

Abstract
STAT3 is constitutively activated in multiple malignant tumors. Compared with regular estrogen receptor (ER)-positive breast cancers, the patients with tamoxifen-resistant breast cancers often exhibit higher levels of STAT3 phosphorylation. Narciclasine (Nar) possesses strong inhibiting effects against a variety of cancer cells; however, the underlying antitumor target(s)/mechanism(s) remains barely understood. In this study, we successfully identified the STAT3 was the direct target of Nar through the combination strategies of connectivity map and drug affinity responsive target stability. In MCF7 cells, Nar could suppress phosphorylation, activation, dimerization, and nuclear translocation of STAT3 by directly binding with the STAT3 SH2 domain. In addition, Nar could specifically degrade total STAT3 via the proteasome pathway in MCF-7/TR (tamoxifen-resistant MCF-7) cells. This distinct mechanism of Nar-targeting STAT3 was mainly attributed to the various levels of reactive oxygen species in regular and tamoxifen-resistant ER-positive breast cancer cells. Meanwhile, Nar-loaded nanoparticles could markedly decrease the protein levels of STAT3 in tumors, resulting in significantly increased MCF-7/TR xenograft tumor regression without obvious toxicity. Our findings successfully highlight the STAT3 as the direct therapeutic target of Nar in ER-positive breast cancer cells, especially, Nar leaded STAT3 degradation as a promising strategy for the tamoxifen-resistant breast cancer treatment.
Keywords: narciclasine, STAT3, MCF-7/TR, ROS, nanoparticles
Graphical abstract
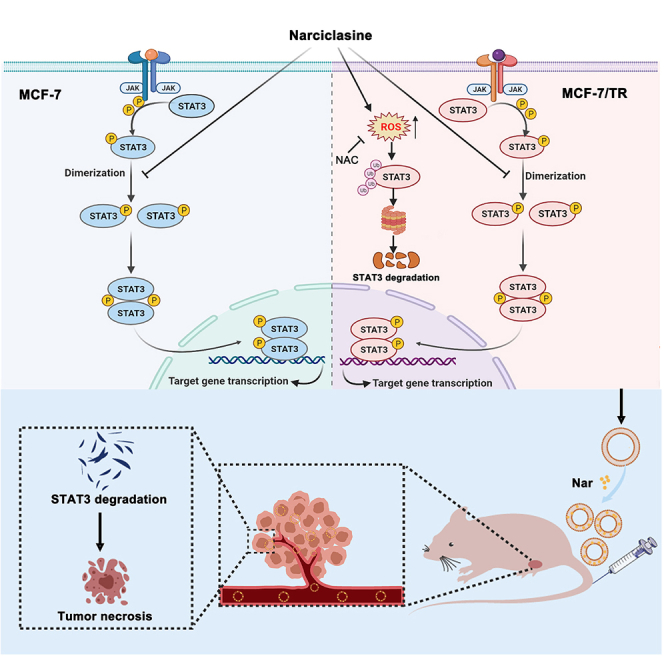
Nar-targeting STAT3 had distinct mechanisms in MCF-7 cells and MCF-7/TR cells. Nar directly bound to STAT3 protein and suppress STAT3 phosphorylation and activation in MCF-7 cells, but in MCF-7/TR cells could specifically promote total STAT3 degradation via a ROS-dependent proteasome pathway.
Introduction
Narciclasine (Nar), a plant growth inhibitor, was first isolated from bulbs of several Narcissus species in 1967, which has been shown to exhibit antitumor, anti-inflammatory, anti-Alzheimer’s disease, and obesity-suppressing activities.1, 2, 3, 4, 5 In the US National Cancer Institute NCI-60 human tumor cell lines screen, Nar displayed broad cytotoxicity against various cancer cell lines.2 Although Nar possesses a marked antitumor effect, the underlying antitumor target(s)/mechanism(s) remains poorly understood. Only a few previous studies showed that Nar inhibited protein synthesis by targeting eEF1A in melanoma cells,6 activated Rho, induced stress fibers in glioblastoma cells,7 and regulated the AMPK-ULK1 axis in triple-negative breast cancer cells.8 In this study, a novel method combination strategy was applied to reveal the new underlying antitumor target for Nar. The connectivity map (CMAP) database, a pattern-matching software that analyzes gene expression profiles, is one of the most effective tools for predicting pharmacological function and mechanism of action in drug discovery and development.9,10 The drug affinity responsive target stability (DARTS) technique, based on drug-binding-incurred changes in protease susceptibility of the target protein, can be used to identify the protein targets of bioactive small-molecule-based agents.11 Thus, we combined this CMAP analysis with DARTS/MS strategy to resolve the molecular targets of Nar.
In this study, we demonstrated that Nar could directly target signal transducer and activator of transcription 3 (STAT3) and significantly inhibit its phosphorylation, which has not yet been reported. The oncogenic transcription factor STAT3 plays a pivotal role in cancer progressions, such as cell proliferation, apoptosis, angiogenesis, metastasis, invasion, immune evasion, and drug resistance.12, 13, 14 Constitutive activation of STAT3 is observed in a broad range of human malignancies, including colorectal, lung, breast, prostate, gastric, glioma, melanoma, ovary, liver, and pancreas cancers.12,15 Therefore, STAT3 is deemed as an attractive therapeutic target for antitumor drug development. In the past two decades, some natural and synthetic compounds have been demonstrated to possess the STAT3 phosphorylation inhibitory effects by directly binding STAT3 domains, such as the DNA and SH2 binding domain.15 In recent years, a few natural products and proteolysis targeting chimera molecules can also exert antitumor effects by targeting total STAT3 degradation.16, 17, 18 In addition, STAT3 remains to be a promising clinical target in breast cancer prevention and therapy.15 STAT3 phosphorylation was also abnormally high in tamoxifen-resistant breast cancer, which resulted in ER-positive breast cancer resistance to tamoxifen.19, 20, 21 In this study, Nar could target STAT3 and inhibit its phosphorylation without affecting the total protein of STAT3 in MCF-7 cells. However, distinct mechanism in tamoxifen-resistant MCF-7 (MCF-7/TR) cells, Nar not only inhibited STAT3 phosphorylation, but also significantly degraded STAT3 total protein via the proteasome pathway and showed better cytotoxicity.
In addition, Nar possessed strong pan-cytotoxicity and poor water solubility,22 which hampered the clinical development. Thus, nanoscale drug delivery was applied in Nar against MCF-7/TR cells in vivo. In this study, we integrated CMAP with DARTS/MS revealed that Nar targeted STAT3 and inhibited its phosphorylation and activation in MCF-7 cell and facilitated the total STAT3 degradation via a reactive oxygen species (ROS)-dependent proteasome pathway in MCF-7/TR cells. Meanwhile, nanoparticulate delivery of Nar significantly suppressed MCF-7/TR cells in vivo.
Results
Potential target of Nar predicted by CMAP-DARTS/MS strategy
Based on strong cytotoxic effects of Nar on cancer cells, the antitumor targets were needed to discover in this study. To investigate the pharmacological functions of Nar, a similarity search against the CMAP was performed. The 940 query genes (>3-fold change; 616 upregulated and 324 downregulated; Data S1) were submitted to the CMAP database for analysis (Figure 1A). The top 20 correlated drugs with lower p values and a positive enrichment score are summarized in Figure 1B (detailed results in Data S2). The most similar of these drugs that positively correlated with Nar were protein synthesis inhibitors (anisomycin, emetine, cycloheximide [CHX], and puromycin). There are also three molecular-targeted drugs, niclosamide (STAT3 inhibitor), 15-delta prostaglandin J2 (PPARγ agonist), and trichostatin A (HDAC inhibitor). Based on the known pharmacological functions of these positively correlated agents, it appeared likely that Nar exerts its antitumor activity by inhibiting protein synthesis, STAT3, HDAC, and/or activating PPARγ. The CMAP results strongly suggested that one pharmacological function of Nar is inhibiting protein synthesis, which corroborated previous studies.22,23 Thus, these previous studies served to further validate both the efficacy and accuracy of CMAP analysis for mechanism of action determination.
Figure 1.

The prediction of antitumor targets of Nar by CMAP-DARTS/MS strategy
(A) The query genes of Nar were submitted to the CMAP database for analysis. (B) The top 20 drugs/agents that were positively correlated with Nar by the CMAP analysis. (C) Following DARTS assay, the samples were subjected to LC-MS/MS for proteomic analysis, and the possible Nar binding proteins were obtained. (D) The KEGG pathway was performed to analysis these potential binding proteins of Nar.
To further explore the CMAP predictions, DARTS/MS proteomic characterization was applied. Following DARTS assay, the samples were subjected to proteomics analysis (Figure 1C). To narrow the scope and improve the accuracy, the top 10 potential target proteins (unique peptides ≥3 and the intensity ratio ≥2) of each molecular-weight region were selected (detailed results in Data S3). Then, Kyoto Encyclopedia of Genes and Genomes pathway was performed to analysis these potential target proteins.24 In these pathways, cell proliferation was the most relevant to an antitumor mechanism (Figure 1D). One of the potential target proteins in cell proliferation pathway was STAT3, which is consistent with CMAP predicting Nar as a STAT3 inhibitor. Thus, combining the direct binding proteins predicted by DARTS/MS proteomics and the CMAP pharmacological function predictions, the antitumor target of Nar was postulated to involve its directly binding STAT3 and inhibiting its activation.
Nar directly binds to STAT3 protein and suppresses its phosphorylation and activation in vitro and in vivo
To determine whether Nar is a potential STAT3 inhibitor, its ability to suppress STAT3 expression was first examined in tumor cells. Among the eight human cancer cell lines examined, SW480, DU145, and MCF-7 expressed STAT3 protein at higher levels (Figure S1). Each of these cell lines was treated with Nar at a range of concentrations for 24 h. As shown in Figures 2A–2C, Nar significantly inhibited STAT3 phosphorylation in a concentration-dependent manner without affecting the total level of STAT3 in all three lines. Moreover, Nar was the most effective at inhibiting STAT3 phosphorylation in MCF-7 cells. We also examined the levels of phosphorylated STAT3 (p-STAT3) and total STAT3 in other breast cancer cell lines, namely T47D (ER-positive breast cancer cells) and MDA-MB231 (ER-negative breast cancer cells) treated with Nar, the results showed that Nar significantly inhibited STAT3 phosphorylation in a concentration-dependent manner without significantly affecting the total level of STAT3 in T47D and MDA-MB-231 cells (Figure S2). The results from an MCF-7 cell-based time course study demonstrated that Nar significantly inhibited the phosphorylation (Tyr705) of STAT3 in a time-dependent manner (Figure 2D). Furthermore, Nar treatment also reduced interleukin-6 (IL-6)-induced STAT3 phosphorylation (Tyr705) and activation in MCF-7 cells (Figure 2E). Interestingly, Nar had no significant effect on another phosphorylation site (Ser727) of STAT3 (Figure S3). Thus, Nar could inhibit STAT3 activity by preventing its phosphorylation (Tyr705) and activation in MCF-7 cells. In addition, murine MCF-7 xenografts were established to investigate the effect of Nar on STAT3 signaling in vivo. Nar treatment effectively reduced the size and weight of the MCF-7 xenograft tumors in comparison with the vehicle control (Figures 2F and 2G). No significant change in body weight or pronounced side effect was observed between the vehicle control and treatment groups (Figure S4). The levels of phosphorylated and total STAT3 proteins were examined in the excised tumor tissues by immunohistochemistry (IHC) staining. As shown in Figures 2H and 2I, Nar treatment significantly decreased phosphorylated STAT3 levels. Moreover, the levels of phosphorylated and total STAT3 proteins were examined in the excised tumor tissues by western blot. As shown in Figures 2J and 2K, Nar treatment decreased phosphorylated STAT3 levels. These observations are consistent with Nar as a STAT3 inhibitor that blocks STAT3 phosphorylation and activation in vitro and in vivo.
Figure 2.

Nar directly binds to STAT3 protein and suppresses its phosphorylation and activation in vitro and in vivo
(A–C) MCF-7, SW480, and DU145 cells were treated with different concentrations of Nar for 24 h followed by western blot analysis of total and phosphorylated STAT3. (D) MCF-7 cells were treated with Nar at 100 nM for various time points. (E) Nar inhibition of IL-6-induced STAT3 phosphorylation and activation in MCF-7 cells. (F) Images of representative MCF-7 xenograft tumors from the vehicle- and Nar-treated groups. (G) The weight of dissected xenograft tumor masses. (H and I) The IHC staining analyzed STAT3 and p-STAT3 in MCF-7 xenograft tumors. (J and K) Total proteins were extracted from excised tumors, and the expression levels of STAT3 and p-STAT3 were determined by western blot. (L) MST analysis of Nar binding to recombinant human STAT3 (Kd = 15 μM). (M) The CETSA assay determined the thermal stabilization of STAT3 interaction with Nar at a series of temperatures from 50°C to 68°C. (N) The thermal stabilization of STAT3 by increasing concentrations of Nar was assayed at 62°C. (O) Cell lysates were incubated with Nar-conjugated Sepharose 4B beads or Sepharose 4B beads alone, and then the pulled down proteins were analyzed by western blot. Data are shown as mean ± SD. ∗∗p < 0.01, ∗∗∗p < 0.001.
To determine whether Nar exerted effects by directly binding STAT3 protein, several binding experiments were carried out. First, microscale thermophoresis (MST) assay was performed to determine the direct binding between Nar and recombinant human STAT3. Expectedly, Nar could readily bind to STAT3 with a Kd estimated at 15 μM (Figure 2L). The DARTS assay, which relying on the reduction in the protease susceptibility of the target protein upon drug binding, was conducted.11 After incubating cell lysate samples with Nar, the STAT3 protein was less susceptible to exogenous pronase at the protein to pronase ratio of 1:300 and 1:200, respectively (Figure S5A). With increasing Nar concentration, the protease susceptibility of STAT3 markedly decreased (Figure S5B). Meanwhile, the cellular thermal shift assay (CETSA), which based on the biophysical principle of ligand-induced thermal stabilization of target proteins, was used to investigate the binding between Nar and STAT3.25 As shown in Figure 2M, Nar markedly increased STAT3 accumulation at temperatures ranging from 60°C to 66°C (relative to the DMSO solvent control). The stability of STAT3 protein during heating depended on the concentration of Nar. As shown in Figure 2N, STAT3 protein accumulation markedly increased as Nar concentration increased. To further evaluate the direct binding of Nar and STAT3, the pull-down assay was performed using Nar-conjugated Sepharose 4B beads with cell lysates of MCF-7 cells. As shown in Figure 2O, Nar-conjugated beads apparently pulled down the STAT3 from cell lysate compared with control beads. All these data together demonstrated that Nar directly bound to STAT3 and thus inhibited its activity.
Nar binds to SH2 domain, decreases STAT3 dimerization, and inhibits STAT3 nuclear translocation
To further characterize the Nar-STAT3 interaction, the binding mode of Nar and STAT3 (PDB: 1BG1) was modeled by docking simulation using Maestro software (Schrödinger, version 9.0). As shown in Figure 3A, Nar docked into the SH2 domain of STAT3 and potentially interacted with the SH2 domain via four pronounced hydrogen bonds (red lines). In general, STAT3 undergoes SH2 domain-mediated dimerization upon phosphorylation at the tyrosine 705 residue and subsequently translocate to the nucleus.26 To determine if Nar binding to SH2 domain would consequently reduce STAT3 dimerization, a native PAGE analysis of dimeric STAT3 following Nar treatment was performed. As shown in Figure 3B, Nar inhibited STAT3 dimerization by decreasing the level of dimeric STAT3 while increasing the level of monomeric STAT3. The luciferase reporter gene assay was performed to assess the effect of Nar on STAT3 transcriptional activity. As shown in Figure 3C, Nar significantly inhibited IL-6-induced STAT3-driven luciferase activities dose dependently. The subcellular localization of STAT3 was examined using immunofluorescence staining. Nuclear STAT3 protein levels were drastically reduced following Nar treatment (Figure 3D). Western blot analysis of nuclear and cytoplasmic extract samples further revealed that Nar reduced nuclear STAT3 level and p-STAT3 protein in both the nuclei and cytoplasm (Figure 3E). Following nuclear translocation, STAT3 controls the transcription of downstream target genes that are critical to tumor growth, angiogenesis, and invasion.27 In MCF-7 cells, Nar treatment suppressed the expression of representative STAT3 target genes (cyclin D1, c-Myc, and survivin) while it increased p53 mRNA and protein levels (Figures 3F and S6).
Figure 3.

Nar binds to SH2 domain, decreases STAT3 dimerization and inhibits STAT3 translocation into the nucleus
(A) The predicted binding complex structure of Nar in the SH2 domain of STAT3, where Nar interacts with the SH2 domain via four pronounced hydrogen bonds (red lines). (B) MCF-7 cells were treated with different concentrations of Nar for 24 h, separated using native PAGE, and STAT3 analyzed by western blot. (C) 293T cells were transfected with STAT3-luc, and then treated with Nar and stattic in the presence of 50 ng/mL IL-6 for 24 h. (D) Immunofluorescence staining for STAT3, p-STAT3, and the nucleus (DAPI, blue) after Nar treatment (100 nM, 24 h) in MCF-7 cells. (E) After treatment with Nar, the nuclear and cytoplasmic proteins were extracted to determine the levels of STAT3 and p-STAT3. (F) After treatment with Nar, STAT3 downstream target proteins (i.e., c-Myc, cyclin D1, surviving, and p53) were analyzed by western blot. Data are shown as mean ± SD. ∗∗p < 0.01, ∗∗∗p < 0.01, ###p < 0.001.
Nar specifically targets degradation of STAT3 via a ROS-dependent proteasome pathway in MCF-7/TR cells
In clinical practice, selective estrogen receptor modulator tamoxifen has been used widely to treat ER-positive breast cancer. However, elevated levels of STAT3 phosphorylation have been observed in tamoxifen-resistant breast cancer.19,20 Here, we observed that basal expression levels of ERα and ERβ were significantly suppressed in MCF-7/TR cells, and also found abnormal high expression of phosphorylated STAT3 in MCF-7/TR cells compared with MCF-7 cells (Figure 4A). The above data suggested that Nar directly bound to STAT3 and inhibited its phosphorylation without affecting the total level of STAT3 in MCF-7 cells. Strikingly and unexpectedly, MCF-7/TR cells treated with Nar, not only the phosphorylation level of STAT3 was reduced, but also the total protein level of STAT3 was significantly reduced (Figure 4B). Nar also significantly inhibited the total protein level and phosphorylation level of STAT3 in a time-dependent manner (Figure 4C). To determine the selectivity of Nar to STAT3, other members of the STAT family were also detected. Except for STAT2 and STAT4 that were not detected, Nar treatment had a slight inhibitory effect on the protein levels of STAT1, STAT5A/B, and STAT6 (Figure S7). Therefore, Nar possessed a certain selective and specific suppression effect on STAT3 protein. Meanwhile, the expression of STAT3 target genes cyclin D1 and c-Myc, which are related to cell cycle and proliferation, were significantly inhibited by Nar treatment (Figure 4D), and the mRNA levels of cyclin D1 also significantly decreased by Nar (Figure 4E).
Figure 4.

Nar specifically targets degradation of STAT3 via a ROS-dependent proteasome pathway in MCF-7/TR cells
(A) Protein expression of p-STAT3, STAT3, ERα, and ERβ in MCF-7 and MCF-7/TR cells were determined by western blot. (B) MCF-7/TR cells were treated with different concentrations of Nar for 24 h followed by western blot analysis of total and phosphorylated STAT3. (C) MCF-7/TR cells were treated with Nar at 100 nM for various time points. (D) The expression of STAT3 target genes cyclin D1 and c-Myc were determined by western blot. (E) MCF-7/TR cells were treated with Nar at 100 nM for various time points followed by real-time RT-PCR analysis of mRNAs encoding STAT3 and cyclin D1. (F) STAT3 protein levels in MCF-7/TR cells treated with Nar in the absence or presence of proteasome inhibitor bortezomib (BTZ) determined by western blot. (G and H) MCF-7/TR cells treatment with Nar and cycloheximide (CHX) alone or in combination for the indicated time points, STAT3 protein were determined by western blot and quantified by degradation kinetic curves. (I) MCF-7/TR and MCF-7 cells with Nar treatment for 1 and 3 h, and cellular ROS levels were detected. (J) MCF-7/TR cells were treated with the ROS scavenger N-acetylcysteine (NAC) for 1 h, followed treatment with or without Nar, and the images of cellular ROS levels were captured. (K) STAT3 and p-STAT3 levels in MCF-7/TR cells treated with NAC, Nar, and NAC + Nar. (L) STAT3 levels in MCF-7/TR cells treated with Nar or Nar+ different concentrations of H2O2. Data are shown as mean ± SD. ∗∗p < 0.01.
To investigate how Nar downregulated the protein level of total STAT3 in MCF-7/TR cells, the levels of STAT3 mRNA transcript were examined. The results showed that there were no significant changes in STAT3 mRNA levels between control and Nar treatment (Figure 4E), suggesting that downregulation of STAT3 occurred post transcriptionally. Thus, we hypothesized that Nar-mediated STAT3 degradation might probably be attributed to the ubiquitin proteasome pathway. As shown in Figure 4F, the reduction in STAT3 protein caused by Nar treatment can be efficiently rescued by proteasome inhibitor bortezomib (BTZ), which indicated that Nar targeted STAT3 to proteasome degradation. Then, MCF-7/TR cells were treated with CHX, blocking protein synthesis, to determine the effect of Nar on the protein stability of STAT3. The results showed that Nar treatment reduced STAT3 protein half-life from 123 to 38.8 min in MCF-7/TR cells (Figures 4G and 4H).
To investigate why Nar degraded STAT3 in MCF-7/TR cells but not in MCF-7 cells, cellular ROS levels were determined. Previous studies showed that the level of ROS in MCF-7/TR cells was higher than that in MCF-7 cells, which prompted us to test whether ROS contribute to STAT3 degradation.28,29 As shown in Figure 4I, we found that the level of ROS in MCF-7/TR cells was significantly higher than that in MCF-7 cells, and that Nar treatment also could significantly increase the ROS level in MCF-7/TR cells. Meanwhile, Nar treatment increased the ROS level (green) in MCF-7/TR cells, and this ROS signal was significantly diminished by pretreating with the N-acetylcysteine (NAC) (Figure 4J). Next, NAC experiment was performed to determine whether degradation of STAT3 was related to ROS levels. Consistent with expectation, treatment with the ROS scavenger NAC efficiently rescued STAT3 from proteasome degradation (Figure 4K). We also detected the effect of NAC on STAT3 in the absence of Nar. The results showed that NAC alone had no effect on STAT3, and NAC had a slight decrease on p-STAT3 (Figure 4K). To further confirm that ROS were involved in the degradation of STAT3, we performed western blot analysis of Nar on STAT3 stability treated with increasing concentrations of H2O2 (ROS generator). The results showed that Nar could significantly decrease STAT3 with increasing concentration of H2O2 (Figure 4L). Taken together, these results demonstrated that Nar targeted STAT3 to proteasome degradation through a ROS-dependent mechanism in MCF-7/TR cells.
Nar inhibits cell proliferation and promotes cell-cycle arrest in G2/M in MCF-7/TR cells
Nar, as a direct STAT3 inhibitor, possessed a role to degrade STAT3 protein via the ROS-dependent proteasome pathway in MCF-7/TR cells. Subsequently, cell counting kit-8 (CCK-8) assays showed that Nar exerted higher cytotoxicity in MCF-7/TR compared with MCF-7 cells (Figure 5A). To further assess the growth inhibition of Nar on MCF-7/TR cells, a colony-formation assay was performed. The results showed that Nar treatment significantly suppressed the colony-formation efficiency of MCF-7/TR cells (Figures 5B and 5C). To confirm whether Nar inhibited cell proliferation by inducing cell-cycle arrest, flow cytometry was performed. Nar treatment significantly induced G2/M phase arrest in MCF-7/TR cells in a concentration-dependent manner (Figures 5D and 5E). Moreover, the annexin V-FITC/PI double-staining assay showed that only higher concentration of Nar could cause an increase in the proportion of apoptotic cells (Figures 5F and 5G). Hence, Nar affects MCF-7/TR cell viability mainly by inhibiting cell proliferation and inducing cell-cycle G2/M phase arrest.
Figure 5.

Nar inhibits cell proliferation, promoting cell-cycle arrest in G2/M in MCF-7/TR cells
(A) Viability of MCF-7 and MCF-7/TR cells was measured using the CCK-8 assay. (B and C) Colony-formation assay was used to validate the effect of Nar on MCF-7/TR cells growth. (D–G) The cell-cycle distribution and percentage of apoptotic cells were analyzed by flow cytometry. Data are shown as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Nar suppresses the growth of MCF-7/TR xenograft tumors
The previously reported toxicity and the poor water solubility of Nar were important obstacles hampering the efficacy in vivo. Thus, the nano-drug delivery system was used to reduce the toxicity and increase therapeutic efficacy. We successfully encapsulated Nar into liposomes using a thin-film hydration method. Nar liposomes (Nar-LPs) were spherical in shape with a smooth surface as observed using cryoelectron microscopy (cryo-EM) (Figure 6A). Nar-LPs exhibited the hydrodynamic size of 176.1 nm (Figure S8A) and zeta potential of −2.8 mV (Figure S8B). Drug loading and encapsulation efficiencies were 90% and 10%, respectively. Nar could be released from liposomes in a sustained manner (Figure S8C). The cytotoxicity of Nar-LPs against MCF-7 and MCF-7/TR cells were further evaluated using free Nar as control. As expected, the cytotoxicity of Nar-LPs was comparable with that of free Nar in MCF-7 and MCF-7/TR cells (Figures 6B and 6C).
Figure 6.

Nar suppresses the growth of MCF-7/TR xenograft tumors
(A) TEM image of Nar-LP nanoparticles. (B and C) The cytotoxicity of Nar-LP and Nar in MCF-7 cells and MCF-7/TR cells was determined using the CCK-8 assay. (D) Schematic plan for the administration of Nar-LP and Nar. (E) Representative image of MCF-7/TR xenograft tumors after treatment. Nar (2 mg/kg) was administered daily via intraperitoneal injection, and Nar-LP (5 mg/kg) was administered daily via tail vein injection. (F) Measurement of tumor volume at indicated time points after different treatments. (G) Weights of xenografted tumors were summarized. (H) The body weights of nude mice in each group during the treatment. (I and J) Total proteins were extracted from excised tumors, and the expression levels of STAT3, p-STAT3, and cyclin D1were determined by western blot. (K and L) The IHC staining analyzed Ki67, STAT3, and p-STAT3 in MCF-7/TR xenograft tumors. Data are shown as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Furthermore, MCF-7/TR xenograft model was performed to evaluate the antitumor effect of Nar-LPs in vivo (Figure 6D). As shown in Figures 6E–6G, liposomal nanoparticles enhanced the water solubility and reduced the toxicity of Nar, and Nar-LPs (5 mg/kg) significantly inhibited tumor growth in vivo. In addition, no significant weight loss (Figure 6H) and no histological differences in lung, heart, liver, kidney, or spleen were found in Nar-LPs treatment groups (Figure 6K). These results indicated that, with Nar-LPs, there were no side effects at 5 mg/kg in vivo. Consistent with the in vitro data, Nar-LPs significantly decreased the protein levels of STAT3, p-STAT3, and cyclin D1 in xenograft tumors (Figures 6I and 6J).
Discussion
Mounting evidence supports the application of Nar as an antitumor agent. Several studies have reported some antitumor mechanisms of Nar, but natural products often hit multiple molecular targets. Therefore, we used two omics strategies to identify STAT3 as another important Nar antitumor target. This STAT3-mediated mechanism of action for Nar was first confirmed in this study. The data in vitro and in vivo suggested that Nar could suppress STAT3 phosphorylation and activation, directly bind to the SH2 domain of STAT3 protein, and inhibit STAT3 dimerization and nuclear translocation in MCF-7 cells.
At present, endocrine therapy is highly effective in treatment with ER-positive breast cancer by suppressing estrogen production and/or targeting the ER directly.30 Thus, selective estrogen receptor modulators, such as tamoxifen, have been used widely to treat ER-dependent tumors. However, de novo resistance or acquired resistance limited the clinical efficacy of tamoxifen. In recent studies, the expression and activation of STAT3 phosphorylation could contribute to tamoxifen resistance in breast cancer.20,21 In addition, overexpression of STAT3 target gene cyclin D1 in breast tumors correlated with refractoriness to tamoxifen clinical treatment,31,32 and blocking cyclin D1 expression or function could inhibit development and growth of tamoxifen-resistant breast tumors.33,34 Thus, Nar-targeting STAT3 may be better for the treatment of tamoxifen-resistant breast cancer. To our surprise, Nar not only inhibited STAT3 phosphorylation but also specifically degraded the level of total STAT3 protein and reduced STAT3 protein stability in MCF-7/TR cells. The level of ROS was the primary factor for distinct mechanism of Nar-targeting STAT3 in MCF-7 cells and MCF-7/TR cells. In previous works, the level of ROS was elevated in tamoxifen treatment of breast cancer, which also could build resistance to treatment.28,29 Moreover, an increase of ROS can modify the cell-signaling proteins, including degradation of the ubiquitination/proteasome system.35 Indeed, we found that the level of ROS in MCF-7/TR cells was higher than that in MCF-7 cells, and Nar also could significantly increase the level of ROS in MCF-7/TR cells. In addition, inhibition of ROS accumulation by the ROS scavenger NAC also restored the level of STAT3 protein in MCF-7/TR cells. Our results strongly suggested that Nar targets STAT3 to cause proteasome degradation through accumulation of ROS, but the specific mechanism still needs to be further explored.
The cytotoxic effect of Nar in MCF-7/TR cells was better than that in MCF-7 cells, suggesting that Nar could be more effective against tamoxifen-resistant breast cancer by targeting degradation of STAT3 in vitro. However, the considerable toxicity and poor water solubility of Nar were important obstacles hampering the in vivo efficacy and clinical development.22,36,37 In previous works, the medicinal chemistry approach generated a large number of Nar derivatives, but the antitumor effect of most of these modifications did not improve.36,38 Therefore, the nanoscale drug targeted delivery system was used to enhance the water solubility of Nar. In the case of non-toxic side effects, a Nar dose of 5 mg/kg had more significant inhibitory effect on MCF-7/TR subcutaneous tumor growth.
In this study, using two omics target identification modalities (CMAP and DARTS/MS), we revealed STAT3 to be an antitumor target of Nar (Figure 7A). Meanwhile, Nar-targeting STAT3 had distinct mechanisms in MCF-7 cells and MCF-7/TR cells. Nar directly bound to STAT3 protein and suppressed STAT3 phosphorylation and activation in MCF-7 cells; however, in MCF-7/TR cells it could specifically promote total STAT3 degradation via a ROS-dependent proteasome pathway (Figure 7B). In addition, Nar-encapsulated liposomal nanoparticles exerted a strong antitumor effect in vivo, which established Nar as a novel therapeutic against MCF7/TR (Figure 7C). Therefore, this research provided an efficient strategy for the discovery of natural product targets, and also highlighted targeting STAT3 degradation as a promising therapeutic strategy for the treatment of tamoxifen-resistant breast cancer.
Figure 7.

Hypothetic model illustrating the antitumor research of Nar
(A) Predicting potential target of Nar. (B) Narciclasine targets STAT3 via distinct mechanisms in breast cancer MCF-7 and tamoxifen-resistant MCF-7 cells. (C) Nar-LPs suppress the growth of MCF-7/TR xenograft tumors.
Materials and methods
Materials
Nar was isolated from bulbs of Narcissus species in our laboratory. Protease inhibitor, phosphatase inhibitor, and pronase were from Roche (Mannhein, Germany). CNBr-activated Sepharose 4B was obtained from Sigma (St. Louis, MO, USA). Recombinant human IL-6 was purchased from PeproTech (Rocky Hill, NJ, USA). BTZ and NAC were from MedChemExpress (Monmouth Junction, NJ, USA). Antibodies specific for cyclin D1 (ab40754), c-Myc (ab32072), STAT1 (ab109320), STAT2 (ab32367), STAT4 (ab68156), STAT5 (ab194898), and STAT6 (ab32520) were from Abcam (Cambridge, UK), and antibodies for STAT3 (9139 and 4904), p-STAT3 (9145), P53 (2527), and survivin (2808) were from Cell Signaling Technology (Danvers, MA, USA).
Cell lines and culture
Hepatocellular carcinoma (Hep-3B and HepG2), colorectal carcinoma (HCT116 and SW480), and lung carcinoma (A549) cells were obtained from the Cell Bank of Shanghai Institute of Cell Biology, Chinese Academy of Sciences. Prostate cancer (DU145), malignant melanoma (SK-Mel-28), breast cancer (MCF-7), and tamoxifen-resistant breast cancer (MCF-7/TR) cells were obtained from ATCC (Manassas, VA, USA). The HCT116 was maintained in McCoy’s 5A medium (Gibco, Carlsbad, CA, USA) supplemented with 10% FBS (Biological Industries, Cromwell, CT, USA). The A549 was maintained in F-12K medium (Gibco) supplemented with 10% FBS. The SW480 cells were maintained in L-15 medium (Gibco) supplemented with 10% FBS. The SK-Mel-28, Hep-3B, HepG2, DU145, and MCF-7 cells were maintained in MEM/EBSS (HyClone, Logan, UT, USA) supplemented with 10% FBS. The MCF-7/TR cells were maintained in DMEM (Gibco) supplemented with 10% FBS, 1 μM 4-hydroxytamoxifen, and 10 μg/mL insulin.
CMAP analysis
The gene expression profile of Nar-treated MCF-7 cells was obtained from our previous data dataset GSE85871 (https://www.ncbi.nlm.nih.gov/geo/).39 The differential expression probes of Nar were selected according to fold change (FC ≥ 3). The 940 gene expression signatures of Nar were represented by two sets (“up-” and “down-” probe sets, saved as .grp files, and used as the inputs for CMAP), which was made up by the significant up/downregulation probes respectively. The query in the CMAP was performed as a “quick query” in the query section of http://portals.broadinstitute.org/cmap/.
DARTS/MS proteomics analysis
The DARTS assay was performed according to the protocol described previously.11 In brief, aliquots of MCF-7 cell lysates were treated with Nar (100 μM) or solvent control (DMSO) for 1 h at room temperature. The samples were digested with pronase at the specified ratios under room temperature for 30 min. Digestion was stopped and samples were boiled for western blot analysis. For proteomic analysis, the samples were separated by SDS-PAGE and stained with Coomassie blue. Each lane was excised and divided into three sections based on molecular weight (<70, 70–100, and >100 kDa). Each sample was subjected to an in-gel digestion procedure, followed by LC-MS/MS.
MST
The MST measurement for binding of Nar to STAT3 was performed as described previously.40 In brief, 5 μL recombinant human STAT3 (Novoprotein, China) labeling the protein NHS-RED dye was mixed with 5 μL Nar in different concentrations. Then, samples were incubated for 10 min at room temperature and analyzed using a Monolith NT.115 MST device (NanoTemper, Germany).
CETSA
The CETSA experiment was carried out as described previously.25 In brief, MCF-7 cells were lysed using liquid nitrogen, and cell lysate samples were subjected to centrifugation and the supernatant collected. Aliquots were incubated with Nar (100 μM) or solvent control (DMSO) for 30 min at room temperature. Samples were divided in a volume of 50 μL/tube and heated at a range of temperatures for 3 min, cooled for 3 min at room temperature, and kept on ice. For concentration-response studies, cell lysate samples were incubated with Nar at specified concentrations, heated at 62°C for 3 min, cooled for 3 min at room temperature, and kept on ice. All samples were subjected to centrifugation and the supernatant (soluble fractions) analyzed by western blot.
Pull-down assay
Pull-down assay using immobilized Nar was performed as described previously.41 In brief, the CNBr-activated Sepharose 4B was washed and swelled in 1 mM HCl for 30 min, then washed with the coupling buffer (0.1 M NaHCO3 and 0.5 M NaCl [pH 7.0]). CNBr-activated Sepharose 4B beads were mixed with the Nar in coupling buffer at 4°C for 24 h. Nar-conjugated and control beads were washed with three cycles of high and low pH buffer solutions (buffer 1, 0.1 M acetate and 0.5 M NaCl [pH 4.0]; buffer 2, 0.1 M Tris-HCl and 0.5 M NaCl [pH 8.0]). Coupled and uncoupled beads were separately incubated with the cell lysate on a rotator for 12 h at 4°C. The beads were then washed three times with the lysis buffer, and the bound proteins were analyzed by western blot.
Molecular docking
The STAT3 X-ray structure (PDB: 1BG1) was obtained from the RSCB Protein DataBank (http://www.rcsb.org/) and prepared using the protein preparation wizard Maestro software package (Schrödinger, version 9.0). The SH2 domain of STAT3 protein was defined as the binding pocket.42 The 3D conformer of Nar was downloaded from PubChem (https://pubchem.ncbi.nlm.nih.gov). Nar was docked into a grid box using the extra precision (XP) mode of the Glide program in the Schrödinger suite. The OPLS_2005 force field and all default parameters were applied during docking calculation. The docking pose with the lowest Glide score was generated using PyMOL (http://www.pymol.org/).
Native PAGE
The native PAGE analysis was performed as described previously.43 In brief, native protein samples of MCF-7 cells lysate were mixed with non-denatured gel sample loading buffer (Beyotime, Haimen, China) and loaded onto SDS-free PAGE gels. Following electrophoresis, proteins were transferred to PVDF membranes and immunoblotted with specific antibody as described for western blot analysis.
Luciferase assay
293T cells were co-transfected with STAT3-Luc and renilla luciferase using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) for 24 h. Following treatment with 50 ng/mL IL-6 and the indicated concentrations of Nar and stattic. Luciferase activity was measured using the Dual-Glo Luciferase Assay Kit (Promega) using a Luminescence Microplate Reader (BioTek, Winooski, VT, USA).
Cell viability assay
Cell viability was tested using CCK-8) (Dojindo). Cells (5,000/well) were seeded in 96-well plates. Overnight, cells were treated with different concentrations of Nar for 24 h. The CCK-8 assay was then carried out to assess the effect of Nar on cell viability, and half-maximal inhibitory concentration values were calculated.
Colony-formation assay
Cells were seeded in 12-well plates at 1 × 103/well, followed by treatment with Nar (5, 10, and 50 nM) for 5 days. Colonies were then subsequently fixed with 4% paraformaldehyde and stained with 0.5% crystal violet. Finally, the numbers of cell colonies were imaged and counted.
Apoptosis and cell-cycle analysis
MCF-7/TR cells seeded in 6-well plates were treated with Nar for 24 h. Both floating and adherent cells were harvested for further analysis. The Annexin V-FITC/PI Apoptosis Detection Kit (BD Biosciences, San Jose, CA, USA) was used to detect apoptosis and the cell cycle and the Apoptosis Analysis Kit (Beyotime) was used for cell-cycle analysis, following the manufacturer’s instructions.
Wound-healing and transwell assays
For the wound-healing assay, MCF-7/TR cells were cultured in 12-well plates and cells at 90% were scratched with a pipette tip. After scratch, cells were treated with 50 and 100 nM Nar, and the healing process of the wound was measured over periods of 24 and 48 h. For the transwell assay, MCF-7/TR cells were seeded in a cell invasion chamber of a 24-well transwell plate. Cells were cultured in the absence or presence of Nar for 24 h, then fixed and stained with crystal violet.
Determination of ROS levels
The levels of intracellular ROS were monitored by using a Reactive Oxygen Species Assay Kit (Beyotime). MCF-7 and MCF-7/TR cells were seeded in 96-well plates at 5 × 103/well and cultured overnight, then followed by treatment with Nar (200 nM) for 1 and 3 h. Cells were stained with 10 μM DCFH-DA at 37°C for 30 min, then detected by a Cytation 5 Cell Imaging Multi-Mode Reader (BioTek) at an excitation wavelength of 488 nm and an emission wavelength of 525 nm. MCF-7/TR cells were treated with the ROS scavenger, 10 mM NAC, for 1 h, followed by treatment with or without Nar (200 nM) for 24 h; subsequently, the cells were gently washed with PBS followed by incubation with 10 μM DCFH-DA at 37°C for 30 min. The images were captured with Operetta CLS.
RNA extraction and real-time PCR analysis
MCF-7/TR cells were treated with Nar (100 nM) for 3, 6, and 12 h, and total mRNA extracted using the TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. RT-PCR amplifications were performed using the FastStart Essential DNA Green Master and LightCycler 96 Instrument (Roche, Basel, Switzerland). Total mRNA was reverse transcribed into cDNA using a PrimeScript RT reagent kit (Takara, Shiga, Japan) following the manufacturer’s protocol. β-Actin was used as the reference gene. The forward and reverse primers used are listed in Table S1.
Western blot analysis
For BTZ experiments, cells were treated with 20 nM BTZ for 12 h before Nar was added for another 8 h treatment, and cells were collected for western blot analysis. For CHX chase studies, cells were treated with Nar for 3 h, then 50 mg/mL of CHX was added, and cells were collected at the indicated time points for western blot analysis. For NAC experiment, cells were treated with the ROS scavenger, 10 mM NAC, for 1 h, followed by treatment with or without Nar, and cells were collected for western blot analysis. Total cellular proteins were extracted with NP-40 lysis buffer, and nuclear and cytoplasmic proteins were extracted with a nuclear and cytoplasmic protein extraction kit (Beyotime). Protein concentrations were determined using a BCA protein assay kit (Beyotime). For western blot analysis, the samples were separated by SDS-PAGE and transferred onto PVDF membranes. After blocking with 5% nonfat dry milk, the membranes were incubated with primary antibodies at the manufacturers’ recommended dilutions at 4°C overnight. A goat anti-rabbit or donkey anti-mouse secondary antibody (IRDye 800, LI-COR Biosciences, Lincoln, NE, USA) was added at the recommended dilutions and the incubation continued for another 1 h at room temperature. The immunoreactive bands were scanned using an Odyssey Infrared Imaging System (LI-COR Biosciences).
Immunofluorescence staining
Breast tumor MCF-7 cells were treated with or without Nar 100 nM for 24 h. Cells were washed and fixed in 4% paraformaldehyde for 20 min at room temperature, and then permeabilized in 0.1% Triton X-100 for 30 min at room temperature. After blocking with 5% nonfat dry milk and incubation with the indicated antibodies overnight at 4°C, cells were incubated with corresponding secondary antibodies for 1 h at room temperature, and then stained with DAPI for 30 min. Images were taken using fluorescence microscopy (Leica Microsystems, Germany).
Preparation and characterization of liposome
Nar-encapsulated liposomal nanoparticles were prepared using a thin-film hydration method. In brief, 1.25 mg Nar was dissolved in 0.5 mL methanol. The lipids, 19.9 mg DPPC, 2.7 mg cholesterol, and 2.4 mg DSPE-MPEG2000, were dissolved in 4.5 mL chloroform. The mixture solutions were placed in a round-bottomed flask and fully removed using a rotary evaporator. The resulting thin film was then hydrated with 2.5 mL PBS for 30 min to form liposomes. The liposomes were extruded using a 200-nm polycarbonate membrane filter to control size homogeneity. Finally, Nar-loaded liposomes was purified to remove unloaded Nar by centrifugation at 12,000 rpm for 5 min. The amount of Nar loaded was quantified using HPLC. Nar loading efficiency was calculated by the following equation: loading efficiency (%) = (encapsulated Nar/initial input of Nar) × 100%. The drug release study was carried out in PBS according to a previously reported method.44 The particle size and zeta potential of prepared liposomes were measured using a Zetasizer Nano ZS instrument (Malvern, UK). The microstructure of Nar-encapsulated liposomes was characterized using cryo-TEM (Talos F200C G2).
Xenograft models
All animal studies were approved by the Committee on the Ethics of Animal Experiment of the Shanghai University of Traditional Chinese Medicine, which complied with the national and international guidelines. Female BALB/c-nu mice (4 weeks old) were obtained from Shanghai Slake Experimental Animal and housed under specific pathogen-free conditions. For the MCF-7 xenograft experiment, the breast pad xenografts were established by subcutaneous injections of 8 × 106 MCF-7 cells. Following a 2-week inoculation period, the tumor-bearing mice were randomly divided into Nar treatment and control groups. The treatment group was injected with 1 mg/kg Nar every 2 days for 4 weeks and those in the control group were injected with the same volume of saline. Body weights and tumor size were recorded two times per week. For MCF-7/TR xenograft experiment, 2 × 106 MCF-7/TR cells were subcutaneously injected into the breast pad of mice. The mice were then randomized into three treatment groups: control, Nar (2 mg/kg), and Nar-LPs (5 mg/kg). Nar was administered daily via intraperitoneal injection, and Nar-LPs was administered daily via tail vein injection. Tumor volume was calculated using the equation: volume (mm3) = length × (width2)/2. After 8 days, Mice were sacrificed, and tumors and other major organs, such as liver, spleen, kidney, and heart, were isolated.
IHC analysis
The IHC analysis was carried out as described previously.45 The paraffin-embedded tissue sections were deparaffinized in xylene, treated with a graded series of alcohol and distilled water. Antigen recovery was performed using microwave boiling treatment in 10 mM sodium citrate buffer (pH 6.0), and the slices were incubated with 3% H2O2 to block endogenous peroxide activity. After blocking, the slides were incubated with primary antibodies against p-STAT3 and STAT3 at 4°C overnight. The slides were incubated with secondary antibody at 37°C for 30 min, followed by incubation with DAB and hematoxylin, and images were captured under a microscope.
Statistical analysis
Statistical analysis was performed with unpaired t test when comparing two different groups or one-way ANOVA with Tukey’s multiple comparison tests. All calculated values are shown as mean ± standard deviation (SD). All statistical analyses were performed using SPSS software (version 16.0, SPSS, Chicago, IL, USA).
Acknowledgments
The work was supported by National Natural Science Foundation of China (81772798 and 81903510) and the National Science and Technology Major Project of China (2019ZX09201004-003-010 and 2018ZX09731016-005), the Program for Professor of Special Appointment (Young Eastern Scholar) at Shanghai Institutions of Higher Learning (QD2018035), the “Chenguang Program” of Education Commission of Shanghai Municipality (18CG46), the Shanghai Engineering Research Center for the Preparation of Bioactive Natural Products (16DZ2280200), and the National Key Research and Development Program of China (2017YFC1700200).
Author contributions
W.Z., X.L., and S.L. designed the research. C.L. and Y.H. carried out the experiments and performed data analysis. Q.W., H.Z., and J.J. participated in part of the experiments. D.L., Y.Z., and Y.S. participated in part of the data analysis. All authors have approved the final article.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2021.12.025.
Contributor Information
Weidong Zhang, Email: wdzhangy@hotmail.com.
Xin Luan, Email: luanxin@shutcm.edu.cn.
Sanhong Liu, Email: liush@shutcm.edu.cn.
Supplemental information
References
- 1.Ceriotti G. Narciclasine: an antimitotic substance from narcissus bulbs. Nature. 1967;213:595–596. doi: 10.1038/213595a0. [DOI] [PubMed] [Google Scholar]
- 2.Pettit G.R., Pettit G.R., 3rd, Backhaus R.A., Boyd M.R., Meerow A.W. Antineoplastic agents, 256. Cell growth inhibitory isocarbostyrils from Hymenocallis. J. Nat. Prod. 1993;56:1682–1687. doi: 10.1021/np50100a004. [DOI] [PubMed] [Google Scholar]
- 3.Fuchs S., Hsieh L.T., Saarberg W., Erdelmeier C.A.J., Wichelhaus T.A., Schaefer L., Koch E., Furst R. Haemanthus coccineus extract and its main bioactive component narciclasine display profound anti-inflammatory activities in vitro and in vivo. J. Cell. Mol. Med. 2015;19:1021–1032. doi: 10.1111/jcmm.12493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim J., Park Y., Chun Y.S., Cha J.W., Kwon H.C., Oh M.S., Chung S., Yang H.O. Effect of Lycoris chejuensis and its active components on experimental models of Alzheimer's disease. J. Agric. Food Chem. 2015;63:6979–6988. doi: 10.1021/acs.jafc.5b00889. [DOI] [PubMed] [Google Scholar]
- 5.Julien S.G., Kim S.Y., Brunmeir R., Sinnakannu J.R., Ge X., Li H., Ma W., Yaligar J., Kn B.P., Velan S.S., et al. Narciclasine attenuates diet-induced obesity by promoting oxidative metabolism in skeletal muscle. PLoS Biol. 2017;15:e1002597. doi: 10.1371/journal.pbio.1002597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Goietsenoven G., Hutton J., Becker J.P., Lallemand B., Robert F., Lefranc F., Pirker C., Vandenbussche G., Van Antwerpen P., Evidente A., et al. Targeting of eEF1A with Amaryllidaceae isocarbostyrils as a strategy to combat melanomas. FASEB J. 2010;24:4575–4584. doi: 10.1096/fj.10-162263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lefranc F., Sauvage S., Van Goietsenoven G., Megalizzi V., Lamoral-Theys D., Debeir O., Spiegl-Kreinecker S., Berger W., Mathieu V., Decaestecker C., et al. Narciclasine, a plant growth modulator, activates Rho and stress fibers in glioblastoma cells. Mol. Cancer Ther. 2009;8:1739–1750. doi: 10.1158/1535-7163.MCT-08-0932. [DOI] [PubMed] [Google Scholar]
- 8.Cao C., Huang W., Zhang N., Wu F., Xu T., Pan X., Peng C., Han B. Narciclasine induces autophagy-dependent apoptosis in triple-negative breast cancer cells by regulating the AMPK-ULK1 axis. Cell Prolif. 2018;51:e12518. doi: 10.1111/cpr.12518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lamb J., Crawford E.D., Peck D., Modell J.W., Blat I.C., Wrobel M.J., Lerner J., Brunet J.P., Subramanian A., Ross K.N., et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 10.Qu X.A., Rajpal D.K. Applications of Connectivity Map in drug discovery and development. Drug Discov. Today. 2012;17:1289–1298. doi: 10.1016/j.drudis.2012.07.017. [DOI] [PubMed] [Google Scholar]
- 11.Lomenick B., Hao R., Jonai N., Chin R.M., Aghajan M., Warburton S., Wang J., Wu R.P., Gomez F., Loo J.A. Target identification using drug affinity responsive target stability (DARTS) Proc. Natl. Acad. Sci. U S A. 2009;106:21984–21989. doi: 10.1073/pnas.0910040106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beebe J.D., Liu J.Y., Zhang J.T. Two decades of research in discovery of anticancer drugs targeting STAT3, how close are we? Pharmacol. Ther. 2018;191:74–91. doi: 10.1016/j.pharmthera.2018.06.006. [DOI] [PubMed] [Google Scholar]
- 13.Zhang T., Li J., Yin F., Lin B., Wang Z., Xu J., Wang H., Zuo D., Wang G., Hua Y., et al. Toosendanin demonstrates promising antitumor efficacy in osteosarcoma by targeting STAT3. Oncogene. 2017;36:6627–6639. doi: 10.1038/onc.2017.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garg M., Shanmugam M.K., Bhardwaj V., Goel A., Gupta R., Sharma A., Baligar P., Kumar A.P., Goh B.C., Wang L., et al. The pleiotropic role of transcription factor STAT3 in oncogenesis and its targeting through natural products for cancer prevention and therapy. Med. Res. Rev. 2020;41:1291–1336. doi: 10.1002/med.21761. [DOI] [PubMed] [Google Scholar]
- 15.Mohan C.D., Rangappa S., Preetham H.D., Chandra Nayaka S., Gupta V.K., Basappa S., Sethi G., Rangappa K.S. Targeting STAT3 signaling pathway in cancer by agents derived from Mother Nature. Semin. Cancer Biol. 2020 doi: 10.1016/j.semcancer.2020.03.016. [DOI] [PubMed] [Google Scholar]
- 16.Alsamri H., El Hasasna H., Al Dhaheri Y., Eid A.H., Attoub S., Iratni R. Carnosol, a natural polyphenol, inhibits migration, metastasis, and tumor growth of breast cancer via a ROS-dependent proteasome degradation of STAT3. Front. Oncol. 2019;9:743. doi: 10.3389/fonc.2019.00743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ru-Xiang X., Xiao-Hu N., Ou-Yang J., Ying X., Dan-Yan L., Xing-Yu D., Ru-En L. Paeoniflorin inhibits human glioma cells via STAT3 degradation by the ubiquitin–proteasome pathway. Drug Des. Dev. Ther. 2015;9:5611. doi: 10.2147/DDDT.S93912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bai L., Zhou H., Xu R., Zhao Y., Chinnaswamy K., McEachern D., Chen J., Yang C.Y., Liu Z., Wang M., et al. A potent and selective small-molecule degrader of STAT3 achieves complete tumor regression in vivo. Cancer Cell. 2019;36:498–511 e417. doi: 10.1016/j.ccell.2019.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xing J., Li J., Fu L., Gai J., Guan J., Li Q. SIRT4 enhances the sensitivity of ER-positive breast cancer to tamoxifen by inhibiting the IL-6/STAT3 signal pathway. Cancer Med. 2019;8:7086–7097. doi: 10.1002/cam4.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bui Q.T., Im J.H., Jeong S.B., Kim Y.M., Lim S.C., Kim B., Kang K.W. Essential role of Notch4/STAT3 signaling in epithelial-mesenchymal transition of tamoxifen-resistant human breast cancer. Cancer Lett. 2017;390:115–125. doi: 10.1016/j.canlet.2017.01.014. [DOI] [PubMed] [Google Scholar]
- 21.Zhu N., Zhang J., Du Y.P., Qin X.D., Miao R.D., Nan J., Chen X., Sun J.J., Zhao R., Zhang X.X., et al. Loss of ZIP facilitates JAK2-STAT3 activation in tamoxifen-resistant breast cancer. Proc. Natl. Acad. Sci. U S A. 2020;117:15047–15054. doi: 10.1073/pnas.1910278117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kornienko A., Evidente A. Chemistry, biology, and medicinal potential of narciclasine and its congeners. Chem. Rev. 2008;108:1982–2014. doi: 10.1021/cr078198u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jimenez A., Santos A., Alonso G., Vazquez D. Inhibitors of protein synthesis in eukaryotic cells: comparative effects of some Amaryllidaceae alkaloids. BBA. 1976;425:342–348. doi: 10.1016/0005-2787(76)90261-6. [DOI] [PubMed] [Google Scholar]
- 24.Sun N.N., Sun W.C., Li S.M., Yang J.B., Yang L.F., Quan G.H., Gao X., Wang Z.J., Cheng X., Li Z.H., et al. Proteomics analysis of cellular proteins co-immunoprecipitated with nucleoprotein of influenza A virus (H7N9) Int. J. Mol. Sci. 2015;16:25982–25998. doi: 10.3390/ijms161125934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Molina D.M., Jafari R., Ignatushchenko M., Seki T., Larsson E.A., Dan C., Sreekumar L., Cao Y.H., Nordlund P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013;341:84–87. doi: 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- 26.Yu H., Kortylewski M., Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007;7:41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y., Shen Y.C., Wang S.N., Shen Q., Zhou X. The role of STAT3 in leading the crosstalk between human cancers and the immune system. Cancer Lett. 2018;415:117–128. doi: 10.1016/j.canlet.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang G., Nowsheen S., Aziz K., Georgakilas A.G. Toxicity and adverse effects of tamoxifen and other anti-estrogen drugs. Pharmacol. Ther. 2013;139:392–404. doi: 10.1016/j.pharmthera.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 29.Cho S.K., Pedram A., Levin E.R., Kwon Y.J. Acid-degradable core-shell nanoparticles for reversed tamoxifen-resistance in breast cancer by silencing manganese superoxide dismutase (MnSOD) Biomaterials. 2013;34:10228–10237. doi: 10.1016/j.biomaterials.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hanker A.B., Sudhan D.R., Arteaga C.L. Overcoming endocrine resistance in breast cancer. Cancer Cell. 2020;37:496–513. doi: 10.1016/j.ccell.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jirstrom K., Stendahl M., Ryden L., Kronblad A., Bendahl P.O., Stal O., Landberg G. Adverse effect of adjuvant tamoxifen in premenopausal breast cancer with cyclin D1 gene amplification. Cancer Res. 2005;65:8009–8016. doi: 10.1158/0008-5472.CAN-05-0746. [DOI] [PubMed] [Google Scholar]
- 32.Stendahl M., Kronblad A., Rydén L., Emdin S., Bengtsson N.O., Landberg G. Cyclin D1 overexpression is a negative predictive factor for tamoxifen response in postmenopausal breast cancer patients. Br. J. Cancer. 2004;90:1942–1948. doi: 10.1038/sj.bjc.6601831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kilker R.L., Planas-Silva M.D. Cyclin D1 is necessary for tamoxifen-induced cell cycle progression in human breast cancer cells. Cancer Res. 2006;66:11478–11484. doi: 10.1158/0008-5472.CAN-06-1755. [DOI] [PubMed] [Google Scholar]
- 34.Shi Q., Li Y., Li S., Jin L., Lai H., Wu Y., Cai Z., Zhu M., Li Q., Li Y., et al. LncRNA DILA1 inhibits cyclin D1 degradation and contributes to tamoxifen resistance in breast cancer. Nat. Commun. 2020;11:5513. doi: 10.1038/s41467-020-19349-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang J.X., Wang X.L., Vikash V., Ye Q., Wu D.D., Liu Y.L., Dong W.G. ROS and ROS-mediated cellular signaling. Oxid Med. Cell. Longev. 2016;2016:4350965. doi: 10.1155/2016/4350965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Furst R. Narciclasine—an Amaryllidaceae alkaloid with potent antitumor and anti-inflammatory properties. Planta Med. 2016;82:1389–1394. doi: 10.1055/s-0042-115034. [DOI] [PubMed] [Google Scholar]
- 37.Van Goietsenoven G., Mathieu V., Lefranc F., Kornienko A., Evidente A., Kiss R. Narciclasine as well as other Amaryllidaceae isocarbostyrils are promising GTP-ase targeting agents against brain cancers. Med. Res. Rev. 2013;33:439–455. doi: 10.1002/med.21253. [DOI] [PubMed] [Google Scholar]
- 38.Ingrassia L., Lefranc F., Dewelle J., Pottier L., Mathieu V., Spiegl-Kreinecker S., Sauvage S., El Yazidi M., Dehoux M., Berger W., et al. Structure-activity relationship analysis of novel derivatives of narciclasine (an Amaryllidaceae isocarbostyril derivative) as potential anticancer agents. J. Med. Chem. 2009;52:1100–1114. doi: 10.1021/jm8013585. [DOI] [PubMed] [Google Scholar]
- 39.Lv C., Wu X., Wang X., Su J., Zeng H., Zhao J., Lin S., Liu R., Li H., Li X., et al. The gene expression profiles in response to 102 Traditional Chinese Medicine (TCM) components: a general template for research on TCMs. Sci. Rep. 2017;7:352. doi: 10.1038/s41598-017-00535-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qu Y., Olsen J.R., Yuan X., Cheng P.F., Levesque M.P., Brokstad K.A., Hoffman P.S., Oyan A.M., Zhang W., Kalland K.H., et al. Small molecule promotes β-catenin citrullination and inhibits Wnt signaling in cancer. Nat. Chem. Biol. 2018;14:94–101. doi: 10.1038/nchembio.2510. [DOI] [PubMed] [Google Scholar]
- 41.Tran P.L., Kim S.A., Choi H.S., Yoon J.H., Ahn S.G. Epigallocatechin-3-gallate suppresses the expression of HSP70 and HSP90 and exhibits anti-tumor activity in vitro and in vivo. BMC Cancer. 2010;10:276. doi: 10.1186/1471-2407-10-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Furtek S.L., Backos D.S., Matheson C.J., Reigan P. Strategies and approaches of targeting STAT3 for cancer treatment. ACS Chem. Biol. 2016;11:308–318. doi: 10.1021/acschembio.5b00945. [DOI] [PubMed] [Google Scholar]
- 43.Shin D.S., Kim H.N., Shin K.D., Yoon Y.J., Kim S.J., Han D.C., Kwon B.M. Cryptotanshinone inhibits constitutive signal transducer and activator of transcription 3 function through blocking the dimerization in DU145 prostate cancer cells. Cancer Res. 2009;69:193–202. doi: 10.1158/0008-5472.CAN-08-2575. [DOI] [PubMed] [Google Scholar]
- 44.Ashrafzadeh M.S., Akbarzadeh A., Heydarinasab A., Ardjmand M. In vivo glioblastoma therapy using targeted liposomal cisplatin. Int. J. Nanomed. 2020;15:7035–7049. doi: 10.2147/IJN.S255902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Y., Wang X., Zeng S., Zhang X., Zhao J., Zhang X., Chen X., Yang W., Yang Y., Dong Z., et al. The natural polyphenol curcumin induces apoptosis by suppressing STAT3 signaling in esophageal squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2018;37:303. doi: 10.1186/s13046-018-0959-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.