

Summary
Neurotrophic factors and their signaling cascades play important roles in synaptic growth, which can be investigated in cultured primary neurons to better control the concentrations and timing of neurotrophic factor treatment. Here, we provide a protocol detailing the preparation of cultured primary mouse neurons and the neurotrophic factor treatment. We then describe electrophysiological recording of synaptic transmission, immunocytochemistry of AMPA receptor expression, and imaging analysis of dendritic spines. This platform enables characterization of synaptic growth at functional and morphological levels.
For complete details on the use and execution of this profile, please refer to Zhou et al. (2021).
Subject areas: Cell Biology, Cell culture, Cell isolation, Microscopy, Molecular Biology, Neuroscience
Graphical abstract
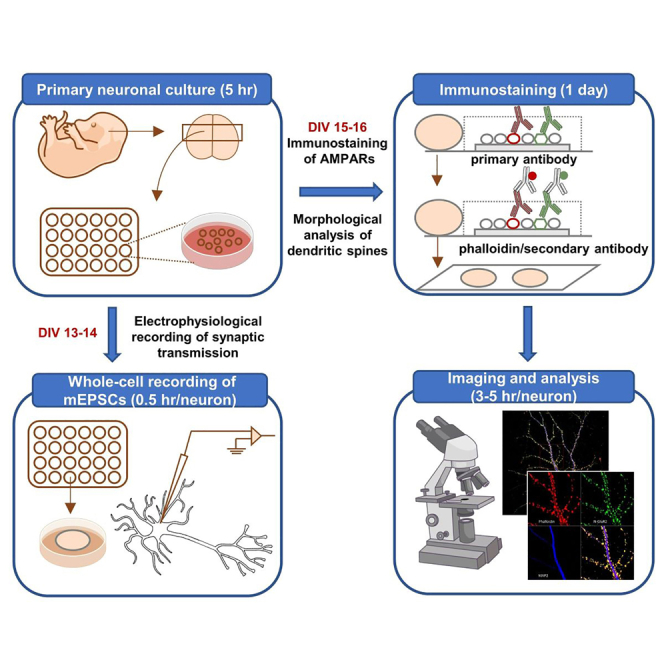
Highlights
-
•
Detailed protocol to characterize synaptic growth in primary mouse neuronal cultures
-
•
Utilizes electrophysiological recording, fluorescence labeling and confocal imaging
-
•
Enables assessment of synaptic growth at both functional and morphological levels
Neurotrophic factors and their signaling cascades play important roles in synaptic growth, which can be investigated in cultured primary neurons to better control the concentrations and timing of neurotrophic factor treatment. Here, we provide a protocol detailing the preparation of cultured primary mouse neurons and the neurotrophic factor treatment. We then describe electrophysiological recording of synaptic transmission, immunocytochemistry of AMPA receptor expression, and imaging analysis of dendritic spines. This platform enables characterization of synaptic growth at functional and morphological levels.
Before you begin
In our previous study, we demonstrated that neurite growth-promoting factor 2 (NGPF2), a neurotrophic-factor, can promote synaptic growth in dissociated primary cortical neuronal cultures at DIV (days in vitro) 13–16 (Zhou et al., 2021). Here, we describe the experimental steps, reagents, equipment, and methods of statistical analysis that were utilized for the characterization of NGPF2-induced synaptic growth from the following three perspectives: dendritic spine morphological changes, surface and synaptic expression of AMPA receptors, and recording of synaptic transmission.
Preparation and culture of primary mouse cortical neurons
-
1.Prepare PDL-coated coverslips:
 Timing: 8 h
Timing: 8 h
-
a.Incubate coverslips (12 mm in diameter for 24-well culture plates) with 0.1 M HCl in a glass bottle at room temperature (20°C–25°C) for 2 h on a platform rocker at 20 rpm.
-
b.Remove the HCl solution in a fume hood following institutional safety guidelines.
-
c.Extensively wash coverslips in a Petri dish by incubating with autoclaved water for 30 min.
-
d.Repeat Step c five times.
-
e.Store coverslips in 100% ethanol for later use within 3 months.
-
f.Use #5 eye surgical forceps to hold coverslips and flame with an alcohol lamp to remove ethanol, then place coverslips in 24-well plate, one per well.
-
g.Sterilize the coverslips by ultraviolet light exposure for 30 min.
-
h.Add 0.5 mL of poly-D-lysine (PDL) solution (0.05 mg/mL, dissolved in H2O) to each well containing coverslips and place in a humidified incubator (5% CO2, 37°C) for 2 h.
-
i.Aspirate the PDL solution, and wash coverslips three times with autoclaved H2O.Note: To prevent coated coverslips from drying, coverslips should be placed in a humidified incubator and used for cell seeding within 3 h.
-
a.
-
2.Preparation of culture medium:Timing: 30 min
-
a.Prepare culture medium: Neurobasal A supplemented with B-27 supplement, GlutaMAX supplement, and Penicillin/streptomycin (see “MATERIALS AND EQUIPMENT” for detailed information).
-
b.Pass the culture medium through a 0.22 μm strainer (vacuum filter/storage bottle system) to sterilize the medium and store at 4°C for later use.
-
c.Transfer culture medium to 50-mL conical tubes with vented caps and place in the humidified incubator (5% CO2, 37°C) at least 30 min before use.
 CRITICAL: Pre-warm culture medium using conical tubes or flasks with vented caps is critical. The pH-balanced medium introduces minimal fluctuations to the microenvironment of cultured neurons when replacing medium.
CRITICAL: Pre-warm culture medium using conical tubes or flasks with vented caps is critical. The pH-balanced medium introduces minimal fluctuations to the microenvironment of cultured neurons when replacing medium.
-
a.
-
3.
Tissue dissociation and cell seeding
Day 1Timing: 5 h
-
a.Prepare dissociation medium by dissolving papain (10 U/mL) and DNase I (125 U/mL) in the culture medium.
-
b.Filter-sterilize (0.22 μm) 3 mL of dissociation medium (for each pup) and place in culture incubator to pre-warm.
-
c.Wipe P0 mouse pups using tissue paper sprayed with 75% ethanol gently and place pups in a sterile Petri dish on an ice pack.
-
d.Following the guidelines approved by the experimenters’ Institutional Animal Care and Use Committee, decapitate the pups.
-
e.Cut the skull along the medulla oblongata with eye surgical scissors, remove the skull and skin.
-
f.Transfer cortical tissue to a Petri dish containing ice-cold Hanks’s Balanced Salt Solution (HBSS) using a stainless-steel micro-spoon.
-
g.Thoroughly remove meninges under dissection microscope with #5 fine forceps.
-
h.Mince the cortical tissue into small pieces (approximate 1 mm3) with fine scissors and transfer using a pipette into a 15-mL centrifuge tube containing 3 mL pre-warmed dissociation medium for cortical tissue of each pup.
-
i.Place the tube on a platform shaker (30 rpm) in a humidified incubator (5% CO2, 37°C) for 20 min.
-
j.Gently pipette the cells up and down using a 1 mL wide bore pipet tip 10 times every 10 min.
-
k.Pass the cell suspension through a 40 μm sterile cell strainer and collect the flow-through in 15-mL centrifuge tube.
-
l.Centrifuge at 300×g (RCF) for 5 min at room temperature.
-
m.Discard the supernatant and resuspend the cells with culture medium.
-
n.Count cell density using a hemocytometer.
-
o.Adjust the cell density to 6×104 cells/mL with culture medium. Seed cells onto PDL-coated coverslips, 0.5 mL in each well. Allow the cells to settle onto the coverslips in the incubator.
-
p.After 4 h, replace all medium with fresh pre-warmed and pH-balanced culture medium, 0.5 mL in each well.CRITICAL: When adding or removing medium, place pipette tips on the side of the well. Do not pipette directly to the cells.Day 4Timing: 12 h
-
q.On DIV 4, in order to inhibit the proliferation of glial cells, add AraC stock solution (1 mg/mL dissolved in Milli-Q water) to the wells containing neurons at the final concentration of 1 μg/mL, incubate for 12 h.
-
r.Replace all medium with fresh pre-warmed and pH-balanced culture medium, 0.5 mL in each well.Day 7–16Timing: 10 min
-
s.Repeat replacing 250 μL medium with fresh pre-warmed culture medium in each well every 3 days.
-
t.Culture cortical neurons until DIV13-14 for recording or DIV15-16 for immunostaining.
-
a.
Preparation of extracellular fluid (ECF) and intracellular solution
-
4.
Prepare ECF by adding the following chemicals to Milli-Q water (in mM): 120 NaCl, 1.2 MgCl2, 3.0 KCl, 2.0 CaCl2, 25 HEPES, 25 D-glucose (see “MATERIALS AND EQUIPMENT” for detailed information).
-
5.
Adjust the pH of ECF to 7.35 with 1 M NaOH.
-
6.
If the osmolarity of ECF is lower than 280–290 mOsm, gradually add and dissolve sucrose powder in the ECF, until the osmolarity reaches 280–290 mOsm.
-
7.
Sterilize the ECF by passing through a 0.22 μm vacuum filter/storage bottle system, store at 4°C. Use within one week.
-
8.
Prepare tetrodotoxin (TTX) stock solution (1 mM) with sterile H2O, aliquot and store at −20°C. Use within three months.
-
9.
Prepare picrotoxin stock solution (100 mM) with sterile H2O, aliquot and store at −20°C. Use within three months.
-
10.
Prepare intracellular solution with the following chemicals (in mM) in Milli-Q water: 130 CsMeS, 5 NaCl, 1 MgCl2, 0.05 EGTA, 10 HEPES, 1 Mg-ATP, 0.3 Na3-GTP, and 5 QX-314. (see “MATERIALS AND EQUIPMENT” for detailed information.)
-
11.
Adjust the pH of intracellular solution to 7.35 with 1 M CsOH.
-
12.
Adjust the osmolarity of intracellular solution to 290–300 mOsm with sucrose.
-
13.
Aliquot the intracellular solution to 1 mL per microcentrifuge tube and store at −20°C. Use within one month.
Note: TTX and picrotoxin stock solution are stored in aliquots. Each aliquot is thawed and added to ECF on the day of electrophysiological recordings. Avoid multiple freeze-thaw cycles.
Confocal imaging setup
Images can be obtained using a Zeiss LSM 700 confocal microscope at room temperature. Images at 2,048 × 2,048 pixels can be acquired using Zen 2010 software (Zeiss) and Zeiss 63 × (NA 1.4, oil immersion) objective with the same settings and configurations for all samples within each experiment. Configure Z-stack images using Acquisition Dimensions and four slices with 0.5 mm interval. Export images files in Tiff file format for analysis.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse GluA2-NTD (1:1000) | Merck Millipore | Cat#MAB397; RRID: AB_2113875 |
| Rabbit MAP2 (1:1000) | Merck Millipore | Cat#MAB3418; RRID: AB_11212326 |
| Rabbit Synapsin 1 (1:1000) | Cell Signaling Technology | Cat#5297; RRID: AB_2616578 |
| Chemicals, peptides, and recombinant proteins | ||
| Tetrodotoxin | Alomone Labs | Cat#T-550 |
| Picrotoxin | Sigma-Aldrich | Cat#R284556 |
| Rhodamine Phalloidin | Thermo Fisher Scientific | Cat#R415 |
| Poly-D-Lysine | Sigma-Aldrich | Cat#P7280 |
| Diamond Antifade Mountant | Thermo Fisher Scientific | Cat#P36965 |
| GlutaMax | Thermo Fisher Scientific | Cat#35050061 |
| Neurobasal A | Thermo Fisher Scientific | Cat#10888022 |
| B27 | Thermo Fisher Scientific | Cat#17504044 |
| Penicillin-Streptomycin | Gibco | Cat#15140122 |
| Papain | Worthington Biochemical Corporation | Cat#A003124 |
| DNase I | Thermo Fisher Scientific | Cat#89836 |
| HBSS | Thermo Fisher Scientific | Cat#88284 |
| AraC | Sigma-Aldrich | Cat#C1768 |
| Triton X-100 | Sigma-Aldrich | Cat#X100 |
| NaCl | Sigma-Aldrich | Cat#S5886 |
| MgCl2 | Sigma-Aldrich | Cat#449172 |
| KCl | Sigma-Aldrich | Cat# P5405 |
| CaCl2 | Sigma-Aldrich | Cat# C5670 |
| HEPES | Sigma-Aldrich | Cat#54457 |
| D-Glucose | Sigma-Aldrich | Cat#G7021 |
| CsMeS | Sigma-Aldrich | Cat#C1426 |
| EGTA | Sigma-Aldrich | Cat#03777 |
| Mg-ATP | Sigma-Aldrich | Cat#A9187 |
| Na-GTP | Sigma-Aldrich | Cat#G8877 |
| QX-314 | Sigma-Aldrich | Cat#L5783 |
| NaOH | Sigma-Aldrich | Cat#S2770 |
| CsOH | Sigma-Aldrich | Cat#232068 |
| Sucrose | Sigma-Aldrich | Cat#V900116 |
| Experimental models: Organisms/strains | ||
| C57BL/6 wildtype mice | Charles River | N/A |
| Software and algorithms | ||
| pCLAMP 10 | Molecular Device | https://www.moleculardevices.com/ |
| Mini Analysis Program 6.0.3 | Synaptosoft Inc | http://www.synaptosoft.com/ |
| Zen 2010 software | Zeiss | https://www.zeiss.com/microscopy/int/home.html |
| ImageJ | National Institutes of Health | https://imagej.nih.gov/ij/index.html |
| Other | ||
| 12 mm coverslip | Assistant | Cat#1001/12 |
| 24-well plate | Corning | Cat#3524 |
| 15-mL conical tubes | Corning | Cat#430790 |
| 50-mL conical tubes | Corning | Cat#430829 |
| Petri dish | Corning | Cat# 430167 |
| Falcon® 40 μm cell strainer, sterile | Corning | Cat#352340 |
| 1-mL wide bore pipet tip | Corning | Cat#T-1005-WB-C |
| 50-mL conical tubes with vented cap | Corning | Cat#CLS431720 |
| #5 forceps | Dumont | Cat#11254-20 |
| Eye surgical scissors | WPI | Cat#500216 |
| Glass pipettes | Sutter | Cat#BF100-78-10 |
| Syringe filter, 0.22 μm | Corning | Cat#431215 |
| Vacuum Filter/Storage Bottle System, 0.22 μm, 500 mL | Corning | Cat#430769 |
| Humidified incubator | Thermo Fisher | Cat#Form 311 |
| Centrifuge | Eppendorf | Cat#5804R |
| Amplifier | Molecular Devices | Cat#700B |
| Digitizer | Molecular Devices | Cat#1440A |
| Micromanipulator | SUTTER | Cat#MP-225 |
| Micropipette puller | SUTTER | Cat#P-1000 |
| Confocal microscope | Zeiss | Cat#LSM 700 |
| pH meter | Thermo Fisher | Cat#AE150 |
Materials and equipment
Culture Medium
| Reagent | Final concentration | Amount |
|---|---|---|
| Neurobasal A | 1× | 480 mL |
| B27 Supplement (50×) | 1× | 10 mL |
| GlutaMax | 1× | 5 mL |
| Penicillin-Streptomycin | 1× | 5 mL |
| Total | 500 mL |
Note: store at 4°C, use within 1 month.
Dissociation Medium
| Reagent | Final concentration | Amount |
|---|---|---|
| Papain | 10 U/mL | 50 μL |
| DNase | 125 U/mL | 40 μL |
| Culture Medium | 1× | 4.91 mL |
| Total | 5 mL |
Note: Prepare before tissue dissociation, use within 30 min.
Extracellular Fluid (ECF)
| Reagent | Final concentration | Amount |
|---|---|---|
| NaCl | 120 mM | 7.013 g |
| MgCl2 | 1.2 mM | 0.114 g |
| KCl | 3.0 mM | 0.224 g |
| CaCl2 | 2.0 mM | 0.222 g |
| HEPES | 25.0 mM | 5.958 g |
| D-Glucose | 25.0 mM | 4.504 g |
| 1 M NaOH | N/A | Adjust pH to 7.35 |
| Milli-Q water | N/A | Fill up to 1000 mL |
| Sucrose | N/A | Adjust osmolarity to 280–290 mOsm |
| Total | 1000 mL |
Note: store at 4°C, use within 1 week.
Intracellular Solution
| Reagent | Final concentration | Amount |
|---|---|---|
| CsMeS | 130 mM | 5.928 g |
| NaCl | 5.0 mM | 0.058 g |
| MgCl2 | 1.0 mM | 0.019 g |
| EGTA | 0.05 mM | 0.004 g |
| HEPES | 10 mM | 0.477 g |
| Mg-ATP | 3.0 mM | 0.304 g |
| Na3GTP | 0.3 mM | 0.031 g |
| QX-314 | 5.0 mM | 0.343 g |
| Milli-Q water | N/A | Fill up to 200 mL |
| 1 M CsOH | N/A | Adjust pH to 7.35 |
| Sucrose | N/A | Adjust osmolarity to 280–290 mOsm |
| Total | 200 mL |
Note: store at −20°C, use within 2 months.
Step-by-step method details
Electrophysiological recording of mEPSCs
Whole-cell patch clamp is the most widely utilized technique to study synaptic transmission in cultured neurons. Miniature postsynaptic currents (mEPSCs) are the results of spontaneous synaptic transmission in the absence of action potentials, thus reflecting the basal-level synaptic transmission strength. We used Multiclamp recording system (700B amplifier, DigiData 1440A digitizer and pClamp software) (Molecular Devices, CA) to record mEPSCs.
-
1.
On DIV13, add purified recombinant NGPF2 (Zhou et al., 2021) or PBS as vehicle to the culture medium at a final concentration of 1.5 ng/mL.
-
2.
On DIV14, 24–36 h post NGPF2 treatment, transfer a coverslip using fine forceps to recording chamber, that contains ECF supplemented with TTX (1 μM) and picrotoxin (100 μM). TTX is a Na+ channel blocker that inhibits the elicitation of action potentials. Picrotoxin is a noncompetitive antagonist of GABAA receptors thus inhibits GABAergic currents in recordings.
-
3.
Recording pipettes were prepared by pulling glass pipettes (Borosilicate glass, Sutter Instrument) on a pipette puller (Sutter Instrument P-1000).
Note: Electrophysiological recording is performed at room temperature (22°C–25°C).
-
4.
Search and locate the target neurons to be recorded under the microscope.
-
5.
Fill the glass recording pipette with intracellular solution.
-
6.
Immerse the tip of the recording pipette in the ECF in the recording chamber, read the tip resistance. Only use pipettes with a tip resistance of around 4–5 MΩ.
Note: Replace ECF in the recording chamber after each recording.
-
7.
Apply slight positive pressure to the pipette through tubing and guide the tip of the pipette to approach the target neuron.
-
8.
By using the micromanipulator, slowly press the pipette tip onto the cell body until an obvious dimple is formed (Methods video S1: Microscope view of whole-cell patch process, and Figure 1B). Release positive pressure and apply a subtle negative pressure to form the seal. In pClamp software, switch from Bath to Patch, and apply −65 mV voltage to facilitate the formation of Giga-ohm seal (Methods video S2: Clampex interface view showing whole-cell patch process). Once formed, apply a slight suction to rupture the cell membrane patch, thus forming whole-cell patch configuration.
-
9.
In voltage-clamp mode, clamp the neurons at −65mV throughout the recordings.
-
10.
Apply a −3 mV step pulse every 20 s to monitor series resistance. Only recordings with a pipette-membrane seal greater than one giga-ohm and a drift of series resistance less than 15% are included for statistical analysis.
-
11.
Analyze mEPSCs using MiniAnalysis Program (Synaptosoft).
Figure 1.

Whole-cell patch recording of mEPSCs in cultured neurons at DIV13-14
(A) Equipment for electrophysiological recording. 1: Microscope. 2: Recording chamber. 3: Micromanipulator. 4: Amplifier. 5: Digitizer. 6: Monitor.
(B) Example phase-contrast photo showing the patching of a cultured pyramidal neuron. Scale bar: 10 μm.
(C) Sample trace of recorded mEPSC events held at −65 mV in voltage clamp configuration. Scale bars: 20 pA/2.5 s (upper) and 20 pA/0.5 s (lower).
1–10 s: Patching pipette approaching neuron. 11–13 s: Formation of giga-ohm seal.
6–10 s: Formation of giga-ohm seal. 33–59 s: Formation of whole-cell patching clamp.
Neurotrophic factor treatment for imaging
-
12.
On DIV14-15, add PBS as vehicle and recombinant NGPF2 protein (Zhou et al., 2021) to culture medium at the final concentration of 1.5 ng/mL.
-
13.
On DIV15-16, at 24–36 h post NGPF2 treatment, remove culture medium, quickly wash coverslips with pre-cold PBS and fix cortical neurons with ice-cold 4% paraformaldehyde and 4% sucrose in PBS for 30 min at 4°C.
-
14.
Remove paraformaldehyde, wash coverslips in PBS for 5 min on a platform shaker (60 rpm).
-
15.
Repeat washing 3 times. Fixed coverslips from this step can be used for immunostaining of surface AMPA receptors (step 16), synaptic AMPA receptors (step 23), and dendrites (step 32).
Immunostaining of surface AMPA receptors
GluA2 is an essential subunit of AMPA receptors. To immunostaining AMPA receptors, use GluA2 as target protein.
-
16.
Block the fixed cells from step 15 with 5% fetal bovine serum (FBS) diluted in PBS for 1 h at room temperature.
-
17.
Wash with PBS for 5 min, 3 times.
-
18.
Incubate the coverslips with an antibody against the extracellular N-terminal domain of GluA2 (GluA2-NTD) (Millipore, Cat#MAB397) at 1:500 dilution in PBS for 12–16 h at 4°C, on a platform shaker (20 rpm).
-
19.
Remove primary antibody solution, and wash the cells with PBS for 5 min, 3 times.
-
20.
Add a secondary antibody (Alexa Fluor 488 donkey anti-mouse IgG) at 1:1000 dilution in PBS, incubate at room temperature for 1 h, on a platform shaker (20 rpm).
-
21.
Remove secondary antibody solution, wash with PBS for 5 min, 5 times.
-
22.
Pipette 8 μL ProLong™ Gold Antifade Mountant (Thermo Fisher) on the surface of a glass slide, and mount coverslips on slides with neurons facing down.
Immunostaining of synaptic AMPA receptors
-
23.
Permeabilize the fixed cells from step 15 with 0.25% Triton X-100 in PBS for 10 min at room temperature, on a platform shaker (20 rpm).
-
24.
Discard the solution and wash with PBS for 5 min, 3 times.
-
25.
Add 500 μL 5% FBS in PBS for 1 h at room temperature.
-
26.
Remove the blocking solution and wash with PBS for 5 min, 3 times.
-
27.
Incubate the coverslips with primary antibodies against GluA2-NTD (Millipore, Cat#MAB397, mouse) and Synapsin 1 (Cell Signaling, Cat#5297, rabbit), both at 1:1000 dilution in PBS for 12–16 h at 4°C, on a platform shaker (20 rpm).
-
28.
Remove primary antibody solution, wash with PBS for 5 min, 3 times.
-
29.
Add secondary antibodies (Alexa Fluor 488 donkey anti-mouse IgG and Alexa Fluor 555 goat anti–rabbit IgG) at 1:1000 dilution in PBS, incubate at room temperature for 1 h, on a platform shaker (20 rpm).
-
30.
Remove secondary antibody solution, wash with PBS for 5 min, 5 times.
-
31.
Pipette 8 μL ProLong™ Gold Antifade Mountant (Thermo Fisher) on the surface of a glass slide, and mount coverslips on slides with neurons facing down.
Fluorescent staining of dendritic spines
-
32.
After Step 26, incubate the coverslips with primary antibodies against microtubule-associated protein 2 (MAP2) (Millipore, Cat#MAB3418, rabbit) at 1:1000 dilution, for 1 h at room temperature, on a platform shaker (20 rpm).
-
33.
Remove primary antibody solution, and wash with PBS for 5 min, 3 times.
-
34.
Add secondary antibody (Alexa Fluor 488 goat anti-rabbit IgG) at 1:1000 dilution and rhodamine phalloidin (Thermo Fisher, Cat#R415) at 1:1500 dilution, incubate at room temperature for 1 h, on a platform shaker (20 rpm).
-
35.
Remove secondary antibody solution, and wash with PBS for 5 min, 5 times.
-
36.
Pipette 8 μL ProLong™ Gold Antifade Mountant (Thermo Fisher) on the surface of a glass slide, and mount coverslips on slides with neurons facing down.
Expected outcomes
For preparation of primary cortical neuronal cultures, it is expected that: (1) at DIV13-14, the day of mEPSC recordings, most neurons appear healthy with clear contours when observed under microscope in phase contrast configuration (Figure 1B). (2) At DIV15-16, ample amount of scope fields contain single neurons, so that the microstructures can be clearly imaged (Figures 2A and3A). Neurons that cluster together are not suitable for imaging studies.
Figure 2.

Immunocytochemistry of surface and synaptic expression of GluA2 AMPA receptor subunit
(A) Surface GluA2 immunostained under non-permeabilizated condition, using an antibody against extracellular domain of GluA2. Scale bar: 10 μm.
(B) Confocal images showing colocalization of GluA2 (green) and Synapsin (red). Arrow: colocalized GluA2 and Synapsin puncta. Arrow head: left, Synapsin puncta not colocalized with GluA2; right, GluA2 puncta not colocalized with Synapsin. Scale bar: 5 μm.
Figure 3.

Fluorescent staining of dendritic spines for morphological analysis
(A) Merged image showing dendritic marker MAP2 (green) and spines labeled by rhodamine phalloidin (red). Scale bar: 20 μm.
(B) Zoom-in image showing various types of dendritic spines: (1) mushroom-shaped spine, (2) stubby spine and (3) thin spine. Scale bar: 5 μm.
(C) Individual spines with distinct head-to-neck ratio. Scale bar: 1 μm.
For mEPSC recordings, most neurons should form stable Giga-ohm seal. Whole-cell voltage patch clamp can last at least 10 min. The drift of series resistance should be less than 15% during the recordings. The mean values of the frequency and amplitude of mEPSC events are expected to be around 2 Hz and 20 pA respectively (Zhou et al., 2021). Neurons that were treated with NGPF2 for over 24 h are expected to show larger mEPSC amplitude than neurons of control group treated with PBS (Zhou et al., 2021) .
For immunocytochemistry of synaptic AMPA receptor subunit GluA2, Synapsin puncta should appear condensed, located on the tip of the dendritic spine or along the dendritic shafts (Figure 2B). Since AMPA receptors distribute in both synaptic and extra-synaptic pools, the fluorescent signals of GluA2 puncta are expected to partially co-localize with Synapsin puncta (Zhou et al., 2011, 2018, 2021). Neurons that were treated with NGPF2 for over 24 h are expected to show higher colocalization ratio of GluA2 and Synapsin puncta (Zhou et al., 2011, 2018, 2021), compared to that of control group treated with PBS.
For dendritic spine imaging and analysis, DIV16 neurons are expected to show ample amount of protrusions along the dendritic shafts. These protrusions consist of different types of morphological characteristics that correlate with different stages of spine development (Zhou et al., 2011, 2021). Neurons that were treated with NGPF2 for over 24 h are expected to show higher proportion of spines with mature morphology (Figures 3B and 3C), i.e., mushroom-shaped spines, compared to that of control group treated with PBS.
Quantification and statistical analysis
For mEPSC analysis, load abf. files in MiniAnalysis program 6.3 (Synaptosoft Inc.). Set the event detection parameters in the Analysis tab applicable to mEPSCs. Manually verify the correct detection and identification of mEPSC events. Save the detected events in evt. format for analysis of inter-event interval and amplitude.
To measure surface GluA2 expression, adjust the brightness and contrast of original images using Aim Image Browser (Zeiss) software with the same settings and configurations for all sample images within experiment. Randomly select and extract subregions covering 100–150 mm linear dendrites in total per neuron. Export and save images in Tiff file format. Open the Tiff file in the ImageJ (Fiji) software, set appropriate configurations for all sample images within experiment, and read total fluorescence intensity values for statistical analysis. To measure synaptic GluA2 expression, randomly select and extract subregions covering 100–150 mm linear dendrites in total per neuron. Export and save in Tiff file format. In ImageJ (Fiji) software, set the configurations so that the Synapsin puncta with an area greater than 0.1 mm2 can be automatically identified and counted. Manually verify the identification of Synapsin puncta. Then, measure the GluA2 fluorescence signal located within these puncta. Read the values for statistical analysis (e.g., using Sigmaplot 11).
For dendritic spine morphological analysis, again, randomly select and extract subregions covering 100–150 mm linear dendrites in total per neuron. Adjust the brightness and contrast of original images using Aim Image Browser (Zeiss) software with the same settings and configurations for all sample images and save in Tiff file format. In ImageJ (Fiji) software, define the phalloidin-labeled protrusion of 0.3–4 μm in length as spines. The spine length was measured from the base of the spine (defined by MAP2 staining) to the tip of the spine head. For spine classification, use the following criteria: mushroom spines: head-to-neck ratio greater than 1.1, head diameter greater than 0.35 mm, and width-length ratio less than 1.0; stubby spines: head-to-neck ratio less than 1.1 and width-length ratio greater than 1.0; thin spines: head-to-neck ratio was less than 1.1, and either (1) length-to-head ratio was greater than 2.5 or (2) head size was less than 0.35 mm. Visual verification by an experimenter blind to the group assignment is necessary to ensure correct classification.
Limitations
The major limitations of this protocol that uses dissociated primary neuronal cultures are predominantly associated with the in vitro culture conditions. Interpretation of the results may not recapitulate all the characteristics of in vivo synaptic growth induced by neurotrophic factors. Given the inherent growth trajectory of neurons in culture conditions, the time window to examine synaptic growth is limited, thus the timing of experimentation is critical. For example, here we recorded mEPSCs on DIV13-14 to study the effects of neurotrophic factor on synaptic transmission as a functional readout. Our observation is that postnatal cortical neurons cultured using Neurobasal A/B27 medium system do not show electrophysiological responses at earlier days, but have large amplitude and frequency of mEPSC events even at basal level after DIV15-16. Thus the recordings may not be suitable to reveal the increased synaptic transmission caused by the exposure to exogenous neurotrophic factors. On the other hand, we examined dendritic spines at DIV15-16 as spines did not appear until DIV15 in this culture system. Thus, we were unable to examine synaptic growth at morphological and functional levels at the same time points. Here we used rhodamine phalloidin to stain filamentous actin that is enriched in the dendritic spine to indicate spine morphology. Alternatively, neuronal cultures can be prepared from transgenic mice expressing green fluorescent protein (GFP), thus intracellular GFP can be used to indicate spine morphology. Moreover, obtaining high-quality whole-cell patch recording is highly skill-dependent. In data analysis, the automatic detection and identification of mEPSC events by the software requires visual verification. An experienced experimenter blind to the group assignment is necessary for all analysis. Similarly, the identification of dendritic spines and synaptic protein puncta also requires visual verification to exclude fluorescence artifacts and contaminants.
Troubleshooting
Problem 1
(Step: preparation and culture of primary mouse cortical neurons, step 3)
Heterogeneous cell growth in neuronal culture.
Potential solution
First, when preparing primary cortical neuronal cultures, be sure to thoroughly remove meninges as much as possible. Cells in residual meanings proliferate rapidly. Second, on DIV4, add proliferation inhibitor AraC (1 μg/mL) to the medium and incubate for 12 h. This effectively inhibits the growth of glial cells, endothelial cells and other proliferative cells. However, exposure to AraC longer than 12 h also affects the survival of primary neurons, especially for mature neurons. Timely replacement with fresh culture medium after AraC treatment is necessary. Third, exclude the use of serum or other supplements with undefined ingredients.
Problem 2
(Step: fluorescent staining of dendritic spines, steps 32–36)
Neurons do not show clear dendritic protrusions when imaged using confocal microscopy.
Potential solution
Since the exact experimental conditions may vary in different laboratories, the exact DIV of spine growth may vary. Our suggestion is to carefully observe dendritic protrusions in phase contrast mode using higher magnification (e.g., 40×) objectives, before adding neurotrophic factors to the culture medium. Ensure that spines are starting to form, and then start the treatment procedure. Second, our experience is that in low density neuronal cultures, spine growth may be sparse. Therefore, increased cell density may facilitate spine formation. Third, addition of 10 ng/mL basic fibroblast growth factor (bFGF) in culture maintenance medium after DIV4 improves neuronal culture growth.
Problem 3
(Step: electrophysiological recording of mEPSCs, steps 8–10)
Encountering poor giga-ohm seal, frequent reseal, and frequent loss of patching when performing whole-cell patch clamp recordings.
Potential solution
Whole-cell patch recording is technically demanding and requires good training and practice. For difficulty in forming a giga-ohm seal, first examine the possible clogging in patch pipette due to dirt or debris. The intracellular solution can be filtered through 0.22 μm filter tip before use. Always apply slight positive pressure once the pipette tip is immersed in the ECF, as the outflowing solution keeps the tip clean. When approaching the cell body, the extracellular solution also cleans the surface of cell membrane thus allows the formation of a clean giga-ohm seal. During the course of giga-ohm seal formation, gradually change the holding potential from 0 mV to −70 mV to facilitate sealing (Xia et al., 2016; Yang et al., 2017; Zhang et al., 2020). Check the size and shape of the tip of the patch pipette, as larger tips (e.g., tip resistance at around 1–3 MΩ) are unlikely to form tight seals.
The problem of frequent reseal is most likely caused by the use of a small sized tip (e.g., tip resistance above 5 MΩ). Ruptured membrane may easily clog the small sized tip thus resealing. If giga-ohm seal and whole cell configuration are achieved, but the cell is quickly lost, the health of cultured neurons should be examined. Under the microscope, the contour of cell body should be clear and debris from dead cells should be minimal (Liu et al., 2016; Xia et al., 2016; Zhou et al., 2011). When forming giga-ohm seal, the release of positive pressure should be followed by rapid recovery of deformed cell membrane and a quick rise in serial resistance. All these are indicators of good cell health. Next, examine the osmolarity of ECF and intracellular solution. Slightly higher osmolality in the ECF (e.g., 280 mOsm/kg in ECF and 300 mOsm/kg in intracellular solution) facilitates the maintenance of good patching. Lastly, always use freshly prepared and filtered solutions.
Problem 4
(Step: electrophysiological recording of mEPSCs, steps 8–10)
Low-quality electrophysiological traces for mEPSC analysis.
Potential solution
In addition to culture “healthy” neurons, experimenters should ensure the correct concentrations and expiry/valid status of TTX and picrotoxin used in recordings, so that they can effectively inhibit electrical currents mediated by sodium channels and GABAA receptors. Proper grounding and shielding of the recording equipment are also very important to eliminate electrical noise. On-site trouble-shooting by highly-experienced electrophysiologist is necessary.
Problem 5
(Step: immunostaining of surface AMPA receptors, steps 23–31)
Fail to detect surface expression of GluA2 AMPA receptor subunit by immunostaining.
Potential solution
The abundance of surface AMPA receptors is generally correlated with the development of neurons, i.e., higher in mature neurons. Similar to problem 2, our suggestion is to use mature cultured neurons. A convenient way to judge whether the cultured neurons are ready for this experiment is to observe dendritic protrusions in phase contrast mode using higher magnification (e.g., 40×) objectives. Visible dendritic protrusions indicate that the neurons are ready for study of surface AMPA receptors.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Zikai Zhou (zhouzikai@simm.ac.cn).
Materials availability
This study did not generate unique materials or reagents. Further information and requests for materials should be directed to and will be fulfilled by the lead contact, Zikai Zhou, upon request.
Acknowledgments
The authors would like to thank all members of Zhou lab and Jia lab. This work was supported by grants from National Natural Science Foundation of China (81971022 Z.Z.), Canadian Institutes of Health Research (PJT155959, PJT168922, Z.P.J.), Canadian Natural Science and Engineering Research Council (RGPIN341498, RGPIN06295, Z.P.J.), China Postdoctoral Science Foundation (No. 2018M642036, G.H.).
Author contributions
Conceptualization, Z.Z. and Z.J.; Investigation, G.H., X.Y.W., and Z.Z.; Writing, Z.Z., G.H., and Z.J. All authors read and approved the final version of the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.101112.
Contributor Information
Zhengping Jia, Email: zhengping.jia@sickkids.ca.
Zikai Zhou, Email: zhouzikai@simm.ac.cn.
Data and code availability
This study did not generate any custom code, software, or algorithms.
References
- Liu A., Zhou Z., Dang R., Zhu Y., Qi J., He G., Leung C., Pak D., Jia Z., Xie W. Neuroligin 1 regulates spines and synaptic plasticity via LIMK1/cofilin-mediated actin reorganization. J. Cell Biol. 2016;212:449–463. doi: 10.1083/jcb.201509023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S., Zhou Z., Leung C., Zhu Y., Pan X., Qi J., Morena M., Hill M.N., Xie W., Jia Z. p21-activated kinase 1 restricts tonic endocannabinoid signaling in the hippocampus. eLife. 2016;5:e14653. doi: 10.7554/eLife.14653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., He G., Zhang X., Chen L., Kong Y., Xie W., Jia Z., Liu W.-T., Zhou Z. Transient inhibition of LIMKs significantly attenuated central sensitization and delayed the development of chronic pain. Neuropharmacology. 2017;125:284–294. doi: 10.1016/j.neuropharm.2017.06.031. [DOI] [PubMed] [Google Scholar]
- Zhang X., Kong Y., He G., Zhou Z. Neonatal exposure to ketamine disrupts developmental synapse unsilencing and predisposes adult mice for stressor-evoked anxiety. Neuropharmacology. 2020;180:108300. doi: 10.1016/j.neuropharm.2020.108300. [DOI] [PubMed] [Google Scholar]
- Zhou Z., He G., Zhang X., Lv X., Zhang X., Liu A., Xia S., Xie H., Dang R., Han L., et al. NGPF2 triggers synaptic scaling up through ALK-LIMK-cofilin-mediated mechanisms. Cell Rep. 2021;36:109515. doi: 10.1016/j.celrep.2021.109515. [DOI] [PubMed] [Google Scholar]
- Zhou Z., Hu J., Passafaro M., Xie W., Jia Z. GluA2 (GluR2) regulates metabotropic glutamate receptor-dependent long-term depression through n-cadherin-dependent and cofilin-mediated actin reorganization. J. Neurosci. 2011;31:819–833. doi: 10.1523/JNEUROSCI.3869-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z., Liu A., Xia S., Leung C., Qi J., Meng Y., Xie W., Park P., Collingridge G.L., Jia Z. The C-terminal tails of endogenous GluA1 and GluA2 differentially contribute to hippocampal synaptic plasticity and learning. Nat. Neurosci. 2018;21:50–62. doi: 10.1038/s41593-017-0030-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1–10 s: Patching pipette approaching neuron. 11–13 s: Formation of giga-ohm seal.
6–10 s: Formation of giga-ohm seal. 33–59 s: Formation of whole-cell patching clamp.
Data Availability Statement
This study did not generate any custom code, software, or algorithms.