

Abstract
The resolution of inflammation (or inflammation-resolution) is an active and highly coordinated process. Inflammation-resolution is governed by several endogenous factors, and specialized pro-resolving mediators (SPMs) are one such class of molecules that have robust biological function. Non-resolving inflammation is associated with a variety of human diseases, including atherosclerosis. Moreover, non-resolving inflammation is a hallmark of ageing, an inevitable process associated with increased risk for cardiovascular disease. Uncovering mechanisms as to why inflammation-resolution is impaired in ageing and in disease and identifying useful biomarkers for non-resolving inflammation are unmet needs. Recent work has pointed to a critical role for balanced ratios of SPMs and pro-inflammatory lipids (i.e. leucotrienes and/or specific prostaglandins) as a key determinant of timely inflammation resolution. This review will focus on the accumulating findings that support the role of non-resolving inflammation and imbalanced pro-resolving and pro-inflammatory mediators in atherosclerosis. We aim to provide insight as to why these imbalances occur, the importance of ageing in disease progression, and how haematopoietic function impacts inflammation-resolution and atherosclerosis. We highlight open questions regarding therapeutic strategies and mechanisms of disease to provide a framework for future studies that aim to tackle this important human disease.
Keywords: Atherosclerosis, Ageing, Inflammation, Immunology, hematopoiesis
1. Introduction
Atherosclerosis is a major killer worldwide and is characterized by the accumulation of immune cells and lipids in medium and large arteries.1 Life-threatening clinical manifestations of atherosclerosis include (but are not limited to) myocardial infarctions and ischaemic strokes and are collectively known as major adverse cardiovascular events (MACEs). These life-threatening events typically occur due to plaque rupture or erosion which results in acute atherothrombosis and vascular occlusion. The pathogenesis of atherosclerosis is linked to persistent inflammation and dyslipidaemia including high levels of low-density lipoprotein (LDL). Current therapeutic strategies to limit LDL such as statins and PCSK9 inhibitors are effective at reducing LDL levels in the blood; however, MACEs are only reduced by ∼50%,2 which is likely due to the pro-inflammatory nature of the disease. Indeed, chronic inflammation (or the residual inflammatory risk, RIR) is thought to be a culprit in the progressive and non-resolving nature of atherosclerotic cardiovascular disease.3 Interestingly, significant LDL lowering with statins and PCSK9 inhibitors as seen in the SPIRE trials, still demonstrated evidence of RIR in individuals, which suggests that dramatic cholesterol lowering may not be enough to fully terminate the progressive nature of this disease.4 Moreover, this finding suggests that inflammation must be mitigated to potentially control the progression of this disease. CANTOS was the first demonstration in humans that blockade of interleukin-1β (IL-1β) reduced MACEs.5 COLCOT was another trial in which anti-inflammatory colchicine also reduced MACEs.6 Collectively, results from these trials provide direct evidence that blockade of pro-inflammatory factors can reduce MACEs. Moreover, they offer much needed momentum towards understanding key mediators and mechanisms associated with failed non-resolving in the arterial wall.
While inflammation is a clear culprit in atherosclerosis, directly targeting/blocking inflammation poses some risks, including immune suppression and an increased likelihood for infection, which was noted in the CANTOS trial. Therefore, identifying factors and cellular programmes that can control inflammation without compromising host defense may be ideal treatments for atherosclerosis that can potentially be used with therapies that already exist. This review will focus on a particular family of ligands called ‘specialized pro-resolving mediators’ or SPMs that function to control inflammation and promote tissue repair in a manner that does not compromise host defense.7 This review will also pinpoint some mechanisms as to why SPMs may not be readily made within atherosclerotic plaques and how restoration of the key defective mediators may help limit the progression of atherosclerosis.
2. SPMs activate inflammation-resolution programmes
While it was previously thought that passive disappearance of pro-inflammatory factors was sufficient for the cessation of inflammation, it is now known that the resolution of acute inflammation (or inflammation-resolution) is an active and highly coordinated process.7 Inflammation-resolution is governed by a panoply of endogenous factors that include SPMs,7 protein/peptide mediators such as annexin A18 and interleukin 10, gases such as and carbon monoxide and hydrogen sulfide,9,10 and nucleotides such as adenosine and inosine.11 Here, we specifically highlight how SPMs impact inflammation-resolution in atherosclerosis and discuss key gaps in our understanding of non-resolving inflammation in this context.
SPMs are actively biosynthesized at the onset of acute inflammation to counterbalance numerous pro-inflammatory signals. This ‘push-and-pull’ of pro-inflammatory and pro-resolving ligands ultimately controls the magnitude and duration of inflammation.7 Recent work has pointed to the balance between SPMs and pro-inflammatory ligands like leucotrienes (LTs) or some of the prostaglandins (PGs) as a key determinant to whether inflammation resolves in a timely manner or persists.12,13 SPMs are biosynthesized from arachidonic acid (AA), eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), or n-3 docosapentaenoic acid via the actions of lipoxygenases (LOXs) and cyclooxygenases (COXs)7,14 (Figure 1). This superfamily of mediators includes lipoxins, resolvins, protectins, their aspirin-triggered isomers, maresins, cysteinyl-conjugated SPMs (CTRs), and 13-series resolvins (RvTs).14–20 SPMs are not immunosuppressive in animal models13,21 and are protective in several disease models,7 including atherosclerosis (Table 1), which will be the focus of this review.
Figure 1.

Schematic of SPM generation and their actions via specific GPCRs on select cellular targets. Precursor fatty acids, including arachidonic acid (AA), eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), or n-3 docosapentaenoic acid (n-3DPA), are biosynthesized into several families of SPMs by the action of enzymes including 5-LOX, 15-LOX, and COX-2. The receptors for many SPMs have been identified, as shown, but many remain to be elucidated. SPMs also have distinct cellular targets, though more work is needed to examine cell-specific expression of receptors under different conditions and in different tissues.
Table 1.
Pro-resolving ligands are protective in atherosclerosis
| Treatment | Animal model | Diet | Context | Outcome |
|---|---|---|---|---|
| Proteins/peptides | ||||
| Ac2-2622 (i.v.) | Ldlr−/− | Western diet | Advanced athero (progression) |
|
| Ac2-2623 (i.v.) | Apoe−/− | Western diet | Early athero (atherogenesis) | ↓ Leucocyte migration into arterial wall |
| IL-1024 (i.v.) | Ldlr−/− | Western diet | Advanced athero (progression) |
|
| Human recombinant annexin 125 (i.p.) | Ldlr−/− | Western diet | Mid-advanced athero (progression) | ↓ Lesion area, lesional necrosis |
| SPMs | ||||
| RvE126 (oral-topical) | Rabbit | High cholesterol | Early athero (atherogenesis) | ↓ Lesion area |
| RvE127 (oral) | ApoE*3 Leiden | Western diet | Mid-advanced athero (progression) | ↓ Lesion area, lesional mRNA of Adam17, CD44, CCR5, CCl2, CD74 |
| RvE1 signalling ChemR2328 |
|
Western diet | Advanced athero (progression) | Removal of ChemR23 lead to ↓lesion and necrotic area |
| RvD129 | Ldlr−/− | Western diet | Advanced athero (progression) |
|
| RvD2 and Mar130 | Apoe−/− | Western diet | Advanced athero (progression) |
|
| ATL31 | Apoe−/− | Western diet | Advanced athero (progression) |
|
SPMs have distinct chemical structures and are potent ligands that bind and activate specific G-protein coupled receptors7,32–36 (Figure 1). As a testament to their potency, the concentration of Resolvin D1 (RvD1) in local, resolving blister exudates from humans is ∼85pM.37 RvD1 enhances the clearance of apoptotic cells (i.e. efferocytosis) in this dose range,38 supporting the idea that low concentrations of SPMs can evoke significant biological activity on human cells. Deletion of key SPM receptors, such as ALX/FPR2, GPR18, or LGR6, leads to exacerbated inflammation and defective resolution.39–42 In the context of atherosclerosis, removal of RvE1’s receptor, ChemR23, resulted in increased lesion size and plaque necrosis, demonstrating a critical role for endogenous RvE1 in thwarting atherosclerosis progression.28 These findings provide compelling evidence that SPMs are essential endogenous mediators of the inflammation-resolution response and therefore critical for limiting disease associated with non-resolving inflammation. Indeed, they are protective in several disease models including injury-induced neointimal hyperplasia,43,44 myocardial infarction,45,46 and atherosclerosis.26,29,30,47 A consequence of ongoing inflammation in atherosclerosis is damage to the arterial wall. In addition to limiting pro-inflammatory lipid mediators and cytokines, SPMs stimulate the host to repair and regenerate tissue. Therefore, SPM therapies may be an attractive new approach towards treatment of atherosclerosis, with the dual properties of both safely limiting inflammation and enhancing repair.
3. Prime suspect: impaired inflammation-resolution in atherosclerosis
The balance of lipid mediators in the vasculature has been studied and appreciated for decades.48 For example, the timely and controlled synthesis of thromboxane activates platelets to prevent blood loss and initiate the recruitment of inflammatory cells required for repair. Prostacyclin limits platelet activation while swiftly restoring flow in the vasculature. Thromboxane and prostacyclin are synthesized through COX and balance between these lipid mediators is critical for maintaining vascular homeostasis. Disruption in this balance (as we have learned from selective COX-2 inhibitors) can lead to catastrophic outcomes such as acute thrombotic events.49 Consequently, it is not surprising that disproportions of other lipid mediators may have maladaptive outcomes in vascular diseases, and, in fact, imbalances in SPMs and LTs or PGs are associated with atherosclerosis.29,30,50 Specifically, we found that SPMs were imbalanced with LTs in highly necrotic regions of human plaques compared with less necrotic regions.29 In an effort to demonstrate causation, we investigated Ldlr−/− mice and found that highly necrotic, advanced lesions had a decreased SPM:LT ratio compared with lesions with minimal necrosis, and RvD1 treatment during advanced atherosclerosis reduced necrosis and increased plaque remodelling in mice.29 The low dose of RvD1 given also increased several other species of intraplaque SPMs and improved the SPM:LT ratio in lesions,29 suggesting that RvD1 stimulates a feed-forward pro-resolution mechanism that can repair damage within the arterial wall. At the same time, a separate group found a decrease in the SPM:LT and SPM:PG ratio in advancing murine plaques. Viola et al.30 found that the SPMs that were the most reduced in advanced plaques were RvD2 and Maresin 1 (MaR1). Exogenous administration of MaR1 and RvD2 to Apoe−/− mice with established atherosclerotic disease limited necrosis and atheroprogression. Several SPMs (and other pro-resolving ligands) limit atherosclerosis in different animal models (summarized in Table 1). Together, these results suggest that the imbalance in SPMs in plaques may drive plaque necrosis and atheroprogression.
Based on the work of several groups around the world the SPM:LT/PG ratio may also be a potential biomarker for atherosclerosis and susceptibility for MACEs in humans. For example, a decreased RvD1:leucotriene B4 (LTB4) in saliva was associated with increased intimal-medial thickness,51 a metric associated with advancing disease. Analysis of human plasma revealed significantly lower RvD1 levels in highly symptomatic stroke patients compared with asymptomatic patients,52 therefore suggesting that low levels of RvD1 may be associated with specific outcomes or disease trajectories. The RvE1:LTB4 ratio was also lower in the context of aortic valve disease in humans,53 again suggesting that this ratio is critical in vascular disease. The ability to predict responses to treatments and/or potential outcomes is clinically relevant and currently lacking. Collectively, the examples used here were determined by different assays, including ELISA and LC-MS/MS suggesting that several different types of analysis can generate meaningful data, which may be leveraged to develop more routine screens for improved patient care.
The SPM:LT ratio in the aforementioned studies were largely determined by decreased levels of SPMs, suggesting that there may be a defect either in the synthesis of key SPMs during atheroprogression or an exuberance of their breakdown. While catabolism of SPMs may be a viable hypothesis, some metabolites of RvD1 and RvD2, as examples, have equipotent biologic actions, at least with regard to limiting monocyte adhesion to adipocytes.54 Therefore, we postulate that reduced or defective synthesis, plays a critical role in SPM:LT imbalances in atherosclerosis. There are several possible mechanisms that may underly this imbalance on a cellular level including factors that affect key enzymes, oxidative stress, and SPM production.55 We previously uncovered a new pathway that explains, in part, why endogenous SPMs could be defective in atherosclerosis.29,55 Under conditions of oxidative stress as seen in inflammation, 5-LOX is phosphorylated and translocates to the nuclear membrane in macrophages, which favours the biosynthesis of the pro-inflammatory mediator, LTB456,57 (Figure 2). In this context, RvD1 promotes nuclear exclusion of 5-LOX and thereby suppresses LTB4 and enhances production of the pro-resolving molecule, lipoxin A4 (LXA4) in macrophages.55 Newer work suggests that the SPM:LT ratio may also involve the interaction between the DAMP high-mobility group box 1 (HMGB1) and with Complement 1q (C1q). Liu et al.58 found that HMGB1 drives LTB4 production via phosphorylation and nuclear translocation of 5-LOX in macrophages. However, when HMGB1 is present with C1q, 5-LOX is cytoplasmic a subsequent increase in SPMs is observed.58 Interestingly, C1q deficiency is associated with systemic lupus erythematosus, a patient population with a well-known predisposition for accelerated atherosclerosis.59 There are likely numerous permutations as to how SPM:LT ratios are regulated at the signalling level and via enzymatic activity and uncovering these pathways may lead to new strategies that can restore the SPM:LT/PG balance.
Figure 2.
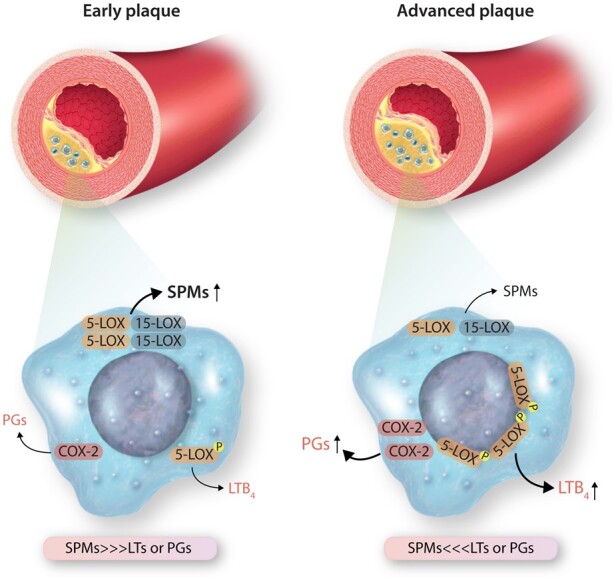
Proposed scheme for SPM:LT/PG imbalances in advanced atherosclerotic plaques. Macrophages are major cellular players in atherosclerosis and can biosynthesize PG, LTs, and SPMs. How a macrophage ‘chooses’ to produce a SPM or an LT for example, may in part be due to localization of 5-LOX. Phosphorylated and nuclear 5-LOX drives LT synthesis, while non-nuclear synthesis promotes the formation of SPMs like lipoxins. Pro-inflammatory factors and oxidative stress drive nuclear 5-LOX in macrophages which may be one of many mechanisms associated with an imbalance in SPM:LT in advanced atherosclerotic plaques.
Other processes that can impact SPM production include transcellular biosynthesis between interacting cells7 and the release of precursors from cellular phospholipids.60 Therefore, the cell composition and the phenotype of cells within plaques may be critical determinants as to whether SPM can be formed. For example, human ‘M2-like’ macrophages are more robust generators of SPMs than ‘M1-like macrophages61,62’. M2-macrophages in atherosclerosis are critical for regression, in part through their ability to synthesize SPMs.63,64 Other reports demonstrate that five-lipoxygenase activating enzyme, a protein that facilitates the synthesis of LTB4, also plays an important role in the production of SPMs.65,66 Leucotriene A4 hydrolase is yet another intriguing target for atherosclerosis and potentially other vascular diseases, because inhibition of this enzyme has been linked to a decrease in LTB4 and an increase in lipoxins.65,67
Lastly, macrophage function, and in particular the ability to carry out efferocytosis, facilitates the synthesis of SPMs over LTs.61,68,69 This feed-forward circuit was first demonstrated in murine systems68 and was further recapitulated in human macrophages that had ingested apoptotic PMN.61 A potential mechanism for the increase in SPMs during efferocytosis is transcellular biosynthesis.61 How apoptotic cell corpses are handled once ingested and whether the enzymes within those cells are recycled, donated, or broken down are not known and would be of immense interest. For example, do dead cells become organelle or enzyme ‘donors’ and if so, does that facilitate the synthesis of SPMs over LTs? Another mechanism that promotes production of SPMs during efferocytosis is through MerTK signalling.69 Cai et al.69 demonstrated that MerTK signalling leads to increased SPMs and that silencing of MerTK resulted in reduced SPMs. Ga6-stimulated MerTK signalling leads to decreased phosphorylation of 5-LOX, increased SPMs and decreased LTs.55,69 MerTK signalling in human macrophages decreased activity of the mitogen-activated protein kinase p38 and the kinase MK2, resulting in increased non-phosphorylated, cytoplasmic 5-LOX, and enhanced SPM biosynthesis.70 The ability to bind target cells for efferocytic clearance is also highly regulated, in part via integrin-dependent adhesion. Endothelial cells and macrophages can secrete the molecule developmental endothelial locus 1 which regulates binding to neutrophils, efficient efferocytosis, and production of SPMs.71 This work suggests that specific signalling pathways and cell-cell interactions associated with dead cell clearance can impact SPM generation.
Ample data show that a decreased SPM:LT or PG ratio is associated with worsened disease, but what about the opposite? Does an increased SPM:LT or PG ratio correlate with plaque resolution/regression in humans? Welty et al.72 began to tackle this question in a recent study in which they monitored coronary artery disease patients given Lovaza (which is an omega-3 EPA and DHA ethyl ester mixture), for 30 months. First, they found that that some individuals had a low plasma omega-3 index and other had a high plasma omega-3 index.72 Those with a high omega-3 fatty acid (FA) index were more likely to be women and to have lower white blood cell and neutrophil counts compared with those with a low omega-3 FA index.72 Most individuals with a high omega-3 index exhibited an increased SPM:LT ratio and coronary plaque regression, which was independent of LDL-C and hsCRP levels. This work suggests that the SPM:LT ratio may be a more precise measure of residual inflammation in the setting of already low hsCRP. Results from the REDUCE-IT and EVAPORATE trials demonstrated that Vascepa (which is an EPA ethyl ester) reduced the incidence of cardiovascular events and reduced plaque volume73,74 again suggesting that omega-3 FAs may aid in plaque regression. Whether Vascepa’s protective actions are via the synthesis of SPMs in not known and a deeper exploration of these patient samples may yield important mechanistic clues as to why this omega-3 regimen showed a benefit. Conversely, the STRENGTH trial, which tested the use of free acid EPA and DHA in cardiovascular patients with hypertriglyceridaemia was stopped early due to no reductions in MACE between the control (corn oil) and the EPA and DHA treatment group.75 There are key differences in REDUCE-IT versus STRENGTH trials and the obvious is EPA ethyl ester vs. free acid EPA+DHA, respectively. On the surface, one would postulate that the addition of DHA may be a reason for these discrepant findings, but until there is a trial with DHA alone, jumping to that conclusion may be premature. A glaring difference in the trials are the oils used in the control groups. REDUCE-IT used mineral oil whereas the STRENGTH trial used corn oil as a control. Corn oil is rich in omega-6 FAs and has small but appreciable amounts of omega-3 alpha-linoleic acid (ALA). ALA can be converted to EPA in humans76 and so the differences between control and experimental groups may be muddled.
Moreover, an intriguing component of the Welty et al.72 study was that there was a small subset of individuals who had a high omega-3 index but still exhibited atherosclerosis progression. These individuals did not have an increased SPM:LT ratio, which suggests that not all individuals respond to omega-3 supplementation and specifically not all individuals can make SPMs in the presence of omega-3’s. Why some humans can synthesize SPMs over LTs in the presence of omega-3’s and why other cannot is not known. One possibility is that confounding factors, like diabetes status, smoking, sedentary lifestyle or unhealthy diet can increase inflammation or oxidative stress may drive localization of biosynthetic enzymes in a manner that is not suitable to synthesize SPMs.29 Another explanation may be hidden in subtle diet-gene interactions. For example, individuals with a specific 5-LOX variant who also ingested a diet rich in AA had increased carotid atherosclerosis compared to individuals with non-variant 5-LOX, while those with the same mutation but who ingested a diet rich in omega-3 FAs had less carotid atherosclerosis compared with the control population.77 These results suggest that certain single-nucleotide polymorphisms (SNPs) may not associate with disease itself but may reveal pathologic or protective changes when in combination with lifestyle choices, such as diet. How other lifestyle choices such as exercise and stress interact with the genetic landscape of certain individuals may reveal specific clues as to who may benefit from specific treatments and who cannot. More experiments will be needed to further understand mechanisms regulating differences in SPM synthesis among individual patients.
4. What drives failed resolution in atherosclerosis?
Smoking, diabetes status, and dyslipidaemia are well-known risk factors for atherosclerosis and cardiovascular diseases, and inflammation-resolution is impaired in these contexts.78,79 Other non-traditional risk factors such as poor diet, stress, and ageing itself all contribute to atherosclerosis, though whether inflammation-resolution programmes are defective in these conditions is vastly underexplored. Inflammation-resolution ‘programmes’ encompass a variety of mediators and cellular processes, which requires a coordinated effort between several cell types, factors, and organ systems. At the same time, cardiovascular disease is a result of the intersection of many cell types and organ systems. One such tissue, the blood, continuously bathes vessels, and plaques. Therefore, the blood and its source, the bone marrow, are critical players in chronic inflammatory diseases and will be a major focus of the remaining part of the review.
Immune cells are important responders to damage and can be recruited to areas of injury, and exuberant immune activity increases MACEs.80 Cells of myeloid origin have been shown to contribute to plaque progression and instability in animal models,81,82 and both increased neutrophils and monocytes have been linked with atheroprogression in humans.83,84 Neutrophils are found in advanced human plaques,85,86 a state also characterized by imbalances in SPM:LT/PG ratios.29 Neutrophils can contribute to impaired inflammation-resolution if hyperactivated and release neutrophil extracellular traps or NETs that further promote inflammation and thrombosis.87 Indeed, platelets have also been linked with atheroprogression and myocardial infarction,88,89 and both platelet production and activity can be impacted by stress, disease status, infection, and ageing. While mature myeloid cells can be recruited into plaques and modulate the local microenvironment, increasing evidence supports the idea that altered blood production, or haematopoiesis, can impact atheroprogression.90 Increases in haematopoietic stem cell (HSC) clones carrying somatic mutations correlates strongly with coronary artery disease, and emerging clinical evidence demonstrating a strong link between clonal haematopoiesis and cardiovascular disease has been extensively reviewed elsewhere.91–94
In addition to the generation of blood cells, the bone marrow is an important site of cell removal, and the homeostatic process of cell clearance regulates bone marrow function, including the niches that maintain HSCs.95 While the regenerative capacity of bone marrow is well-established as evidenced by the success of HSC transplantation, mechanisms of inflammation-resolution at this site have not been clearly defined. Lipidomic analysis in mice revealed higher SPM levels in the bone marrow, relative to the spleen, and efferocytosis of bone forming cells is important for homeostatic bone turnover.96,97 Thus, inflammation-resolution processes operate in a variety of tissues, and these programmes may contribute to atheroprogression both locally and from a distance. The plasticity among immune cell types, the intersection of various systems, and the relatively slow progression of disease highlights the complexity of understanding mechanisms driving of cardiovascular disease. Many questions remain regarding how SPMs and SPM:LT/PG balances impact immune cell generation and myeloid function, and in turn, how haematopoietic output may influence the balance of SPMs and LTs/PGs in different tissues.
4.1 Poor diet and a sedentary lifestyle drive atherosclerosis: healthy diet and regular exercise boost endogenous inflammation-resolution
Beyond genetics, an important driver of atherosclerosis is poor diet. This is particularly important in terms of inflammation-resolution as SPMs are biosynthesized from essential FAs. Diets rich in omega-3 FAs have been well documented to thwart the progression atherosclerosis98 and whether that is due to the synthesis and action of SPMs is still not known but is a topic of great interest. As mentioned above, atherosclerosis is a complex disease that involves lipids, inflammation, and genetic factors, and effective treatments in the future will likely be multi-pronged. It would be harmful to suggest that a single pill can act as a ‘magic bullet’ to quell atherosclerosis. However, maintaining a healthy lifestyle, which includes a diet rich in omega-3 FAs and regular exercise is beneficial. Indeed, leisurely exercise limits all cause and cardiovascular mortality in humans.99 One possible mechanism is related to the bone marrow and the production of inflammatory cells. Exercise protects mice from chronic leucocytosis which ultimately resulted in reduced atherosclerosis in mice.100 Whether SPMs or resolution mechanisms are involved in exercise-induced reduction in haematopoietic output of inflammatory cells is not known. In other contexts, exercise was shown to increase RvD1 and promote efferocytosis.101 Therefore, it is possible that exercise-induced RvD1 could limit atherosclerosis, given the already known protective roles of RvD1 in murine models of atherosclerosis.29
Increased dietary omega-3 FAs are appreciated to reduce systemic inflammation, though clear mechanisms are poorly understood. Mice fed a fish oil diet where there was a higher omega-3:omega-6 ratio exhibited increased primitive myeloid progenitors in the bone marrow, but when cultured ex vivo, reduced myeloid cell production was observed.102,103 Fish oil did not impact circulating white blood cells, but the findings suggested that increased dietary omega-3 FAs suppressed the differentiation of myeloid progenitors and support the notion that regulating myeloid cell production may be one mechanism whereby diet changes reduce atherosclerosis. In contrast, mice fed a diet laden with saturated fat and cholesterol (i.e. the Western diet) exhibited immune activation and enhanced myelopoiesis, similar responses seen after exposure to pathogens, and these responses were mediated by the intracellular Nod-like receptor NLRP3.104 These NLRP3-dependent responses result in epigenetic reprogramming in myeloid progenitors and long-lived immune dysfunction that may facilitate the chronic immune responses observed in patients with unhealthy diets. Recent evidence in a murine model of JAK2V617F-induced myeloproliferation and Western diet has linked DNA-damage and the DNA-sensing inflammasome, AIM2, as an essential driver of atheroprogression via the production of IL-1β.105 Thus, in combination with underlying genetic perturbations, diet may be particularly important for disease progression.
Dietary FAs can be metabolized to generate LTs, PGs, and SPMs, and the precise mechanisms regulating the systemic and haematopoietic effects of dietary changes are important questions. Recent evidence from a human study demonstrated that enriched marine oil supplement could induce changes to blood SPM levels and impact the transcriptomic profile of circulating cells, as well as improve phagocytic capacity by neutrophils and monocytes.106 This proof-of-concept study reveals the relatively rapid changes to immune system function and systemic levels of SPMs occurs upon supplementation of omega-3 FAs. One caveat of this study is that all volunteers were healthy and relatively young. In an entirely different scenario, where diet is restricted by fasting, blood production is also modulated and, notably, fasting limits the number and inflammatory function of monocytes.107 In humans, obesity has been shown to correlate with increased circulating monocytes, again supporting the critical role of diet in regulating atheroprogression and linking atheroprogression to changes in haematopoiesis. The intersection of diet and overall caloric consumption likely plays an important role in the synthesis and balance of SPMs and PGs/LTs, and therefore atheroprogression.
4.2 Disrupted sleep and stress promote atherosclerosis: restoring circadian rhythms may boost endogenous resolution programmes
Natural sleep patterns that are also important to health and circadian rhythms are now well-appreciated to control inflammation and atheroprogression.108 These daily rhythms that regulate our sleep cycle also regulate the bone marrow and its release of leucocytes and the ability of the kidney to filter blood and regulate blood pressure. Thus, disturbances in sleep caused by work or stress are poised to impact the very cells and organs that can contribute to progression of cardiovascular disease and incidence of MACEs. The release of HSCs and progenitors from the bone marrow into circulation is also regulated by internal clock genes109 and this daily homeostatic fluctuation is thought to provide cells to distal tissues for repair and immunosurveillance.110 Also, insufficient sleep is linked to increased myelopoiesis and atherosclerotic plaques in Apoe−/− mice.111 Similar to the impact of sleep disturbances, exposure to stressful stimuli increased haematopoietic function in Apoe−/− mice and revealed enhanced development of unstable plaques.112 Beyond systemic responses, evidence also suggests that circadian-dependent mechanisms are also at play within plaques themselves, wherein the timing of cell death and clearance regulates the growth of the necrotic core.113 A prevailing biological function of sleep is one of restoration and repair and indeed, efferocytosis and phagocytosis in the bone marrow and other tissues, such as the brain, are dependent on circadian rhythms.110,114 Collectively, sufficient sleep and reduced stress are beneficial to thwart atherosclerosis progression, but how these processes intersect with inflammation-resolution programmes is less understood. One clue is a recent paper that described diurnal synthesis of key SPMs.115 These data support the notion that resolution programmes and/or resolution therapy may be optimal at specific times of day, and disturbances in normal sleep patterns may impair inflammation-resolution.
4.3 Does the aged immune system set the stage for atherosclerosis?
In general, increasing age is a well-known and major risk factor for atherosclerosis. The impact of advanced age on the development and progression of atherosclerosis, as well as the treatment approaches for elderly patients with atherosclerosis, is a critically underexplored arena.116 Chronological ageing involves, genetic, environmental, and metabolic changes that impact all organs systems.117 Animal models suggest that ageing drives atherosclerosis in part through changes in the bone marrow.118 Monocytosis was associated with increased atherosclerosis in ageing, and bone marrow transfers from aged mice into young Ldlr−/− mice produced larger plaques than Ldlr−/− recipients that received young marrow.118 These findings suggest that the bone marrow from aged mice promoted atherosclerosis. Indeed, ageing is associated with profound changes to blood production, and a key feature of haematopoietic dysfunction in this context is the strong bias in myeloid cell production due to an expanded pool of myeloid-biased HSCs and progenitors.119,120 At the same time, ageing is associated with reduced lymphoid cell generation that has been linked to a loss of lymphoid progenitor cells, in part, due to changes in the aged bone marrow microenvironment.121,122 The function of mature cells is also compromised in ageing and linked with both poor immunity and more severe disease in response to infection.123–125 Increased pro-inflammatory responses are observed in ageing and suggest that dysfunctional inflammation-resolution programmes are central to declining host defense and more pathological outcomes in response to infection in the elderly. Though no data exist to demonstrate that resolution impairments that arise during ageing cause atherosclerosis, evidence supports that the ageing landscape may set the scene for disease.
The aged immune system is vastly different than in younger individuals and new work in animal models suggests that haematopoietic cell senescence drives organ inflammation and damage.126 Cellular senescence is a programme in which cells are halted in their proliferation while acquiring a secretory phenotype characterized by production of pro-inflammatory factors, referred to as the senescence-associated secretory phenotype or SASP.127 Transient emergence of senescent cells upon injury is important for wound healing and can promote healing in arthritis.128 However, in the context of ageing senescent cells are associated with ongoing inflammation or inflammaging.129 Senescent cells exacerbate inflammation129 and can also contribute to inefficient clearance of dead cells.130 Briefly, we found that released factors from senescent cells decreased efferocytosis through enhanced MerTK cleavage.130 RvD1 efferocytosis in this context rescued efferocytosis. Most of the senescence literature in the atherosclerosis arena has focused on smooth muscle and endothelial cells131,132 and little attention has been given to haematopoietic-derived cells.133 We recently found that haematopoietic cells promoted plaque necrosis and that removal of senescent cells during advanced atherosclerosis limited plaque necrosis and promoted the synthesis of key SPMs.134 Moreover, we observed that senescent macrophages themselves were highly pro-inflammatory and were poor efferocytes,134 therefore suggesting that senescent macrophages cannot carry out a key programme of inflammation-resolution. Moreover, these findings suggest that the accumulation of senescent macrophages or other haematopoietic cells in plaques may be a key driver of deranged resolution programmes in atherosclerosis.
Overall, we still do not fully understand why the aged immune system does not function as optimally as the young. SPMs,130,135 drugs that target senescent cells,136 and a restricted calorie diet137 can limit inflammaging, which suggests that inflammaging is a ‘modifiable’ risk factor. As mentioned above, proper sleep cycles, a diet enriched in omega-3 FAs and regular exercise promote endogenous SPMs and resolution in mice.101,106,115 Indeed, there is well documented literature that there is a lower cardiovascular disease risk for middle aged or elderly individuals who engage in regular exercise compared with their sedentary counterparts.138 Whether SPMs are boosted in elderly humans who engage in routine exercise, or whether SPMs themselves can act in the bone marrow to correct age-related haematopoietic changes is not known and may provide important missing details for how to optimally treat elderly individuals.
Along these lines, ageing is associated with imbalances in the SPM:LT ratio in mice and humans in.135,139 Mechanisms and consequences associated with these imbalances are underexplored. Efferocytosis, which is a major cellular programme of inflammation-resolution, is widely known to be impaired in ageing.135,140–145 An efferocytosis receptor called TIM-4, was decreased on macrophages from elderly humans in the context of self-limited inflammation.146 P38 signalling was attributed to decreased levels of TIM-4 and blockade of p38 restored efferocytosis in the elderly.146 This work provides a rationale for therapeutic strategies that target p38 to promote efferocytosis in ageing.147 Because p38 signalling is associated with several pro-inflammatory pathways, direct inhibition of the kinase over time may lead to immune suppression. Of note, RvD1 does not inhibit, but rather controls p38 activation and promotes a pro-resolution circuit in macrophages.55 As mentioned above, MerTK promotes efferocytosis and increases SPMs.69 Indeed, Gas6, which is a ligand for MerTK promotes SPM over LTs.70 Interestingly, Frisch et al.143 found that not only was efferocytosis impaired the bone marrow from aged mice but that levels of Gas6 were also significantly reduced the bone marrow from aged mice, compared with young controls. Whether SPMs were also decreased in aged bone marrow was not explored but would provide important mechanistic information. A deeper exploration of efferocytosis mechanisms in ageing may help inform the development of new tissue-reparative therapies for the elderly.
As mentioned above, chronological ageing is complex and includes cellular metabolic and epigenetic alterations that impact all organs and tissue systems,117 and, from a practical standpoint, studying ageing is challenging and time-consuming. Therefore, the use of models that ‘accelerate’ or mimic ageing of the bone marrow compartment may be of use to home in on mechanisms that drive haematopoietic ageing. Sublethal radiation is a well-known model for accelerated ageing, particularly within the bone marrow.148–150 This model leads to HSC senescence and mimics several features of ageing such as alterations in HSC functions like reduced clonogenicity and skewed differentiation towards myeloid lineages.149 Senescence is associated with ageing and atherosclerosis and so the use of sub lethal radiation to understand mechanisms associated with haematopoietic and immune cell senescence may be reveal important mechanistic clues as to how senescence of immune cells impacts inflammation-resolution and/or diseases like atherosclerosis. Moreover, this model can accelerate the pace of research towards uncovering novel mechanistic based therapies and provide a more rapid platform for testing new strategies for age-related diseases. And so in addition to chronological ageing, more work should be focused on proof-of-concept models to augment our understanding of how ageing of the bone marrow and thus the birthplace of our immune cells can impact atherosclerosis.
5. Closing remarks: therapeutic opportunities and areas of future study
Pro-resolving mediators actively counteract inflammation without compromising host-defense.7 In animal models, SPMs lower the threshold for antibiotic therapy and enhance host defense mechanisms regarding viral and bacterial infections and.13,151 Because atherosclerosis is a long-term progressive disease, directly blocking inflammation for an extended time may lead to unintended host defense issues. A therapeutic strategy that activates the host to repair an injury (e.g. through boosting efferocytosis) may be a promising approach towards controlling inflammation and aiding regeneration of damaged tissue. It may be an approach that ultimately prevents atherosclerotic progression and/or can be utilized to improve current therapies aimed at driving plaque regression. Several important unanswered questions exist, however, and addressing the fundamental mechanisms balancing inflammation-resolution programmes in unique contexts and in distinct tissues may help refine strategies to improve health and limit cardiovascular disease.
First, the inflammation-resolution field needs more ‘accessible’ biomarkers. The discovery of SPMs paved a previously unchartered path towards our understanding of how the body resolves inflammation.7 Identification of SPMs in tissue by LC-MS/MS is expensive due to the required equipment and expertise. SPM ELISAs may be helpful to profile inflammation-resolution patterns in individuals and if used in a multiplexing format, may yield a more high-throughput readout for patient care. Quantitative PCR (qPCR) and RNAseq have become standard laboratory techniques. And so we should be asking the question, as to whether there are inflammation-resolution RNA signatures. Data could be interrogated with RNA sequencing in archived databases and validated in newly generated data sets, and potentially reveal a ‘resolution status’. Simple proof-of-concept experiments that directly compare self-limited vs. non-resolving inflammation can be profiled and used as a starting point to address this gap.
An additional open question is whether specific SNPs or epigenetic changes are associated with efficient inflammation resolution. Genetic and genomic patterns that may control inflammation-resolution are not known and if uncovered can provide strategies to (i) tailor current drugs for better use in humans or (ii) develop new personalized therapeutics to specifically enhance endogenous resolution. In humans, the innate inflammatory response can be assessed in vivo using models that include a systemic inflammatory reaction following endotoxin152,153 or a contained, sterile inflammatory reaction following a cantharidin-induced skin blister.154,155 Human skin blisters were used to stimulate self-limited acute inflammation in humans and this model revealed that there are individuals who resolve more swiftly than others in a manner that is associated with the presence of key SPMs and their receptor expression on cells within the exudate.156–158 A deeper exploration of the DNA within the cells may reveal important genetic or epigenetic clues regarding inflammation-resolution mechanisms in humans.
Another hypothetical question is, if SPMs are a new drug, then which SPM should we choose to treat atherosclerosis? SPMs have common and distinct functions. One of the most widely studied common functions is the ability of nearly all SPMs to enhance efferocytosis. A distinct function, as an example, is in the context of platelets. EPA-derived RvE1 limits ADP-induced platelet aggregation whereas, DHA-derived PD1 does not.159 However, DHA-derived RvD4 limits thrombosis is murine models,160 and whether other SPMs carry out this function is not known. Therefore, perhaps a combination of SPMs that can mitigate atherosclerotic plaque formation and limit platelet activation/thrombosis may be ideal.
Overall, atherosclerosis was previously thought to be an inevitable steady and degenerative accompaniment to ageing.161 Instead, we now appreciate that atherosclerosis develops episodically, can regress, and that lifestyle and medical measures can dramatically modulate the course of disease.161 Moreover, atherosclerosis is associated with defense mechanisms that have gone awry that ultimately lead to arterial damage.162,163 The discovery of SPMs has shed light on an entirely new way to control inflammation, enhance host defense and promote repair and so this is an exciting time for inflammation-resolution and atherosclerosis research. Nevertheless, the continued identification of biomarkers, cellular processes, and factors associated with inflammation-resolution may: (i) reveal new ways to treat atherosclerosis and (ii) help inform treatment in a highly patient-specific manner.
Conflict of interest: none declared.
Funding
This work was supported by the National Institutes of Health or NIH (HL119587 and HL141127 to G.F., and R35GM131842 to K.C.M.).
Contributor Information
Gabrielle Fredman, The Department of Molecular and Cellular Physiology, Albany Medical College, Albany, NY 12208, USA.
Katherine C MacNamara, The Department of Immunology and Infectious Disease, Albany Medical College, Albany, NY 12208, USA.
This article is part of the Spotlight Issue on Cardiovascular Immunology.
References
- 1. Libby P. The biology of atherosclerosis comes full circle: lessons for conquering cardiovascular disease. Nat Rev Cardiol 2021;18:683–684. [DOI] [PubMed] [Google Scholar]
- 2. Sampson UK, Fazio S, Linton MF. Residual cardiovascular risk despite optimal LDL cholesterol reduction with statins: the evidence, etiology, and therapeutic challenges. Curr Atheroscler Rep 2012;14:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ridker PM. Residual inflammatory risk: addressing the obverse side of the atherosclerosis prevention coin. Eur Heart J 2016;37:1720–1722. [DOI] [PubMed] [Google Scholar]
- 4. Pradhan AD, Aday AW, Rose LM, Ridker PM. Residual inflammatory risk on treatment with PCSK9 inhibition and statin therapy. Circulation 2018;138:141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ, Group CT. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 2017;377:1119–1131. [DOI] [PubMed] [Google Scholar]
- 6. Tardif JC, Kouz S, Waters DD, Bertrand OF, Diaz R, Maggioni AP, Pinto FJ, Ibrahim R, Gamra H, Kiwan GS, Berry C, Lopez-Sendon J, Ostadal P, Koenig W, Angoulvant D, Gregoire JC, Lavoie MA, Dube MP, Rhainds D, Provencher M, Blondeau L, Orfanos A, L'Allier PL, Guertin MC, Roubille F. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med 2019;381:2497–2505. [DOI] [PubMed] [Google Scholar]
- 7. Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014;510:92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Perretti M, Chiang N, La M, Fierro IM, Marullo S, Getting SJ, Solito E, Serhan CN. Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A4 receptor. Nat Med 2002;8:1296–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wallace JL, Ianaro A, Flannigan KL, Cirino G. Gaseous mediators in resolution of inflammation. Semin Immunol 2015;27:227–233. [DOI] [PubMed] [Google Scholar]
- 10. Shinohara M, Serhan CN. Novel endogenous proresolving molecules: essential fatty acid-derived and gaseous mediators in the resolution of inflammation. J Atheroscler Thromb 2016;23:655–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cronstein BN, Montesinos MC, Weissmann G. Salicylates and sulfasalazine, but not glucocorticoids, inhibit leukocyte accumulation by an adenosine-dependent mechanism that is independent of inhibition of prostaglandin synthesis and p105 of NFkappaB. Proc Natl Acad Sci USA 1999;96:6377–6381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fredman G, Li Y, Dalli J, Chiang N, Serhan CN. Self-limited versus delayed resolution of acute inflammation: temporal regulation of pro-resolving mediators and microRNA. Sci Rep 2012;2:639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chiang N, Fredman G, Backhed F, Oh SF, Vickery T, Schmidt BA, Serhan CN. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature 2012;484:524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chiang N, Serhan CN. Specialized pro-resolving mediator network: an update on production and actions. Essays Biochem 2020;64:443–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Serhan CN, Hamberg M, Samuelsson B. Lipoxins: novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc Natl Acad Sci USA 1984;81:5335–5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Serhan CN, Clish CB, Brannon J, Colgan SP, Chiang N, Gronert K. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J Exp Med 2000;192:1197–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, Moussignac RL. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J Exp Med 2002;196:1025–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hong S, Gronert K, Devchand PR, Moussignac RL, Serhan CN. Novel docosatrienes and 17S-resolvins generated from docosahexaenoic acid in murine brain, human blood, and glial cells. Autacoids in anti-inflammation. J Biol Chem 2003;278:14677–14687. [DOI] [PubMed] [Google Scholar]
- 19. Dalli J, Chiang N, Serhan CN. Identification of 14-series sulfido-conjugated mediators that promote resolution of infection and organ protection. Proc Natl Acad Sci USA 2014;111:E4753–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dalli J, Chiang N, Serhan CN. Elucidation of novel 13-series resolvins that increase with atorvastatin and clear infections. Nat Med 2015;21:1071–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Spite M, Norling LV, Summers L, Yang R, Cooper D, Petasis NA, Flower RJ, Perretti M, Serhan CN. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature 2009;461:1287–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fredman G, Kamaly N, Spolitu S, Milton J, Ghorpade D, Chiasson R, Kuriakose G, Perretti M, Farokzhad O, Tabas I. Targeted nanoparticles containing the proresolving peptide Ac2-26 protect against advanced atherosclerosis in hypercholesterolemic mice. Sci Transl Med 2015;7:275ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Drechsler M, de Jong R, Rossaint J, Viola JR, Leoni G, Wang JM, Grommes J, Hinkel R, Kupatt C, Weber C, Doring Y, Zarbock A, Soehnlein O. Annexin A1 counteracts chemokine-induced arterial myeloid cell recruitment. Circ Res 2015;116:827–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kamaly N, Fredman G, Fojas JJ, Subramanian M, Choi WI, Zepeda K, Vilos C, Yu M, Gadde S, Wu J, Milton J, Carvalho Leitao R, Rosa Fernandes L, Hasan M, Gao H, Nguyen V, Harris J, Tabas I, Farokhzad OC. Targeted interleukin-10 nanotherapeutics developed with a microfluidic chip enhance resolution of inflammation in advanced atherosclerosis. ACS Nano 2016;10:5280–5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kusters DH, Chatrou ML, Willems BA, De Saint-Hubert M, Bauwens M, van der Vorst E, Bena S, Biessen EA, Perretti M, Schurgers LJ, Reutelingsperger CP. Pharmacological treatment with annexin A1 reduces atherosclerotic plaque burden in LDLR-/- mice on western type diet. PLoS One 2015;10:e0130484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hasturk H, Abdallah R, Kantarci A, Nguyen D, Giordano N, Hamilton J, Van Dyke TE. Resolvin E1 (RvE1) attenuates atherosclerotic plaque formation in diet and inflammation-induced atherogenesis. Arterioscler Thromb Vasc Biol 2015;35:1123–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Salic K, Morrison MC, Verschuren L, Wielinga PY, Wu L, Kleemann R, Gjorstrup P, Kooistra T. Resolvin E1 attenuates atherosclerosis in absence of cholesterol-lowering effects and on top of atorvastatin. Atherosclerosis 2016;250:158–165. [DOI] [PubMed] [Google Scholar]
- 28. Laguna-Fernandez A, Checa A, Carracedo M, Artiach G, Petri MH, Baumgartner R, Forteza MJ, Jiang X, Andonova T, Walker ME, Dalli J, Arnardottir H, Gisterå A, Thul S, Wheelock CE, Paulsson-Berne G, Ketelhuth DFJ, Hansson GK, Bäck M. ERV1/ChemR23 signaling protects against atherosclerosis by modifying oxidized low-density lipoprotein uptake and phagocytosis in macrophages. Circulation 2018;138:1693–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fredman G, Hellmann J, Proto JD, Kuriakose G, Colas RA, Dorweiler B, Connolly ES, Solomon R, Jones DM, Heyer EJ, Spite M, Tabas I. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat Commun 2016;7:12859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Viola JR, Lemnitzer P, Jansen Y, Csaba G, Winter C, Neideck C, Silvestre-Roig C, Dittmar G, Doring Y, Drechsler M, Weber C, Zimmer R, Cenac N, Soehnlein O. Resolving lipid mediators maresin 1 and resolvin D2 prevent atheroprogression in mice. Circ Res 2016;119:1030–1038. [DOI] [PubMed] [Google Scholar]
- 31. Petri MH, Laguna-Fernandez A, Arnardottir H, Wheelock CE, Perretti M, Hansson GK, Back M. Aspirin-triggered lipoxin inhibits atherosclerosis progression in ApoE-/- mice. Br J Pharmacol 2017;174:4043–4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fiore S, Maddox JF, Perez HD, Serhan CN. Identification of a human cDNA encoding a functional high affinity lipoxin A4 receptor. J Exp Med 1994;180:253–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Arita M, Bianchini F, Aliberti J, Sher A, Chiang N, Hong S, Yang R, Petasis NA, Serhan CN. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J Exp Med 2005;201:713–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Krishnamoorthy S, Recchiuti A, Chiang N, Fredman G, Serhan CN. Resolvin D1 receptor stereoselectivity and regulation of inflammation and proresolving microRNAs. Am J Pathol 2012;180:2018–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chiang N, Serhan CN. Structural elucidation and physiologic functions of specialized pro-resolving mediators and their receptors. Mol Aspects Med 2017;58:114–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Flak MB, Koenis DS, Sobrino A, Smith J, Pistorius K, Palmas F, Dalli J. GPR101 mediates the pro-resolving actions of RvD5n-3 DPA in arthritis and infections. J Clin Invest 2020;130:359–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rathod KS, Kapil V, Velmurugan S, Khambata RS, Siddique U, Khan S, Van Eijl S, Gee LC, Bansal J, Pitrola K, Shaw C, D'Acquisto F, Colas RA, Marelli-Berg F, Dalli J, Ahluwalia A. Accelerated resolution of inflammation underlies sex differences in inflammatory responses in humans. J Clin Invest 2017;127:169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Krishnamoorthy S, Recchiuti A, Chiang N, Yacoubian S, Lee CH, Yang R, Petasis NA, Serhan CN. Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc Natl Acad Sci USA 2010;107:1660–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dufton N, Hannon R, Brancaleone V, Dalli J, Patel HB, Gray M, D'Acquisto F, Buckingham JC, Perretti M, Flower RJ. Anti-inflammatory role of the murine formyl-peptide receptor 2: ligand-specific effects on leukocyte responses and experimental inflammation. J Immunol 2010;184:2611–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gobbetti T, Coldewey SM, Chen J, McArthur S, Le Faouder P, Cenac N, Flower RJ, Thiemermann C, Perretti M. Nonredundant protective properties of FPR2/ALX in polymicrobial murine sepsis. Proc Natl Acad Sci USA 2014;111:18685–18690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chiang N, Dalli J, Colas RA, Serhan CN. Identification of resolvin D2 receptor mediating resolution of infections and organ protection. J Exp Med 2015;212:1203–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chiang N, Libreros S, Norris PC, de la Rosa X, Serhan CN. Maresin 1 activates LGR6 receptor promoting phagocyte immunoresolvent functions. J Clin Invest 2019;129:5294–5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Miyahara T, Runge S, Chatterjee A, Chen M, Mottola G, Fitzgerald JM, Serhan CN, Conte MS. D-series resolvin attenuates vascular smooth muscle cell activation and neointimal hyperplasia following vascular injury. FASEB J 2013;27:2220–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wu B, Mottola G, Chatterjee A, Lance KD, Chen M, Siguenza IO, Desai TA, Conte MS. Perivascular delivery of resolvin D1 inhibits neointimal hyperplasia in a rat model of arterial injury. J Vasc Surg 2017;65:207–217.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kain V, Ingle KA, Colas RA, Dalli J, Prabhu SD, Serhan CN, Joshi M, Halade GV. Resolvin D1 activates the inflammation resolving response at splenic and ventricular site following myocardial infarction leading to improved ventricular function. J Mol Cell Cardiol 2015;84:24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Keyes KT, Ye Y, Lin Y, Zhang C, Perez-Polo JR, Gjorstrup P, Birnbaum Y. Resolvin E1 protects the rat heart against reperfusion injury. Am J Physiol Heart Circ Physiol 2010;299:H153– H164. [DOI] [PubMed] [Google Scholar]
- 47. Merched AJ, Ko K, Gotlinger KH, Serhan CN, Chan L. Atherosclerosis: evidence for impairment of resolution of vascular inflammation governed by specific lipid mediators. FASEB J 2008;22:3595–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bunting S, Moncada S, Vane JR. The prostacyclin–thromboxane A2 balance: pathophysiological and therapeutic implications. Br Med Bull 1983;39:271–276. [DOI] [PubMed] [Google Scholar]
- 49. Vane JR. Biomedicine. Back to an aspirin a day? Science 2002;296:474–475. [DOI] [PubMed] [Google Scholar]
- 50. Brezinski DA, Nesto RW, Serhan CN. Angioplasty triggers intracoronary leukotrienes and lipoxin A4. Impact of aspirin therapy. Circulation 1992;86:56–63. [DOI] [PubMed] [Google Scholar]
- 51. Thul S, Labat C, Temmar M, Benetos A, Back M. Low salivary resolvin D1 to leukotriene B4 ratio predicts carotid intima media thickness: a novel biomarker of non-resolving vascular inflammation. Eur J Prev Cardiol 2017;24:903–906. [DOI] [PubMed] [Google Scholar]
- 52. Bazan HA, Lu Y, Jun B, Fang Z, Woods TC, Hong S. Circulating inflammation-resolving lipid mediators RvD1 and DHA are decreased in patients with acutely symptomatic carotid disease. Prostaglandins Leukot Essent Fatty Acids 2017;125:43–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Artiach G, Carracedo M, Plunde O, Wheelock CE, Thul S, Sjövall P, Franco-Cereceda A, Laguna-Fernandez A, Arnardottir H, Bäck M. Omega-3 polyunsaturated fatty acids decrease aortic valve disease through the resolvin E1 and ChemR23 Axis. Circulation 2020;142:776–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Claria J, Dalli J, Yacoubian S, Gao F, Serhan CN. Resolvin d1 and resolvin d2 govern local inflammatory tone in obese fat. J Immunol 2012;189:2597–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fredman G, Ozcan L, Spolitu S, Hellmann J, Spite M, Backs J, Tabas I. Resolvin D1 limits 5-lipoxygenase nuclear localization and leukotriene B4 synthesis by inhibiting a calcium-activated kinase pathway. Proc Natl Acad Sci USA 2014;111:14530–14535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Woods JW, Evans JF, Ethier D, Scott S, Vickers PJ, Hearn L, Heibein JA, Charleson S, Singer II. 5-lipoxygenase and 5-lipoxygenase-activating protein are localized in the nuclear envelope of activated human leukocytes. J Exp Med 1993;178:1935–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Serhan CN, Haeggstrom JZ, Leslie CC. Lipid mediator networks in cell signaling: update and impact of cytokines. FASEB J 1996;10:1147–1158. [DOI] [PubMed] [Google Scholar]
- 58. Liu T, Xiang A, Peng T, Doran AC, Tracey KJ, Barnes BJ, Tabas I, Son M, Diamond B. HMGB1-C1q complexes regulate macrophage function by switching between leukotriene and specialized proresolving mediator biosynthesis. Proc Natl Acad Sci USA 2019;116:23254–23263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Skaggs BJ, Hahn BH, McMahon M. Accelerated atherosclerosis in patients with SLE–mechanisms and management. Nat Rev Rheumatol 2012;8:214–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Norris PC, Gosselin D, Reichart D, Glass CK, Dennis EA. Phospholipase A2 regulates eicosanoid class switching during inflammasome activation. Proc Natl Acad Sci USA 2014;111:12746–12751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dalli J, Serhan CN. Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood 2012;120:e60–e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Werz O, Gerstmeier J, Libreros S, De la Rosa X, Werner M, Norris PC, Chiang N, Serhan CN. Human macrophages differentially produce specific resolvin or leukotriene signals that depend on bacterial pathogenicity. Nat Commun 2018;9:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sharma M, Schlegel MP, Afonso MS, Brown EJ, Rahman K, Weinstock A, Sansbury BE, Corr EM, van Solingen C, Koelwyn GJ, Shanley LC, Beckett L, Peled D, Lafaille JJ, Spite M, Loke P, Fisher EA, Moore KJ. Regulatory T cells license macrophage pro-resolving functions during atherosclerosis regression. Circ Res 2020;127:335–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rahman K, Vengrenyuk Y, Ramsey SA, Vila NR, Girgis NM, Liu J, Gusarova V, Gromada J, Weinstock A, Moore KJ, Loke P, Fisher EA. Inflammatory Ly6Chi monocytes and their conversion to M2 macrophages drive atherosclerosis regression. J Clin Invest 2017;127:2904–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lehmann C, Homann J, Ball AK, Blocher R, Kleinschmidt TK, Basavarajappa D, Angioni C, Ferreiros N, Hafner AK, Radmark O, Proschak E, Haeggstrom JZ, Geisslinger G, Parnham MJ, Steinhilber D, Kahnt AS. Lipoxin and resolvin biosynthesis is dependent on 5-lipoxygenase activating protein. FASEB J 2015;29:5029–5043. [DOI] [PubMed] [Google Scholar]
- 66. Elias I, Ferre T, Vila L, Munoz S, Casellas A, Garcia M, Molas M, Agudo J, Roca C, Ruberte J, Bosch F, Franckhauser S. Alox5ap overexpression in adipose tissue leads to LXA4 production and protection against diet-induced obesity and insulin resistance. Diabetes 2016;65:2139–2150. [DOI] [PubMed] [Google Scholar]
- 67. Qiu H, Gabrielsen A, Agardh HE, Wan M, Wetterholm A, Wong C-H, Hedin U, Swedenborg J, Hansson GK, Samuelsson B, Paulsson-Berne G, Haeggström JZ. Expression of 5-lipoxygenase and leukotriene A4 hydrolase in human atherosclerotic lesions correlates with symptoms of plaque instability. Proc Natl Acad Sci USA 2006;103:8161–8166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 2007;447:869–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cai B, Thorp EB, Doran AC, Subramanian M, Sansbury BE, Lin CS, Spite M, Fredman G, Tabas I. MerTK cleavage limits proresolving mediator biosynthesis and exacerbates tissue inflammation. Proc Natl Acad Sci USA 2016;113:6526–6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cai B, Kasikara C, Doran AC, Ramakrishnan R, Birge RB, Tabas I. MerTK signaling in macrophages promotes the synthesis of inflammation resolution mediators by suppressing CaMKII activity. Sci Signal 2018;11:eaar3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kourtzelis I, Li X, Mitroulis I, Grosser D, Kajikawa T, Wang B, Grzybek M, von Renesse J, Czogalla A, Troullinaki M, Ferreira A, Doreth C, Ruppova K, Chen LS, Hosur K, Lim JH, Chung KJ, Grossklaus S, Tausche AK, Joosten LAB, Moutsopoulos NM, Wielockx B, Castrillo A, Korostoff JM, Coskun U, Hajishengallis G, Chavakis T. DEL-1 promotes macrophage efferocytosis and clearance of inflammation. Nat Immunol 2019;20:40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Welty FK, Schulte F, Alfaddagh A, Elajami TK, Bistrian BR, Hardt M. Regression of human coronary artery plaque is associated with a high ratio of (18-hydroxy-eicosapentaenoic acid + resolvin E1) to leukotriene B4. FASEB J 2021;35:e21448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bhatt DL, Steg PG, Miller M, Brinton EA, Jacobson TA, Ketchum SB, Doyle RT, Juliano RA, Jiao L, Granowitz C, Tardif J-C, Ballantyne CM; REDUCE-IT Investigators. Cardiovascular risk reduction with icosapent ethyl for hypertriglyceridemia. N Engl J Med 2019;380:11–22. [DOI] [PubMed] [Google Scholar]
- 74. Budoff MJ, Bhatt DL, Kinninger A, Lakshmanan S, Muhlestein JB, Le VT, May HT, Shaikh K, Shekar C, Roy SK, Tayek J, Nelson JR. Effect of icosapent ethyl on progression of coronary atherosclerosis in patients with elevated triglycerides on statin therapy: final results of the EVAPORATE trial. Eur Heart J 2020;41:3925–3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Nicholls SJ, Lincoff AM, Garcia M, Bash D, Ballantyne CM, Barter PJ, Davidson MH, Kastelein JJP, Koenig W, McGuire DK, Mozaffarian D, Ridker PM, Ray KK, Katona BG, Himmelmann A, Loss LE, Rensfeldt M, Lundstrom T, Agrawal R, Menon V, Wolski K, Nissen SE. Effect of high-dose omega-3 fatty acids vs corn oil on major adverse cardiovascular events in patients at high cardiovascular risk: the STRENGTH Randomized Clinical Trial. JAMA 2020;324:2268–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gerster H. Can adults adequately convert alpha-linolenic acid (18:3n-3) to eicosapentaenoic acid (20:5n-3) and docosahexaenoic acid (22:6n-3)? Int J Vitam Nutr Res 1998;68:159–173. [PubMed] [Google Scholar]
- 77. Dwyer JH, Allayee H, Dwyer KM, Fan J, Wu H, Mar R, Lusis AJ, Mehrabian M. Arachidonate 5-lipoxygenase promoter genotype, dietary arachidonic acid, and atherosclerosis. N Engl J Med 2004;350:29–37. [DOI] [PubMed] [Google Scholar]
- 78. Hsiao HM, Sapinoro RE, Thatcher TH, Croasdell A, Levy EP, Fulton RA, Olsen KC, Pollock SJ, Serhan CN, Phipps RP, Sime PJ. A novel anti-inflammatory and pro-resolving role for resolvin D1 in acute cigarette smoke-induced lung inflammation. PLoS One 2013;8:e58258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hellmann J, Tang Y, Kosuri M, Bhatnagar A, Spite M. Resolvin D1 decreases adipose tissue macrophage accumulation and improves insulin sensitivity in obese-diabetic mice. FASEB J 2011;25:2399–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Murphy AJ, Tall AR. Disordered haematopoiesis and athero-thrombosis. Eur Heart J 2016;37:1113–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest 2007;117:195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Koltsova EK, Hedrick CC, Ley K. Myeloid cells in atherosclerosis: a delicate balance of anti-inflammatory and proinflammatory mechanisms. Curr Opin Lipidol 2013;24:371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Stiekema LCA, Willemsen L, Kaiser Y, Prange KHM, Wareham NJ, Boekholdt SM, Kuijk C, de Winther MPJ, Voermans C, Nahrendorf M, Stroes ESG, Kroon J. Impact of cholesterol on proinflammatory monocyte production by the bone marrow. Eur Heart J 2021;doi: 10.1093/eurheartj/ehab465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kawaguchi H, Mori T, Kawano T, Kono S, Sasaki J, Arakawa K. Band neutrophil count and the presence and severity of coronary atherosclerosis. Am Heart J 1996;132:9–12. [DOI] [PubMed] [Google Scholar]
- 85. Ionita MG, van den Borne P, Catanzariti LM, Moll FL, de Vries JP, Pasterkamp G, Vink A, de Kleijn DP. High neutrophil numbers in human carotid atherosclerotic plaques are associated with characteristics of rupture-prone lesions. Arterioscler Thromb Vasc Biol 2010;30:1842–1848. [DOI] [PubMed] [Google Scholar]
- 86. Silvestre-Roig C, Braster Q, Ortega-Gomez A, Soehnlein O. Neutrophils as regulators of cardiovascular inflammation. Nat Rev Cardiol 2020;17:327–340. [DOI] [PubMed] [Google Scholar]
- 87. Doring Y, Libby P, Soehnlein O. Neutrophil extracellular traps participate in cardiovascular diseases: recent experimental and clinical insights. Circ Res 2020;126:1228–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Vinholt PJ, Hvas AM, Frederiksen H, Bathum L, Jorgensen MK, Nybo M. Platelet count is associated with cardiovascular disease, cancer and mortality: a population-based cohort study. Thromb Res 2016;148:136–142. [DOI] [PubMed] [Google Scholar]
- 89. Murphy AJ, Bijl N, Yvan-Charvet L, Welch CB, Bhagwat N, Reheman A, Wang Y, Shaw JA, Levine RL, Ni H, Tall AR, Wang N. Cholesterol efflux in megakaryocyte progenitors suppresses platelet production and thrombocytosis. Nat Med 2013;19:586–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Poller WC, Nahrendorf M, Swirski FK. Hematopoiesis and cardiovascular disease. Circ Res 2020;126:1061–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Mozzini C, Pagani M. Clonal hematopoiesis and cardiovascular diseaseas: the connection. Curr Probl Cardiol 2021;100962. [DOI] [PubMed] [Google Scholar]
- 92. Fawaz S, Mansier O, Pucheu Y, Marti S, Leroy H, Gaufroy A, Broitman J, James C, Couffinhal T. Clonal haematopoiesis and cardiovascular diseases: a growing relationship. Arch Cardiovasc Dis 2021;114:316–324. [DOI] [PubMed] [Google Scholar]
- 93. Jung C, Evans MA, Walsh K. Genetics of age-related clonal hematopoiesis and atherosclerotic cardiovascular disease. Curr Opin Cardiol 2020;35:219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Amorós-Pérez M, Fuster JJ. Clonal hematopoiesis driven by somatic mutations: a new player in atherosclerotic cardiovascular disease. Atherosclerosis 2020;297:120–126. [DOI] [PubMed] [Google Scholar]
- 95. Casanova-Acebes M, Pitaval C, Weiss LA, Nombela-Arrieta C, Chèvre R, A-González N, Kunisaki Y, Zhang D, van Rooijen N, Silberstein LE, Weber C, Nagasawa T, Frenette PS, Castrillo A, Hidalgo A. Rhythmic modulation of the hematopoietic niche through neutrophil clearance. Cell 2013;153:1025–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. McCauley LK, Dalli J, Koh AJ, Chiang N, Serhan CN. Cutting edge: parathyroid hormone facilitates macrophage efferocytosis in bone marrow via proresolving mediators resolvin D1 and resolvin D2. J Immunol 2014;193:26–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Michalski MN, Koh AJ, Weidner S, Roca H, McCauley LK. Modulation of osteoblastic cell efferocytosis by bone marrow macrophages. J Cell Biochem 2016;117:2697–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Jimenez-Torres J, Alcalá-Diaz JF, Torres-Peña JD, Gutierrez-Mariscal FM, Leon-Acuña A, Gómez-Luna P, Fernández-Gandara C, Quintana-Navarro GM, Fernandez-Garcia JC, Perez-Martinez P, Ordovas JM, Delgado-Lista J, Yubero-Serrano EM, Lopez-Miranda J. Mediterranean diet reduces atherosclerosis progression in coronary heart disease: an analysis of the CORDIOPREV randomized controlled trial. Stroke 2021;doi:10.1161/STROKEAHA.120.033214. [DOI] [PubMed] [Google Scholar]
- 99. Lee DC, Pate RR, Lavie CJ, Sui X, Church TS, Blair SN. Leisure-time running reduces all-cause and cardiovascular mortality risk. J Am Coll Cardiol 2014;64:472–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Frodermann V, Rohde D, Courties G, Severe N, Schloss MJ, Amatullah H, McAlpine CS, Cremer S, Hoyer FF, Ji F, van Koeverden ID, Herisson F, Honold L, Masson GS, Zhang S, Grune J, Iwamoto Y, Schmidt SP, Wojtkiewicz GR, Lee IH, Gustafsson K, Pasterkamp G, de Jager SCA, Sadreyev RI, MacFadyen J, Libby P, Ridker P, Scadden DT, Naxerova K, Jeffrey KL, Swirski FK, Nahrendorf M. Exercise reduces inflammatory cell production and cardiovascular inflammation via instruction of hematopoietic progenitor cells. Nat Med 2019;25:1761–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zheng JJ, Pena Calderin E, Hill BG, Bhatnagar A, Hellmann J. Exercise promotes resolution of acute inflammation by catecholamine-mediated stimulation of resolvin D1 biosynthesis. J Immunol 2019;203:3013–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Varney ME, Buchanan JT, Dementieva Y, Hardman WE, Sollars VE. A high omega-3 fatty acid diet has different effects on early and late stage myeloid progenitors. Lipids 2011;46:47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Varney ME, Hardman WE, Sollars VE. Omega 3 fatty acids reduce myeloid progenitor cell frequency in the bone marrow of mice and promote progenitor cell differentiation. Lipids Health Dis 2009;8:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Christ A, Günther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, Scholz CJ, Oosting M, Haendler K, Baßler K, Klee K, Schulte-Schrepping J, Ulas T, Moorlag SJCFM, Kumar V, Park MH, Joosten LAB, Groh LA, Riksen NP, Espevik T, Schlitzer A, Li Y, Fitzgerald ML, Netea MG, Schultze JL, Latz E. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell 2018;172:162–175.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Fidler TP, Xue C, Yalcinkaya M, Hardaway B, Abramowicz S, Xiao T, Liu W, Thomas DG, Hajebrahimi MA, Pircher J, Silvestre-Roig C, Kotini AG, Luchsinger LL, Wei Y, Westerterp M, Snoeck HW, Papapetrou EP, Schulz C, Massberg S, Soehnlein O, Ebert B, Levine RL, Reilly MP, Libby P, Wang N, Tall AR. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature 2021;592:296–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Souza PR, Marques RM, Gomez EA, Colas RA, De Matteis R, Zak A, Patel M, Collier DJ, Dalli J. Enriched marine oil supplements increase peripheral blood specialized pro-resolving mediators concentrations and reprogram host immune responses: a randomized double-blind placebo-controlled study. Circ Res 2020;126:75–90. [DOI] [PubMed] [Google Scholar]
- 107. Jordan S, Tung N, Casanova-Acebes M, Chang C, Cantoni C, Zhang D, Wirtz TH, Naik S, Rose SA, Brocker CN, Gainullina A, Hornburg D, Horng S, Maier BB, Cravedi P, LeRoith D, Gonzalez FJ, Meissner F, Ochando J, Rahman A, Chipuk JE, Artyomov MN, Frenette PS, Piccio L, Berres ML, Gallagher EJ, Merad M. Dietary intake regulates the circulating inflammatory monocyte pool. Cell 2019;178:1102–1114.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Steffens S, Winter C, Schloss MJ, Hidalgo A, Weber C, Soehnlein O. Circadian control of inflammatory processes in atherosclerosis and its complications. Arterioscler Thromb Vasc Biol 2017;37:1022–1028. [DOI] [PubMed] [Google Scholar]
- 109. Scheiermann C, Kunisaki Y, Lucas D, Chow A, Jang JE, Zhang D, Hashimoto D, Merad M, Frenette PS. Adrenergic nerves govern circadian leukocyte recruitment to tissues. Immunity 2012;37:290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Massberg S, Schaerli P, Knezevic-Maramica I, Kollnberger M, Tubo N, Moseman EA, Huff IV, Junt T, Wagers AJ, Mazo IB, von Andrian UH. Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell 2007;131:994–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. McAlpine CS, Kiss MG, Rattik S, He S, Vassalli A, Valet C, Anzai A, Chan CT, Mindur JE, Kahles F, Poller WC, Frodermann V, Fenn AM, Gregory AF, Halle L, Iwamoto Y, Hoyer FF, Binder CJ, Libby P, Tafti M, Scammell TE, Nahrendorf M, Swirski FK. Sleep modulates haematopoiesis and protects against atherosclerosis. Nature 2019;566:383–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Heidt T, Sager HB, Courties G, Dutta P, Iwamoto Y, Zaltsman A, von Zur Muhlen C, Bode C, Fricchione GL, Denninger J, Lin CP, Vinegoni C, Libby P, Swirski FK, Weissleder R, Nahrendorf M. Chronic variable stress activates hematopoietic stem cells. Nat Med 2014;20:754–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Schober A, Blay RM, Saboor Maleki S, Zahedi F, Winklmaier AE, Kakar MY, Baatsch IM, Zhu M, Geißler C, Fusco AE, Eberlein A, Li N, Megens RTA, Banafsche R, Kumbrink J, Weber C, Nazari-Jahantigh M. MicroRNA-21 controls circadian regulation of apoptosis in atherosclerotic lesions. Circulation 2021;144:1059–1073. [DOI] [PubMed] [Google Scholar]
- 114. Griffin P, Sheehan PW, Dimitry JM, Guo C, Kanan MF, Lee J, Zhang J, Musiek ES. REV-ERBα mediates complement expression and diurnal regulation of microglial synaptic phagocytosis. Elife 2020;9:e58765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Colas RA, Souza PR, Walker ME, Burton M, Zasłona Z, Curtis AM, Marques RM, Dalli J. Impaired production and diurnal regulation of vascular RvDn-3 DPA increase systemic inflammation and cardiovascular disease. Circ Res 2018;122:855–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Tyrrell DJ, Goldstein DR. Ageing and atherosclerosis: vascular intrinsic and extrinsic factors and potential role of IL-6. Nat Rev Cardiol 2021;18:58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Ehrhardt N, Cui J, Dagdeviren S, Saengnipanthkul S, Goodridge HS, Kim JK, Lantier L, Guo X, Chen YI, Raffel LJ, Buchanan TA, Hsueh WA, Rotter JI, Goodarzi MO, Peterfy M. Adiposity-independent effects of aging on insulin sensitivity and clearance in mice and humans. Obesity (Silver Spring) 2019;27:434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Du W, Wong C, Song Y, Shen H, Mori D, Rotllan N, Price N, Dobrian AD, Meng H, Kleinstein SH, Fernandez-Hernando C, Goldstein DR. Age-associated vascular inflammation promotes monocytosis during atherogenesis. Aging Cell 2016;15:766–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Beerman I, Bhattacharya D, Zandi S, Sigvardsson M, Weissman IL, Bryder D, Rossi DJ. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc Natl Acad Sci USA 2010;107:5465–5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Challen GA, Boles NC, Chambers SM, Goodell MA. Distinct hematopoietic stem cell subtypes are differentially regulated by TGF-beta1. Cell Stem Cell 2010;6:265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Young K, Borikar S, Bell R, Kuffler L, Philip V, Trowbridge JJ. Progressive alterations in multipotent hematopoietic progenitors underlie lymphoid cell loss in aging. J Exp Med 2016;213:2259–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Young K, Eudy E, Bell R, Loberg MA, Stearns T, Sharma D, Velten L, Haas S, Filippi MD, Trowbridge JJ. Decline in IGF1 in the bone marrow microenvironment initiates hematopoietic stem cell aging. Cell Stem Cell 2021;28:1473–1482.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Montgomery RR, Shaw AC. Paradoxical changes in innate immunity in aging: recent progress and new directions. J Leukoc Biol 2015;98:937–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Nikolich-Zugich J, Bradshaw CM, Uhrlaub JL, Watanabe M. Immunity to acute virus infections with advanced age. Curr Opin Virol 2021;46:45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Channappanavar R, Perlman S. Age-related susceptibility to coronavirus infections: role of impaired and dysregulated host immunity. J Clin Invest 2020;130:6204–6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Yousefzadeh MJ, Flores RR, Zhu Y, Schmiechen ZC, Brooks RW, Trussoni CE, Cui Y, Angelini L, Lee KA, McGowan SJ, Burrack AL, Wang D, Dong Q, Lu A, Sano T, O'Kelly RD, McGuckian CA, Kato JI, Bank MP, Wade EA, Pillai SPS, Klug J, Ladiges WC, Burd CE, Lewis SE, LaRusso NF, Vo NV, Wang Y, Kelley EE, Huard J, Stromnes IM, Robbins PD, Niedernhofer LJ. An aged immune system drives senescence and ageing of solid organs. Nature 2021;594:100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007;8:729–740. [DOI] [PubMed] [Google Scholar]
- 128. Montero-Melendez T, Nagano A, Chelala C, Filer A, Buckley CD, Perretti M. Therapeutic senescence via GPCR activation in synovial fibroblasts facilitates resolution of arthritis. Nat Commun 2020;11:745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol 2013;75:685–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Rymut N, Heinz J, Sadhu S, Hosseini Z, Riley CO, Marinello M, Maloney J, MacNamara KC, Spite M, Fredman G. Resolvin D1 promotes efferocytosis in aging by limiting senescent cell-induced MerTK cleavage. FASEB J 2020;34:597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Wang JC, Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res 2012;111:245–259. [DOI] [PubMed] [Google Scholar]
- 132. Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation 2002;105:1541–1544. [DOI] [PubMed] [Google Scholar]
- 133. Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016;354:472–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Sadhu S, Decker C, Sansbury BE, Marinello M, Seyfried A, Howard J, Mori M, Hosseini Z, Arunachalam T, Finn AV, Lamar JM, Jourd’heuil D, Guo L, MacNamara KC, Spite M, Fredman G. Radiation-induced macrophage senescence impairs resolution programs and drives cardiovascular inflammation. J Immunol 2021;207:1812–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Arnardottir HH, Dalli J, Colas RA, Shinohara M, Serhan CN. Aging delays resolution of acute inflammation in mice: reprogramming the host response with novel nano-proresolving medicines. J Immunol 2014;193:4235–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Kirkland JL, Tchkonia T, Zhu Y, Niedernhofer LJ, Robbins PD. The clinical potential of senolytic drugs. J Am Geriatr Soc 2017;65:2297–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Flanagan EW, Most J, Mey JT, Redman LM. Calorie restriction and aging in humans. Annu Rev Nutr 2020;40:105–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Ferraro RA, Pallazola VA, Michos ED. Physical activity, CVD, and older adults. Aging (Albany NY) 2019;11:2545–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Gangemi S, Pescara L, D'Urbano E, Basile G, Nicita-Mauro V, Davì G, Romano M. Aging is characterized by a profound reduction in anti-inflammatory lipoxin A4 levels. Exp Gerontol 2005;40:612–614. [DOI] [PubMed] [Google Scholar]
- 140. Aprahamian T, Rifkin I, Bonegio R, Hugel B, Freyssinet JM, Sato K, Castellot JJ Jr, Walsh K. Impaired clearance of apoptotic cells promotes synergy between atherogenesis and autoimmune disease. J Exp Med 2004;199:1121–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Aprahamian T, Takemura Y, Goukassian D, Walsh K. Ageing is associated with diminished apoptotic cell clearance in vivo. Clin Exp Immunol 2008;152:448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Linehan E, Dombrowski Y, Snoddy R, Fallon PG, Kissenpfennig A, Fitzgerald DC. Aging impairs peritoneal but not bone marrow-derived macrophage phagocytosis. Aging Cell 2014;13:699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Frisch BJ, Hoffman CM, Latchney SE, LaMere MW, Myers J, Ashton J, Li AJ, Saunders J 2nd, Palis J, Perkins AS, McCabe A, Smith JN, McGrath KE, Rivera-Escalera F, McDavid A, Liesveld JL, Korshunov VA, Elliott MR, MacNamara KC, Becker MW, Calvi LM. Aged marrow macrophages expand platelet-biased hematopoietic stem cells via interleukin1B. JCI Insight 2019;4:e124213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Kulkarni U, Zemans RL, Smith CA, Wood SC, Deng JC, Goldstein DR. Excessive neutrophil levels in the lung underlie the age-associated increase in influenza mortality. Mucosal Immunol 2019;12:545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Wong CK, Smith CA, Sakamoto K, Kaminski N, Koff JL, Goldstein DR. Aging impairs alveolar macrophage phagocytosis and increases influenza-induced mortality in mice. J Immunol 2017;199:1060–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. De Maeyer RPH, van de Merwe RC, Louie R, Bracken OV, Devine OP, Goldstein DR, Uddin M, Akbar AN, Gilroy DW. Blocking elevated p38 MAPK restores efferocytosis and inflammatory resolution in the elderly. Nat Immunol 2020;21:615–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Gilroy DW, De Maeyer RPH, Tepper M, O’Brien A, Uddin M, Chen J, Goldstein DR, Akbar AN. Treating exuberant, non-resolving inflammation in the lung; Implications for acute respiratory distress syndrome and COVID-19. Pharmacol Ther 2021;221:107745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Wang Y, Schulte BA, LaRue AC, Ogawa M, Zhou D. Total body irradiation selectively induces murine hematopoietic stem cell senescence. Blood 2006;107:358–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Shao L, Feng W, Li H, Gardner D, Luo Y, Wang Y, Liu L, Meng A, Sharpless NE, Zhou D. Total body irradiation causes long-term mouse BM injury via induction of HSC premature senescence in an Ink4a- and Arf-independent manner. Blood 2014;123:3105–3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W, Luo Y, Wang X, Aykin-Burns N, Krager K, Ponnappan U, Hauer-Jensen M, Meng A, Zhou D. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med 2016;22:78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Morita M, Kuba K, Ichikawa A, Nakayama M, Katahira J, Iwamoto R, Watanebe T, Sakabe S, Daidoji T, Nakamura S, Kadowaki A, Ohto T, Nakanishi H, Taguchi R, Nakaya T, Murakami M, Yoneda Y, Arai H, Kawaoka Y, Penninger JM, Arita M, Imai Y. The lipid mediator protectin D1 inhibits influenza virus replication and improves severe influenza. Cell 2013;153:112–125. [DOI] [PubMed] [Google Scholar]
- 152. Michel O, Nagy AM, Schroeven M, Duchateau J, Neve J, Fondu P, Sergysels R. Dose-response relationship to inhaled endotoxin in normal subjects. Am J Respir Crit Care Med 1997;156:1157–1164. [DOI] [PubMed] [Google Scholar]
- 153. Patel PN, Shah RY, Ferguson JF, Reilly MP. Human experimental endotoxemia in modeling the pathophysiology, genomics, and therapeutics of innate immunity in complex cardiometabolic diseases. Arterioscler Thromb Vasc Biol 2015;35:525–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Day RM, Harbord M, Forbes A, Segal AW. Cantharidin blisters: a technique for investigating leukocyte trafficking and cytokine production at sites of inflammation in humans. J Immunol Methods 2001;257:213–220. [DOI] [PubMed] [Google Scholar]
- 155. Jenner WJ, Gilroy DW. Assessment of leukocyte trafficking in humans using the cantharidin blister model. JRSM Cardiovasc Dis 2012;1:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Morris T, Stables M, Colville-Nash P, Newson J, Bellingan G, de Souza PM, Gilroy DW. Dichotomy in duration and severity of acute inflammatory responses in humans arising from differentially expressed proresolution pathways. Proc Natl Acad Sci USA 2010;107:8842–8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Morris T, Stables M, Hobbs A, de Souza P, Colville-Nash P, Warner T, Newson J, Bellingan G, Gilroy DW. Effects of low-dose aspirin on acute inflammatory responses in humans. J Immunol 2009;183:2089–2096. [DOI] [PubMed] [Google Scholar]
- 158. Motwani MP, Colas RA, George MJ, Flint JD, Dalli J, Richard-Loendt A, De Maeyer RP, Serhan CN, Gilroy DW. Pro-resolving mediators promote resolution in a human skin model of UV-killed Escherichia coli-driven acute inflammation. JCI Insight 2018;3:e94463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Dona M, Fredman G, Schwab JM, Chiang N, Arita M, Goodarzi A, Cheng G, von Andrian UH, Serhan CN. Resolvin E1, an EPA-derived mediator in whole blood, selectively counterregulates leukocytes and platelets. Blood 2008;112:848–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Cherpokova D, Jouvene CC, Libreros S, DeRoo EP, Chu L, de la Rosa X, Norris PC, Wagner DD, Serhan CN. Resolvin D4 attenuates the severity of pathological thrombosis in mice. Blood 2019;134:1458–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Libby P. The changing landscape of atherosclerosis. Nature 2021;592:524–533. [DOI] [PubMed] [Google Scholar]
- 162. Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature 1993;362:801–809. [DOI] [PubMed] [Google Scholar]
- 163. Ross R. Rous-Whipple Award Lecture. Atherosclerosis: a defense mechanism gone awry. Am J Pathol 1993;143:987–1002. [PMC free article] [PubMed] [Google Scholar]