

Abstract
Top-down proteomics is a key mass spectrometry-based technology for comprehensive analysis of proteoforms. Proteoforms exhibit multiple high charge states and isotopic forms in full MS scans. The dissociation behavior of proteoforms in different charge states and subjected to different collision energies is highly variable. The current widely employed data-dependent acquisition (DDA) method selects a narrow m/z range (corresponding to a single proteoform charge state) for dissociation from the most abundant precursors. We describe here Mesh, a novel dissociation strategy, to dissociate multiple charge states of one proteoform with multiple collision energies. We show that the Mesh strategy has the potential to generate fragment ions with improved sequence coverage and improve identification ratios in top-down proteomic analyses of complex samples. The strategy is implemented within an open-source instrument control software program named MetaDrive to perform real time deconvolution and precursor selection.
Graphical Abstract
INTRODUCTION
Interest in top-down proteomics for targeted protein analyses and protein post-translational modification analyses has dramatically increased in the last decade.1-3 In contrast to the more widely used bottom-up proteomic analyses, in which proteins are proteolytically digested into peptides prior to mass spectrometric analysis, in top-down proteomics, intact protein ions are directly subjected to gas-phase fragmentation within the mass spectrometer, retaining protein-level information.4,5 The technologies for top-down proteomics have improved greatly in recent years, such that thousands of proteoforms can be identified in large scale studies.6-9 However, limitations still exist for the current fragmentation methods used for top-down proteomics. In top-down proteomics, ESI (electrospray ionization) generates proteoforms with multiple high charge states across the m/z range of a typical full MS spectrum. The dissociation behavior of proteoforms is related to both the charge state and specific activation conditions, as reported in previous studies with collision-induced dissociation (CID)10,11, HCD12,13, activated ion-electron transfer dissociation (AIETD)14, and electron capture dissociation (ECD)15. The fragment ions obtained can differ for the various charge states of one proteoform with the same collision energy (CE), or for the same charge state of one proteoform subjected to various CEs. For example, Reid et al. showed that extensive nonspecific fragmentation of the bovine ubiquitin backbone occurs only for intermediate charge states (+7, +8, +9)10.
Proteoforms amenable to top-down proteomics methodology can range in mass from a few kDa to a few hundreds of kDa16 and can differ substantially in composition, hydrophobicity, and ionizability2. The data dependent acquisition (DDA) method extensively employed in current top-down proteomics is similar to that used for the bottom-up strategy, which depends on selecting highly abundant precursors for dissociation. With this strategy, it is challenging to obtain optimal dissociation conditions suited to every proteoform. To address this problem, advanced precursor ion selection algorithms and collision energy optimizations have been applied for bottom-up and top-down proteomics.17-20 In 2014, Durbin et al. developed Autopilot to improve data acquisition for top-down proteomics.17 Autopilot performs intelligent data collection with online mass detection, and real-time searching to guide precursor selection and fragmentation. Their results showed an improvement to the overall sequence coverage of many proteins, demonstrating that intelligent data acquisition implemented on advanced instruments could increase unique protein identifications. In Autopilot, charge state selection and CE are sequentially adjusted whenever fragmentation yields a low confidence identification.
Dissociation with multiple CEs (or Stepped-HCD) has also been shown to be able to increase the diversity of fragment ions of regular peptides21, cross-linked peptides22 and glycopeptides.23,24 Each different collision energy provides different fragmentation of the labile peptide bonds along the peptide backbone.23,25 The stepped-HCD method has not been used for top-down proteomics to the best of our knowledge.
Here, we report a new, open-source software program, MetaDrive, which performs real-time deconvolution to reveal proteoform charge states and isotopologues in a precursor (MS1) scan. This information is then utilized for precursor selection so that multiple charge states from a single proteoform can be combined for a single dissociation event resulting in increased number of matched fragmentation and sequence coverage. We also show that applying multiple collision energies (stepped HCD) improves fragmentation for intact proteoforms. We then developed a new dissociation method called Mesh, which combines several charge states of one proteoform for fragmentation with multiple CEs. Mesh has the potential to provide more fragment ions with increased sequence coverage and increases the identification ratio for top-down proteomics.
METHODS
Sample Preparation
Bovine Ubiquitin (UniProt Accession P0CG53), horse Cytochrome C (UniProt Accession L7MRG1), and horse myoglobin (UniProt Accession P68082) were purchased as standards from Sigma. Each sample was dissolved to yield solutions of ~10 pmol/μL in 49.9:49.9:0.2 acetonitrile/water/formic acid for infusion studies.
Yeast cells (Saccharomyces cerevisiae strain Y1788) were grown to log phase (OD600 = 0.7) at which time they were washed, pelleted, snap-frozen in liquid nitrogen, and stored at −80 °C until use. Yeast cells were lysed by heat and proteins were precipitated in acetone. The proteins were then dissolved in 1% sodium dodecyl sulfate (SDS) and separated by molecular weight (MW) using a GELFrEE system (Expedeon).7,26 Approximately 400 μg of protein were collected into 11 fractions. Two of the fractions were selected for top-down analyses. Prior to mass spectrometric analyses, sodium dodecyl sulfate was removed from the fractions via methanol–chloroform precipitation and proteins were reconstituted with 5% acetonitrile (ACN) and 0.2% formic acid in water.
Mass Spectrometry
All the mass spectrometry experiments are processed using the Q Exactive HF mass spectrometer (ThermoFisher Scientific). Standard proteins were subjected to direct infusion electrospray ionization mass spectrometry for top-down analysis. Full scans were performed in the Orbitrap between 375 and 1,500 m/z at a resolution of 120 000 at m/z 200, followed by MS/MS HCD scans at a resolution of 60 000 and a m/z range of 400-2000 m/z, where the parent ion selection was controlled by MetaDrive. Four different types of MetaDrive controlled fragmentation schemes were performed separately, including: Top method (the traditional method to fragment a single charge state with a single normalized collision energy (NCE)), Multi-charge method (to fragment multiple charge states with a single NCE), Stepped HCD method (to fragment single charge states with multiple NCEs) and Mesh method (to fragment multiple charge states with multiple NCEs). The Top method or the Stepped HCD method can both be applied using the Thermo Fisher Scientific standard instrument control software program Xcalibur. Note that Xcalibur selects the most abundant charge states by default. It is also possible to sort the precursors by charge state during DDA for the Thermo Fisher Tribrid instrument. We programmed MetaDrive to select one low charge state for the Top/Stepped-HCD method or three low charge states for the Multi-charge/Mesh method rather than the most abundant charge states.
The implementation for fragmenting multiple charge states in one spectrum is based on the “build-in multiple-ion injection” method of the quadrupole Qrbitrap mass spectrometer.27-29 For the Multi-charge/Mesh scan, the following processes are conducted in the instrument after a series of charge states have been picked by MetaDrive for fragmentation: (1) The precursor ions with the highest selected charge state (lowest m/z range) is isolated and collected until the maximum ion injection time is reached or until the AGC target value is reached. The collected precursor ions are then fragmented and stored. (2) This process is repeated until precursor ions of all selected charge states are fragmented and stored. (3) The combined fragment ion population is then injected into the Orbitrap cell and analyzed, resulting in a single fragmentation spectrum for merged fragmented ions.
Top-down proteomic analyses of yeast samples (~2 μg protein each injection) were performed using HPLC-ESI-MS/MS26 (NanoAcquity, Waters coupled to Q Exactive HF). Four different types of MetaDrive controlled fragmentation schemes were performed separately as described above. Note that we did not use vendor software for either precursor deconvolution or precursor exclusion. For all fragmentation schemes, the full scans were performed in the Orbitrap between 375 and 1,500 m/z at a resolution of 120,000, followed by MS/MS HCD scans of the top two highest intensity parent ions and 60,000 resolution, with a mass range of 400-2000 m/z. Dynamic exclusion was enabled with an exclusion window of 30 s.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium30 via the PRIDE partner repository with the data set identifier PXD022941. (Reviewer account details: Username: reviewer_pxd022941@ebi.ac.uk; Password: t6xfoiLi)
Instrument control software program
We developed a new software program, MetaDrive, that performs real-time deconvolution coupled with new methods for precursor selection and fragmentation. The deconvolution algorithm was adapted from MS-Deconv.31 MetaDrive controls an Orbitrap Q-Exactive through Thermo IAPI (Instrument Application Programming Interface) software (an IAPI license is available from Thermo Scientific at no cost, and is required to use MetaDrive).27 We use MetaDrive to optimize proteoform precursor selection in this study. It could also be used as a platform to develop novel data acquisition methods. MetaDrive is written in C# and is freely available as open-source code at https://github.com/smith-chem-wisc/MetaDrive.
Data analysis
The pTop2 software program (http://pfind.ict.ac.cn/software.html) was used to perform top-down analysis of the raw files.32 This software reports the number of matched fragment ions generated for each spectrum. Most of the default parameter settings were used, except that the max charge was changed to 50, mixture spectra was not checked, and variable modifications including oxidation at methionine, dehydro at cysteine (loss of a hydrogen), and acetylation at protein N-terminal were added. Detailed parameter settings used in pTop2 are described in Supplementary Table 1. A FASTA file of the standard proteins and yeast (2019.09) from UniProt database was employed.
RESULTS AND DISCUSSION
The effects of charge state and NCE
Proteoforms appear in multiple charge states in top-down precursor mass spectra. The current, widely adopted data-dependent analysis (DDA) scheme to identify proteoforms dissociates a single charge state with a specified CE. Because proteoform fragmentation efficiency varies with both charge state and collision energy, this approach often does not automatically produce optimal fragmentation for confident identification.2,17
We evaluated the yield of matched fragment ions for the same proteoform across different charge states and different normalized collision energies (NCE) using higher-energy collisional dissociation (HCD). NCE is defined as a single energy value provided in Orbitrap mass spectrometer HCD MS/MS experiments.33 DDA is often coupled with HCD, one of the most commonly used fragmentation methods in top-down proteomics.2,6,8 Three standard proteins including ubiquitin, cytochrome C and myoglobin, each exhibiting unique charge state distributions, were examined for this study (Figure 1). To simplify the evaluation, we only considered the number of matched b and y-ions identified by pTop in each MS/MS spectrum, which is a widely used matching function.32,34
Figure 1. Effects of charge state and NCE upon proteoform dissociation.

Three standard proteins (ubiquitin, cytochrome C and myoglobin) were used to study fragmentation efficiency. The number of matched fragment ions varied with the charge states selected and collisional energies employed. No unique condition works for every standard protein.
Different charge states of the same proteoform yielded significantly different numbers of matched fragment ions, while the effects of varying NCE values differed for different proteoforms. Ubiquitin in the +9 charge state produced the highest number of matched fragment ions for all NCEs. Cytochrome C produced twice the number of matched fragment ions for charge +10 than for charge +16 at NCE 20, while this was reversed at NCE 10. Myoglobin produced more matched fragment ions from the +15 charge state than from higher charge states for most collision energies (Figure 1). For each proteoform, our results showed that the charge state with the highest intensity did not yield the highest number of matched fragment ions, in agreement with previous studies.14,35 Different charge states of a selected proteoform are optimally fragmented with different NCEs. For a selected charge state of a protein, varying NCEs not only generated different numbers of matched fragment ions (Figure 1), but they also produced different b ions and y ions along the protein sequence (data not shown). For all three proteins, a particular low charge state produced the most matched fragment ions, but that low charge state is not necessarily the lowest charge state for the protein, as shown for ubiquitin and cytochrome C. If the current, widely adopted DDA scheme were employed for analysis of these proteoforms, the most abundant, high charge state peaks would have been more likely to be selected for fragmentation.
Comparison of different dissociation methods
We developed a new data acquisition method to improve dissociation by fragmenting several different charge states while applying multiple NCEs. The new method is implemented in MetaDrive (Figure 2), which is a software program that can control the QE-HF via IAPI. Each time MetaDrive receives a full scan from the instrument, it performs a real-time deconvolution to keep track of precursors. We designed and compared four different type of fragmentation schemes in this study, including: Top method (the traditional method to fragment a single charge state with a single NCE), Multi-charge method (to fragment multiple charge states with a single NCE), Stepped HCD method (to fragment a single charge state with multiple NCEs) and Mesh method (to fragment multiple charge states with multiple NCEs). Note that all the four fragmentation schemes each only generates one spectrum for any selected proteoform. In the Mesh Method, a fragment ion could originate from any selected charge state or energy (Supplement Figure 1).
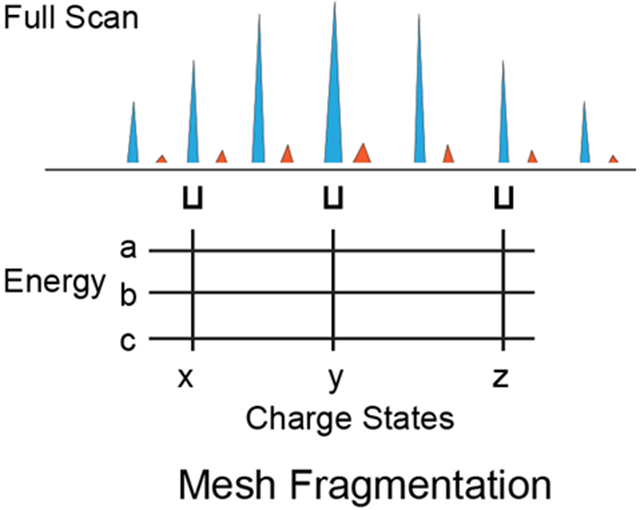
Figure 2. Workflow of Mesh dissociation method in MetaDrive.

The instrument control software program MetaDrive performs real-time deconvolution of a full MS spectrum. The deconvolution results contain unknown proteoforms with defined charge states. MetaDrive can choose a proteoform and apply the Mesh dissociation method. Mesh selected multiple charge states (marked x, y and z) of the proteoform and used stepped HCD with different energies (marked a, b and c) to generate one single spectrum.
We first applied these four fragmentation methods to the three standard proteins (see Figure 3). The instrument selects the most abundant peaks for fragmentation in DDA. The charge state with the highest intensity is selected for each proteoform in the Top method. For the Multi-charge and Mesh methods, we used MetaDrive to dissociate ions in several charge states near the highest intensity charge state with NCE 25, as this energy is commonly used in top-down proteomics studies.8,36 For the Stepped HCD and Mesh methods, we applied NCEs 15, 25 and 35.
Figure 3. Fragmentation results obtained using different fragmentation schemes.

Three standard proteins (ubiquitin, cytochrome C and myoglobin) were subjected to four distinct fragmentation schemes. For each protein, the charge state with the highest intensity was selected for the Top method, and multiple charge states near it were selected for the Multi-charge and Mesh methods. Charge states selected are labeled on each column; For example, ‘C7-11-13’ means charge state +9, +11, +13. Values are average numbers of matched fragment ions in MS/MS scans with the same fragmentation condition collected in one single direct infusion run. Error bars show one standard deviation.
Fragmenting with multiple NCEs always improved the dissociation efficiency. For each of the three standard proteins, the multiple NCE methods produced more matched fragment ions than the single NCE methods. The number of matched fragment ions almost doubled for cytochrome C and myoglobin when multiple NCEs are applied. Fragmenting multiple charge states has the potential to increase the matched fragmentation. The average numbers of matched peaks increased for ubiquitin when fragmenting multiple charge states (+7, +9, +10, +11 and +13) together compared to when fragmenting only charge state +11 using NCE 25. The Mesh method generated the most matched fragment ions for myoglobin (Figure 3). The number of matched fragment ions doubling when fragmenting charge states +17, +21 and +25 together compared to fragmenting only charge state +21 using stepped HCD. There was limited improvement for the cytochrome C sample with the Multi-charge method; the matched fragment ions using the Mesh method were even fewer than when using the stepped HCD method. Fragmenting multiple charge state is not a simplified process of fragmenting each charge state sequentially. Further studies are needed to illustrate the observation.
From these data, we observed that for most applied NCEs the largest numbers of matching fragment ions commonly come from a charge state that is of lower intensity than the most abundant charge state. Further studies to enable effective selection of charge states are needed. In contrast, the improvement from using multiple NCEs was clear for all three standard proteins. For cytochrome C and myoglobin, use of multiple fragmentation energies doubled the number of matched fragment ions. Overall, applying multiple NCEs did significantly improve the protein dissociation.
Yeast sample analysis
We applied the four fragmentation schemes to collect complex proteoform data from size-separated (GELFrEE) yeast cell lysates.7 Two intermediate fractions of high complexity were selected for analysis. Based on the results from the standard protein experiments, we designed the Mesh method to fragment proteoforms, which fragments multiple charges with stepped HCD and generates one spectrum. The Mesh method performs isotope deconvolution and then charge state deconvolution of an MS1 scan in real-time to identify precursors with charge states larger than 5, provided that they exist. One of the deconvoluted proteoforms is selected and all of the m/z values for that proteoform are ordered from low charge state value to high charge state value. The algorithm then selects three m/z values, corresponding to three charge states, for fragmentation by skipping the lowest charge state (the intensity of which is very low), and then selecting the 2nd, 4th and 6th low charge state m/z values. Less than three are selected if not all are present. After selecting these three low charge states for each proteoform, stepped HCD with NCEs of 15, 20 and 25 are applied to fragment the precursors.
We observed an increase in identification ratio with selection of multiple charge states and/or multiple collision energies for both fractions (see Figure 4). The identification ratio is calculated as the number of identified spectra divided by the number of MS2 scans in each file. Note that we instruct the program to select for lower intensity charge states first as we found that the charge state with highest intensity does not always generate the most matched fragment ions (Figure 3). Fragmenting multiple charge states increased the average identification ratio by ~2% in this set of studies, although this improvement is not statistically significant (Figure 4). Applying multiple NCEs can increase the average identification ratio by ~10%. The Mesh method combines contributions from fragmenting multiple charge states with multiple NCEs and thereby yielded the highest average identification ratio (Figure 4). Applying multiple NCE energies yielded more improvement than did selecting multiple charge states (Figure 4). However, the current implementation in MetaDrive slows the data acquisition when multiple NCE energies are applied or when fragmenting multiple charge states (Supplement Figure 3). The relative identification yield improved when applying multiple NCE energies but not when fragmenting multiple charge states (Supplement Figure 4). In a comparison of the sequence coverage for proteoforms identified by the methods, we observed improved sequence coverage for the Stepped-HCD method and limited improvement for the Mesh method (Supplement Figure 2). The selection of multiple charge states brings challenges for the computation process. First, deconvolution errors, which occur frequently in the processing of MS1 mass spectra of intact proteoforms2,32, can lead to improper selection of multiple charge states. Second, fragmenting multiple charge states in one dissociation event yielded an MS2 spectrum that was not a simple summation of the fragments that would have been observed had each of the individual charge states been fragmented separately. We do not yet understand the mechanism of co-fragmentation. We intend to explore this process in future studies. Third, the computation time for the real-time charge deconvolution algorithm is a source of delay in the decision-making process. The current implementation showed a ~5% decrease in the total number of MS2 scans gathered for each analysis compared with Xcalibur (data not shown). Future efforts to improve the speed of the deconvolution algorithm or to apply novel algorithms such as FLASHDeconv37 will seek to allow the scan rates to be increased. Further improvements in the performance of the Mesh strategy should be possible by addressing these issues.
Figure 4. Identification ratio of different fragmentation schemes.

Two yeast Gel-Free fractions (frac3 and frac4) were used to compare the four fragmentation schemes. The Stepped HCD and Mesh methods significantly improved identification ratio. For each method, three technical replicates were performed. Values are averages of triplicate injections (3 separate files, shown in dot plot format), and error bars show one standard deviation.
Conclusions
We showed that real-time deconvolution of proteoform MS1 spectra allows improved and more versatile selection of proteoform charge states and dissociation energies for MS2 analysis. The current widely adopted DDA method often selects a single charge state of a proteoform. We developed Mesh, a new dissociation strategy to fragment multiple charge states with stepped HCD. The results showed cases with increased identifications and improved sequence coverage for the stepped HCD and the Mesh methods for complex protein samples. Developing deconvolution algorithms with higher accuracy and efficiency could further improve the Mesh method. The new dissociation strategies are implemented in the real-time instrument control software program MetaDrive, which can be used as an open source platform to develop precursor selection and data acquisition methods.
Supplementary Material
Supplementary Figures 1. Spectra comparison of the four different fragmentation methods.
Supplementary Figures 2. Fragmentation comparison of the four fragmentation methods of shared proteoforms in yeast fraction. Supplementary Table 1. Parameters used of pTop2 (PDF)
ACKNOWLEDGEMENTS
We greatly appreciate discussions with Brian L. Frey, Leah V. Schaffer and other Smith group members to provide suggestions and address challenges in implementing ideas. This work was supported by National Institute of Health (NIH) Grant R35GM126914 awarded to L.M.S.
Footnotes
The authors declare no competing financial interest.
REFERENCE
- (1).Toby TK; Fornelli L; Kelleher NL Progress in top-down proteomics and the analysis of proteoforms. Annu. Rev. Anal. Chem 2016, 9, 499–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Chen B; Brown KA; Lin Z; Ge Y Top-down proteomics: ready for prime time? Anal. Chem 2017, 90 (1), 110–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Schaffer LV; Millikin RJ; Miller RM; Anderson LC; Fellers RT; Ge Y; Kelleher NL; LeDuc RD; Liu X; Payne SH; et al. Identification and quantification of proteoforms by mass spectrometry. Proteomics 2019, 19 (10), 1800361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Smith LM; Kelleher NL; The Consortium for Top Down Proteomics. Proteoform: a single term describing protein complexity. Nat. Methods 2013, 10 (3), 186–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Smith LM; Kelleher NL Proteoforms as the next proteomics currency. Science (80-.). 2018, 359 (6380), 1106–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Catherman AD; Durbin KR; Ahlf DR; Early BP; Fellers RT; Tran JC; Thomas PM; Kelleher NL Large-scale top-down proteomics of the human proteome: membrane proteins, mitochondria, and senescence. Mol. Cell. Proteomics 2013, 12 (12), 3465–3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Tran JC; Zamdborg L; Ahlf DR; Lee JE; Catherman AD; Durbin KR; Tipton JD; Vellaichamy A; Kellie JF; Li M Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature 2011, 480 (7376), 254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Durbin KR; Fornelli L; Fellers RT; Doubleday PF; Narita M; Kelleher NL Quantitation and identification of thousands of human proteoforms below 30 kDa. J. Proteome Res 2016, 15 (3), 976–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Dai Y; Buxton KE; Schaffer LV; Miller RM; Millikin RJ; Scalf M; Frey BL; Shortreed MR; Smith LM Constructing Human Proteoform Families Using Intact-Mass and Top-Down Proteomics with a Multi-Protease Global Post-Translational Modification Discovery Database. J. Proteome Res 2019, 18 (10), 3671–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Reid GE; Wu J; Chrisman PA; Wells JM; McLuckey SA Charge-state-dependent sequence analysis of protonated ubiquitin ions via ion trap tandem mass spectrometry. Anal. Chem 2001, 73 (14), 3274–3281. [DOI] [PubMed] [Google Scholar]
- (11).Chanthamontri C; Liu J; McLuckey SA Charge state dependent fragmentation of gaseous α-synuclein cations via ion trap and beam-type collisional activation. Int. J. Mass Spectrom 2009, 283 (1–3), 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Bashyal A; Sanders JD; Holden DD; Brodbelt JS Top-down analysis of proteins in low charge states. J. Am. Soc. Mass Spectrom 2019, 30 (4), 704–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Haverland NA; Skinner OS; Fellers RT; Tariq AA; Early BP; LeDuc RD; Fornelli L; Compton PD; Kelleher NL Defining gas-phase fragmentation propensities of intact proteins during native top-down mass spectrometry. J. Am. Soc. Mass Spectrom 2017, 28 (6), 1203–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Riley NM; Westphall MS; Coon JJ Activated ion-electron transfer dissociation enables comprehensive top-down protein fragmentation. J. Proteome Res 2017, 16 (7), 2653–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Shaw JB; Malhan N; Vasil’ev YV; Lopez NI; Makarov A; Beckman JS; Voinov VG Sequencing Grade Tandem Mass Spectrometry for Top–Down Proteomics Using Hybrid Electron Capture Dissociation Methods in a Benchtop Orbitrap Mass Spectrometer. Anal. Chem 2018, 90 (18), 10819–10827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Cai W; Tucholski T; Chen B; Alpert AJ; McIlwain S; Kohmoto T; Jin S; Ge Y Top-down proteomics of large proteins up to 223 kDa enabled by serial size exclusion chromatography strategy. Anal. Chem 2017, 89 (10), 5467–5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Durbin KR; Fellers RT; Ntai I; Kelleher NL; Compton PD Autopilot: an online data acquisition control system for the enhanced high-throughput characterization of intact proteins. Anal. Chem 2014, 86 (3), 1485–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kreimer S; Belov ME; Danielson WF; Levitsky LI; Gorshkov MV; Karger BL; Ivanov AR Advanced precursor ion selection algorithms for increased depth of bottom-up proteomic profiling. J. Proteome Res 2016, 15 (10), 3563–3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hebert AS; Thoöing C; Riley NM; Kwiecien NW; Shiskova E; Huguet R; Cardasis HL; Kuehn A; Eliuk S; Zabrouskov V Improved precursor characterization for data-dependent mass spectrometry. Anal. Chem 2018, 90 (3), 2333–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Révész Á; Milley MG; Nagy K; Szabó D; Kalló G; Csősz É; Vékey K; Drahos L Tailoring to Search Engines: Bottom-Up Proteomics with Collision Energies Optimized for Identification Confidence. J. Proteome Res 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Diedrich JK; Pinto AFM; Yates JR III Energy dependence of HCD on peptide fragmentation: stepped collisional energy finds the sweet spot. J. Am. Soc. Mass Spectrom 2013, 24 (11), 1690–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Stieger CE; Doppler P; Mechtler K Optimized fragmentation improves the identification of peptides cross-linked by MS-cleavable reagents. J. Proteome Res 2019, 18 (3), 1363–1370. [DOI] [PubMed] [Google Scholar]
- (23).Liu M-Q; Zeng W-F; Fang P; Cao W-Q; Liu C; Yan G-Q; Zhang Y; Peng C; Wu J-Q; Zhang X-J pGlyco 2.0 enables precision N-glycoproteomics with comprehensive quality control and one-step mass spectrometry for intact glycopeptide identification. Nat. Commun 2017, 8 (1), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Yang H; Yang C; Sun T Characterization of glycopeptides using a stepped higher-energy C-trap dissociation approach on a hybrid quadrupole orbitrap. Rapid Commun. Mass Spectrom 2018, 32 (16), 1353–1362. [DOI] [PubMed] [Google Scholar]
- (25).Wysocki VH; Tsaprailis G; Smith LL; Breci LA Mobile and localized protons: a framework for understanding peptide dissociation. J. Mass Spectrom 2000, 35 (12), 1399–1406. [DOI] [PubMed] [Google Scholar]
- (26).Dai Y; Shortreed MR; Scalf M; Frey BL; Cesnik AJ; Solntsev S; Schaffer LV; Smith LM Elucidating Escherichia coli proteoform families using intact-mass proteomics and a global PTM discovery database. J. Proteome Res 2017, 16 (11), 4156–4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Meier F; Geyer PE; Virreira Winter S; Cox J; Mann M BoxCar acquisition method enables single-shot proteomics at a depth of 10,000 proteins in 100 minutes. Nat. Methods 2018, 15, 440–448. [DOI] [PubMed] [Google Scholar]
- (28).Egertson JD; Kuehn A; Merrihew GE; Bateman NW; MacLean BX; Ting YS; Canterbury JD; Marsh DM; Kellmann M; Zabrouskov V Multiplexed MS/MS for improved data-independent acquisition. Nat. Methods 2013, 10 (8), 744–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Gallien S; Bourmaud A; Kim SY; Domon B Technical considerations for large-scale parallel reaction monitoring analysis. J. Proteomics 2014, 100, 147–159. [DOI] [PubMed] [Google Scholar]
- (30).Perez-Riverol Y; Csordas A; Bai J; Bernal-Llinares M; Hewapathirana S; Kundu DJ; Inuganti A; Griss J; Mayer G; Eisenacher M; et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019, 47 (D1), D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Liu X; Inbar Y; Dorrestein PC; Wynne C; Edwards N; Souda P; Whitelegge JP; Bafna V; Pevzner PA Deconvolution and database search of complex tandem mass spectra of intact proteins: a combinatorial approach. Mol. Cell. Proteomics 2010, 9 (12), 2772–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Sun R-X; Luo L; Wu L; Wang R-M; Zeng W-F; Chi H; Liu C; He S-M pTop 1.0: a high-accuracy and high-efficiency search engine for intact protein identification. Anal. Chem 2016, 88 (6), 3082–3090. [DOI] [PubMed] [Google Scholar]
- (33).Hartel NG; Liu CZ; Graham NA Improved discrimination of asymmetric and symmetric arginine dimethylation by optimization of the normalized collision energy in LC-MS proteomics. J. Proteome Res 2020. [DOI] [PubMed] [Google Scholar]
- (34).Park J; Piehowski PD; Wilkins C; Zhou M; Mendoza J; Fujimoto GM; Gibbons BC; Shaw JB; Shen Y; Shukla AK Informed-Proteomics: open-source software package for top-down proteomics. Nat. Methods 2017, 14 (9), 909–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Fabris D; Kelly M; Murphy C; Wu Z; Fenselau C High-energy collision-induced dissociation of multiply charged polypeptides produced by electrospray. J. Am. Soc. Mass Spectrom 1993, 4 (8), 652–661. [DOI] [PubMed] [Google Scholar]
- (36).Schaffer LV; Shortreed MR; Cesnik AJ; Frey BL; Solntsev SK; Scalf M; Smith LM Expanding Proteoform identifications in top-down proteomic analyses by constructing Proteoform families. Anal. Chem 2018, 90 (2), 1325–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Jeong K; Kim J; Gaikwad M; Hidayah SN; Heikaus L; Schlüter H; Kohlbacher O FLASHDeconv: Ultrafast, high-quality feature deconvolution for top-down proteomics. Cell Syst. 2020, 10 (2), 213–218. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures 1. Spectra comparison of the four different fragmentation methods.
Supplementary Figures 2. Fragmentation comparison of the four fragmentation methods of shared proteoforms in yeast fraction. Supplementary Table 1. Parameters used of pTop2 (PDF)