

Abstract
A divergent approach to C-C bond forming macrocycle construction is described. Modular sulfonylhydrazone and derived pyridotriazole substrates with three key building blocks have been constructed and cyclized to afford diverse macrocyclic frameworks. Broad substrate scope and functional group tolerance have been demonstrated. In addition, site-selective post-functionalization allowed for further diversification of macrocyclic cores.
Graphical Abstract

INTRODUCTION
Macrocyclic compounds, long prevalent among bioactive natural products,1 play increasingly critical roles in drug discovery2 particularly against the broad spectrum of protein targets considered “non-druggable” by conventional small molecules such as protein-protein interactions (PPIs).3 Accordingly, development of new methods for the production of diverse libraries of new macrocyclic chemotypes has attracted considerable interest from organic and medicinal chemists, particularly in the field of diversity-oriented synthesis (DOS).4
Macrocyclic ring closure can be accomplished through a wide number of known conjunctive, intramolecular reactions. The most common of such reactions are macrolactonizations and macrolactamizations,5 frequently used in both solid-supported and solution-phase, combinatorial assembly of libraries of cyclic peptides, cyclic depsipeptides and other peptidomimetics, as well as non-peptidic macrolactams/macrolactones.6 Yudin and coworkers have pioneered use of both amphoteric molecules7a–c and heterocyclic grafts7d–e for macrocyclization and diversification of linear peptides. Other cyclization strategies commonly utilized in the context of solution phase, diversity-oriented synthesis of macrocycles include ring-closing and enyne metatheses,8 Huisgen (“click”)-type azide-alkyne cycloadditions,9 isocyanide-based multicomponent reactions (Ugi, Passerini, aziridine/aldehyde)10 nucleophilic aromatic substitutions,11 and combinations thereof.12 Beyond the highly prevalent Huisgen cycloaddition, other variants of Cu-catalyzed alkyne macrocyclizations12e, 13 and cycloadditions9b, 14 are very commonly utilized, as are a diverse array of intramolecular palladium-catalyzed C-C and C-X couplings.15, 12e Beyond these prevalent transformations, there are also reported examples of library generation using photocatalyzed macrocyclization,16 reductive amination,17 nucleophilic substitution,18 Friedel-Crafts alkylations,19 the intramolecular Ullmann reaction,20 Pauson-Khand reactions,12e Rhodium-catalyzed O-H/N-H insertions,21 condensation reactions,22 and cyclopropanations.23 While many other synthetic methods for macrocyclic ring closure have been utilized in target-oriented syntheses,5a, 24 these methodologies are often limited by reaction efficiency, substrate scope, and functional group tolerance, thereby presenting a key challenge for DOS.25
Given the limited scope of transformations available for macrocyclic ring closure, we have employed a reaction screening approach to identify new C-C bond forming macrocyclizations that are tolerant of diverse substitutions, ring sizes, and functional groups. Inspired by the multifunctional utility of alkynes in several of the aforementioned conjunctive chemistries,8, 9, 13 as well as recent examples including Harran’s use of cinnamyl carbonates as divergently-reactive functional handles for MC synthesis,15p,q,s, 19 we considered whether alkyne-tethered sulfonylhydrazones26 could serve as useful substrates for divergent macrocyclizations. Sulfonylhydrazones are versatile reaction partners27 and have been used as vinyl synthons,28 reaction partners for palladium-catalyzed couplings,29 copper-catalyzed couplings,30 and transition-metal carbene-based cross-couplings.31 Petasis-type reactions of sulfonylhydrazones and boronate reagents have also been reported.32 Owing to their synthetic versatility, N-tosylhydrazones have been recognized as useful synthons for construction of diverse ring systems via cyclization chemistry.26a In this study, we describe our initial efforts to utilize alkyne-tethered sulfonylhydrazones as prototype substrates for divergent C-C bond forming macrocycle (MC) construction.
RESULTS AND DISCUSSION
Initially, we envisioned executing a copper-catalyzed macrocyclization using modular N-tosylhydrazone substrates of the general design 1 which can be conveniently assembled by amide couplings of three key building blocks: the “A” block (N-tosylhydrazone-tethered carboxylic acid), the “B” block (amino acid linker), and the “C” block (alkyne-tethered amine) (Figure 1). We envisioned that, in the presence of a base, intramolecular addition of an in situ-generated copper acetylide to the tosylhydrazone followed by extrusion of the toluenesulfinate leaving group and nitrogen gas should afford either internal alkyne 2 or the macrocyclic allene 3.30
Figure 1.

Original plan for macrocycle construction using modular alkynyl N-tosylhydrazone substrates 1.
The model substrate 9 was designed to test this hypothesis (Scheme 1) and was constructed from building blocks that were envisioned to template macrocyclization by enforcing proximity of the reactive groups.33 A meta-formylbenzoic acid-derived hydrazone and a gem-di-methylated terminal alkyne were used for the A-block and C-block fragments, respectively, flanking a gabapentin B-block which was envisioned to enforce a GABA-type turn.34 A representative substrate synthesis is illustrated in Scheme 1. Amidation of Fmoc-protected gabapentin 4 with alkynylamine 5 followed by Fmoc deprotection afforded alkynylamine 6. Compound 8, prepared by condensation of 3-formylbenzoic acid 7 and p-toluenesulfonyl hydrazide, was amidated with amine 6 to provide the alkynyl tosylhydrazone substrate 9 in good yield.
Scheme 1.

Representative Synthesis of Modular Model Substrate 9a
aReagents and conditions: (i) EDCI, HOBt, DIEA, THF, 85%; (ii) Piperidine, DMF, 85%; (iii) TsNHNH2, 1:1 CHCl3:MeOH, quantitative; (iv) EDCI, HOBt, 6, DIEA, THF, 71%.
Preliminary experiments using copper (I) catalysts and weak organic bases such as triethylamine failed to generate macrocyclic products from substrate 9. After extensive screening of catalysts, bases, solvents, and temperatures, we found that copper(I) triflate-catalyzed treatment (10 mol%) of 9 in isopropanol (4 mM) under microwave irradiation at 90 °C using sodium tert-butoxide as base provided a 10% yield of the 16-membered macrocyclic alkyne 11 (Scheme 2A). The major side products observed (12–15) were derived from decomposition of tosylhydrazone 9 as previously reported.35a–c Interestingly, we discovered that significant amounts of pyrazole macrocycle 17 were formed in a control experiment run in the absence of the transition metal catalyst (Scheme 2B).26a Following subsequent optimization, we determined that with three equivalents of 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene (MTBD) used as base, an improved 59% yield of 17 could be obtained, along with smaller amounts of the side products 12, 14–15, and 18.35 Mechanistically, we postulated that 9 undergoes base-mediated, in situ generation of diazo intermediate 16 followed by intramolecular 1,3-dipolar cycloaddition to afford head-to-tail cycloaddition product 17 as the sole regioisomer isolated.35f
Scheme 2.

Macrocycle Construction through C-C Bond-Formation
With these optimized conditions in hand, we next investigated substrate scope and functional group tolerance for the intramolecular dipolar cycloaddition. First, we examined the effects of incorporating varied C-block alkynes to the generic sulfonylhydrazone scaffold represented by substrate 19 (Table 1). While a single regioisomer was isolated in the cyclization of 9, we presumed that expanded ring sizes and varied substitutions may lead to formation of both regioisomeric pyrazoles 20 and 21 as possible macrocyclic products. Indeed, tosylhydrazone 19a bearing an aliphatic-linked alkyne was cyclized to provide the 19-membered pyrazole 20a in 13% yield, as well as the regioisomeric 18-membered product 21a in 21% yield. Ethylene glycol-linked alkyne 19b afforded the 20-membered macrocycle 20b and the 19-membered macrocycle 21b in 18% and 34% yields, respectively. Substrates bearing propargyl ethers, 19c–e were also cyclized in good yield as fully separable product mixtures. For phenol ether 19c, a 23% yield of macrocycle 20c was obtained, the structure of which was confirmed by X-ray crystallographic analysis (Figure 2). In addition, the ring-opened phenol 20c’ was produced in 16% yield, presumably from decomposition of 20c under basic conditions. Branched substrate 19d gave a 75% overall yield of separable macrocyclic products 20d and 21d. Interestingly, the base-mediated macrocyclization also showed impressive functional group tolerance. For example, alkynyl substrate 19e bearing an unprotected primary amine was cyclized and further acetylated (for purposes of purification) to afford products 20e and 21e in 67% and 6% yields, respectively. Despite relatively low reactivity, the internal alkyne substrate 19f was also converted to macrocyclic pyrazoles 20–21f. The presence of conjugated methyl and allyl ester moieties was also tolerated, in which case moderate-to-good yields of macrocycles 20g-h were obtained. For all macrocycle products, regioisomer assignments were made based on a combination of 2D NMR data and comparison to compound 20c, which was unambiguously assigned by X-ray crystallography and exhibited a diagnostic nOe signal between Ha and Hb (Figure 2A). As a representative example, Figure 2B depicts a similar proximity between protons Ha and Hb in the DFT-optimized structure of 20a, which also exhibited the same diagnostic Ha-Hb nOe interaction. In contrast, Figure 2C depicts a DFT-optimized structure of compound 21a, which did not exhibit any nOe interactions between Ha and the pyrazole Hb, but did sho a strong nOe signal between Ha and Hc. Assignments for all other regioisomeric pairs were made using analogous diagnostic nOe signals. Notably, in both the DFT-optimized and X-ray crystallographic structures for 20a/21a/20c, we observed an intramolecular H-bond reminiscent of a γ-amino (“GABA”) hairpin turn,34 underscoring the utility of gabapentin as a templating B-block to facilitate macrocyclizations.
Table 1.
Formation of Pyrazole Macrocycles Containing Varied C-Blocks
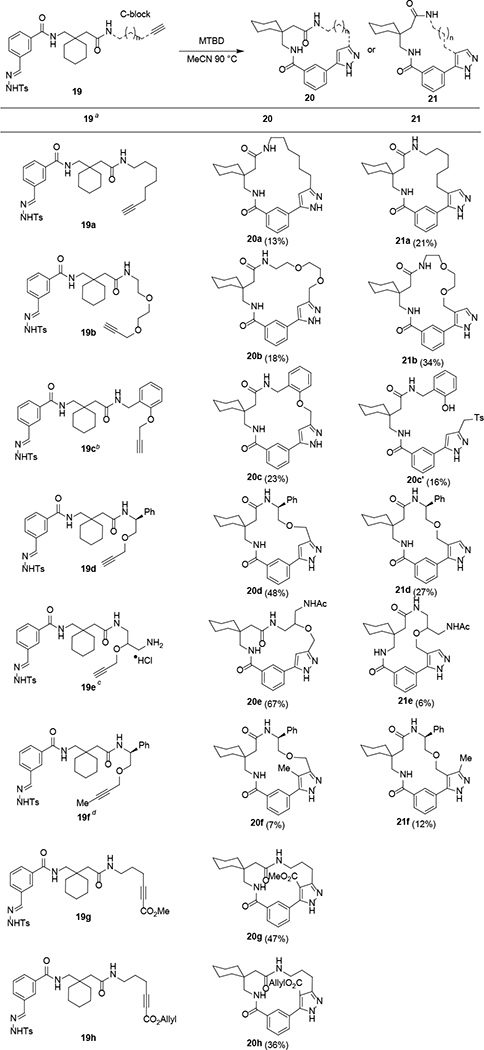
|
19, 3 equiv MTBD, 4 mM in MeCN, 90 °C
side product 20c’ was isolated in 16% yield
4 equiv of MTBD used followed by acetylation using polystyrene-DIEA and AcOSu
100 °C.
Figure 2.

(A) X-ray crystal structure of compound 20c depicting observed diagnostic nOe signals between Ha & Hb which were used for subsequent regioisomer assignment of other substrates. (B) DFT-optimized structure of 20a. For this regioisomer, a diagnostic nOe signal between Ha & Hb was observed, similarly to 20c. (C) DFT-optimized structure of 21a. For this regioisomer, a diagnostic nOe signal between Ha & Hc was observed.
We next designed substrates with varied A- and B-blocks to explore additional scope for the macrocyclization (Scheme 3). A heterocyclic gabapentin-mimetic B-block was used in substrate 22a which afforded macrocycle 23a in 35% yield (Scheme 3a). Consistent with our earlier observations with substrate 9, head-to-tail regioisomers were solely observed as products from linear precursors incorporating gem-dimethylated pentyn-4-yl-1-amine as the C-block, presumably due to severe conformational restrictions induced by the gem-dimethyl groups. The isoserine-derived substrate 22b was cyclized to macrocyclic pyrazoles 23–24b in low yield (Scheme 3b), a result that we attributed to the absence of a turn-inducing B-block element. When hydroxyproline was utilized as an alternate turn-inducing group, substrate 22c underwent cyclization to afford 13-membered ring macrocycle 24c in 46% yield (Scheme 3c). Similarly, substrate 22d, bearing a biphenylsubstituted tosylhydrazone A-block and a chiral alkyne C-block, produced separable mixtures of pyrazole regioisomers 23d and 24d in 52% and 11% yields, respectively (Scheme 3d).
Scheme 3.

Construction of Pyrazole Macrocycles Containing Varied A-, B-, and C-Blocks
While the scope of the macrocyclization was broadly tolerant of several functionalities, we did identify some limitations (Scheme 4). For example, substrates 25a and 25b both gave intractable complex mixtures under the cyclization conditions. The phenol-containing substrate 25c, a close analog of substrate 9, failed to cyclize, affording sulfone 26 in 28% yield as the only cleanly isolated product. Likewise, the pyrrole-containing sulfonylhydrazone substrate 25d decomposed under the basic conditions required for macrocyclization.
Scheme 4.

Challenging Substrates for Macrocyclization
With a successful route to pyrazolo-macrocycles in hand, we next explored other reactivities that could intramolecularly pair the tethered p-tosylhydrazone and alkyne functionalities with the goal of generating multiple macrocyclic chemotypes from a single substrate design (“divergent” synthesis).36 Somewhat serendipitously, we discovered that picolinic acid derivative 27 (Scheme 5A) under optimized pyrazole formation conditions produced pyridotriazole 28 in nearly quantitative yield. It has been demonstrated that, in solution, pyridotriazoles such as 28 exist in closed/open form equilibrium with the valence tautomer diazo pyridine, serving as stabilized rhodium carbene precursors.37 Inspired by the scope of reactions possible with the tautomeric pyridotriazole substrates,37b–h we next explored the reactive utility of this motif as a macrocyclization linchpin. Firstly, we found that 28 can be cyclized to give the originally desired pyrazoles 29 and 30 under thermolytic conditions (mesitylene, 200 °C). Treating the same substrate using Rh(II) catalysis led to the formation of two macrocycles, assigned by 2D NMR analysis to be cyclopentane 31 and diene 32, in 81% and 9% yields, respectively. Our proposed mechanism for the formation of macrocycles 31–32 is shown in Scheme 5B. Rhodium carbenoid 33 can be generated from pyridotriazole 28 after Rh-catalyzed diazo decomposition. From this intermediate, formation of a four-membered rhodacycle via alkyne addition can occur in either a head-to-head (path a) or head-to-tail (path b) fashion. For path a, ring opening of the rhodacycle 34 affords carbenoid 35, which can be converted to cyclopentane 31 through sp3 C-H insertion.38 For path b, the head-to-tail rhodacycle 36 may undergo retro-[2+2] ring opening to give conjugated rhodium carbenoid 37. Finally, β-hydride elimination of 37 gives rise to macrocyclic diene 32.39
Scheme 5.

. (A) Divergent Macrocyclization of Pyridotriazole Substrates (B) Proposed Mechanism for Formation of 30a and 30b
Interestingly, rhodium (II) catalysis of hydroxyproline-containing pyridotriazole substrate 38 afforded the piperidyl amide 39 in 31% yield and indolizine 40 in 11% yield respectively (Scheme 6). Attempted oxidation of piperidyl amide 39 failed to give even trace amounts of indolizine 40 which indicates two likely independent pathways for macrocyclization. Our proposed mechanism for formation of 39–40 is shown in Scheme 6B. Rhodium carbenoid 41, generated from pyridotriazole 38, may be converted to the four-membered rhodacycle 42 via alkyne addition in a head-to-head manner. Retro-[2+2] ring opening would afford carbenoid 43. Interestingly, we did not observe any formation of the expected C-H insertion product from this carbenoid; we postulate that resonance of the vinyl carbene species 43 may allow equilibration to the double bond isomer 44, likely driven by chelation to one or more macrocycle nitrogens.40 From this isomer, either N-H insertion would afford piperidyl amide 39 (path a) or 6π-electrocyclization would produce 46 (path b).37b Subsequent reductive elimination may afford 47 which we postulate may undergo further oxidation to indolizine 40. Interestingly, for this substrate, products derived from the other head-to-tail [2+2] rhodacycle formation were not observed which may be a consequence of the smaller, more constrained ring (compared to analogous substrate 28) preventing proper orbital overlap for C-H insertion.
Scheme 6.

(A) Rh(II)-Catalyzed Macrocyclization of Alkynyl Pyridotriazole (B) Proposed Mechanism for Formation of 39 and 40
In order to test the efficiency and functional group tolerance of this divergent pathway, alkynyl pyridotriazole 48 was chosen for investigation (Scheme 7A). Under thermal conditions, substrate 48 was converted to pyrazoles 49–50 in 54% yield, while rhodium (II) catalysis produced the macrocyclic diene ether 51 in 71% yield. We next explored whether the rhodium-catalyzed cyclization strategy was further applicable to non-triazolo Rh-carbenoids generated from other N-tosylhydrazone substrates (Scheme 7B). To this end, we confirmed that substrates 19a and 19c could be converted to macrocyclic dienes 52 (15% yield) and 53 (31% yield), respectively under Rh(II) catalysis in the presence of a weak base. Interestingly, we found macrocycle dienes 52 and 53 to be air sensitive, likely contributing to their low isolated yields.41
Scheme 7.

(A) Divergent Macrocyclization of Alkynyl Pyridotriazole (B) Rh(II)-Catalyzed Macrocyclization of Alkynyl Tosylhydrazones
Finally, as a means to further diversify macrocyclic scaffolds produced by divergent processes, we also explored the potential for site-selective post-functionalization to append substituent groups to the newly-formed macrocyclic core. First, we explored the propensity for regioselective post-functionalization of the pyrazole ring (Scheme 8). N-Arylation of pyrazole 20d using Buchwald conditions42 selectively afforded aryl pyrazole macrocycle 56 in 68% yield (Scheme 8A). Base-mediated epoxide opening afforded a 63% yield of 58 and 59 in 1:2.6 ratio (Scheme 8A). For the free hydroxyl group-containing macrocycle 24c, Chan-Lam reaction43 provided the mono-arylated product 61 (Scheme 8B).
Scheme 8.

Post-Functionalization of Pyrazole Macrocycles
As a second means of post-functionalization, we also explored the reactivity of the macrocyclic diene produced via Rh-catalyzed cyclization (Scheme 9). Upon treatment of diene 51 with dienophiles such as N-phenylmaleimide or DMAD under thermal conditions, the desired [4+2] cycloadduct was not observed even under harsh conditions. DFT calculations for 51 showed that the s-trans-diene is thermodynamically favored in comparison to the s-cis-diene, which could explain the failure of the Diels-Alder cycloaddition (Scheme 9B). In contrast, inverse electrondemand Diels-Alder (IEDDA) reaction44 of 51 and tetrazine 62 followed by nitrogen extrusion afforded pyridazine 63 and the ring-opened hydropyridazine 64 (Scheme 9A). Further experimentation including hydrogenation of enol ether 63 to pyridazine 65 and silylation of 64 to 66 further confirmed the structures of 63 and 64.
Scheme 9.

(A) Post-Functionalization of Macrocyclic Dienes (B) DFT Optimization (B3LYP/6–31G**++) of diene 51 in both s-trans and s-cis configurations
CONCLUSIONS
In summary, we have described a new C-C bond forming strategy for macrocyclization using modular tosylhydrazone/alkyne substrates which has afforded a diverse array of new macrocyclic chemotypes from single substrates under divergent reaction conditions. The broad substrate scope and good functional group tolerance of the base-mediated dipolar cycloaddition allows access to pyrazole-containing macrocycles which can be further diversified using reactions including pyrazole arylation and epoxide opening. In addition, Rh-catalyzed macrocyclization of the same substrates led to diverse macrocyclic diene/cyclopentane/piperidyl amide/indolizine frameworks. Further diversification of the dienyl macrocycles has also been demonstrated via sequential, inverse electron demand Diels-Alder cycloadditions. Further studies to expand the functionality, reaction scope, and divergent reactions of modular substrates to produce new macrocyclic chemotypes is currently in progress and will be reported in future publications.
EXPERIMENTAL SECTION
General Information.
1H NMR spectra were recorded at 400 MHz or 500 MHz at ambient temperature with CDCl3 or CD3OD (Cambridge Isotope Laboratories, Inc.) as solvents unless otherwise stated. Chemical shifts are reported in parts per million relative to the internal solvent peak (CDCl3: δ 7.26 for 1H, δ 77.16 for 13C; CD3OD: δ 3.31 for 1H, δ 49.00 for 13C). Data for 1H NMR are reported as follows: chemical shift, multiplicity (br = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constants and integration. All 13C NMR spectra were recorded with complete proton decoupling. Infrared spectra were recorded on a Nicolet Nexus 670 FT-IR spectrophotometer. High-resolution mass spectra were obtained in the Boston University Chemical Instrumentation Center using a Waters Q-TOF mass spectrometer. Melting points were recorded on a Mel-temp (Laboratory Devices). Analytical thin layer chromatography was performed using 0.25 mm silica gel 60-F plates. Preparative TLC was conducted with glass-backed 250 μm silica gel 60-F plates (Merck KGaA). Flash chromatography was performed using 200–400 mesh silica gel (Sorbent Technologies, Inc.) or using pre-packed columns (SI-HC by puriFlash®, SNAP Ultra C18 by Biotage or disposable columns packed with C18-derivatized Silica, Luer Lock Type, Pore Size 120Å, Particle Size 40–60μm by Yamazen) on the Interchim puriFlash®450 or Yamazen Smart Flash EPCLC W-Prep2XY systems. Yields refer to chromatographically and spectroscopically pure materials, unless otherwise stated. Optical rotations were recorded on an AUTOPOL III digital polarimeter at 589 nm and specific rotations are recorded as [α]D22 (concentration in grams/100 mL solvent at 22 °C). All reactions were carried out in oven-dried glassware under an argon atmosphere unless otherwise noted. Thermolyses were conducted in Teflon® capped, heavy wall sealed vials (Chemglass CG-1880, 15 mL or CG-4920-01 Complete Package as stated) to minimize solvent/reagent evaporation. Microwave reactions were performed in sealed vessels in a CEM Discover reactor with reaction temperature monitored using an external surface sensor. All other reactions were carried out in oven-dried glassware under an argon/nitrogen atmosphere unless otherwise noted. The Scilligence Electronic Laboratory Notebook (ELN) was used for experimental procedure planning. HPLC grade tetrahydrofuran, methylene chloride, diethyl ether, and toluene were purchased from Fisher and were purified and dried by passing through a PURE SOLV® solvent purification system (Innovative Technology, Inc.). Other ACS grade solvents for chromatography were purchased from Fisher. Anhydrous 1,2-dichloroethane was purchased from Sigma Aldrich and was used as received. Anhydrous acetonitrile was purchased from Acros and was used as received. IBX-polystyrene resin was purchased from Novabiochem. Polystyrene-DIEA resin was purchased from Argonaut Technologies Inc. All other reagents and relevant catalysts were purchased from Sigma-Aldrich, Acros, Oakwood Chemical, Alfa Aesar, and Strem Chemicals.
Experimental Procedures.
Synthesis of linear macrocyclization substrate 9.
2,2-Dimethylpent-4-yn-1-amine (5).
To a 1 L round-bottomed flask were added 2,2-dimethylpent-4-ynenitrile45 (9.8 g, 91.46 mmol) and Et2O (400 mL). The reaction solution was cooled to 0 °C, and LiAlH4 (214.75 mL, 201.21 mmol, 4% in THF) was added slowly. The slurry was stirred at rt for 8 h before being cooled to 0 °C. Sodium sulfate decahydrate was added slowly until no bubbling was observed. The slurry was stirred at room temperature for 30 min before filtration through a Celite plug. The filtrate was concentrated to afford alkynyl amine 5 (5.09 g, 70.92 mmol, 78% yield) as a yellow oil. Spectroscopic data was in agreement with the literature.46
(9H-Fluoren-9-yl)methyl ((1-(2-((2,2-dimethylpent-4-yn-1-yl)amino)-2-oxoethyl)cyclohexyl)methyl)-carbamate (6a).
To a solution of Fmoc-1-aminomethyl-cyclohexane acetic acid (0.29 g, 0.74 mmol) and HOBt (0.11 g, 0.81 mmol) in THF (3 mL) at 0 °C were added EDCI (0.16 g, 0.81 mmol) and DIPEA (0.23 mL, 1.35 mmol). After 10 min at room temperature, 2,2-dimethylpent-4-yn-1-amine (75 mg, 0.34 mmol) was added at 0 °C. The reaction slurry was warmed to room temperature and stirred for 2 h. The reaction mixture was diluted with CH2Cl2 (60 mL) and was washed with sat. NaHCO3 (10 mL) and brine (10 mL). The organic layer was dried over sodium sulfate, filtered, and concentrated. The residue was purified by flash chromatography (silica gel, 25% acetone in hexanes) to afford product 6a (0.28 g, 0.33 mmol, 85% yield) as a yellow solid. Rf = 0.56 (33% acetone in hexanes); m.p. = 117–119 °C (acetone); IR (thin film): νmax 3304.73, 3062.90, 2929.11, 2866.09, 1705.15, 1646.91, 1527.52, 1451.08, 1264.90, 738.11, 703.67 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.76 (d, J = 7.5 Hz, 2H), 7.61 (d, J = 7.5 Hz, 2H), 7.40 (t, J = 7.5 Hz, 2H), 7.31 (td, J = 7.4, 1.2 Hz, 2H), 6.73 (t, J = 6.4 Hz, 1H), 5.67 (t, J = 6.9 Hz, 1H), 4.42 (d, J = 6.9 Hz, 2H), 4.21 (t, J = 6.9 Hz, 1H), 3.22 (t, J = 6.4 Hz, 4H), 2.15 (d, J = 2.6 Hz, 2H), 2.12 (s, 2H), 2.04 (t, J = 2.6 Hz, 1H), 1.59 – 1.24 (m, 10H), 1.02 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 171.63, 157.59, 144.02, 141.39, 127.75, 127.13, 125.19, 120.04, 81.89, 70.70, 66.76, 48.76, 47.81, 47.46, 43.82, 37.46, 34.78, 34.48, 29.91, 26.07, 25.02, 21.62; HRMS-ESI (m/z): [M+Na]+ calcd. for C31H38N2O3Na+, 509.2780; found, 509.2776.
2-(1-(Aminomethyl)cyclohexyl)-N-(2,2-dimethylpent-4-yn-1-yl)acetamide (6).
To a solution of 6a (0.24 g, 0.49 mmol) in DMF (5 mL) was added piperidine (1 mL). After 2 h, the reaction mixture was concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, 6:1:0.1 CH2Cl2:MeOH:10% NH4OH (aq)) to afford product 6 (90 mg, 0.34 mmol, 69% yield) as a yellowish oil. Rf = 0.05 (10% MeOH in CH2Cl2); IR (thin film): νmax 3309.61, 3058.22, 2927.61, 2856.28, 1688.01, 1555.93, 1452.04, 1368.71, 1329.63, 1268.86, 735.27, 628.96 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.18 (t, J = 6.4 Hz, 1H), 3.16 (d, J = 6.4Hz, 2H), 2.68 (s, 2H), 2.32 (br s, 2H), 2.29 (s, 2H), 2.09 (d, J = 2.6 Hz, 2H), 2.00 (t, J = 2.6 Hz, 1H), 1.56 – 1.21 (m, 10H), 0.97 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 172.06, 81.69, 70.45, 49.31, 48.27, 42.81, 36.87, 34.65, 34.53, 29.71, 26.11, 24.83, 21.47. HRMS-ESI (m/z): [M+Na]+ calcd for C16H28N2ONa+, 287.2099; found, 287.2101.
(E)-3-((2-Tosylhydrazineylidene)methyl)benzoic acid (8).
A mixture of 3-formylbenzoic acid (0.11 g, 0.73 mmol) and p-toluenesulfonyl hydrazide (0.16 g, 0.88 mmol) in CHCl3/MeOH (v/v 1:1, 2.4 mL) was stirred under reflux at 70 °C for 12 h before cooling to room temperature. Solvents were removed in vacuo and the resulting crude material was purified by flash column chromatography (silica gel, 11:89:10 MeOH/CH2Cl2/H2O) to afford product 8 (0.17 g, 0.53 mmol, 73% yield) as a light yellow solid. Rf = 0.31 (10% MeOH in CH2Cl2); m.p. = 178–180 °C (MeOH); IR (thin film): νmax 3528.74, 3185.89, 2922.81, 1698.38, 1597.86, 1323.96, 1165.12, 1058.89, 889.89, 672.24 cm−1; 1H NMR (400 MHz, CD3OD) δ 8.18 (s, 1H), 7.99 (dt, J = 7.8, 1.4 Hz, 2H), 7.87 (s, 1H), 7.83 (d, J = 8.0 Hz, 2H), 7.83 – 7.76 (m, 1H), 7.45 (t, J = 7.7 Hz, 1H), 7.36 (d, J = 8.0 Hz, 2H), 2.37 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 169.18, 147.37, 145.41, 137.36, 135.77, 132.64, 132.06, 131.87, 130.65, 129.92, 129.32, 128.77, 21.48; HRMS-ESI (m/z): [M+Na]+ calcd. for C15H14N2O4SNa+, 341.0572; found, 341.0570.
(E)-N-((1-(2-((2,2-Dimethylpent-4-yn-1-yl)amino)-2-oxoethyl)cyclohexyl)methyl)-3-((2-tosylhydraz-ineylidene) methyl)benzamide (9).
To a solution of acid 8 (0.17 g, 0.53 mmol) and HOBt (72 mg, 0.53 mmol) in THF (4 mL) at 0 °C were added EDCI (0.1 g, 0.53 mmol) and DIPEA (0.19 mL, 1.07 mmol). After 10 min, amine (0.11 g, 0.41 mmol) in THF (1 mL) was added. The reaction was complete after 3 h. The reaction mixture was diluted with CH2Cl2 (50 mL) and was washed with sat. NaHCO3 (10 mL) and brine (10 mL). The organic layer was dried over sodium sulfate, filtered, and concentrated in vacuo. Purification by flash chromatography (silica gel, 32% acetone in hexanes) afforded product 9 (164 mg, 0.29 mmol, 71% yield) as a white foam. Rf = 0.26 (33% acetone in hexanes); m.p. = 174–176 °C (acetone); IR (thin film): νmax 3390.75, 3304.53, 3069.40, 2928.62, 2865.43, 1641.74, 1537.80, 1455.03, 1327.04, 1185.01, 1166.45, 1061.14 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.88 (s, 1H), 8.48 (t, J = 5.7 Hz, 1H), 8.03 (s, 1H), 7.89 (s, 1H), 7.83 (d, J = 8.0 Hz, 3H), 7.79 (d, J = 8.1 Hz, 1H), 7.37 (t, J = 7.8 Hz, 1H), 7.20 (d, J = 8.0 Hz, 2H), 6.46 (t, J = 6.0 Hz, 1H), 3.55 (d, J = 5.7 Hz, 2H), 3.25 (d, J = 6.0 Hz, 2H), 2.31 (s, 5H), 2.13 (d, J = 2.6 Hz, 2H), 2.03 (t, J = 2.6 Hz, 1H), 1.68 – 1.33 (m, 10H), 1.00 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 172.49, 166.89, 146.74, 143.59, 135.66, 134.29, 134.07, 129.35, 128.64, 128.58, 127.73, 127.52, 81.55, 70.67, 48.71, 47.70, 45.05, 37.34, 34.53, 34.44, 29.71, 25.75, 24.79, 21.44, 21.34; HRMS-ESI (m/z): [M+Na]+ calcd. for C31H40N4O4SNa+, 587.2668; found, 587.2673.
Macrocyclizations of alkynyl sulfonyl hydrazone substrate 9 to produce 11 and 17.
10,10-Dimethyl-3,8-diazaspiro[bicyclo[13.3.1]nonadecane-5,1'-cyclohexane]-1(18),15(19),16-trien-12-yne-2,7-dione (11).
A mixture of alkynyl tosylhydrazone 9 (5 mg, 8.85 μmol), 2-propanol (2.2 mL), NaOt-Bu (13 μL, 2 M in THF) and copper(I) trifluoromethanesulfonate toluene complex (0.46 mg, 0.89 μmol) was irradiated using a microwave at 90 °C for 5 min. The mixture was filtered through a short plug of silica gel and the filtrate was concentrated in vacuo. The crude material was purified using preparative TLC (35% acetone in hexanes) to afford product 11 (0.35 mg, 0.92 μmol, 10% yield) as a colorless oil along with side products 12–15 (see Section IIIE). Rf = 0.4 (35% acetone in hexanes); IR (thin film): νmax 3300.36, 3069.09, 2930.58, 2867.72, 2232.76, 2199.15, 1076.02, 1643.92, 1541.94, 1257.10, 1029.78, 736.55 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.17 (t, J = 5.4 Hz, 1H), 8.08 (s, 1H), 7.81 (d, J = 7.7 Hz, 1H), 7.32 (t, J = 7.6 Hz, 1H), 7.21 (d, J = 7.7 Hz, 1H), 5.75 (s, J = 6.3 Hz, 1H), 3.67 (s, 2H), 3.59 (d, J = 5.7 Hz, 2H), 3.46 (d, J = 6.3 Hz, 2H), 2.35 (s, 2H), 2.17 (t, J = 2.2 Hz, 2H), 1.62 – 1.29 (m, 10H), 1.02 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 173.45, 168.13, 137.14, 136.15, 130.34, 128.42, 126.57, 126.03, 81.21, 79.39, 48.74, 48.50, 47.47, 37.55, 34.65, 34.56, 29.33, 26.12, 25.96, 24.86, 21.87; HRMS-ESI (m/z): [M+H]+ calcd. for C24H32N2O2H+, 381.2542; found, 381.2544. DFT-optimized structure of 11 is consistent with key 2D NMR interactions for structural assignment of 11 (see Figure S1 in the Supporting Information for details).
(Z)-11',11'-Dimethylspiro[cyclohexane-1,6'-4,9-diaza-1(3,5)-pyrazola-2(1,3)-benzenacyclododecaph-ane]-3',8'-dione (17).
A mixture of alkynyl tosylhydrazone 9 (25 mg, 44.27 μmol), MeCN (11 mL), and MTBD (7-Methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene, 19 μL, 0.13 mmol) in sealed tube was stirred at 90 °C for 13 h. The solvent was concentrated and the crude material was purified by preparative TLC (40% acetone in CH2Cl2) to afford macrocyclic pyrazole 17 (15 mg, 36.72 μmol, 59% yield) as a white solid along with side products 12, 14–15 and 18. Rf = 0.25 (40% acetone in hexanes); m.p. = 209–210 °C (acetone); IR (thin film): νmax 3243.81, 2925.23, 2850.14, 1643.25, 1534.81, 1454.72, 1378.08, 1326.37, 1295.85, 1251.45, 813.77, 737.84 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.09 (s, 1H), 7.95 (s, 1H), 7.78 (d, J = 7.8 Hz, 1H), 7.73 (d, J = 7.6 Hz, 1H), 7.46 (t, J = 7.7 Hz, 1H), 7.02 (s, 1H), 5.96 (s, 1H), 3.52 (d, J = 5.6 Hz, 2H), 3.50 – 3.45 (m, 2H), 2.62 (s, 2H), 2.37 (s, 2H), 1.59 – 1.27 (m, 10H), 0.92 (s, 6H); 13C NMR (101 MHz, CD3OD/CDCl3=1:1) δ 173.83, 169.33, 151.63, 144.55, 135.16, 133.54, 133.11, 129.31, 126.51, 125.93, 107.03, 49.16, 46.07, 38.18, 37.52, 35.37, 34.34, 26.75, 25.75, 21.41; HRMS-ESI (m/z): [M+H]+ calcd. for C24H32N4O2H+, 409.2604; found, 409.2622. DFT-optimized structure of 17 is consistent with key 2D NMR interactions for structural assignment of 17 (see Figure S2 in the Supporting Information for details).
Characterization data for macrocyclization side products 12–15, 18.
During the Cu(I)-catalyzed macrocyclization of 9, side products 12–15 were isolated from the reaction mixtures. During MTBD-mediated macrocyclization of 9, side products 12, 14–15, and 18 were isolated. For each set of reaction conditions, the side products were repurified to afford material suitable for characterization, and accurate yields were not determined.
N-((1-(2-((2,2-Dimethylpent-4-yn-1-yl)amino)-2-oxoethyl)cyclohexyl)methyl)-3-formylbenzamide (12).
Rf = 0.25 (25% acetone in hexanes); IR (thin film): νmax 3305.05, 3077.60, 2929.41, 2865.63, 1703.64, 1643.08, 1548.75, 1469.38, 1209.25, 737.05 cm−1; 1H NMR (400 MHz, CDCl3) δ 10.08 (s, 1H), 8.40 (t, J = 6.3 Hz, 1H), 8.39 (s, 2H), 8.16 (d, J = 7.7 Hz, 1H), 8.01 (d, J = 7.6 Hz, 1H), 7.61 (t, J = 7.7 Hz, 1H), 6.27 (t, J = 6.3 Hz, 1H), 3.53 (d, J = 6.3 Hz, 2H), 3.28 (d, J = 6.3 Hz, 2H), 2.28 (s, 2H), 2.15 (d, J = 2.6 Hz, 2H), 2.06 (t, J = 2.6 Hz, 1H), 1.64 – 1.32 (m, 10H), 1.02 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 191.82, 172.50, 166.28, 136.36, 135.58, 132.94, 131.35, 129.24, 129.20, 81.65, 70.73, 48.76, 47.68, 45.60, 37.26, 34.74, 34.69, 29.79, 25.90, 24.88, 21.57; HRMS-ESI (m/z): [M+Na]+ calcd. for C24H32N2O3Na+, 419.2311; found, 419.2313.
N-((1-(2-((2,2-Dimethylpent-4-yn-1-yl)amino)-2-oxoethyl)cyclohexyl)methyl)-3-(isopropoxymethyl)-benzamide (13).
Rf = 0.6 (60% EtOAc in hexanes); IR (thin film): νmax 3380.18, 3079.87, 2968.99, 2928.93, 1640.77, 1548.33, 1468.94, 1368.82, 1296.53, 1211.12, 1125.24, 737.01, 702.69 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.82 (d, J = 1.9 Hz, 1H), 7.77 – 7.70 (t, J = 7.6 Hz, 1H), 7.70 (t, J = 5.9 Hz, 1H), 7.49 (d, J = 7.6 Hz, 1H), 7.40 (t, J = 7.6 Hz, 1H), 6.71 (t, J = 5.9 Hz, 1H), 4.54 (s, 2H), 3.69 (hept, J = 6.0 Hz, 1H), 3.49 (d, J = 6.4 Hz, 2H), 3.25 (d, J = 6.2 Hz, 2H), 2.23 (s, 2H), 2.16 (d, J = 2.7 Hz, 2H), 2.04 (t, J = 2.7 Hz, 1H), 1.65 – 1.32 (m, 10H), 1.22 (d, J = 6.1 Hz, 6H), 1.02 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 172.11, 167.80, 139.76, 134.65, 130.60, 128.70, 126.42, 126.04, 81.88, 71.35, 70.83, 69.78, 48.94, 46.98, 45.25, 37.81, 34.98, 34.80, 30.02, 26.11, 25.13, 22.26, 21.77; HRMS-ESI (m/z): [M+H]+ calcd. for C27H40N2O3H+, 441.3117; found, 41.3135.
N-((1-(2-((2,2-Dimethylpent-4-yn-1-yl)amino)-2-oxoethyl)cyclohexyl)methyl)-3-(tosylmethyl)-benz-amide (14).
Rf = 0.50 (50% EtOAc in hexanes); m.p. = 95–97 °C; IR (thin film): νmax 3291.02, 3061.64, 2927.96, 2864.90, 1641.65, 1541.29, 1453.62, 1315.22, 1151.20, 1086.27, 814.29, 733.47, 704.46 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.97 (t, J = 6.2 Hz, 1H), 7.80 (d, J = 7.7 Hz, 1H), 7.54 (s, 1H), 7.49 (d, J = 7.9 Hz, 2H), 7.33 (t, J = 7.7 Hz, 1H), 7.26 (d, J = 7.7 Hz, 1H), 7.22 (d, J = 9.3 Hz, 2H), 6.67 (t, J = 6.4 Hz, 1H), 4.32 (s, 2H), 3.43 (d, J = 6.2 Hz, 2H), 3.24 (d, J = 6.2 Hz, 2H), 2.38 (s, 3H), 2.20 (s, 2H), 2.14 (d, J = 2.6 Hz, 2H), 2.05 (t, J = 2.6 Hz, 1H), 1.63 – 1.28 (m, 10H), 1.01 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 172.22, 166.84, 144.99, 135.06, 134.85, 133.58, 129.85, 129.73, 128.84, 128.81, 128.66, 127.45, 81.83, 70.90, 62.69, 48.90, 47.13, 45.56, 37.66, 34.91, 34.79, 29.99, 26.04, 25.09, 21.77, 21.72; HRMS-ESI (m/z): [M+H]+ calcd. for C31H40N2O4SH+, 537.2787; found, 537.2786.
3,3'-((1E,1'E)-Dydrazine-1,2-diylidenebis(methaneylylidene))bis(N-((1-(2-((2,2-dimethylpent-4-yn-1-yl)amino)-2-oxoethyl)cyclohexyl)methyl)benzamide) (15).
Rf = 0.33 (30% EtOAc in CH2Cl2); m.p. = 168–170 °C; IR (thin film): νmax 3300.80, 3116.19, 2926.48, 2868.83, 1640.86, 1542.13, 1470.16, 1455.95, 1368.28, 1296.67, 1247.78, 1212.66 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.71 (s, 2H), 8.29 (s, 2H), 8.03 (d, J = 7.8 Hz, 2H), 8.00 (t, J = 6.2 Hz, 2H), 7.96 (d, J = 7.8 Hz, 2H), 7.53 (t, J = 7.7 Hz, 2H), 6.49 (t, J = 6.4 Hz, 2H), 3.54 (d, J = 6.2 Hz, 4H), 3.28 (d, J = 6.3 Hz, 4H), 2.27 (s, 4H), 2.16 (d, J = 2.7 Hz, 4H), 2.07 (t, J = 2.7 Hz, 2H), 1.69 – 1.61 (m, 4H), 1.56 – 1.35 (m, 16H), 1.03 (s, 12H); 13C NMR (101 MHz, CDCl3) δ 172.31, 167.06, 161.62, 135.34, 134.45, 130.77, 129.82, 129.13, 127.95, 81.83, 70.92, 48.94, 47.32, 45.54, 37.70, 34.96, 34.81, 30.01, 26.08, 25.12, 21.76; HRMS-ESI (m/z): [M+H]+ calcd. for C48H64N6O4H+, 789.5067; found, 789.5052.
N-((1-(2-((2,2-Dimethylpent-4-yn-1-yl)amino)-2-oxoethyl)cyclohexyl)methyl)-3-(hydroxymethyl)-benzamide (18).
Rf = 0.33 (35% acetone in hexanes); IR (thin film): νmax 3302.46, 2929.00, 1635.91, 1528.18, 1453.42, 1264.85, 1209.18, 1035.69, 732.95, 700.90, 634.99 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.88 (t, J = 6.4 Hz, 1H), 7.81 (s, 1H), 7.73 (d, J = 7.6 Hz, 1H), 7.47 (d, J = 7.6 Hz, 1H), 7.39 (t, J = 7.6 Hz, 1H), 6.73 (s, 1H), 4.70 (s, 2H), 3.47 (d, J = 6.3 Hz, 2H), 3.23 (d, J = 6.2 Hz, 2H), 2.48 (br, 1H), 2.22 (s, 2H), 2.14 (d, J = 2.7 Hz, 2H), 2.04 (t, J = 2.7 Hz, 1H), 1.66 – 1.28 (m, 10H), 1.01 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 172.33, 167.83, 141.64, 134.77, 129.94, 128.81, 126.25, 125.63, 81.86, 70.89, 64.85, 48.96, 47.05, 45.31, 37.77, 34.94, 34.78, 30.01, 26.08, 25.12, 21.75; HRMS-ESI (m/z): [M+Na]+ calcd. for C24H34N2O3Na+, 421.2467; found, 421.2455.
Preparation of macrocyclization substrates 19a-19h.
2,5-Dioxopyrrolidin-1-yl (E)-3-((2-tosylhydrazineylidene)methyl)benzoate (8a).
To a solution of acid 8 (3 g, 9.42 mmol) and N-hydroxysuccinimide (1 g, 9.43 mmol) in CH2Cl2 (100 mL) at 0 °C was added EDCI (1.81 g, 9.43 mmol). The resulting mixture was stirred at room temperature for 2 h. Additional N-hydroxysuccinimide (0.11 g, 0.94 mmol) and EDCI (0.18 g, 0.94 mmol) were then added. After another 4 h, the reaction solution was diluted with CH2Cl2 (200 mL). The organic solution was washed with water (40 mL), 2 N HCl (40 mL), sat. NaHCO3 (40 mL) and brine (40 mL) and wasthen dried over sodium sulfate. The solution was decanted and concentrated in vacuo. The crude material was purified by flash column chromatography (silica gel, 6% EtOAc in CH2Cl2) to afford product 8a (3.16 g, 7.61 mmol, 81% yield) as a white solid. Rf = 0.25 (40% acetone in hexanes); m.p. = 208–210 °C (EtOAc); IR (thin film): νmax 3611.90, 3180.34, 2922.31, 1801.29, 1772.24, 1365.73, 1207.15, 1184.82, 1166.42, 1069.93 cm−1; 1H NMR (400 MHz, (CD3)2SO) δ 11.75 (s, 1H), 8.28 – 8.22 (m, 1H), 8.08 (d, J = 7.5 Hz, 1H), 8.04 (s, 1H), 8.00 (d, J = 8.4 Hz, 1H), 7.76 (d, J = 8.1 Hz, 2H), 7.73 – 7.62 (m, 1H), 7.40 (d, J = 8.1 Hz, 2H), 2.90 (s, 4H), 2.35 (s, 3H); 13C NMR (101 MHz, (CD3)2SO) δ 170.72, 161.85, 145.59, 144.08, 136.45, 135.41, 133.76, 131.49, 130.66, 130.19, 128.07, 127.59, 125.52, 26.01, 21.45; HRMS-ESI (m/z): [M+Na]+ calcd. for C19H17N3O6SNa+, 438.0736; found, 438.0722.
2,5-Dioxopyrrolidin-1-yl (E)-2-(1-((3-((2-tosylhydrazineylidene)methyl)benzamido)methyl)cyclohex-yl)acetate (8b).
To a solution of activated acid 8a (10 g, 24.07 mmol) and gabapentin (4.53 g, 26.48 mmol) in MeCN (200 mL) was added DIPEA (8.8 mL, 50.55 mmol) at 0 °C. The reaction mixture was stirred at room temperature for 12 h before concentrating in vacuo. The resulting solid was suspended in THF (200 mL). HOSu (3.05 g, 26.48 mmol) and EDCI (5.08 g, 26.48 mmol) were added in three3 portions at room temperature. The reaction mixture was stirred for 12 h before addition of another portion of HOSu (0.92 g, 7.94 mmol) and EDCI (1.52 g, 7.94 mmol). After another 2 h, the reaction solution was concentrated in vacuo. Water (100 mL) was added and the aqueous solution was extracted with EtOAc (300 mL) three times. The combined organic layers were washed with 2 N HCl (80 mL), sat. NaHCO3 (80 mL) and brine (80 mL), then dried over sodium sulfate, decanted and concentrated in vacuo. The crude material was purified by flash column chromatography (silica gel, 40% acetone in hexanes) to afford product 8b (13.5 g, 23.74 mmol, 99% yield). Rf = 0.75 (60% acetone in hexanes); m.p. = 97–98 °C (acetone); IR (thin film): νmax 3399.66, 2934.56, 2864.98, 1811.82, 1781.20, 1735.24, 1647.27,1540.27, 1364.84, 1210.31, 1166.53, 1064.84 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.06 (s, 1H), 8.04 (br s, 1H), 7.89 (d, J = 8.0 Hz, 1H), 7.82 (d, J = 8.0 Hz, 2H), 7.75 (s, 1H), 7.59 (d, J = 7.6 Hz, 1H), 7.42 (t, J = 7.7 Hz, 1H), 7.29 (d, J = 8.0 Hz, 2H), 3.54 (d, J = 6.6 Hz, 2H), 2.90 (s, 4H), 2.68 (s, 2H), 2.39 (s, 3H), 1.77 – 1.39 (m, 10H); 13C NMR (101 MHz, CDCl3) δ 170.06, 167.63, 146.53, 144.07, 135.42, 134.51, 133.79, 130.13, 129.64, 128.89, 127.70, 125.40, 46.41, 39.37, 34.34, 25.75, 25.66, 21.55, 21.43; HRMS-ESI (m/z): [M+Na]+ calcd. for C28H32N4O7SNa+, 591.1889; found, 591.1868.
(E)-N-((1-(2-(Oct-7-yn-1-ylamino)-2-oxoethyl)cyclohexyl)methyl)-3-((2-tosylhydrazineylidene)meth-yl)benzamide (19a).
To a solution of activated acid 8b (2 g, 3.52 mmol) in THF (35 mL) at 0 °C was added oct-7-yn-1-amine47 (0.53 g, 4.22 mmol) followed by DIPEA (0.74 mL, 4.22 mmol). The reaction solution was stirred at room temperature for 19 h and was then concentrated in vacuo. The residue was dissolved in EtOAc (500 mL) and the solution was washed with water (100 mL), sat. NaHCO3 (100 mL), 2 N HCl (100 mL) and brine (100 mL), then dried over sodium sulfate, decanted, and concentrated in vacuo. Purification by flash column chromatography (silica gel, 58% EtOAc in hexanes) afforded product 19a (1.26 g, 2.19 mmol, 62% yield) as a white foam. Rf = 0.26 (65% EtOAc in hexanes); m.p. = 89–91 °C (EtOAc); IR (thin film): νmax 3287.46, 3076.02, 2929.60, 2859.10, 1639.34, 1535.06, 1454.56, 1326.34, 1165.04, 1060.59, 691.88 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.96 (s, 1H), 8.30 (d, J = 6.1 Hz, 1H), 7.94 (d, J = 8.6 Hz, 2H), 7.85 (t, J = 9.2 Hz, 4H), 7.41 (t, J = 7.8 Hz, 1H), 7.26 (d, J = 7.9 Hz, 2H), 6.05 (t, J = 5.7 Hz, 1H), 3.54 (d, J = 6.1 Hz, 2H), 3.28 (q, J = 6.7 Hz, 2H), 2.36 (s, 3H), 2.24 (s, 2H), 2.16 (td, J = 6.9, 2.6 Hz, 2H), 1.94 (t, J = 2.6 Hz, 1H), 1.57 – 1.27 (m, 18H); 13C NMR (101 MHz, CDCl3) δ 172.36, 167.07, 146.91, 143.78, 135.79, 134.47, 134.21, 129.52, 128.83, 128.73, 127.94, 127.63, 127.52, 84.51, 68.42, 39.71, 37.42, 34.57, 29.35, 28.26, 28.23, 26.41, 25.91, 21.57, 21.49, 18.25; HRMS-ESI (m/z): [M+Na]+ calcd. for C32H42N4O4SNa+, 601.2824; found, 601.2817.
(E)-N-((1-(2-Oxo-2-((2-(2-(prop-2-yn-1-yloxy)ethoxy)ethyl)amino)ethyl)cyclohexyl)methyl)-3-((2-tosylhydrazineylidene)methyl)benzamide (19b).
To a solution of 8b (0.75 g, 1.59 mmol) and alkynyl amine (0.27 g, 1.91 mmol) in THF (15 mL) at 0 °C was added DIPEA (0.69 mL, 3.98 mmol). The reaction solution was stirred at room temperature for 4 h and was then concentrated in vacuo. The residue was dissolved in EtOAc (500 mL) and the solution was washed with water (100 mL), sat. NaHCO3 (100 mL), 2 N HCl (100 mL) and brine (100 mL) and finally dried over sodium sulfate, decanted, and concentrated in vacuo. Purification by flash column chromatography (silica gel, 80% EtOAc in hexanes) afforded product 19b (0.55 g, 0.92 mmol, 58% yield) as a white foam. Rf = 0.64 (11% acetone in EtOAc); m.p. = 73–75 °C (acetone); IR (thin film): νmax 328.21, 3062.38, 2924.46, 2864.03, 1637.18, 1529.66, 1453.68, 1326.22, 1163.67, 1090.79, 813.77, 660.59, 581.43, 547.51 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.80 (s, 1H), 8.54 (s, 1H), 7.98 (s, 1H), 7.92 (s, 1H), 7.90 – 7.84 (m, 2H), 7.82 (d, J = 8.1 Hz, 2H), 7.41 (t, J = 7.5 Hz, 1H), 6.39 (s, 1H), 4.19 (d, J = 2.5 Hz, 2H), 3.71 – 3.62 (m, 4H), 3.62 – 3.44 (m, 6H), 2.45 (d, J = 2.4 Hz, 1H), 2.36 (s, 3H), 2.29 (s, 2H), 1.58 – 1.30 (m, 10H); 13C NMR (101 MHz, CDCl3) δ 172.55, 166.79, 146.91, 143.69, 135.85, 134.54, 134.22, 129.48, 128.78, 128.69, 127.87, 127.68, 127.52, 79.39, 74.95, 69.93, 69.56, 68.97, 58.34, 53.80, 39.33, 37.46, 34.58, 29.25, 25.91, 21.57, 21.48; HRMS-ESI (m/z): [M+Na]+ calcd. for C31H40N4O6SNa+, 619.2566; found, 619.2556.
tert-Butyl (2-(prop-2-yn-1-yloxy)benzyl)carbamate (19cb).
To a 250 mL round-bottomed flask were added nitrile 19ca (1.7 g, 10.82 mmol) and Et2O (70 mL) at 0 °C. 4% LiAlH4 in THF (28.86 mL, 27.04 mmol) was added slowly. The reaction solution was stirred at room temperature for 14 h. Sodium sulfate decahydrate was added slowly until no bubbling was observed. The slurry was stirred at room temperature for 30 min before filtration through a Celite plug. The filtrate was concentrated and the crude oil was dissolved in THF (30 mL). The solution was cooled to 0 °C, then di-tert-butyl dicarbonate (2.70 g, 12.36 mmol) and NEt3 (3.45 mL, 24.71 mmol) were added. After 18 h, the reaction solution was diluted with EtOAc (150 mL), washed with 2 N HCl (30 mL), sat. NaHCO3 (30 mL) and brine (30 mL), then decanted and concentrated in vacuo. The crude material was purified by flash column chromatography (silica gel, 8% EtOAc/hexanes) to afford product 19cb (0.74 g, 2.83 mmol, 26% yield) as a light yellowish solid. Rf = 0.24 (10% EtOAc in hexanes); m.p. = 72–73 °C (EtOAc); IR (thin film): νmax 3427.87, 3381.77, 3292.97, 2977.37, 2931.80, 1703.80, 1505.05, 1366.35, 1264.94, 1170.23, 1025.88, 753.27 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.35 – 7.21 (m, 2H), 7.01 – 6.93 (m, 2H), 4.99 (s, 1H), 4.74 (d, J = 2.3 Hz, 2H), 4.32 (d, J = 6.0 Hz, 2H), 2.52 (d, J = 2.3 Hz, 1H), 1.44 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 155.87, 155.39, 129.45, 128.46, 127.74, 121.56, 111.81, 79.14, 78.53, 75.74, 55.91, 40.17, 28.44; HRMS-ESI (m/z): [M+Na]+ calcd. for C15H19NO3Na+, 284.1263; found, 284.1254.
(2-(Prop-2-yn-1-yloxy)phenyl)methanamine (19cc).
To a solution of substrate 19cb (0.25 g, 0.96 mmol) in CH2Cl2 (6 mL) at 0 °C was added TFA (2 mL). The reaction solution was stirred at room temperature for 30 min before concentration in vacuo. The crude oil was dissolved in CH2Cl2 (50 mL), washed with 2 N NaOH (5 mL), dried over sodium sulfate, decanted, and concentrated to afford product 19cc (0.133 g, 0.83 mmol, 86% yield) as a light yellowish oil. The product was carried forward without further purification. Rf = 0.05 (15% MeOH in CH2Cl2); IR (thin film): νmax 3292.40, 2920.57, 1676.10, 1601.69, 1493.67, 1457.57, 1200.76, 1133.22, 1023.56, 753.82, 679.18 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.32 – 7.20 (m, 2H), 7.02 – 6.93 (m, 2H), 4.75 (d, J = 2.4 Hz, 2H), 3.89 (s, 2H), 3.48 (s, 2H), 2.51 (t, J = 2.4 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 155.34, 129.85, 129.28, 128.53, 121.57, 111.82, 78.52, 75.80, 55.76, 41.42; HRMS-ESI (m/z): [M+Na]+ calcd. for C10H11NONa+, 184.0738; found, 184.0729.
(E)-N-((1-(2-Oxo-2-((2-(prop-2-yn-1-yloxy)benzyl)amino)ethyl)cyclohexyl)methyl)-3-((2-tosylhydra-zineylidene)methyl)benzamide (19c).
To a solution of activated acid 8b (70 mg, 0.12 mmol) in THF (1.8 mL) at 0 °C were added 19cc (24 mg, 0.15 mmol) and DIPEA (25 μL, 0.15 mmol). The reaction solution was stirred at room temperature for 17 h, then diluted with EtOAc (30 mL), washed with water (5 mL), 2 N HCl (5 mL), sat. NaHCO3 (5 mL) and brine (5 mL), and finally dried over sodium sulfate, decanted and concentrated. The crude material was purified by flash column chromatography (silica gel, 58% EtOAc in hexanes) to afford product 19c (60 mg, 0.1 mmol, 79% yield) as a white foam. Rf = 0.25 (60% EtOAc in hexanes); m.p. = 142–143 °C (EtOAc); IR (thin film): νmax 3366.32, 3288.69, 2926.68, 2851.52, 1636.44, 1577.49, 1455.87, 1326.38, 1263.76, 1163.88, 1020.38, 750.82, 583.51 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.68 (s, 1H), 8.59 (t, J = 5.9 Hz, 1H), 8.00 (s, 1H), 7.89 (s, 1H), 7.90 – 7.78 (m, 4H), 7.39 (t, J = 7.7 Hz, 1H), 7.30 – 7.22 (m, 2H), 7.20 (d, J = 8.1 Hz, 2H), 6.97 (d, J = 8.1 Hz, 1H), 6.90 (t, J = 7.5 Hz, 1H), 6.37 (t, J = 5.9 Hz, 1H), 4.73 (d, J = 2.4 Hz, 2H), 4.47 (d, J = 5.9 Hz, 2H), 3.53 (d, J = 5.9 Hz, 2H), 2.53 (t, J = 2.4 Hz, 1H), 2.31 (s, 3H), 2.29 (s, 2H), 1.68 – 1.29 (m, 10H); 13C NMR (101 MHz, CDCl3) δ 172.33, 166.71, 155.44, 146.92, 143.66, 135.90, 134.43, 134.24, 129.90, 129.49, 128.95, 128.78, 128.51, 127.97, 127.77, 127.70, 126.35, 121.60, 111.83, 78.36, 75.91, 55.94, 47.82, 45.55, 39.78, 37.71, 34.61, 25.93, 21.62, 21.50; HRMS-ESI (m/z): [M+Na]+ calcd. for C34H38N4O5SNa+, 637.2461; found, 637.2469.
(S,E)-N-((1-(2-oxo-2-((1-phenyl-2-(prop-2-yn-1-yloxy)ethyl)amino)ethyl)cyclohexyl)methyl)-3-((2-tosylhydrazineylidene)methyl)benzamide (19d).
To a solution of activated acid 8b (0.59 g, 1.04 mmol) in THF (11 mL) at 0 °C was added 19da (0.2 g, 1.14 mmol) and DIPEA (0.2 mL, 1.14 mmol). The reaction solution was stirred at room temperature for 17 h and was concentrated in vacuo. The residue was dissolved in EtOAc (500 mL) and the solution was washed with water (100 mL), sat. NaHCO3 (100 mL), 2 N HCl (100 mL) and brine (100 mL), then dried over sodium sulfate, decanted and concentrated in vacuo. Purification by flash column chromatography (silica gel, 50% EtOAc in hexanes) gave product 19d (0.54 g, 0.86 mmol, 83%) as a white solid. Rf = 0.15 (50% EtOAc in hexanes); m.p. = 150–151 °C (EtOAc); IR (thin film): νmax 3282.70, 3064.32, 2927.29, 2862.65, 1737.78, 1637.45, 1529.05, 1326.01, 1164.73, 1091.72, 1064.69, 734.86 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.39 (s, 1H), 8.28 (t, J = 6.1 Hz, 1H), 7.89 (s, 1H), 7.86 (t, J = 1.8 Hz, 1H), 7.85 – 7.81 (m, 2H), 7.79 (dt, J = 7.9, 1.3 Hz, 1H), 7.70 (ddd, J = 7.8, 1.9, 1.1 Hz, 1H), 7.35 – 7.27 (m, 4H), 7.22 – 7.18 (m, 2H), 6.76 (d, J = 7.8 Hz, 1H), 5.25 (ddd, J = 7.9, 6.0, 4.3 Hz, 1H), 4.22 – 4.08 (m, 2H), 3.85 (dd, J = 9.9, 4.3 Hz, 1H), 3.77 (dd, J = 9.9, 6.1 Hz, 1H), 3.63 (dd, J = 13.9, 6.6 Hz, 1H), 3.45 (dd, J = 13.9, 5.7 Hz, 1H), 2.90 (d, J = 23.6 Hz, 1H), 2.44 (t, J = 2.4 Hz, 1H), 2.34 (s, 2H), 2.32 (s, 3H), 1.67 – 1.34 (m, 10H); 13C NMR (101 MHz, CDCl3) δ 172.23, 167.10, 146.91, 143.78, 139.17, 135.76, 134.47, 134.12, 129.53, 128.78, 128.58, 128.04, 127.69, 127.63, 127.38, 126.81, 79.21, 75.25, 72.14, 58.29, 53.05, 37.74, 34.70, 34.39, 29.28, 25.93, 21.61, 21.51; HRMS-ESI (m/z): [M+Na]+ calcd. for C35H40N4O5SNa+, 651.2617; found, 651.2605; [α]D21 = + 45.6 (c = 0.1, MeOH).
Di-tert-butyl (2-(prop-2-yn-1-yloxy)propane-1,3-diyl)dicarbamate (19eb).
To a suspension of NaH (0.13 g, 5.37 mmol) in THF (20 mL) at 0 °C was added alcohol 19ea (1.2 g, 4.13 mmol) in THF (20 mL). The resulting slurry was stirred at room temperature for 30 min before cooling to 0 °C. Propargyl bromide (0.69 mL, 6.2 mmol, 80% in toluene) was added and the reaction solution was stirred at room temperature for 20 h, then cooled to 0 °C and quenched with water (5 mL). The aqueous layer was extracted with EtOAc (20 mL) three times. The combined organic layers were washed with sat. NH4Cl (10 mL) and brine (10 mL), then dried over sodium sulfate, decanted, and concentrated in vacuo. The crude residue was purified by flash column chromatography (silica gel, 20% EtOAc in hexanes) to afford product 19eb (0.22 g, 0.68 mmol, 16% yield) as a colorless oil. Rf = 0.66 (30% EtOAc in hexanes); IR (thin film): νmax 3360.16, 2976.88, 2935.08, 1696.91, 1512.66, 1366.89, 1251.49, 1167.85, 1096.74, 867.66, 663.31 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.06 (s, 1H), 4.82 (br s, 1H), 4.23 (d, J = 2.4 Hz, 2H), 3.61 (p, J = 5.1 Hz, 1H), 3.37 (dt, J = 12.6, 5.1 Hz, 2H), 3.14 (dt, J = 14.5, 5.7 Hz, 2H), 2.45 (d, J = 2.5 Hz, 1H), 1.44 (s, 18H); 13C NMR (101 MHz, CDCl3) δ 156.26, 79.75, 79.13, 76.51, 74.69, 56.75, 40.18, 28.25; HRMS-ESI (m/z): [M+Na]+ calcd. for C16H28N2O5Na+, 351.1896; found, 351.1893.
tert-Butyl (E)-(2-(prop-2-yn-1-yloxy)-3-(2-(1-((3-((2-tosylhydrazineylidene)methyl)benzamido)meth-yl)cyclohexyl)acetamido)propyl)carbamate (19ec).
To a solution of 19eb (0.7 g, 2.13 mmol) in CH2Cl2 (5.5 mL) at 0 °C was added 4 N HCl in dioxane (5.5 mL). The reaction mixture was stirred at room temperature for 2.5 h before concentration in vacuo to give yellow solid. The crude solid was dissolved in MeOH/H2O (2:1, 6 mL). The reaction solution was cooled to 0 °C before slow addition of di-tert-butyl dicarbonate (0.56 g, 2.56 mmol) in MeOH (4 mL) and 2 N NaOH (1.33 mL). The reaction solution was warmed to room temperature for 17 h before being concentrated in vacuo. 2 N NaOH (3 mL) was added and the aqueous solution was saturated with solid NaCl before extracting with EtOAc (15 mL) three times. The combined organic layers were washed with brine (5 mL), dried over sodium sulfate, then filtered and concentrated in vacuo. The crude oil was dissolved in THF (12 mL) and the resulting solution was cooled to 0 °C. Activated acid 8b (0.53 g, 0.93 mmol) and DIPEA (0.24 mL, 1.4 mmol) were added. The reaction solution was stirred at room temperature for 13 h, then quenched with water (6 mL). The aqueous layer was extracted with EtOAc (20 mL) three times. The combined organic layers were washed with 2 N HCl (10 mL), sat. NaHCO3 (10 mL) and brine (10 mL), then dried over sodium sulfate, decanted, and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, 50% EtOAc in hexanes) to provide product 19ec (0.36 g, 0.53 mmol, 24% yield) as a white foam. Rf = 0.25 (60% EtOAc in hexanes); m.p. = 172–173 °C (EtOAc); IR (thin film): νmax 3370.95, 3294.82, 2929.50, 2865.50, 1692.95, 1641.14, 1529.36, 1365.13, 1165.95, 1094.83, 569.00 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.05 (s, 1H), 8.57 (s, 1H), 7.94 (d, J = 6.9 Hz, 2H), 7.87 (d, J = 8.1 Hz, 2H), 7.83 (m, 7.85 – 7.80, 2H), 7.41 (t, J = 7.8 Hz, 1H), 7.26 (d, J = 7.8 Hz, 2H), 6.86 (s, 1H), 5.00 (t, J = 7.1 Hz, 1H), 4.28 – 4.15 (m, 2H), 3.55 (ddt, J = 46.2, 19.7, 4.9 Hz, 4H), 3.42 – 3.15 (m, 3H), 2.46 (s, 1H), 2.36 (s, 3H), 2.31 (d, J = 3.5 Hz, 2H), 1.79 – 1.48 (m, 10H), 1.44 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 172.91, 166.86, 156.75, 146.82, 143.71, 137.62, 135.87, 134.61, 134.19, 129.49, 128.78, 128.10, 127.72, 127.38, 79.87, 79.72, 76.19, 74.94, 56.84, 47.76, 45.52, 40.72, 39.03, 37.53, 34.68, 34.54, 28.35, 25.89, 21.58, 21.48; HRMS-ESI (m/z): [M+Na]+ calcd. for C35H47N5O7SNa+, 704.3094; found, 704.3092.
(E)-2-(Prop-2-yn-1-yloxy)-3-(2-(1-((3-((2-tosylhydrazineylidene)methyl)benzamido)methyl)cyclo-hexyl)acetamido)propan-1-aminium chloride (19e).
To a solution of 19ec (13 mg, 0.02 mmol) in CH2Cl2 (0.18 mL) at 0 °C was added 4 N HCl in dioxane (0.06 mL). The reaction mixture was stirred at room temperature for 2 h before being concentrated in vacuo to afford product 19e (11.8 mg, 0.02 mmol) as a yellow solid. The product was carried forward to the next step without further purification. Rf = 0.05 (10% MeOH in CH2Cl2); m.p. = 183–184 °C (MeOH); IR (thin film): νmax 3386.86, 3270.58, 3053.96, 2930.44, 1641.97, 1457.17, 1362.82, 1326.52, 1165.57, 1093.10 cm−1; 1H NMR (400 MHz, CD3OD) δ 8.13 (d, J = 1.8 Hz, 1H), 7.90 (s, 1H), 7.88 – 7.81 (m, 3H), 7.70 (d, J = 7.7 Hz, 1H), 7.48 (t, J = 7.8 Hz, 1H), 7.36 (d, J = 8.0 Hz, 2H), 4.36 (d, J = 2.4 Hz, 2H), 3.96 (dt, J = 8.5, 4.2 Hz, 1H), 3.61 – 3.42 (m, 4H), 3.17 (dd, J = 13.3, 3.7 Hz, 1H), 3.05 – 2.98 (m, 1H), 2.97 (t, J = 2.2 Hz, 1H), 2.39 (s, 3H), 2.37 (s, 2H), 1.67 – 1.38 (m, 10H); 13C NMR (101 MHz, CD3OD) δ 174.03, 168.52, 146.28, 144.04, 135.95, 134.56, 134.44, 129.95, 129.31, 128.76, 128.52, 127.42, 125.11, 78.85, 75.73, 73.43, 56.44, 43.00, 40.74, 38.67, 37.23, 33.85, 25.67, 21.30, 20.18; HRMS-ESI (m/z): [M+H]+ calcd. for C30H39N5O5SH+, 582.2750; found, 582.2767.
(S,E)-N-((1-(2-((2-(But-2-yn-1-yloxy)-1-phenylethyl)amino)-2-oxoethyl)cyclohexyl)methyl)-3-((2-tosylhydrazineylidene)methyl)benzamide (19f).
(S)-2-Phenylglycinol (0.64 g, 4.67 mmol) was dissolved in dry THF (30 mL) and the solution was added dropwise to a stirred suspension of NaH (0.22 g, 9.33 mmol) in dry THF (2 mL) at 0 °C. The slurry was stirred for 1 h at room temperature before being cooled to 0 °C. A solution of 1-bromo-2-butyne (0.43 mL, 4.9 mmol) in THF (4 mL) was added dropwise and the reaction solution was stirred at room temperature for 1 h and then heated to reflux under N2 for 12 h. The slurry was cooled to room temperature which was followed by the addition of brine (15 mL). The aqueous layer was extracted with diethyl ether (50 mL) three times. The combined organic layers were dried over sodium sulfate, filtered, and concentrated in vacuo. The crude oil was dissolved in THF (36 mL) and the resulting solution was cooled to 0 °C before addition of 8b (2.05 g, 3.61 mmol) and DIPEA (0.75 mL, 4.33 mmol). The reaction solution was stirred at room temperature for 17 h and was then quenched with water (15 mL) and extracted with EtOAc (50 mL) three times. The combined organic layers were washed with 2 N HCl (20 mL), sat. NaHCO3 (20 mL), and brine (20 mL), then dried over sodium sulfate, decanted, and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, 45% EtOAc in hexanes) to provide product 19f (0.78 g, 1.21 mmol, 33% yield) as a yellow solid. Rf = 0.26 (55% EtOAc in hexanes); m.p. = 153–154 °C (EtOAc); IR (thin film): νmax 3371.12, 3063.44, 2925.19, 2858.27, 1640.57, 1535.60, 1361.64, 1166.66, 1088.92, 736.51, 701.70 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.74 (s, 1H), 8.40 (t, J = 6.1 Hz, 1H), 7.92 (s, 1H), 7.87 – 7.75 (m, 4H), 7.70 – 7.64 (m, 1H), 7.36 – 7.26 (m, 5H), 7.24 – 7.20 (m, 1H), 7.18 (d, J = 8.2 Hz, 2H), 6.79 (d, J = 7.6 Hz, 1H), 5.23 (td, J = 6.6, 4.3 Hz, 1H), 4.11 (dq, J = 4.7, 2.4 Hz, 2H), 3.82 (dd, J = 10.0, 4.2 Hz, 1H), 3.73 (dd, J = 10.0, 6.2 Hz, 1H), 3.63 (dd, J = 13.9, 6.5 Hz, 1H), 3.46 (dd, J = 13.9, 5.6 Hz, 1H), 2.35 (s, 2H), 2.30 (s, 3H), 1.81 (t, J = 2.3 Hz, 3H), 1.69 – 1.34 (m, 10H); 13C NMR (101 MHz, CDCl3) δ 172.17, 166.72, 146.84, 143.71, 139.29, 135.81, 134.46, 134.09, 129.50, 128.75, 128.57, 128.55, 127.88, 127.74, 127.70, 127.62, 126.71, 83.21, 74.54, 71.81, 58.94, 53.07, 47.64, 45.54, 37.82, 34.83, 34.49, 25.95, 21.66, 21.50, 3.59; HRMS-ESI (m/z): [M+Na]+ calcd. for C36H42N4O5SNa+, 665.2774; found, 665.2795; [α]D25 = + 34.6 (c = 0.1, CHCl3).
Methyl 6-((tert-butoxycarbonyl)amino)hex-2-ynoate (19gb).
To a solution of carboxylic acid47 19ga (0.31 g, 1.36 mmol) in MeOH/PhMe (1:1, 10 mL) at 0 °C was added trimethylsilyldiazomethane (2M solution in diethyl ether, 1.36 mL, 2.73 mmol). The reaction was stirred at 0 °C for 2 h and was then concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, 16% EtOAc in hexanes) to afford product 19gb (0.26 g, 1.09 mmol, 80% yield) as a white wax. Rf = 0.2 (15% EtOAc in hexanes); m.p. = 55–57 °C (EtOAc); IR (thin film): νmax 3353.30, 2976.10, 2236.74, 1690.48, 1516.07, 1435.31, 1365.70, 1247.03, 1164.55, 1076.22, 752.74 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.61 (s, 1H), 3.76 (s, 3H), 3.22 (q, J = 6.6 Hz, 2H), 2.39 (t, J = 7.1 Hz, 2H), 1.77 (p, J = 6.9 Hz, 2H), 1.43 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 155.92, 153.90, 88.58, 78.90, 73.02, 52.40, 39.37, 28.21, 27.80, 15.96; HRMS-ESI (m/z): [M+Na]+ calcd. for C12H19NO4Na+, 264.1212; found, 264.1205.
Methyl (E)-6-(2-(1-((3-((2-tosylhydrazineylidene)methyl)benzamido)methyl)cyclohexyl)-acetamido)-hex-2-ynoate (19g).
To a solution of 19gb (0.33 g, 1.35 mmol) in CH2Cl2 (5.3 mL) at 0 °C was added 4 N HCl in dioxane (5.3 mL). The reaction mixture was stirred at room temperature for 2 h before being concentrated in vacuo. The crude solid was dissolved in MeCN (12 mL). The solution was cooled to 0 °C followed by addition of 8b (0.7 g, 1.23 mmol) and DIPEA (0.4 mL, 2.7 mmol). After 14 h at room temperature, the reaction solution was concentrated in vacuo and was dissolved in EtOAc (50 mL). The organic layer was washed with water (10 mL), 2 N HCl (10 mL), sat. NaHCO3 (10 mL) and brine (10 mL) and was subsequently dried over sodium sulfate, decanted, and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, 60% EtOAc in hexanes) to provide product 19g (0.55 g, 0.92 mmol, 75% yield) as a white foam. Rf = 0.25 (60% EtOAc in hexanes); m.p. = 148–149 °C (EtOAc); IR (thin film): νmax 3370.98, 3063.95, 2927.37, 2852.55, 2238.17, 1712.20, 1637.39, 1532.56, 1434.75, 1257.96, 1164.06, 1074.31, 735.66, 584.10 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.54 (s, 1H), 8.35 (t, J = 6.0 Hz, 1H), 7.97 (s, 1H), 7.91 (s, 1H), 7.83 (t, J = 8.5 Hz, 4H), 7.39 (t, J = 7.8 Hz, 1H), 7.23 (d, J = 8.2 Hz, 2H), 6.59 (t, J = 6.0 Hz, 1H), 3.75 (s, 3H), 3.52 (d, J = 5.9 Hz, 2H), 3.39 (q, J = 6.6 Hz, 2H), 2.39 (t, J = 7.0 Hz, 2H), 2.33 (s, 3H), 2.27 (s, 2H), 1.82 (q, J = 8.9, 7.9 Hz, 2H), 1.66 – 1.33 (m, 10H); 13C NMR (101 MHz, CDCl3) δ 172.58, 167.16, 154.24, 146.87, 143.89, 135.68, 134.50, 134.14, 129.55, 128.87, 128.15, 127.66, 127.24, 88.79, 73.32, 52.75, 38.69, 37.32, 34.55, 27.33, 25.89, 21.55, 21.50, 16.38; HRMS-ESI (m/z): [M+Na]+ calcd. for C31H38N4O6SNa+, 617.2410; found, 617.2405.
Allyl 6-((tert-butoxycarbonyl)amino)hex-2-ynoate (19ha).
To a solution of acid 19ga (4.3 g, 17.41 mmol) and DMAP (0.21 g, 1.74 mmol) in CH2Cl2 (70 mL) at 0 °C was added allyl alcohol (1.3 mL, 19.15 mmol) followed by DCC (3.95, 19.15 mmol). The reaction mixture was stirred at room temperature for 4 h and was then filtered through a short plug of silica gel and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, 15% EtOAc in hexanes) to give product 19ha (4.6 g, 17.21 mmol, 99% yield) as a light yellow oil. Rf = 0.45 (20% EtOAc in hexanes); IR (thin film): νmax 3362.32, 2976.61, 2234.91, 1705.01, 1515.19, 1448.25, 1365.77, 1242.88, 1166.29, 1073.54, 992.38, 936.51, 751.72 cm−1; 1H NMR (500 MHz, CDCl3) δ 5.92 (ddt, J = 16.6, 11.1, 5.9 Hz, 1H), 5.36 (d, J = 17.1 Hz, 1H), 5.28 (d, J = 10.3 Hz, 1H), 4.65 (d, J = 5.9 Hz, 2H), 4.61 (s, 1H), 3.22 (q, J = 6.7 Hz, 2H), 2.39 (t, J = 7.1 Hz, 2H), 1.78 (p, J = 7.0 Hz, 2H), 1.44 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 155.90, 153.22, 131.16, 119.18, 88.63, 79.19, 73.23, 66.26, 39.47, 28.32, 27.85, 16.09; HRMS-ESI (m/z): [M+Na]+ calcd. for C14H21NO4Na+, 290.1368; found, 290.1374.
Allyl (E)-6-(2-(1-((3-((2-tosylhydrazineylidene)methyl)benzamido)methyl)cyclohexyl)acetamido)hex-2-ynoate (19h).
To a solution of 19ha (3.45 g, 12.93 mmol) in CH2Cl2 (50 mL) at 0 °C was added 4 N HCl in dioxane (50 mL). The reaction mixture was stirred at room temperature for 3 h before being concentrated in vacuo. The crude residue was dissolved in THF (120 mL) and was cooled to 0 °C which was followed by addition of activated acid 8b (7 g, 12.31 mmol) and DIPEA (4.93 mL, 28.31 mmol). The reaction mixture was stirred at room temperature for 24 h, then concentrated in vacuo and dissolved in EtOAc (200 mL). The organic layer was washed with 2 N HCl (40 mL), sat. NaHCO3 (40 mL) and brine (40 mL) and was finally dried over sodium sulfate, decanted, and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, 55% EtOAc in hexanes) to give product 19h (2.1 g, 3.39 mmol, 28% yield) as a white solid. Rf = 0.25 (60% EtOAc in hexanes); m.p. = 165–167 °C (EtOAc); IR (thin film): νmax 3274.20, 3077.87, 2925.80, 2866.37, 2234.25, 1711.19, 1640.36, 1537.22, 1453.68, 1246.12, 1165.64,1067.27, 750.64 cm−1; 1H NMR (400 MHz, CDCl3) δ 10.04 (s, 1H), 8.52 (s, 1H), 7.98 (s, 1H), 7.88 (s, 1H), 7.79 (d, J = 7.6 Hz, 3H), 7.74 (d, J = 8.2 Hz, 1H), 7.33 (t, J = 7.8 Hz, 1H), 7.18 (d, J = 7.9 Hz, 2H), 5.87 (ddt, J = 16.6, 11.1, 5.9 Hz, 1H), 5.31 (d, J = 17.2 Hz, 1H), 5.23 (d, J = 10.4 Hz, 1H), 4.60 (d, J = 5.9 Hz, 2H), 3.47 (d, J = 5.8 Hz, 2H), 3.35 (q, J = 6.6 Hz, 2H), 2.36 (t, J = 7.2 Hz, 2H), 2.29 (s, 3H), 2.26 (s, 2H), 1.78 (p, J = 7.1 Hz, 2H), 1.61 – 1.25 (m, 10H); 13C NMR (101 MHz, CDCl3) δ 172.69, 167.36, 153.44, 146.86, 143.87, 135.69, 134.39, 134.16, 131.05, 129.54, 128.96, 128.87, 128.24, 127.67, 127.19, 119.37, 88.85, 73.37, 66.47, 47.70, 44.70, 38.80, 37.35, 34.50, 27.27, 25.86, 21.51, 16.42; HRMS-ESI (m/z): [M+Na]+ calcd. for C33H40N4O6SNa+, 643.2566; found, 643.2551.
Macrocyclizations of substrates 19 to produce macrocycles 20 and 21.
A solution of 19a (40 mg, 69 μmol) and MTBD (30 μL, 0.21 mmol) in MeCN (17.2 mL) was stirred at 90 °C for 48 h before being concentrated in vacuo. Purification by preparative TLC (63% acetone in hexanes) afforded 20a (3.7 mg, 9 μmol, 13% yield) as a yellow solid and 21a (6.3 mg, 15 μmol, 21% yield) as a yellow solid.
(Z)-Spiro[cyclohexane-1,6'-4,9-diaza-1(5,3)-pyrazola-2(1,3)-benzenacyclopentadecaphane]-3',8'-dione (20a).
Rf = 0.5 (50% acetone in hexanes); m.p. = 190–191 °C (acetone); IR (thin film): νmax 3226.70, 2928.28, 2854.12, 2400.32, 1996.22, 1628.56, 1582.87, 1456.53, 1310.64, 1262.39, 737.17 cm−1; 1H NMR (500 MHz, CD3OD) δ 10.32 (s, 1H), 8.54 (s, 1H), 8.44 (d, J = 1.7 Hz, 1H), 7.98 (s, 1H), 7.89 (dt, J = 7.8, 1.5 Hz, 1H), 7.51 (t, J = 7.8 Hz, 1H), 6.81 (s, 1H), 3.51 (s, 2H), 3.27 – 3.20 (m, 2H), 2.79 – 2.74 (m, 2H), 2.45 (d, J = 1.9 Hz, 2H), 1.80 (d, J = 9.0 Hz, 2H), 1.67 – 1.58 (m, 2H), 1.58 – 1.37 (m, 14H); 13C NMR (101 MHz, CD3OD/CDCl3=1:1) δ 173.18, 167.47, 149.56, 147.47, 134.17, 132.82, 128.94, 127.27, 127.19, 124.57, 100.77, 49.13, 47.35, 38.78, 36.19, 34.14, 27.61, 25.97, 25.85, 25.23, 24.85, 21.46; HRMS-ESI (m/z): [M+Na]+ calcd. for C25H34N4O2H+, 445.2579; found, 445.2559. DFT-optimized structure of 20a is consistent with key 2D NMR interactions for structural assignment of 20a (see Figure S3 in the Supporting Information for details).
Spiro[cyclohexane-1,6'-4,9-diaza-1(5,4)-pyrazola-2(1,3)-benzenacyclopentadecaphane]-3',8'-dione (21a).
Rf = 0.5 (50% acetone in hexanes); m.p. = 186–187 °C (acetone); IR (thin film): νmax 3326.51, 2923.42, 2850.71, 1975.75, 1957.34, 1737.28, 1633.49, 1456.14 cm−1; 1H NMR (500 MHz, CD3OD) δ 8.55 (s, 1H), 7.99 (s, 1H), 7.90 (d, J = 7.7 Hz, 1H), 7.73 (s, 1H), 7.56 (t, J = 7.7 Hz, 1H), 7.50 (s, 1H), 3.51 (s, 2H), 3.16 (t, J = 6.3 Hz, 2H), 2.63 (t, J = 7.6 Hz, 2H), 2.31 (s, 2H), 1.66 – 1.34 (m, 16H), 1.34 – 1.24 (m, 2H); 13C NMR (101 MHz, CD3OD) δ 173.14, 167.99, 148.90, 139.17, 134.52, 130.82, 128.74, 128.14, 126.79, 125.77, 118.53, 44.86, 37.13, 36.77, 34.38, 29.67, 28.85, 27.10, 25.70, 24.61, 23.24, 21.27; HRMS-ESI (m/z): [M+H]+ calcd. for C25H34N4O2H+, 423.2760; found, 423.2774. DFT-optimized structure of 21a is consistent with key 2D NMR interactions for structural assignment of 21a (see Figure S4 in the Supporting Information for details).
A solution of 19b (100 mg, 0.17 mmol) and MTBD (72 μL, 0.5 mmol) in MeCN (41 mL) was stirred at 90 °C for 17 h after which time the reaction mixture was concentrated in vacuo. Purification by flash column chromatography (silica gel, 25% acetone in CH2Cl2) gave 20b (13 mg, 30 μmol, 18% yield) as a white solid and 21b (25 mg, 57 μmol, 34% yield) as a yellow solid. The regioisomers were assigned by key nOe interactions in analogy to 20a/21a/20c (see Table S1 in the Supporting Information for details).
(Z)-Spiro[cyclohexane-1,6'-12,15-dioxa-4,9-diaza-1(5,3)-pyrazola-2(1,3)-benzenacyclohexadecaph-ane]-3',8'-dione (20b).
Rf = 0.16 (30% acetone in CH2Cl2); m.p. = 225–226 °C (acetone); IR (thin film): νmax 3243.54, 2926.62, 2859.69, 2371.00, 1638.12, 1556.64, 1453.44, 1352.57, 1311.58, 1119.49, 813.36, 735.43 cm−1; 1H NMR (500 MHz, CD3OD) δ 8.48 (d, J = 2.8 Hz, 1H), 7.94 (s, 1H), 7.90 (d, J = 7.6 Hz, 1H), 7.52 (dt, J = 7.7, 4.6 Hz, 1H), 7.05 (s, 1H), 4.66 (s, 2H), 3.74 – 3.66 (m, 4H), 3.63 (t, J = 7.1 Hz, 2H), 3.48 (t, J = 7.0 Hz, 2H), 3.43 (s, 2H), 2.44 (s, 2H), 1.63 – 1.34 (m, 10H); 13C NMR (101 MHz, CD3OD) δ 173.63, 167.28, 134.44, 128.94, 127.30, 126.81, 124.28, 102.18, 71.01, 69.40, 69.05, 49.20, 45.63, 39.22, 35.99, 34.04, 25.71, 21.37; HRMS-ESI (m/z): [M+H]+ calcd. for C24H32N4O4H+, 441.2502; found, 441.2495.
Spiro[cyclohexane-1,6'-12,15-dioxa-4,9-diaza-1(5,4)-pyrazola-2(1,3)-benzenacyclohexa-decaphane]-3',8'-dione (21b).
Rf = 0.25 (30% acetone in CH2Cl2); m.p. = 224–225 °C (acetone); IR (thin film): νmax 3250.61, 3066.23, 2926.10, 2864.66, 2389.23, 1636.92, 1551.63, 1456.36, 1434.87, 1349.76, 1265.00, 1092.82, 734.81 cm−1; 1H NMR (500 MHz, CD3OD) δ 8.36 (t, J = 1.7 Hz, 1H), 7.87 (d, J = 7.9 Hz, 1H), 7.85 (s, 1H), 7.76 (s, 1H), 7.55 (t, J = 7.8 Hz, 1H), 4.47 (s, 2H), 3.77 (dd, J = 5.7, 3.3 Hz, 2H), 3.72 (dd, J = 5.8, 3.5 Hz, 2H), 3.56 (dd, J = 5.5, 4.2 Hz, 2H), 3.45 (s, 2H), 3.38 (dd, J = 5.5, 4.2 Hz, 2H), 2.19 (s, 2H), 1.64 – 1.47 (m, 8H), 1.47 – 1.36 (m, 2H); 13C NMR (101 MHz, CD3OD) δ 172.34, 169.38, 134.73, 129.97, 128.56, 127.88, 127.87, 126.32, 114.70, 70.17, 70.09, 69.33, 63.07, 46.84, 41.45, 39.23, 37.32, 34.15, 25.81, 21.26; HRMS-ESI (m/z): [M+H]+ calcd. for C24H32N4O4H+, 441.2502; found, 441.2520.
A solution of 19c (100 mg, 0.16 mmol) and MTBD (70 μL, 0.49 mmol) in MeCN (40 mL) was stirred at 90 °C for 48 h. The reaction mixture was cooled and concentrated in vacuo. Purification by preparative TLC (62% acetone in hexanes) afforded 20c (17 mg, 37 μmol, 23% yield) as a white solid and 20c' (16 mg, 26 μmol, 16% yield) as a white solid.
(Z)-Spiro[cyclohexane-1,10'-4-oxa-7,12-diaza-2(5,3)-pyrazola-1(1,3),5(1,2)-dibenzenacyclotrideca-phane]-8',13'-dione (20c).
Rf = 0.5 (75% acetone in hexanes); m.p. = 234–235 °C (acetone); IR (thin film): νmax 3242.63, 2916.82, 2848.73, 2462.41, 2160.22, 1633.23, 1558.68, 1488.80, 1456.31 cm−1; 1H NMR (500 MHz, CD3OD/CDCl3=1:1) δ 10.52 (s, 1H), 8.84 (d, J = 5.8 Hz, 1H), 8.51 (t, J = 1.8 Hz, 1H), 7.80 (dt, J = 7.8, 1.4 Hz, 1H), 7.73 (d, J = 7.7 Hz, 1H), 7.41 (t, J = 7.8 Hz, 1H), 7.21 (d, J = 7.2 Hz, 1H), 7.20 (s, 1H), 7.08 – 7.03 (m, 1H), 6.92 (d, J = 8.3 Hz, 1H), 6.85 (t, J = 7.5 Hz, 1H), 5.45 (s, 2H), 4.61 (d, J = 5.2 Hz, 2H), 3.44 – 3.36 (m, 2H), 2.59 (s, 2H), 1.59 – 1.36 (m, 10H); 13C NMR (101 MHz, CD3OD/CDCl3=1:1) δ 173.16, 167.18, 154.13, 133.98, 130.21, 128.71, 128.37, 127.14, 126.70, 125.76, 125.70, 125.52, 120.95, 112.64, 103.07, 62.81, 49.90, 46.00, 37.45, 36.10, 33.78, 25.68, 21.36; HRMS-ESI (m/z): [M+H]+ calcd. for C27H30N4O3H+, 459.2396; found, 459.2416. DFT-optimized structure of 20c is consistent with key 2D NMR interactions for structural assignment of 20c (see Figure S5 in the Supporting Information for details).
N-((1-(2-((2-hydroxybenzyl)amino)-2-oxoethyl)cyclohexyl)methyl)-3-(5-(tosylmethyl)-1H-pyrazol-3-yl)benzamide (20c').
Rf = 0.69 (75% acetone in hexanes); m.p. = 145–147 °C (acetone); IR (thin film): νmax 3230.98, 2929.78, 1639.45, 1537.00, 1487.32, 1456.70, 1313.56, 1302.24, 1149.40, 755.26 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.51 (br s, 1H), 8.14 (t, J = 1.8 Hz, 1H), 7.99 (t, J = 6.3 Hz, 1H), 7.83 – 7.76 (m, 2H), 7.76 – 7.72 (m, 1H), 7.62 (d, J = 8.2 Hz, 2H), 7.48 (t, J = 7.7 Hz, 1H), 7.23 (d, J = 8.2 Hz, 2H), 7.17 (td, J = 7.7, 1.7 Hz, 1H), 7.11 (dd, J = 7.5, 1.7 Hz, 1H), 6.91 (dd, J = 8.1, 1.2 Hz, 1H), 6.79 (td, J = 7.4, 1.2 Hz, 1H), 6.61 (s, 1H), 4.50 (s, 2H), 4.35 (d, J = 6.3 Hz, 2H), 3.42 (d, J = 6.3 Hz, 2H), 2.36 (s, 3H), 2.24 (s, 2H), 1.52 – 1.25 (m, 10H); 13C NMR (101 MHz, CDCl3) δ 174.30, 168.04, 155.61, 145.53, 145.14, 139.33, 135.29, 134.93, 130.93, 130.45, 130.01, 129.84, 129.49, 129.00, 128.53, 127.12, 124.48, 124.01, 120.10, 117.75, 104.87, 55.67, 47.65, 43.83, 40.61, 37.66, 34.59, 25.96, 21.77, 21.63; HRMS-ESI (m/z): [M+H]+ calcd. for C34H38N4O5SH+, 615.2641; found, 615.2649.
A solution of 19d (100 mg, 0.16 mmol) and MTBD (68 μL, 0.48 mmol) in MeCN (39 mL) was stirred at 90 °C for 22 h. The reaction mixture was cooled to rt and was then concentrated in vacuo. Purification by flash column chromatography (silica gel, 30% acetone in hexanes) afforded 20d (36 mg, 76 μmol, 48% yield) as a yellow solid and 21d (20 mg, 42 μmol, 27% yield) as a yellow solid. Regioisomers were assigned by key nOe interactions in analogy to 20a/21a/20c (see Table S1 in the Supporting Information for details).
(S,Z)-10'-Phenylspiro[cyclohexane-1,6'-12-oxa-4,9-diaza-1(5,3)-pyrazola-2(1,3)-benzenacyclotride-caphane]-3',8'-dione (20d).
Rf = 0.4 (50% acetone in hexanes); m.p. = 220–221 °C (acetone); IR (thin film): νmax 3294.18, 2925.44, 2854.09, 2152.71, 1642.12, 1553.85, 1454.06, 1384.72, 1263.60, 1084.64 cm−1; 1H NMR (500 MHz, CD3OD) δ 10.22 (s, 0H), 8.46 – 8.33 (m, 1H), 7.84 (s, 1H), 7.79 (ddd, J = 8.0, 2.0, 1.0 Hz, 1H), 7.56 (t, J = 7.7 Hz, 1H), 7.36 – 7.31 (m, 3H), 7.31 – 7.26 (m, 2H), 5.10 (dd, J = 10.5, 3.9 Hz, 1H), 4.72 (d, J = 14.3 Hz, 1H), 4.56 (d, J = 14.3 Hz, 1H), 4.09 (dd, J = 10.2, 3.9 Hz, 1H), 3.96 (t, J = 10.2 Hz, 1H), 3.70 (d, J = 13.7 Hz, 1H), 3.17 (d, J = 13.7 Hz, 1H), 2.58 – 2.42 (m, 2H), 1.64 – 1.25 (m, 10H); 13C NMR (101 MHz, CD3OD) δ 173.23, 168.41, 140.74, 139.15, 135.10, 129.72, 129.18, 128.16, 127.56, 127.40, 126.63, 126.39, 105.35, 70.28, 62.02, 53.89, 49.24, 45.44, 37.03, 34.72, 33.27, 25.67, 21.38, 21.16; HRMS-ESI (m/z): [M+H]+ calcd. for C28H32N4O3H+, 473.2553; found, 473.2555; [α]D25 = + 86.0 (c = 0.05, MeOH).
(S)-10'-Phenylspiro[cyclohexane-1,6'-12-oxa-4,9-diaza-1(5,4)-pyrazola-2(1,3)-benzenacyclotrideca-phane]-3',8'-dione (21d).
Rf = 0.45 (50% acetone in hexanes); m.p. = 214–215 °C (acetone); IR (thin film): νmax 3271.76, 2925.83, 2853.77, 1638.72, 1535.08, 1453.75, 1364.46, 1311.01, 1078.48, 699.75 cm−1; 1H NMR (500 MHz, CD3OD) δ 8.76 (d, J = 9.3 Hz, 1H), 8.45 (s, 1H), 7.93 (br s, 1H), 7.91 (s, 1H), 7.90 (s, 1H), 7.77 (s, 1H), 7.59 (t, J = 7.8 Hz, 1H), 7.34 – 7.22 (m, 5H), 5.24 (td, J = 9.3, 4.2 Hz, 1H), 4.51 (d, J = 12.9 Hz, 1H), 4.44 (d, J = 12.9 Hz, 1H), 4.24 (dd, J = 13.7, 9.0 Hz, 1H), 3.82 – 3.66 (m, 2H), 3.02 (dd, J = 13.9, 3.1 Hz, 1H), 2.36 – 2.21 (m, 2H), 1.81 – 1.23 (m, 10H); 13C NMR (101 MHz, CDCl3) δ 172.55, 167.11, 146.46, 138.90, 136.15, 134.95, 129.47, 129.01, 128.73, 128.50, 127.78, 126.70, 126.05, 113.57, 71.63, 62.96, 52.40, 46.23, 45.68, 37.99, 35.74, 34.39, 26.01, 21.69, 21.49; HRMS-ESI (m/z): [M+H]+ calcd. for C28H32N4O3H+, 473.2553, found, 473.2565; [α]D21 = + 47.2 (c = 0.05, MeOH).
A solution of 19e (120 mg, 0.21 mmol) and MTBD (118 μL, 0.83 mmol) in MeCN (51 mL) was stirred at 90 °C for 14 h. The reaction was cooled to rt, and AcOH (41 μL, 0.73 mmol) was added, and the reaction mixture was concentrated in vacuo. The crude residue was dissolved in THF (4 mL) then polystyrene-DIEA (155 mg, 4 mmol/g, 0.62 mmol) and AcOSu (71 mg, 0.45 mmol) were added and the resulting suspension was stirred at room temperature for 22 h before filtration. The filtrate was concentrated in vacuo and purified by flash column chromatography (silica gel, 6% then 10% MeOH in CH2Cl2) and preparative TLC (10% MeOH in CHCl3) to afford 20e (65 mg, 0.14 mmol, 67% yield) as a yellow solid and 21e (6 mg, 13 μmol, 6% yield) as a yellow solid. Regioisomers were assigned by key nOe interactions in analogy to 20a/21a/20c (see Table S1 in the Supporting Information for details).
(Z)-N-((3',8'-Dioxospiro[cyclohexane-1,6'-12-oxa-4,9-diaza-1(5,3)-pyrazola-2(1,3)-benzenacyclotri-decaphan]-11'-yl)methyl)acetamide (20e).
Rf = 0.36 (10% MeOH in CH2Cl2); m.p. >250 °C (MeOH); IR (thin film): νmax 3262.08, 2927.30, 2854.20, 2396.57, 1636.67, 1560.26, 1454.20, 1373.57, 1310.87, 1077.65, 736.46 cm−1; 1H NMR (400 MHz, CD3OD) δ 10.29 (t, J = 5.1 Hz, 1H), 8.51 (t, J = 1.8 Hz, 1H), 8.23 (t, J = 6.1 Hz, 1H), 8.15 (t, J = 6.0 Hz, 1H), 7.86 – 7.74 (m, 2H), 7.53 (t, J = 7.7 Hz, 1H), 7.45 (s, 1H), 4.82 – 4.63 (m, 2H), 3.97 (dd, J = 10.7, 4.3 Hz, 1H), 3.82 (ddd, J = 13.7, 7.3, 3.4 Hz, 1H), 3.52 (dd, J = 13.7, 5.7 Hz, 1H), 3.49 – 3.32 (m, 3H), 2.67 (ddd, J = 13.4, 10.5, 4.8 Hz, 1H), 2.56 – 2.39 (m, 2H), 2.00 (s, 3H), 1.64 – 1.30 (m, 10H); 13C NMR (101 MHz, CD3OD) δ 174.26, 172.59, 167.83, 146.10, 134.77, 132.09, 129.32, 129.12, 126.44, 105.52, 74.09, 62.66, 49.18, 46.06, 41.30, 39.89, 36.95, 33.87, 33.79, 25.68, 21.33, 21.29, 21.14; HRMS-ESI (m/z): [M+H]+ calcd. for C25H33N5O4H+, 468.2611; found, 468.2629.
N-((3',8'-Dioxospiro[cyclohexane-1,6'-12-oxa-4,9-diaza-1(5,4)-pyrazola-2(1,3)-benzenacyclotrideca-phan]-11'-yl)methyl)acetamide (21e).
Rf = 0.23 (10% MeOH in CH2Cl2); m.p. >250 °C (MeOH); IR (thin film): νmax 3419.30, 2928.49, 2851.08, 2254.34, 2131.03, 1646.92, 1542.53, 1454.33, 1377.06, 1298.94, 1026.03, 824.40 cm−1; 1H NMR (400 MHz, CD3OD) δ 8.15 (s, 1H), 7.82 (d, J = 7.8 Hz, 2H), 7.76 (s, 1H), 7.57 (t, J = 7.8 Hz, 1H), 4.68 (d, J = 12.6 Hz, 1H), 4.42 (d, J = 12.6 Hz, 1H), 4.04 (d, J = 13.8 Hz, 1H), 3.89 (dd, J = 14.5, 3.1 Hz, 1H), 3.72 (d, J = 6.8 Hz, 1H), 3.42 (dd, J = 13.8, 7.4 Hz, 1H), 3.33 (d, J = 3.3, 1H), 3.12 (d, J = 13.7 Hz, 1H), 2.93 (dd, J = 14.4, 2.5 Hz, 1H), 2.44 (d, J = 13.5 Hz, 1H), 2.24 (d, J = 13.5 Hz, 1H), 1.97 (s, 3H), 1.74 – 1.38 (m, 10H); 13C NMR (126 MHz, (CD3)2SO) δ 173.21, 170.21, 167.04, 149.64, 136.42, 131.98, 129.76, 129.38, 127.58, 125.95, 114.81, 76.39, 60.94, 47.03, 46.27, 36.98, 36.26, 32.74, 26.19, 23.08, 21.61, 21.45; HRMS-ESI (m/z): [M+H]+ calcd. for C25H33N5O4H+, 468.2611; found, 468.2618.
A solution of 19f (200 mg, 0.31 mmol) and MTBD (0.13 mL, 0.93 mmol) in MeCN (77 mL) was stirred at 100 °C for 90 h, cooled to rt, and concentrated in vacuo. Purification by flash column chromatography (C18 cartridge, 1–28% MeCN in H2O with 0.1% formic acid) twice gave 20f (11 mg, 23 μmol, 7% yield) as a white solid and 21f (18 mg, 37 μmol, 12% yield) as a white solid. Regioisomers were assigned by key nOe interactions in analogy to 20a/21a/20c (see Table S1 in the Supporting Information for details).
(S)-3'-Methyl-10'-phenylspiro[cyclohexane-1,6'-12-oxa-4,9-diaza-1(5,4)-pyrazola-2(1,3)-benzenacy-clotridecaphane]-3',8'-dione (21f).
Rf = 0.27 (52% acetone in hexanes); m.p. = 188–190 °C (acetone); IR (thin film): νmax 3277.78, 3059.77, 2926.27, 2862.52, 1644.25, 1533.75, 1453.45, 1296.94, 1085.29, 1027.09, 736.41, 700.23 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.99 (s, 1H), 7.89 (d, J = 5.5 Hz, 1H), 7.46 – 7.28 (m, 8H), 5.60 – 5.47 (m, 1H), 4.33 (d, J = 11.8 Hz, 1H), 4.24 (d, J = 12.5 Hz, 1H), 4.18 (d, J = 11.3 Hz, 1H), 4.00 – 3.89 (m, 1H), 3.77 (d, J = 7.6 Hz, 1H), 2.97 (d, J = 13.6 Hz, 1H), 2.45 (d, J = 13.6 Hz, 1H), 2.33 (d, J = 13.8 Hz, 1H), 2.00 (s, 3H), 1.75 – 1.29 (m, 10H); 13C NMR (101 MHz, CDCl3) δ 172.62, 167.27, 147.55, 144.03, 139.30, 134.83, 131.02, 129.64, 129.15, 128.93, 128.60, 127.89, 126.98, 125.99, 111.70, 71.83, 62.72, 52.02, 46.46, 45.48, 38.24, 35.61, 34.84, 26.18, 21.82, 21.67, 10.52; HRMS-ESI (m/z): [M+H]+ calcd. for C29H34N4O3H+, 487.2709; found, 487.2714; [α]D25 = + 13.0 (c = 0.2, CHCl3).
(S,Z)-4'-Methyl-10'-phenylspiro[cyclohexane-1,6'-12-oxa-4,9-diaza-1(5,3)-pyrazola-2(1,3)-benzen-acyclotridecaphane]-3',8'-dione (20f).
Note: 20f exists as a mixture of conformational isomers. See Figure S7 and S8 in the Supporting Information for details. Rf = 0.23 (52% acetone in hexanes); m.p. = 188–190 °C (acetone); IR (thin film): νmax 3241.44, 3057.40, 2926.45, 2866.78, 1641.36, 1530.66, 1454.33, 1311.36, 1079.81, 1026.35, 736.34, 700.50 cm−1; 1H NMR (400 MHz, (CD3)2SO at 100 °C, mixture of conformers) δ 12.66 (s, 0.5H), 9.46 (s, 0.1H), 9.27 (d, J = 8.1 Hz, 0.3H), 8.51 (s, 0.7H), 8.15 (d, J = 7.2 Hz, 0.2H), 7.79 (s, 1H), 7.70 (s, 0.2H), 7.65 (d, J = 7.7 Hz, 1.6H), 7.54 (q, J = 8.8, 7.6 Hz, 1H), 7.37 (d, J = 7.8 Hz, 0.7H), 7.30 (d, J = 6.5 Hz, 0.2H), 7.24 (t, J = 7.4 Hz, 1.4H), 7.19 (d, J = 6.9 Hz, 0.6H), 7.13 (d, J = 7.5 Hz, 1.5H), 4.90 (s, 0.2H), 4.83 (d, J = 13.1 Hz, 0.3H), 4.56 (s, 1.5H), 4.40 (d, J = 13.3 Hz, 0.3H), 4.14 (q, J = 7.4 Hz, 0.8H), 3.85 – 3.64 (m, 2.3H), 3.53 (s, 0.8H), 3.45 – 3.32 (m, 0.4H), 3.32 – 3.21 (m, 0.4H), 2.57 – 2.39 (m, 1.4H), 2.20 (d, J = 14.4 Hz, 1.4H), 2.10 (s, 2.2H), 1.60 – 1.14 (m, 10H); 13C NMR (101 MHz, CDCl3, mixture of conformers) δ 172.73, 172.65, 167.87, 167.79, 167.10, 167.01, 148.55, 145.69, 143.23, 139.01, 138.95, 135.88, 135.27, 132.01, 131.36, 129.38, 129.10, 128.78, 128.66, 128.42, 128.09, 128.00, 127.63, 127.33, 127.21, 115.40, 114.35, 70.37, 66.41, 65.09, 63.52, 55.68, 54.93, 49.57, 48.62, 47.37, 46.52, 38.08, 36.97, 34.89, 34.71, 34.18, 25.84, 21.73, 21.60, 9.46, 8.47; HRMS-ESI (m/z): [M+H]+ calcd. for C29H34N4O3H+, 487.2709; found, 487.2718; [α]D25 = + 4.8 (c = 0.1, CHCl3/MeOH = 1:1).
Methyl (E)-3',8'-dioxospiro[cyclohexane-1,6'-4,9-diaza-1(5,3)-pyrazola-2(1,3)-benzenacyclododeca-phane]-4'-carboxylate (20g).
A solution of 19g (100 mg, 0.17 mmol) and MTBD (26 μL, 0.18 mmol) in MeCN (42 mL) was stirred at 90 °C for 14 h, then concentrated in vacuo. Purification by flash column chromatography (silica gel, 38% acetone in hexanes) gave 20g (35 mg, 80 μmol, 47% yield) as a yellow solid. Rf = 0.34 (50% acetone in hexanes); m.p. = 210–212 °C (acetone); IR (thin film): νmax 3201.05, 3090.32, 2928.38, 2381.88, 1707.17, 1631.08, 1607.54, 1456.21, 1308.10, 1220.20, 1108.72, 734.77 cm−1; 1H NMR (400 MHz, CDCl3/CD3OD=1:1) δ 9.75 (s, 1H), 7.97 (d, J = 6.5 Hz, 1H), 7.74 (s, 1H), 7.73 (d, J = 8.6 Hz, 1H), 7.68 (d, J = 7.5 Hz, 1H), 7.49 (t, J = 7.6 Hz, 1H), 3.68 (d, J = 13.6 Hz, 1H), 3.49 (s, 3H), 3.34 – 3.23 (m, 1H), 3.07 (d, J = 13.7 Hz, 1H), 2.92 (t, J = 11.2 Hz, 1H), 2.70 – 2.51 (m, 2H), 2.28 (d, J = 14.3 Hz, 1H), 2.17 (d, J = 14.3 Hz, 1H), 2.14 – 1.92 (m, 2H), 1.60 – 1.25 (m, 10H); 13C NMR (101 MHz, CD3OD) δ 173.36, 169.60, 164.97, 157.52, 151.83, 136.44, 135.20, 129.32, 128.16, 127.15, 110.27, 51.14, 49.56, 47.87, 38.61, 36.84, 36.19, 33.03, 29.96, 26.50, 25.29, 22.15, 22.07; HRMS-ESI (m/z): [M+H]+ calcd. for C24H30N4O4H+, 439.2345; found, 439.2332. DFT-optimized structure of 20g is consistent with key 2D NMR interactions for structural assignment of 20g (see Figure S9 in the Supporting Information for details).
Allyl (E)-3',8'-dioxospiro[cyclohexane-1,6'-4,9-diaza-1(5,3)-pyrazola-2(1,3)-benzenacyclododecaph-ane]-4'-carboxylate (20h).
A solution of 19h (100 mg, 0.16 mmol) and MTBD (25 μL, 0.18 mmol) in MeCN (40 mL) was stirred at 90 °C for 14 h, then concentrated in vacuo. Purification by preparative TLC (62% acetone in hexanes) afforded 20h (27 mg, 58 μmol, 36% yield) as a yellow solid. Rf = 0.25 (40% acetone in hexanes); m.p. = 190–192 °C (acetone); IR (thin film): νmax 3265.28, 3084.13, 2929.20, 1705.20, 1644.34, 1537.84, 1484.68, 1300.47, 1115.99, 1030.66, 919.25, 737.11, 702.35 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.19 (s, 1H), 7.76 (d, J = 7.6 Hz, 1H), 7.64 (s, 1H), 7.59 (d, J = 7.3 Hz, 1H), 7.40 (t, J = 7.6 Hz, 1H), 6.13 (s, 1H), 5.53 (ddt, J = 16.1, 10.5, 5.3 Hz, 1H), 4.96 (d, J = 10.4 Hz, 1H), 4.86 (d, J = 17.2 Hz, 1H), 4.42 (h, J = 7.0, 5.8 Hz, 2H), 3.57 (dd, J = 13.3, 6.2 Hz, 1H), 3.28 (d, J = 13.8 Hz, 1H), 3.22 (d, J = 11.2 Hz, 1H), 2.89 (d, J = 8.6 Hz, 1H), 2.72 – 2.54 (m, 1H), 2.48 (t, J = 11.6 Hz, 1H), 2.16 (s, 2H), 2.15 – 2.01 (m, 1H), 1.91 (s, 1H), 1.56 – 1.16 (m, 10H); 13C NMR (126 MHz, CDCl3) δ 172.52, 167.72, 163.23, 154.06, 153.15, 135.24, 132.45, 131.66, 128.99, 128.60, 127.26, 127.16, 117.56, 110.71, 64.40, 48.60, 48.06, 38.07, 37.18, 34.91, 33.42, 28.92, 25.83, 25.00, 21.66; HRMS-ESI (m/z): [M+H]+ calcd. for C26H32N4O4H+, 465.2502; found, 465.2516. DFT-optimized structure of 20h is consistent with key 2D NMR interactions for structural assignment of 20h (see Figure S10 in the Supporting Information for details).
Preparation of substrates 22a-d and 25a-c.
tert-Butyl (E)-4-(2-((2,5-dioxopyrrolidin-1-yl)oxy)-2-oxoethyl)-4-((3-((2-tosylhydrazineylidene)meth-yl)benzamido)methyl)piperidine-1-carboxylate (22ab).
A vial containing lactam 22aa (0.5 g, 1.97 mmol) and barium hydroxide octahydrate (1.24 g, 3.93 mmol) in dioxane/water (1:1, 20 mL) was stirred at 90 °C for 22 h. The reaction mixture was cooled to room temperature before addition of sat. NaHCO3 (7.9 mL) and 8a (0.28 g, 0.67 mmol). The mixture was stirred at room temperature for 32 h before being extracted with EtOAc (30 mL) three times. The combined organic layers were washed with brine (10 mL), dried over sodium sulfate, filtrated, and concentrated in vacuo. To the crude residue was added HOSu (77 mg, 0.67 mmol) and THF (6.6 mL) followed by EDCI (0.13 g, 0.67 mmol). The mixture was stirred at room temperature for 14 h before addition of water (10 mL). The aqueous layers were extracted with EtOAc (50 mL) three times. The combined organic phases were washed with 2 N HCl (20 mL), sat. NaHCO3 (20 mL) and brine (20 mL), then dried over sodium sulfate, decanted, and concentrated in vacuo. Purification by flash column chromatography (silica gel, 60% EtOAc in hexanes) afforded product 22ab (0.2 g, 0.30 mmol, 45% yield) as a white foam. Rf = 0.25 (70% EtOAc in hexanes); m.p. = 165–166 °C (EtOAc); IR (thin film): νmax 1783.06, 1736.21, 1544.07, 1430.36, 1366.16, 1209.13, 1164.44, 1065.48, 817.94, 678.01 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.61 (s, 1H), 7.94 (s, 1H), 7.80 (s, 1H), 7.76 (d, J = 7.9 Hz, 1H), 7.72 (d, J = 8.0 Hz, 2H), 7.52 (d, J = 7.7 Hz, 1H), 7.41 (t, J = 6.6 Hz, 1H), 7.30 (t, J = 7.7 Hz, 1H), 7.18 (d, J = 8.1 Hz, 2H), 3.67 – 3.48 (m, 5H), 3.42 (dd, J = 15.1, 6.2 Hz, 2H), 2.94 – 2.74 (m, 5H), 2.66 (s, 2H), 2.28 (s, 3H), 1.69 (dd, J = 11.9, 4.9 Hz, 2H), 1.63 – 1.50 (m, 2H), 1.39 (s, 9H); 13C NMR (126 MHz, CDCl3) δ 170.09, 167.74, 167.00, 154.80, 146.29, 144.00, 135.49, 134.22, 133.87, 130.41, 129.61, 128.99, 128.92, 127.60, 125.00, 79.70, 53.81, 44.46, 38.07, 37.75, 33.47, 28.40, 25.75, 21.50; HRMS-ESI (m/z): [M+Na]+ calcd. for C32H39N5O9SNa+, 692.2366; found, 692.2345.
tert-Butyl (E)-4-(2-((2,2-dimethylpent-4-yn-1-yl)amino)-2-oxoethyl)-4-((3-((2-tosylhydrazineylid-ene)methyl)benzamido)methyl)piperidine-1-carboxylate (22ac).
To a solution of 22ab (0.26 g, 0.39 mmol) in THF (3 mL) at 0 °C was added 5 (47 mg, 0.43 mmol) in THF (0.9 mL) followed by DIPEA (0.1 mL, 0.58 mmol). The reaction solution was stirred at room temperature for 14 h and condensed in vacuo. The residue was dissolved in EtOAc (500 mL) and the solution was washed with water (100 mL), sat. NaHCO3 (100 mL), 2 N HCl (100 mL) and brine (100 mL), then dried over sodium sulfate, decanted, and concentrated in vacuo. Purification by flash column chromatography (silica gel, 60% EtOAc in hexanes) gave product 22ac (0.25 g, 96% yield) as a light yellow solid. Rf = 0.24 (70% EtOAc in hexanes); m.p. = 175–176 °C (acetone); IR (thin film): νmax 3297.60, 3068.18, 2970.90, 2933.98, 1649.34, 1544.26, 1430.31, 1327.30, 1164.72, 1063.65 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.33 (s, 1H), 8.32 (s, 1H), 7.95 (d, J = 9.3 Hz, 2H), 7.82 (dd, J = 11.8, 8.1 Hz, 3H), 7.41 (t, J = 7.8 Hz, 1H), 7.25 (d, J = 8.1 Hz, 2H), 6.65 (s, 1H), 3.77 – 3.35 (m, 6H), 3.26 (d, J = 6.2 Hz, 2H), 2.35 (s, 3H), 2.30 (s, 2H), 2.14 (d, J = 2.7 Hz, 2H), 2.04 (s, 1H), 1.74 – 1.46 (m, 4H), 1.44 (s, 9H), 1.00 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 171.64, 167.09, 154.90, 146.55, 143.90, 135.66, 134.18, 129.59, 129.04, 128.91, 128.08, 127.74, 127.48, 81.61, 79.67, 70.87, 48.88, 46.01, 45.13, 36.38, 34.64, 33.80, 29.89, 28.45, 24.99, 24.87, 21.53; HRMS-ESI (m/z): [M+Na]+ calcd. for C35H47N5O6SNa+, 688.3145; found, 688.3120.
(E)-N-((4-(2-((2,2-Dimethylpent-4-yn-1-yl)amino)-2-oxoethyl)-1-tosylpiperidin-4-yl)methyl)-3-((2-tosylhydrazineylidene)methyl)benzamide (22a).
To a solution of 22ac (88 mg, 0.13 mmol) in CH2Cl2 (0.9 mL) at 0 °C was added 4 N HCl in dioxane (0.3 mL). The reaction mixture was stirred at room temperature for 2 h before concentration in vacuo. The crude solid was dissolved in CH2Cl2 (0.5 mL) and THF (0.4 mL). The resulting solution was cooled to 0 °C, then sat. NaHCO3 (0.1 mL) and TsCl (30 mg, 0.16 mmol) were added. After stirring at room temperature for 28 h, the reaction solution was cooled to 0 °C and acidified with 2 N HCl until pH < 2 before concentration in vacuo. CH2Cl2 (20 mL) was added to dissolve the crude solid. The CH2Cl2 solution was washed with water (5 mL), sat. NH4Cl (5 mL) and brine (5 mL), then dried over sodium sulfate, decanted and concentrated in vacuo. Purification by flash column chromatography (silica gel, 70% EtOAc in hexanes) gave product 22a (55 mg, 76 μmol, 58% yield) as a white solid. Rf = 0.24 (70% EtOAc in hexanes); m.p. = 195–197 °C (EtOAc); IR (thin film): νmax 3381.33, 3289.36, 3063.52, 2924.46, 1644.16, 1540.74, 1325.82, 1163.55, 1091.56, 931.76, 814.53, 725.40, 548.71 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.17 (s, 1H), 8.26 (t, J = 6.4 Hz, 1H), 7.86 (d, J = 12.1 Hz, 2H), 7.82 – 7.75 (m, 4H), 7.63 (d, J = 8.0 Hz, 2H), 7.38 (t, J = 7.8 Hz, 1H), 7.32 (d, J = 8.0 Hz, 2H), 7.21 (d, J = 8.0 Hz, 2H), 7.20 – 7.17 (m, 1 H), 3.47 (d, J = 6.2 Hz, 2H), 3.27 (dt, J = 11.4, 5.0 Hz, 2H), 3.21 (d, J = 6.1 Hz, 2H), 3.10 (dt, J = 12.2, 6.0 Hz, 2H), 2.41 (s, 3H), 2.33 (s, 3H), 2.25 (s, 2H), 2.11 (d, J = 2.7 Hz, 2H), 1.98 (t, J = 2.6 Hz, 1H), 1.70 (d, J = 5.6 Hz, 4H), 0.98 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 171.46, 167.30, 146.57, 144.09, 144.00, 135.51, 134.06, 134.01, 132.68, 130.02, 129.61, 129.12, 128.95, 128.29, 127.66, 127.49, 127.37, 81.61, 70.69, 48.77, 45.42, 44.47, 42.24, 35.81, 34.64, 33.50, 29.78, 24.86, 21.60, 21.52; HRMS-ESI (m/z): [M+Na]+ calcd. for C37H45N5O6S2Na+, 742.2709; found, 742.2711.
(E)-2-Hydroxy-3-(3-((2-tosylhydrazineylidene)methyl)benzamido)propanoic acid (22ba).
To a solution of 8a (1 g, 2.41 mmol) and isoserine (0.25 g, 2.41 mmol) in THF (12 mL) and water (12 mL) was added sat. NaHCO3 (4.4 mL). The resulting mixture was stirred at room temperature for 22 h before cooling to 0 °C. 2 N HCl (2.4 mL) was added and the reaction mixture was concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, 20:10:2 MeOH/CH2Cl2/H2O) to give product 22ba (0.89 g, 2.19 mmol, 91% yield) as a white solid. Rf = 0.05 (15% MeOH in CH2Cl2); m.p. (decomposed at 220 °C); IR (thin film): νmax 3337.57, 3063.67, 1644.23, 1598.13, 1545.00, 1445.59, 1325.95, 1165.73, 1061.53, 1020.51, 814.28, 587.45 cm−1; 1H NMR (400 MHz, CD3OD) δ 8.00 (s, 1H), 7.87 (s, 1H), 7.85 – 7.79 (m, 3H), 7.76 (d, J = 7.8 Hz, 1H), 7.46 (t, J = 7.8 Hz, 1H), 7.39 (d, J = 8.0 Hz, 2H), 4.28 – 4.18 (m, 1H), 3.74 (dd, J = 13.4, 3.8 Hz, 2H), 3.70 – 3.56 (m, 1H), 2.40 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 178.14, 169.62, 147.72, 145.22, 136.97, 135.63, 135.31, 130.57, 130.47, 129.91, 129.74, 128.56, 127.13, 71.92, 45.15, 21.49; HRMS-ESI (m/z): [M+Na]+ calcd. for C18H19N3O6SNa+, 428.0892; found, 428.0893.
(E)-N-(3-((2,2-Dimethylpent-4-yn-1-yl)amino)-2-hydroxy-3-oxopropyl)-3-((2-tosylhydrazineylid-ene)methyl)benzamide (22b).
To a solution of 22ba (0.69 g, 1.7 mmol) and HOBt (0.25 g, 1.87 mmol) in THF (14 mL) at 0 °C were added EDCI (0.36 g, 1.87 mmol) and DIPEA (0.59 mL, 3.39 mmol). After 10 min, primary amine (0.28 g, 1.87 mmol) in THF (2 mL) was added. The reaction solution was stirred at room temperature for 18 h. The reaction mixture was diluted with CH2Cl2 (50 mL), washed with sat. NaHCO3 (10 mL) and brine (10 mL). The organic layer was dried over sodium sulfate, filtered, and concentrated in vacuo.Purification by flash column chromatography (silica gel, 58% EtOAc in CH2Cl2) gave product 22b (0.28 g, 0.56 mmol, 33% yield) as a white solid. Rf = 0.26 (60% EtOAc in CH2Cl2); m.p. = 178–180 °C (EtOAc); IR (thin film): νmax 3381.13, 3302.07, 3064.49, 2961.32, 1650.16, 1538.84, 1469.11, 1325.76, 1165.38, 1061.03, 736.59, 584.36 cm−1; 1H NMR (400 MHz, CD3OD) δ 8.02 (t, J = 1.7 Hz, 1H), 7.87 (s, 1H), 7.85 – 7.78 (m, 3H), 7.74 (d, J = 7.8 Hz, 1H), 7.46 (t, J = 7.8 Hz, 1H), 7.37 (d, J = 8.0 Hz, 2H), 4.29 (dd, J = 7.1, 4.0 Hz, 1H), 3.74 (dd, J = 13.8, 4.0 Hz, 1H), 3.61 (dd, J = 13.7, 7.1 Hz, 1H), 3.17 (d, J = 1.6 Hz, 2H), 2.39 (s, 3H), 2.28 (t, J = 2.7 Hz, 1H), 2.08 (d, J = 2.7 Hz, 2H), 0.94 (s, 6H); 13C NMR (101 MHz, CD3OD) δ 174.86, 170.10, 147.43, 145.34, 137.35, 135.83, 135.71, 130.86, 130.67, 129.97, 129.79, 128.70, 126.99, 82.30, 72.31, 71.90, 45.21, 35.92, 30.36, 24.93, 24.91, 21.51; HRMS-ESI (m/z): [M+Na]+ calcd. for C25H30N4O5SNa+, 521.1835; found, 521.1831.
(9H-Fluoren-9-yl)methyl (2S,4R)-2-((2,2-dimethylpent-4-yn-1-yl)carbamoyl)-4-hydroxypyrrolidine-1-carboxylate (22ca).
To a solution of Fmoc-Hyp-OH (1 g, 2.83 mmol) in CH2Cl2 (25 mL) at 0 °C were added HOBt (0.46 g, 3.4 mmol) and EDCI (0.65 g, 3.4 mmol) followed by DIPEA (0.99 mL, 5.66 mmol). After 5 min, 5 (0.38 g, 1.7 mmol) was added. The reaction solution was stirred at room temperature for 14 h, The reaction mixture was diluted with CH2Cl2 (50 mL) and was washed with sat. NaHCO3 (10 mL) and brine (10 mL). The organic layer was dried over sodium sulfate, filtered, and concentrated in vacuo. Purification by flash column chromatography (silica gel, 50% aceteone in hexanes) gave product 22ca (1.03 g, 2.32 mmol, 82% yield) as a white foam. Rf = 0.26 (40% acetone in hexanes); m.p. = 165–167 °C (acetone); IR (thin film): νmax 3370.06, 3318.48, 2965.64, 2922.06, 1692.28, 1678.98, 1665.41, 1561.88, 1435.83, 1355.27, 1136.75, 765.76, 739.69 cm−1; 1H NMR (400 MHz, CDCl3, 60 °C) δ 7.75 (d, J = 7.6 Hz, 2H), 7.57 (t, J = 6.1 Hz, 2H), 7.39 (t, J = 7.5 Hz, 2H), 7.30 (t, J = 7.5 Hz, 2H), 6.74 (s, 1H), 4.57 – 4.47 (m, 2H), 4.47 – 4.34 (m, 2H), 4.23 (t, J = 6.7 Hz, 1H), 3.54 (s, 2H), 3.17 (d, J = 6.3 Hz, 2H), 2.43 (s, 1H), 2.23 – 2.00 (m, 2H), 2.08 (t, J = 1.9 Hz, 2H), 1.97 (s, 1H), 0.95 (s, 6H); 13C NMR (101 MHz, CDCl3, 60 °C) δ 171.69, 156.37, 144.07, 143.98, 141.57, 127.94, 127.32, 125.15, 120.15, 81.80, 70.82, 70.12, 68.00, 59.80, 55.00, 48.99, 47.58, 37.78, 34.91, 30.12, 24.98; HRMS-ESI (m/z): [M+Na]+ calcd. for C27H30N2O4Na+, 469.2103; found, 469.2120; [α]D25 = − 5.1 (c = 0.1, CHCl3/MeOH = 1:1).
(2S,4R)-N-(2,2-Dimethylpent-4-yn-1-yl)-4-hydroxypyrrolidine-2-carboxamide (22cb).
To a solution of 22ca (0.2 g, 0.45 mmol) in CH2Cl2 (2 mL) at room temperature was added Et2NH (2 mL). The reaction solution was stirred for 1 h before concentration in vacuo. Water (10 mL) was added and the aqueous layer was washed with Et2O (5 mL) two times. The aqueous phase was concentrated to give product 22cb (0.1 g, 0.45 mmol, 100% yield) as a white solid. Rf = 0.04 (10% MeOH in CH2Cl2); m.p. = 120–121 °C (MeOH); IR (thin film): νmax 3296.95, 2963.05, 2929.61, 1647.07, 1532.27, 1469.56, 1390.40, 1323.96, 1207.14, 1095.89, 1063.41, 642.29 cm−1; 1H NMR (400 MHz, CD3OD) δ 4.41 – 4.29 (m, 1H), 3.89 (t, J = 8.3 Hz, 1H), 3.15 (s, 2H), 2.99 (dd, J = 12.0, 4.0 Hz, 1H), 2.90 (dt, J = 12.0, 1.8 Hz, 1H), 2.32 (t, J = 2.7 Hz, 1H), 2.16 (ddt, J = 14.0, 8.6, 1.9 Hz, 1H), 2.11 (d, J = 2.7 Hz, 2H), 1.81 (ddd, J = 13.6, 8.6, 5.0 Hz, 1H), 0.97 (s, 6H); 13C NMR (101 MHz, CD3OD) δ 177.29, 82.20, 73.59, 71.84, 60.78, 56.02, 41.08, 35.95, 30.40, 24.97, 24.91; HRMS-ESI (m/z): [M+H]+ calcd. for C12H20N2O2H+, 225.1603; found, 225.1614. HRMS-ESI (m/z): [M+H]+ calcd. for C12H20N2O2H+, 225.1603; found, 225.1614; [α]D21 = − 13.0 (c = 0.1, MeOH).
(2S,4R)-N-(2,2-Dimethylpent-4-yn-1-yl)-4-hydroxy-1-(3-((E)-(2-tosylhydrazineylidene)methyl)benz-oyl)pyrrolidine-2-carboxamide (22c).
To a solution of carboxylic acid 8 (30 mg, 94 μmol) and HOBt (13 mg, 99 μmol) in THF (1 mL) at 0 °C were added EDCI (19 mg, 99 μmol) and DIPEA (33 μL, 0.19 mmol). After 10 min, 22cb (22 mg, 99 μmol) was added and then the reaction solution was stirred at room temperature for 22 h. The reaction mixture was diluted with CH2Cl2 (50 mL), washed with sat. NaHCO3 (10 mL) and brine (10 mL). The organic layer was dried over sodium sulfate, filtered, and concentrated in vacuo.. Purification by flash column chromatography (silica gel, 18% acetone in EtOAc) gave product 22c (30 mg, 57.2 μmol, 61% yield) as a white solid. Rf = 0.4 (25% acetone in EtOAc); m.p. = 173–175 °C (acetone); IR (thin film): νmax 3299.44, 3087.67, 2964.75, 2922.13, 1666.47, 1617.09, 1437.36, 1327.52, 1165.25, 1066.08, 580.68 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.75 (s, 1H), 7.79 (d, J = 7.9 Hz, 2H), 7.67 (s, 1H), 7.57 (d, J = 7.8 Hz, 1H), 7.41 (s, 1H), 7.36 (d, J = 7.8 Hz, 1H), 7.22 (d, J = 8.1 Hz, 2H), 7.20 – 7.13 (m, 2H), 4.89 (t, J = 8.2 Hz, 1H), 4.45 (s, 1H), 3.65 (d, J = 11.0 Hz, 1H), 3.37 (d, J = 11.4 Hz, 1H), 3.26 (dd, J = 13.6, 6.5 Hz, 1H), 3.13 (dd, J = 13.5, 6.1 Hz, 1H), 2.40 (d, J = 9.1 Hz, 1H), 2.32 (s, 3H), 2.33 – 2.19 (m, 1H), 2.02 – 2.14 (m, 2H), 2.00 (t, J = 2.6 Hz, 1H), 0.95 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 172.35, 170.65, 146.72, 144.18, 135.78, 135.53, 133.93, 129.78, 128.90, 128.80, 127.78, 126.58, 81.82, 70.95, 70.26, 59.60, 58.82, 48.64, 37.75, 35.18, 29.70, 24.77, 21.61; HRMS-ESI (m/z): [M+H]+ calcd. for C27H32N4O5SH+, 525.2172; found, 525.2167; [α]D21 = − 18.3 (c = 0.1, CHCl3/MeOH=1:1).
(9H-Fluoren-9-yl)methyl (2S,4R)-4-hydroxy-2-(((S)-1-phenyl-2-(prop-2-yn-1-yloxy)ethyl)carbamo-yl)pyrrolidine-1-carboxylate (22da).
To a solution of Fmoc-Hyp-OH (5.5 g, 15.56 mmol) in CH2Cl2 (75 mL) at 0 °C were added HOBt (2.31 g, 17.12 mmol) and EDCI (3.28 g, 17.12 mmol) followed by DIPEA (5.42 mL, 31.13 mmol). After 5 min, 19da (3 g, 17.12 mmol) in CH2Cl2 (10 mL) was added. The reaction solution was stirred at room temperature for 19 h before diluted with CH2Cl2 (150 mL). The solution was washed with water (40 mL), 2 N HCl (40 mL), sat. NaHCO3 (40 mL) and brine (40 mL), then dried over sodium sulfate, decanted and concentrated in vacuo. Purification by flash column chromatography (silica gel, 80% EtOAc in hexanes) gave product 22da (5.5 g, 10.77 mmol, 69% yield) as a white foam. Rf = 0.17 (80% EtOAc in hexanes); m.p. = 110–112 °C (EtOAc); IR (thin film): νmax 3404.22, 3291.73, 3060.84, 2946.91, 1676.49, 1542.96, 1451.55, 1424.08, 1354.67, 1091.22, 757.98, 740.11 cm−1; 1H NMR (400 MHz, CDCl3, 60 °C) δ 7.76 (d, J = 7.6 Hz, 2H), 7.57 (t, J = 7.0 Hz, 2H), 7.39 (t, J = 7.5 Hz, 2H), 7.33 – 7.24 (m, 4H), 7.24 – 7.13 (m, 3H), 5.15 (dt, J = 7.7, 5.3 Hz, 1H), 4.62 – 4.42 (m, 3H), 4.35 (s, 1H), 4.20 (t, J = 7.0 Hz, 1H), 4.08 (s, 2H), 3.76 (q, J = 7.7, 5.8 Hz, 2H), 3.56 (s, 2H), 2.40 (br s, 1H), 2.36 (t, J = 2.4 Hz, 1H), 2.28 – 2.00 (m, 2H); 13C NMR (101 MHz, CDCl3, 60 °C) δ 171.19, 156.20, 144.07, 141.51, 139.51, 128.60, 127.89, 127.60, 127.31, 127.27, 126.87, 125.26, 125.20, 120.09, 79.44, 75.04, 72.23, 70.02, 68.09, 59.74, 58.51, 55.04, 53.00, 47.52, 37.30; HRMS-ESI (m/z): [M+H]+ calcd. for C31H30N2O5H+, 511.2233; found, 511.2240; [α]D25 = − 3.6 (c = 0.1, CHCl3).
(2S,4R)-4-Hydroxy-N-((S)-1-phenyl-2-(prop-2-yn-1-yloxy)ethyl)pyrrolidine-2-carboxamide (22db).
To a solution of 22da (2.3 g, 4.5 mmol) in CH2Cl2 (16 mL) at room temperature was added Et2NH (16 mL). The reaction solution was stirred for 1.5 h and was then concentrated in vacuo. Water (20 mL) was added and the aqueous layer was washed with Et2O (20 mL) three times. The aqueous layer was concentrated in vacuo to provide 22db (1.26 g, 4.36 mmol, 97% yield) as a yellowish oil. Rf = 0.05 (30% MeOH in CH2Cl2); IR (thin film): νmax 3291.20, 2935.79, 2865.50, 1651.32, 1520.10, 1453.98, 1356.88, 1094.37, 959.83, 760.66, 701.00 cm−1; 1H NMR (500 MHz, CD3OD) δ 7.32 (d, J = 4.4 Hz, 4H), 7.28 – 7.21 (m, 1H), 5.09 (dd, J = 7.1, 4.8 Hz, 1H), 4.35 (dt, J = 4.3, 2.4 Hz, 1H), 4.23 – 4.11 (m, 2H), 3.97 (t, J = 8.3 Hz, 1H), 3.82 – 3.71 (m, 2H), 3.03 (dd, J = 12.0, 4.1 Hz, 1H), 2.91 (dt, J = 12.0, 1.8 Hz, 1H), 2.86 (t, J = 2.4 Hz, 1H), 2.17 (ddt, J = 13.5, 8.0, 1.8 Hz, 1H), 1.82 (ddd, J = 13.6, 8.7, 5.0 Hz, 1H); 13C NMR (101 MHz, CD3OD) δ 176.15, 140.79, 129.52, 128.55, 127.84, 80.27, 76.28, 73.36, 73.21, 60.68, 58.99, 55.98, 53.99, 40.93; HRMS-ESI (m/z): [M+Na]+ calcd. for C16H20N2O3Na+, 311.1372; found, 311.1384; [α]D21 = −106.6 (c = 0.1, MeOH).
(2S,4R)-4-hydroxy-N-((S)-1-phenyl-2-(prop-2-yn-1-yloxy)ethyl)-1-(2'-((E)-(2-tosylhydrazineylidene)methyl)-[1,1'-biphenyl]-3-carbonyl)pyrrolidine-2-carboxamide (22d).
A mixture of aldehyde 22dc (0.11 g, 0.5 mmol) and tosylhydrazide (93 mg, 0.5 mmol) in CHCl3/MeOH (1:1, 1.2 mL) was refluxed at 70 °C for 13 h, cooled to rt, and concentrated in vacuo to give a yellowish solid. To the crude solid was added THF (4.5 mL) and HOBt (67 mg, 0.5 mmol). The reaction mixture was cooled to 0 °C before addition of EDCI (96 mg, 0.5 mmol) and DIPEA (0.14 mL, 0.83 mmol). After 5 min, 22db (0.12 g, 0.42 mmol) was added and the reaction mixture was stirred at room temperature for 18 h before concentration in vacuo. The residue was dissolved in CH2Cl2 (120 mL) and the organic solution was washed with water (30 mL), 2 N HCl (30 mL), sat. NaHCO3 (30 mL), brine (30 mL), and dried over sodium sulfate, decanted, and concentrated in vacuo. Purification by flash column chromatography (silica gel, 60% EtOAc in CH2Cl2) gave product 22d (0.2 g, 0.29 mmol, 70% yield) as a white solid. Rf = 0.73 (25% acetone in EtOAc); m.p. = 172–174 °C (acetone); IR (thin film): νmax 3335.36, 3294.89, 3064.00, 2924.89, 1668.02, 1614.21, 1442.12, 1329.08, 1165.80, 1093.40, 959.46, 759.22, 587.35 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.92 (s, 0.5H), 9.63 (s, 0.5H), 7.90 (d, J = 7.5 Hz, 0.5H), 7.85 (d, J = 7.8 Hz, 1H), 7.77 (d, J = 7.1 Hz, 2H), 7.61 (d, J = 8.1 Hz, 0.5H), 7.40 – 7.27 (m, 5H), 7.25 – 7.10 (m, 5H), 7.01 – 6.87 (m, 1H), 6.80 (d, J = 8.2 Hz, 1.5H), 6.70 (s, 0.5H), 5.14 (q, J = 5.8, 5.1 Hz, 0.5H), 4.83 (t, J = 8.3 Hz, 1H), 4.38 (d, J = 16.9 Hz, 1H), 4.12 (d, J = 17.7 Hz, 1.5H), 4.03 (s, 1H), 3.90 (d, J = 12.7 Hz, 0.5H), 3.82 – 3.68 (m, 1.5H), 3.68 – 3.55 (m, 1H), 3.48 (d, J = 12.0 Hz, 2H), 2.53 (s, 0.5H), 2.41 (d, J = 5.7 Hz, 1H), 2.35 (s, 2H), 2.32 (s, 1.5H), 2.16 (t, J = 11.8 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 171.68, 171.39, 170.97, 170.92, 147.79, 146.28, 143.86, 143.62, 140.96, 140.53, 139.58, 139.26, 139.10, 138.32, 136.16, 135.81, 135.49, 135.21, 131.91, 131.50, 131.09, 130.80, 130.62, 129.96, 129.75, 129.66, 128.77, 128.63, 128.46, 128.32, 128.04, 127.82, 127.67, 127.41, 127.13, 126.78, 126.47, 126.39, 126.21, 126.10, 124.43, 79.39, 79.27, 75.44, 75.32, 72.29, 70.25, 68.58, 60.67, 59.21, 58.57, 58.38, 58.30, 56.14, 53.77, 53.05, 41.17, 37.29, 21.63; HRMS-ESI (m/z): [M+Na]+ calcd. for C37H36N4O6SNa+, 687.2253; found, 687.2269; [α]D25 = + 31.2 (c = 0.1, MeOH).
(E)-N-(1-((2,2-dimethylpent-4-yn-1-yl)amino)-1-oxo-3-phenylpropan-2-yl)-3-((2-tosylhydrazineylid-ene)methyl)benzamide (25a).
To a solution of 8a (1 g, 2.41 mmol) and DL-phenylalanine (0.4 g, 2.41 mmol) in THF/H2O (1:1, 18 mL) was added sat. NaHCO3 (4.4 mL). The reaction mixture was stirred at room temperature for 14 h before being cooled to 0 °C. The mixture was acidified with 2 N HCl to pH <2 and was then extracted with CHCl3/iPrOH (3:1, 30 mL) three times. The combined organic layers were dried over sodium sulfate, decanted, and concentrated in vacuo. The crude acid was suspended in THF (20 mL) before being cooled to 0 °C. HOBt (0.36 g, 2.65 mmol), EDCI (0.51 g, 2.65 mmol), and DIPEA (0.84 mL, 4.82 mmol) were added. After 10 min, amine 5 (0.29 g, 2.65 mmol) in THF (3 mL) was added. The reaction mixture was stirred at room temperature for 14 h before concentration in vacuo. The residue was dissolved in EtOAc (100 mL) and organic solution was washed with water (20 mL), 2 N HCl (20 mL), sat. NaHCO3 (20 mL), and brine (20 mL), then dried over sodium sulfate, decanted, and concentrated in vacuo. Purification by flash column chromatography (silica gel, 55% EtOAc in hexanes) gave product 25a (0.36 g, 0.65 mmol, 27% yield) as a white solid. Rf = 0.22 (60% EtOAc in hexanes); m.p. = 165–166 °C (EtOAc); IR (thin film): νmax 3303.76, 3067.18, 2964.69, 1637.20, 1543.52, 1325.72, 1163.67, 1062.91, 753.89 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.63 (s, 1H), 8.02 (s, 1H), 7.93 (d, J = 7.9 Hz, 1H), 7.91 – 7.80 (m, 3H), 7.68 (d, J = 7.8 Hz, 1H), 7.42 (t, J = 7.8 Hz, 1H), 7.34 – 7.27 (m, 6H), 7.25 – 7.20 (m, 2H), 7.14 (d, J = 8.2 Hz, 1H), 6.38 (s, 1H), 5.09 (q, J = 7.7 Hz, 1H), 3.30 – 3.11 (m, 3H), 3.06 (dd, J = 13.6, 5.9 Hz, 1H), 2.38 (s, 3H), 2.01 – 1.84 (m, 3H), 0.85 (d, J = 2.6 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ 172.28, 166.73, 146.97, 143.89, 136.70, 135.57, 134.08, 133.01, 129.64, 129.38, 128.72, 128.55, 128.09, 127.60, 126.90, 81.54, 70.72, 55.58, 48.72, 38.56, 34.67, 29.59, 24.61, 24.54, 21.51; HRMS-ESI (m/z): [M+H]+ calcd. for C31H34N4O4SH+, 559.2379; found, 559.2380.
2,5-Dioxopyrrolidin-1-yl (E)-3-phenyl-4-(3-((2-tosylhydrazineylidene)methyl)benzamido)butanoate (25ba).
To a solution of 8a (1 g, 2.41 mmol) and 4-amino-3-phenylbutyric acid (0.52 g, 2.89 mmol) in MeCN (20 mL) at 0 °C was added DIPEA (0.92 mL, 5.3 mmol). The reaction mixture was stirred at room temperature for 16 h before being concentrated in vacuo. The residue was dissolved in THF (23 mL), then HOSu (0.33 g, 2.88 mmol) and EDCI (0.55 g, 2.88 mmol) were added. The reaction mixture was stirred for 24 h before being concentrated in vacuo. The residue was dissolved in EtOAc (120 mL) and the solution was washed with water (20 mL), 2 N HCl (20 mL), brine (20 mL), then dried over sodium sulfate, decanted, and concentrated in vacuo. Purification by flash column chromatography (silica gel, 20% EtOAc in CH2Cl2) gave product 25ba (0.16 g, 0.28 mmol, 12% yield) as a yellow solid. Rf = 0.16 (25% EtOAc in CH2Cl2); m.p. = 110–112 °C (EtOAc); IR (thin film): νmax 3375.70, 3066.30, 2926.14, 1815.38, 1738.11, 1642.12, 1541.57, 1364.61, 1210.29, 1166.91, 1069.17, 703.28 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.87 (s, 1H), 7.82 (d, J = 8.2 Hz, 2H), 7.78 (d, J = 7.1 Hz, 2H), 7.65 (d, J = 7.8 Hz, 1H), 7.60 (d, J = 7.7 Hz, 1H), 7.39 – 7.28 (m, 5H), 7.24 (s, 1H), 6.83 (t, J = 6.1 Hz, 1H), 3.88 (dt, J = 14.0, 6.0 Hz, 1H), 3.70 (dt, J = 13.2, 6.0 Hz, 1H), 3.65 – 3.54 (m, 1H), 3.05 (qd, J = 16.1, 7.1 Hz, 2H), 2.78 (s, 4H), 2.36 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 169.52, 167.61, 167.46, 146.75, 144.37, 140.32, 135.44, 134.74, 133.82, 129.91, 129.85, 129.15, 129.00, 128.80, 127.95, 127.78, 127.62, 126.24, 45.02, 41.86, 35.45, 25.72, 21.72; HRMS-ESI (m/z): [M+H]+ calcd. for C29H28N4O7SH+, 577.1757; found, 577.1763.
(E)-N-(4-((2,2-Dimethylpent-4-yn-1-yl)amino)-4-oxo-2-phenylbutyl)-3-((2-tosylhydrazineylidene)-methyl)benzamide (25b).
To a solution of activated acid 25ba (0.18 g, 0.31 mmol) in THF (3.5 mL) at 0 °C were added primary amine 6 (38 mg, 0.34 mmol) and DIPEA (60 μL, 0.34 mmol). The reaction solution was stirred at room temperature for 40 h and was concentrated in vacuo. The residue was dissolved in EtOAc (500 mL) and the solution was washed with water (100 mL), sat. NaHCO3 (100 mL), 2 N HCl (100 mL) and brine (100 mL), then dried over sodium sulfate, decanted and concentrated in vacuo Purification by flash column chromatography (silica gel, 70% EtOAc in hexanes) gave product 25b (0.11 g, 0.19 mmol, 59% yield) as a white solid. Rf = 0.17 (80% EtOAc in hexanes); m.p. = 110–112 °C (EtOAc); IR (thin film): νmax 3302.45, 3062.43, 2961.27, 2927.19, 1638.71, 1537.10, 1468.44, 1366.84, 1305.12, 1162.89, 1019.76, 953.72, 813.17, 700.72 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.41 (s, 1H), 7.92 (s, 1H), 7.87 – 7.82 (m, 3H), 7.80 (d, J = 8.0 Hz, 1H), 7.63 (d, J = 7.7 Hz, 1H), 7.41 – 7.28 (m, 6H), 7.23 (m, 2H), 7.05 (t, J = 5.7 Hz, 1H), 6.09 (t, J = 5.9 Hz, 1H), 3.86 (dt, J = 14.2, 7.1 Hz, 1H), 3.69 (dt, J = 13.7, 5.5 Hz, 1H), 3.54 (p, J = 6.4 Hz, 1H), 3.18 (dd, J = 13.6, 6.8 Hz, 1H), 3.00 (dd, J = 13.6, 5.9 Hz, 1H), 2.77 (dd, J = 14.6, 7.5 Hz, 1H), 2.62 (dd, J = 14.5, 6.5 Hz, 1H), 2.35 (s, 3H), 1.98 – 1.96 (m, 1H), 1.93 (dd, J = 4.4, 2.6 Hz, 2H), 0.84 (s, 3H), 0.82 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 172.46, 167.43, 146.87, 144.07, 141.65, 135.81, 134.35, 134.32, 129.75, 129.24, 129.06, 128.98, 128.08, 127.85, 127.75, 127.45, 127.38, 81.77, 70.77, 48.74, 45.68, 42.44, 41.38, 34.71, 29.62, 24.74, 21.67; HRMS-ESI (m/z): [M+H]+ calcd. for C32H36N4O4SH+, 573.2536; found, 573.2518.
(E)-N-((1-(2-((2,2-Dimethylpent-4-yn-1-yl)amino)-2-oxoethyl)cyclohexyl)methyl)-2-hydroxy-5-((2-tosylhydrazineylidene)methyl)benzamide (25c).
A mixture of aldehyde 25ca (0.15 g, 0.91 mmol) and tosylhydrazide (0.17 g, 0.91 mmol) in CHCl3/MeOH (1:1, 2 mL) was refluxed at 70 °C for 19 h before being cooled to rt and concentrated in vacuo. To the residue in THF (7 mL) at 0 ℃ were added HOBt (0.13 g, 0.97 mmol) and EDCI (0.19 g, 0.97 mmol). After 5 min, primary amine 6 (0.2 g, 0.75 mmol) and DIPEA (0.34 mL, 1.94 mmol) were added and the resulting mixture was stirred at room temperature for 44 h before being concentrated in vacuo. The residue was dissolved in CH2Cl2 (100 mL) and the solution was washed with 2 N HCl (15 mL), dried over sodium sulfate, decanted and concentrated in vacuo. Purification by flash column chromatography (silica gel, 30% EtOAc in hexanes) gave product 25c (0.38 g, 0.66 mmol, 88% yield) as a white foam. Rf = 0.5 (60% EtOAc in hexanes); m.p. = 178–179 °C (EtOAc); IR (thin film): νmax 3305.05, 3054.47, 2930.94, 1643.94, 1583.92, 1525.47, 1365.34, 1265.40, 1167.44, 738.20, 704.55 cm−1; 1H NMR (400 MHz, CDCl3) δ 13.10 (s, 1H), 8.84 (t, J = 5.9 Hz, 1H), 8.27 (t, J = 5.3 Hz, 1H), 7.88 (d, J = 7.9 Hz, 2H), 7.74 (s, 2H), 7.65 (d, J = 8.8 Hz, 1H), 7.28 (d, J = 8.8 Hz, 2H), 6.90 (d, J = 8.7 Hz, 1H), 6.34 (s, 1H), 3.48 (d, J = 5.8 Hz, 2H), 3.27 (d, J = 6.0 Hz, 2H), 2.38 (s, 3H), 2.29 (s, 2H), 2.12 (s, 2H), 2.07 – 1.97 (m, 1H), 1.67 – 1.27 (m, 10H), 0.97 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 172.65, 169.71, 163.71, 147.65, 144.29, 135.32, 131.95, 129.77, 128.04, 126.51, 124.23, 119.05, 114.49, 81.81, 71.09, 60.57, 48.99, 46.89, 45.95, 37.54, 34.88, 34.71, 29.99, 25.95, 25.14, 21.72; HRMS-ESI (m/z): [M+Na]+ calcd. for C31H40N4O5SNa+, 603.2617; found, 603.2618.
(E)-N-(2,2-Dimethylpent-4-yn-1-yl)-2-(1-((2-(2-((2-tosylhydrazineylidene)methyl)-1H-pyrrol-1-yl)-acetamido)methyl)cyclohexyl)acetamide (25d).
A mixture of aldehyde 25da (67 mg, 0.44 mmol) and tosylhydrazide (81 mg, 0.44 mmol) in CHCl3/MeOH (1:1, 3 mL) was refluxed at 70 °C for 4 h before being concentrated in vacuo. The crude was suspended in THF (3.6 mL) at 0 °C, then HOBt (64 mg, 0.47 mmol) and EDCI (90 mg, 0.47 mmol) were added. After 5 min, primary amine 6 (96 mg, 0.36 mmol) and DIPEA (0.15 mL, 0.84 mmol) were added to the reaction mixture and the resulting mixture was stirred at room temperature for 41 h before being concentrated in vacuo. The crude product was dissolved in EtOAc (100 mL) and the solution was washed with water (15 mL), 2 N HCl (15 mL), sat. NaHCO3 (15 mL), brine (15 mL), dried over sodium sulfate, decanted, and concentrated in vacuo. Purification by flash column chromatography (silica gel, 25% EtOAc in CH2Cl2) gave product 25d (54 mg, 95 μmol, 26% yield) as a colorless oil. Rf = 0.5 (65% EtOAc in hexanes); IR (thin film): νmax 3287.40, 3084.83, 2928.22, 2852.65, 1644.68, 1541.68, 1356.61, 1165.47, 1081.71, 946.05, 735.09, 551.64 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.05 (s, 1H), 7.88 (s, 1H), 7.80 (d, J = 8.0 Hz, 2H), 7.32 (d, J = 8.0 Hz, 2H), 7.13 – 7.03 (m, 1H), 6.92 (s, 1H), 6.90 (s, 1H), 6.50 (d, J = 3.9 Hz, 1H), 6.21 (t, J = 3.2 Hz, 1H), 4.73 (s, 2H), 3.21 (d, J = 6.2 Hz, 2H), 3.15 (d, J = 6.7 Hz, 2H), 2.42 (s, 3H), 2.15 (d, J = 2.6 Hz, 2H), 2.04 (t, J = 2.6 Hz, 1H), 1.92 (s, 2H), 1.51 – 1.33 (m, 6H), 1.26 – 1.15 (m, 4H), 1.02 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 171.66, 168.84, 144.53, 144.49, 135.42, 129.92, 129.47, 128.05, 125.68, 119.25, 110.30, 82.06, 70.72, 52.89, 48.88, 45.99, 43.47, 37.46, 34.88, 34.47, 29.96, 26.03, 25.07, 21.78, 21.62; HRMS-ESI (m/z): [M+H]+ calcd. for C30H41N5O4SH+, 568.2958; found, 568.2961.
Macrocyclization of substrates 22 and 25 to produce MCs 23 and 24.
(Z)-11',11'-Dimethyl-1-tosylspiro[piperidine-4,6'-4,9-diaza-1(3,5)-pyrazola-2(1,3)-benzenacyclodo-decaphane]3',8'-dione (23a).
A solution of 22a (18 mg, 25 μmol) and MTBD (11 μL, 0.75 mmol) in MeCN (6 mL) was stirred at 90 °C for 40 h, cooled to rt, and concentrated in vacuo. Purification by flash column chromatography (C18, 1–60% MeCN in H2O with 0.1% formic acid) gave 23a (5 mg, 9 μmol, 35% yield) as a white solid. The structure of 23a, was assigned based on a key nOe interaction in analogy to compounds 20a and 20c (see Table S1 in the Supporting Information for details). Rf = 0.40 (55% acetone in hexanes); m.p. >240 °C (acetone); IR (thin film): νmax 3294.38, 2929.37, 2862.72, 1634.99, 1537.80, 1453.97, 1325.38, 1157.27, 1089.88, 928.78, 816.54, 729.98 cm−1; 1H NMR (500 MHz, CDCl3/CD3OD =1:1) δ 8.53 (br s, 1H), 7.91 (s, 1H), 7.75 (d, J = 7.5 Hz, 1H), 7.63 (d, J = 7.6 Hz, 1H), 7.59 (d, J = 8.1 Hz, 2H), 7.46 (t, J = 7.7 Hz, 1H), 7.34 (d, J = 8.0 Hz, 2H), 6.94 (s, 1H), 3.35 (s, 4H), 3.29 – 3.21 (m, 2H), 2.86 – 2.75 (m, 2H), 2.60 (s, 2H), 2.43 (s, 2H), 2.42 (s, 3H), 1.69 – 1.53 (m, 4H), 0.91 (s, 6H); 13C NMR (126 MHz, CD3OD) δ 172.61, 169.34, 143.95, 135.00, 133.50, 133.26, 132.99, 129.75, 129.44, 127.35, 127.28, 126.60, 125.89, 106.89, 47.70, 46.13, 41.81, 37.42, 36.16, 35.23, 33.20, 26.81, 20.94; HRMS-ESI (m/z): [M+H]+ calcd. for C30H37N5O4SH+, 564.2645; found, 564.2643.
A solution of substrate (0.2 g, 0.4 mmol) and MTBD (0.17 mL, 1.2 mmol) in MeCN (100 mL) was stirred at 90 °C for 36 h, cooled to rt, and then concentrated in vacuo. Purification by flash column chromatography (silica gel, 9.5% MeOH in CH2Cl2; then C18, 1–20-40% MeCN in water with 0.1% formic acid) gave 23b (11.4 mg, 33 μmol, 8% yield) as a yellow solid and 24b (8.7 mg, 25 μmol, 6% yield) as a yellow solid. Regioisomers were assigned in analogy to 20a, 21a, and 20c (see Table S1 in the Supporting Information for details).
(Z)-6-Hydroxy-10,10-dimethyl-11H-4,8-diaza-1(5,3)-pyrazola-2(1,3)-benzenacycloundecaphane-3,7-dione (23b).
Rf = 0.12 (10% MeOH in CH2Cl2); m.p. >250 °C (MeOH); IR (thin film): νmax 3257.84, 2957.02, 2522.85, 2215.75, 1642.08, 1607.65, 1467.60, 1394.98, 1347.64, 1119.86, 814.70, 758.82 cm−1; 1H NMR (400 MHz, CD3OD) δ 8.61 (s, 1H), 8.36 (s, 1H), 7.76 (d, J = 7.6 Hz, 1H), 7.59 (d, J = 7.8 Hz, 1H), 7.51 (t, J = 7.6 Hz, 1H), 6.59 (s, 1H), 4.30 (dd, J = 11.0, 5.1 Hz, 1H), 3.62 (dd, J = 14.8, 11.0 Hz, 1H), 3.47 (dd, J = 14.9, 5.0 Hz, 1H), 3.40 (d, J = 14.0 Hz, 1H), 3.01 (d, J = 13.9 Hz, 1H), 2.87 (d, J = 14.7 Hz, 1H), 2.67 (d, J = 14.7 Hz, 1H), 1.04 (s, 3H), 0.87 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 172.87, 150.10, 144.60, 132.69, 132.28, 131.69, 129.18, 127.39, 126.27, 107.29, 69.35, 50.16, 46.64, 37.37, 34.81, 26.54, 25.90; HRMS-ESI (m/z): [M+H]+ calcd. for C18H22N4O3H+, 343.1770; found, 343.1776.
6-Hydroxy-10,10-dimethyl-11H-4,8-diaza-1(5,4)-pyrazola-2(1,3)-benzenacycloundecaphane-3,7-dione (24b).
Rf = 0.15 (10% MeOH in CH2Cl2); m.p. >250 °C (MeOH); IR (thin film): νmax 3276.54, 2964.84, 2021.12, 1652.79, 1607.70, 1539.22, 1430.74, 1389.95, 1354.72, 1309.66, 1114.87, 749.87 cm−1; 1H NMR (400 MHz, CD3OD) δ 8.53 (s, 1H), 8.28 (d, J = 2.3 Hz, 1H), 7.95 (s, 1H), 7.66 (d, J = 7.7 Hz, 1H), 7.56 (s, 1H), 7.52 (t, J = 7.8 Hz, 1H), 4.32 (s, 1H), 3.76 (d, J = 3.3 Hz, 2H), 3.55 (d, J = 13.4 Hz, 1H), 3.10 (d, J = 14.9 Hz, 1H), 2.94 (d, J = 13.3 Hz, 1H), 2.61 (d, J = 14.9 Hz, 1H), 1.01 (s, 3H), 0.91 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 173.81, 171.07, 135.17, 132.01, 129.07, 128.57, 126.88, 125.82, 114.81, 71.54, 49.91, 42.95, 37.20, 31.34, 25.10, 23.09; HRMS-ESI (m/z): [M+H]+ calcd. for C18H22N4O3H+, 343.1770; found, 343.1784.
(42S,44R)-44-Hydroxy-8,8-dimethyl-11H-6-aza-1(5,4)-pyrazola-4(1,2)-pyrrolidina-2(1,3)-benzena-cyclononaphane-3,5-dione (24c).
A solution of 22c (0.1 g, 0.19 mmol) and MTBD (82 μL, 0.57 mmol) in MeCN (44 mL) was stirred at 90 °C for 40 h, cooled to rt, and then concentrated in vacuo. Purification by flash column chromatography (silica gel, 8% MeOH in CH2Cl2) gave product 24c (32 mg, 87 μmol, 46% yield) as a white solid. The structure of 24c was assigned based on a key nOe interaction in analogy to 21a, 21b, 21d, and 21e (see Table S1 in the Supporting Information for details). Rf = 0.15 (8% MeOH in CH2Cl2); m.p. = 235–236 °C (MeOH); IR (thin film): νmax 3280.31, 2953.53, 1660.68, 1622.07, 1429.24, 1367.56, 1200.73, 1079.95, 721.36 cm−1; 1H NMR (500 MHz, CD3OD) δ 7.56 (t, J = 7.6 Hz, 1H), 7.54 – 7.47 (m, 2H), 7.40 (d, J = 7.4 Hz, 1H), 7.29 (s, 1H), 4.60 – 4.52 (m, 1H), 4.16 (dd, J = 8.6, 5.5 Hz, 1H), 3.94 (ddd, J = 12.3, 3.5, 1.5 Hz, 1H), 3.72 (dd, J = 12.3, 5.0 Hz, 1H), 3.36 (d, J = 15.5 Hz, 1H), 2.96 (d, J = 14.9 Hz, 1H), 2.35 (d, J = 13.2 Hz, 1H), 2.28 – 2.18 (m, 2H), 2.08 (dt, J = 13.2, 5.4 Hz, 1H), 0.98 (s, 3H), 0.38 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 174.22, 171.88, 136.62, 129.92, 129.18, 125.83, 124.80, 115.50, 68.19, 60.94, 54.16, 49.23, 39.45, 36.32, 30.05, 26.47, 22.82; HRMS-ESI (m/z): [M+Na]+ calcd. for C20H24N4O3Na+, 391.1746; found, 391.1759; [α]D21 = + 171.2 (c = 0.05, MeOH).
A solution of substrate (60 mg, 90 μmol) and MTBD (39 μL, 0.27 mmol) in MeCN (21 mL) was stirred at 90 °C for 16 h, cooled to rt, and then concentrated in vacuo. Purification by preparative TLC (8% MeOH in CHCl3) afforded compound 23d (24 mg, 47 μmol, 52% yield) as a white solid and 24d (5 mg, 10 μmol, 11% yield) as a white solid. The structure of compound 23d was assigned in analogy to compounds 20a-e and 23a-b based on the characteristic 13C chemical shift of the pyrrole methine (see Table S1 in the Supporting Information for details). The structure of compound 24d, which exhibited slowly interconverting rotamers that prevented 2D NMR analysis at room temperature, was then assigned by exclusion. Key 13C NMR shift is used in structural assignment of regioisomers 23d and 24d (see Figure S11 in the Supporting Information for details).
(52S,54R,8S,Z)-54-Hydroxy-8-phenyl-11H-10-oxa-7-aza-1(5,3)-pyrazola-5(1,2)-pyrrolidina-2(1,2),3(1,3)-dibenzenacycloundecaphane-4,6-dione (23d).
Rf = 0.33 (7.5% MeOH in CHCl3); m.p. = 190–191 °C (MeOH); IR (thin film): νmax 3417.96, 3229.07, 2924.15, 2855.61, 2542.77, 1656.14, 1403.83, 1316.88, 1082.72, 765.75, 734.84, 701.36 cm−1; 1H NMR (500 MHz, CD3OD) δ 7.70 (d, J = 7.5 Hz, 1H), 7.68 – 7.58 (m, 3H), 7.58 – 7.50 (m, 2H), 7.48 (dt, J = 9.0, 4.6 Hz, 1H), 7.36 – 7.19 (m, 6H), 7.11 (d, J = 6.4 Hz, 1H), 6.07 (s, 1H), 4.93 (d, J = 5.1 Hz, 1H), 4.61 (dd, J = 10.6, 7.3 Hz, 1H), 4.40 – 4.26 (m, 3H), 3.87 (dd, J = 11.5, 3.6 Hz, 1H), 3.76 (qd, J = 9.2, 4.7 Hz, 2H), 3.49 (d, J = 11.4 Hz, 1H), 2.27 (dd, J = 13.4, 7.5 Hz, 1H), 1.99 (ddd, J = 13.8, 10.8, 3.9 Hz, 1H); 13C NMR (126 MHz, CD3OD) δ 172.44, 171.70, 140.22, 139.91, 139.51, 133.84, 132.33, 130.36, 129.38, 129.19, 128.96, 128.23, 127.97, 127.79, 127.28, 126.89, 126.20, 106.24, 72.27, 69.55, 64.06, 61.09, 58.39, 53.48, 37.60; HRMS-ESI (m/z): [M+H]+ calcd. for C30H28N4O4H+, 509.2189; found, 509.2208; [α]D21 = − 80.8 (c = 0.05, MeOH).
(52S,54R,8S)-54-Hydroxy-8-phenyl-11H-10-oxa-7-aza-1(5,4)-pyrazola-5(1,2)-pyrrolidina-2(1,2),3(1,3)-dibenzenacycloundecaphane-4,6-dione (24d). Note: variable temperature NMR experiments showed that rotamers of compound 24d coalesced at 85 °C in (CD3)2SO.
Rf = 0.23 (7.5% MeOH in CHCl3); m.p. = 198–200 °C (MeOH); IR (thin film): νmax 3416.66, 3269.04, 3059.70, 2925.43, 2863.86, 1654.22, 1512.98, 1398.85, 1085.24, 1007.30, 762.78, 704.72 cm−1; 1H NMR (500 MHz, CD3OD, mixture of rotamers) δ 7.69 (s, 0.5H), 7.65 (t, J = 8.6 Hz, 1H), 7.54 (d, J = 8.1 Hz, 3H), 7.50 – 7.42 (m, 2H), 7.39 (d, J = 7.6 Hz, 1H), 7.35 – 7.28 (m, 4H), 7.26 – 7.20 (m, 2H), 7.20 – 7.11 (m, 1.5H), 4.99 – 4.89 (m, 1H), 4.70 – 4.54 (m, 1.5H), 4.43 (s, 0.5H), 4.43 – 4.32 (s, 1H), 4.29 (s, 0.5H), 3.98 – 3.84 (m, 1.5H), 3.84 – 3.76 (m, 0.5H), 3.72 (dd, J = 11.6, 3.4 Hz, 0.5H), 3.62 (q, J = 7.9, 6.9 Hz, 0.5H), 3.46 (s, 0.5H), 3.38 – 3.32 (m, 0.5H), 3.14 (d, J = 11.6 Hz, 0.5H), 2.31 (t, J = 11.0 Hz, 0.5H), 2.14 (s, 0.5H), 2.01 – 1.84 (m, 1H); 13C NMR (101 MHz, CD3OD, mixture of rotamers) δ 172.60, 142.56, 141.61, 141.38, 139.77, 139.02, 135.89, 135.37, 131.36, 131.00, 130.09, 129.63, 128.72, 128.07, 127.95, 127.13, 126.93, 126.44, 126.28, 124.94, 123.80, 116.63, 115.67, 72.39, 71.14, 69.50, 67.84, 63.07, 62.83, 59.15, 57.99, 54.11, 53.62, 39.49, 38.13, 29.33; HRMS-ESI (m/z): [M+H]+ calcd. for C30H28N4O4H+, 509.2189; found, 509.2186; [α]D25 = − 3.6 (c = 0.1, CHCl3/MeOH = 1:1).
Characterization of the undesired side-product 26.
N-((1-(2-((2,2-Dimethylpent-4-yn-1-yl)amino)-2-oxoethyl)cyclohexyl)methyl)-2-hydroxy-5-(tosyl-methyl)benzamide (26).
A solution of alkynyl tosylhydrazone 25c (30 mg, 52 μmol) and MTBD (22 μL, 0.15 mmol) in MeCN (12 mL) was stirred at 90 °C for 15 h, cooled to rt, then concentrated in vacuo. Purification by flash column chromatography (silica gel, 30% EtOAc in hexanes) gave 26 (8 mg, 14 μmol, 28% yield) as a colorless oil. Rf = 0.17 (30% EtOAc in hexanes); IR (thin film): νmax 3375.46, 3299.01, 2926.96, 2868.94, 1645.38, 1591.27, 1498.87, 1313.37, 1265.93, 1150.50, 1086.26, 736.64, 528.46 cm−1; 1H NMR (400 MHz, CDCl3) δ 12.85 (s, 1H), 8.75 (d, J = 6.0 Hz, 1H), 7.51 (d, J = 7.8 Hz, 2H), 7.37 (d, J = 2.1 Hz, 1H), 7.23 (d, J = 8.0 Hz, 2H), 7.00 (dd, J = 8.7, 2.0 Hz, 1H), 6.82 (d, J = 8.5 Hz, 1H), 6.17 (t, J = 6.5 Hz, 1H), 4.24 (s, 2H), 3.43 (d, J = 5.8 Hz, 2H), 3.30 (d, J = 6.4 Hz, 2H), 2.39 (s, 3H), 2.24 (s, 2H), 2.16 (d, J = 2.7 Hz, 2H), 2.11 (d, J = 2.6 Hz, 1H), 1.66 – 1.29 (m, 10H), 1.02 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 172.21, 169.54, 162.01, 144.60, 135.74, 134.76, 129.55, 128.95, 128.68, 118.48, 118.24, 114.69, 81.62, 71.05, 62.22, 48.83, 46.93, 46.25, 37.25, 34.77, 34.68, 29.96, 29.69, 25.86, 25.08, 21.66; HRMS-ESI (m/z): [M+H]+ calcd. for C31H40N2O5SH+, 553.2736; found, 553.2745.
Preparation and divergent cyclizations of pyridotriazole 28.
N-((1-(2-((2,2-Dimethylpent-4-yn-1-yl)amino)-2-oxoethyl)cyclohexyl)methyl)-6-(hydroxymethyl)-picolinamide (27b).
To a solution of acid 27a48 (21 mg, 0.14 mmol) and HOBt (20 mg, 0.15 mmol) in THF (1.3 mL) at 0 °C were added EDCI (29 mg, 0.15 mmol) and DIPEA (48 μL, 0.27 mmol). After 5 min, primary amine 6 (40 mg, 0.15 mmol) was added and the reaction mixture was stirred at room temperature for 13 h. The reaction mixture was diluted with CH2Cl2 (50 mL), washed with sat. NaHCO3 (10 mL) and brine (10 mL). The organic layer was dried over sodium sulfate, filtered,and concentrated in vacuo. Purification by flash column chromatography (silica gel, 50% EtOAc in hexanes) gave product 27b (25 mg, 63 μmol, 46% yield) as a colorless oil. Rf = 0.25 (50% EtOAc in hexanes); IR (thin film): νmax 3302.31, 2927.32, 2864.66, 1649.05, 1530.39, 1452.49, 1368.63, 1211.00, 1066.15, 996.17, 826.52, 755.42, 631.60 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.67 (t, J = 6.4 Hz, 1H), 8.04 (d, J = 7.6 Hz, 1H), 7.80 (t, J = 7.7 Hz, 1H), 7.35 (d, J = 7.8 Hz, 1H), 6.88 (t, J = 6.3 Hz, 1H), 4.81 (s, 2H), 4.53 (s, 1H), 3.48 (d, J = 6.4 Hz, 2H), 3.25 (d, J = 6.3 Hz, 2H), 2.27 (s, 2H), 2.14 (d, J = 2.7 Hz, 2H), 2.01 (t, J = 2.7 Hz, 1H), 1.65 – 1.30 (m, 10H), 1.00 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 172.19, 164.85, 158.58, 149.15, 137.91, 122.79, 120.83, 81.97, 70.71, 64.33, 48.96, 47.41, 45.73, 37.44, 34.82, 34.73, 29.92, 26.08, 24.97, 21.74; HRMS-ESI (m/z): [M+H]+ calcd. for C23H33N3O3H+, 400.2600; found, 400.2588.
(E)-N-((1-(2-((2,2-Dimethylpent-4-yn-1-yl)amino)-2-oxoethyl)cyclohexyl)methyl)-6-((2-tosylhydra-zineylidene)methyl)picolinamide (27).
A mixture of 27b (40 mg, 0.1 mmol) and IBX-polystyrene (1.1 mmol/g, 0.3 g, 0.33 mmol) in EtOAc (2 mL) was stirred at 90 °C for 2 h, cooled to rt, and filtered through a short pipette packed with Celite, and concentration in vacuo. The crude oil and tosylhydrazide (21 mg, 0.11 mmol) were dissolved in in CHCl3/MeOH (1:1, 2 mL) and were refluxed at 70 °C for 12 h before being cooled to rt and concentrated in vacuo. Purification by flash column chromatography (silica gel, 40% EtOAc in hexanes) gave product 27 (30 mg, 53 μmol, 53% yield) as a white foam. Rf = 0.23 (45% EtOAc in hexanes); m.p. = 162–164 °C (EtOAc); IR (thin film): νmax 3326.81, 3119.54, 2923.90, 2865.48, 1662.16, 1535.11, 1449.21, 1361.44, 1325.63, 1167.82, 1079.89, 654.90 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.62 (s, 1H), 8.50 (t, J = 7.1 Hz, 1H), 8.10 (d, J = 7.6 Hz, 1H), 7.94 (d, J = 7.9 Hz, 1H), 7.90 – 7.83 (m, 3H), 7.80 (t, J = 7.8 Hz, 1H), 7.33 (d, J = 8.0 Hz, 2H), 3.49 (d, J = 6.7 Hz, 2H), 3.26 (d, J = 6.1 Hz, 2H), 2.41 (s, 3H), 2.19 (d, J = 2.7 Hz, 2H), 2.17 (s, 2H), 2.05 – 2.01 (d, J = 2.7 Hz, 1H), 1.57 – 1.32 (m, 10H), 1.04 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 171.88, 164.82, 151.54, 149.13, 146.46, 144.34, 137.75, 135.70, 129.78, 127.98, 122.73, 122.61, 81.99, 70.72, 49.06, 46.30, 43.36, 37.86, 34.78, 34.65, 29.95, 26.08, 25.05, 21.74, 21.66; HRMS-ESI (m/z): [M+H]+ calcd. for C30H39N5O4SH+, 566.2801; found, 566.2813.
N-((1-(2-((2,2-Dimethylpent-4-yn-1-yl)amino)-2-oxoethyl)cyclohexyl)methyl)-[1,2,3]triazolo-[1,5-a]pyridine-7-carboxamide (28).
A solution of alkynyl tosylhydrazone 27 (5 mg, 8.84 μmol) and MTBD (3.8 μL, 26.51 μmol) in MeCN was stirred at 90 °C for 15 h, cooled to rt, and concentrated in vacuo. Purification by flash column chromatography (silica gel, 50% EtOAc in hexanes) gave product 28 (3.4 mg, 8.3 μmol, 94% yield) as a white solid. Rf = 0.23 (50% EtOAc in hexanes); m.p. = 155–156 °C (EtOAc); IR (thin film): νmax 3301.84, 3080.54, 2929.22, 1665.74, 1548.66, 1457.81, 1316.89, 1209.62, 1107.39, 822.82, 734.97, 676.30 cm−1; 1H NMR (400 MHz, CDCl3) δ 10.30 (d, J = 6.8 Hz, 1H), 8.26 (s, 1H), 8.16 (d, J = 7.0 Hz, 1H), 7.98 (d, J = 8.8 Hz, 1H), 7.68 (t, J = 6.0 Hz, 1H), 7.53 – 7.44 (m, 1H), 3.66 (d, J = 6.6 Hz, 2H), 3.27 (d, J = 6.0 Hz, 2H), 2.24 (s, 2H), 2.21 (d, J = 2.6 Hz, 2H), 2.01 (t, J = 2.7 Hz, 1H), 1.68 – 1.34 (m, 10H), 1.07 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 170.93, 160.36, 134.90, 129.85, 126.71, 125.63, 121.17, 120.94, 81.99, 70.28, 48.73, 47.60, 42.09, 37.45, 34.74, 34.41, 29.76, 25.97, 24.86, 21.50; HRMS-ESI (m/z): [M+H]+ calcd. for C23H31N5O2H+, 410.2556; found, 410.2575.
To a high-pressure flask were added alkynyl pyridotriazole 28 (50 mg, 0.12 mmol) and mesitylene (30 mL). The reaction mixture was stirred at 200 °C for 18 h before being cooled to room temperature. The slurry was filtered through a short plug of silica gel, which was washed with hexanes (30 mL) followed by acetone (50 mL). The acetone filtrate was concentrated in vacuo and the crude residue was purified by flash column chromatography (silica gel, 30% then 45% acetone in hexanes) to afford compound 29 (13 mg, 32 μmol, 26%) as a white solid and 30 (12 mg, 29 μmol, 24%) as a white solid. Structural assignments were made by analogy (see Table S1 in the Supporting Information for details).
(Z)-4',4'-Dimethylspiro[cyclohexane-1,9'-6,11-diaza-1(2,6)-pyridina-2(5,3)-pyrazolacyclododecaph-ane]-7',12'-dione (29).
Rf = 0.18 (50% acetone in hexanes); m.p. = 225–226 °C (acetone); IR (thin film): νmax 3312.33, 2925.23, 2865.73, 1658.53, 1566.60, 1528.34, 1454.99, 1386.74, 262.71, 1215.04, 828.70, 737.23 cm−1; 1H NMR (500 MHz, CDCl3, major rotamer) δ 9.29 (t, J = 6.3 Hz, 1H), 7.91 (dd, J = 7.3, 1.3 Hz, 1H), 7.87 – 7.80 (m, 2H), 7.01 (s, 1H), 5.93 (t, J = 5.3 Hz, 1H), 3.64 (d, J = 6.4 Hz, 2H), 3.33 (d, J = 5.3 Hz, 2H), 2.66 (s, 2H), 2.27 (s, 2H), 1.66 – 1.35 (m, 10H), 0.96 (s, 6H); 13C NMR (101 MHz, CDCl3, major rotamer) δ 172.01, 164.61, 152.93, 149.72, 149.67, 142.21, 138.83, 120.47, 119.66, 106.95, 49.07, 45.07, 44.77, 38.76, 38.15, 34.86, 34.13, 27.47, 25.75, 21.56; HRMS-ESI (m/z): [M+H]+ calcd. for C23H31N5O2H+, 410.2556; found, 410.2563. DFT-optimized structure of 29 is consistent with key 2D NMR interactions for structural assignment of 29 (see Figure S12 in the Supporting Information for details).
4',4'-Dimethylspiro[cyclohexane-1,9'-6,11-diaza-1(2,6)-pyridina-2(5,4)-pyrazolacyclododeca-phane]-7',12'-dione (30).
Rf = 0.33 (50% acetone in hexanes); m.p. = 220–221 °C (acetone); IR (thin film): νmax 3384.72,3284.92, 2925.66, 2854.10, 1653.77, 1592.01, 1531.41, 1452.62, 1319.44, 1252.29, 752.99, 736.07 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.34 (t, J = 7.1 Hz, 1H), 8.14 – 8.06 (m, 2H), 7.84 (td, J = 7.8, 1.4 Hz, 1H), 7.50 (s, 1H), 5.94 (t, J = 6.7 Hz, 1H), 3.59 (d, J = 7.0 Hz, 2H), 3.20 (d, J = 6.6 Hz, 2H), 3.06 (s, 2H), 2.25 (s, 2H), 1.73 – 1.34 (m, 10H), 0.90 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 172.58, 165.07, 152.31, 148.79, 146.76, 138.03, 131.79, 124.30, 120.73, 116.47, 50.28, 46.67, 44.59, 38.44, 36.42, 36.09, 32.59, 25.94, 24.61, 21.66; HRMS-ESI (m/z): [M+H]+ calcd. for C23H31N5O2H+, 410.2556; found, 410.2563. DFT-optimized structure of 30 is consistent with key 2D NMR interactions for structural assignment of 30 (see Figure S13 in the Supporting Information for details).
To a vial containing tetrakis(triphenylacetato)dirhodium(II) (6.6 mg, 4.88 μmol) was added pyridotriazole 28 (20 mg, 49 μmol) in DCE (12 mL). The reaction solution was stirred at 90 °C for 3 h before addition of DMSO (0.14 mL, 1.95 mmol). After 5 min at 90 °C, the reaction solution was cooled to room temperature and was concentrated in vacuo. Purification by preparative TLC (55% EtOAc in hexanes) afforded 31 (15 mg, 39 μmol, 81% yield) as a white solid and 32 (1.7 mg, 4.46 μmol, 9% yield) as a white solid.
(Z)-3'-Methylspiro[cyclohexane-1,8'-5,10-diaza-1(2,6)-pyridina-3(1,3)-cyclopentanacycloundecaph-ane]-6',11'-dione (31).
Rf = 0.28 (25% acetone in hexanes); m.p. = 230–231 °C (acetone); IR (thin film): νmax 3311.76, 3055.68, 2926.49, 2856.05, 2517.61, 2474.80, 1647.27, 1584.98, 1468.15, 1435.61, 1179.52, 995.82, 735.82 cm−1; 1H NMR (500 MHz, CD3OD/CDCl3=1:1) δ 8.69 (d, J = 10.9 Hz, 1H), 8.19 (dd, J = 9.1, 4.4 Hz, 1H), 7.71 (d, J = 4.4 Hz, 2H), 7.17 (p, J = 4.2 Hz, 1H), 6.35 (t, J = 2.4 Hz, 1H), 4.31 (dd, J = 13.8, 11.0 Hz, 1H), 3.86 (dd, J = 13.0, 8.9 Hz, 1H), 3.74 (dt, J = 19.3, 2.9 Hz, 1H), 3.16 (d, J = 13.9 Hz, 1H), 2.74 (q, J = 9.0 Hz, 1H), 2.63 (td, J = 12.7, 12.2, 4.7 Hz, 2H), 2.51 (d, J = 19.3 Hz, 1H), 2.32 (d, J = 12.7 Hz, 1H), 2.09 (d, J = 12.7 Hz, 1H), 1.75 – 1.62 (m, 2H), 1.52 (dddd, J = 39.9, 19.1, 10.6, 4.6 Hz, 6H), 1.38 – 1.24 (m, 4H), 1.02 (s, 3H); 13C NMR (126 MHz, CD3OD) δ 174.10, 166.04, 156.26, 154.60, 148.75, 137.26, 125.47, 118.21, 117.99, 49.82, 49.70, 45.00, 44.76, 44.52, 37.79, 37.63, 36.38, 33.68, 31.88, 25.66, 23.53, 21.38, 21.27; HRMS-ESI (m/z): [M+H]+ calcd. for C23H31N3O2H+, 382.2495; found, 382.2494. DFT-optimized structure of 31 is consistent with key 2D NMR interactions for structural assignment of 31 (see Figure S14 in the Supporting Information for details).
(11'Z,13'E)-10',10'-Dimethylspiro[cyclohexane-1,5'-3,8-diaza1(2,6)-pyridinacyclotetradecaphanene]-11',13'-diene-2',7'-dione (32). Note: Compound 32 is stable in the solid state, but shows air sensitivity and slow decomposition in solution.
Rf = 0.32 (25% acetone in hexanes); m.p. = 175–177 °C (decomposed, acetone); IR (thin film): νmax 3293.07, 2244.85, 2864.31, 1653.16, 1518.71, 1445.93, 1367.10, 1296.74, 1183.92, 993.69, 870.00, 745.16 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.27 (t, J = 6.9 Hz, 1H), 8.29 (t, J = 13.6 Hz, 1H), 7.91 (d, J = 7.7 Hz, 1H), 7.74 (t, J = 7.7 Hz, 1H), 7.19 (d, J = 7.7 Hz, 1H), 6.51 (d, J = 14.7 Hz, 1H), 6.19 (t, J = 12.0 Hz, 1H), 5.96 (t, J = 6.3 Hz, 1H), 5.57 (d, J = 11.6 Hz, 1H), 3.66 (d, J = 6.6 Hz, 2H), 3.61 (d, J = 6.1 Hz, 2H), 2.32 (s, 2H), 1.66 – 1.30 (m, 10H), 1.19 (s, 6H); 13C NMR (126 MHz, CDCl3) δ 172.32, 164.29, 153.35, 149.86, 143.54, 138.23, 131.08, 129.47, 126.81, 124.59, 119.87, 48.46, 45.04, 44.86, 38.07, 37.67, 35.00, 29.05, 25.99, 21.57; HRMS-ESI (m/z): [M+H]+ calcd. for C23H31N3O2H+, 382.2495; found, 382.2489. DFT-optimized structure of 32 is consistent with key 2D NMR interactions for structural assignment of 32 (see Figure S15 in the Supporting Information for details).
Preparation and divergent cyclizations of pyridotriazole 38.
Methyl [1,2,3]triazolo[1,5-a]pyridine-7-carboxylate (38b).
To a round-bottomed flask equipped with a reflux condenser were added aldehyde 38a (1 g, 6.06 mmol), tosylhydrazide (1.18, 6.36 mmol), K2CO3 (2.51 g, 18.17 mmol), and PhMe (20 mL). The resulting slurry was stirred at 80 °C for 13 h. Water (50 mL) was added and the aqueous layer was extracted with CH2Cl2 (150 mL) three times. The combined organic layers were washed with brine (50 mL), dried over sodium sulfate, decanted and concentrated in vacuo. Purification by flash column chromatography (silica gel, 25% EtOAc in CH2Cl2) gave product 38b (0.60 g, 3.41 mmol, 56% yield) as a white solid. Rf = 0.33 (30% acetone in hexanes); m.p. = 103–105 °C (acetone); IR (thin film): νmax 3115.64, 3097.36, 3036.07, 2951.83, 2849.11, 1707.88, 1539.56, 1438.95, 1358.42, 1289.88, 1214.13, 969.26,7 52.28 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.23 (s, 1H), 7.96 (d, J = 8.8 Hz, 1H), 7.78 (d, J = 6.9 Hz, 1H), 7.33 (dd, J = 8.8, 7.0 Hz, 1H), 4.12 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 160.42, 134.66, 127.00, 126.41, 124.11, 122.47, 120.89, 53.05; HRMS-ESI (m/z): [M+Na]+ calcd. for C8H7N3O2, 200.0436; found, 200.0443.
[1,2,3]Triazolo[1,5-a]pyridine-7-carboxylic acid (38c).
To a solution of 38b (2.6 g, 14.68 mmol) in MeOH/H2O (1:2, 60 mL) was added 2 N NaOH (15 mL) at 0 °C. The reaction mixture was stirred at room temperature for 3.5 h, was cooled to 0 °C, and neutralized with 2 N HCl (15 mL). The mixture was filtered over a short plug of C8 silica gel (40–63 μm, 60Å from SiliCycle). The solid was rinsed with water (5 mL) twice. The obtained solid was dried in a desiccator under vacuum to give product 38c (1.58 g, 9.69 mmol, 66% yield) as a pink solid. Rf = 0.05 (15% MeOH in CH2Cl2); m.p. = 150 °C (decomposed, MeOH); IR (thin film): νmax 3087.81, 2922.94, 1724.33, 1593.94, 1453.94, 1368.06, 1267.20, 1227.39, 1146.46, 1102.12, 754.30 cm−1; 1H NMR (500 MHz, CD3OD) δ 8.32 (s, 1H), 8.18 (dd, J = 8.8, 1.2 Hz, 1H), 7.94 (dd, J = 7.0, 1.2 Hz, 1H), 7.49 (dd, J = 8.9, 7.0 Hz, 1H); 13C NMR (101 MHz, (CD3)2SO) δ 161.90, 135.04, 128.64, 126.72, 125.36, 122.62, 120.52; HRMS-ESI (m/z): [M+H]+ calcd. for C7H5N3O2H+, 164.0460; found, 164.0465.
(2S,4R)-1-([1,2,3]Triazolo[1,5-a]pyridine-7-carbonyl)-N-(2,2-dimethylpent-4-yn-1-yl)-4-hydroxy-pyrrolidine-2-carboxamide (38).
To a solution of acid 38c (0.15 g, 0.92 mmol) and HOBt (0.12 g, 0.92 mmol) in THF/CH2Cl2 (1:1, 4 mL) at 0 °C was added EDCI (0.18 g, 0.92 mmol). After 5 min, the reaction solution were added amine 22cb (0.19 g, 0.84 mmol) in THF/CH2Cl2 (1:1, 2 mL) and DIPEA (0.29 mL, 1.67 mmol). The reaction solution was stirred at room temperature for 18 h. The reaction mixture was diluted with CH2Cl2 (50 mL) and was washed with sat. NaHCO3 (10 mL) and brine (10 mL). The organic layer was dried over sodium sulfate, filtered, and concentrated in vacuo. Purification by flash column chromatography (silica gel, 60% acetone in hexanes) afforded product 38 (0.17 g, 0.45 mmol, 54% yield) as a white solid. Rf = 0.24 (50% acetone in hexanes); m.p. = 173–175 °C (acetone); IR (thin film): νmax 3307.54, 3082.91, 2963.06, 1653.55, 1548.86, 1432.67, 1318.65, 1208.52, 1101.62, 822.53 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.18 (s, 1H), 7.86 (d, J = 8.8 Hz, 1H), 7.53 (br s, 1H), 7.39 (dd, J = 8.9, 6.8 Hz, 1H), 7.27 (d, J = 7.1 Hz, 1H), 5.08 (t, J = 8.2 Hz, 1H), 4.47 (s, 1H), 3.65 (dd, J = 11.5, 4.0 Hz, 1H), 3.34 (dd, J = 13.4, 6.8 Hz, 1H), 3.26 (dd, J = 13.5, 6.1 Hz, 1H), 3.15 (d, J = 11.5 Hz, 1H), 2.51 (dd, J = 13.4, 9.4 Hz, 1H), 2.46 – 2.36 (m, 1H), 2.23 – 2.10 (m, 2H), 1.99 (t, J = 2.7 Hz, 1H), 1.03 (s, 3H), 1.03 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 171.39, 161.96, 133.85, 132.04, 126.53, 125.90, 119.40, 115.55, 81.91, 70.53, 69.92, 59.45, 56.31, 48.67, 38.88, 35.29, 29.77, 24.76, 24.72; HRMS-ESI (m/z): [M+Na]+ calcd. for C19H23N5O3Na+, 392.1699; found, 392.1689; [α]D25 = − 20.9 (c = 0.1, CHCl3/MeOH = 1:1).
To a vial containing tetrakis(triphenylacetato)dirhodium(II) (7 mg, 5.4 μmol) was added pyridotriazole 38 (20 mg, 54 μmol) in DCE (13 mL). The reaction solution was stirred at 90 °C for 13 h before addition of DMSO (38 μL, 0.54 mmol). After 5 min at 90 °C, the reaction solution was cooled to room temperature and was concentrated in vacuo. Purification by preparative TLC (60% acetone in hexanes) afforded macrocycle 39 (5.7 mg, 16.70 μmol, 31% yield) as a yellow oil and 40 (2 mg, 5.89 μmol, 11% yield) as a greenish oil.
(16R,17aS,Z)-16-Hydroxy-4,4-dimethyl-4,5,15,16,17,17a-hexahydro-1H,3H,13H-8,12-(azeno)-2,6-methanopyrrolo[1,2-a][1,4]diazacyclopentadecine-1,13-dione (39).
Rf = 0.50 (60% acetone in hexanes); IR (thin film): νmax 3404.79, 2955.08, 1629.52, 1581.36, 1464.11, 1430.63, 1285.20, 1211.47, 1153.68, 1076.62, 1034.86, 754.69 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.88 (d, J = 7.7 Hz, 1H), 7.74 (t, J = 7.8 Hz, 1H), 7.25 (d, J = 8.3 Hz, 1H), 6.35 (s, 1H), 5.46 (dd, J = 8.4, 3.5 Hz, 1H), 4.98 (d, J = 12.7 Hz, 1H), 4.87 (p, J = 5.6 Hz, 1H), 4.18 (dd, J = 12.7, 2.8 Hz, 2H), 3.95 (dd, J = 12.6, 5.7 Hz, 1H), 3.30 (d, J = 12.7 Hz, 1H), 2.60 (d, J = 13.0 Hz, 1H), 2.34 (d, J = 11.3 Hz, 1H), 2.20 (ddd, J = 12.9, 6.0, 3.5 Hz, 1H), 2.16 – 2.11 (m, 1H), 2.08 (d, J = 11.5 Hz, 1H), 1.06 (s, 3H), 0.87 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 174.13, 165.70, 153.81, 150.89, 142.20, 137.58, 124.97, 124.07, 122.47, 68.92, 58.18, 55.71, 54.35, 50.20, 49.63, 41.58, 37.97, 28.49, 24.27; HRMS-ESI (m/z): [M+H]+ calcd. for C19H23N3O3H+, 342.1818; found, 342.1804; [α]D25 = + 113.8 (c = 0.2, CHCl3/MeOH = 1:1). DFT-optimized structure of 39 is consistent with key 2D NMR interactions for structural assignment of 39 (see Figure S16 in the Supporting Information for details).
(10R,11aS)-10-Hydroxy-2,2-dimethyl-2,3,9,10,11,11a-hexahydro-1H,8H,12H-4a1,8a,12a-triazacy-clopenta[6,7]cycloocta[1,2,3,4-def]fluorene-8,12-dione (40).
Rf = 0.74 (60% acetone in hexanes); IR (thin film): νmax 3411.66, 2954.97, 2925.43, 1677.61, 1636.30, 1423.38, 1283.56, 1195.34, 1128.52, 971.11, 735.56, 694.42 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.34 (d, J = 8.8 Hz, 1H), 7.07 (d, J = 6.8 Hz, 1H), 6.63 (t, J = 7.8 Hz, 1H), 6.36 (s, 1H), 5.23 (d, J = 7.6 Hz, 1H), 4.96 – 4.85 (m, 1H), 4.51 (d, J = 12.4 Hz, 1H), 3.80 (d, J = 4.5 Hz, 2H), 2.84 (dd, J = 13.2, 7.1 Hz, 1H), 2.72 (d, J = 12.4 Hz, 1H), 2.61 (s, 2H), 2.16 (br s, 1H), 1.95 (dt, J = 13.1, 6.5 Hz, 2H), 1.10 (s, 3H), 0.97 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 169.56, 163.04, 132.68, 129.97, 121.96, 119.68, 117.73, 117.69, 115.44, 100.35, 69.32, 57.13, 55.43, 53.80, 37.03, 36.48, 31.63, 27.88, 25.69; HRMS-ESI (m/z): [M+H]+ calcd. for C19H21N3O3H+, 340.1661; found, 340.1653; [α]D25 = + 79.6 (c = 0.1, CHCl3). DFT-optimized structure of 40 is consistent with key 2D NMR interactions for structural assignment of 40 (see Figure S17 in the Supporting Information for details).
Preparation and divergent cyclizations of pyridotriazole 48.
(9H-Fluoren-9-yl)methyl (2S,4R)-4-hydroxy-2-(((R)-1-phenyl-2-(prop-2-yn-1-yloxy)ethyl)carba-moyl)pyrrolidine-1-carboxylate (48b)
. To a solution of Fmoc-Hyp-OH (5 g, 14.15 mmol) in CH2Cl2 (55 mL) at 0 °C were added HOBt (2.1 g, 15.56 mmol) and EDCI (2.98 g, 15.56 mmol) followed by DIPEA (4.93 mL, 28.3 mmol). After 5 min, amine 48a (3.29 g, 15.56 mmol) in CH2Cl2 (15 mL) was added. The reaction mixture was stirred at room temperature for 15 h before being diluted with CH2Cl2 (100 mL). The solution was washed with water (40 mL), 2 N HCl (40 mL), sat. NaHCO3 (40 mL), brine (40 mL), dried over sodium sulfate, decanted, and concentrated in vacuo. Purification by flash column chromatography (silica gel, 75% EtOAc in hexanes) gave product 48b (5.28 g, 10.34 mmol, 73% yield) as a white foam. Rf = 0.17 (80% EtOAc in hexanes); m.p. = 105–107 °C (EtOAc); IR (thin film): νmax 3411.77, 3302.50, 2933.42, 1675.52, 1528.40, 1425.08, 1351.12, 1097.99, 740.90 cm−1; 1H NMR (400 MHz, CDCl3, 60 °C) δ 7.76 (d, J = 7.6 Hz, 2H), 7.58 (t, J = 8.3 Hz, 2H), 7.39 (t, J = 7.5 Hz, 2H), 7.34 – 7.24 (m, 6H), 7.21 (dd, J = 7.8, 3.8 Hz, 1H), 5.16 (dt, J = 8.2, 5.3 Hz, 1H), 4.56 – 4.51 (m, 1H), 4.52 – 4.44 (m, 2H), 4.44 – 4.34 (m, 1H), 4.28 – 4.16 (m, 1H), 4.06 (t, J = 2.0 Hz, 2H), 3.79 (dd, J = 9.9, 4.7 Hz, 1H), 3.73 (dd, J = 9.9, 5.8 Hz, 1H), 3.66 – 3.48 (m, 1H), 3.61 (d, J = 4.8 Hz, 1H), 2.52 – 2.33 (m, 1H), 2.31 (t, J = 2.5 Hz, 1H), 2.23 – 1.84 (m, 2H); 13C NMR (101 MHz, CDCl3, 60 °C) δ 171.18, 156.25, 144.19, 144.03, 141.56, 139.56, 128.68, 127.94, 127.92, 127.69, 127.32, 127.00, 125.31, 125.23, 120.13, 79.51, 74.94, 72.21, 70.13, 68.14, 59.78, 58.51, 55.10, 52.97, 47.55; HRMS-ESI (m/z): [M+H]+ calcd. for C31H30N2O5H+, 511.2233; found, 511.2219; [α]D25 = − 6.0 (c = 0.05, CHCl3/MeOH = 1:1).
(2S,4R)-4-Hydroxy-N-((R)-1-phenyl-2-(prop-2-yn-1-yloxy)ethyl)pyrrolidine-2-carboxamide (48c).
To a solution of 48b (10.35 g, 20.27 mmol) in CH2Cl2 (70 mL) at room temperature was added Et2NH (70 mL). The reaction solution was stirred for 3 h and was concentrated in vacuo. Water (100 mL) was added and the aqueous layer was washed with Et2O (100 mL) three times. The aqueous layer was concentrated in vacuo to provide compound 48c (5.71 g, 19.83 mmol, 98% yield) as a yellowish oil. Rf = 0.05 (30% MeOH in CH2Cl2); IR (thin film): νmax 3291.47, 2937.97, 2116.98, 1654.27, 1523.37, 1454.14, 1358.49, 1094.33, 701.46 cm−1; 1H NMR (500 MHz, CD3OD) δ 7.33 (d, J = 4.3 Hz, 4H), 7.26 (h, J = 4.3 Hz, 1H), 5.10 (dd, J = 7.1, 4.8 Hz, 1H), 4.39 – 4.33 (m, 1H), 4.17 (t, J = 2.8 Hz, 2H), 3.99 (t, J = 8.4 Hz, 1H), 3.82 – 3.70 (m, 2H), 3.07 (dd, J = 12.0, 4.0 Hz, 1H), 2.94 (dt, J = 11.9, 1.9 Hz, 1H), 2.87 (t, J = 2.4 Hz, 1H), 2.19 (ddt, J = 13.5, 8.0, 1.9 Hz, 1H), 1.85 (ddd, J = 13.5, 8.7, 4.9 Hz, 1H); 13C NMR (101 MHz, CD3OD) δ 175.77, 140.66, 129.53, 128.57, 127.83, 80.29, 76.29, 73.24, 73.17, 60.58, 58.95, 55.86, 54.01, 40.85; HRMS-ESI (m/z): [M+Na]+ calcd. for C16H20N2O3Na+, 311.1372; found, 311.1363; [α]D25 = − 59.8 (c = 0.1, MeOH).
(2S,4R)-1-([1,2,3]Triazolo[1,5-a]pyridine-7-carbonyl)-4-hydroxy-N-((R)-1-phenyl-2-(prop-2-yn-1-yloxy)ethyl)pyrrolidine-2-carboxamide (48).
To a solution of acid 38c (0.34 g, 2.08 mmol) and HOBt (0.28 g, 2.08mmol) in THF/CH2Cl2 (1:1, 14 mL) at 0 °C was added EDCI (0.4 g, 208 mmol). After 5 min, amine 48c (0.5 g, 1.73 mmol) in THF/CH2Cl2 (1:1, 2 mL) and DIPEA (0.6 mL, 3.47 mmol) were added. The reaction solution was stirred at room temperature for 23 h before concentration in vacuo. The residue was dissolved in CH2Cl2 (120 mL) and the solution was washed with water (30 mL), sat. NaHCO3 (30 mL) and brine (30 mL), then dried over sodium sulfate, decanted, and concentrated in vacuo. Purification by flash column chromatography (silica gel, 95% EtOAc in CH2Cl2) gave product 48 (0.57 g, 1.31 mmol, 75% yield) as a white solid. Rf = 0.33 (25% acetone in EtOAc); m.p. = 142–143 °C (acetone); IR (thin film): νmax 3298.49, 3083.78, 2932.14, 2003.72, 1650.86, 1545.57, 1430.10, 1087.77, 827.45, 747.24 cm−1; 1H NMR (400 MHz, CDCl3, rotamer ratio of 10:1) δ 8.33 (d, J = 7.6 Hz, 1H), 8.15 (s, 1H), 7.81 (d, J = 8.7 Hz, 1H), 7.34 (dd, J = 10.6, 7.6 Hz, 3H), 7.30 – 7.25 (m, 1H), 7.22 (t, J = 7.4 Hz, 1H), 7.17 (d, J = 7.2 Hz, 1H), 5.21 (q, J = 7.0 Hz, 1H), 5.05 (t, J = 8.1 Hz, 1H), 4.41 (s, 1H), 4.37 – 4.22 (m, 2H), 3.90 (d, J = 6.6 Hz, 2H), 3.60 (dt, J = 11.4, 3.0 Hz, 1H), 3.17 (d, J = 11.3 Hz, 1H), 2.49 (dd, J = 14.0, 8.4 Hz, 1H), 2.41 (t, J = 2.5 Hz, 1H), 2.38 – 2.26 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 170.76, 161.80, 138.91, 133.75, 132.03, 128.47, 127.41, 126.87, 126.36, 125.86, 119.14, 115.09, 79.68, 74.75, 72.60, 69.80, 59.07, 58.26, 55.80, 53.54, 38.70; HRMS-ESI (m/z): [M+Na]+ calcd. for C23H23N5O4Na+, 456.1648; found, 456.1642; [α]D25 = − 18.9 (c = 0.1, CHCl3/MeOH = 1:1).
To a high-pressure flask were added pyridotriazole substrate 48 (0.1 g, 0.23 mmol) and mesitylene (57 mL). The reaction mixture was stirred at 200 °C for 13 h before aqueous workup. Purification by preparative TLC (10% MeOH in CHCl3) followed flash column chromatography (C18, 16–35% MeCN in water with 0.1% formic acid) afforded macrocycles 49 (19 mg, 44 μmol, 19% yield) and 50 (35 mg, 81 μmol, 35% yield).
(42S,44R,7R,Z)-44-Hydroxy-7-phenyl-11H-9-oxa-6-aza-2(2,6)-pyridina-1(5,3)-pyrazola-4(1,2)-pyrrol-idinacyclodecaphane-3,5-dione (49).
Rf = 0.25 (10% MeOH in CH2Cl2); m.p. = 213–215 °C; IR (thin film): νmax 3292.98, 2938.85, 2871.53, 2409.47, 1603.52, 1569.56, 1444.69, 1408.41, 1090.62, 1021.74, 823.46, 700.08 cm−1; 1H NMR (400 MHz, CD3OD/CDCl3=1:1) δ 8.35 (d, J = 8.2 Hz, 1H), 8.01 (d, J = 7.8 Hz, 1H), 7.89 (t, J = 7.8 Hz, 1H), 7.81 (d, J = 7.4 Hz, 1H), 7.38 – 7.27 (m, 5H), 7.27 – 7.17 (m, 1H), 5.55 (d, J = 6.7 Hz, 1H), 5.08 (s, 1H), 4.81 (d, J = 14.3 Hz, 1H), 4.54 (s, 1H), 4.40 – 4.28 (m, 1H), 3.87 (dd, J = 12.6, 5.2 Hz, 1H), 3.80 (dd, J = 12.6, 6.5 Hz, 1H), 3.68 (s, 2H), 2.33 – 2.12 (m, 2H); 13C NMR (101 MHz, CD3OD/CDCl3=1:1) δ 172.33, 165.28, 151.46, 149.02, 140.55, 137.97, 128.42, 128.17, 127.15, 126.34, 123.59, 120.69, 107.28, 72.23, 66.73, 64.19, 62.57, 55.11, 53.50, 40.84; HRMS-ESI (m/z): [M+H]+ calcd. for C23H23N5O4H+, 434.1828; found, 434.1831; [α]D25 = + 9.9 (c = 0.2, CHCl3/MeOH = 1:1). DFT-optimized structure of 49 is consistent with key 2D NMR interactions for structural assignment of 49 (see Figure S18 in the Supporting Information for details).
(42S,44R,7R)-44-Hydroxy-7-phenyl-11H-9-oxa-6-aza-2(2,6)-pyridina-1(5,4)-pyrazola-4(1,2)-pyrrol-idinacyclodecaphane-3,5-dione (50).
Compound 50 showed two sets of peaks in the 1H and 13C NMR at room temperature in all solvent systems employed presumably due to the presence of rotamers, tentatively assigned as the cis/trans rotamers at the hydroxyproline tertiary amide based on DFT analysis (see Figure S19 in the Supporting Information for details). Coalescence was observed in the 1H NMR spectrum in (CD3)2SO upon heating to 90 °C (see Figure S20 in the Supporting Information for details). Rf = 0.25 (10% MeOH in CH2Cl2); m.p. = 210–212 °C (MeOH); IR (thin film): νmax 3359.65, 3262.92, 2943.98, 1656.54, 1591.46, 1453.31, 1412.29, 1090.19, 1050.22, 1024.87, 830.36, 761.68, 702.75 cm−1; 1H NMR (400 MHz, CDCl3/CD3OD=1:3) δ 8.33 (d, J = 7.5 Hz, 0.5H), 8.21 (s, 0.5H), 8.04 (t, J = 8.8 Hz, 1H), 7.95 (t, J = 7.8 Hz, 0.5H), 7.89 (t, J = 7.8 Hz, 0.5H), 7.77 (d, J = 7.9 Hz, 0.5H), 7.66 – 7.53 (m, 2H), 7.18 (dq, J = 15.2, 7.5 Hz, 3H), 7.07 (d, J = 7.6 Hz, 1.5H), 5.20 (t, J = 8.0 Hz, 0.5H), 5.03 (dd, J = 9.2, 6.5 Hz, 0.5H), 4.94 (s, 0.5H), 4.87 (d, J = 12.6 Hz, 0.5H), 4.75 (s, 0.5H), 4.47 (d, J = 9.7 Hz, 0.5H), 4.37 (d, J = 9.5 Hz, 1.5H), 4.29 (d, J = 12.6 Hz, 0.5H), 4.02 (dd, J = 11.3, 3.0 Hz, 0.5H), 3.92 (dd, J = 11.3, 4.3 Hz, 0.5H), 3.88 – 3.68 (m, 1.5H), 3.52 (dd, J = 10.4, 2.9 Hz, 0.5H), 3.38 (t, J = 9.9 Hz, 0.5H), 3.20 (d, J = 11.8 Hz, 0.5H), 2.46 (t, J = 11.8 Hz, 0.5H), 2.21 (ddd, J = 22.1, 14.7, 7.1 Hz, 1H), 1.92 (ddd, J = 13.1, 8.3, 4.7 Hz, 0.5H); 13C NMR (101 MHz, CD3OD) δ 173.02, 172.94, 172.76, 172.68, 171.21, 168.13, 154.27, 153.17, 151.66, 148.15, 140.62, 139.89, 139.14, 138.34, 134.20, 133.79, 129.17, 129.08, 128.07, 127.74, 126.89, 126.72, 125.94, 123.83, 123.54, 120.52, 117.21, 116.44, 75.12, 72.67, 70.37, 68.46, 66.16, 62.53, 61.86, 58.80, 57.98, 57.11, 53.83, 53.74, 40.99, 39.06; HRMS-ESI (m/z): [M+H]+ calcd. for C23H23N5O4H+, 434.1828; found, 434.1827; [α]D25 = − 17.3 (c = 0.2, CHCl3/MeOH = 1:1).
(32S,34R,6R,9Z,11E)-34-Hydroxy-6-phenyl-8-oxa-5-aza-1(2,6)-pyridina-3(1,2)-pyrrolidinacyclodode-caphane-9,11-diene-2,4-dione (51).
To a vial containing tetrakis(triphenylacetato)dirhodium(II) (19 mg, 14 μmol) was added pyridotriazole 48 (60 mg, 0.14 mmol) in DCE (35 mL). The reaction solution was stirred at 90 °C for 14 h before addition of DMSO (0.1 mL, 1.4 mmol). After 5 min at 90 °C, the reaction solution was cooled to room temperature and was concentrated in vacuo. Purification by flash column chromatography (silica gel, 60% EtOAc in CH2Cl2) gave 51 (80 mg, 0.1 mmol, 71% yield) as a white solid. Rf = 0.56 (10% MeOH in CH2Cl2); m.p. = 215–217 °C (MeOH); IR (thin film): νmax 3375.39, 3300.90, 2942.17, 1668.42, 1614.64, 1572.64, 1466.11, 1427.32, 1254.69, 1076.85, 994.29, 753.84, 702.53 cm−1; 1H NMR (400 MHz, CD3OD/CHCl3=1:1) δ 8.72 (d, J = 6.8 Hz, 1H), 7.96 – 7.88 (dd, J = 15.7, 11.0 Hz, 1H), 7.87 (d, J = 7.7 Hz, 1H), 7.71 (t, J = 7.8 Hz, 1H), 7.35 – 7.23 (m, 5H), 7.20 (d, J = 7.8 Hz, 1H), 6.67 (d, J = 15.7 Hz, 1H), 6.35 (d, J = 5.2 Hz, 1H), 5.75 (dd, J = 11.0, 5.2 Hz, 1H), 5.59 (d, J = 7.9 Hz, 1H), 5.13 (q, J = 6.8 Hz, 1H), 4.27 (p, J = 6.6 Hz, 1H), 3.90 (d, J = 6.8 Hz, 2H), 3.84 – 3.71 (m, 2H), 2.38 (dd, J = 13.0, 6.3 Hz, 1H), 2.28 (dt, J = 12.7, 8.1 Hz, 1H); 13C NMR (101 MHz, CD3OD/CHCl3=1:1) δ 172.75, 165.63, 154.55, 151.97, 147.93, 138.75, 137.27, 128.58, 128.14, 128.01, 127.56, 126.23, 124.12, 123.22, 115.45, 73.97, 66.74, 61.84, 54.80, 54.00, 40.77; HRMS-ESI (m/z): [M+H]+ calcd. for C23H23N3O4H+, 406.1767; found, 406.1779; [α]D25 = + 8.8 (c = 0.05, CHCl3/MeOH = 1:1). DFT-optimized structure, key 2D NMR interactions, and coupling constants were used for structural assignment of 51 (see Figure S21 and Figure S22 in the Supporting Information for details).
(14'Z,16'E)-Spiro[cyclohexane-1,5'-3,8-diaza-1(1,3)-benzenacycloheptadecaphanene]-14',16'-diene-2',7'-dione (52). Note: The product 52 is stable in the solid state, but in solution is very sensitive to air. Solvent removal and chromatography were carried out under N2. Eluents for purification were degassed by N2 before use.
To a vial containing tetrakis(triphenylacetato)dirhodium(II) (23 mg, 17 μmol) was added alkynyl tosylhydrazone 19a (0.1 g, 0.17 mmol) in DCE (40 mL). The reaction solution was stirred at 90 °C for 14 h before being cooled to room temperature. The solvent was carefully removed in vacuo with N2 backfill. Purification by flash column chromatography with degassed solvents (silica gel, 30% EtOAc in hexanes; then C18, 1–75% MeCN in water) gave product 52 (10 mg, 25 μmol, 15% yield) as a yellow solid. Rf = 0.66 (60% EtOAc in hexanes); m.p. = 205–210 °C(decomposed, water); IR (thin film): νmax 3282.69, 2926.29, 2853.35, 1637.77, 1545.88, 1453.63, 1377.76, 1301.14, 984.37, 945.18, 732.04 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.79 (t, J = 4.70 Hz, 1H), 8.35 (s, 1H), 7.97 (d, J = 7.8 Hz, 1H), 7.44 – 7.34 (m, 2H), 7.30 – 7.21 (m, 1H), 6.58 (d, J = 15.5 Hz, 1H), 6.21 (t, J = 10.9 Hz, 1H), 5.89 (t, J = 6.5 Hz, 1H), 5.58 (q, J = 8.5 Hz, 1H), 3.52 (d, J = 4.8 Hz, 2H), 3.34 – 3.25 (m, 2H), 2.38 (s, 2H), 2.37 – 2.29 (m, 2H), 1.69 – 1.34 (m, 16H); 13C NMR (101 MHz, CDCl3) δ 172.65, 167.08, 137.56, 134.92, 133.07, 131.91, 131.75, 129.43, 128.93, 127.85, 124.90, 121.04, 49.86, 48.29, 38.93, 36.15, 34.60, 28.09, 27.52, 26.74, 26.15, 25.09, 21.96; HRMS-ESI (m/z): [M+H]+ calcd. for C25H34N2O2H+, 395.2699; found, 395.2687. DFT-optimized structure, key 2D NMR interactions, and coupling constants were used for structural assignment of 52 (see Figure S23 in the Supporting Information for details).
(3'Z,5'E)-Spiro[cyclohexane-1,11'-2-oxa-9,14-diaza-1(1,2),7(1,3)-dibenzenacyclopentadecaphanene]-3',5'-diene-8',13'-dione (53). Note: The product 53 is stable in the solid state, but in solution is very sensitive to air. Solvent removal and chromatography were carried out under N2. Eluents for purification were degassed by N2 before use.
To a vial containing tetrakis(triphenylacetato)dirhodium(II) (11 mg, 8 μmol) was added alkynyl tosylhydrazone 19c (50 mg, 81 μmol) in DCE (20 mL). The reaction solution was stirred at 90 °C for 19 h before cooling to room temperature. The solvent was carefully removed in vacuo with N2 backfill. Purification by flash column chromatography using degassed solvents (C18, 1–60% MeCN in water with 0.01% formic acid) gave product 53 (11 mg, 26 μmol, 31% yield) as a yellow solid. Rf = 0.33 (50% EtOAc in hexanes); m.p. = 214–216 °C(decomposed, water); IR (thin film): νmax 3270.78, 2925.71, 2855.91, 1639.71, 1546.02, 1454.17, 1277.43, 1243.40, 1138.07, 1024.41, 752.22 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.87 (t, J = 4.85 Hz, 1H), 8.59 (s, 1H), 7.98 – 7.85 (m, 2H), 7.35 (t, J = 7.6 Hz, 1H), 7.30 (d, J = 7.5 Hz, 1H), 7.26 (m, 2H), 7.03 (t, J = 7.5 Hz, 1H), 6.98 (d, J = 8.2 Hz, 1H), 6.69 (d, J = 5.5 Hz, 2H), 6.66 (t, J = 6.2 Hz, 1H), 6.58 (d, J = 16.0 Hz, 1H), 5.84 (dd, J = 11.1, 5.5 Hz, 1H), 4.69 (d, J = 6.3 Hz, 2H), 3.47 (d, J = 4.7 Hz, 2H), 2.42 (s, 2H), 1.61 – 1.28 (m, 10H); 13C NMR (101 MHz, CDCl3) δ 171.95, 167.04, 154.21, 140.67, 137.46, 134.60, 130.97, 129.94, 129.20, 129.01, 128.86, 127.22, 126.93, 122.98, 122.53, 121.56, 113.89, 113.21, 49.60, 47.48, 36.66, 36.37, 34.58, 25.95, 21.81; HRMS-ESI (m/z): [M+H]+ calcd. for C27H30N2O3H+, 431.2335; found, 431.2348. DFT-optimized structure, key 2D NMR interactions, and coupling constants were used for structural assignment of 53 (see Figure S24 in the Supporting Information for details).
Post-functionalization reactions of macrocycles 20d, 24c, and 51.
(S,Z)-1'-(4-Methoxyphenyl)-10'-phenylspiro[cyclohexane-1,6'-12-oxa-4,9-diaza-1(3,5)-pyrazola-2(1,3)-benzenacyclotridecaphane]-3',8'-dione (56).
To a vial containing 20d (6 mg, 12.7 μmol), CuI (0.5 mg, 2.5 μmol) and K3PO4 (8 mg, 38 μmol) were added a solution of 4-iodoanisole 54 (4.5 mg, 19 μmol) and trans-N,N'-dimethylcyclo-hexane-1,2-diamine 55 (1.4 mg, 10 μmol) in dioxane (0.1 mL). The resulting mixture was stirred at 110 °C for 18 h before cooling to room temperature. The mixture was filtered through a short plug of silica gel using acetone (10 mL) and the filtrate was concentrated in vacuo. Purification by flash column chromatography (silica gel, 65% EtOAc in hexanes) gave product 56 (5 mg, 8.64 μmol, 68% yield) as a white solid. Rf = 0.34 (75% EtOAc in hexanes); m.p. = 190–191 °C (EtOAc); IR (thin film): νmax 3266.70, 3059.77, 2926.63, 2853.88, 2383.37, 1631.32, 1517.05, 1449.23, 1252.18, 1086.13, 1025.42, 734.50, 700.25 cm−1; 1H NMR (400 MHz, CD3OD) δ 10.24 (s, 1H), 8.48 (s, 1H), 7.92 (d, J = 7.6 Hz, 1H), 7.82 (d, J = 7.8 Hz, 1H), 7.62 – 7.51 (m, 1H), 7.49 (s, 1H), 7.42 – 7.34 (m, 2H), 7.36 – 7.25 (m, 5H), 7.04 – 6.94 (m, 2H), 5.18 (dd, J = 10.5, 3.8 Hz, 1H), 4.67 (dd, J = 14.8, 4.3 Hz, 1H), 4.51 (dd, J = 14.8, 6.4 Hz, 1H), 4.21 – 4.08 (m, 1H), 4.01 (q, J = 7.4, 4.5 Hz, 1H), 3.82 (d, J = 3.1 Hz, 3H), 3.68 (dd, J = 13.4, 5.8 Hz, 1H), 3.25 (d, J = 14.0 Hz, 1H), 2.51 (d, J = 4.6 Hz, 2H), 1.68 – 1.21 (m, 10H); 13C NMR (101 MHz, CD3OD) δ 173.36, 168.29, 159.72, 152.51, 141.52, 138.90, 134.90, 133.56, 131.85, 129.59, 129.18, 128.25, 127.73, 127.54, 127.00, 126.53, 125.90, 114.01, 108.07, 70.16, 62.49, 54.63, 53.79, 49.18, 45.61, 37.07, 34.54, 33.40, 25.68, 21.36, 21.17; HRMS-ESI (m/z): [M+H]+ calcd. for C35H38N4O4H+, 579.2971; found, 579.2969; [α]D21 = + 82.8 (c = 0.05, MeOH). Key 2D NMR interactions and coupling constants were used for structural assignment of 56 (see Figure S25 in the Supporting Information for details).
A mixture of 20d (26 mg, 55 μmol), K2CO3 (15 mg, 0.11 mmol), and isobutene oxide 57 (0.25 mL) in DMF (0.25 mL) was stirred at 90 °C for 13 h. The reaction mixture was then cooled to room temperature before concentration in vacuo. Purification using preparative TLC (55% EtOAc in CH2Cl2) afforded compound 58 (5.4 mg, 10 μmol, 18% yield) as a white solid and compound 59 (14 mg, 26 μmol, 47% yield) as a white solid.
(S,Z)-1'-(2-Hydroxy-2-methylpropyl)-10'-phenylspiro[cyclohexane-1,6'-12-oxa-4,9-diaza-1(5,3)-pyrazola-2(1,3)-benzenacyclotridecaphane]-3',8'-dione (58).
Rf = 0.34 (40% acetone in hexanes); m.p. = 170–171 °C (EtOAc); IR (thin film): νmax 3267.91, 3057.59, 2926.65, 2850.04, 1641.54, 1605.89, 1527.01, 1304.49, 1119.71, 1092.69, 913.94, 735.06 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.83 (dd, J = 6.9, 3.4 Hz, 1H), 8.10 (s, 1H), 8.02 – 7.95 (m, 1H), 7.61 – 7.54 (m, 2H), 7.40 (td, J = 11.0, 10.5, 7.6 Hz, 5H), 7.00 (s, 1H), 5.91 (d, J = 6.6 Hz, 1H), 5.10 (ddd, J = 10.1, 6.4, 3.3 Hz, 1H), 4.85 (d, J = 14.3 Hz, 1H), 4.50 (d, J = 14.3 Hz, 1H), 4.19 – 4.11 (m, 1H), 4.08 (d, J = 11.3 Hz, 2H), 3.77 (t, J = 9.5 Hz, 1H), 3.68 (dd, J = 13.8, 3.5 Hz, 1H), 3.25 (dd, J = 13.8, 3.5 Hz, 1H), 2.39 (d, J = 3.1 Hz, 2H), 1.63 – 1.27 (m, 10H), 1.16 (s, 3H), 1.02 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 172.60, 167.22, 149.66, 146.36, 138.61, 136.35, 132.14, 130.06, 129.20, 129.17, 128.60, 128.56, 128.09, 127.57, 107.84, 70.91, 68.99, 65.83, 58.13, 55.20, 49.27, 47.34, 37.16, 34.65, 34.05, 26.90, 26.87, 25.84, 21.70, 21.60; HRMS-ESI (m/z): [M+H]+ calcd. for C32H40N4O4H+, 545.3128; found, 545.3137; [α]D21 = + 137.6 (c = 0.05, MeOH). DFT-optimized structure consistent with key 2D NMR interactions were used for the structural assignment of 58 (see Figure S26 in the Supporting Information for details).
(S,Z)-1'-(2-Hydroxy-2-methylpropyl)-10'-phenylspiro[cyclohexane-1,6'-12-oxa-4,9-diaza-1(3,5)-pyrazola-2(1,3)-benzenacyclotridecaphane]-3',8'-dione (59).
Rf = 0.4 (40% acetone in hexanes); m.p. = 195–196 °C (EtOAc); IR (thin film): νmax 3264.12, 3063.12, 2928.53, 2862.65, 1641.36, 1527.28, 1497.05, 1302.41, 1118.88, 1073.61, 912.52, 735.65, 700.67 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.61 (t, J = 5.0 Hz, 1H), 8.43 (d, J = 2.2 Hz, 1H), 7.90 (d, J = 7.8 Hz, 1H), 7.87 (d, J = 7.7 Hz, 1H), 7.50 (t, J = 7.7 Hz, 1H), 7.43 – 7.32 (m, 6H), 6.14 (d, J = 6.6 Hz, 1H), 5.17 (ddd, J = 10.3, 6.5, 3.6 Hz, 1H), 4.77 (d, J = 14.6 Hz, 1H), 4.49 (d, J = 14.6 Hz, 1H), 4.26 (br s, 1H), 4.22 (dd, J = 9.9, 3.4 Hz, 1H), 3.99 – 3.89 (m, 2H), 3.87 (t, J = 10.2 Hz, 1H), 3.63 – 3.58 (m, 1H), 3.33 (dd, J = 13.8, 3.8 Hz, 1H), 2.39 (s, 2H), 1.59 – 1.26 (m, 10H), 1.14 (s, 3H), 1.13 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 172.95, 167.25, 152.33, 140.82, 138.24, 135.03, 133.42, 129.37, 129.34, 129.16, 128.57, 127.65, 127.17, 126.90, 107.38, 70.83, 69.30, 62.39, 58.72, 54.23, 49.13, 47.30, 37.42, 34.40, 34.26, 26.93, 26.86, 25.86, 21.71, 21.58; HRMS-ESI (m/z): [M+H]+ calcd. for C32H40N4O4H+, 545.3128; found, 545.3141; [α]D21 = + 106.0 (c = 0.05, MeOH). DFT-optimized structure consistent with key 2D NMR interactions were used for the structural assignment of 59 (see Figure S27 in the Supporting Information for details).
(42S,44R)-44-Hydroxy-11-(4-methoxyphenyl)-8,8-dimethyl-11H-6-aza-1(3,4)-pyrazola-4(1,2)-pyrroli-dina-2(1,3)-benzenacyclononaphane-3,5-dione (61).
To a solution of 24c (10 mg, 27 μmol), 3Å MS (10 mg), and (4-methoxyphenyl)boronic acid 60 (10 mg, 68 μmol) in THF (0.3 mL) was added Cu(OAc)2 (7.4 mg, 41 μmol) followed by pyridine (22 μL, 0.27 mmol). The mixture was stirred room temperature for 12 h before being filtered through a short plug of silica gel. The filtrate was concentrated in vacuo and purified by preparative TLC (6.5% MeOH in CH2Cl2) to give product 61 (9 mg, 19 μmol, 70% yield) as a white solid. Rf = 0.4 (10% MeOH in CH2Cl2); m.p. = 240–241 °C (MeOH); IR (thin film): νmax 3415.37, 3321.71, 2953.18, 2919.89, 2850.18, 1658.63, 1622.14, 1517.64, 1466.32, 1435.70, 1249.54 cm−1; 1H NMR (400 MHz, CD3OD) δ 8.02 (s, 1H), 7.70 – 7.63 (m, 3H), 7.59 (t, J = 7.7 Hz, 1H), 7.45 – 7.39 (m, 1H), 7.36 (s, 1H), 7.07 – 7.00 (m, 2H), 4.57 (t, J = 4.5 Hz, 1H), 4.19 (dd, J = 8.6, 5.5 Hz, 1H), 4.00 – 3.90 (m, 1H), 3.74 (dd, J = 12.3, 5.0 Hz, 1H), 3.44 (d, J = 13.3 Hz, 1H), 3.08 (d, J = 15.1 Hz, 1H), 2.40 – 2.22 (m, 3H), 2.09 (dt, J = 13.2, 5.5 Hz, 1H), 1.01 (s, 3H), 0.39 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 174.20, 171.99, 158.53, 152.14, 136.55, 134.29, 133.47, 130.13, 129.21, 127.69, 125.75, 124.71, 120.49, 118.14, 114.20, 68.20, 60.97, 54.59, 54.18, 49.33, 39.48, 36.61, 30.13, 26.49, 22.54; HRMS-ESI (m/z): [M+H]+ calcd. for C27H30N4O4H+, 475.2345; found, 475.2332; [α]D21 = + 114.8 (c = 0.05, MeOH). DFT-optimized structure consistent with key 2D NMR interactions were used for the structural assignment of 61 (see Figure S28 in the Supporting Information for details).
A high-pressure round-bottomed flask containing macrocyclic diene 51 (50 mg, 0.12 mmol) and 3,6-dimethyl-1,2,4,5-tetrazine 62 (68 mg, 0.62 mmol) was stirred at 110 °C for 27 h. The solution was cooled to room temperature and was concentrated in vacuo. Purification by preparative TLC (10% MeOH in CHCl3) gave unreacted diene 51 (6 mg, 15 μmol, 12% sm recovered) as a reddish solid, pyridazine 63 (31 mg, 64 μmol, 52% yield) as a yellow solid and pyridazine alcohol 64 (12 mg, 24 μmol, 19% yield) as a yellow solid.
(42S,44R,7R,Z)-44-Hydroxy-13,16-dimethyl-7-phenyl-9-oxa-6-aza-1(4,5)-pyridazina-2(2,6)-pyridina-4(1,2)-pyrrolidinacycloundecaphan-10-ene-3,5-dione (63).
Rf = 0.45 (10% MeOH in CH2Cl2); m.p. = 205–207 °C (MeOH); IR (thin film): νmax 3461.61, 3376.27, 2926.30, 1660.16, 1618.35, 1586.02, 1570.44, 1433.08, 1393.23, 1101.39, 757.22, 700.50 cm−1; 1H NMR (400 MHz, CD3OD) δ 8.69 (d, J = 6.5 Hz, 1H), 8.00 (t, J = 7.8 Hz, 1H), 7.93 (d, J = 7.8 Hz, 1H), 7.62 (d, J = 7.6 Hz, 1H), 7.23 (dq, J = 13.8, 7.1 Hz, 3H), 7.13 (d, J = 7.3 Hz, 2H), 6.42 (d, J = 6.5 Hz, 1H), 5.62 (d, J = 6.8 Hz, 1H), 5.57 (d, J = 8.6 Hz, 1H), 4.59 (dt, J = 10.5, 5.4 Hz, 1H), 4.37 (s, 1H), 3.89 – 3.80 (m, 1H), 3.78 – 3.66 (m, 2H), 2.67 (s, 2H), 2.46 (s, 3H), 2.42 (d, J = 7.7 Hz, 1H), 1.92 – 1.79 (m, 1H); 13C NMR (101 MHz, CD3OD) δ 173.75, 168.76, 160.35, 158.27, 154.96, 153.95, 148.05, 139.67, 139.51, 138.77, 135.38, 129.62, 128.80, 127.98, 127.59, 125.02, 101.80, 74.07, 68.84, 61.57, 58.33, 54.89, 41.87, 20.99, 20.65; HRMS-ESI (m/z): [M+H]+ calcd. for C27H27N5O4H+, 486.2141; found, 486.2151; [α]D25 = − 20.4 (c = 0.05, MeOH). DFT-optimized structure, key 2D NMR interactions and coupling constants were used for the structural assignment of pyridazine 63 (see Figure S29 in the Supporting Information for details).
(12S,14R,9R,E)-14-hydroxy-63,66-dimethyl-9-phenyl-61,64-dihydro-7-oxa-10-aza-6(5,4)-pyridazi-na-3(2,6)-pyridina-1(1,2)-pyrrolidinacycloundecaphan-4-ene-2,11-dione (64).
Rf = 0.36 (10% MeOH in CH2Cl2); m.p. = 178–180 °C (MeOH); IR (thin film): νmax 3302.45, 3063.96, 2929.27, 1666.46, 1625.07, 1585.17, 1567.10, 1432.73, 1084.94, 759.55, 702.17 cm−1; 1H NMR (400 MHz, CD3OD, rotamer ratio of 1.5:1) δ 7.98 (t, J = 7.8 Hz, 0.6H), 7.92 (d, J = 15.8 Hz, 0.4H), 7.91 (d, J = 16.2 Hz, 0.6H), 7.87 – 7.80 (m, 1.6H), 7.77 (d, J = 7.7 Hz, 0.4H), 7.73 – 7.67 (m, 1H), 7.63 (d, J = 16.2 Hz, 0.4H), 7.58 (d, J = 15.8 Hz, 0.6H), 7.38 (d, J = 12.1 Hz, 0.6H), 7.36 – 7.30 (m, 2.4H), 7.28 – 7.22 (m, 0.6H), 7.15 – 7.08 (m, 1.2H), 7.09 – 7.03 (m, 0.8H), 5.43 (t, J = 7.9 Hz, 0.4H), 5.04 (t, J = 6.2 Hz, 0.6H), 4.87 (t, J = 8.7 Hz, 0.6H), 4.64 (t, J = 6.3 Hz, 0.4H), 4.53 – 4.41 (m, 1H), 4.16 (dd, J = 12.1, 4.0 Hz, 0.6H), 4.00 (dt, J = 12.1, 1.9 Hz, 0.6H), 3.93 (dt, J = 12.8, 2.2 Hz, 0.4H), 3.86 (dd, J = 12.8, 4.4 Hz, 0.4H), 3.82 (d, J = 6.2 Hz, 1.2H), 3.52 – 3.43 (m, 0.8H), 2.77 (s, 3H), 2.67 (s, 3H), 2.41 (td, J = 9.5, 8.8, 4.2 Hz, 0.4H), 2.36 – 2.26 (m, 0.6H), 2.19 – 2.02 (m, 1H); 13C NMR (101 MHz, CD3OD, rotamer ratio of 1.5:1) δ 174.47, 174.08, 168.56, 168.50, 160.34, 160.18, 157.55, 157.50, 154.41, 154.28, 154.24, 154.08, 141.08, 140.99, 139.48, 139.21, 137.42, 137.20, 137.09, 136.43, 129.50, 129.32, 128.42, 128.26, 128.00, 127.95, 127.53, 127.43, 126.33, 125.17, 125.04, 124.79, 124.73, 124.36, 71.31, 68.84, 66.23, 65.54, 63.21, 61.61, 59.73, 57.53, 57.14, 57.03, 41.79, 38.66, 21.44, 21.42, 19.95, 19.89; HRMS-ESI (m/z): [M+H]+ calcd. for C27H29N5O4H+, 488.2298; found, 488.2297; [α]D25 = − 11.5 (c = 0.1, MeOH).
(42S,44R,7R)-44-Hydroxy-13,16-dimethyl-7-phenyl-9-oxa-6-aza-1(4,5)-pyridazina-2(2,6)-pyridi-na-4(1,2)-pyrrolidinacycloundecaphane-3,5-dione (65).
A mixture of pyridazine 63 (44 mg, 91 μmol), AcOH (10 μL, 0.18 mmol), MeOH/THF (1:1, 8 mL) and 10% Pd/C (48 mg) was stirred under an atmosphere of H2 (1 atm) for 40 h before filtration through a short plug of C8 silica gel (40–63 μm, 60Å from SiliCycle). The filtrate was basified with 2 N NaOH until pH>12. After concentration in vacuo, the residue was purified using a C18 cartridge (1–50% MeCN in water) to give product 65 (24.8 mg, 51 μmol, 56% yield) as a white solid. Rf = 0.45 (10% MeOH in CH2Cl2); m.p. = 188–190 °C (MeOH); IR (thin film): νmax 3445.67, 3349.98, 2934.34, 2863.39, 1668.77, 1618.39, 1584.44, 1432.58, 1394.54, 1339.06, 1116.00, 760.28, 701.36 cm−1; 1H NMR (400 MHz, CD3OD) δ 8.70 (d, J = 8.3 Hz, 1H), 8.13 – 8.05 (m, 1H), 8.10 (s, 1H), 7.68 (dd, J = 6.9, 1.9 Hz, 1H), 7.29 – 7.16 (m, 3H), 7.15 – 7.06 (m, 2H), 5.68 (t, J = 8.6 Hz, 1H), 4.36 (t, J = 4.8 Hz, 1H), 3.85 (d, J = 13.0 Hz, 1H), 3.74 (dd, J = 12.9, 3.8 Hz, 1H), 3.46 (ddt, J = 12.9, 8.7, 4.8 Hz, 2H), 3.37 (d, J = 11.4 Hz, 1H), 3.35 – 3.23 (m, 3H), 3.13 (ddd, J = 14.7, 10.2, 4.7 Hz, 1H), 2.76 (s, 3H), 2.52 – 2.41 (m, 1H), 2.39 (s, 3H), 1.89 (ddd, J = 17.6, 8.3, 3.7 Hz, 1H); 13C NMR (101 MHz, CD3OD) δ 173.64, 168.26, 161.06, 158.55, 155.23, 153.43, 140.57, 139.71, 139.40, 138.12, 129.57, 128.57, 127.64, 127.51, 125.41, 73.09, 68.67, 67.40, 61.64, 58.44, 52.74, 42.04, 29.53, 21.24, 20.87; HRMS-ESI (m/z): [M+H]+ calcd. for C27H29N5O4H+, 488.2298; found, 488.2284; [α]D25 = − 38.0 (c = 0.3, MeOH).
(2S,4R)-4-((tert-butyldimethylsilyl)oxy)-N-((R)-2-((tert-butyldimethylsilyl)oxy)-1-phenylethyl)-1-(6-((E)-2-(3,6-dimethylpyridazin-4-yl)vinyl)picolinoyl)pyrrolidine-2-carboxamide (66).
To a solution of pyridazine 64 (4.5 mg, 9 μmol) in anhydrous CH2Cl2 (0.6 mL) at 0 °C were added TBSOTf (42 μL, 0.18 mmol) and DIPEA (32 μL, 0.18 mmol). The reaction solution was stirred at room temperature for 10 min before concentration in vacuo. Purification by flash column chromatography (C18, 1–90% MeCN in H2O with 0.1% formic acid) provided compound 66 (5 mg, 7 μmol, 76% yield) as a light yellow oil. Rf = 0.40 (33% acetone in CH2Cl2); IR (thin film): νmax 3290.83, 2954.02, 2927.77, 2856.24, 1673.19, 1629.65, 1463.21, 1411.83, 1256.98, 1093.81, 836.88, 777.92, 756.56; 1H NMR (500 MHz, CDCl3, rotamer ratio of 1:1) δ 8.16 (d, J = 7.9 Hz, 0.5H), 7.90 (t, J = 8.9 Hz, 1H), 7.86 (d, J = 7.9 Hz, 1H), 7.79 (d, J = 7.6 Hz, 0.5H), 7.74 (d, J = 15.8 Hz, 0.5H), 7.54 (s, 0.5H), 7.53 – 7.48 (m, 1H), 7.36 – 7.29 (m, 3H), 7.21 (d, J = 16.3 Hz, 1H), 7.00 (d, J = 7.8 Hz, 0.5H), 6.95 (d, J = 16.2 Hz, 0.5H), 6.93 – 6.84 (m, 2H), 5.28 (t, J = 6.7 Hz, 0.5H), 5.04 – 4.97 (m, 1H), 4.96 – 4.90 (m, 0.5H), 4.55 (q, J = 5.3 Hz, 1H), 4.10 (ddd, J = 17.6, 12.2, 4.8 Hz, 1H), 3.91 – 3.81 (m, 1.5H), 3.77 (td, J = 10.3, 8.9, 4.5 Hz, 1H), 3.62 (dd, J = 10.4, 4.6 Hz, 0.5H), 2.83 (s, 1.5H), 2.79 (s, 3H), 2.75 (s, 1.5H), 2.55 (dt, J = 12.1, 5.5 Hz, 0.5H), 2.38 (t, J = 5.7 Hz, 1H), 2.08 – 1.98 (m, 0.5H), 0.87 (s, 4.5H), 0.81 (s, 4.5H), 0.80 (s, 4.5H), 0.71 (s, 4.5H), 0.11 (s, 1.5H), 0.10 (s, 1.5H), 0.04 (s, 1.5H), −0.05 (s, 1.5H), −0.07 (s, 1.5H), −0.14 (s, 1.5H), −0.17 (s, 1.5H), −0.18 (s, 1.5H); 13C NMR (126 MHz, CDCl3, rotamer ratio of 1:1) δ 173.02, 170.32, 167.70, 165.91, 158.35, 157.64, 157.25, 156.46, 153.71, 152.23, 152.13, 151.96, 140.24, 140.18, 138.20, 138.02, 137.86, 128.46, 128.44, 128.10, 127.43, 127.14, 127.11, 127.05, 126.72, 125.77, 125.69, 125.26, 125.11, 125.07, 124.75, 124.01, 71.18, 68.59, 66.77, 66.44, 63.42, 59.95, 58.32, 56.30, 55.50, 54.88, 41.42, 36.23, 25.87, 25.83, 25.80, 25.76, 21.23, 20.29, 20.16, 19.57, 18.26, 18.24, 18.12, 18.02, −4.54, −4.62, −4.68, −4.77, −5.52, −5.59, −5.62; HRMS-ESI (m/z): [M+H]+ calcd. for C39H57N5O4Si2H+, 716.4027; found, 716.; [α]D27 = + 0.01 (c = 0.2, CHCl3).
Supplementary Material
ACKNOWLEDGMENT
We thank the National Institutes of Health (R24 GM-111625) for research funding. We thank Profs. Scott Schaus and John Snyder (Boston University) for helpful discussions, Dr. Jeffrey Bacon (Boston University) for X-ray crystallographic analyses, Dr. Paul Ralifo for assistance with NMR analysis, and Dr. Norman Lee for mass spectrometry analysis. NMR (CHE-0619339) and MS (CHE-0443618) facilities at Boston University are supported by the NSF.
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information.
This material is available free of charge via the Internet at http://pubs.acs.org.
Experimental details and computational data (PDF)
X-ray crystallographic data for 20c (CIF)
REFERENCES
- (1).(a) Madsen CM; Clausen MH, Biologically Active Macrocyclic Compounds – from Natural Products to Diversity-Oriented Synthesis. Eur. J. Org. Chem 2011, 17, 3107–3115. [Google Scholar]; (b) Wessjohann LA; Ruijter E; Garcia-Rivera D; Brandt W What Can A Chemist Learn From Nature’s Macrocycles-A Brief, Conceptual View. Mol. Divers 2005, 9, 171–186.15789564 [Google Scholar]
- (2).(a) Wolfson W Grabbing tor the Ring: Macrocycles Tweak the Conventions of Drug Making. Chem. Biol 2012, 19 (11), 1356–1357. [DOI] [PubMed] [Google Scholar]; (b) Yu X; Sun D Macrocyclic Drugs and Synthetic Methodologies toward Macrocycles. Molecules 2013, 18 (6), 6230–6268. [DOI] [PubMed] [Google Scholar]; (c) Marsault E; Peterson ML Macrocycles Are Great Cycles: Applications, Opportunities, and Challenges of Synthetic Macrocycles in Drug Discovery. J. Med. Chem 2011, 54 (7), 1961–2004. [DOI] [PubMed] [Google Scholar]; (d) Driggers EM; Hale SP; Lee J; Terrett N, The K Exploration of Macrocycles for Drug Discovery-An Underexploited Structural Class. Nat. Rev. Drug Discov 2008, 7 (7), 608–624. [DOI] [PubMed] [Google Scholar]; (e) Vendeville S; Cummings MD Synthetic Macrocycles in Small-Molecule Drug Discovery. Annu. Rep. Med. Chem 2013, 48, 371–386. [DOI] [PubMed] [Google Scholar]; (f) Mallinson J; Collins I Macrocycles in New Drug Discovery. Future Med. Chem 2012, 4 (11), 1409–1438. [DOI] [PubMed] [Google Scholar]; (g) Giordanetto F; Kihlberg J Macrocyclic Drugs and Clinical Candidates: What Can Medicinal Chemists Learn from Their Properties? J. Med. Chem 2014, 57 (2), 278–295. [DOI] [PubMed] [Google Scholar]; (h) You L; An R; Liang K; Cui B; Wang X Macrocyclic Compounds: Emerging Opportunities for Current Drug Discovery. Curr. Pharm. Des 2016, 22 (26), 4086–4093. [DOI] [PubMed] [Google Scholar]; (i) Whitty A; Zhou L Horses for Courses: Reaching Outside Drug-Like Chemical Space for Inhibitors of Challenging Drug Targets. Future Med. Chem 2015, 7 (9), 1093–1095. [DOI] [PubMed] [Google Scholar]; (j) Ermert P Design, Properties and Recent Application of Macrocycles in Medicinal Chemistry. CHIMIA Int. J. Chem 2017, 71 (10), 678–702. [DOI] [PubMed] [Google Scholar]; (k) Abdelraheem EMM; Shaabani S; Domling A Artificial Macrocycles. Synlett. 2018, 29 (9), 1136–1151. [DOI] [PubMed] [Google Scholar]; (l) Alihodzic S; Bukvic M; Elenkov IJ; Hutinec A; Kostrun S; Pesic D; Saxty G; Tomaskovic L; Ziher D Current Trends in Macrocyclic Drug Discovery and beyond-Ro5. Prog. Med. Chem 2018, 57, 113–233. [DOI] [PubMed] [Google Scholar]; (m) Yudin AK Macrocycles: Lessons from the Distant Past, Recent Developments, and Future Directions. Chem. Sci 2015, 6, 30–49. [DOI] [PubMed] [Google Scholar]; (n) Viarengo-Baker LA; Brown LE; Rzepiela AA; Whitty A Defining and navigating macrocycle chemical space. Chem. Sci 2021. 12, 4309–4328. [DOI] [PubMed] [Google Scholar]
- (3).(a) Villar EA; Beglov D; Chennamadhavuni S; Porco JA Jr.; Kozakov D; Vajda S; Whitty A How Proteins Bind Macrocycles. Nat. Chem. Biol 2014, 10 (9), 723–731. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dougherty PG; Qian Z; Pei D Macrocycles as Protein-Protein Interaction Inhibitors. Biochem. J 2017, 474 (7), 1109–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Whitty A; Viarengo LA; Zhong M Progress towards the Broad Use of NonPeptide Synthetic Macrocycles in Drug Discovery. Org. Biomol. Chem 2017, 15 (37), 7729–7735. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Whitty A; Zhong M; Viarengo L; Beglov D; Hall DR; Vajda S Quantifying the Chameleonic Properties of Macrocycles and Other High-MolecularWeight Drugs. Drug Discov. Today 2016, 21 (5), 712–717. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Jin L; Wang W; Fang G Targeting Protein-Protein Interaction by Small Molecules. Annu. Rev, Pharmacol. Toxicol 2014, 54, 435–456. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Aeluri M; Chamakuri S; Dasari B; Guduru SKR; Jimmidi R; Jogula S; Arya P Small Molecule Modulators of Protein-Protein Interactions: Selected Case Studies. Chem. Rev 2014, 114 (9), 4640–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Doak BC; Zheng J; Dobritzsch D; Kihlberg J How Beyond Rule of 5 Drugs and Clinical Candidates Bind to Their Targets. J. Med. Chem 2016, 59 (6), 2312–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Villoutreix BO; Kuenemann MA; Poyet JL; Bruzzoni-Giovanelli H; Labbe C; Lagorce D; Sperandio O; Miteva MA Drug-Like Protein Protein Interaction Modulators: Challenges and Opportunities for Drug Discovery and Chemical Biology. Mol. Inf 2014, 33, 414–437. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Sheng C; Dong G; Miao Z; Zhang W; Wang W State-of-the-art Strategies for Targeting Protein-Protein Interactions by Small-Molecule Inhibitors. Chem. Soc. Rev 2015, 44, 8238–8259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Terrett NK Methods for the Synthesis of Macrocycle Libraries for Drug Discovery. Drug Discov. Today Technol 2010, 7 (2), e97–e104. [DOI] [PubMed] [Google Scholar]; (b) Collins S; Bartlett S; Nie F; Sore HF; Spring DR Oriented Synthesis of Macrocycle Libraries for Drug Discovery and Chemical Biology. Synthesis 2016, 48 (10), 1457–1473. [DOI] [PubMed] [Google Scholar]; (c) Mortensen KT; Osberger TJ; King TA; Sore HF; Spring DR Strategies for the Diversity-Oriented Synthesis of Macrocycles. Chem. Rev 2019, 119 (17), 10288–10317. [DOI] [PubMed] [Google Scholar]; (d) Nie F; Kunciw DL; Wilcke D; Stokes JE; Galloway WRJD; Bartlett S; Sore HF; Spring DR A Multidimensional Diversity-Oriented Synthesis Strategy for Structurally Diverse and Complex Macrocycles. Angew. Chem. Int. Ed 2016, 55 (37), 11139–11143. [DOI] [PubMed] [Google Scholar]
- (5).(a) Li Y; Yin X; Dai M Catalytic Macrolactonizations for Natural Product Synthesis. Nat. Prod. Rep 2017, 34, 1185–1192. [DOI] [PubMed] [Google Scholar]; (b) White CJ; Yudin AK Contemporary Strategies for Peptide Macrocyclization. Nature Chem. 2011, 3, 509–524. [DOI] [PubMed] [Google Scholar]; (c) Chow HY; Zhang Y; Matheson E; Li X Ligation Technologies for the Synthesis of Cyclic Peptides. Chem. Rev 2019, 119 (17), 9971–10001. [DOI] [PubMed] [Google Scholar]; (d) White AM; Craik DJ Discovery and Optimization of Peptide Macrocycles. Expert Opin. Drug Discov 2016, 11 (12), 1151–1163. [DOI] [PubMed] [Google Scholar]; (e) Alehashem MS; Ariffin AB; Savage PB; Dabdawb WAY; Thomas NF Treasures Old and New: What We Can Learn regarding the Macrocyclic Problem from Past and Present Efforts in Natural Product Total Synthesis. RSC Adv. 2020, 10, 10989–11012. [DOI] [PubMed] [Google Scholar]
- (6).(a) Zorzi A; Deyle K; Heinis C Cyclic Peptide Therapeutics: Past, Present and Future. Curr. Opin. Chem. Biol 2017, 38, 24–29. [DOI] [PubMed] [Google Scholar]; (b) Russo A; Aiello C; Grieco P; Marasco D Targeting Undruggable Proteins: Design of Synthetic Cyclopeptides. Curr. Med. Chem 2016, 23 (8), 748–762. [DOI] [PubMed] [Google Scholar]; (c) Bhat A; Roberts LR; Dwyer JJ Lead Discovery and Optimization Strategies for Peptide Macrocycles. Eur. J. Med. Chem 2015, 94, 471–479. [DOI] [PubMed] [Google Scholar]; (d) Passioura T; Katoh T; Goto Y; Suga H Selection-Based Discovery of Druglike Macrocyclic Peptides. Ann. Rev. Biochem 2014, 83, 727–752. [DOI] [PubMed] [Google Scholar]; (e) Thapa P; Espiritu MJ; Cabalteja C; Bingham J-P The Emergence of Cyclic Peptides: The Potential of Bioengineered Peptide Drugs. Int. J. Pept. Res. Ther 2014, 20, 545–551. [DOI] [PubMed] [Google Scholar]; (f) Leenheer D; ten Dijke P; Hipolito CJ A Current Perspective on Applications of Macrocyclic-Peptide-Based High-Affinity Ligands. Peptide Science 2016, 106, 889–900. [DOI] [PubMed] [Google Scholar]; (g) White AM; Craik DJ Discovery and Optimization of Peptide Macrocycles. Expert Opin. Drug Discov 2016, 11 (12), 1151–1163. [DOI] [PubMed] [Google Scholar]; (h) Lau YH; de Andrade P; Wu Y; Spring DR Peptide Stapling Techniques Based on Different Macrocyclisation Chemistries. Chem. Soc. Rev 2015, 44, 91−102. [DOI] [PubMed] [Google Scholar]
- (7).(a) Zaretsky S; Hickey JL; Tan J; Pichugin D; Denis MAS; Ler S; Chung BKW; Scully CCG; Yudin AK Mechanistic Investigation of Aziridine Aldehyde-Driven Peptide Macrocyclization: the Imidoanhydride Pathway. Chem. Sci 2015, 6, 5446–5455. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hili R; Rai V; Yudin AK Macrocyclization of Linear Peptides Enabled by Amphoteric Molecules. J. Am. Chem. Soc 2010, 132 (9), 2889–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) He Z; Zajdlik A; Yudin AK Air- and Moisture-Stable Amphoteric Molecules: Enabling Reagents in Synthesis. Acc. Chem. Res 2014, 47 (4), 1029–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Frost JR; Scully CCG; Yudin AK Oxadiazole Grafts in Peptide Macrocycles. Nat. Chem 2016, 8, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Kaldas SJ; Yudin AK Achieving Skeletal Diversity in Peptide Macrocycles through The Use of Heterocyclic Grafts. Chem. Eur. J 2018, 24 (28), 7074–7082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Estrada-Ortiz N; Neochoritis CG; Twarda-Clapa A; Musielak B; Holak TA; Dömling A Artificial Macrocycles as Potent p53-MDM2 Inhibitors. ACS Med. Chem. Lett 2017, 8 (10), 1025–1030; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Goud EY; Rao BK; Thirupahi G; Hemasri Y; Rao CP; Kumar PV; Rao YJ Synthesis of Highly Z-Selective Coumarin Annulated Dioxocine, Dioxacindione and Macrocycles Using Grubbs’ Second-Generation Catalyst. ChemistrySelect 2017, 2 (3), 1170–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Granger BA; Brown DG Design and Synthesis of Peptide-based Macrocyclic Cyclophilin Inhibitors. Bioorganic Med. Chem. Lett 2016, 26 (21), 5304–5307. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Javed S; Bodugam M; Torres J; Ganguly A; Hanson PR Modular Synthesis of Novel Macrocycles Bearing α,β-Unsaturated Chemotypes through a Series of One-Pot, Sequential Protocols. Chem. Eur. J 2016, 22 (20), 6755–6758. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Grossmann A; Bartlett S; Janecek M; Hodgkinson JT; Spring DR Diversity-Oriented Synthesis of Drug-Like Macrocyclic Scaffolds Using an Orthogonal Organo- and Metal Catalysis Strategy. Angew. Chem. Int. Ed 2014, 53 (48), 13093–13097. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Cohrt AE; Nielsen TE Solid-Phase Synthesis of Peptide Thioureas and Thiazole-Containing Macrocycles through Ru-Catalyzed Ring-Closing Metathesis. ACS Comb. Sci 2014, 16 (2), 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Dandapani S; Lowe JT; Comer E; Marcaurelle LA Diversity-Oriented Synthesis of 13- to 18-Membered Macrolactams via Ring-Closing Metathesis. J. Org. Chem 2011, 76 (19), 8042–8048. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Tannert R; Milroy L-G; Ellinger B; Hu T-S; Arndt H-D; Waldmann H Synthesis and Structure-Activity Correlation of Natural-Product Inspired Cyclodepsipeptides Stabilizing F-Actin. J. Am. Chem. Soc 2010, 132 (9), 3063–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Oikawa M; Naito S; Sasaki M Skeletal Diversity by Allylation/RCM On Ugi Four-Component Coupling Reaction Products. Tetrahedron Lett. 2006, 47 (27), 4763–4767. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Maurya SK; Rana R An Eco-Compatible Strategy For The Diversity-Oriented Synthesis Of Macrocycles Exploiting Carbohydrate-Derived Building Blocks. Beilstein J. Org. Chem 2017, 13, 1106–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Yu M; Lou S; Gonzalez-Bobes F Ring-Closing Metathesis in Pharmaceutical Development: Fundamentals, Applications, and Future Directions. Org. Process Res. Dev 2018, 22 (8), 918–946. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Chamakuri S; Guduru SKR; Pamu S; Chandrasekar G; Kitambi SS; Arya P A Modular Approach to Build Macrocyclic Diversity in Aminoindoline Scaffolds Identifies Antiangiogenesis Agents from a Zebrafish Assay. Eur. J. Org. Chem 2013, 19, 3959–3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Isidro-Llobet A; Hadje Georgiou K; Galloway WRJD; Giacomini E; Hansen MR; Mendez-Abt G; Tan YS; Carro L; Sore HF; Spring DR A Diversity-Oriented Synthesis Strategy Enabling The Combinatorial-Type Variation of Macrocyclic Peptidomimetic Scaffolds. Org. Biomol. Chem 2015, 13 (15), 4570–4580. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) O'Connell KMG; Beckmann HSG; Laraia L; Horsley HT; Bender A; Venkitaraman AR; Spring DR A Two-Directional Strategy for the Diversity-Oriented Synthesis of Macrocyclic Scaffolds. Org. Biomol. Chem 2012, 10 (37), 7545–7551. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Isidro-Llobet A; Murillo T; Bello P; Cilibrizzi A; Hodgkinson JT; Galloway WRJD; Bender A; Welch M; Spring DR Diversity-Oriented Synthesis of Macrocyclic Peptidomimetics. Proc. Natl. Acad. Sci. U.S.A 2011, 108 (17), 6793–6798. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Pasini D The Click Reaction as an Efficient Tool for the Construction of Macrocyclic Structures. Molecules 2013, 18 (8), 9512–9530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) Rivera DG; Vercillo OE; Wessjohann LA, Rapid Generation of Macrocycles with Natural-Product-like Side Chains by Multiple multicomponent Macrocyclizations (Mibs). Org. Biomol. Chem 2008, 6 (10), 1787–1795. [DOI] [PubMed] [Google Scholar]; (b) Rivera DG; Vercillo OE; Wessjohann LA, Combinatorial Synthesis of Macrocycles by Multiple Multicomponent Macrocyclization Including Bifunctional Building Blocks (MiB). Synlett 2007, 2007 (2), 0308–0312. [DOI] [PubMed] [Google Scholar]; (c) Reguera L; Rivera DG Multicomponent Reaction Toolbox for Peptide Macrocyclization and Stapling. Chem. Rev 2019, 119 (17), 9836–9860. [DOI] [PubMed] [Google Scholar]; (d) Zaretsky S; Tan J; Hickey JL; Yudin AK, Macrocyclic Templates for Library Synthesis of Peptido-Conjugates. In Peptide Libraries: Methods and Protocols, Derda R, Ed. Springer New York: New York, NY, 2015; pp 67–80. [DOI] [PubMed] [Google Scholar]; (e) Wessjohann LA; Neves RAW; Puentes AR; Morejon MC Macrocycles from Multicomponent Reactions. In Multicomponent Reactions in Organic Synthesis; Zhu J; Wang Q; WAng M-X Ed.; Wiley-VCH Verlag GmbH & Co. KGaA, 2015; pp 231–264. [DOI] [PubMed] [Google Scholar]; (f) Wessjohann LA; Rivera DG; Vercillo OE Multiple Multicomponent Macrocyclizations (MiBs): A Strategic Development Toward Macrocycle Diversity. Chem. Rev 2009, 109 (2), 796–814. [DOI] [PubMed] [Google Scholar]; (g) Madhavachary R; Abdelraheem EMM; Rossetti A; Twarda-Clapa A; Musielak B; Kurpiewska K; Kalinowska-Tluscik J; Holak TA; Domling A Two-Step Synthesis of Complex Artificial Macrocyclic Compounds. Angew. Chem. Int. Ed 2017, 56 (36), 10725–10729. [DOI] [PubMed] [Google Scholar]; (h) Liao GP; Abdelraheem EMM; Neochoritis CG; Kurpiewska K; Kalinowska-Tluscik J; McGowan DC; Domling A Versatile Multicomponent Reaction Macrocycle Synthesis Using alpha-Isocyano-omega-carboxylic Acids. Org. Lett 2015, 17 (20), 4980–4983. [DOI] [PubMed] [Google Scholar]; (i) Abdelraheem EMM; Khaksar S; Kurpiewska K; Kalinowska-Tluscik J; Shaabani S; Domling A Two-Step Macrocycle Synthesis by Classical Ugi Reaction. J. Org. Chem 2018, 83 (3), 1441–1447. [DOI] [PubMed] [Google Scholar]; (j) Ricardo MG; Morales FE; Garay H; Reyes O; Vasilev D; Wessjohann LA; Rivera DG Bidirectional macrocyclization of peptides by double multicomponent reactions. Org. Biomol. Chem 2015, 13, 438–446. [DOI] [PubMed] [Google Scholar]; (k) Abdelraheem EMM; Kurpiewska K; Kalinowska-Tluscik J; Domling A Artificial Macrocycles by Ugi Reaction and Passerini Ring Closure. J. Org. Chem 2016, 81 (19), 8789–8795. [DOI] [PubMed] [Google Scholar]
- (11).(a) Lin H; Bruhn DF; Maddox MM; Singh AP; Lee RE; Sun D Synthesis and Antibacterial Evaluation of Macrocyclic Diarylheptanoid Derivatives. Bioorganic Med. Chem. Lett 2016, 26 (16), 4070–4076. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Comer E; Liu H; Joliton A; Clabaut A; Johnson C; Akella LB; Marcaurelle LA Fragment-Based Domain Shuffling Approach for The Synthesis of Pyran-Based Macrocycles. Proc. Natl. Acad. Sci. U.S.A 2011, 108 (17), 6751–6756. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jefferson EA; Arakawa S; Blyn LB; Miyaji A; Osgood SA; Ranken R; Risen LM; Swayze EE New Inhibitors of Bacterial Protein Synthesis from A Combinatorial Library of Macrocycles. J. Med. Chem 2002, 45 (16), 3430–3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).(a) Ciardiello JJ; Stewart HL; Sore HF; Galloway WRJD; Spring DR A Novel Complexity-to-Diversity Strategy for the Diversity-Oriented Synthesis of Structurally Diverse and Complex Macrocycles from Quinine. Bioorganic Med. Chem 2017, 25 (11), 2825–2843. [DOI] [PubMed] [Google Scholar]; (b) Ciardiello JJ; Galloway WRJD; O'Connor CJ; Sore HF; Stokes JE; Wu Y; Spring DR An Expedient Strategy for the Diversity-Oriented Synthesis of Macrocyclic Compounds with Natural Product-Like Characteristics. Tetrahedron 2016, 72 (25), 3567–3578. [DOI] [PubMed] [Google Scholar]; (c) Beckmann HSG; Nie F; Hagerman CE; Johansson H; Tan YS; Wilcke D; Spring DR A Strategy for the Diversity-Oriented Synthesis of Macrocyclic Scaffolds Using Multidimensional Coupling. Nat. Chem 2013, 5 (10), 861–867. [DOI] [PubMed] [Google Scholar]; (d) Marcaurelle LA; Comer E; Dandapani S; Duvall JR; Gerard B; Kesavan S; Lee MD; Liu H; Lowe JT; Marie J-C; Mulrooney CA; Pandya BA; Rowley A; Ryba TD; Suh B-C; Wei J; Young DW; Akella LB; Ross NT; Zhang Y-L; Fass DM; Reis SA; Zhao W-N; Haggarty SJ; Palmer M; Foley MA An Aldol-Based Build/Couple/Pair Strategy for the Synthesis of Medium- and Large-Sized Rings: Discovery of Macrocyclic Histone Deacetylase Inhibitors. J. Am. Chem. Soc 2010, 132 (47), 16962–16976. [DOI] [PubMed] [Google Scholar]; (e) Nie F; Kunciw DL; Wilcke D; Stokes JE; Galloway WRJD; Bartlett S; Sore HF; Spring DR A Multidimensional Diversity-Oriented Synthesis Strategy for Structurally Diverse and Complex Macrocycles. Angew. Chem. Int. Ed 2016, 55 (37), 11139–11143. [DOI] [PubMed] [Google Scholar]; (f) Zakharova EA; Shmatova OI; Kutovaya IV; Khrustalev VN; Nenajdenko VG Synthesis of Macrocyclic Peptidomimetics via the Ugi-Click-Strategy. Org. Biomol. Chem 2019, 17, 3433–3445. [DOI] [PubMed] [Google Scholar]; (g) Salvador CEM; Pieber B; Neu PM; Torvisco A; Andrade CKZ; Kappe CO A Sequential Ugi Multicomponent/Cu-Catalyzed Azide-Alkyne Cycloaddition Approach for the Continuous Flow Generation of Cyclic Peptoids. J. Org. Chem 2015, 80 (9), 4590–4602. [DOI] [PubMed] [Google Scholar]; (h) Chavez-Acevedo L; Miranda LD Synthesis of Novel Tryptamine-Based Macrocycles Using an Ugi 4-CR/Microwave Assisted Click-Cycloaddition Reaction Protocol. Org. Biomol. Chem 2015, 13, 4408–4412. [DOI] [PubMed] [Google Scholar]; (i) Pehere AD; Zhang X; Abell AD Macrocyclic Peptidomimetics Prepared by Ring-Closing Metathesis and Azide-Alkyne Cycloaddition. Aust. J. Chem 2016, 70 (2), 138–151. [DOI] [PubMed] [Google Scholar]; (j) Fitzgerald ME; Mulrooney CA; Duvall JR; Wei J; Suh B-C; Akella LB; Vrcic A; Marcaurelle LA Build/Couple/Pair Strategy for the Synthesis of Stereochemically Diverse Macrolactams via Head-to-Tail Cyclization. ACS Comb. Sci 2012, 14 (2), 89–96. [DOI] [PubMed] [Google Scholar]
- (13).(a) Bedard AC; Collins SK Microwave Accelerated Glaser-Hay Macrocyclizations at High Concentrations. Chem. Commun 2012, 48, 6420–6422. [DOI] [PubMed] [Google Scholar]; (b) Godin E; Thanh SN; Guerrero-Morales J; Santandrea J; Caron A; Minozzi C; Beaucage N; Rey B; Morency M; Abel-Snape X; Collins SK Synthesis and Diversification of Macrocyclic Alkynediyl Sulfide Peptides. Chem. Eur. J 2020, 26 (64), 14575–14579. [DOI] [PubMed] [Google Scholar]
- (14).(a) Dong X; Wang Q; Zhang Q; Xu S; Wang Z Construction of Dihydropyran-Bridged Macrocycles by Inverse-Electron-Demand Diels-Alder Reaction. Tetrahedron 2013, 69 (52), 11144–11154. [Google Scholar]; (b) Acharya A; Eickhoff JA; Chen K; Catalano VJ; Jeffrey CS Access to Bicyclic Hydroxamate Macrocycles via Intramolecular Aza-(4 + 3) Cyloaddition Reactions of Aza-Oxyallylic Cation Intermediates. Org. Chem. Front 2016, 3, 330–334. [Google Scholar]; (c) Prasanna R; Purushothaman S; Raghunathan R Rapid Assembly of Heterocycle Grafted Macrocycles via Tandem One-Pot Double 1,3-Dipolar Cycloaddition Reaction. Org. Biomol. Chem 2014, 12, 9375–9383.25318016 [Google Scholar]; (d) Prabhakaran P; Rajakumar P Regio- and Stereoselective Synthesis of Spiropyrrolidine-Oxindole and Bis-Spiropyrrolizidine-Oxindole Grafted Macrocycles through [3+2] Cycloaddition of Azomethine Ylides. RSC Adv. 2020, 10, 10263–10276. [Google Scholar]
- (15).(a) Dong H; Limberakis C; Liras S; Price D; James K, Peptidic Macrocyclization via Palladium-Catalyzed Chemoselective Indole C-2 Arylation. Chem. Commun 2012, 48 (95), 11644–11646; [DOI] [PubMed] [Google Scholar]; (b) Li B; Li X; Han B; Chen Z; Zhang X; He G; Cheng G Construction of Natural-Product-Like Cyclophane-Braced Peptide Macrocycles via sp3 C−H Arylation. J. Am. Chem. Soc 2019, 141 (23), 9401−9407; [DOI] [PubMed] [Google Scholar]; (c) Li X; Qi Liping; Li B; Zhao Z; He G; Chen G. Synthesis of Cyclophane-Braced Peptide Macrocycles via Palladium-Catalyzed Intramolecular C(sp3)-H Arylation of N-Methyl Alanine at C-Termini. Org. Lett 2020, 22 (15), 6209–6213. [DOI] [PubMed] [Google Scholar]; (d) Krieger J-P; Ricci G; Lesuisse D; Meyer C; Cossy J, Harnessing C−H Activation of Benzhydroxamates as a Macrocyclization Strategy: Synthesis of Structurally Diverse Macrocyclic Isoquinolones. Chem. Eur. J 2016, 22 (38), 13469–13473. [DOI] [PubMed] [Google Scholar]; (e) Han B; Li B; Qi L; Yang P; He G; Chen G Construction of Cyclophane-Braced Peptide Macrocycles via Palladium-Catalyzed Picolinamide-Directed Intramolecular C(sp2)-H Arylation. Org. Lett 2020, 22 (17), 6879–6883. [DOI] [PubMed] [Google Scholar]; (f) Zhang X; Lu G; Sun M; Mahankali M; Ma Y; Zhang M; Hua W; Hu Y; Wang Q; Chen J; He G; Qi X; Shen W; Liu P; Chen G A General Strategy for Synthesis of Cyclophane-Braced Peptide Macrocycles via Palladium-Catalysed Intramolecular sp3 C-H Arylation. Nature Chem. 2018, 10, 540–548. [DOI] [PubMed] [Google Scholar]; (g) Sengupta S; Mehta G Macrocyclization via C-H Functionalization: A New Paradigm in Macrocycle Synthesis. Org. Biomol. Chem 2020, 18, 1851–1876. [DOI] [PubMed] [Google Scholar]; (h) Zeng Q; Dong K; Pei C; Dong S; Hu W; Qiu L; Xu X Divergent Construction of Macrocyclic Alkynes via Catalytic Metal Carbene C(sp2)-H Insertion and the Buchner Reaction. ACS Catal. 2019, 9 (12), 10773–10779. [DOI] [PubMed] [Google Scholar]; (i) Krieger J-P; Lesuisse D; Ricci G; Perrin M-A; Meyer C; Cossy J Rhodium(III)-Catalyzed C-H Activation/Heterocyclization as a Macrocyclization Strategy. Synthesis of Macrocyclic Pyridones. Org. Lett 2017, 19 (10), 2706–2709. [DOI] [PubMed] [Google Scholar]; (j) Tang J; He Y; Chen H; Sheng W; Wang H Synthesis of Bioactive And Stabilized Cyclic Peptides by Macrocyclization Using C(sp3)-H Activation. Chem. Sci 2017, 8, 4565–4570. [DOI] [PubMed] [Google Scholar]; (k) Niu T-F; Sun M; Lv M-F; Yi W-B; Cai C Synthesis of Highly Functionalized Macrocycles by Tandem Multicomponent Reactions and Intramolecular Sonogashira Cross-Coupling. Org. Biomol. Chem 2013, 11, 7232–7238. [DOI] [PubMed] [Google Scholar]; (l) Perez-Labrada K; Cruz-Mendoza MA; Chavez-Riveros A; Hernandez-Vazquez E; Torroba T; Miranda LD Diversity-Oriented Synthesis and Cytotoxic Activity Evaluation of Biaryl-Containing Macrocycles. Org. Biomol. Chem 2017, 15, 2450–2458. [DOI] [PubMed] [Google Scholar]; (m) Bai Z; Cai C; Yu Z; Wang H Backbone-Enabled Directional Peptide Macrocyclization through Late-Stage Palladium-Catalyzed delta-C(sp2)-H Olefination. Angew. Chem. Int. Ed 2018, 57 (42), 13912–13916. [DOI] [PubMed] [Google Scholar]; (n) Aeluri M; Gaddam J; Trinath DVKS; Chandrasekar G; Kitambi SS; Arya P An Intramolecular Heck Approach To Obtain 17-Membered Macrocyclic Diversity and the Identification of an Antiangiogenesis Agent from a Zebrafish Assay. Eur. J. Org. Chem 2013, 19, 3955–3958. [DOI] [PubMed] [Google Scholar]; (o) Hussain A; Yousuf SK; Kumar D; Lambu M; Singh B; Maity S; Mukherjee D Intramolecular Base-Free Sonogashira Reaction for the Synthesis of Benzannulated Chiral Macrocycles Embedded in Carbohydrate Templates. Adv. Synth. Catal 2012, 354 (10), 1933–1940. [DOI] [PubMed] [Google Scholar]; (p) Lawson KV; Rose TE; Harran PG Template-Induced Macrocycle Diversity through Large Ring-Forming Alkylations of Tryptophan. Tetrahedron 2013, 69, 7683−7691. [DOI] [PubMed] [Google Scholar]; (q) Lawson KV; Rose TE; Harran PG Template-Constrained Macrocyclic Peptides Prepared from Native, Unprotected Precursors. Proc. Natl. Acad. Sci. U. S. A 2013, 110, E3753−E3760. [DOI] [PubMed] [Google Scholar]; (r) Hopkins BA; Smith GF; Sciammetta N Synthesis of Cyclic Peptidomimetics via a Pd-Catalyzed Macroamination Reaction. Org. Lett 2016, 18 (16), 4072–4075. [DOI] [PubMed] [Google Scholar]; (s) Zhao H; Negash L; Wei Q; LaCour TG; Estill SJ; Capota E; Pieper AA; Harran PG Acid Promoted Cinnamyl Ion Mobility within Peptide Derived Macrocycles. J. Am. Chem. Soc 2008, 130, 13864−13866. [DOI] [PubMed] [Google Scholar]; (t) Hu CH; Valente MWN; Halpern OS; Jusuf S; Khan JA; Locke GA; Duke GJ; Liu X; Duclos FJ; Wexler RR; Kick EK; Smallheer JM Small molecule and macrocyclic pyrazole derived inhibitors of myeloperoxidase (MPO). Bioorg. Med. Chem. Lett 2021, 42, 128010. [DOI] [PubMed] [Google Scholar]
- (16).(a) Raynal L; Rose NC; Donald JR; Spicer CD Photochemical Methods for Peptide Macrocyclisation. Chem. Eur. J 2021, 27 (1), 69–88. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lee H; Boyer NC; Deng Q; Kim H-Y; Sawyer TK; Sciammetta N Photoredox Ni-Catalyzed Peptide C(sp2)-O Cross-Coupling: from Intermolecular Reactions to Side Chain-to-Tail Macrocyclization. Chem. Sci 2019, 10, 5073–5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).(a) Gueret SM; Meier P; Roth H-J Cyclic Carbo-Isosteric Depsipeptides and Peptides as a Novel Class of Peptidomimetics. Org. Lett 2014, 16 (5), 1502–1505. [DOI] [PubMed] [Google Scholar]; (b) Zhou J; Reidy M; O'Reilly C; Jarikote DV; Negi A; Samali A; Szegezdi E; Murphy PV Decorated Macrocycles via Ring-Closing Double-Reductive Amination. Identification of an Apoptosis Inducer of Leukemic Cells That at Least Partially Antagonizes a 5-HT2 Receptor. Org. Lett 2015, 17 (7), 1672–1675. [DOI] [PubMed] [Google Scholar]
- (18).Mothukuri GK; Kale SS; Stenbratt CL; Zorzi A; Vesin J; Chapalay JB; Deyle K; Turcatti G; Cendron L; Angelini A; Heinis C Macrocycle Synthesis Strategy Based on Stepwise Adding and Reacting Three Components Enables Screening of Large Combinatorial Libraries. Chem. Sci 2020, 11, 7858–7863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Rose TE; Lawson KV; Harran PG Large Ring-Forming Alkylations Provide Facile Access to Composite Macrocycles. Chem. Sci 2015, 6, 2219–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Collins JC; Farley KA; Limberakis C; Liras S; Price D; James K Macrocyclizations for Medicinal Chemistry: Synthesis of Druglike Macrocycles by High-Concentration Ullmann Coupling. J. Org. Chem 2012, 77 (24), 11079–11090. [DOI] [PubMed] [Google Scholar]
- (21).Muthusamy S; Karikalan T Rhodium(II) Catalyzed Synthesis of Macrocycles Incorporating Oxindole via O-H/N-H Insertion Reactions. Org. Biomol. Chem 2014, 12, 9243–9256. [DOI] [PubMed] [Google Scholar]
- (22).(a) Satyanarayana M; Vitali F; Frost JR; Fasan R, Diverse Organo-Peptide Macrocycles via A Fast and Catalyst-Free Oxime/Intein-Mediated Dual Ligation. Chem. Commun 2012, 48 (10), 1461–1463; [DOI] [PubMed] [Google Scholar]; (b) Roberts KD; Lambert JN; Ede NJ; Bray AM, Preparation of Cyclic Peptide Libraries Using Intramolecular Oxime Formation. J. Pept. Sci 2004, 10 (11), 659–665. [DOI] [PubMed] [Google Scholar]; (c) Sharma VR,; Mehmood A; Janesko BG; Simanek EE. Efficient Syntheses of Macrocycles Ranging From 22–28 Atoms through Spontaneous Dimerization to Yield Bis-Hydrazones. RSC Adv. 2020, 10, 3217–3220. [DOI] [PubMed] [Google Scholar]; (d) Hubert JG; Stepek IA; Noda H; Bode JW Synthetic Fermentation of Beta-Peptide Macrocycles by Thiadiazole-Forming Ring-Closing Reactions. Chem. Sci 2018, 9, 2159–2167. [DOI] [PubMed] [Google Scholar]; (e) Nefzi A; Arutyunyan S; Fenwick JE Two-Step Hantzsch Based Macrocyclization Approach for the Synthesis of Thiazole-Containing Cyclopeptides. J. Org. Chem 2010, 75 (22), 7939–7941. [DOI] [PubMed] [Google Scholar]; (f) Yuan T; Ye X; Zhao P; Teng S; Yi Y; Wang J; Shan C; Wojtas L; Jean J; Chen H; Shi X Regioselective Crossed Aldol Reactions under Mild Conditions via Synergistic Gold-Iron Catalysis. Chem. 2020, 6 (6), 1420–1431. [DOI] [PubMed] [Google Scholar]
- (23).Zeng Q; He C; Zhou S; Dong K; Qiu L; Xu X Dirhodium(II)-Catalyzed Cyclopropanation of Alkyne-Containing alpha-Diazoacetates for the Synthesis of Cycloalkynes. Adv. Synth. Catal 2020, 362 (15), 3137–3141. [Google Scholar]
- (24).(a) Ronson TO; Taylor RJK; Fairlamb IJS Palladium-Catalysed Macrocyclisations in the Total Synthesis of Natural Products. Tetrahedron, 2015, 71 (7), 989–1009. [Google Scholar]; (b) Zheng K; Hong R Stereoconfining Macrocyclizations in the Total Synthesis of Natural Products. Nat. Prod. Rep 2019, 36, 1546–1575.30758359 [Google Scholar]; (c) Manda SLK; Tripathi S; Ghoshal A; Ambule MD; Srivastava AK; Panda G A Comparative Synthetic Strategy Perspective on alpha-Amino Acid- and Non-Amino Acid-Derived Synthons towards Total Syntheses of Selected Natural Macrolides. Chem. Eur. J 2020, 26 (23), 5131–5156.31846112 [Google Scholar]
- (25).Shang S; Tan DS, Advancing Chemistry and Biology Through Diversity-Oriented Synthesis of Natural Product-Like Libraries. Curr. Opin. Chem. Biol 2005, 9 (3), 248–258. [DOI] [PubMed] [Google Scholar]
- (26).(a) Yang L-L; Ouyang J; Zou H-N; Zhu S-F; Zhou Q-L Enantioselective Insertion of Alkynyl Carbenes into Si–H Bonds: An Efficient Access to Chiral Propargylsilanes and Allenylsilanes. J. Am. Chem. Soc 2021, 143 (17), 6401–6406. [DOI] [PubMed] [Google Scholar]; (b) Choi K; Park H; Lee C Rhodium-Catalyzed Tandem Addition-Cyclization-Rearrangement of Alkynylhydrazones with Organoboronic Acids. J. Am. Chem. Soc 2018, 140 (33), 10407–10411. [DOI] [PubMed] [Google Scholar]
- (27).(a) Barluenga J; Valdes C Tosylhydrazones: New Uses for Classic Reagents in Palladium-Catalyzed Cross-Coupling and Metal-Free Reactions. Angew. Chem. Int. Ed 2011, 50 (33), 7486–7500. [DOI] [PubMed] [Google Scholar]; (b) Arunprasath D; Devi Bala B; Sekar G Luxury of N-Tosylhydrazones in Transition-Metal-Free Transformations. Adv. Synth. Catal 2019, 361 (6), 1172–1207. [DOI] [PubMed] [Google Scholar]; (c) Wang H; Deng Y-H; Shao Z An Update of N-Tosylhydrazones: Versatile Reagents for Metal-Catalyzed and Metal-Free Coupling Reactions. Synthesis 2018, 50 (12), 2281–2306. [DOI] [PubMed] [Google Scholar]; (d) Xia Y; Wang J N-Tosylhydrazones: Versatile Synthons in the Construction of Cyclic Compounds. Chem. Soc. Rev 2017, 46, 2306–2362. [DOI] [PubMed] [Google Scholar]; (e) Shao Z; Zhang H N-Tosylhydrazones: Versatile Reagents for Metal-Catalyzed and Metal-Free Cross-Coupling Reactions. Chem. Soc. Rev 2012, 41 (2), 560–572. [DOI] [PubMed] [Google Scholar]
- (28).(a) Shapiro RH; Heath MJ Tosylhydrazones. V. Reaction of Tosylhydrazones with Alkyllithium Reagents. A New Olefin Synthesis. J. Am. Chem. Soc 1967, 89 (22), 5734–5735. [Google Scholar]; (b) Zhang Y; Wang J Alkene Synthesis Through Transition Metal-Catalyzed Cross-Coupling of N-Tosylhydrazones. In Stereoselective Alkene Synthesis. Topics in Current Chemistry; Wang J Ed.; Springer, Berlin, Heidelberg, 2012, pp 239–269. [Google Scholar]
- (29).(a) Mundal DA; Lutz KE; Thomson RJ A Direct Synthesis of Allenes by a Traceless Petasis Reaction. J. Am. Chem. Soc 2012, 134 (13), 5782−5785. [DOI] [PubMed] [Google Scholar]; (b) Barroso R; Cabal MP; Valdes C Pd-catalyzed Auto-Tandem Cascades Based on N-Sulfonylhydrazones: Hetero- and Carbocyclization Processes. Synthesis, 2017, 49 (19), 4434–4447. [DOI] [PubMed] [Google Scholar]
- (30).Jadhav AP; Ray D; Rao VUB; Singh RP Copper-Catalyzed Direct Cross-Coupling of Compounds Containing Activated C–H/Heteroatom–H Bonds with N-Tosylhydrazones. Eur. J. Org. Chem 2016, 14, 2369–2382. [Google Scholar]
- (31).(a) Xia Y; Wang J Transition-Metal-Catalyzed Cross-Coupling with Ketones or Aldehydes via N-Tosylhydrazones. J. Am. Chem. Soc 2020, 142 (24), 10592–10605. [DOI] [PubMed] [Google Scholar]; (b) Qiu D; Mo F; Zhang Y; Wang J Chapter Two - Recent Advances in Transition-Metal-Catalyzed Cross-Coupling Reactions With N-Tosylhydrazones. Adv. Organomet. Chem 2017, 67, 151–219. [DOI] [PubMed] [Google Scholar]; (c) Liu Z; Wang J Cross-Coupling Reactions Involving Metal Carbene: From C=C/C-C Bond Formation to C-H Bond Functionalization. J. Org. Chem 2013, 78 (20), 10024–10030. [DOI] [PubMed] [Google Scholar]; (d) Xiao Q; Zhang Y; Wang J Diazo Compounds and N-Tosylhydrazones: Novel Cross-Coupling Partners in Transition-Metal-Catalyzed Reactions. Acc. Chem. Res 2013, 46 (2), 236–247. [DOI] [PubMed] [Google Scholar]
- (32).(a) Mundal DA; Lutz KE; Thomson RJ A Direct Synthesis of Allenes by a Traceless Petasis Reaction. J. Am. Chem. Soc 2012, 134 (13), 5782–5785. [DOI] [PubMed] [Google Scholar]; (b) Allwood DM; Blakemore DC; Brown AD; Ley SV Metal-Free Coupling of Saturated Heterocyclic Sulfonylhydrazones with Boronic Acids. J. Org. Chem 2014, 79 (1), 328–338. [DOI] [PubMed] [Google Scholar]; (c) Jiang Y; Thomson RJ; Schaus SE Asymmetric Traceless Petasis Borono-Mannich Reactions of Enals: Reductive Transposition of Allylic Diazenes. Angew. Chem. Int. Ed 2017, 56 (52), 16631–16635. [DOI] [PubMed] [Google Scholar]; (d) Wang J When Diazo Compounds Meet with Organoboron Compounds. Pure Appl. Chem 2018, 90 (4), 617–623. [DOI] [PubMed] [Google Scholar]; (e) Merchant RR; Lopez JA A General C(sp3)–C(sp3) Cross-Coupling of Benzyl Sulfonylhydrazones with Alkyl Boronic Acids. Org. Lett 2020, 22 (6), 2271–2275. [DOI] [PubMed] [Google Scholar]; (f) Yang Y; Tsien J; David AB; Hughes JME; Merchant RR; Qin T. Practical and Modular Construction of C(sp3)-Rich Alkyl Boron Compounds. J. Am. Chem. Soc 2021, 143 (1), 471–480. [DOI] [PubMed] [Google Scholar]
- (33).(a) Martí-Centelles V; Pandey MD; Burguete MI; Luis SV Macrocyclization Reactions: The Importance of Conformational, Configurational, and Template-Induced Preorganization. Chem. Rev 2015, 115 (16), 8736–8834. [DOI] [PubMed] [Google Scholar]; (b) Jung ME; Piizzi G gem-Disubstituent Effect: Theoretical Basis and Synthetic Applications. Chem. Rev 2005, 105 (5), 1735–1766. [DOI] [PubMed] [Google Scholar]; (c) Blankenstein J; Zhu J Conformation-Directed Macrocyclization Reactions. Eur. J. Org. Chem 2005, 10, 1949–1964. [DOI] [PubMed] [Google Scholar]; (d) Gadais C; Van holsbeeck K; Moors SLC; Buyst D; Feher K; Van Hecke K; Tourwe ED; Brigaud T; Martin C; De Proft F; Pytowicz J; Martins JC; Chaume G; Ballet S Trifluoromethylated Proline Surrogates as Part of “Pro–Pro” Turn Inducing Templates. ChemBioChem 2019, 20 (19), 2513–2518. [DOI] [PubMed] [Google Scholar]
- (34).(a) Mrksich M; Parks ME; Dervan PB, Motif Hairpin Peptide. A New Class of Oligopeptides for Sequence-Specific Recognition in the Minor Groove of Double-Helical DNA. J. Am. Chem. Soc 1994, 116 (18), 7983–7988. [Google Scholar]; (b) Herman DM; Baird EE; Dervan PB Stereochemical Control of the DNA Binding Affinity, Sequence Specificity, and Orientation Preference of Chiral Hairpin Polyamides in the Minor Groove. J. Am. Chem. Soc 1998, 120 (7), 1382–1391. [Google Scholar]; (c) Li BC; Montgomery DC; Puckett JW; Dervan PB Synthesis of Cyclic Py-Im Polyamide Libraries. J. Org. Chem, 2013, 78 (1), 124–133.23106218 [Google Scholar]; (d) Meier JL; Montgomery DC; Dervan PB Enhancing the cellular uptake of Py–Im polyamides through next-generation aryl turns. Nucleic Acids Res. 2012, 40 (5), 2345–2356.22080545 [Google Scholar]; (e) Chenoweth DM; Dervan PB Allosteric modulation of DNA by small molecules. Proc. Natl. Acad. Sci. U. S. A 2009, 106 (32), 13175–13179.19666554 [Google Scholar]; (f) Chenoweth DM; Harki DA; Phillips JW; Dose C; Dervan PB Cyclic Pyrrole-Imidazole Polyamides Targeted to the Androgen Response Element. J. Am. Chem. Soc 2009, 131 (20), 7182–7188.19413319 [Google Scholar]
- (35).(a) Barluenga J; Tomas-Gamasa M; Aznar F; Valdes C Synthesis of Sulfones by Iron-Catalyzed Decomposition of Sulfonylhydrazones. Eur. J. Org. Chem 2011. (8), 1520–1526. [Google Scholar]; (b) Zhang J-L; Chan PWH; Che C-M Ruthenium(II) Porphyrin Catalyzed Cyclopropanation of Alkenes with Tosylhydrazones. Tetrahedron Lett. 2003, 44 (48), 8733–8737. [Google Scholar]; (c) Feng XW; Wang J; Zhang J; Yang J; Wang N; Yu X-Q Copper-Catalyzed Nitrogen Loss of Sulfonylhydrazones: A Reductive Strategy for the Synthesis of Sulfones from Carbonyl Compounds. Org. Lett 2010, 12 (19), 4408–4411.20812669 [Google Scholar]; (d) Chandrasekhar S; Rajaiah G; Chandraiah L; Swamy DN Direct Conversion of Tosylhydrazones to tert-Butyl Ethers under Bamford-Stevens Reaction Conditions. Synlett 2001. (11), 1779–1780. [Google Scholar]; (e) Barluenga J; Tomas-Gamasa M; Aznar F; Valdes C Straightforward Synthesis of Ethers: Metal-Free Reductive Coupling of Tosylhydrazones with Alcohols or Phenols. Angew. Chem. Int. Ed 2010, 49 (29), 4993–4996. [Google Scholar]; (f) Merchant RR; Allwood DM; Blakemore DC; Ley SV Regioselective Preparation of Saturated Spirocyclic and Ring-Expanded Fused Pyrazoles. J. Org. Chem 2014, 79 (18), 8800–8811.25148419 [Google Scholar]
- (36).(a) Comer E; Rohan E; Deng L; Porco JA An Approach to Skeletal Diversity Using Functional Group Pairing of Multi-functional Scaffolds. Org. Lett 2007, 9 (11), 2123–2126. [DOI] [PubMed] [Google Scholar]; (b) Serba C; Winssinger N Following the Lead from Nature: Divergent Pathways in Natural Product Synthesis and Diversity-Oriented Synthesis. Eur. J. Org. Chem 2013, 20, 4195–4214. [DOI] [PubMed] [Google Scholar]; (c) Muthusamy S; Selvaraj K; Suresh E Demonstration of 11–21-Membered Intramolecular Sul-fonium Ylides: Regio- and Diastereoselective Synthesis of Spiro-Oxindole-Incorporated Macro-cycles. Eur. J. Org. Chem 2016, 10, 1849–1859. [DOI] [PubMed] [Google Scholar]; (d) Zeng Q; Dong K; Pei C; Dong S; Hu W; Qiu L; Xu X Divergent Construction of Macrocyclic Alkynes via Catalytic Metal Carbene C(sp2)-H Insertion and the Buchner Reaction. ACS Catal. 2019, 9(12), 10773–10779. [DOI] [PubMed] [Google Scholar]; (e) Lawson KV; Rose TE; Harran PG Template-Induced Macrocycle Diversity through Large Ring-Forming Alkylations of Tryptophan. Tetrahedron 2013, 69, 7683–7691. [DOI] [PubMed] [Google Scholar]; (f) Shareef A-R; Sherman DH; Montgomery, Nickel-Catalyzed Regiodivergent Approach to Macrolide Motifs. Chem. Sci 2012, 3(3), 892–895. [DOI] [PubMed] [Google Scholar]
- (37).(a) Dehaen W; Bakulev VA Chemistry of 1,2,3-triazoles, Springer, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chuprakov S; Hwang FW; Gevorgyan V Transannulation of Pyridotriazoles with Alkynes and Nitriles, Angew. Chem. Int. Ed 2007, 46 (25), 4757–4759. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yadagiri D; Rivas M; Gevorgyan V Denitrogenative Transformations of Pyridotriazoles and Related Compounds: Synthesis of N-Containing Heterocyclic Compounds and Beyond. J. Org. Chem 2020, 85 (17), 11030–11046. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Shi Y; Gulevich AV; Gevorgyan V Rhodium-Catalyzed NH Insertion of Pyridyl Carbenes Derived from Pyridotriazoles: A General and Efficient Approach to 2-Picolylamines and Imidazo[1,5-a]pyridines. Angew. Chem. Int. Ed 2014, 53 (51), 14191–14195. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zhang Z; Gevorgyan V Co-Catalyzed Transannulation of Pyridotriazoles with Isothiocyanates and Xanthate Esters. Org. Lett 2020, 22 (21), 8500–8504. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Shi. Y.; Gevorgyan, V. Cu-catalyzed transannulation reaction of pyridotriazoles: general access to fused polycyclic indolizines. Chem. Commun 2015, 51, 17166–17169. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Helan V; Gulevich AV; Gevorgyan V Cu-catalyzed transannulation reaction of pyridotriazoles with terminal alkynes under aerobic conditions: efficient synthesis of indolizines. Chem. Sci 2015, 6, 1928–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Zhang Z; Yadagiri D; Gevorgyan V Light-induced metal-free transformations of unactivated pyridotriazoles. Chem. Sci 2019, 10, 8399–8404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).:(a) Wei W-X; Li Y; Wen Y-T; Li M; Li X-S; Wang C-T; Liu H-C; Xia Y; Zhang B-S; Jiao R-Q; Liang Y-M Experimental and Computational Studies of Palladium-Catalyzed Spirocyclization via a Narasaka–Heck/C(sp3 or sp2)–H Activation Cascade Reaction. J. Am. Chem. Soc 2021, 143 (20), 7868–7875. For Rh-catalyzed spirocyclization C-H activation, see:33974798(b) Le PQ; May JA Hydrazone-Initiated Carbene/Alkyne Cascades to Form Polycyclic Products: Ring-Fused Cyclopropenes as Mechanistic Intermediates. J. Am. Chem. Soc 2015, 137 (38), 12219–12222.26394217
- (39).:(a) Wenkert E; Davis LL; Mylari BL; Solomon MF; da Silva RR; Shulman S; Wamet RJ Cyclopentanone Synthesis by Intramolecular Carbon-Hydrogen Insertion of Diazo Ketones. A Diterpene-to-Steroid Skeleton Conversion. J. Org. Chem 1982, 47 (17), 3242–3247.(b) Taber DF; Petty EH General Route to Highly Functionalized Cyclopentane Derivatives by Intramolecular C-H Insertion. J. Org. Chem 1982, 47 (24), 4808–4809.(c) Taber DF; Hennessy MJ; Louey JP Rh-Mediated Cyclopentane Construction Can Compete with β-Hydride Elimination: Synthesis of (±)-Tochuinyl Acetate. J. Org. Chem 1992, 57 (2), 436–441.Representive example of intermolecular C-H insertion:(d) Liu W; Ren Z; Bosse AT; Liao K; Goldstein EL; Bacsa J; Musaev DG; Stoltz BM; Davies HML Catalyst-Controlled Selective Functionalization of Unactivated C–H Bonds in the Presence of Electronically Activated C–H Bonds. J. Am. Chem. Soc 2018, 140 (38), 12247–12255.30222321
- (40).Diver ST Ruthenium Vinyl Carbene Intermediates in Enyne Metathesis. Coord. Chem. Rev 2007, 251 (5), 671–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41). See supporting information for details.
- (42).Antilla JC; Baskin JM; Barder TE; Buchwald SL Copper−Diamine-Catalyzed N-Arylation of Pyrroles, Pyrazoles, Indazoles, Imidazoles, and Triazoles. J. Org. Chem 2004, 69 (17), 5578–5587. [DOI] [PubMed] [Google Scholar]
- (43).Lam PYS; Clark CG; Saubern S; Adams J; Winters MP; Chan DMT; Combs A New Aryl/Heteroaryl C-N Bond Cross-Coupling Reactions via Arylboronic Acid/Cupric Acetate Arylation. Tetrahedron Lett. 1998, 39 (19), 2941–2944. [Google Scholar]
- (44).(a) Soenen DR; Zimpleman JM; Boger DL Synthesis and Inverse Electron Demand Diels−Alder Reactions of 3,6-Bis(3,4-dimethoxybenzoyl)-1,2,4,5-tetrazine. J. Org. Chem 2003, 68 (9), 3593–3598. [DOI] [PubMed] [Google Scholar]; (b) Benson SC; Palabrica CA; Snyder JK Indole as A Dienophile in Inverse Electron Demand Diels-Alder Reactions. 5H-Pyridazino[4,5-B]Indoles as Cycloadducts with 3,6-Dicarbomethoxy-1,2,4,5-Tetrazine. J. Org. Chem 1987, 52 (20), 4610–4614. [DOI] [PubMed] [Google Scholar]
- (45).Karmakar R; Yun SY; Chen J; Xia Y; Lee D Benzannulation of Triynes to Generate Functionalized Arenes by Spontaneous Incorporation of Nucleophiles. Angew. Chem. Int. Ed 2015, 54, 6582–6586. [DOI] [PubMed] [Google Scholar]
- (46).Jacobi PA; Liu H Synthesis of 1,2,3,7,8,9-Hexahydrodipyrrins and Secocorrins: Important Precursors for the Construction of Corrins. Org. Lett 1999, 1, 341–344. [DOI] [PubMed] [Google Scholar]
- (47).Vadola PA; Sames D Catalytic Coupling of Arene C–H Bonds and Alkynes for the Synthesis of Coumarins: Substrate Scope and Application to the Development of Neuroimaging Agents. J. Org. Chem 2012, 77 (18), 7804–7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Breschi MC; Calderone V; Digiacomo M; Macchia M; Martelli A; Martinotti E; Minutolo F; Rapposelli S; Rossello A; Testai L; Balsamo A New NO-Releasing Pharmacodynamic Hybrids of Losartan and Its Active Metabolite: Design, Synthesis, and Biopharmacological Properties. J. Med. Chem 2006, 49 (8), 2628–2639. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.