

Abstract
Efforts to enhance wellness and ameliorate disease via nutritional, chronobiological, and pharmacological interventions have markedly intensified interest in ketone body metabolism. The two ketone body redox partners, acetoacetate (AcAc) and D-β-hydroxybutyrate (βOHB) serve distinct metabolic and signaling roles in biological systems. A highly efficient, specific, and reliable approach to simultaneously quantify AcAc and D-βOHB in biological specimens is lacking, due to challenges of separating the structural isomers and enantiomers of βOHB, and to the chemical instability of AcAc. Here we present a single UPLC-MS/MS method that simultaneously quantifies both AcAc and βOHB using independent stable isotope internal standards for both ketones. This method incorporates one sample preparation step requiring only seven minutes of analysis per sample. The output is linear over three orders of magnitude, shows very low limits of detection and quantification, is highly specific, and shows favorable recovery yields from mammalian serum and tissue samples. Tandem MS discriminates D-βOHB from structural isomers 2- or 4-hydroxybutyrate as well as 3-hydroxyisobutyrate (3-HIB). Finally, a simple derivatization distinguishes D- and L-enantiomers of βOHB, 3-HIB, and 2-OHB, using the same rapid chromatographic platform. Together, this simple, efficient, reproducible, scalable, and all-encompassing method will support basic and clinical research laboratories interrogating ketone metabolism and redox biochemistry.
Keywords: Quantification of ketone bodies, D-beta-hydroxybutyrate structural and stereo-isomers, Acetoacetate instability, Acetoacetate internal standard, UPLC-MS/MS
Graphical Abstract
1. Introduction
Ketone bodies are evolutionarily conserved and endogenously synthetized metabolites that become significant contributors to energy metabolism in mammals during multiple physiological periods such as long-term starvation, short-term fasting, the neonatal period, pregnancy, or adherence to low-carbohydrate, high-fat diets [1–4]. Attention to ketone body metabolism has intensified recently, as intermittent fasting, time restricted eating, adherence to low carbohydrate ketogenic diets, or application of ingested exogenous ketones are all under investigation in many pre-clinical and clinical studies, with the objective of enhancing wellness and performance, improving health, combatting disease, and offsetting the effects of aging [5–10].
At a biochemical level ketone bodies play imperative roles in many mammalian species, as their metabolism is interwoven with a myriad of crucial biochemical pathways such as fatty acid β-oxidation, the tricarboxylic acid cycle (TCA), gluconeogenesis, and de novo lipogenesis. The generation of ketone bodies is driven through an inter-organ shuttle that initiates with adipose tissue lipolysis of triacylglycerol to free fatty acids, which are transported to the liver for mitochondrial β-oxidation [11]. In hepatic mitochondria, fatty acid β-oxidation-derived acetyl-CoA is the prime ketone body substrate through the action of the fate committing enzyme 3-hydroxymethylglutyryl-CoA synthase 2 (HMGCS2) [12]. In mammals, liver hepatocytes and gut enterocytes robustly express HMGCS2, supporting the ultimate production of acetoacetate (AcAc) and β-hydroxybutyrate (βOHB), the most abundant ketone bodies. AcAc is the oxidized nicotinamide adenine dinucleotide (NAD+)/NADH-dependent redox partner and precursor of D-βOHB, which is generated through a reaction catalyzed by β-hydroxybutyrate dehydrogenase 1 (BDH1) [13, 14]. Hepatically generated ketones are exported through circulating blood to extrahepatic tissues such as heart, skeletal muscle, and brain, where D-βOHB is converted by BDH1 back to AcAc, and mitochondrial succinyl-CoA:3-oxoacid-CoA transferase (SCOT) ushers ketone bodies toward acetyl-CoA to support production of high energy phosphates via TCA cycle oxidation [15]. When applying the equilibrium constant for the reaction catalyzed by BDH1, the concentration ratio of AcAc/D-βOHB within a tissue provides an index of that tissue’s mitochondrial NAD+/NADH ratio [16]. AcAc can also spontaneously decarboxylate to acetone, which can be exploited to estimate the level of ketone bodies in biological fluids and exhaled breath [17–20].
Ketone metabolism is classically engaged during states of carbohydrate restriction. Circulating levels of ketone bodies in normally fed humans oscillate during the day between 50–200 μM, but exercise or a 24h fast can increase ketone levels up to 1 mM, and in pathological states like diabetic ketoacidosis, in which circulating concentrations reach up to 20 mM, β-oxidation and ketogenesis reduce blood pH [3, 15, 21]. Thus, approaches to reliably and efficiently quantify ketone bodies in the circulation and in tissues are of great importance. Ketone bodies are typically measured using narrow dynamic range-spectrophotometric methods, detecting either a derivatized product of acetone, or through the addition of exogenous BDH1, tracking the NADH produced through amplifying redox cycles [18, 22]. More sensitive, selective, dynamic, and rapid methods using gas chromatography (GC)-MS, or liquid chromatography (LC) separations with tandem mass spectrometry (MS) detection have been employed to interrogate a variety of biological specimens [23–29]. While these MS-based methods are sensitive, selective and relatively fast, they measure D- and L- enantiomers of βOHB indiscriminately, requiring indirect fractionation using a cumbersome application of exogenous BDH1 that leverages this enzyme’s selectivity for D-βOHB [30]. While mechanisms supporting L-βOHB biological generation are incompletely understood, and its metabolic and signaling roles are poorly defined, L-βOHB has been detected in biological specimens through methods requiring time-consuming derivatization and chromatographic separation approaches [31–33]. In addition, the chemical instability of decarboxylation-prone AcAc in extracted biospecimens poses an additional analytic challenge. Current LC-MS/MS techniques approaches estimate AcAc, or its derivatized products, based upon external calibration curves, or a surrogate internal standard, rather than an authentic isotopically-labeled AcAc as internal standard [34, 35]. Taken together, a single method that rapidly, reproducibly, and accurately quantifies D-βOHB, discriminating its structural isomers and stereoisomers, and AcAc, using stable isotope labeled internal standards for both D-βOHB and AcAc, in biological material is lacking and thus poses an unmet need.
Here, we present an efficient, selective, sensitive, and scalable method for the quantitative estimation of ketone bodies in serum and tissue extracts, based upon the use of authentic stable isotope-labeled internal standards for both βOHB and AcAc. Quantifications of ketones in mouse serum and liver are achieved using authentic stable isotope labeled internal standards. Synthetized [U-13C4]AcAc is used as an internal standard for endogenous AcAc that is independently quantified by LC-MS/MS following NaBD4 reduction to [3-D1, U-13C4]βOHB. Deuterated [3,4,4,4-D4]βOHB is used as an internal standard for endogenous βOHB, collectively allowing quantification of ketone bodies using discrete mass shift transitions. This rapid UPLC separation method also separates D- and L-βOHB enantiomers after a simple derivatization. Finally, tandem MS readily allows this method to discriminate both βOHB enantiomers from their structural isomers, 3-hydroxyisobutyrate (3-HIB), 2-hydroxybutyrate (2-OHB) and 4-hydroxybutyrate (4-OHB). Thus, this efficient, specific, and reproducible method for quantitative estimation of all ketone bodies is expected to be of widespread application to basic and clinical laboratories that routinely apply LC-MS/MS techniques to measure ketone bodies.
2. Methods
2.1. Chemicals and Materials
LC-MS grade methanol (MeOH), acetonitrile (ACN) water, and (S)-2-Hydroxybutyric acid (S-2-OHB) were purchased from Thermo Fisher Scientific. Acetic acid (AA), (S)-(+)-1-(2-Pyrrolidinylmethyl)-pyrrolidine (PMP), Triphenylphosphine (TPP), sodium borodeuteride (NaBD4), ethyl-acetoacetate, sodium D-, L- or DL- 3-hydroxybutyrate, DL-2-hydroxybutyrate (2-OHB), DL-3-hydroxyisobutyrate (3-HIB), sodium (S)-hydroxyisobutyrate (S-3-HIB), and Dowex powder (50WX8 hydrogen form) were obtained from Sigma Aldrich. Sodium DL-3-hydroxybutyrate ([3,4,4,4-D4]βOHB, 98%), and [U-13C4]AcAc (synthetized from ethyl-[U-13C4]AcAc) purchased from Cambridge isotope laboratories were used as internal standards (I.S.). 2,2′- dipyridyl disulfide (DPDS) was obtained from Merck, and 4-hydroxybutyrate methyl ester was purchased from Santa Cruz Biotechnology.
2.2. Synthesis of AcAc and 4-hydroxybutyrate (4-OHB)
AcAc, [U-13C4]AcAc, or 4-OHB were synthetized by a base-catalyzed hydrolysis of ethyl-acetoacetate (ethyl-AcAc), ethyl-[U-13C4]AcAc, or 4-hydroxybutyrate methyl ester (4-OHB-Me), respectively, as previously described [36]. Briefly, 1 mL of ethyl-AcAc, ethyl-[U-13C4]AcAc, or 4-OHB-Me was mixed with 8 mL of 1M NaOH, while stirring at 60 C. After 30 min, the reaction was neutralized with 50% HCl to pH 7–8. AcAc, [U-13C4]AcAc, and 4-OHB aliquots were stored at −80 C unless otherwise stated.
2.3. AcAc stability
Three independently synthetized AcAc samples were diluted in water or in extraction solution [ACN:MeOH:water (2:2:1, v/v/v)] at 100 μM, aliquoted and left either at room temperature (+21 C), +4 C, −20 C, −80 C or in liquid nitrogen tank (LN2, estimated temperature is −200 C) for indicated duration of time. In order to control the stability of AcAc each sample was spiked with 20 μM βOHB. To study the stability of AcAc in serum, wild type control mice were starved for 24h, bled, and serum was obtained using the approach described in Biospecimen preparation, below. Mouse sera were pooled to obtain enough of representative samples. Three to six independently obtained serum samples were spiked with 20 μM [3,4,4,4-D4]βOHB (I.S.), aliquoted and left at either room temperature (+21 C), +37 C, +4 C, −20 C, −80 C or in liquid nitrogen (LN2) for indicated durations. Prior to analysis, serum samples were extracted by the addition of 4 volumes of cold ACN:MeOH (1:1, v/v), vortex, and centrifuged at 4 C, 15,000 × g for 10 min. Extracted serum or AcAc sample in water or extraction solution were analyzed using optimized UPLC-MS/MS system where signals of AcAc, βOHB, and [3,4,4,4-D4]βOHB were monitored using PRM scan modes. For all stability studies, one aliquot of each sample was analyzed immediately, whereas the rest were stored for indicated times in various conditions. Prior to our experiments, the effects of storage duration and temperature on synthetized I.S. [U-13C4]AcAc were unknown, and in fact were a measured outcome of our experiments. Furthermore, the formal quantification of yield of I.S. [U-13C4]AcAc from ethyl-[U-13C4]AcAc requires NaBD4-driven reduction of [U-13C4]AcAc to [3-D1,U-13C4]βOHB (measured against I.S. [3,4,4,4-D4]βOHB, see Section 2.5, below). Therefore, to control for these two variables, AcAc stability was estimated by comparing AcAc/βOHB signal ratios among conditions, based upon the previously shown long term stability of βOHB against challenging matrices [38].
2.4. Biospecimen preparation
Mice were maintained on a standard low-fat chow diet (2016 Teklad global 16% protein rodent diet) and given autoclaved water ad libitum. Lights were off between 2000 and 0600 in a room maintained at 21 C. Serum was obtained from submandibular bleed of 9- to 16-week-old male or female wild-type (WT) mice from the C57BL/6NJ background. To create HMGCS2 knockdown mice, antisense oligonucleotide (ASO) treatment was initiated in six week-old mice by injecting (25 mg/kg, intraperitoneal) of Hmgcs2-targeted ASO (Ionis 191229) or scrambled sequence control ASOs (Ionis 141923; Control ASO) biweekly for 4 weeks, continued through the duration of the experiment [39]. To stimulate ketogenesis for fasting studies, mice were fasted for indicated durations of time, where food was removed from the animals, but water was available ad libitum. To synchronize the onset of the fast, mice were injected intra-peritoneally with 2 g/kg of 20% glucose in saline as food was removed. Blood from mice was allowed to sit on ice for 30 min and was centrifuged at 4 C, 8,000 × g for 10 min. Separated serum was aliquoted and stored at −80 C. LN2-chilled Wollenberger tongs were used to harvest liver tissues via freeze-clamp from animals sacrificed by cervical dislocation. All animal experiments were performed through protocols formally approved by the Institutional Animal Care and Use Committee at the University of Minnesota.
2.5. Quantitation of [U-13C4]AcAc internal standard (I.S.)
Estimation of the concentration of synthetized [U-13C4]AcAc is adapted from reference [37] and is based on a stable isotope labeled internal standard method. To 25 μL of synthetized [U-13C4]AcAc sample (diluted 1000–4000 times to fit the calibration curve), 10 μL of [3,4,4,4-D4]βOHB as I.S. at known concentration (recommended final concentration is 50 μM) was added. To externally quantify the [U-13C4]AcAc I.S. used for spiking into biological specimens, [U-13C4]AcAc I.S. was reduced to [3-D1,U-13C4]βOHB by using 30 μL of freshly prepared 1.8 M NaBD4 in 0.1M NaOH. Samples were left for 5 minutes at room temperature, and 55 μL of ACN was added. In parallel, cation exchange dowex powder was loaded into an empty column scaffold installed in the solid phase extraction manifold. Loaded columns were cleaned with deionized water. After sample centrifugation at 4 C, 15,000 × g, samples were loaded on prepared cation exchange column in order to desalt the sample from an excess of Na+, eluted using 3 mL of LCMS grade water, and dried via SpeedVac. These additional desalting steps, using dowex column, drying, and extraction, are necessary to minimize ESI ion-suppression via Na+ after NaBD4 reduction. Absence of the Dowex desalting step after the reduction with 1.8M NaBD4 overwhelms ESI, abrogating any signal.
Dried samples were resuspended in 60 μL of 98% water/2% MeOH with 0.0125% AA, vortex, centrifuged at 15,000 × g for 10 min, and the supernatant injected in the LC-MS/MS system (see Instrumentation, below). Concentration of [3-D1, U-13C4]βOHB [parallel reaction monitoring (PRM) 108.0598→61.0203] obtained after reduction of [U-13C4]AcAc by NaBD4 was calculated based on the area of PRM signals and known concentration of [3,4,4,4-D4]βOHB (PRM 107.0646→59.0133). Similarly, quantification of unlabeled synthetized AcAc can be performed based on the area of [3-D1]βOHB signal obtained after reduction of AcAc by NaBD4 (PRM 104.0464→59.0133) and calculated based on known concentration of I.S. [3,4,4,4-D4]βOHB (PRM 107.0646→59.0133, see Fig.1A). This approach allowed the use of two independent I.S., each spiked into biospecimens: [U-13C4]AcAc (externally quantified) and [3,4,4,4-D4]βOHB, together used to estimate endogenous AcAc and βOHB abundances, respectively, as described immediately below.
Figure 1. UPLC-MS/MS identification of AcAc, βOHB, and βOHB structural isomers in serum extracts.

(A) Summary of compound names, precursor → fragment transition in Parallel Reaction Monitoring (PRM) negative mode, retention time (RT) in minutes, compound structures, and predicted fragments to quantify AcAc and βOHB ketone bodies (green boxes, top left and right), synthetized AcAc standards (gray box, lower left) after reduction with NaBD4, and structural isomers of βOHB (orange box, lower right) using Cortecs UPLC T3 column. Stable isotope internal standard (I.S.) used for the quantification of AcAc was [U-13C4]AcAc, whose concentration was externally quantified prior to its spiking into biospecimens by reduction to [3-D1,U-13C4]βOHB, and comparison against [3,4,4,4-D4]βOHB. Stable isotope I.S. for quantification of βOHB was [3,4,4,4-D4]βOHB, which was spiked into biospecimens. Lines indicate where the molecules are fragmented upon higher-energy collisional dissociation [HCD using 30 N(CE)], while dots indicate 13C atomic positions. (B) Ionizations of βOHB, 2-OHB, 4-OHB, and 3-HIB are more efficient in negative mode, as determined by the ratio of the area of Full MS scan signal of 20 μM standards detected in negative versus positive mode under same chromatographic conditions (n=3/group). (C) Utilization of MS/MS mode to quantify relative contributions of structural isomers βOHB in mouse serum. In extracted serum from mice fasted for 24h, m/z 103.0401 is comprised of 85% βOHB in control mice, but only 35% in ketogenesis insufficient mice (n>10/group). Data are expressed as the mean standard error of the mean (SEM).
2.6. Quantification of ketone bodies in serum
To fit within the method’s dynamic range, serum samples were diluted 5x with water before extraction. Diluted serum samples (5 μL) were extracted by the addition of 20 μL of cold ACN:MeOH (1:1, v/v) containing known concentrations of internal standards [3,4,4,4-D4]βOHB and [U-13C4]AcAc (recommended final concentration of each I.S. is 50 μM). Serum samples were vortex, and centrifuged at 4 C, 15,000 × g for 10 min. Supernatant was analyzed using UPLC-MS/MS quantification analysis setup described below. For a high throughput extraction analysis, 60 μL of freshly prepared extraction solution of cold ACN:MeOH (1:1, v/v) with appropriate amount of [3,4,4,4-D4]βOHB and [U-13C4]AcAc (I.S. to yield 50 μM concentrations of each) was added to 96 well protein precipitation plate (Thermo Fisher Scientific, 60304–201), and to each well 15 μL of serum was added. Covered precipitation plates were placed onto 96 well microplate for autosampler, shaken for 3 min at 4 C, and centrifuged at 4 C, 500 × g for 3 min. Microplate for autosampler was directly used for the UPLC-MS/MS quantification analysis. For simple and high throughput analysis, the concentrations of AcAc (PRM, 101.0244→57.0340) or βOHB (PRM, 103.0401→59.0133) were calculated using stable isotope labeled internal standard method, which were based on the areas of PRM signals and known concentrations of I.S. [U-13C4]AcAc (PRM, 105.0378→60.0441) or [3,4,4,4-D4]βOHB (PRM 107.0646→59.0133), respectively.
2.7. Quantification of ketone bodies in liver tissue
Freeze clamped liver specimens (~20 mg) were homogenized using a zinc-bead homogenizer (Bead Mill, Omni International) in 500 μL of −20°C-chilled ACN:MeOH:water (2:2:1 v/v/v) containing known concentrations of [3,4,4,4-D4]βOHB and [U-13C4]AcAc (recommended final concentration of each I.S. is 20 μM for liver extracts). Homogenized livers were subjected to three cycles of vortexing (10 sec), freeze-thawing (30 sec), and water bath sonication (5 min), and centrifuged at 4 C, 15,000 × g for 10 min to remove protein pellets from the solvent. Supernatant was used for a UPLC-MS/MS quantification analysis using identical PRM transition signals as in the serum.
2.8. Sample Derivatization
Derivatization protocol was adapted from reference [40]. Extracted serum samples were SpeedVac dried, and equal volumes (100 μL) of freshly prepared 1 mM PMP, 10 mM TPP and 10 mM DPDS, all dissolved in ACN, were added and incubated for 90 min at room temperature. Samples were then SpeedVac dried, resuspended in a quarter of initial volume in 2% MeOH/98% water with 0.0125% AA, vortexed, sonicated for 5 min, centrifuged at 15,000 × g for 10 min, and the supernatant injected in the LC-MS/MS system using positive ionization mode. In order to quantify the enantiomeric distributions of βOHB and 2-OHB, 0.5 μL injection volumes were used, whereas 1 μL was used for 3-HIB enantiomers due to its low serum concentration.
2.9. Instrumentation
Analysis of non-reduced and reduced standards, or derivatized D- and L-ketones, was performed using Vanquish liquid chromatography (LC) system. Our first analysis was performed on Atlantis T3 column (150 × 1 mm 3 μm) as previously described [37], which was then optimized using Cortecs UPLC T3 column (100 × 2.1 mm 1.6 μm), used for the remainder of the analyses. For all analyses, the following mobile phases were used: A) 98% water/2% methanol with 0.0125% AA and B) 60% water/40% methanol with 0.0125% AA. Analysis using Atlantis column was performed using following binary gradient: 0–20% B for 10 min, 20–90% B for 0.5 min, 90% B for 4 min, 90–0% B for 0.5 min, and 0% B for 4 min at 0.1 mL/min flow rate. Samples analyzed using Cortecs UPLC column were separated using following binary gradient: 0–20% B for 3 min, 20–90% B for 0.5 min, 90% B for 1.5, 90–0% B for 0.5 min, and 0% B for 2 min at 0.3 mL/min. Columns were maintained at 30 C and the injection volume was 2 μL for Atlantis or 0.5 μL for the Cortecs column. LC system was hyphenated to Thermo Q Exactive Plus MS equipped with heated electrospray ionization (ESI) source. The MS system was operated in Full Scan MS or PRM modes using negative or positive ionization modes during optimization of the method. In final quantitative targeted analysis, only PRM mode was used with appropriate inclusion list (see Fig. 1A) and resolution and automatic gain control (AGC) targets in PRM mode were set to 70,000 and 1e5, respectively. For PRM mode the isolation window was set to m/z 1.0 and collision energy 30 (arbitrary units). For enantiomer separation MS scan range was 50–300 m/z in Full MS scan mode, operated solely in positive mode. The resolution was set to 35,000 with 2e5 AGC target. For PRM scan mode, resolution was 17,500 with AGC target 2e5, isolation window 1.0 m/z and optimal collision energy was 50 (arbitrary units). Common ESI parameters were the same in both approaches: auxiliary gas: 10, sheath gas flow 35, sweep gas 1, spray voltage 3 kV, capillary temperature 275 C, S-lens 50, and auxiliary gas temperature 150 C.
2.10. Calibration, quantification, and validation
To quantify AcAc and βOHB, [3,4,4,4-D4]βOHB (1 mg/mL) or synthetized [U-13C4]AcAc [concentration determined empirically as described in 2.5, Quantitation of [U-13C4]AcAc internal standard (I.S.), above] stored as aliquots at −20 C or −80 C, respectively, were used as internal standards (I.S.) (for details about quantification of ketones in serum or tissues, see Sections 2.5, 2.6, or Fig. 1A). To study linearity three independent external calibration curves of AcAc or βOHB were constructed from 0.1–250 μmoles/L with fixed amount of internal standards ([U-13C4]AcAc and [3,4,4,4-D4]βOHB at 20 μmoles/L). Slope, intercept and correlation coefficient (R2) were obtained by linear regression analysis where AcAc/[U-13C4]AcAc or βOHB/[3,4,4,4-D4]βOHB peak area ratios were plotted against the concentration. Limit of detection and quantification (LOD and LOQ) were calculated as the minimum concentration yielding a signal to noise ratio (S/R) equal to 3 and 10, respectively. LOD and LOQ were calculated for three independently prepared calibration curves. Repeatability (technical error) was estimated as relative standard deviation (%RSD) at three AcAc and βOHB concentrations levels (250 μmoles/L, 30 μmoles/L and 4 μmoles/L) by five repeated injection of standard solutions (n=5/group). The precision of the method was determined by evaluating inter-day and inter-sample precision. Inter-day precision was performed by the triplicate injection of extracted serum in two consecutive days (n=6) obtained from fed or 24h fasted mice. Inter-sample precision was estimated by the triplicate injection of five individual extracts in same day (n=15) from fed or 24 h fasted mice. Recovery was evaluated by spiking two or three independent serum or tissue samples in triplicate, obtained from fed or 24h fasted mice with known amount of AcAc and βOHB before the extraction procedure.
2.11. Statistical analysis
Data were plotted and statistical analysis was performed using Prism (GraphPad v8.2.1). Numbers of observations, assessments of normal distributions and statistical tests applied are provided in Figure Legends.
3. Results and discussion
3.1. Separation and identification of β-hydroxybutyrate and acetoacetate in serum
To profile βOHB in serum, we analyzed a mouse serum extract spiked with 50 μM [3,4,4,4-D4]βOHB, first using an Atlantis T3 C18 column hyphenated to a Q-Exactive Plus MS system in Full MS and PRM scan modes using negative ionization. While endogenous βOHB from the serum extract (m/z 103.0401) was retained on this C18 column for 3.5 min, peak shouldering was evident on the extracted ion chromatogram (XIC) signal in Full MS scan mode (Fig. S1A, black XIC, top). Scrutiny of the tandem MS/MS spectrum for the precursor ion 103.0401 revealed the predicted βOHB m/z fragmentation ion 59.0133, but also two additional m/z fragments 57.0344 and 73.0295, suggesting co-elution of βOHB isomers on this platform (Fig. S1A, upper right spectrum). Using public databases (Metlin and HMDB [41, 42]) we putatively assigned these molecules to 3-hydroxyisobutyrate (3-HIB; transition 103.0401→73.0290) and either 4-hydroxybutyrate (4-OHB) or 2-hydroxybutyrate (2-OHB), which share same 103.0401→57.0340 transition. Thus, to improve LC separation, the Cortecs UPLC T3 column was implemented, which compressed analytical time to only 7 minutes, while separating putative 3-HIB (Fig. S1A, blue XIC) from other βOHB isomers in serum extract. Retention time (RT), m/z and MS/MS signal comparisons from serum extract to authentic standards confirmed the identities of βOHB, and 3-HIB (Fig. S1B–C). Moreover, use of internal standards allowed XIC assignment of the endogenous signal for the 103.0401→ 57.0340 transition to 2-OHB, and not 4-OHB (Fig. S1D). Fig. 1A summarizes the RT, m/z and dominant MS/MS fragments used for the analysis of unlabeled and labeled I.S. analytes on UPLC Cortecs column with negative PRM scan mode on Q-Exactive MS.
Discrimination of 3-HIB (a metabolite of the amino acid valine) in serum was previously described using GC-MS, and differentiation of 4-OHB (γ-hydroxybutyrate) from βOHB was previously developed for forensic purposes using LC-MS/MS in blood and urine samples [28, 43]. However, isomeric 2-OHB (α-hydroxybutyrate, a by-product of threonine and methionine catabolism, and of glutathione biosynthesis [44]) has not been reported as an interfering signal in serum. As expected, due to the carboxylate groups in βOHB, 3-HIB, 2-OHB, and 4-OHB molecules, injection in negative mode results in 5–20 fold more efficient ionization than positive mode (Fig. 1B, Table S1). Although we did not formally quantify βOHB structural isomers, to assess the relative contributions of βOHB, 3-HIB, or 2/4-OHB to the precursor ion with m/z 103.0401, we analyzed the fractional contribution of βOHB, 3-HIB, or 2/4-OHB fragments in PRM mode to the total pool of the 103.0401 signal in mouse serum from ketogenesis insufficient mice (which had been treated with Hmgcs2 ASO) or scrambled control ASO-treated mice after a 24h fast (see Table S1) [39]. In serum of ketogenesis insufficient mice, we expect less contribution of βOHB to the m/z 103.0401 due to its impaired production, and indeed the relative contributions of 3-HIB and 2-OHB to the total pool exceeded 64.3±1.0% of the total signal for the m/z 103.0401. In the serum of control mice, these isomers comprised only 15.3±1.5% of the signal for this ion, together underscoring the necessity to employ MS/MS to segregate structural isomers of βOHB in biological samples (Fig. 1C).
Finally, the RT, m/z and tandem MS/MS spectrum of AcAc standard synthetized from ethyl-AcAc by base hydrolysis matched those observed within the serum extract, confirming AcAc identity (Fig. 1A, Fig. S1E–F).
3.2. Acetoacetate stability
βOHB measurement often serves as a proxy of ketogenesis, because AcAc poses challenges in its quantification due to its propensity to decarboxylate to acetone [45–48]. Spontaneous AcAc decarboxylation proceeds through a cyclic transition state in which the carboxylate proton is transferred to the β-carbonyl oxygen, generating a keto-enol tautomer that readily yields acetone and CO2 [17, 49]. Catalytic decarboxylation mechanisms proceed through the formation of a Schiff base intermediate from the β-carbonyl carbon [50]. The absence of a β-carbonyl group (and presence instead of a β-hydroxyl) within βOHB prevents its decarboxylation and thus supports its long-term stability in serum, plasma, and whole blood [38]. Indeed, older methodologies provide a surrogate for AcAc by tracking its decarboxylation to acetone as an indirect measure to estimate ketone concentrations in various matrices [22, 25, 51]. To evaluate the stability of synthesized AcAc under conditions commonly relevant to biological samples, we first tested its tolerance to storage at different temperatures, by preparing three individual 100 μM AcAc standard samples in water, spiking them with 20 μM βOHB, and storing aliquots for up to 35 days at room temperature (+21 C), +4 C, −20 C, −80 C, or LN2. βOHB is stable in various challenging matrices and storage conditions such as serum, plasma, and even whole blood stored at 4°C and room temperature for at least 7 days [38]. Therefore, the AcAc/βOHB ion intensity ratio was used as an index of AcAc stability in fresh and stored samples solutions thawed immediately before analysis. In water, synthesized AcAc standard was remarkably stable in all studied conditions, as signal diminished only at room temperature after 28 days (Fig. 2A). Using a similar approach, we evaluated the stability of synthesized AcAc in a common AcN/MeOH/water (2:2:1) biological sample extraction solution. Decomposition of AcAc in this extraction solution at room temperature was much faster than in water, with 21.8±2.1% loss of exogenous AcAc relative signal at room temperature in only 6h (Fig. 2B). Decomposition of AcAc in this extraction solution was attenuated, but nonetheless observed at +4°C, at which more significant loss of AcAc was observed after 7-day period. Lower temperatures preserved synthesized AcAc in extraction solution for at least 35 days.
Figure 2. Variations of AcAc stability are matrix and temperature dependent, but AcAc is tolerant to freeze thaw cycles.

Stability of synthesized AcAc (n=3/group) in (A) water or (B) AcN/MeOH/water (2:2:1) extraction solution (n=3/group), stored at +4 C, 21 C (room temperature), +37 C, −20 C, −80 C, or LN2, quantified by the AcAc/βOHB ratio (100 μM AcAc and 20 μM βOHB were added simultaneously during sample preparation; βOHB does not undergo decarboxylation and is stable). (C) Stability of endogenous AcAc in serum (n=3–6/group), stored at +4 C, room temperature, +37 C, −20 C, −80 C, or LN2, quantified by AcAc/[3,4,4,4-D4]βOHB (20 μM [3,4,4,4-D4]βOHB was added to serum immediately after collection from mice fasted for 24h). (D) Stability of AcAc as calculated by the AcAc/βOHB (in water or AcN/MeOH/water) or AcAc/[3,4,4,4-D4]βOHB (in serum) signal ratios in fresh samples and samples subjected to three freeze/thaw cycles, all relative to unthawed sample (n=6/group). Freeze/thaw cycles were performed within first nine hours of storage after adding 100 μM AcAc and 20 μM βOHB (in water or AcN/MeOH/water) or 20 μM [3,4,4,4-D4]βOHB, serum from mice in the fasted state. Data expressed as the mean standard error of the mean (SEM).
We next evaluated the stability of endogenous AcAc in serum obtained from control mice fasted for 24h. Three to six individual serum samples were spiked with 20 μM [3,4,4,4-D4]βOHB, aliquoted and stored for 35 days at room temperature, +4 C, +37 C, −20 C, −80 C or LN2. Signal for endogenous AcAc in serum stored at +37 C was unstable, with 65.9±3.9% loss observed within 6h (Fig. 2C). Relative AcAc signal also decayed significantly in serum stored at room temperature after 2 days (42.0±1.9%), and at +4 C after 14 days (71.1±1.4%) but remained stable at −80 C or in LN2 after 35 days. Other reports also have suggested lability AcAc signal in plasma or serum [35, 45, 47]. Our results underscore that endogenous AcAc is stable in serum samples for at least 35 days when stored either at −80 C or LN2, standard temperatures for storage of biological samples. Notably, endogenous AcAc derived from serum extracted immediately after collection, followed by storage in AcN:MeOH:water extraction solution at +4 C, a common storage condition for extracted samples in the LC autosampler, was unstable (Fig. S2).
Finally, to test tolerance to freeze thaw cycles, we prepared synthesized AcAc standards (100 μM, diluted in either water or AcN:MeOH:water extraction solution, along with 20 μM βOHB). We also tracked the relative endogenous AcAc signal in serum from mice in the fasted state (20 μM [3,4,4,4-D4]βOHB spiked at collection time). Samples were stored at −80 C and subjected to three freeze/thaw cycles within nine hours of storage. AcAc signal was tolerant to freeze thaw cycles in all matrices (Fig. 2D). Taken together, while AcAc is stable in water at all temperatures for at least 35 days, AcAc is unstable at higher temperatures in serum and in AcN:MeOH:water-extracted samples. While AcAc is remarkably tolerant to freeze/thaw cycles, interrogation of AcAc in serum analysis will be most accurate when data are collected immediately after extraction, or when serum or exacted samples are stored ≤ −80 C.
3.3. Development and use of authentic internal standards for rapid quantification of ketone bodies in biological samples
Although AcAc synthetized from its ethyl ester is remarkably stable in water, an internal standard [U-13C4]AcAc is not commercially available. Moreover, external calibration curves, or alternative standard addition methods are typically tedious and time-consuming methodologies, commonly subject to varying impacts of matrix effects between the samples and standards. Thus, in order to develop a stable isotope internal standard method for AcAc quantitation in biological samples, we synthetized [U-13C4]AcAc from ethyl-[U-13C4]AcAc using base-catalyzed hydrolysis, as we have previously described [36]. To estimate synthetized [U-13C4]AcAc concentration, we reduced it to [3-D1, U-13C4]βOHB using the reducing agent NaBD4 [37], and quantified its concentration using the commercially available [3,4,4,4-D4]βOHB I.S. (Fig. 3A). AcAc reduction to βOHB using different reduction durations (n=3/group) showed this reaction to be rapid and effective at AcAc concentrations ranging from 5 to 250 μM (Fig. 3B). We observed an average of 5.2% inaccuracy between predicted and calculated AcAc concentration. Injection of reduced [U-13C4]AcAc (i.e., [3-D1, U-13C4]βOHB) together with [3,4,4,4-D4]βOHB I.S. confirmed the absence of any residual unreacted [U-13C4]AcAc, and also allowed quantitation of the % yield of synthetized [U-13C4]AcAc derived from ethyl-[U-13C4]AcAc (Fig. 3C). To support a high-throughput assay for ketone body quantification, we use this approach to quantify [U-13C4]AcAc in large batch quantities that are aliquoted and stored in water at −80 C (conditions in which AcAc is highly stable) for further use as an internal standard.
Figure 3. Preparation and quantification of [U-13C4]AcAc stable isotope internal standard by its synthesis and reduction to [3-D1, U-13C4]βOHB.
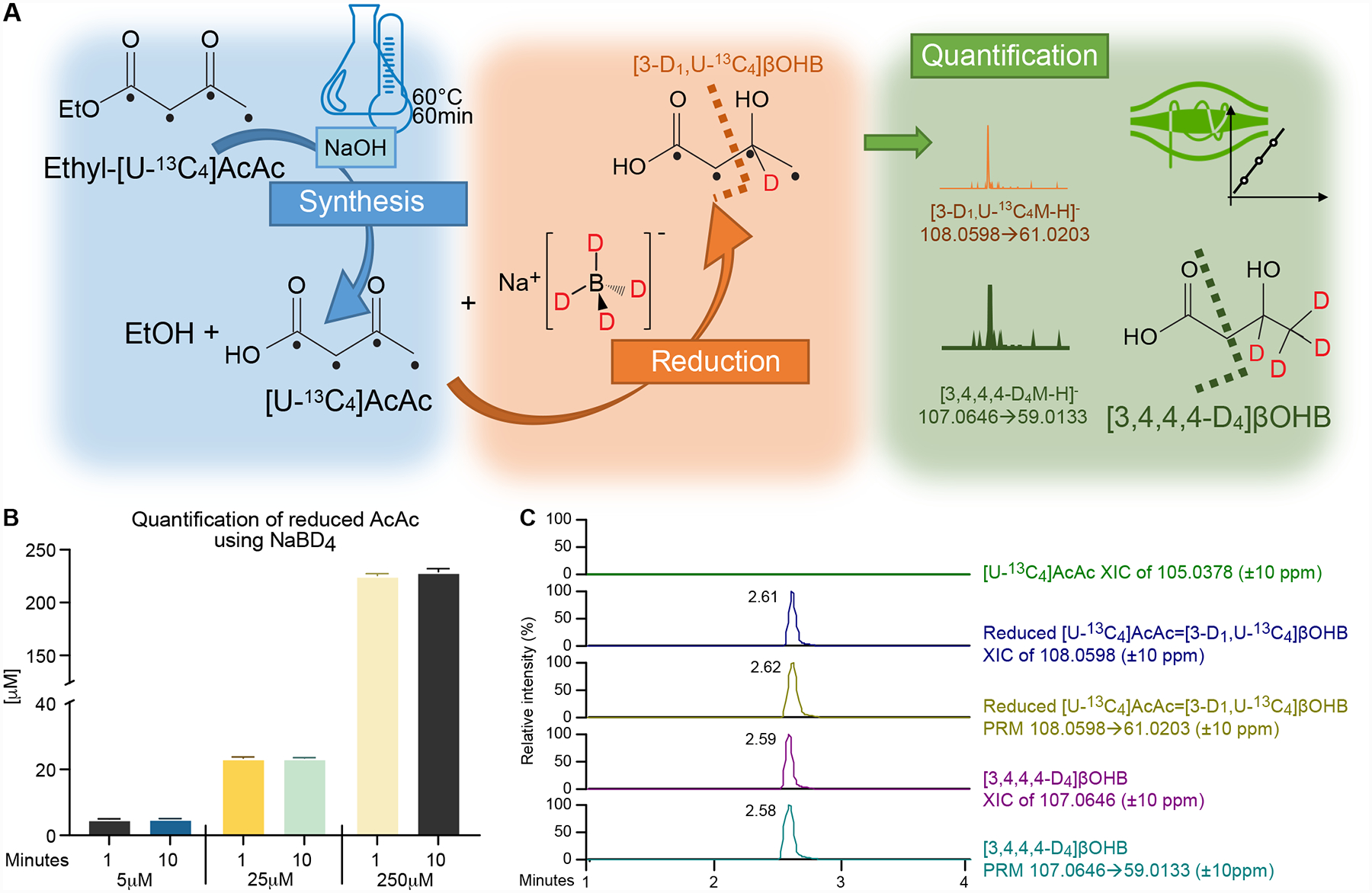
(A) [U-13C4]AcAc is synthetized from ethyl-[U-13C4]AcAc using base-catalyzed hydrolysis. To formally quantify the [U-13C4]AcAc internal standard, synthetized [U-13C4]AcAc is reduced by the reducing agent NaBD4 to [3-D1, U-13C4]βOHB, and quantified using commercially available deuterated [3,4,4,4-D4]βOHB (I.S.) by UPLC-MS/MS method using indicated PRM transitions. Black dots indicate 13C atomic positions. Dashed lines indicate where the molecules are fragmented in HCD chamber. (B) Quantification of [3-D1, U-13C4]βOHB formed from NaBD4-reduction of [U-13C4]AcAc indicates the reaction is rapid and complete across [U-13C4]AcAc starting concentrations (5–250 μM) (n=3/group). (C) NaBD4-reduced extract lacks any residual signal from [U-13C4]AcAc substrate (green XIC of 105.0378, ±10 ppm), while signal attributable to reduced [3-D1, U-13C4]βOHB was observed in Full MS scan (blue XIC of 108.0598, ±10 ppm) and PRM (yellow XIC of 108.0598→61.0203, ±10 ppm) modes. Data was compared to Full MS scan (purple XIC of 107.0646, 10 ppm) and PRM (light blue XIC of 107.0646 → 69.0133, 10 ppm) corresponding to commercially available deuterated [3,4,4,4-D4]βOHB (I.S.). Data are expressed as the mean standard error of the mean (SEM).
Validation of AcAc and βOHB analysis using UPLC-MS/MS showed excellent linearity (R2≥0.997) in the studied dynamic range that exceeded three orders of magnitude (0.1 – 250 μmol/L) (Table S2). LOD and LOQ were below 11.7±2.1 and 39.5±7.6 nmol/L, respectively. Instrument repeatability (technical error) was determined at three concentrations (4, 30 and 250 μmol/L) and was always below 3.3%. Since the matrix in fed and fasted serum could vary, inter-sample precision, inter-day precision and recovery studies were performed in samples acquired from fed or 24h fasted mice. Inter-sample precision in various days was lower than 6.5% for βOHB and 7.5% for AcAc in either fed or fasted serum, while inter-day precision calculated by injecting same extract in three consecutive days was 3.3% for βOHB and 2.7% for AcAc (Table S3). Recovery was determined by spiking three individual serum samples with known amounts of AcAc or βOHB and ranged from 95.1±1.0% to 110.9±5.7% (Table S4).
This developed methodology was used to quantify AcAc and βOHB in serum samples obtained from wild type mice, showing predicted incremental increases of concentration of both ketone bodies with duration of fasting, and as expected the magnitude of circulating βOHB increase with fasting was greater compared to those of AcAc (Fig. 4A–B) [52]. As expected, circulating ketone concentrations were significantly blunted after 24 hours of fasting in serum of ketogenesis insufficient mice that had received Hmgcs2 ASO, compared to mice that had received scrambled control ASO (Fig. 4C), which is consistent with our prior analysis of this model using a traditional spectrophotometric assay [39].
Figure 4. Application of novel UPLC-MS/MS method to quantify AcAc and βOHB concentrations in serum and liver tissue extracts.
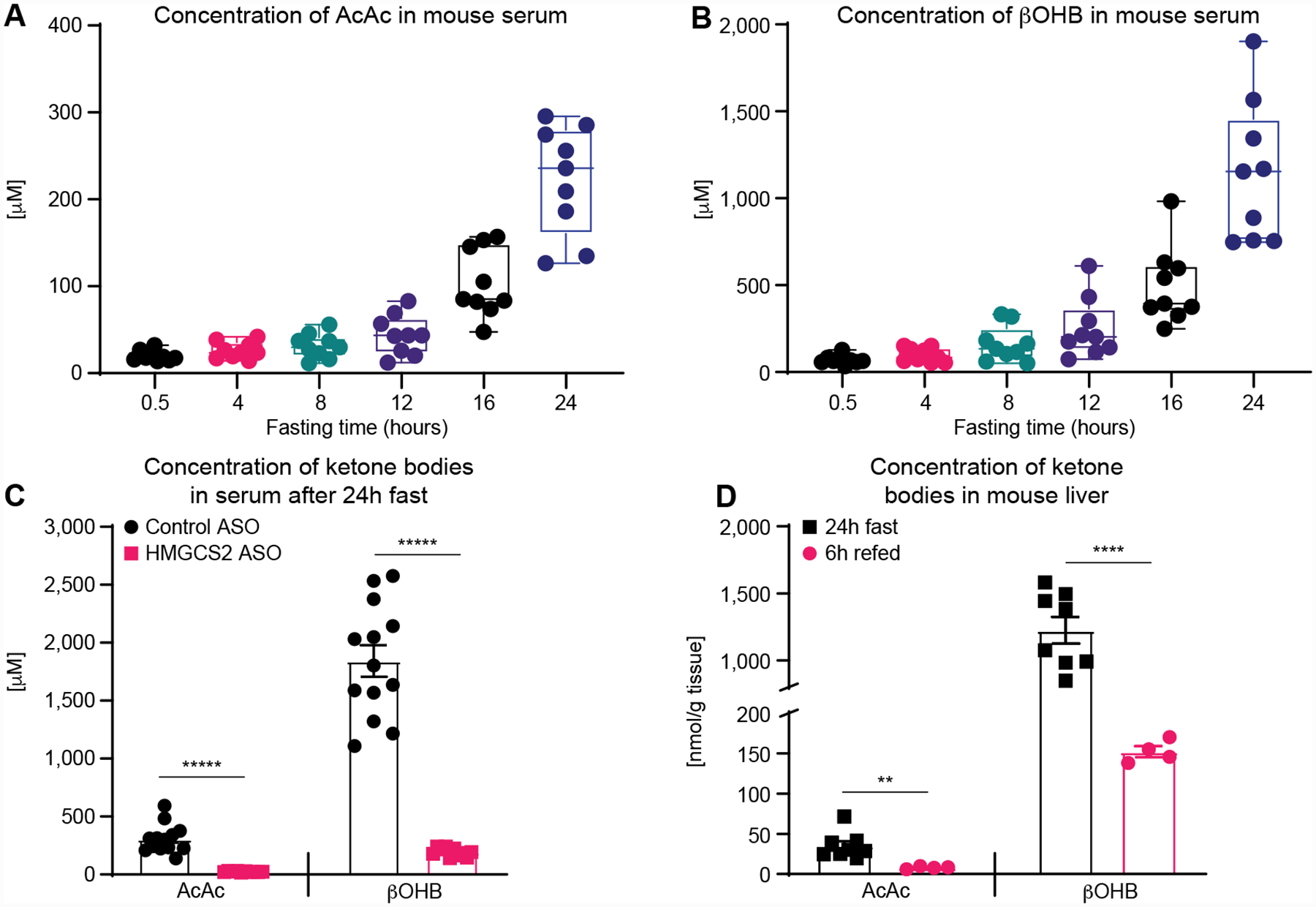
Concentrations of (A) AcAc, and (B) βOHB in extracts of serum collected from wild type mice after increasing fasting durations (n=9/group) show the expected incremental rises in both ketones, with expected greater increases in βOHB concentrations. (C) After a 24h fast, concentrations of AcAc and βOHB are markedly blunted in ketogenesis insufficient mice (i.e., those receiving Hmgcs2 ASO), compared to mice receiving a scrambled control ASO (n=11/group). (D) AcAc and βOHB concentrations in liver tissue extracts from wild type mice, showing expected decreases in concentration after 6 h of refed period, compared to liver concentrations after a 24h fast (n>4/group). Data are expressed as the mean standard error of the mean (SEM). Significant differences in (C) and (D) were determined by multiple Student’s t test with Holm-Šidák correction when compared with control; **p < 0.001, ****p < 0.0001, *****p < 0.000001.
As AcAc was tolerant to freeze/thaw cycles, we also developed an extraction method for liver tissue samples spiked with stable isotope standards, by applying three cycles of LN2 and sonication cycles. This method is similar to a commonly used metabolomics extraction method where final extraction buffer composition is the same to serum extraction buffer [53]. Indeed, [U-13C4]AcAc I.S. signal was comparable among samples analyzed directly in extraction buffer, analyzed in extraction buffer after three cycles of freeze/thaw, or freshly spiked within tissue extract samples (Fig. S3). Applying this method, concentrations of AcAc and βOHB both showed expected ~10-fold increases in extracts from livers of wild type mice fasted for 24h, compared to those from wild type mouse livers harvested 6h after initiation of refeeding following a 24h fast (Fig. 4D). Recovery studies, performed using spiked AcAc and βOHB internal standards in three individual tissue liver samples, showed a recovery range from 84.6±12.0% to 111.9±1.0% (Table S5), which suggest no matrix effect.
3.4. Discrimination of βOHB enantiomers and structural isomers
Although liver hepatocytes predominantly produce D-βOHB (and not L-βOHB), a fraction of the βOHB pool within the circulation and tissues is comprised by the L-βOHB enantiomer [32]. While multiple reports underscore physiological and cellular roles for βOHB beyond energy transfer [54–56], the full scope of enantioselectivity for these responses has been incompletely defined. Because spectrophotometric measurement methods employ exogenous BDH1 enzyme reagent, they only detect D-βOHB, and not L-βOHB. MS-based methodologies fail to distinguish D-βOHB from L-βOHB, measuring both together, and chromatographic separations described heretofore are complex and inefficient, requiring multiple columns or extreme chromatographic conditions with prolonged separation times [31–33]. Therefore, we sought to differentiate the βOHB enantiomers by incorporating an efficient approach compatible with our UPLC-MS/MS workflow. Derivatization of D- or L-βOHB with stereoselective S-PMP renders βOHB into chromatographically separable diastereomers (Fig. 5A) [40]. Direct infusion of DL-βOHB standard derivatized in this manner revealed effective derivatization reactions, in which m/z 241.1910 was observed in positive mode full MS scan when reactants were incubated in the presence of DL-βOHB, but not when incubated in the absence of DL-βOHB (Fig. S4A–B). Moreover, fragmentation of the DL-βOHB-S-PMP m/z 241.1910 derivatized precursor showed the expected m/z fragment 170.1170 in PRM mode (Fig. 5B). As expected, injection of unreacted DL-βOHB, or derivatized DL-βOHB, D-βOHB, or L-βOHB standards, using same UPLC Cortecs chromatographic workflow employed for quantification, showed chromatographic separation of D- from L-βOHB signals to the baseline (Rs=1.8) only when βOHB is derivatized (compare Fig. S1B, Fig. 5C and Fig. S4C–D). As expected, full scan MS and MS/MS spectra of S-PMP-derivatized D- or L-βOHB standards were identical (data not shown).
Figure 5. MS/MS detection, identification and UPLC separation of DL-βOHB enantiomers and their structural isomers 2-OHB, 3-HIB, and 4-OHB.
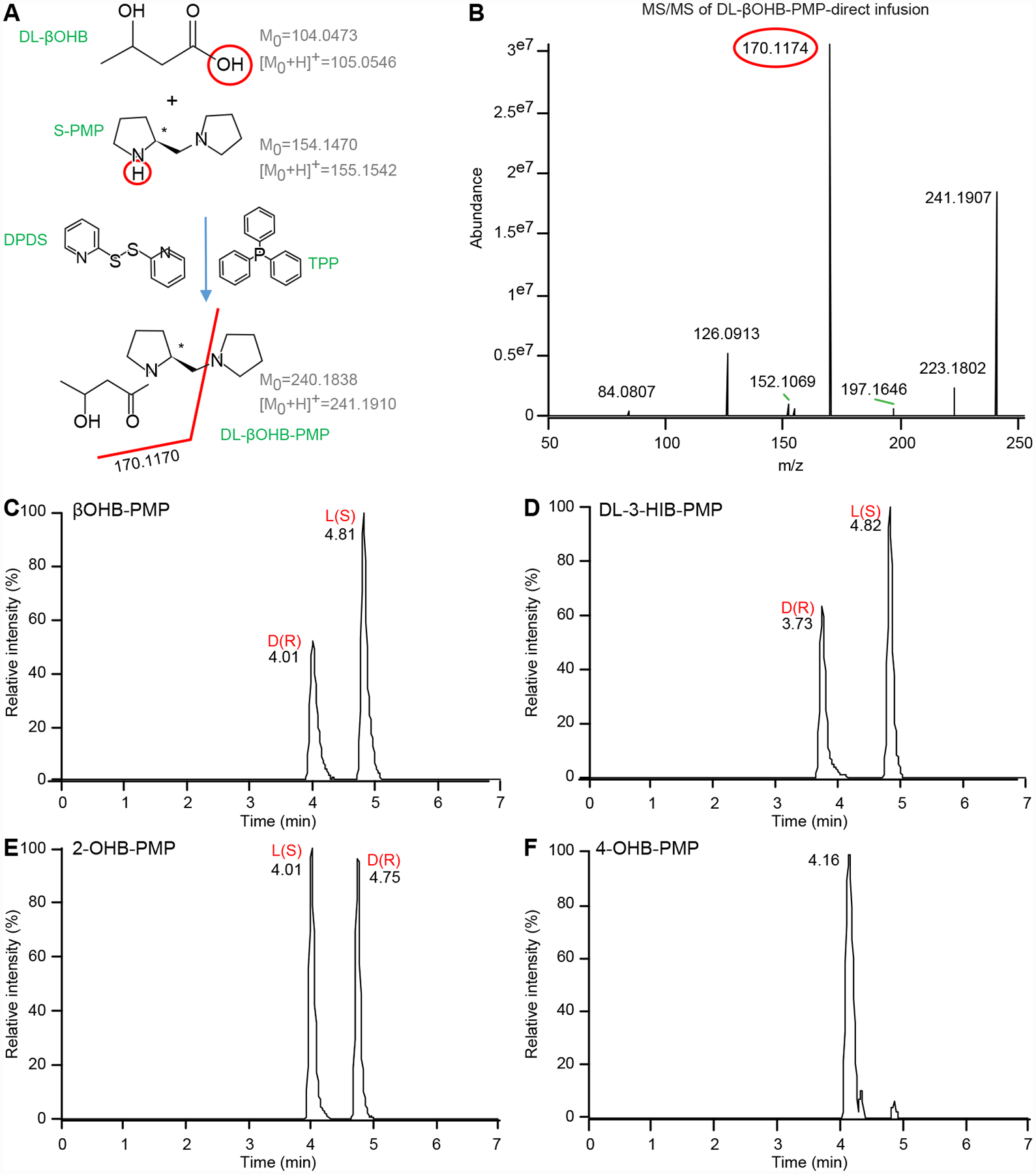
(A) Scheme of βOHB derivatization using S-PMP in the presence of TPP and DPDS catalysts with monoisotopic mass of uncharged and positively charged molecules. Red line indicates where the molecules are fragmented in HCD chamber. (B) MS/MS of derivatized 20 μM DL-βOHB-S-PMP precursor in positive ionization mode, m/z 241.1910, ± 10 ppm with predicted m/z fragment 170.1170 using direct infusion. Separation of 20 μM of derivatized (C) DL-βOHB-S-PMP, (D) DL-3-HIB-PMP, (E) DL-2-OHB-PMP showing baseline separation of its D(R)- and L(S)- enantiomers and (F) 4-OHB-PMP (XIC for all are m/z 241.1910, ±10 ppm). For separation of each enantiomer individually, see supplementary Figure S4.
We also evaluated whether this derivatization approach allows efficient separation of 3-HIB and 2-OHB enantiomers (4-OHB has no chiral center). Indeed, derivatization of DL-3-HIB and DL-2-OHB standards showed baseline separation of D- and L-derived S-PMP diastereomers of both DL-3-HIB and DL-2-OHB, but not 4-OHB (Fig. 5D–F, S4E–F). Since S-PMP-derivatized βOHB, 3-HIB, 2-OHB and 4-OHB all produce isobaric precursor ions in full MS scan, with similar RTs among various diastereomers, we also used tandem MS to reveal MS/MS spectra of the S-PMP derivatized standards’ precursor ions that would distinguish each standard’s distribution of enantiomers. Direct infusion of S-PMP-derivatized DL-βOHB, DL-3-HIB, DL-2-OHB, or 4-OHB at 35 and 50 N(CE) showed that the MS/MS spectra of m/z 241.1910 precursor ion at 35 N(CE) for all derivatized standards were similar, producing m/z 84.0807, 126.0913, 170.1174 and 223.1802 fragments, while not breaking entire signal of ionized precursors (Fig. S5A–B). However, MS/MS spectra obtained at a higher N(CE) of 50 significantly diminished the abundance of the m/z 241.1910 precursor ion’s intensity, while intensifying its fragments (Fig. S5C–D). All selected precursor → fragment transitions selective for S-PMP-derivatized DL-βOHB; DL-3-HIB; DL-2-OHB; or 4-OHB isomers, at both 35 and 50 N(CE) enabled selective detection of each molecule (Table 1, Fig. S6).
Table 1. Optimized UPLC-MS/MS parameters for detection of DL-analytes derivatized with S-PMP.
RT, retention time in minutes on UPLC Cortecs column using optimal chromatographic separation; FS, Full MS scan mode; N(CE), Normalize collision energy; PRM, Parallel Reaction Monitoring. 4-OHB is achiral therefore only one signal is produced. Tandem mass spectra of derivatized standards yielded specific low intensity fragments at 35 and 50 N(CE) for DL-βOHB, DL-3-HIB, DL-2-OHB, or 4-OHB with calculated putative formulas of those fragments. RTs corresponding to each enantiomer were confirmed with stereoselective standards.
| S-PMP-derivatized analyte | RT (min) | FS (m/z) Positive Mode | (N)CE | PRM transition | |
|---|---|---|---|---|---|
| D (R) enantiomer | L (S) enantiomer | ||||
| β-hydroxybutyrate (βOHB) | 4.0 | 4.8 | 241.1910 | 35 | 197.1654 (C11H21N2O) |
| 50 | 197.1654 (C11H21N2O) | ||||
| 3-hydroxyisobutyrate (3-HIB) | 3.7 | 4.7 | 35 | 211.1810 (C12H23N2O) | |
| 50 | 139.0997 (C8H13NO) | ||||
| 2-hydroxybutyrate (2-OHB) | 4.0 | 4.7 | 35 | 112.0762 (C6H10NO) | |
| 50 | 142.1232 (C8H16NO) | ||||
| 4-hydroxybutyrate (4-OHB)- achiral | 4.1 | 35 | 153.1392 (C9H17N2) | ||
| 50 | 154.1232 (C9H16NO) | ||||
To apply this enantiomeric discrimination workflow to serum of ketogenesis insufficient mice and their control ASO-treated counterparts, animals were starved for 24h and relative contributions of each enantiomer for βOHB, 3-HIB, and 2-OHB were quantified. As predicted, the D-βOHB enantiomer in the serum of control mice was predominant (96.8±0.4%), while in ketogenesis insufficient mice, the D-βOHB enantiomer comprised only 65.4±9.6% of the βOHB pool (Fig. 6A). Enantiomeric separation of serum 3-HIB supported exclusive contribution of the L-3-HIB enantiomer in serum from both control and ketogenesis insufficient mice fasted for 24h (Fig. 6B). This is consistent with 3-HIB generation exclusively from L-valine in mammals [57]. Finally, 2-OHB was also present almost exclusively as the L- enantiomer (97.0±1%) in the serum of mice fasted for 24h, and its D- and L- distribution was unaffected by ketogenic insufficiency (Fig. 6C).
Figure 6. Enantiomeric distributions of βOHB, 3-HIB, and 2-OHB in mouse serum.
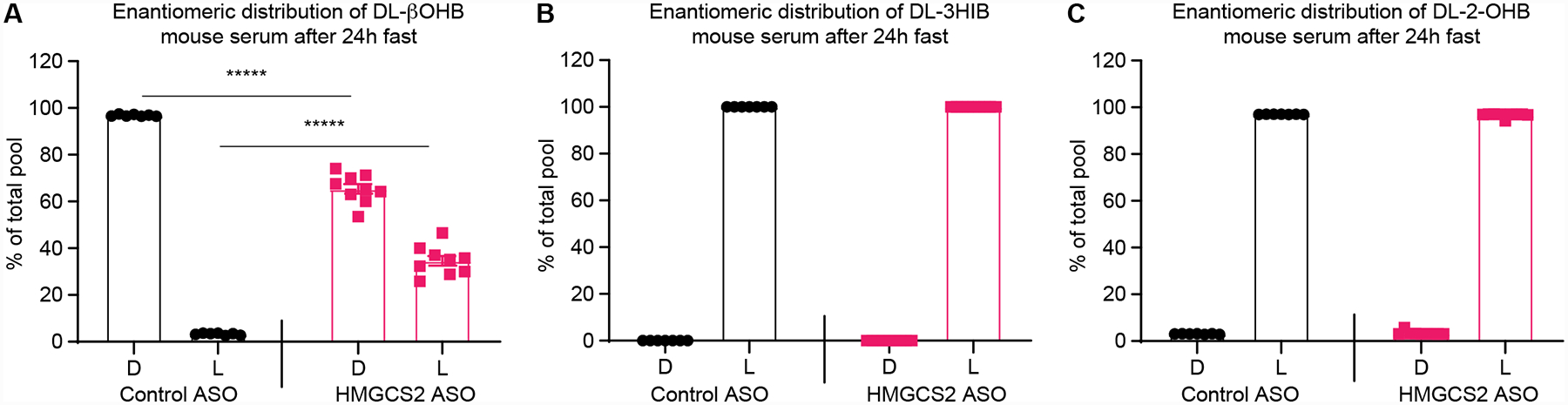
Enantiomeric distributions of (A) βOHB, (B) 3-HIB, and (C) 2-OHB in serum collected after a 24h fast from scrambled control ASO- and ketogenesis insufficient Hmgcs2 ASO-treated mice (n=7–9/group). Data expressed as the mean standard error of the mean (SEM). Significant differences in were determined by multiple Student’s t test with Holm-Šidák correction when compared with control. *****, p < 0.000001.
4. Conclusions
A UPLC-MS/MS methodology using stable isotope internal standard additions was developed for rapid and reliable quantification of both ketone bodies AcAc and βOHB in serum and tissues. The method allows reliable discrimination from βOHB structural isomers 3-HIB, 2-OHB, and 4-OHB, as well as separation of D- from L-enantiomers of each of these structural isomers. This method is readily scalable to high throughput platforms, which will be useful in interrogation of the increasing span of dietary interventions for wellness and disease that modulate ketone metabolism; assessment of ketones as biomarkers of metabolic state; monitoring the responses to therapeutic approaches that modulate ketone metabolism or deliver various forms of exogenous ketone bodies; and evaluation of pre-clinical models.
Supplementary Material
One-step UPLC-MS/MS method to quantify AcAc and D-βOHB in serum and tissue
Novel application of 13C-labeled internal standards, conferring broad dynamic range
Differentiates βOHB, 2-/4-hydroxybutyrate or 3-hydroxyisobutyrate enantiomers
Acknowledgements
The authors are grateful for support from NIH (DK091538). The authors thank Yi-Cheng Sin for technical support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
The authors declare no conflict of interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online.
References
- [1].Robinson AM, Williamson DH, Physiological roles of ketone bodies as substrates and signals in mammalian tissues, Physiol Rev 60(1) (1980) 143–87. [DOI] [PubMed] [Google Scholar]
- [2].Koeslag JH, Noakes TD, Sloan AW, Post-exercise ketosis, J Physiol 301 (1980) 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cahill GF Jr., Fuel metabolism in starvation, Annual review of nutrition 26 (2006) 1–22. [DOI] [PubMed] [Google Scholar]
- [4].Cotter DG, d’Avignon DA, Wentz AE, Weber ML, Crawford PA, Obligate role for ketone body oxidation in neonatal metabolic homeostasis, J Biol Chem 286(9) (2011) 6902–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Puchalska P, Crawford PA, Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics, Cell Metab 25(2) (2017) 262–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].de Cabo R, Mattson MP, Effects of Intermittent Fasting on Health, Aging, and Disease, N Engl J Med 381(26) (2019) 2541–2551. [DOI] [PubMed] [Google Scholar]
- [7].Mujica-Parodi LR, Amgalan A, Sultan SF, Antal B, Sun X, Skiena S, Lithen A, Adra N, Ratai EM, Weistuch C, Govindarajan ST, Strey HH, Dill KA, Stufflebeam SM, Veech RL, Clarke K, Diet modulates brain network stability, a biomarker for brain aging, in young adults, Proc Natl Acad Sci U S A 117(11) (2020) 6170–6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Caffa I, Spagnolo V, Vernieri C, Valdemarin F, Becherini P, Wei M, Brandhorst S, Zucal C, Driehuis E, Ferrando L, Piacente F, Tagliafico A, Cilli M, Mastracci L, Vellone VG, Piazza S, Cremonini AL, Gradaschi R, Mantero C, Passalacqua M, Ballestrero A, Zoppoli G, Cea M, Arrighi A, Odetti P, Monacelli F, Salvadori G, Cortellino S, Clevers H, De Braud F, Sukkar SG, Provenzani A, Longo VD, Nencioni A, Fasting-mimicking diet and hormone therapy induce breast cancer regression, Nature 583(7817) (2020) 620–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hopkins BD, Pauli C, Du X, Wang DG, Li X, Wu D, Amadiume SC, Goncalves MD, Hodakoski C, Lundquist MR, Bareja R, Ma Y, Harris EM, Sboner A, Beltran H, Rubin MA, Mukherjee S, Cantley LC, Suppression of insulin feedback enhances the efficacy of PI3K inhibitors, Nature 560(7719) (2018) 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Stubbs BJ, Cox PJ, Evans RD, Santer P, Miller JJ, Faull OK, Magor-Elliott S, Hiyama S, Stirling M, Clarke K, On the Metabolism of Exogenous Ketones in Humans, Front Physiol 8 (2017) 848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].McGarry JD, Foster DW, Regulation of hepatic fatty acid oxidation and ketone body production, Annual review of biochemistry 49 (1980) 395–420. [DOI] [PubMed] [Google Scholar]
- [12].Ferre P, Satabin P, Decaux JF, Escriva F, Girard J, Development and regulation of ketogenesis in hepatocytes isolated from newborn rats, Biochem J 214(3) (1983) 937–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lehninger AL SH, Wise JB D-beta-Hydroxybutyric dehydrogenase of mitochondria, J Biol Chem 235 (1960) 2450–5. [PubMed] [Google Scholar]
- [14].Bock H, Fleischer S, Preparation of a homogeneous soluble D-beta-hydroxybutyrate apodehydrogenase from mitochondria, J Biol Chem 250(15) (1975) 5774–61. [PubMed] [Google Scholar]
- [15].Balasse EO, Fery F, Ketone body production and disposal: effects of fasting, diabetes, and exercise, Diabetes/metabolism reviews 5(3) (1989) 247–70. [DOI] [PubMed] [Google Scholar]
- [16].Williamson DH, Lund P, Krebs HA, The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver, Biochem J 103(2) (1967) 514–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Pedersen KJ, The ketonic decomposition of beta-keto carboxylic acids, Journal of the American Chemical Society 51(7) (1929) 2098–2107. [Google Scholar]
- [18].Galan A, Hernandez J, Jimenez O, Measurement of blood acetoacetate and beta-hydroxybutyrate in an automatic analyser, J Autom Methods Manag Chem 23(3) (2001) 69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kupari M, Lommi J, Ventila M, Karjalainen U, Breath acetone in congestive heart failure, Am J Cardiol 76(14) (1995) 1076–8. [DOI] [PubMed] [Google Scholar]
- [20].Yokokawa T, Sato T, Suzuki S, Oikawa M, Yoshihisa A, Kobayashi A, Yamaki T, Kunii H, Nakazato K, Suzuki H, Saitoh SI, Ishida T, Shimouchi A, Takeishi Y, Elevated exhaled acetone concentration in stage C heart failure patients with diabetes mellitus, BMC Cardiovasc Disord 17(1) (2017) 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Koeslag JH, Daily blood ketone body concentrations after acute exercise, S Afr Med J 57(4) (1980) 125–7. [PubMed] [Google Scholar]
- [22].Goschke H, Estimation of ketone bodies in blood, cerebrospinal fluid and urine, Clin Chim Acta 28(2) (1970) 359–64. [DOI] [PubMed] [Google Scholar]
- [23].Kimura M, Kobayashi K, Matsuoka A, Hayashi K, Kimura Y, Head-space gas-chromatographic determination of 3-hydroxybutyrate in plasma after enzymic reactions, and the relationship among the three ketone bodies, Clin Chem 31(4) (1985) 596–8. [PubMed] [Google Scholar]
- [24].Kimura M, Ogasahara N, Kobayashi K, Hitoi A, Matsuoka A, Kimura Y, Improvement in the head-space gas-chromatographic method for determination of three ketone bodies in plasma, Clin Chem 36(1) (1990) 160–1. [PubMed] [Google Scholar]
- [25].Lopez-Soriano FJ, Argiles JM, Simultaneous determination of ketone bodies in biological samples by gas chromatographic headspace analysis, J Chromatogr Sci 23(3) (1985) 120–3. [DOI] [PubMed] [Google Scholar]
- [26].Felby S, Nielsen E, Determination of ketone bodies in postmortem blood by head-space gas chromatography, Forensic Sci Int 64(2–3) (1994) 83–8. [DOI] [PubMed] [Google Scholar]
- [27].Hassan HM, Cooper GA, Determination of beta-hydroxybutyrate in blood and urine using gas chromatography- mass spectrometry, J Anal Toxicol 33(8) (2009) 502–7. [DOI] [PubMed] [Google Scholar]
- [28].Dahl SR, Olsen KM, Strand DH, Determination of gamma-hydroxybutyrate (GHB), beta-hydroxybutyrate (BHB), pregabalin, 1,4-butane-diol (1,4BD) and gamma-butyrolactone (GBL) in whole blood and urine samples by UPLC-MSMS, J Chromatogr B Analyt Technol Biomed Life Sci 885–886 (2012) 37–42. [DOI] [PubMed] [Google Scholar]
- [29].Sorensen LK, Rittig NF, Holmquist EF, Jorgensen KA, Jorgensen JO, Moller N, Johannsen M, Simultaneous determination of beta-hydroxybutyrate and beta-hydroxy-beta-methylbutyrate in human whole blood using hydrophilic interaction liquid chromatography electrospray tandem mass spectrometry, Clin Biochem 46(18) (2013) 1877–83. [DOI] [PubMed] [Google Scholar]
- [30].Webber RJ, Edmond J, Utilization of L(+)-3-hydroxybutyrate, D(−)-3-hydroxybutyrate, acetoacetate, and glucose for respiration and lipid synthesis in the 18-day-old rat, J Biol Chem 252(15) (1977) 5222–6. [PubMed] [Google Scholar]
- [31].Tsai YC, Chou YC, Wu AB, Hu CM, Chen CY, Chen FA, Lee JA, Stereoselective effects of 3-hydroxybutyrate on glucose utilization of rat cardiomyocytes, Life Sci 78(12) (2006) 1385–91. [DOI] [PubMed] [Google Scholar]
- [32].Hsu WY, Kuo CY, Fukushima T, Imai K, Chen CM, Lin PY, Lee JA, Enantioselective determination of 3-hydroxybutyrate in the tissues of normal and streptozotocin-induced diabetic rats of different ages, J Chromatogr B Analyt Technol Biomed Life Sci 879(29) (2011) 3331–6. [DOI] [PubMed] [Google Scholar]
- [33].Calderon C, Lammerhofer M, Chiral separation of short chain aliphatic hydroxycarboxylic acids on cinchonan carbamate-based weak chiral anion exchangers and zwitterionic chiral ion exchangers, J Chromatogr A 1487 (2017) 194–200. [DOI] [PubMed] [Google Scholar]
- [34].Zeng M, Cao H, Fast quantification of short chain fatty acids and ketone bodies by liquid chromatography-tandem mass spectrometry after facile derivatization coupled with liquid-liquid extraction, J Chromatogr B Analyt Technol Biomed Life Sci 1083 (2018) 137–145. [DOI] [PubMed] [Google Scholar]
- [35].Peng M, Cai Y, Fang X, Liu L, Rapid quantification of metabolic intermediates in blood by liquid chromatography-tandem mass spectrometry to investigate congenital lactic acidosis, Anal Chim Acta 942 (2016) 50–57. [DOI] [PubMed] [Google Scholar]
- [36].Puchalska P, Martin SE, Huang X, Lengfeld JE, Daniel B, Graham MJ, Han X, Nagy L, Patti GJ, Crawford PA, Hepatocyte-Macrophage Acetoacetate Shuttle Protects against Tissue Fibrosis, Cell Metab 29(2) (2019) 383–398 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sunny NE, Satapati S, Fu X, He T, Mehdibeigi R, Spring-Robinson C, Duarte J, Potthoff MJ, Browning JD, Burgess SC, Progressive adaptation of hepatic ketogenesis in mice fed a high-fat diet, Am J Physiol Endocrinol Metab 298(6) (2010) E1226–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Custer EM, Myers JL, Poffenbarger PL, Schoen I, The storage stability of 3-hydroxybutyrate in serum, plasma, and whole blood, Am J Clin Pathol 80(3) (1983) 375–80. [DOI] [PubMed] [Google Scholar]
- [39].Cotter DG, Ercal B, Huang X, Leid JM, d’Avignon DA, Graham MJ, Dietzen DJ, Brunt EM, Patti GJ, Crawford PA, Ketogenesis prevents diet-induced fatty liver injury and hyperglycemia, J Clin Invest 124(12) (2014) 5175–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Tsutsui H, Mochizuki T, Maeda T, Noge I, Kitagawa Y, Min JZ, Todoroki K, Inoue K, Toyo’oka T, Simultaneous determination of DL-lactic acid and DL-3-hydroxybutyric acid enantiomers in saliva of diabetes mellitus patients by high-throughput LC-ESI-MS/MS, Anal Bioanal Chem 404(6–7) (2012) 1925–34. [DOI] [PubMed] [Google Scholar]
- [41].Domingo-Almenara X, Montenegro-Burke JR, Ivanisevic J, Thomas A, Sidibe J, Teav T, Guijas C, Aisporna AE, Rinehart D, Hoang L, Nordstrom A, Gomez-Romero M, Whiley L, Lewis MR, Nicholson JK, Benton HP, Siuzdak G, XCMS-MRM and METLIN-MRM: a cloud library and public resource for targeted analysis of small molecules, Nat Methods 15(9) (2018) 681–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wishart DS, Feunang YD, Marcu A, Guo AC, Liang K, Vazquez-Fresno R, Sajed T, Johnson D, Li C, Karu N, Sayeeda Z, Lo E, Assempour N, Berjanskii M, Singhal S, Arndt D, Liang Y, Badran H, Grant J, Serra-Cayuela A, Liu Y, Mandal R, Neveu V, Pon A, Knox C, Wilson M, Manach C, Scalbert A, HMDB 4.0: the human metabolome database for 2018, Nucleic Acids Res 46(D1) (2018) D608–D617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Des Rosiers C, Montgomery JA, Desrochers S, Garneau M, David F, Mamer OA, Brunengraber H, Interference of 3-hydroxyisobutyrate with measurements of ketone body concentration and isotopic enrichment by gas chromatography-mass spectrometry, Anal Biochem 173(1) (1988) 96–105. [DOI] [PubMed] [Google Scholar]
- [44].Gall WE, Beebe K, Lawton KA, Adam KP, Mitchell MW, Nakhle PJ, Ryals JA, Milburn MV, Nannipieri M, Camastra S, Natali A, Ferrannini E, Group RS, alpha-hydroxybutyrate is an early biomarker of insulin resistance and glucose intolerance in a nondiabetic population, PLoS One 5(5) (2010) e10883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Fritzsche I, Buhrdel P, Melcher R, Bohme HJ, Stability of ketone bodies in serum in dependence on storage time and storage temperature, Clin Lab 47(7–8) (2001) 399–403. [PubMed] [Google Scholar]
- [46].McNeil CA, Pramfalk C, Humphreys SM, Hodson L, The storage stability and concentration of acetoacetate differs between blood fractions, Clin Chim Acta 433 (2014) 278–83. [DOI] [PubMed] [Google Scholar]
- [47].Price CP, Llyod B, Alberti GM, A kinetic spectrophotometric assay for rapid determination of acetoacetate in blood, Clin Chem 23(10) (1977) 1893–7. [PubMed] [Google Scholar]
- [48].Yamanishi H, Iyama S, Yamaguchi Y, Amino N, Stability of acetoacetate after venesection, Clin Chem 39(5) (1993) 920. [PubMed] [Google Scholar]
- [49].Ouellette RJ, Rawn JD, Carboxylic Acids, in: Ouellette RJ, Rawn JD (Eds.), Organic Chemistry, Academic Press; 2018, pp. 625–663. [Google Scholar]
- [50].Hamilton GA, Westheimer FH, ON THE MECHANISM OF THE ENZYMATIC DECARBOXYLATION OF ACETOACETATE1, Journal of the American Chemical Society 81(23) (1959) 6332–6333. [Google Scholar]
- [51].Musa-Veloso K, Likhodii SS, Cunnane SC, Breath acetone is a reliable indicator of ketosis in adults consuming ketogenic meals, Am J Clin Nutr 76(1) (2002) 65–70. [DOI] [PubMed] [Google Scholar]
- [52].Cahill GF Jr., Herrera MG, Morgan AP, Soeldner JS, Steinke J, Levy PL, Reichard GA Jr., Kipnis DM, Hormone-fuel interrelationships during fasting, J Clin Invest 45(11) (1966) 1751–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ivanisevic J, Zhu ZJ, Plate L, Tautenhahn R, Chen S, O’Brien PJ, Johnson CH, Marletta MA, Patti GJ, Siuzdak G, Toward ‘omic scale metabolite profiling: a dual separation-mass spectrometry approach for coverage of lipid and central carbon metabolism, Anal Chem 85(14) (2013) 6876–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Xie Z, Zhang D, Chung D, Tang Z, Huang H, Dai L, Qi S, Li J, Colak G, Chen Y, Xia C, Peng C, Ruan H, Kirkey M, Wang D, Jensen LM, Kwon OK, Lee S, Pletcher SD, Tan M, Lombard DB, White KP, Zhao H, Li J, Roeder RG, Yang X, Zhao Y, Metabolic Regulation of Gene Expression by Histone Lysine beta-Hydroxybutyrylation, Mol Cell 62(2) (2016) 194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD, Newgard CB, Farese RV Jr., de Cabo R, Ulrich S, Akassoglou K, Verdin E, Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor, Science 339(6116) (2013) 211–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, D’Agostino D, Planavsky N, Lupfer C, Kanneganti TD, Kang S, Horvath TL, Fahmy TM, Crawford PA, Biragyn A, Alnemri E, Dixit VD, The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease, Nat Med 21(3) (2015) 263–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Linster CL, Noel G, Stroobant V, Vertommen D, Vincent MF, Bommer GT, Veiga-da-Cunha M, Van Schaftingen E, Ethylmalonyl-CoA decarboxylase, a new enzyme involved in metabolite proofreading, J Biol Chem 286(50) (2011) 42992–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.