

Abstract
Inflammation orchestrates each stage of the life cycle of atherosclerotic plaques. Indeed, inflammatory mediators likely link many traditional and emerging risk factors with atherogenesis. Atheroma initiation involves endothelial activation with recruitment of leucocytes to the arterial intima, where they interact with lipoproteins or their derivatives that have accumulated in this layer. The prolonged and usually clinically silent progression of atherosclerosis involves periods of smouldering inflammation, punctuated by episodes of acute activation that may arise from inflammatory mediators released from sites of extravascular injury or infection or from subclinical disruptions of the plaque. Smooth muscle cells and infiltrating leucocytes can proliferate but also undergo various forms of cell death that typically lead to formation of a lipid-rich ‘necrotic’ core within the evolving intimal lesion. Extracellular matrix synthesized by smooth muscle cells can form a fibrous cap that overlies the lesion’s core. Thus, during progression of atheroma, cells not only procreate but perish. Inflammatory mediators participate in both processes. The ultimate clinical complication of atherosclerotic plaques involves disruption that provokes thrombosis, either by fracture of the plaque’s fibrous cap or superficial erosion. The consequent clots can cause acute ischaemic syndromes if they embarrass perfusion. Incorporation of the thrombi can promote plaque healing and progressive intimal thickening that can aggravate stenosis and further limit downstream blood flow. Inflammatory mediators regulate many aspects of both plaque disruption and healing process. Thus, inflammatory processes contribute to all phases of the life cycle of atherosclerotic plaques, and represent ripe targets for mitigating the disease.
Keywords: Atherosclerosis, Coronary artery disease, Leucocytes, Endothelium, Macrophages, Lipids
We now recognize that inflammation contributes to all phases of atherosclerosis. This essay aims to place inflammatory pathways into the broader context of the life cycle of atherosclerotic lesions from initiation to the ultimate thrombotic complications. While we can take justifiable pride in much of the progress in elucidating inflammatory mechanisms in atherosclerosis over the last decades, we need to learn much more and we should remain humble regarding these important knowledge gaps.
1. Inflammation and lesion initiation
Inflammation appears to accompany the earliest phase in generation of atherosclerotic plaques, fatty streak formation (Figure 1A and B). Experimentalists typically provoke atherosclerotic lesion formation in animals by initiating a diet rich in cholesterol and saturated fat, and/or by introducing genetic modifications that elevate atherogenic lipoproteins. Under hyperlipidaemic conditions, lipoprotein particles, often in aggregates, accumulate in the intima. The expression of leucocyte adhesion molecules on the endothelial apical surface ensues, preceding the accumulation of leucocytes in the intima.1
Figure 1.
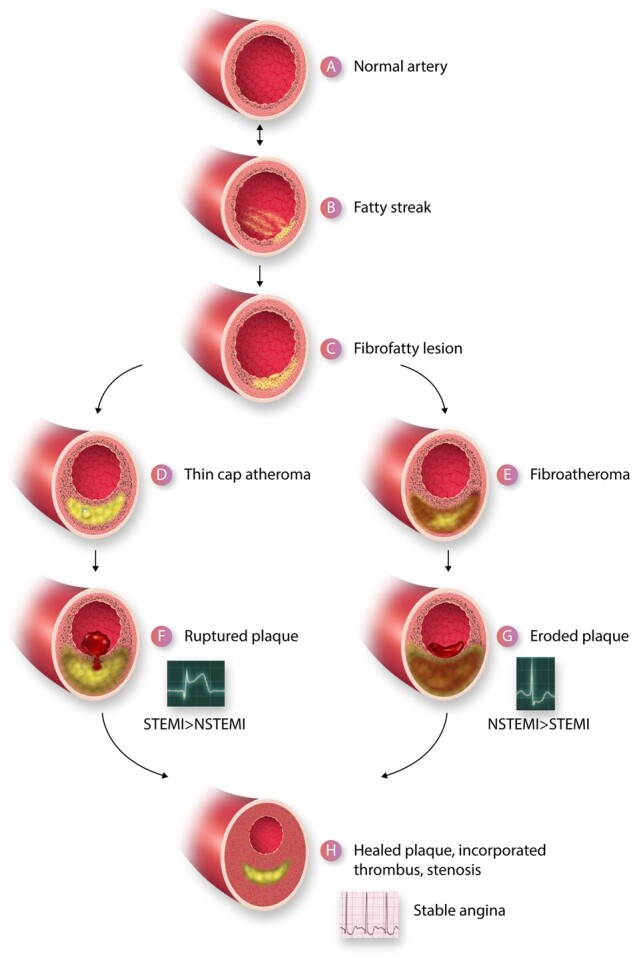
A depiction of the life cycle of an atherosclerotic plaque. (A) Depicts a normal artery. (B) Portrays the initiation of a fatty streak with mononuclear phagocyte recruitment and foam cell formation. The fatty streak may be reversible. (C) A fibrofatty lesion forms when smooth muscle cells lay down extracellular matrix in the intima that enrobes the foam cells. Some smooth muscle cells also accumulate lipid and take on the appearance and markers of foam cells derived from leucocytes of the myeloid series. With further evolution of the plaque, foam cells and smooth muscle cells undergo death and extracellular lipid accumulates from the debris of dying or dead cells and accumulated lipoproteins, forming a lipid core forms in many plaques. A fibrous cap typically forms above the lipid core. Due to decreased synthesis and increased breakdown of extracellular matrix macromolecules such as collagen, this fibrous cap thins creating a thin-capped atheroma (D). Other plaques may evolve to accumulate more matrix and less lipid. For example, the typical substrate of a plaque that has undergone thrombosis due to superficial erosion lacks an organized lipid core, but contains abundant proteoglycan and glycosaminoglycans. This type of plaque does not have a thin fibrous cap and may harbour many smooth muscle cells (E). The thin-capped fibroatheroma can rupture and provoke thrombosis (F). The more fibrous atheromata can undergo erosion (G). The thrombus that complicates eroded plaques is generally an eccentrically located sessile mural thrombus that is more platelet-rich (white thrombus) that the fibrin and erythrocyte rich red thrombus typically provoked by plaque rupture. The ruptured plaque more often gives rise to an ST segment elevation myocardial infarction (STEMI) than a non-ST segment elevation myocardial infarction (NSTEMI) whereas superficial erosion more often gives rise to NSTEMI than STEMI. Regardless of the mechanism of plaque disruption, the resultant thrombi provoke a wound healing response due to elaboration of mediators such as PDGF and TGF-β that augment extracellular matrix synthesis and promote smooth muscle cell migration. Thrombin itself is a mitogen for smooth muscle cells. The incorporation of thrombus provides a provisional matrix replicating within the previously disrupted intima recapitulating the well-known stages of wound healing. The accumulation of extracellular matrix in the healing plaque can lead to increasing luminal stenosis. Such healed, layered plaques tend to have a thicker fibrous cap, less likely to rupture, but these stenotic lesions can give rise to chronic stable angina as depicted by the electrocardiogram characteristic of a positive stress test (H).
In the commonly used animal preparations for study of atherosclerosis, the intimal layer consists of a monolayer of endothelial cells abutting a basement membrane bordered ablumenally by the internal elastic lamina, which forms the border with the tunica media, the artery’s middle layer. Human arteries, however, have a more complex intimal structure preceding the development of atherosclerotic plaques. Even in utero, certain regions of the arterial tree display intimal cushions consisting of smooth muscle cells and extracellular matrix (ECM). Indeed, the human arterial intima generally contains resident smooth muscle cells well before atheroma formation. The proximal left anterior descending coronary artery and carotid siphon, common sites for atheroma formation, may harbour such intimal cushions early in life.2,3 Thus, the human tunica intima furnishes a prepared ‘soil’ for atherogenesis, as pointed out sagely by the late Stephen M. Schwartz.4
Although hyperlipidaemia generally triggers experimental atherosclerosis, in human plaques, we must plead ignorance regarding the stimuli incite endothelial activation and hence the initiation of leucocyte recruitment and the local inflammatory response. Evidence abounds supporting the causality of the key atherogenic lipoprotein low-density lipoprotein (LDL). Indeed, exposure to LDL likely plays a permissive role in human atherosclerosis. Individuals with lifetime lower levels of LDL determined by genetic variants enjoy relative protection from atherosclerosis.5,6 Yet, many atherosclerotic events occur in humans who have LDL concentrations considered ‘average’ or within the accepted normal range.
The mechanisms by which LDL provokes lesion formation remain unsettled, however. In the protected environment of the intima, sequestered from plasma antioxidants, LDL can undergo oxidative modification. Oxidized LDL accumulates in both experimental and human atherosclerotic plaques.7 Oxidized LDL elicits a myriad of pro-atherogenic and pro-inflammatory functions of cells involved in atherogenesis, and can promote atherosclerosis in mice.8 Despite this abundant and convincing body of experimental work and human observations, numerous strategies for combatting oxidative stress in human arteries have not mitigated atherosclerotic events in clinical trials. Perhaps, such interventions come too late in the life cycle of the atherosclerotic plaque to exert a measurable beneficial function.
Other than oxidative modification, LDL can exert a pro-inflammatory function in other ways. Aggregates of LDL, often decorating ECM macromolecules in the intima, may undergo phagocytosis by leucocytes contributing to foam cell formation. Curiously, native LDL appears to activate T lymphocytes more readily than the oxidized lipoprotein.9 Moreover, human observations suggest that LDL itself does not strongly stimulate inflammation, at least as disclosed by elevated concentrations of the inflammatory biomarker C-reactive protein, measured with a high-sensitivity assay (hsCRP).10,11 In contrast, elevated concentrations of triglyceride-rich lipoproteins or remnant lipoprotein particles associate with hsCRP elevations much more strongly than LDL. Thus, while the apolipoprotein B-bearing lipoprotein particles doubtless contribute causally to atherogenesis, remnant lipoproteins appear more pro-inflammatory than LDL itself. These human observations have particular importance in the context of the pandemic of type 2 diabetes, a condition that often associates with elevated concentrations of triglyceride-rich lipoproteins. These triglyceride-rich particles may account for a greater proportion of atherosclerotic risk in people with diabetes type 2 in the current era of increasingly effective control of LDL with the introduction of potent and now inexpensive and generally well-tolerated therapies. How triglyceride-rich lipoproteins exert their pro-inflammatory effect likewise remains unsettled. Apolipoprotein C-III appears to elicit inflammatory responses from cells involved in atherosclerosis, providing one hint in this regard.12–14
In any case, the initiation of atherosclerotic lesions likely depends on more than increased concentration of circulating risk factors such as lipoproteins or systemic risk factors ranging from hypertension, tobacco smoking, air pollution, obesity, disturbed sleep, poor nutrition, and the like. While these factors associated with atherosclerosis risk expose the entire arterial tree, atherosclerotic lesions develop focally. Part of this regionality may reflect the anatomical distribution of the intimal cushions that form early in life in humans.3 The focal formation of incipient atherosclerotic plaques also reflects the local hydrodynamic environment. Laminar shear stress maintains homeostatic and atherothrombosis resistant properties of the endothelium. Disturbed flow activates many functions of endothelial cells that may favour lesion formation.15–17
In addition to expression of adhesion molecules that recruit leucocytes and the elaboration of chemokines that can direct their migration, other endothelial behaviours can contribute to early atherogenesis. In particular, endothelial cells can acquire characteristics of mesenchymal cells, a process known as endothelial-mesenchymal transition (EMT). Endothelial cells that have undergone EMT can migrate into the intima, no longer residing quiescent in an ordered monolayer on the intimal surface.18,19 These endothelial cells that have acquired mesenchymal characteristic may contribute to intimal thickening and inflammation as the atherosclerotic process takes root in the intima.
In sum, despite over a century of experimental probing and exquisite dissection of mechanisms of lesion initiation in experimental animals and in cell culture, we must admit that we lack certitude regarding the mechanisms of initiation of the human atherosclerotic plaque. The optimistic slant on this situation is that we have much to learn about potential targets for intervention that could instruct us in primordial prevention as a way to forestall the ravages of advanced atherosclerosis. Despite acknowledged agnosticism regarding the detailed mechanisms of atherosclerosis initiation, we can today strive to implement healthier lifestyles particularly in youth. Regular physical activity, adopting healthier dietary patterns, avoiding smoking, sugar sweetened beverages, and avoiding obesity could slow or limit atherosclerosis initiation particularly in younger individuals in whom initiating pharmacotherapy presents many challenges. Inheritance places a sketch on the canvas of an individual’s atherosclerotic risk, but behaviours and exposures during life complete the portrait of propensity to develop atherosclerotic events. Thus, the development of polygenic risk scores for predicting atherosclerotic potential early in life may help to direct the intensity of primordial prevention interventions in youth.20
2. Progression of atheroma
2.1 Human atherosclerosis progresses discontinuously in time
Tradition holds that atherosclerosis is a chronic ‘degenerative’ process that progresses continuously and inevitably during ageing. Current evidence suggests a more complex, and perhaps more optimistic view. Yes, atherosclerotic plaques do typically form over many years or even many decades. Yet, a number of lines of evidence suggest that the process by no means progresses monotonically upward.21 Periods of acute evolution appear to punctuate the chronic indolent inflammatory and proliferative process within the typical atherosclerotic plaque. Human arterial imaging studies indicate episodic rather than continuous lesion evolution. Early studies with contrast angiography performed serially indicated an uneven progression of stenoses with time.22,23 Serial intravascular ultrasound studies have likewise demonstrated discontinuous evolution of human atherosclerotic lesions.21
2.2 Triggers for episodic progression of plaques
The triggers for episodes of more rapid progression of established atherosclerotic plaques have become better understood.24 We know that inflammatory processes remote from the atheroma itself can evoke ‘echoes’ within the plaque.25 Infectious processes and a number of stimuli associated with cardiovascular risk can augment haematopoiesis and leucocyte recruitment to the plaque as well as activation of intrinsic vascular cells and leucocytes resident within atheroma.26 Pathogen-associated molecular patterns (PAMPs) and damage associated molecular patterns (DAMPs) elaborated from non-vascular sites of inflammation, infection, or injury can impinge on quiescent arterial intimal lesions and arouse them from and incite a round of inflammatory activation. For example, a remote Gram-negative infection (e.g. of the urinary tract) can cause release into blood bacterial endotoxin (a PAMP) that can elicit an ‘echo’ of local inflammation in a distant atheroma.27 Myocardial cells injured by ischaemic injury or leucocytes recruited to the can elicit the release of soluble mediators such as IL-1β or IL-6 that can travel to arterial lesions and amplify arterial inflammation. Pain and anxiety during an infarction can activate the sympathetic nervous system and cause catecholamine release that can stimulate leucopoiesis in the bone marrow that can foster further accumulation of inflammatory cells in atheroma.28,29 Inflammatory diseases such as rheumatoid arthritis, systemic lupus erythematosis, and psoriasis can likewise release pro-inflammatory mediators from joints and other sites that can augment arterial inflammation and potentially potentiate progression of atherosclerosis.30
2.3 Cellular proliferation and death during atheroma progression
The recognition of inflammation as fundamental to atherogenesis did not always prevail, however. The ability to grow smooth muscle and endothelial cells in fairly homogeneous culture ushered in an era of the cell biological analysis of atherosclerotic plaques. A good deal of atherosclerosis research then focused on the proliferation of arterial smooth muscle cells as a key to lesion growth and evolution, rather than inflammation.31–34 The identification of platelet-derived growth factor (PDGF) galvanized the field in the last quarter of the last century. Initial formulations of the response to injury hypothesis of atherogenesis envisaged denuding injury to the endothelium followed by platelet accumulation and release of PDGF as the initiating event in atherosclerosis. This viewpoint considered lipid accumulation largely secondary, and ascribed a major role to a bland accumulation of smooth muscle cells.32,33 According to this formulation, atherosclerotic plaques resembled a leiomyoma of the artery wall. Evidence for monotypia of smooth muscle cells in human atheromata supported the possibility of clonal expansion of these mesenchymal cells.35–37 Defining that the simian sarcoma virus oncogene v-sis encoded the B chain of PDGF supported this focus on smooth muscle proliferation as primordial in atherogenesis.38 Eventually, the recognition of inflammatory cells by microscopic study and the use of specific monoclonal antibodies characterized more rigorously various cell types in lesions and paved the way for a greater recognition of inflammatory contributions to atherogenesis.
We now view cellular accumulation not merely as the result of proliferation, but as the net sum of proliferation, migration, and cell death. Smooth muscle cells can migrate into the intima from the tunica media, joining the resident smooth muscle cells found in the typical human intimal layer, adding to their accumulation. Smooth muscle proliferation and migration clearly participate in the hyperplastic complications of arterial interventions such as angioplasty and stenting. Mononuclear phagocytes, the most abundant leucocyte in the atherosclerotic plaque, also undergo proliferation and death as well as ongoing recruitment. Experimental studies in mice suggest that monocyte recruitment predominates in the initial phases of atherogenesis while local proliferation typifies the established atherosclerotic plaque.39 Knowledge of leucocyte kinetics in human plaques is much less complete.
2.4 Inflammation modulates extracellular matrix metabolism during atheroma progression
Much of the volume of human atherosclerotic plaque consists of ECM. Smooth muscle cells elaborate much of the plaque’s ECM. The composition of the matrix and its regulation have undergone vast study. Transforming growth factor beta (TGF-β) and allied mediators strongly augment the production of interstitial collagens and other matrix macromolecules from smooth muscle cells.40 In addition to collagen, elastin, proteoglycan, and glycosaminoglycans contribute to the intimal ECM. These matrix constituents retard the emigration of lipoprotein particles and can contribute to their aggregation and modification. As with cellular accumulation, the build-up of intimal ECM depends not only on its synthesis but its breakdown as well. The mechanisms of ECM breakdown and its regulation by inflammatory processes have undergone extensive study.41–43 Breakdown of interstitial collagen contributes to the thinning of the plaque’s fibrous cap (Figure 1D). A trio of specialized interstitial collagenases catalyse the breakdown of this usually very stable molecule. These enzymes belong to the matrix metalloproteinase (MMP) family: MMPs 1,8, and 13 (Figure 2). After initial cleavage by these collagenases, gelatinases such as MMP-2 and MMP-9 continue collagen catabolism. Pro-inflammatory cytokines induce the expression and in some cases activation of the zymogens of these collagen-degrading MMPs.44–46 The growth of human atherosclerotic plaques for much of the life history is outward. Luminal encroachment occurs later in the history of the plaque. The outward remodelling likely involves ECM breakdown as well.
Figure 2.

Contrasting mechanisms of plaque disruption due to fibrous cap rupture vs. superficial erosion. These two forms of plaque disruption involve distinct vascular cellular protagonists: Smooth muscle cells produce the interstitial collagens that lend strength to the plaque’s fibrous cap. Plaque regions depleted of smooth muscle cells have impaired ability to repair and maintain the collagenous extracellular matrix of the plaque’s protective fibrous cap. In contrast, endothelial cell death and desquamation proves pivotal in plaque disruption due to superficial erosion. The interstitial collagenases, matrix metalloproteinases (MMPs) -1, 8, and -13, attack the fibrillar collagen (types I and III) mostly produced by smooth muscle cells that protect the plaque from rupture. In contrast, the Type IV collagenases, MMPs -2 and -9, degrade the non-fibrillar Type IV collagen in the basement membrane underlying the endothelial cell monolayer thus dissolving the substrate on which endothelial cells attach to the intimal surface. Deprived of their extracellular matrix substrate, endothelial cells can undergo death by anoikis and slough more readily. Oxidative stress due to hypochlorous acid (HOCl) generated by myeloperoxidase or superoxide anion ( produced by NADPH oxidase can damage the endothelial monolayer and promote desquamation. Pro-inflammatory cytokines including interleukin-1β, tumour necrosis factor, and CD40 ligand participate in plaque rupture by inducing the interstitial collagenases and tissue factor, the instigator of thrombosis in ruptured plaques. In superficial erosion, NETs contribute to thrombosis and can amplify and propagate endothelial damage through NET-associated interleukin-1α. The main cellular effectors of rupture vs. erosion differ as well, foam cells derived from monocytes or smooth muscle cells predominate in the pathophysiology of fibrous cap rupture. These cells can eventually die from apoptosis or oncosis. In superficial erosion, polymorphonuclear leucocytes appear prominent and can undergo NET formation. Activated platelets contribute to thrombus formation and growth in both forms of plaque disruption. However, thrombi produced by plaque rupture appear more fibrin rich, entrapping erythrocytes, forming ‘red’ thrombi. In contrast, plaques disrupted by erosion tend to generate platelet-rich ‘white’ thrombi. As noted in Figure 1, fibrous cap rupture causes ST segment elevation (STEMI) more commonly than non-ST segment myocardial infarction (NSTEMI) whereas eroded lesions more frequently associate with NSTEMI than STEMI.
A complication of atherosclerosis, aneurysm formation—outward remodelling taken to an extreme—doubtless entails elastinolysis. Fragmentation of elastic lamina characterizes human abdominal aortic aneurysms. In addition to matrix metalloelastase (MMP-12) and neutrophil elastase (a serine proteinase, also expressed by macrophages47), a separate series of elastolytic enzymes regulate elastin breakdown. The cysteinyl proteinases cathepsins S, K, and L possess potent elastinolytic properties and participate in atherogenesis and aneurysm formation.48 Pro-inflammatory cytokines elaborated by vascular cells and lesional leucocytes can stimulate not only the production of members of the MMP family specialized in catabolism of collagen but also of elastin. For example, interferon-gamma elicits the overproduction of members of the sulfhydryl cathepsin family particularly adept at elastin breakdown. The balance between the endogenous inhibitors of these various classes of proteolytic enzymes and the proteinases themselves ultimately regulates all of these aspects of arterial remodelling involved in atherosclerosis.
As in the case of smooth muscle cells and matrix, the contributions of inflammatory cells during progression of atheroma involves a tightly orchestrated balance between pro-inflammatory and anti-inflammatory functions. In addition to the well-known menu of pro-inflammatory cytokines and chemokines, vascular cells and lesional leucocytes can elaborate mediators that mitigate inflammation. TGF-β itself can exert anti-inflammatory actions. Among anti-inflammatory cytokines, Interleukin (IL)-4 and IL-10 can mute inflammation. While endothelial cell nitric oxide can exhibit anti-inflammatory actions, uncoupling of endothelial nitric oxide synthase or induction of high-capacity forms of nitric oxide synthase in leucocytes, particularly in the presence of certain reactive oxygen species, can give rise to highly pro-oxidant species such as peroxynitrate (ONOO-).
2.5 An inflammatory balance prevails during atheroma progression
The adaptive immune cells in plaques can prove pro-inflammatory or anti-inflammatory.49,50 ‘Classical’ mononuclear phagocytes elaborate typically pro-inflammatory cytokines such as IL-1 and tumour necrosis factor while reparative macrophages can elaborate IL-4 and IL-10. T-helper 1 lymphocytes (Th1) typically produce interferon gamma (IFNγ), which can activate mononuclear phagocytes and thus promote inflammation. The role of Th17 cells in atherosclerosis remains controversial.51 Various leucocyte subclasses can mute inflammation by production of anti-inflammatory mediators including ILs-4, 5, 9, 10, TGF-β, and specialized pro-resolving lipid molecules.52 For example, regulatory T and B cells produce TGF-β and IL-10. Th2 lymphocytes typically secrete some of the anti-inflammatory cytokines mentioned above.53,54 B lymphocytes as well can augment or moderate lesion growth and evolution.55,56 B1 lymphocytes produce natural antibodies, which can combat atherogenesis. B2 cells on the balance can aggravate atherosclerosis.
In addition to anti-inflammatory mediators, a class of well-characterized lipid mediators may promote resolution of inflammation distinct from an anti-inflammatory aspect.57,58 This distinction has importance as anti-inflammatory mediators may interfere with host defences and augment susceptibility to infection and interfere with tumour surveillance. Boosting pro-resolving mediators could mitigate atherosclerosis with less liability for impaired host defences.59
As noted above, cell death opposes proliferation of both smooth muscle cells and mononuclear phagocytes. Current understanding suggests that smooth muscle cells may also give rise to foam cells within atherosclerotic plaques that masquerade as macrophages.60,61 Dead and dying cells and their debris coalesce in the core of the plaque underlying the fibrous cap made of extracellular matrix molecules elaborated by smooth muscle cells (Figure 1D). The clearance of dead and dying cells, a process known as efferocytosis, can limit the size of the necrotic core according to mouse experiments.62 Defective clearance of dead cells can favour the expansion of the plaque’s necrotic or lipid core.
Plaque disruption, often due to rupture of the fibrous cap that overlies the lipid core, can cause many acute thrombotic complications of atherosclerosis as discussed below (Figures 1F and G and 2). Yet, many and perhaps most plaque disruptions will not cross the threshold of clinical manifestation but will remain silent. Foci of formation of thrombus, platelet degranulation, neutrophil recruitment and activation can however stimulate a ‘crisis’ in the history of a given atherosclerotic plaque. Even without causing occlusive thrombus formation or even eliciting clinical symptoms, such events can elicit a healing response.63 Thrombin can stimulate smooth muscle cell proliferation and matrix production. As noted above, during thrombosis, platelets release TFG-β and PDGF that can stimulate further smooth muscle cell migration and matrix elaboration.
Many advanced atherosclerotic plaques in humans show evidence for a prior plaque disruption as evidenced by a ‘buried cap’ (Figures 1H and 3).64 Often a layer of ECM, bearing markers of more recently laid-down collagen, overlies the site of prior disruption. In some cases, a neo-lipid core can form above the site of prior fibrous cap fracture. Imaging with optical coherence tomography has demonstrated the presence of such ‘layered’ plaques in humans in vivo as well.63,65,66 Such cycles of disruption followed by healing can account for some of the discontinuities in the evolution of human atherosclerotic plaques observed in imaging studies described above. The concept of incorporated thrombus harkens back to the concept of Rokitansky in the mid-nineteenth century, a viewpoint that opposed the concept promulgated by Virchow regarding inflammation and cellular proliferation.67 We now recognize that both processes participate in the progression of human atherosclerotic plaques.68
Figure 3.

Shows a healed plaque with different strata of an intima without a lipid-rich core but plates of calcium, areas of neovascularization, and a layer of extracellular matrix probably laid down after a plaque disruption (labelled Healing Intima, surrounded by a corona of microvessels), yielding a substantial narrowing of the coronary arterial lumen. Indicated are components of this healed plaque. There are also calcium plates at 5 and 7 O’clock in the intima. Richard N. Mitchell MD, PhD, Department of Pathology, Brigham and Women’s Hospital, generously provided this photograph..
Within the atheroma, cells can undergo senescence, a process associated with inflammatory mediators as part of the senescence-associated secretory pathway (SASP).69,70 In particular, IL-6, a pro-inflammatory cytokine implicated causally in human atherogenesis, typifies the SASP. Genetic manipulation to forestall cellular senescence or the administration of ‘senolytics’ may mitigate the progression of atherosclerotic plaques. This concept provides a relatively new wedge into control of plaque progression.71
Indeed, among the risk factors for atherosclerosis, ageing ranks first. The recent recognition that somatic mutations in bone marrow stem cells can give rise to clones of mutant myeloid cells in the peripheral blood provides a new link between ageing and atherosclerosis.72 The genes mutated in this condition, denoted clonal haematopoiesis of indeterminate potential, are known driver genes for leukaemia. Individuals who harbour these mutant clones, however, have a much higher risk of cardiovascular disease than of developing acute leukaemia.73,74 A number of lines of evidence link inflammatory mediators to the accelerated atherosclerosis associated with clonal haematopoiesis.73,75,76
As plaques progress, they often develop calcification. In experimental atheromata, foci of calcification co-localize with indices of inflammation.77 Current concepts accord a seeding role to microparticles secreted by smooth muscle cells to nucleate calcium mineral accumulation.78 Initially distributed as punctate microscopic accumulations of calcium mineral, sometimes attributed macrophage death, such smaller accumulation of calcium mineral can coalesce, and form calcified plaques. Punctate calcification as visualized on computed tomographic studies associates with clinical events along with imaging evidence of outward remodelling and accumulation of lipid cores. Assessment of coronary artery calcification by radiographic techniques provides a powerful prospective marker of cardiovascular events.79 Yet we currently possess no tools for reducing calcification. Indeed, the statin class of drugs which markedly lower LDL and strikingly decrease cardiovascular events actually increase coronary calcification.80,81 The study of the mechanisms of calcification and its potential modulation provides yet another opportunity for intervention into plaque progression, which is currently unexploited.
3. Inflammation and the acute complications of atherosclerosis
The acute manifestations of coronary atherosclerosis bring patients to the attention of physicians most dramatically. These events arise typically because of thrombosis. Until recently, the acute coronary syndromes encompassed unstable angina pectoris, non-ST-segment elevation myocardial infarction (NSTEMI), and ST-segment elevation myocardial infarction (STEMI) (Figure 1F and G). It is curious that we use a technology that emerged in the early twentieth century to classify myocardial infarction based on the pattern of repolarization on the scalar electrocardiogram introduced in 1904, and for which, Einthoven received a Nobel Prize in 1924, nearly a century ago.82 This situation illustrates that we have considerable room for improvement in our classification and diagnosis of acute coronary syndromes. The categorization of unstable angina has undergone re-evaluation in the current era and is near extinction.83 With assays of biomarkers of myocardial injury of increasing sensitivity, notably the more recent fifth-generation troponin assays, episodes of accelerated ischaemic chest discomfort (angina pectoris) now most often meet criteria for NSTEMI (detection of an elevated cardiac troponin value above the 99th percentile upper reference limit). Moreover, in the current era, patients with accelerating angina pectoris generally undergo revascularization which typically alleviates these symptoms.
Based on autopsy studies, much attention has focused on the rupture of the plaque’s fibrous cap as a trigger to most coronary thrombosis. Indeed, a fracture or fissure of the plaque’s fibrous cap permits the blood with its latent coagulation proteins contact with thrombogenic material in the lipid core, including the potent pro-coagulant tissue factor, thus triggering thrombus formation (Figure 1F). Such events occur most often in plaques with a thin fibrous cap (< 65 µ) overlying the lipid core.84 Plaques with such morphology have been denoted thin-capped fibroatheroma (TCFA), often referred to as ‘vulnerable plaques.’ Autopsy studies consistently identified plaques of this morphology as cause of most fatal myocardial infarctions. Yet, autopsy studies cannot disclose how many plaques with the thin-capped morphology do not cause a fatal event. Such information has emerged from human intravascular imaging studies. In the landmark PROSPECT study, plaques with the thin-capped morphology did not cause a clinical event more than 95% of the time during a greater than three-year follow-up period.85 This result challenges the very nomenclature ‘vulnerable plaque’ to describe thin-capped lesions, as less than 5% of such lesions would provoke an event during follow-up. Moreover, the autopsy studies that disclosed fracture of the fibrous cap as a cause of fatal myocardial infarction were conducted in a period before widespread use of effective preventive therapies such as statins.86
Indeed, we have argued that human atherosclerosis is changing in the current era of increasingly effective control of LDL, hypertension, smoking cessation, and other preventive interventions.87 Human atherosclerotic plaques now show less lipid accumulation and fewer inflammatory cells than in the past.88 Human imaging studies likewise show that effective LDL lowering can shrink lipid cores, and in some cases reduce lesion volume, yielding actual plaque regression.89–91 Studies with magnetic resonance imaging of human carotid plaques show that LDL reduction associates with an increased proportion of plaques made up of ECM, an alteration that corresponds to reinforcement of the fibrous cap demonstrated in experimental studies of rabbits and mice subjected to lipid lowering interventions.92,93 Thus, as therapeutics advance and dissemination of preventive therapies increase, plaque rupture may be on the wane.
Beyond rupture of the plaque’s fibrous cap, pathologists have long recognized that another mechanism of disruption denoted superficial erosion causes a large minority of fatal acute coronary events.94,95 Recent observations have highlighted the growing significance of superficial erosion as a mechanism of plaque disruption (Figures 1G and 2).96–100 Lesions that have caused coronary events due to erosion have morphologic characteristics in some ways diametrically opposed to the TCFA (Figure 2). Eroded plaques show an intact fibrous cap, and indeed are matrix rich rather than depleted of collagen as TCFAs. They contain smooth muscle cells but relatively few leucocytes. The thrombi associated with superficial erosion display characterization of ‘white’ or platelet-rich thrombi as opposed to the ‘red’ fibrin and erythrocyte-rich thrombi more often associated with plaque rupture.
Contemporary studies using optical coherence tomography (OCT), which can readily classify culprit lesions of acute coronary syndromes as ruptured indicate that erosion causes approximately a third of acute coronary syndromes in the current era.101 In addition, plaques classified by OCT as not ruptured or as definite erosion associate more frequently with NSTEMI rather than STEMI.88 The clinical presentation of acute coronary syndromes has under gone a shift in recent decades. NSTEMI has surpassed STEMI. This crossover began before the introduction of the current generation of highly sensitive component assay, so it does not result solely from a reclassification of unstable angina to NSTEMI. ACS due to erosion associates more commonly with NSTEMI than STEMI (Figures 1E and 4). Thus, the waning proportion of ACS due to STEMI may reflect more erosion in the current era of effective preventive therapies that can improve characteristics of plaques associated with rupture.
Figure 4.

This specimen of a human atherosclerotic aorta shows that ulcerated lesions harbouring a thrombus can coexist with raised fibrous lesions shown in pale yellow and atheromata that more resemble a fatty streak. This aorta also developed a small saccular aneurysm. These simple morphological observations show the idea that an individual’s plaques share the same stage in the life cycle is erroneous, as plaques of different characteristics can coexist in close proximity as demonstrated here. Richard N. Mitchell MD, PhD, Department of Pathology, Brigham and Women’s Hospital, generously provided by this photomicrograph.
Recent work has augmented understanding of the mechanisms of plaque disruption and its links to inflammation. Heightened activity of collagenolytic enzymes and reduced ability of smooth muscle cells to synthesize new collagen due to exposure to IFNγ secreted by Th1 lymphocytes in the plaque a thin and friable fibrous cap liable to rupture.41 We have demonstrated that lipid lowering reduces expression of collagenolytic enzymes and reduces tissue factor pro-coagulant expression by cells found in atheromata.
The mechanisms of superficial erosion likely involve an initial activation of endothelial cells to sensitize them to death or desquamation. We have explored one pathway of endothelial activation, ligation of the pattern recognition receptor Toll-like receptor 2 (TLR2) perhaps in response to endogenous ligands such as lower molecular weight fragments of hyaluronic acid. Plaques that have undergone superficial erosion exhibit particularly high concentrations of this glycosaminoglycan.102 In vitro, TLR2 stimulation sensitizes endothelial cells to apoptosis, impairs their ability to repair a wounded monolayer, and absence of TLR2 prevents impairment of endothelial integrity in experimental preparations.103
Our observations in human atherosclerotic plaques associate neutrophil extracellular traps (NETs) with plaques with erosive morphology more commonly than those with the rupture-prone characteristics.102 NETs can augment endothelial injury, amplifying and extending erosive damage in experimental preparations, and promote thrombosis.104,105 NETs present myeloperoxidase at the intimal surface providing a generator of the pro-oxidant species hypochlorous acid which can provoke endothelial cell apoptosis and induce tissue factor expression.106,107 Interleukin-1 alpha (IL-1α) associated with NETs can likewise induce tissue factor expression on endothelial cells and elicit leucocyte adhesion molecule expression (Figure 4).108 We and others have implicated cathepsin G in augmenting local IL-1α activity associated with NETs.108,109 This granulocyte-derived serine proteinase itself associates with NETs.
The enzyme peptidyl arginine deiminase-4 (PAD4) participates in NET formation by inverting basic arginine to neutral citrulline, disturbing the ionic bonds that restrain DNA to the histone multimers in nucleosomes.110 Our studies in experimental preparations devised to replicate some of the characteristics of plaque superficial erosion show that genetic or chemical inhibition of PAD4 activity can limit endothelial damage in arterial regions that replicate features of superficial erosion.111,112
Adaptive immunity may also contribute to superficial erosion. In human studies led by Ulf Landmesser, CD8 lymphocytes appear more commonly associated with lesions characterized as non-rupture or erosion by OCT vs. those classified as culprits that have undergone plaque rupture.113 Local concentrations of the effector enzymes granzyme B and perforin increase at sites of ACS culprit lesions classified as erosion by OCT. Thus, mediators derived from CD8 ‘killer’ T lymphocytes specialized in cell damage may likewise propagate endothelial injury and promote erosive complications of human plaques.
4. Therapeutic implications
We have made much progress in therapeutics to modify atherogenesis and its complications. I have summarized some of these contemporary therapeutic interventions above. Despite current preventive measures and increasingly effective revascularization interventions, the residual risk in individuals with manifest atherosclerosis remains unacceptably high. Particularly in individuals with advanced age or multiple comorbidities, recurrent acute coronary syndromes can occur in a fifth of individuals despite optimum standard of care therapy in the first year.114 While we take satisfaction in our current armamentarium for combatting atherosclerosis, much remains undone.115 The introduction of newer therapies that can drive LDL concentrations even lower than statins alone and anti-diabetic medications such as the glucagon-like peptide-1 receptor agonists and the sodium-glucose cotransporter-2 inhibitors (SGLT-2i) may provide enhanced protection from first ever or recurrent atherosclerotic complications.
The advent of an era of anti-inflammatory therapies provides another potential avenue for interventions orthogonal to LDL lowering that may improve further the risk that persists despite current standard of care.116,117 The demonstration that IL-1 beta neutralization reduced recurrent cardiovascular events in individuals with atherosclerosis provided the first proof in humans of the inflammatory contribution to this disease.118,119 The subsequent demonstration in two large independent studies that colchicine can reduce recurrent atherothrombotic events reinforces this opportunity.120,121 Yet, finding the ‘sweet spot’ of limiting pathways of inflammation relevant to atherosclerosis, while avoiding undue interruption of host defences or producing unwanted actions, provides an opportunity for further innovation in this regard. Anti-inflammatory interventions under consideration for atherosclerosis include neutralization of IL-6 and the inflammasome.122–125
In the lipid arena, targeting triglyceride-rich lipoproteins with novel therapeutics such as monoclonal antibodies, RNA therapeutics, or selective PPARα modulators merits evaluation as additional strategies for reducing residual risk. As noted above, primordial prevention depends largely on public health measures. Perhaps, atherosclerosis would become an orphan disease were lifestyle measures adopted more stringently and if targeted pharmacotherapy were deployed. Indeed, physical activity and diet may exert some of their benefits on cardiovascular outcomes by muting inflammation.126,127 It matters little if we have effective therapies to prevent atherosclerosis and its complications if they do not penetrate into practice. We need to learn from modern behavioural economics and implementation science to overcome barriers in this regard. Moreover, access to preventive therapies encounters many barriers due to health disparities and societal inequities. In addition to our scientific mission, health professionals bear a responsibility to strive to close these gaps to bring the fruits of our research in an equitable fashion to all segments of society.
5. Conceptual implications for moving forward
As in therapeutics, we can take considerable pride in the success of our enterprise in unravelling the pathogenesis of atherosclerosis and its complications. Yet, even with contemporary powerful methodology and investigative approaches, much remains to be done. This essay has focused on inflammatory aspects that govern the life cycle of an atherosclerotic plaque, but in reality, the human arterial tree typically contains lesions at all stages dealt with herein (Figure 4). One can commonly see a fatty streak adjacent to a raised plaque nearby an ulcerated lesion with evidence of thrombosis. Thus, plaques at different points in their life cycle coexist even within centimetres in a human arterial tree. This observation indicates how oversimplified is the categorization into phases of initiation, progression, and complication used to organize this discussion. Such schemata that imply a linear progression of atherosclerosis seriously underestimate the complexity of the clinical disease in a human individual. In our experimental preparations, we often turn on the stimulus for atherogenesis like throwing a light switch. Yet, atherogenic risk factors, behaviours, and exposures bombard humans in a highly dispersed distribution in time.
The discussion above describes subtypes of various lesions. But single-cell technology and high-dimensional -omics including proteomics and cyTOF have revealed a much greater heterogeneity than generally acknowledged and appreciated in the past.128,129 The application of these technologies is providing finer and finer divisions of subpopulations of various cells and has even blurred the identity of different cell types. Examples include the ability of endothelial to undergo EMT, and of smooth muscle cells to undergo metaplasia into foam cells with characteristics of mononuclear phagocytic leucocytes. This fine splitting of cell types is currently in a primarily descriptive phase. We have much work to do to ascribe functions to the various subtypes of cells revealed by high-dimensional-omic technologies. We have previously noted that the reductio ad absurdum of this rush to catalogue cellular heterogeneity would classify each cell as its own subtype.68 As remarked, while quite possibly true as a limiting case, this concept is not particularly helpful or actionable from a therapeutic perspective. We face the challenge of harnessing these novel and powerful technologies to ascribe functions to various cell types and unravel ways in which to address the balance for their pro- and anti-atherosclerotic actions.
We also must strive to avoid glib extrapolations of animal data to human atherosclerosis.130,131 We previously highlighted a tendency to gloss over issues of expression of various functions of cells involved in atherogenesis in space and in time. Moreover, many of our animal preparations involve manipulations that create degrees of hyperlipidaemia far greater than those encountered in contemporary clinical practice. While practically necessary to achieve results within a reasonable period of time, we should remain cognizant that such exaggerated hyperlipidaemia may not reflect the rather indolent inflammatory process underway in the human atherosclerotic plaque. Mouse and cell culture experiments provide necessary and extremely useful avenues to probing various mechanisms and principles. However, we must always strive to acknowledge the complexity of human atherosclerosis and exercise humility in interpreting the results of our experiments to the human situation. Combining the best of laboratory science with reference to the human disease as a touchstone provides a potentially productive path forward to close the gaps in our understanding and improve cardiovascular wellness. Reduction to practice of the burgeoning knowledge of inflammation in the life cycle of atherosclerotic plaques promises to help achieve this goal.117
Funding
Dr. Libby receives funding support from the National Heart, Lung, and Blood Institute (1R01HL134892), the American Heart Association (18CSA34080399), the RRM Charitable Fund, and the Simard Fund.
Conflict of interest: P.L. is an unpaid consultant to, or involved in clinical trials for, Amgen, AstraZeneca, Baim Institute, Beren Therapeutics, Esperion Therapeutics, Genentech, Kancera, Kowa Pharmaceuticals, Medimmune, Merck, Novo Nordisk, Novartis, Pfizer, and Sanofi-Regeneron. P.L. is a member of scientific advisory board for Amgen, Caristo, Cartesian, Corvidia Therapeutics, CSL Behring, DalCor Pharmaceuticals, Dewpoint, Kowa Pharmaceuticals, Olatec Therapeutics, Medimmune, Novartis, PlaqueTec, and XBiotech, Inc. P.L.’s laboratory has received research funding in the last 2 years from Novartis. P.L. is on the Board of Directors of XBiotech, Inc. P.L. has a financial interest in Xbiotech, a company developing therapeutic human antibodies. P.L.'s interests were reviewed and are managed by Brigham and Women's Hospital and Partners HealthCare in accordance with their conflict-of-interest policies.
Data availability
No new data were generated or analysed in support of this research.
This article is part of the Spotlight Issue on Cardiovascular Immunology.
References
- 1. Li H, Cybulsky MI, Gimbrone MA Jr, Libby P. An atherogenic diet rapidly induces VCAM-1, a cytokine regulatable mononuclear leukocyte adhesion molecule, in rabbit endothelium. Arterioscler Thromb 1993;13:197–204. [DOI] [PubMed] [Google Scholar]
- 2. Weninger WJ, Müller GB, Reiter C, Meng S, Rabl SU. Intimal hyperplasia of the infant parasellar carotid artery. Circ Res 1999;85:970–975. [DOI] [PubMed] [Google Scholar]
- 3. Nakashima Y, Chen Y-X, Kinukawa N, Sueishi K. Distributions of diffuse intimal thickening in human arteries: preferential expression in atherosclerosis-prone arteries from an early age. Virchows Archiv 2002;441:279–288. [DOI] [PubMed] [Google Scholar]
- 4. Schwartz SM, deBlois D, O'Brien ER. The intima: soil for atherosclerosis and restenosis. Circ Res 1995;77:445–465. [DOI] [PubMed] [Google Scholar]
- 5. Cohen JC, Boerwinkle E, Mosley TH, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 2006;354:1264–1272. [DOI] [PubMed] [Google Scholar]
- 6. Borén J, Chapman MJ, Krauss RM, Packard CJ, Bentzon JF, Binder CJ, Daemen MJ, Demer LL, Hegele RA, Nicholls SJ, Nordestgaard BG, Watts GF, Bruckert E, Fazio S, Ference BA, Graham I, Horton JD, Landmesser U, Laufs U, Masana L, Pasterkamp G, Raal FJ, Ray KK, Schunkert H, Taskinen M-R, van de Sluis B, Wiklund O, Tokgozoglu L, Catapano AL, Ginsberg HN. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J 2020;41:2313–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ylä-Herttuala S, Palinski W, Rosenfeld ME, Parthasarathy S, Carew TE, Butler S, Witztum JL, Steinberg D. Evidence for the presence of oxidatively modified low density lipoprotein in atherosclerotic lesions of rabbit and man. J Clin Invest 1989;84:1086–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Que X, Hung M-Y, Yeang C, Gonen A, Prohaska TA, Sun X, Diehl C, Määttä A, Gaddis DE, Bowden K, Pattison J, MacDonald JG, Ylä-Herttuala S, Mellon PL, Hedrick CC, Ley K, Miller YI, Glass CK, Peterson KL, Binder CJ, Tsimikas S, Witztum JL. Oxidized phospholipids are proinflammatory and proatherogenic in hypercholesterolaemic mice. Nature 2018;558:301–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gistera A, Klement ML, Polyzos KA, Mailer RKW, Duhlin A, Karlsson MCI, Ketelhuth DFJ, Hansson GK. Low-density lipoprotein-reactive T cells regulate plasma cholesterol levels and development of atherosclerosis in humanized hypercholesterolemic mice. Circulation 2018;138:2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Varbo A, Benn M, Tybjærg-Hansen A, Nordestgaard BG. Elevated remnant cholesterol causes both low-grade inflammation and ischemic heart disease, whereas elevated low-density lipoprotein cholesterol causes ischemic heart disease without inflammation. Circulation 2013;128:1298–1309. [DOI] [PubMed] [Google Scholar]
- 11. Hansen SEJ, Madsen CM, Varbo A, Nordestgaard BG. Low-grade inflammation in the association between mild-to-moderate hypertriglyceridemia and risk of acute pancreatitis: a study of more than 115000 individuals from the general population. Clin Chem 2019;65:321–332. [DOI] [PubMed] [Google Scholar]
- 12. Libby P. Fat fuels the flame: triglyceride-rich lipoproteins and arterial inflammation. Circ Res 2007;100:299–301. [DOI] [PubMed] [Google Scholar]
- 13. Huff MW, Hegele RA. Apolipoprotein C-III: going back to the future for a lipid drug target. Circ Res 2013;112:1405–1408. [DOI] [PubMed] [Google Scholar]
- 14. Norata GD, Tsimikas S, Pirillo A, Catapano AL. Apolipoprotein C-III: from pathophysiology to pharmacology. Trends Pharmacol Sci 2015;36:675–687. [DOI] [PubMed] [Google Scholar]
- 15. Gimbrone MA, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res 2016;118:620–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baeyens N, Bandyopadhyay C, Coon BG, Yun S, Schwartz MA. Endothelial fluid shear stress sensing in vascular health and disease. J Clin Invest 2016;126:821–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Souilhol C, Serbanovic-Canic J, Fragiadaki M, Chico TJ, Ridger V, Roddie H, Evans PC. Endothelial responses to shear stress in atherosclerosis: a novel role for developmental genes. Nat Rev Cardiol 2020;17:52–63. [DOI] [PubMed] [Google Scholar]
- 18. Helmke A, Casper J, Nordlohne J, David S, Haller H, Zeisberg E, von Vietinghoff S. Endothelial-to-mesenchymal transition shapes the atherosclerotic plaque and modulates macrophage function. FASEB J 2019;33:2278–2289. fj201801238R. [DOI] [PubMed] [Google Scholar]
- 19. Wesseling M, Sakkers TR, de Jager SCA, Pasterkamp G, Goumans MJ. The morphological and molecular mechanisms of epithelial/endothelial-to-mesenchymal transition and its involvement in atherosclerosis. Vascul Pharmacol 2018;106:1–8. [DOI] [PubMed] [Google Scholar]
- 20. Aragam KG, Natarajan P. Polygenic scores to assess atherosclerotic cardiovascular disease risk. Circ Res 2020;126:1159–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhao Z, Witzenbichler B, Mintz GS, Jaster M, Choi SY, Wu X, He Y, Margolis MP, Dressler O, Cristea E, Parise H, Mehran R, Stone GW, Maehara A. Dynamic nature of nonculprit coronary artery lesion morphology in STEMI: a serial IVUS analysis from the HORIZONS-AMI trial. JACC Cardiovasc Imaging 2013;6:86–95. [DOI] [PubMed] [Google Scholar]
- 22. Nobuyoshi M, Kimura T, Nosaka H, Mioka S, Ueno K, Yokoi H, Hamasaki N, Horiuchi H, Ohishi H. Restenosis after successful percutaneous transluminal coronary angioplasty: serial angiographic follow-up of 229 patients. J Am Coll Cardiol 1988;12:616–623. [DOI] [PubMed] [Google Scholar]
- 23. Bruschke AV, Kramer J Jr, Bal ET, Haque IU, Detrano RC, Goormastic M. The dynamics of progression of coronary atherosclerosis studied in 168 medically treated patients who underwent coronary arteriography three times. Am Heart J 1989;117:296–305. [DOI] [PubMed] [Google Scholar]
- 24. Libby P, Nahrendorf M, Swirski FK. Leukocytes link local and systemic inflammation in ischemic cardiovascular disease. J Am Coll Cardiol 2016;67:1091–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science 2013;339:161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schloss MJ, Swirski FK, Nahrendorf M. Modifiable cardiovascular risk, hematopoiesis, and innate immunity. Circ Res 2020;126:1242–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Libby P, Loscalzo J, Ridker PM, Farkouh ME, Hsue PY, Fuster V, Hasan AA, Amar S. Inflammation, immunity, and infection in atherothrombosis: JACC Review Topic of the Week. J Am Coll Cardiol 2018;72:2071–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dutta P, Courties G, Wei Y, Leuschner F, Gorbatov R, Robbins CS, Iwamoto Y, Thompson B, Carlson AL, Heidt T, Majmudar MD, Lasitschka F, Etzrodt M, Waterman P, Waring MT, Chicoine AT, van der Laan AM, Niessen HWM, Piek JJ, Rubin BB, Butany J, Stone JR, Katus HA, Murphy SA, Morrow DA, Sabatine MS, Vinegoni C, Moskowitz MA, Pittet MJ, Libby P, Lin CP, Swirski FK, Weissleder R, Nahrendorf M. Myocardial infarction accelerates atherosclerosis. Nature 2012;487:325–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sager HB, Heidt T, Hulsmans M, Dutta P, Courties G, Sebas M, Wojtkiewicz GR, Tricot B, Iwamoto Y, Sun Y, Weissleder R, Libby P, Swirski FK, Nahrendorf M. Targeting interleukin-1beta reduces leukocyte production after acute myocardial infarction. Circulation 2015;132:1880–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mason JC, Libby P. Cardiovascular disease in patients with chronic inflammation: mechanisms underlying premature cardiovascular events in rheumatologic conditions. Eur Heart J 2015;36:482–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ross R, Glomset JA. Atherosclerosis and the arterial smooth muscle cells. Science 1973;180:1332–1339. [DOI] [PubMed] [Google Scholar]
- 32. Ross R, Glomset JA. The pathogenesis of atherosclerosis I. N Eng J Med 1976;295:369–377. [DOI] [PubMed] [Google Scholar]
- 33. Ross R, Glomset JA. The pathogenesis of atherosclerosis II. N Engl J Med 1976;295:420–425. [DOI] [PubMed] [Google Scholar]
- 34. Chamley-Campbell J, Campbell GR, Ross R. The smooth muscle cell in culture. Physiol Rev 1979;59:1–61. [DOI] [PubMed] [Google Scholar]
- 35. Benditt EP. Evidence for a monoclonal origin of human atherosclerotic plaques and some implications. Circulation 1974;50:650–652. [DOI] [PubMed] [Google Scholar]
- 36. Benditt EP. Implications of the monoclonal character of human atherosclerotic plaques. Am J Pathol 1977;86:693–702. [PMC free article] [PubMed] [Google Scholar]
- 37. Murry CE, Gipaya CT, Bartosek T, Benditt EP, Schwartz SM. Monoclonality of smooth muscle cells in human atherosclerosis. Am J Pathol 1997;151:697–705. [PMC free article] [PubMed] [Google Scholar]
- 38. Doolittle RF, Hunkapiller MW, Hood LE, Devare SG, Robbins KC, Aaronson SA, Antoniades HN. Simian sarcoma virus onc gene, v-sis, is derived from the gene (or genes) encoding a platelet-derived growth factor. Science 1983;221:275–277. [DOI] [PubMed] [Google Scholar]
- 39. Robbins CS, Hilgendorf I, Weber GF, Theurl I, Iwamoto Y, Figueiredo JL, Gorbatov R, Sukhova GK, Gerhardt LMS, Smyth D, Zavitz CCJ, Shikatani EA, Parsons M, van Rooijen N, Lin HY, Husain M, Libby P, Nahrendorf M, Weissleder R, Swirski FK. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med 2013;19:1166–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Amento EP, Ehsani N, Palmer H, Libby P. Cytokines and growth factors positively and negatively regulate interstitial collagen gene expression in human vascular smooth muscle cells. Arterioscler Thromb J Vasc Biol 1991;11:1223–1230. [DOI] [PubMed] [Google Scholar]
- 41. Libby P. Mechanisms of acute coronary syndromes and their implications for therapy. N Eng J Med 2013;368:2004–2013. [DOI] [PubMed] [Google Scholar]
- 42. Newby AC. Proteinases and plaque rupture: unblocking the road to translation. Curr Opin Lipidol 2014;25:358–366. [DOI] [PubMed] [Google Scholar]
- 43. Lindsey ML, Libby P. Matrix metalloproteinases in health and disease. In Brinckerhoff CE (ed.). Matrix Metalloproteinases in Cardiovascular Diseases. Singapore: World Scientific Publishing, 2017. pp. 187–225. [Google Scholar]
- 44. Galis ZS, Muszynski M, Sukhova GK, Simon-Morrissey E, Unemori EN, Lark MW, Amento E, Libby P. Cytokine-stimulated human vascular smooth muscle cells synthesize a complement of enzymes required for extracellular matrix digestion. Circ Res 1994;75:181–189. [DOI] [PubMed] [Google Scholar]
- 45. Galis ZS, Muszynski M, Sukhova GK, Simon-Morrissey E, Libby P. Enhanced expression of vascular matrix metalloproteinases induced in vitro by cytokines and in regions of human atherosclerotic lesions. Ann NY Acad Sci 1995;748:501–507. [DOI] [PubMed] [Google Scholar]
- 46. Mach F, Schönbeck U, Bonnefoy JY, Pober JS, Libby P. Activation of monocyte/macrophage functions related to acute atheroma complication by ligation of CD40. Induction of collagenase, stromelysin, and tissue factor. Circulation 1997;96:396–399. [DOI] [PubMed] [Google Scholar]
- 47. Dollery CM, Owen CA, Sukhova GK, Krettek A, Shapiro SD, Libby P. Neutrophil elastase in human atherosclerotic plaques: production by macrophages. Circulation 2003;107:2829–2836. [DOI] [PubMed] [Google Scholar]
- 48. Liu CL, Guo J, Zhang X, Sukhova GK, Libby P, Shi GP. Cysteine protease cathepsins in cardiovascular disease: from basic research to clinical trials. Nat Rev Cardiol 2018;15:351–370. [DOI] [PubMed] [Google Scholar]
- 49. Gistera A, Hansson GK. The immunology of atherosclerosis. Nat Rev Nephrol 2017;13:368–380. [DOI] [PubMed] [Google Scholar]
- 50. Zhao TX, Mallat Z. Targeting the immune system in atherosclerosis: JACC State-of-the-Art Review. J Am Coll Cardiol 2019;73:1691–1706. [DOI] [PubMed] [Google Scholar]
- 51. Taleb S, Tedgui A, Mallat Z. IL-17 and Th17 cells in atherosclerosis: subtle and contextual roles. Arterioscler Thromb J Vasc Biol 2015;35:258–264. [DOI] [PubMed] [Google Scholar]
- 52. Libby P, Tabas I, Fredman G, Fisher EA. Inflammation and its resolution as determinants of acute coronary syndromes. Circ Res 2014;114:1867–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ait-Oufella H, Salomon BL, Potteaux S, Robertson A-KL, Gourdy P, Zoll J, Merval R, Esposito B, Cohen JL, Fisson S, Flavell RA, Hansson GK, Klatzmann D, Tedgui A, Mallat Z. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med 2006;12:178–180. [DOI] [PubMed] [Google Scholar]
- 54. Taleb S, Tedgui A, Mallat Z. Regulatory T-cell immunity and its relevance to atherosclerosis. J Intern Med 2008;263:489–499. [DOI] [PubMed] [Google Scholar]
- 55. Tsiantoulas D, Sage AP, Mallat Z, Binder CJ. Targeting B cells in atherosclerosis: closing the gap from bench to bedside. Arterioscler Thromb J Vasc Biol 2015;35:296–302. [DOI] [PubMed] [Google Scholar]
- 56. Sage AP, Tsiantoulas D, Binder CJ, Mallat Z. The role of B cells in atherosclerosis. Nat Rev Cardiol 2019;16:180–196. [DOI] [PubMed] [Google Scholar]
- 57. Serhan CN, Levy BD. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J Clin Invest 2018;128:2657–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fredman G, Tabas I. Boosting inflammation resolution in atherosclerosis: the next frontier for therapy. Am J Pathol 2017;187:1211–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Viola JR, Lemnitzer P, Jansen Y, Csaba G, Winter C, Neideck C, Silvestre-Roig C, Dittmar G, Doring Y, Drechsler M, Weber C, Zimmer R, Cenac N, Soehnlein O. Resolving lipid mediators maresin 1 and resolvin d2 prevent atheroprogression in mice. Circ Res 2016;119:1030–1038. [DOI] [PubMed] [Google Scholar]
- 60. Owsiany KM, Alencar GF, Owens GK. Revealing the origins of foam cells in atherosclerotic lesions. Arterioscler Thromb J Vasc Biol 2019;39:836–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang Y, Nanda V, Direnzo D, Ye J, Xiao S, Kojima Y, Howe KL, Jarr K-U, Flores AM, Tsantilas P, Tsao N, Rao A, Newman AAC, Eberhard AV, Priest JR, Ruusalepp A, Pasterkamp G, Maegdefessel L, Miller CL, Lind L, Koplev S, Björkegren JLM, Owens GK, Ingelsson E, Weissman IL, Leeper NJ. Clonally expanding smooth muscle cells promote atherosclerosis by escaping efferocytosis and activating the complement cascade. Proc Nat Acad Sci 2020;117:15818–15826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yurdagul A, Doran AC, Cai B, Fredman G, Tabas IA. Mechanisms and consequences of defective efferocytosis in atherosclerosis. Front Cardiovasc Med 2017;4:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Vergallo R, Crea F. Atherosclerotic plaque healing. N Engl J Med 2020;383:846–857. [DOI] [PubMed] [Google Scholar]
- 64. Mann J, Davies MJ. Mechanisms of progression in native coronary artery disease role of healed plaque disruption. Heart 1999;82:265–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fracassi F, Crea F, Sugiyama T, Yamamoto E, Uemura S, Vergallo R, Porto I, Lee H, Fujimoto J, Fuster V, Jang I-K. Healed culprit plaques in patients with acute coronary syndromes. J Am Coll Cardiol 2019;73:2253–2263. [DOI] [PubMed] [Google Scholar]
- 66. Vergallo R, Porto I, D'Amario D, Annibali G, Galli M, Benenati S, Bendandi F, Migliaro S, Fracassi F, Aurigemma C, Leone AM, Buffon A, Burzotta F, Trani C, Niccoli G, Liuzzo G, Prati F, Fuster V, Jang IK, Crea F. Coronary atherosclerotic phenotype and plaque healing in patients with recurrent acute coronary syndromes compared with patients with long-term clinical stability: an in vivo optical coherence tomography study. JAMA Cardiol 2019;4:321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mayerl C, Lukasser M, Sedivy R, Niederegger H, Seiler R, Wick G. Atherosclerosis research from past to present–on the track of two pathologists with opposing views, Carl von Rokitansky and Rudolf Virchow. Virchows Arch 2006;449:96–103. [DOI] [PubMed] [Google Scholar]
- 68. Libby P, Hansson GK. From focal lipid storage to systemic inflammation. J Am Coll Cardiol 2019;74:1594–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016;354:472–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kale A, Sharma A, Stolzing A, Desprez P-Y, Campisi J. Role of immune cells in the removal of deleterious senescent cells. Immun Ageing 2020;17:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Libby P. Assisted living in the atheroma: elderly macrophages promote plaques. Cell Metab 2016;24:779–781. [DOI] [PubMed] [Google Scholar]
- 72. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, Higgins JM, Moltchanov V, Kuo FC, Kluk MJ, Henderson B, Kinnunen L, Koistinen HA, Ladenvall C, Getz G, Correa A, Banahan BF, Gabriel S, Kathiresan S, Stringham HM, McCarthy MI, Boehnke M, Tuomilehto J, Haiman C, Groop L, Atzmon G, Wilson JG, Neuberg D, Altshuler D, Ebert BL. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371:2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, Baber U, Mehran R, Fuster V, Danesh J, Frossard P, Saleheen D, Melander O, Sukhova GK, Neuberg D, Libby P, Kathiresan S, Ebert BL. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med 2017;377:111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Jaiswal S, Libby P. Clonal haematopoiesis: connecting ageing and inflammation in cardiovascular disease. Nat Rev Cardiol 2020;17:137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C, Vuong J, Jacob S, Muralidhar V, Robertson AA, Cooper MA, Andres V, Hirschi KK, Martin KA, Walsh K. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017;355:842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Fidler TP, Xue C, Yalcinkaya M, Hardaway B, Abramowicz S, Xiao T, Liu W, Thomas DG, Hajebrahimi MA, Pircher J, Silvestre-Roig C, Kotini AG, Luchsinger LL, Wei Y, Westerterp M, Snoeck H-W, Papapetrou EP, Schulz C, Massberg S, Soehnlein O, Ebert B, Levine RL, Reilly MP, Libby P, Wang N, Tall AR. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature 2021;592:296–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Aikawa E, Nahrendorf M, Figueiredo JL, Swirski FK, Shtatland T, Kohler RH, Jaffer FA, Aikawa M, Weissleder R. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation 2007;116:2841–2850. [DOI] [PubMed] [Google Scholar]
- 78. Rogers MA, Aikawa E. Cardiovascular calcification: artificial intelligence and big data accelerate mechanistic discovery. Nat Rev Cardiol 2019;16:261–274. [DOI] [PubMed] [Google Scholar]
- 79. Miedema MD, Dardari ZA, Nasir K, Blankstein R, Knickelbine T, Oberembt S, Shaw L, Rumberger J, Michos ED, Rozanski A, Berman DS, Budoff MJ, Blaha MJ. Association of coronary artery calcium with long-term, cause-specific mortality among young adults. JAMA Network Open 2019;2:e197440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Henein M, Granasen G, Wiklund U, Schmermund A, Guerci A, Erbel R, Raggi P. High dose and long-term statin therapy accelerate coronary artery calcification. Int J Cardiol 2015;184:581–586. [DOI] [PubMed] [Google Scholar]
- 81. Puri R, Nicholls SJ, Shao M, Kataoka Y, Uno K, Kapadia SR, Tuzcu EM, Nissen SE. Impact of statins on serial coronary calcification during atheroma progression and regression. J Am Coll Cardiol 2015;65:1273–1282. [DOI] [PubMed] [Google Scholar]
- 82. Crea F, Libby P. Acute coronary syndromes. Circulation 2017;136:1155–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Braunwald E, Morrow DA. Unstable angina: is it time for a requiem? Circulation 2013;127:2452–2457. [DOI] [PubMed] [Google Scholar]
- 84. Burke A, Farb A, Malcom G, Liang Y-H, Smialek J, Virmani R. Coronary risk factors and plaque morphology in men with coronary disease who died suddenly. N Engl J Med 1997;336:1276–1282. [DOI] [PubMed] [Google Scholar]
- 85. Stone GW, Maehara A, Lansky AJ, de Bruyne B, Cristea E, Mintz GS, Mehran R, McPherson J, Farhat N, Marso SP, Parise H, Templin B, White R, Zhang Z, Serruys PW. A prospective natural-history study of coronary atherosclerosis. N Engl J Med 2011;364:226–235. [DOI] [PubMed] [Google Scholar]
- 86. Libby P. The changing landscape of atherosclerosis. Nature 2021;592:524–533. [DOI] [PubMed] [Google Scholar]
- 87. Libby P, Pasterkamp G. Requiem for the ‘vulnerable plaque’. Eur Heart J 2015;36:2984–2987. [DOI] [PubMed] [Google Scholar]
- 88. Pasterkamp G, den Ruijter HM, Libby P. Temporal shifts in clinical presentation and underlying mechanisms of atherosclerotic disease. Nat Rev Cardiol 2017;14:21–29. [DOI] [PubMed] [Google Scholar]
- 89. Puri R, Libby P, Nissen SE, Wolski K, Ballantyne CM, Barter PJ, Chapman MJ, Erbel R, Raichlen JS, Uno K, Kataoka Y, Tuzcu EM, Nicholls SJ. Long-term effects of maximally intensive statin therapy on changes in coronary atheroma composition: insights from SATURN. Eur Heart J Cardiovasc Imaging 2014;15:380–388. [DOI] [PubMed] [Google Scholar]
- 90. Puri R, Nissen SE, Shao M, Ballantyne CM, Barter PJ, Chapman MJ, Erbel R, Libby P, Raichlen JS, Uno K, Kataoka Y, Nicholls SJ. Antiatherosclerotic effects of long-term maximally intensive statin therapy after acute coronary syndrome: insights from Study of Coronary Atheroma by Intravascular Ultrasound: effect of Rosuvastatin Versus Atorvastatin. Arterioscler Thromb J Vasc Biol 2014;34:2465–2472. [DOI] [PubMed] [Google Scholar]
- 91. Nicholls SJ, Puri R, Anderson T, Ballantyne CM, Cho L, Kastelein JJ, Koenig W, Somaratne R, Kassahun H, Yang J, Wasserman SM, Scott R, Ungi I, Podolec J, Ophuis AO, Cornel JH, Borgman M, Brennan DM, Nissen SE. Effect of evolocumab on progression of coronary disease in statin-treated patients: the GLAGOV Randomized Clinical Trial. JAMA 2016;316:2373–2384. [DOI] [PubMed] [Google Scholar]
- 92. Zhao X-Q, Dong L, Hatsukami T, Phan BA, Chu B, Moore A, Lane T, Neradilek MB, Polissar N, Monick D, Lee C, Underhill H, Yuan C. MR imaging of carotid plaque composition during lipid-lowering therapy: a prospective assessment of effect and time course. J Am Coll Cardiol Img 2011;4:977–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Sun J, Lepor N, Canton G, Contreras L, Hippe D, Isquith D, Balu N, Kedan I, Simonini A, Yuan S, Zhao X-Q, Hatsukami T. Carotid Plaque Lipid Content Is Reduced After 6 Months of PCSK9 Inhibition with Alirocumab. 2019.
- 94. Farb A, Burke A, Tang A, Liang Y, Mannan P, Smialek J, Virmani R. Coronary plaque erosion without rupture into a lipid core: a frequent cause of coronary thrombosis in sudden coronary death. Circulation 1996;93:1354–1363. [DOI] [PubMed] [Google Scholar]
- 95. Arbustini E, Dal Bello B, Morbini P, Burke AP, Bocciarelli M, Specchia G, Virmani R. Plaque erosion is a major substrate for coronary thrombosis in acute myocardial infarction. Heart 1999;82:269–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Braunwald E. Coronary plaque erosion: recognition and management. JACC Cardiovasc Imaging 2013;6:288–289. [DOI] [PubMed] [Google Scholar]
- 97. Holmes DR Jr, Lerman A, Moreno PR, King SB 3rd, Sharma SK. Diagnosis and management of STEMI arising from plaque erosion. JACC Cardiovasc Imaging 2013;6:290–296. [DOI] [PubMed] [Google Scholar]
- 98. Luscher TF. Substrates of acute coronary syndromes: new insights into plaque rupture and erosion. Eur Heart J 2015;36:1347–1349. [DOI] [PubMed] [Google Scholar]
- 99. Libby P, Pasterkamp G, Crea F, Jang IK. Reassessing the mechanisms of acute coronary syndromes. Circ Res 2019;124:150–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kolte D, Libby P, Jang I-K. New insights into plaque erosion as a mechanism of acute coronary syndromes. JAMA 2021;325:1043–1044. [DOI] [PubMed] [Google Scholar]
- 101. Partida RA, Libby P, Crea F, Jang IK. Plaque erosion: a new in vivo diagnosis and a potential major shift in the management of patients with acute coronary syndromes. Eur Heart J 2018;39:2070–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Quillard T, Araujo HA, Franck G, Shvartz E, Sukhova G, Libby P. TLR2 and neutrophils potentiate endothelial stress, apoptosis and detachment: implications for superficial erosion. Eur Heart J 2015;36:1394–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Quillard T, Franck G, Mawson T, Folco E, Libby P. Mechanisms of erosion of atherosclerotic plaques. Curr Opin Lipidol 2017;28:434–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Martinod K, Wagner DD. Thrombosis: tangled up in NETs. Blood 2014;123:2768–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Doring Y, Soehnlein O, Weber C. Neutrophil extracellular traps in atherosclerosis and atherothrombosis. Circ Res 2017;120:736–743. [DOI] [PubMed] [Google Scholar]
- 106. Sugiyama S, Kugiyama K, Aikawa M, Nakamura S, Ogawa H, Libby P. Hypochlorous acid, a macrophage product, induces endothelial apoptosis and tissue factor expression: involvement of myeloperoxidase-mediated oxidant in plaque erosion and thrombogenesis. Arterioscler Thromb J Vasc Biol 2004;24:1309–1314. [DOI] [PubMed] [Google Scholar]
- 107. Ferrante G, Nakano M, Prati F, Niccoli G, Mallus MT, Ramazzotti V, Montone RA, Kolodgie FD, Virmani R, Crea F. High levels of systemic myeloperoxidase are associated with coronary plaque erosion in patients with acute coronary syndromes: a clinicopathological study. Circulation 2010;122:2505–2513. [DOI] [PubMed] [Google Scholar]
- 108. Folco EJ, Mawson TL, Vromman A, Bernardes-Souza B, Franck G, Persson O, Nakamura M, Newton G, Luscinskas FW, Libby P. Neutrophil extracellular traps induce endothelial cell activation and tissue factor production through interleukin-1α and cathepsin G. Arterioscler Thromb Vasc Biol 2018;38:1901–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Clancy DM, Henry CM, Sullivan GP, Martin SJ. Neutrophil extracellular traps can serve as platforms for processing and activation of IL-1 family cytokines. FEBS J 2017;284:1712–1725. [DOI] [PubMed] [Google Scholar]
- 110. Lewis HD, Liddle J, Coote JE, Atkinson SJ, Barker MD, Bax BD, Bicker KL, Bingham RP, Campbell M, Chen YH, Chung CW, Craggs PD, Davis RP, Eberhard D, Joberty G, Lind KE, Locke K, Maller C, Martinod K, Patten C, Polyakova O, Rise CE, Rudiger M, Sheppard RJ, Slade DJ, Thomas P, Thorpe J, Yao G, Drewes G, Wagner DD, Thompson PR, Prinjha RK, Wilson DM. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat Chem Biol 2015;11:189–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Franck G, Mawson TL, Folco EJ, Molinaro R, Ruvkun V, Engelbertsen D, Liu X, Tesmenitsky Y, Shvartz E, Sukhova GK, Michel JB, Nicoletti A, Lichtman A, Wagner D, Croce KJ, Libby P. Roles of PAD4 and NETosis in experimental atherosclerosis and arterial injury: implications for superficial erosion. Circ Res 2018;123:33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Molinaro R, Yu M, Sausen G, Bichsel CA, Corbo C, Folco EJ, Lee GY, Liu Y, Tesmenitsky Y, Shvartz E, Sukhova GK, Kloss F, Croce KJ, Farokhzad OC, Shi J, Libby P. Targeted delivery of protein arginine deiminase-4 inhibitors to limit arterial intimal NETosis and preserve endothelial integrity. Cardiovasc Res 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Leistner DM, Kränkel N, Meteva D, Abdelwahed YS, Seppelt C, Stähli BE, Rai H, Skurk C, Lauten A, Mochmann H-C, Fröhlich G, Rauch-Kröhnert U, Flores E, Riedel M, Sieronski L, Kia S, Strässler E, Haghikia A, Dirks F, Steiner JK, Mueller DN, Volk H-D, Klotsche J, Joner M, Libby P, Landmesser U. Differential immunological signature at the culprit site distinguishes acute coronary syndrome with intact from acute coronary syndrome with ruptured fibrous cap: results from the prospective translational OPTICO-ACS study. Eur Heart J 2020;41:3549–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Jernberg T, Hasvold P, Henriksson M, Hjelm H, Thuresson M, Janzon M. Cardiovascular risk in post-myocardial infarction patients: nationwide real world data demonstrate the importance of a long-term perspective. Eur Heart J 2015;36:1163–1170. [DOI] [PubMed] [Google Scholar]
- 115. Pradhan AD, Aday AW, Rose LM, Ridker PM. Residual inflammatory risk on treatment with PCSK9 inhibition and statin therapy. Circulation 2018;138:141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Libby P. Inflammation in atherosclerosis—no longer a theory. Clin Chem 2021;67:131–142. [DOI] [PubMed] [Google Scholar]
- 117. Libby P. Targeting inflammatory pathways in cardiovascular disease: the inflammasome, interleukin-1, interleukin-6 and beyond. Cells 2021;10:951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ, CANTOS Trial Group. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 2017;377:1119–1131. [DOI] [PubMed] [Google Scholar]
- 119. Libby P. Interleukin-1 beta as a target for atherosclerosis therapy: biological basis of CANTOS and beyond. J Am Coll Cardiol 2017;70:2278–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Tardif JC, Kouz S, Waters DD, Bertrand OF, Diaz R, Maggioni AP, Pinto FJ, Ibrahim R, Gamra H, Kiwan GS, Berry C, Lopez-Sendon J, Ostadal P, Koenig W, Angoulvant D, Gregoire JC, Lavoie MA, Dube MP, Rhainds D, Provencher M, Blondeau L, Orfanos A, L'Allier PL, Guertin MC, Roubille F. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med 2019;381:2497–2505. [DOI] [PubMed] [Google Scholar]
- 121. Nidorf SM, Fiolet ATL, Mosterd A, Eikelboom JW, Schut A, Opstal TSJ, The SHK, Xu X-F, Ireland MA, Lenderink T, Latchem D, Hoogslag P, Jerzewski A, Nierop P, Whelan A, Hendriks R, Swart H, Schaap J, Kuijper AFM, van Hessen MWJ, Saklani P, Tan I, Thompson AG, Morton A, Judkins C, Bax WA, Dirksen M, Alings M, Hankey GJ, Budgeon CA, Tijssen JGP, Cornel JH, Thompson PL, the LoDoCo Trial Investigators. Colchicine in patients with chronic coronary disease. N Engl J Med 2020;383:1838–1847. [DOI] [PubMed] [Google Scholar]
- 122. Toprover M, Pillinger MH. Dapansutrile: a new hope? Lancet Rheumatol 2020;2:e247–e249. [DOI] [PubMed] [Google Scholar]
- 123. Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov 2018;17:688. [DOI] [PubMed] [Google Scholar]
- 124. Ridker PM, Devalaraja M, Baeres FMM, Engelmann MDM, Hovingh GK, Ivkovic M, Lo L, Kling D, Pergola P, Raj D, Libby P, Davidson M. IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): a double-blind, randomised, placebo-controlled, phase 2 trial. The Lancet 2021;397:2060–2069. [DOI] [PubMed] [Google Scholar]
- 125. Soehnlein O, Libby P. Targeting inflammation in atherosclerosis—from experimental insights to the clinic. Nat Rev Drug Disc 2021;20:589–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Tchernof A, Nolan A, Sites CK, Ades PA, Poehlman ET. Weight loss reduces C-reactive protein levels in obese postmenopausal women. Circulation 2002;105:564–569. [DOI] [PubMed] [Google Scholar]
- 127. Lavin KM, Perkins RK, Jemiolo B, Raue U, Trappe SW, Trappe TA. Effects of aging and lifelong aerobic exercise on basal and exercise-induced inflammation. J Appl Physiol 2020;129:1493–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir ED, Amadori L, Khan NS, Wong CK, Shamailova R, Hill CA, Wang Z, Remark R, Li JR, Pina C, Faries C, Awad AJ, Moss N, Bjorkegren JLM, Kim-Schulze S, Gnjatic S, Ma'ayan A, Mocco J, Faries P, Merad M, Giannarelli C. Single-cell immune landscape of human atherosclerotic plaques. Nat Med 2019;25:1576–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Zernecke A, Winkels H, Cochain C, Williams JW, Wolf D, Soehnlein O, Robbins CS, Monaco C, Park I, McNamara CA, Binder CJ, Cybulsky Myron I, Scipione CA, Hedrick CC, Galkina Elena V, Kyaw T, Ghosheh Y, Dinh HQ, Ley K. Meta-analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ Res 2020;127:402–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Libby P. Murine "model" monotheism: an iconoclast at the altar of mouse. Circ Res 2015;117:921–925. [DOI] [PubMed] [Google Scholar]
- 131. Drucker DJ. Never waste a good crisis: confronting reproducibility in translational research. Cell Metab 2016;24:348–360. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new data were generated or analysed in support of this research.