierannunzio D., Licitra L., Hackl M., Zielonke N., Oberaigner W., Van Eycken E., et al. Prognoses and improvement for head and neck cancers diagnosed in Europe in early 2000s: the EUROCARE-5 population-based study. Eur. J. Cancer. 2015;51:2130–2143. doi: 10.1016/j.ejca.2015.07.043. [DOI] [PubMed] [Google Scholar]
- 3.Marur S., Forastiere A.A. Head and neck squamous cell carcinoma: update on epidemiology, diagnosis, and treatment. Mayo Clin. Proc. 2016;91:386–396. doi: 10.1016/j.mayocp.2015.12.017. [DOI] [PubMed] [Google Scholar]
- 4.Robert B.M., Dakshinamoorthy M., Ganapathyagraharam Ramamoorthy B., Dhandapani M., Thangaiyan R., Muthusamy G., Nirmal R.M., Prasad N.R. Predicting tumor sensitivity to chemotherapeutic drugs in oral squamous cell carcinoma patients. Sci. Rep. 2018;8:15545. doi: 10.1038/s41598-018-33998-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Colevas A.D., Yom S.S., Pfister D.G., Spencer S., Adelstein D., Adkins D., Brizel D.M., Burtness B., Busse P.M., Caudell J.J., et al. NCCN guidelines insights: head and neck cancers, version 1.2018. J. Natl. Compr. Canc. Netw. 2018;16:479–490. doi: 10.6004/jnccn.2018.0026. [DOI] [PubMed] [Google Scholar]
- 6.Ferlay J., Soerjomataram I., Dikshit R., Eser S., Mathers C., Rebelo M., Parkin D.M., Forman D., Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 7.Florea A., Büsselberg D. Cisplatin as an anti-tumor drug: cellular mechanisms of activity, drug resistance and induced side effects. Cancers. 2011;3:1351–1371. doi: 10.3390/cancers3011351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu L., Zhang Y., Zhang P., Dai X., Gao Y., Lv Y., Qin S., Xu F. Integrated metabolomics and network pharmacology strategy-driven active traditional Chinese medicine ingredients discovery for the alleviation of cisplatin nephrotoxicity. Chem. Res. Toxicol. 2019;32:2411–2421. doi: 10.1021/acs.chemrestox.9b00180. [DOI] [PubMed] [Google Scholar]
- 9.Liang H., Zhao X., Wang C., Sun J., Chen Y., Wang G., Fang L., Yang R., Yu M., Gu Y., et al. Systematic analyses reveal long non-coding RNA (PTAF)-mediated promotion of EMT and invasion-metastasis in serous ovarian cancer. Mol. Cancer. 2018;17:96. doi: 10.1186/s12943-018-0844-7. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10.Wilusz J.E., Sunwoo H., Spector D.L. Long noncoding RNAs: functional surprises from the RNA world. Gene Dev. 2009;23:1494–1504. doi: 10.1101/gad.1800909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang G., Lu X., Yuan L. LncRNA: a link between RNA and cancer. Biochim. Biophys. Acta. 2014;1839:1097–1109. doi: 10.1016/j.bbagrm.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 12.Wu X., Zheng Y., Han B., Dong X. Long noncoding RNA BLACAT1 modulates ABCB1 to promote oxaliplatin resistance of gastric cancer via sponging miR-361. Biomed. Pharmacother. 2018;99:832–838. doi: 10.1016/j.biopha.2018.01.130. [DOI] [PubMed] [Google Scholar]
- 13.Tang T., Guo C., Xia T., Zhang R., Zen K., Pan Y., Jin L. LncCCAT1 promotes breast cancer stem cell function through activating WNT/beta-catenin signaling. Theranostics. 2019;9:7384–7402. doi: 10.7150/thno.37892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu B., Liu Q., Pan S., Huang Y., Qi Y., Li S., Xiao Y., Jia L. The HOTAIR/miR-214/ST6GAL1 crosstalk modulates colorectal cancer procession through mediating sialylated c-Met via JAK2/STAT3 cascade. J. Exp. Clin. Cancer Res. 2019;38:455. doi: 10.1186/s13046-019-1468-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hua K., Deng X., Hu J., Ji C., Yu Y., Li J., Wang X., Fang L. Long noncoding RNA HOST2, working as a competitive endogenous RNA, promotes STAT3-mediated cell proliferation and migration via decoying of let-7b in triple-negative breast cancer. J. Exp. Clin. Cancer Res. 2020;39:58. doi: 10.1186/s13046-020-01561-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu L., Yang N., Du G., Li C., Liu G., Liu S., Xu Y., Di Y., Pan W., Li X. LncRNA CRNDE promotes the epithelial-mesenchymal transition of hepatocellular carcinoma cells via enhancing the Wnt/beta-catenin signaling pathway. J. Cell. Biochem. 2018 doi: 10.1002/jcb.26762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang K.C., Diermeier S.D., Yu A.T., Brine L.D., Russo S., Bhatia S., Alsudani H., Kostroff K., Bhuiya T., Brogi E., et al. MaTAR25 lncRNA regulates the Tensin1 gene to impact breast cancer progression. Nat. Commun. 2020;11:6438. doi: 10.1038/s41467-020-20207-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boon R.A., Jae N., Holdt L., Dimmeler S. Long noncoding RNAs: from clinical genetics to therapeutic targets? J. Am. Coll. Cardiol. 2016;67:1214–1226. doi: 10.1016/j.jacc.2015.12.051. [DOI] [PubMed] [Google Scholar]
- 19.Chen Y.G., Satpathy A.T., Chang H.Y. Gene regulation in the immune system by long noncoding RNAs. Nat. Immunol. 2017;18:962–972. doi: 10.1038/ni.3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang C., Li Y., Yan S., Wang H., Shao X., Xiao M., Yang B., Qin G., Kong R., Chen R., et al. Interactome analysis reveals that lncRNA HULC promotes aerobic glycolysis through LDHA and PKM2. Nat. Commun. 2020;11:3162. doi: 10.1038/s41467-020-16966-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie J., Zheng Y., Xu X., Sun C., Lv M. Long noncoding RNA CAR10 contributes to melanoma progression by suppressing miR-125b-5p to induce RAB3D expression. Onco Targets Ther. 2020;13:6203–6211. doi: 10.2147/OTT.S249736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun L., Wang L., Chen T., Shi Y., Yao B., Liu Z., Wang Y., Li Q., Liu R., Niu Y., et al. LncRNA RUNX1-IT1 which is downregulated by hypoxia-driven histone deacetylase 3 represses proliferation and cancer stem-like properties in hepatocellular carcinoma cells. Cell Death Dis. 2020;11:95. doi: 10.1038/s41419-020-2274-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu S., Wang H., Li J., Zhang J., Wu J., Li Y., Piao Y., Pan L., Xiang R., Yue S. FZR1 as a novel biomarker for breast cancer neoadjuvant chemotherapy prediction. Cell Death Dis. 2020;11:804. doi: 10.1038/s41419-020-03004-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams G.T., Farzaneh F. Are snoRNAs and snoRNA host genes new players in cancer? Nat. Rev. Cancer. 2012;12:84–88. doi: 10.1038/nrc3195. [DOI] [PubMed] [Google Scholar]
- 25.Zimta A., Tigu A.B., Braicu C., Stefan C., Ionescu C., Berindan-Neagoe I. An emerging class of long non-coding RNA with oncogenic role arises from the snoRNA host genes. Front. Oncol. 2020;10:389. doi: 10.3389/fonc.2020.00389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xuan Y., Wang Y. Long non-coding RNA SNHG3 promotes progression of gastric cancer by regulating neighboring MED18 gene methylation. Cell Death Dis. 2019;10:694. doi: 10.1038/s41419-019-1940-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun Y., Wei G., Luo H., Wu W., Skogerbo G., Luo J., Chen R. The long noncoding RNA SNHG1 promotes tumor growth through regulating transcription of both local and distal genes. Oncogene. 2017;36:6774–6783. doi: 10.1038/onc.2017.286. [DOI] [PubMed] [Google Scholar]
- 28.Yue X., Dong C., Ye Z., Zhu L., Zhang X., Wang X., Mo F., Li Z., Pan B. LncRNA SNHG7 sponges miR-449a to promote pituitary adenomas progression. Metab. Brain Dis. 2021;36:123–132. doi: 10.1007/s11011-020-00611-5. [DOI] [PubMed] [Google Scholar]
- 29.Zhao Y., Qin Z.S., Feng Y., Tang X.J., Zhang T., Yang L. Long non-coding RNA (lncRNA) small nucleolar RNA host gene 1 (SNHG1) promote cell proliferation in colorectal cancer by affecting P53. Eur. Rev. Med. Pharmacol. Sci. 2018;22:976–984. doi: 10.26355/eurrev_201802_14379. [DOI] [PubMed] [Google Scholar]
- 30.Jiang H., Li T., Qu Y., Wang X., Li B., Song J., Sun X., Tang Y., Wan J., Yu Y., et al. Long non-coding RNA SNHG15 interacts with and stabilizes transcription factor Slug and promotes colon cancer progression. Cancer Lett. 2018;425:78–87. doi: 10.1016/j.canlet.2018.03.038. [DOI] [PubMed] [Google Scholar]
- 31.Mccarrey J.R., Thomas K. Human testis-specific PGK gene lacks introns and possesses characteristics of a processed gene. Nature. 1987;326:501–505. doi: 10.1038/326501a0. [DOI] [PubMed] [Google Scholar]
- 32.He Y., Luo Y., Zhang D., Wang X., Zhang P., Li H., Ejaz S., Liang S. PGK1-mediated cancer progression and drug resistance. Am. J. Cancer Res. 2019;9:2280–2302. [PMC free article] [PubMed] [Google Scholar]
- 33.Huang B., Cai W., Wang Q., Liu F., Xu M., Zhang Y. Gankyrin drives malignant transformation of gastric cancer and alleviates oxidative stress via mTORC1 activation. Oxid. Med. Cell. Longev. 2018;2018:1–10. doi: 10.1155/2018/9480316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Z., Zhuang W., Wang Z., Xiao W., Don W., Li X., Chen X. MicroRNA-450b-3p inhibits cell growth by targeting phosphoglycerate kinase 1 in hepatocellular carcinoma. J. Cell. Biochem. 2019;120:18805–18815. doi: 10.1002/jcb.29196. [DOI] [PubMed] [Google Scholar]
- 35.Dong W., Li H., Wu X. Rab11-FIP2 suppressed tumor growth via regulation of PGK1 ubiquitination in non-small cell lung cancer. Biochem. Biophys. Res. Commun. 2019;508:60–65. doi: 10.1016/j.bbrc.2018.11.108. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Y., Kwok-Shing Ng P., Kucherlapati M., Chen F., Liu Y., Tsang Y.H., de Velasco G., Jeong K.J., Akbani R., Hadjipanayis A., et al. A pan-cancer proteogenomic Atlas of PI3K/AKT/mTOR pathway alterations. Cancer Cell. 2017;31:820–832. doi: 10.1016/j.ccell.2017.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qiao C., Qiao T., Jin H., Liu L., Zheng M., Wang Z. LncRNA KCNQ1OT1 contributes to the cisplatin resistance of tongue cancer through the KCNQ1OT1/miR-124-3p/TRIM14 axis. Eur. Rev. Med. Pharmacol. Sci. 2020;24:200–212. doi: 10.26355/eurrev_202001_19912. [DOI] [PubMed] [Google Scholar]
- 38.Zheng Y., Zheng B., Meng X., Yan Y., He J., Liu Y. LncRNA DANCR promotes the proliferation, migration, and invasion of tongue squamous cell carcinoma cells through miR-135a-5p/KLF8 axis. Cancer Cell Int. 2019;19:302. doi: 10.1186/s12935-019-1016-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang S., Ma H., Zhang D., Xie S., Wang W., Li Q., Lin Z., Wang Y. LncRNA KCNQ1OT1 regulates proliferation and cisplatin resistance in tongue cancer via miR-211-5p mediated Ezrin/Fak/Src signaling. Cell Death Dis. 2018;9:742. doi: 10.1038/s41419-018-0793-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu H., Zhu W., Qin J., Chen M., Gong L., Li L., Liu X., Tao Y., Yin H., Zhou H., et al. Acetylation of PGK1 promotes liver cancer cell proliferation and tumorigenesis. Hepatology. 2016;65:515–528. doi: 10.1002/hep.28887. [DOI] [PubMed] [Google Scholar]
- 41.Li J., Cheng D., Zhu M., Yu H., Pan Z., Liu L., Geng Q., Pan H., Yan M., Yao M. OTUB2 stabilizes U2AF2 to promote the Warburg effect and tumorigenesis via the AKT/mTOR signaling pathway in non-small cell lung cancer. Theranostics. 2019;9:179–195. doi: 10.7150/thno.29545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mabuchi S., Kuroda H., Takahashi R., Sasano T. The PI3K/AKT/mTOR pathway as a therapeutic target in ovarian cancer. Gynecol. Oncol. 2015;137:173–179. doi: 10.1016/j.ygyno.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 43.Wu X.S., Wang F., Li H.F., Hu Y.P., Jiang L., Zhang F., Li M.L., Wang X.A., Jin Y.P., Zhang Y.J., et al. LncRNA-PAGBC acts as a microRNA sponge and promotes gallbladder tumorigenesis. EMBO Rep. 2017;18:1837–1853. doi: 10.15252/embr.201744147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu J., Xiao Y., Liu B., Pan S., Liu Q., Shan Y., Li S., Qi Y., Huang Y., Jia L. Exosomal MALAT1 sponges miR-26a/26b to promote the invasion and metastasis of colorectal cancer via FUT4 enhanced fucosylation and PI3K/Akt pathway. J. Exp. Clin. Cancer Res. 2020;39:54. doi: 10.1186/s13046-020-01562-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen X., Zeng K., Xu M., Hu X., Liu X., Xu T., He B., Pan Y., Sun H., Wang S. SP1-induced lncRNA-ZFAS1 contributes to colorectal cancer progression via the miR-150-5p/VEGFA axis. Cell Death Dis. 2018;9:982. doi: 10.1038/s41419-018-0962-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang Y., Xiang B., Liu Y., Wang Y., Kan H. LncRNA CDKN2B-AS1 promotes tumor growth and metastasis of human hepatocellular carcinoma by targeting let-7c-5p/NAP1L1 axis. Cancer Lett. 2018;437:56–66. doi: 10.1016/j.canlet.2018.08.024. [DOI] [PubMed] [Google Scholar]
- 47.Deitersen J., Berning L., Stuhldreier F., Ceccacci S., Schlütermann D., Friedrich A., Wu W., Sun Y., Böhler P., Berleth N., et al. High-throughput screening for natural compound-based autophagy modulators reveals novel chemotherapeutic mode of action for arzanol. Cell Death Dis. 2021;12:560. doi: 10.1038/s41419-021-03830-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen J., Na R., Xiao C., Wang X., Wang Y., Yan D., Song G., Liu X., Chen J., Lu H., et al. The loss of SHMT2 mediates 5-fluorouracil chemoresistance in colorectal cancer by upregulating autophagy. Oncogene. 2021;40:3974–3988. doi: 10.1038/s41388-021-01815-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen Z., Qin Z., Li L., Wo Q., Chen X. HOXA13, negatively regulated by miR-139-5p, decreases the sensitivity of gastric cancer to 5-fluorouracil possibly by targeting ABCC4. Front. Oncol. 2021;11:645979. doi: 10.3389/fonc.2021.645979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Han D., Zhang N., Zhao S., Liu H., Wang X., Yang M., Wang S., Li Y., Liu Z., Teng L. AKIP1 promotes glioblastoma viability, mobility and chemoradiation resistance via regulating CXCL1 and CXCL8 mediated NF-kappaB and AKT pathways. Am. J. Cancer Res. 2021;11:1185–1205. [PMC free article] [PubMed] [Google Scholar]
- 51.Maier H.J., Wirth T., Beug H. Epithelial-mesenchymal transition in pancreatic carcinoma. Cancers. 2010;2:2058–2083. doi: 10.3390/cancers2042058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu Y., Ho J., Yu C., Cho C., Wu C., Huang C., Gao H., Yu D. Ellagic acid resensitizes gemcitabine-resistant bladder cancer cells by inhibiting epithelial-mesenchymal transition and gemcitabine transporters. Cancers. 2021;13:2032. doi: 10.3390/cancers13092032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jing C., Li X., Zhou M., Zhang S., Lai Q., Liu D., Ye B., Li L., Wu Y., Li H., et al. The PSMD14 inhibitor Thiolutin as a novel therapeutic approach for esophageal squamous cell carcinoma through facilitating SNAIL degradation. Theranostics. 2021;11:5847–5862. doi: 10.7150/thno.46109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baulida J., García De Herreros A. Snail1-driven plasticity of epithelial and mesenchymal cells sustains cancer malignancy. Biochim. Biophys. Acta. 2015;1856:55–61. doi: 10.1016/j.bbcan.2015.05.005. [DOI] [PubMed] [Google Scholar]
- 55.Roberts C.M., Tran M.A., Pitruzzello M.C., Wen W., Loeza J., Dellinger T.H., Mor G., Glackin C.A. TWIST1 drives cisplatin resistance and cell survival in an ovarian cancer model, via upregulation of GAS6, L1CAM, and Akt signalling. Sci. Rep. 2016;6:37652. doi: 10.1038/srep37652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hanrahan K., O'Neill A., Prencipe M., Bugler J., Murphy L., Fabre A., Puhr M., Culig Z., Murphy K., Watson R.W. The role of epithelial-mesenchymal transition drivers ZEB1 and ZEB2 in mediating docetaxel-resistant prostate cancer. Mol. Oncol. 2017;11:251–265. doi: 10.1002/1878-0261.12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen W., Wang P., Lu Y., Jin T., Lei X., Liu M., Zhuang P., Liao J., Lin Z., Li B., et al. Decreased expression of mitochondrial miR-5787 contributes to chemoresistance by reprogramming glucose metabolism and inhibiting MT-CO3 translation. Theranostics. 2019;9:5739–5754. doi: 10.7150/thno.37556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yu G., Wang L., Han Y., He Q. clusterProfiler: an R Package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.