

Abstract
Previous studies have shown that inhibition of L-type Ca2+ current (ICa) by cytosolic free Mg2+ concentration ([Mg2+]i) is profoundly affected by activation of cAMP-dependent protein kinase pathways. To investigate the mechanism underlying this counterregulation of ICa, rat cardiac myocytes and tsA201 cells expressing L-type Ca2+ channels were whole cell voltage-clamped with patch pipettes in which [Mg2+] ([Mg2+]p) was buffered by citrate and ATP. In tsA201 cells expressing wild-type Ca2+ channels (α1C/β2A/α2δ), increasing [Mg2+]p from 0.2 mM to 1.8 mM decreased peak ICa by 76 ± 4.5% (n = 7). Mg2+-dependent modulation of ICa was also observed in cells loaded with ATP-γ-S. With 0.2 mM [Mg2+]p, manipulating phosphorylation conditions by pipette application of protein kinase A (PKA) or phosphatase 2A (PP2A) produced large changes in ICa amplitude; however, with 1.8 mM [Mg2+]p, these same manipulations had no significant effect on ICa. With mutant channels lacking principal PKA phosphorylation sites (α1C/S1928A/β2A/S478A/S479A/α2δ), increasing [Mg2+]p had only small effects on ICa. However, when channel open probability was increased by α1C-subunit truncation (α1CΔ1905/β2A/S478A/S479A/α2δ), increasing [Mg2+]p greatly reduced peak ICa. Correspondingly, in myocytes voltage-clamped with pipette PP2A to minimize channel phosphorylation, increasing [Mg2+]p produced a much larger reduction in ICa when channel opening was promoted with BAY K8644. These data suggest that, around its physiological concentration range, cytosolic Mg2+ modulates the extent to which channel phosphorylation regulates ICa. This modulation does not necessarily involve changes in channel phosphorylation per se, but more generally appears to depend on the kinetics of gating induced by channel phosphorylation.
Keywords: voltage-gated Ca2+ channel, cardiac myocytes, human embryonic kidney cells, protein kinase A, protein phosphatase 2A
Changes in cytosolic free Mg2+ concentration ([Mg2+]i) have been found to have marked inhibitory effects on L-type Ca2+ current (ICa) (1, 18, 31, 41–43). Our recent study (37) also showed large changes in ICa around physiologically relevant [Mg2+]i. Mg2+ inhibition of ICa could occur by alterations of ion permeation rate and/or modulation of channel gating kinetics. Nonetheless, at this point, evidence for significant block of divalent cation permeation through L-type channels by physiologically relevant [Mg2+]i is lacking (25, 42, 44). Thus, at these concentrations, cytosolic Mg2+ is thought to modulate ICa predominantly by altering channel gating kinetics.
[Mg2+]i effects on ICa are very much dependent on channel phosphorylation state (1, 31, 37, 41, 44, 45). Although the mechanism by which channel phosphorylation affects [Mg2+]i actions on ICa is not clear, two alternatives have been proposed. [Mg2+]i could affect the level of channel phosphorylation by altering the activity of Mg2+-dependent kinases and phosphatases, and it could also modulate the effect of channel phosphorylation on gating kinetics. Previous studies (31) have shown that the former mechanism is possible. In addition, the latter has been discussed as an explanation for the results of Yamaoka and Seyama (44); however, direct experimental support for this mechanism is lacking.
The purpose of this study is to investigate the relationship between channel phosphorylation and [Mg2+]i modulation of L-type Ca2+ channel currents. We have studied [Mg2+]i actions on ICa under different phosphorylation conditions in cardiac ventricular myocytes and tsA201 cells expressing wild-type L-type Ca2+ channels and mutant channels lacking putative protein kinase A (PKA) phosphorylation sites. Our data demonstrate that [Mg2+]i regulation of ICa was pronounced under conditions that promote channel phosphorylation but was reduced under conditions antagonizing channel phosphorylation. However, changes in channel phosphorylation level were not necessary for Mg2+ actions, and even phosphorylation itself was not a prerequisite for [Mg2+]i-dependent modulation of channel gating. Thus, while our data provide direct support for the hypothesis that cytosolic Mg2+ modulates the effect of channel phosphorylation on gating kinetics, the results also suggest that Mg2+ actions are, in general, more prominent under conditions that promote channel opening, irrespective of the specific mechanism that alters channel gating kinetics.
METHODS
Isolation of Ventricular Myocytes
Adult rat cardiac ventricular myocytes were isolated enzymatically from male Sprague-Dawley rats (200–225 g), as described previously (28). Animals received an intraperitoneal injection of sodium pentobarbitone (50–100 mg/kg), and after full anesthesia was achieved, a thoracotomy was performed to rapidly remove the heart, in accordance with the procedures approved by the Institutional Animal Care and Use Committee of the University of Medicine and Dentistry of New Jersey. After isolation, myocytes were stored in a refrigerator and used within 1 to 8 h.
Molecular Biology
Expression plasmids encoding the rabbit α1C/pcDNA (GenBank accession no. X15539 ) (40), rabbit α1C/S1928A/pCR3 (11), rat β2A/S478A/S479A/pRBG (3), rat β2A/pGW (GenBank accession no. M80545 ) (33), and rat α2δ/pcDNA3 (GenBank accession no. NM-012929 ) (36) were supplied by Drs. M. Hosey and D. T. Yue. A truncated α1-subunit (α1CΔ1905), which consisted of 5,715 bases, was constructed using a standard PCR overlap extension technique with α1C/S1928A/pCR3. In brief, α1C/S1928A/pCR3 was first digested with restriction enzymes XbaI and EcoRV to produce a linear subfragment of α1C/pCR3 containing 4,542 bases of α1C. The complementary sequence between bases 4,543 and 5,715 of α1C was created by standard PCR overlap extension to include a unique XbaI restriction site. The PCR product was then ligated with the linear subfragment of α1C/pCR3 to yield α1CΔ1905. This construct was verified by sequence analysis. The pCR3 vector carried both ampicillin- and kanamycin-resistant genes for bacterial selection.
Cell Culture
tsA201 cells, a subclone of the human embryonic kidney cell line (HEK)-293 expressing the simian virus-40 T antigen (a gift of Dr. Roman Shirokov), were maintained in a monolayer culture in DMEM/F-12 Ham’s medium (Sigma, St. Louis, MO), supplemented with 10% fetal bovine serum (Hyclone, Logan, UT), 100 U/ml penicillin G, and 100 μg/ml streptomycin at 37°C in a humidified atmosphere of air with 5% CO2.
Expression of Channel Subunits
The tsA201 cells were used for 12–14 passages and were transferred every 4 days. Seventy percent confluent cultures of tsA201 cells were transiently cotransfected with a mixture of plasmids containing 20 μg α1C (wild type/mutant), 20 μg β2A (wild type/mutant), 20 μg α2δ Ca2+ channel subunits, and 1 μg enhanced green fluorescent protein cDNA using the Ca2+ phosphate method (7). Twenty-four hours after transfection, the cells were replated onto coverslips at low density for an additional 2 days before electrophysiological recording to allow for optimal transient expression of heterologous proteins. Enhanced green fluorescent protein was used as a visual indicator for successful transfection of cells with 485-nm light.
Electrophysiology
Cells were voltage-clamped with an Axopatch 1C amplifier using standard patch-clamp techniques, as previously reported (37). Pipettes were pulled to resistances of 1.5–2.0 MΩ for tsA201 cells and 1.0–1.5 MΩ for myocytes when filled with a buffered salt solution (see below). After gigaohm seal formation and patch breakthrough, whole cell currents were elicited by depolarizing voltage steps of 200-ms duration to various test potentials from the holding potential. Transfected tsA201 cells were held at −80 mV and the cell membrane was directly depolarized to various test potentials. Myocytes were held at −70 mV and the cell membrane was depolarized to −40 mV with a 1-s ramp to suppress Na+ current before the test depolarization (30). In some cases, 30 μM TTX was also included in bathing solutions to block Na+ current. Outward K+ currents were blocked by Cs+ and tetraethylammonium (TEA) ions in the pipette solution. Experiments were conducted when whole cell voltage-clamps had time constants <300 μs without series resistance or capacitance compensation. Cell capacitance was estimated by integrating current elicited with 5-mV depolarizations from the holding potential.
In general, cells were depolarized every 30 s to a test potential of +20 mV in tsA201 cells and 0 mV in myocytes. Current-voltage (I-V) relationships were also obtained periodically by varying the test potential between −30 and +90 mV (in 10-mV increments) in tsA201 cells and between −30 and +60 mV in myocytes. In this case, depolarizations were elicited every 5 s. Current records were acquired by sampling at 10 kHz and applying a low-pass filter at 5 kHz. Data were analyzed using pCLAMP software, version 8.0 (Axon Instruments, Union City, CA). L-type channel current was measured as a 200 μM Cd2+-sensitive difference current. Membrane currents, dis played without linear leak subtraction, were obtained after 5 min in the whole cell configuration, as previously published (37).
Drugs and Chemicals
Solutions.
The pipette solution was composed of (in mM) 100 cesium gluconate, 10 PIPES (cesium salt), 15 TEA-Cl, 0.5 NaH2PO4, 0.1 Tris-GTP, 5 EGTA along with ATP, Mg-ATP, citric acid, magnesium citrate, MgCl2, and CaCl2 to produce free [Mg2+] ([Mg2+]p) of 0.2 mM and 1.8 mM, pH 7.2, at a free [Ca2+] of 100 nM (WinMAXC 2.40, obtained at http://www.stanford.edu/~cpatton/maxc.html). The superfusion solution in experiments with cardiac myocytes was a modified Tyrode solution containing (in mM) 145 NaCl, 4 KCl, 2 (or 0.5) CaCl2, 10 HEPES, 1 MgCl2, and 10 glucose, pH 7.4, or a high-TEA bathing solution for tsA201 cells containing (in mM) 150 TEA-Cl, 10 CaCl2, 10 HEPES, 1 MgCl2, and 10 glucose, pH 7.4. Experiments were performed at room temperature (22–25°C).
Reagents.
Unless specified, reagents were obtained from Sigma. Adenosine 5′-O-(2-thiodiphosphate) tetralithium salt (ATP-γ-S; Calbiochem, San Diego, CA), catalytic subunit of PKA (Sigma), and protein phosphatase 2A (PP2A; Upstate Biochemicals, Lake Placid, NY) were prepared as concentrated stock solutions that were directly added to pipette solutions before experiments. Several reagents purchased from Calbiochem (BAY K8644, IBMX, and TTX) and forskolin (Biomol, Plymouth Meeting, PA) were prepared as concentrated stock solutions that were applied to bathing solutions 30 min before experiments.
Data Analysis
Data are expressed as means ± SE for the number of cells indicated. Statistical significance was determined with the use of ANOVA and Student’s t-tests in commercial software (SigmaPlot, Jandel Scientific and JMP IN, Duxbury, Pacific Grove, CA). A P value <0.05 was considered statistically significant. The percent change, confidence intervals (95%), and SE for current density ratios were calculated using Fieller’s theorem (13).
RESULTS
Effect of [Mg2+]i on Wild-Type L-Type ICa in tsA201 Cells
In studying mechanisms by which changes in [Mg2+]i around its physiological level of 0.6–1.3 mM (6, 21, 38) might affect ICa in cardiac myocytes, most of our experiments have been performed with [Mg2+]p in the range of 0.2 to 1.8 mM (37). These same conditions were used in experiments with tsA201 cells heterologously expressing L-type Ca2+ channel subunits (α1C, β2A, α2δ). A time diary of these experiments is shown in Fig. 1A.
Fig. 1.

L-type Ca2+ current (ICa) recorded in tsA201 cells expressing wild-type channels (α1C/β2A/α2δ) dialyzed with low and high pipette free [Mg2+] ([Mg2+]p). A: time diaries of ICa in tsA201 cells depolarized to +20 mV from a holding potential of −80 mV during dialysis with pipette solutions containing 0.2 mM (●) and 1.8 mM [Mg2+] (▲). Time 0 coincides with patch breakthrough. Data are means ± SE. Eight experiments were performed for each [Mg2+]p. Inset, superimposed currents recorded with 0.2 mM (a) and 1.8 mM (b) [Mg2+]p. Horizontal and vertical calibration bar values are 50 ms and 5 pA/pF, respectively. B: current-voltage (I-V) relationships of ICa in tsA201 cells dialyzed with 0.2 mM [Mg2+]p (●) and 1.8 mM [Mg2+]p (▲). Currents were recorded 5 min after whole cell patch-clamp configuration was established. Experimental replicates are 7 and 10 for 0.2 and 1.8 mM [Mg2+]p, respectively.
In the tsA201 cells dialyzed with 0.2 mM [Mg2+]p, ICa increased for 1–4 min after patch breakthrough and then slowly decreased for the remainder of the recording period. The pattern of the time diary of ICa with 0.2 mM [Mg2+]p in tsA201 cells was quite similar to that in cardiac myocytes (37). In the tsA201 cells dialyzed with 1.8 mM [Mg2+]p, the initial current density at the first recording point (10 s) (5.3 ± 1.8 pA/pF, n = 8) was not different from that with 0.2 mM [Mg2+]p (8.7 ± 2.6 pA/pF, n = 8). However, at all time points beyond 30 s after patch breakthrough, ICa was much smaller than that in cells patch-clamped with 0.2 mM [Mg2+]p. Although ICa in the tsA201 cells dialyzed with 1.8 mM [Mg2+]p also increased for 1–2 min after patch breakthrough, the initial increase of ICa was much smaller than that with 0.2 mM [Mg2+]p. The relative amplitude of ICa measured with 1.8 mM vs. 0.2 mM [Mg2+]p also changed rapidly within 3–5 min after patch breakthrough and changed slowly thereafter (ranging from 28% to 24% of ICa with 0.2 mM [Mg2+]p at times >5 min). These results are consistent with our previous data in cardiac myocytes (37). As a consequence, results reported in subsequent experiments were recorded 5 min after establishing whole cell voltage clamps to allow adequate solution dialysis between pipette and cytosol. Representative currents recorded 5 min after patch breakthrough are shown in Fig. 1A, inset.
The I-V relationship was determined by a series of voltage pulses from −30 to +90 mV, as described in METHODS. With 0.2 mM [Mg2+]p, the threshold for ICa activation occurred at −20 mV and peak current was observed at 30 mV (Fig. 1B). The membrane potential dependence of this current is shifted approximately +20 mV compared with ICa observed in cardiac myocytes, consistent with other reports, in which ICa has been measured in heterologous expression systems (e.g., Ref. 11). When [Mg2+]p was increased from 0.2 to 1.8 mM, peak ICa amplitude was decreased from 22.7 ± 3.5 pA/pF (n = 7) to 5.4 ± 0.9 pA/pF (n = 10), a 76 ± 4.5% (n = 7) reduction (Table 1), and the peak of the I-V relationship was shifted 5–10 mV in the negative direction (Fig. 1B).
Table 1.
Effect of [Mg2+]i on ICa in tsA201 cells expressing heterologous channel subunits
| Channels | Phosphorylation Conditions | ICa, pA/pF | %Reduction of ICa | |
|---|---|---|---|---|
| 0.2 mM [Mg2+]p | 1.8 mM [Mg2+]p | |||
| Control | 22.7±3.5 (7) | 5.4±0.9*(10) | 76±4.5 | |
| PKA | 36.4±4.0 (7) | 5.9±0.8*(7) | 83±3.1 | |
| α1C/β2A/α2δ | PP2A | 12.9±1.7 (12) | 6.9±0.5*(7) | 46±7.8† |
| PKA+ATP-γ-S | 34.5±4.2 (8) | 8.7±1.2*(8) | 73±4.8 | |
| α1C/S1928A/β2A/S478A/S479A/α2δ | NA | 15.5±2.0 (10) | 11.1±1.1*(9) | 21±12.5† |
| α1CΔ19Q5/β2A/S478A/S479A/α2δ | NA | 28.4±2.4 (11) | 8.6±1.6*(12) | 69±4.7 |
Values are means ± SE. Numbers in parentheses indicate replicates for each experimental condition. ICa, L-type Ca2+ current; PP2A, protein phosphatase 2A; [Mg2+]p, pipette free [Mg2+]; [Mg2+]i; cytosolic free [Mg2+]; NA, not applicable because PKA did not affect currents carried by these mutant channels. ICa density in tsA201 cells voltage-clamped with patch electrodes containing the indicated [Mg2+]p. Currents were recorded at 5 min after breakthrough into the whole cell patch-clamp configuration.
P < 0.05, statistically significant changes of ICa, comparing high (1.8 mM) vs. low (0.2 mM) [Mg2+]p.
A reduction of ICa by [Mg2+]i that is significantly different (P < 0.05) than that produced under control conditions with α1C/β2A/α2δ expression.
Effects of [Mg2+]i on Wild-Type L-Type Channels in tsA201 Cells Under Low and High Phosphorylation Conditions
Previous studies (1, 31, 37, 41, 44, 45) have shown that [Mg2+]i effects on ICa are dependent on channel phosphorylation state in cardiac myocytes. To determine whether these effects could be reproduced in tsA201 cells, phosphorylating conditions were manipulated by adding protein kinases and phosphatases to the patch pipette solution. Inclusion of PKA in the pipette solution increased ICa amplitude by ~50% (Fig. 2A, middle) compared with control conditions (Fig. 2A, left) with 0.2 mM [Mg2+]p, an indication that inclusion of PKA promoted channel phosphorylation. Under these conditions, increasing [Mg2+]p from 0.2 to 1.8 mM reduced peak ICa amplitude by 83 ± 3.1% (Table 1). Conversely, inclusion of PP2A, a Ca2+- and Mg2+-independent phosphatase (19), in the patch pipette solution to promote channel dephosphorylation yielded a 40% decrease in ICa amplitude with 0.2 mM [Mg2+]p (Fig. 2A, right vs. left panels). Furthermore, increasing [Mg2+]p to 1.8 mM decreased peak ICa amplitude by 46 ± 7.8% (Table 1), significantly smaller than that observed under control conditions. Figure 2B shows the I-V relationships for these experiments. In all cases, increasing [Mg2+]p caused a leftward shift in the peak of the I-V relationship. Taken together, the data showed that manipulating channel phosphorylation conditions in the presence of 0.2 mM [Mg2+]i allowed the amplitude of ICa to be changed over a threefold range, whereas the same maneuvers with 1.8 mM [Mg2+]i resulted in no significant change in ICa amplitude. This demonstration that [Mg2+]i modulation of ICa is dependent on channel phosphorylation conditions is consistent with published effects of [Mg2+]i in cardiac myocytes (37, 41). Thus the tsA201 expression system would appear to be a good model to study [Mg2+]i-dependent effects on ICa.
Fig. 2.

Effect of [Mg2+]p on ICa in tsA201 cells expressing wild-type channels (α1C/β2A/α2δ) under low and high phosphorylation conditions. A: typical ICa records at a test potential of +20 mV in cells dialyzed with 0.2 mM (solid symbols) and 1.8 mM [Mg2+]p (open symbols) in control conditions (left) and with 30 U/ml PKA (middle) and 1 U/ml phosphatase 2A (PP2A; right) added to the pipette solution. B: I-V relationships for ICa in cells dialyzed with 0.2 mM (solid symbols) and 1.8 mM [Mg2+]p (open symbols) with control (circle) and PKA (triangle)- and PP2A (square)-containing pipette solutions. Currents were recorded at 5 min after breakthrough into the whole cell patch-clamp configuration. The number of experiments ranges from 7 to 10.
Effects of [Mg2+]i on Wild-Type L-Type Channels in tsA201 Cells Under Thiophosphorylation Conditions
Our previous studies in cardiac myocytes (37) and our present data above in tsA201 cells demonstrated that under conditions promoting channel phosphorylation, increasing [Mg2+]p from 0.2 to 1.8 mM has a profoundly antagonistic effect on ICa amplitude, whereas conditions promoting channel dephosphorylation reduce [Mg2+]i effects on ICa amplitude. This phosphorylation dependence of [Mg2+]i effects on ICa could be explained by a change in the channel phosphorylation level and/or a modulation of the effects of channel phosphorylation on gating kinetics. To distinguish between these two possible mechanisms, tsA201 cells expressing wild-type L-type channels were voltage-clamped with patch electrodes containing solutions that included the catalytic subunit of PKA and 4 mM ATP-γ-S. Inclusion of ATP-γ-S with PKA should promote the formation of thiophosphorylated proteins (14) that are insensitive to potential changes in the activities of Mg2+-dependent phosphatases (19).
As expected, ICa amplitude in cells voltage-clamped with 0.2 mM [Mg2+]p and PKA was significantly increased (Fig. 3A). Under these conditions, increasing [Mg2+]p from 0.2 mM to 1.8 mM decreased peak ICa amplitude by 73 ± 4.8% (Table 1) and shifted the peak of the I-V relationship in the negative direction (Fig. 3B). These effects are comparable to those under basal (Fig. 1B) and high phosphorylation (Fig. 2B) conditions. Thus the data suggest that Ca2+ channel phosphorylation need not change in order for intracellular Mg2+ to strongly modulate gating kinetics.
Fig. 3.
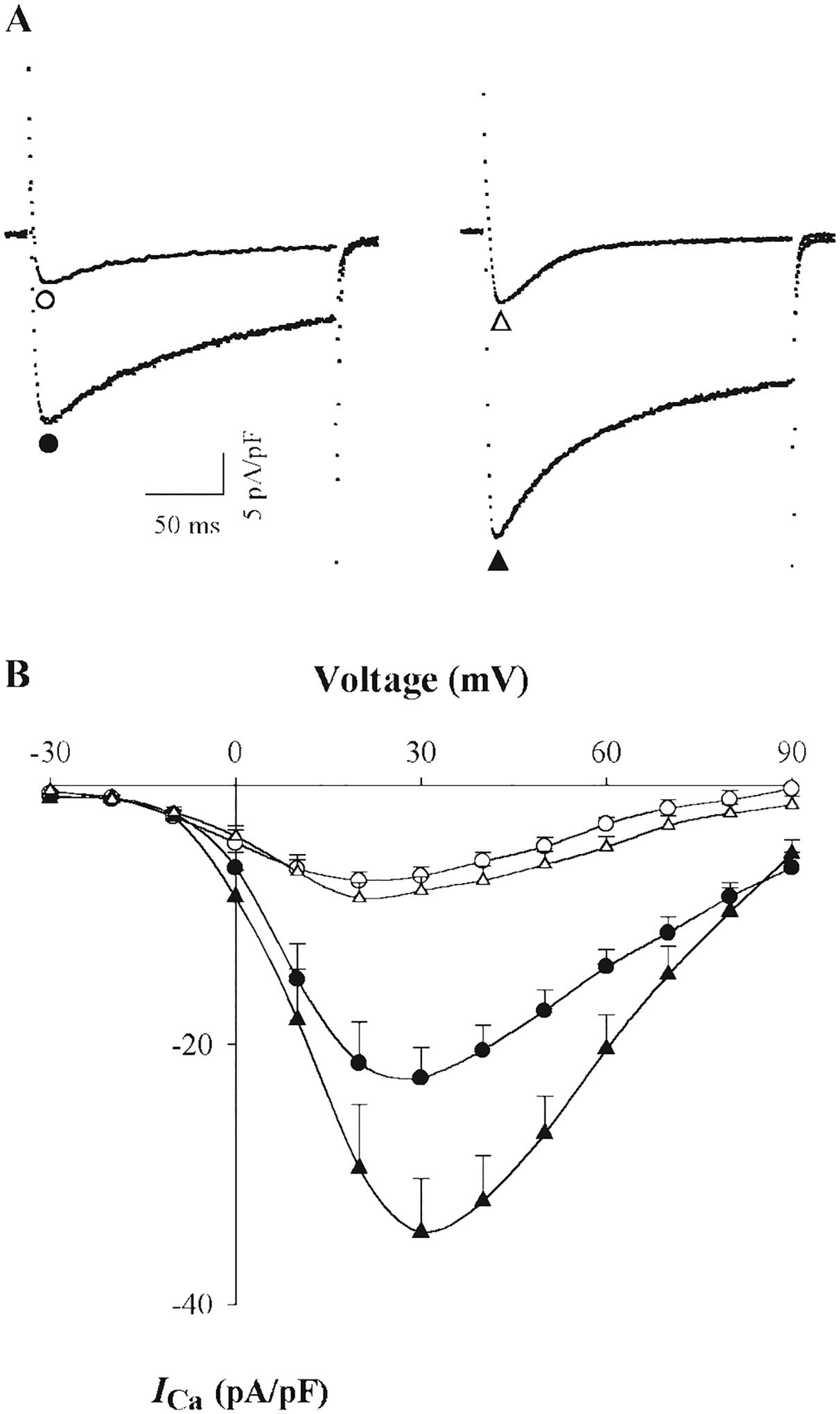
Effect of [Mg2+]p on ICa in tsA201 cells expressing wild-type channels (α1C/β2A/α2δ) under thiophosphorylation conditions. A: typical ICa records at a test potential of +20 mV in cells dialyzed with 0.2 mM (●, ▲) and 1.8 mM [Mg2+]p (○, △) in absence (left) and presence of 4 mM adenosine 5′-O-(2-thiodiphosphate) tetralithium salt (ATP-γ-S) plus 30 U/ml PKA (right). B: I-V relationships for ICa in cells dialyzed with 0.2 mM (solid symbol) and 1.8 mM [Mg2+]p (open symbols) in the absence (●, ○) and presence of ATP-γ-S and PKA (▲, △), respectively. Currents were recorded at 5 min after breakthrough into the whole cell patch-clamp configuration. The number of experiment replicates is 7 or 8.
Effect of [Mg2+]i on Mutant L-Type Channels Lacking Putative PKA Phosphorylation Sites in tsA201 Cells
Is channel phosphorylation an absolute requirement for Mg2+ modulation on the L-type Ca2+ channels? To answer this question, Ca2+ channels lacking putative PKA-sensitive phosphorylation sites were expressed in tsA201 cells to determine whether Mg2+ would modulate gating in these channels. Although the L-type Ca2+ channel contains many potential Ser/Thr kinase sites, only three serines actually appear to become phosphorylated by PKA in situ, Ser1928 in the α-subunit and Ser478, Ser479 in the β-subunit (3, 11, 18, 29, 32). For this reason, α- and β-subunits with all three PKA-sensitive Ser mutated to Ala (α1C/S1928A, β2A/S478A/S479A) were expressed with the α2δ-subunit in tsA201 cells. This triple Ser to Ala mutant channel was designed to mimic low phosphorylation conditions induced by PP2A in wild-type channels.
With the triple Ser to Ala mutant channel (Fig. 4A, left), current densities (15.5 ± 2.0 pA/pF, n = 10) were lower than those observed with wild-type channels (22.7 ± 3.5 pA/pF, n = 7) when cells were voltage-clamped with patch pipettes containing 0.2 mM Mg2+. Inclusion of PKA in the pipette solution had no effect on ICa amplitude at 0.2 mM [Mg2+]p (Fig. 4B), confirmation that the Ala mutations rendered the channel insensitive to this kinase. Peak ICa amplitude was 11.1 ± 1.1 pA/pF (n = 9) in cells expressing this mutant channel and voltage-clamped with 1.8 mM [Mg2+]p. This current density was calculated to be only 21 ± 12.5% (n = 9) less than that observed in cells voltage-clamped with electrodes containing 0.2 mM [Mg2+]p (Fig. 4B; Table 1), a much smaller decrease in current than observed with wild-type channels expressed in these cells. Thus these data show that the lack of PKA-sensitive phosphorylation sites resulted in a significantly reduced effect of [Mg2+]i on channel function, analogous to the effect of PP2A on [Mg2+]p modulation of ICa.
Fig. 4.

Effect of [Mg2+]p on ICa in tsA201 cells expressing mutated Ca2+ channels. A: typical ICa records at a test potential of +20 mV with 0.2 mM (solid symbols) and 1.8 mM (open symbols) [Mg2+]p in cells expressing α1C/S1928A/β2A/S478A/S479A/α2δ (left) and α1CΔ1905/β2A/S478A/S479A/α2δ (right) channel subunits. B: I-V relationships in cells expressing α1C/S1928A/β2A/S478A/S479A/α2δ (●, ○) and α1CΔ1905/β2A/S478A/S479A/α2δ (▲, △) channel subunits with 0.2 mM (solid symbol) and 1.8 mM [Mg2+]p (open symbol). Experiments in which PKA was included in the patch pipette solution are indicated for cells expressing α1C/S1928A/β2A/S478A/S479A/α2δ (■) and α1CΔ1905/β2A/S478A/S479A/α2δ (♦) channel subunits with 0.2 mM [Mg2+]p. Currents were recorded at 5 min after breakthrough into the whole cell patch-clamp configuration. The number of experiments ranged from 7 to 12.
In a second series of experiments, an α-subunit truncated proximal to Ser1928 (α1CΔ1905) was expressed with the β-subunit containing the double Ser to Ala substitution (β2A/S478A/S479A) and α2δ. This truncation of the α-subunit has been reported previously to dramatically increase channel opening (10, 39), analogous to the effects of PKA phosphorylation on channel gating. In tsA201 cells expressing channels with α1CΔ1905 (Fig. 4A, right), Ca2+ current densities with 0.2 mM [Mg2+]p (28.4 ± 2.4 pA/pF, n = 11) were larger than those observed with wild-type channels. Inclusion of PKA in the pipette solution had no effects on ICa amplitude at 0.2 mM [Mg2+]p (Fig. 4B), again confirming that removal of phosphorylation sites in α- and β-subunits was effective at abolishing the action of PKA on channel function. Interestingly, increasing [Mg2+]p from 0.2 to 1.8 mM reduced the peak ICa amplitude by 69 ± 4.7% (Table 1). The magnitude of this [Mg2+]i-dependent reduction was not significantly different than that observed with wild-type channels. As a consequence, these data directly demonstrate that PKA-dependent channel phosphorylation need not occur in L-type channels for [Mg2+]i to have a pronounced effect on ICa.
Relevance of Intracellular Mg2+ Effects in Cardiac Myocytes
A pertinent question at this point is whether the results obtained in tsA201 cells are applicable to cardiac myocytes. To determine whether a change in channel phosphorylation is necessary for [Mg2+]i effects, thiophosphorylation experiments were repeated in myocytes preincubated for 30 min with 10 μM forskolin and 300 μM IBMX in modified Tyrode solution containing 0.5 mM Ca2+ (37). ICa was then measured 5 min after patch breakthrough with a pipette solution containing 4 mM ATP-γ-S. Under these conditions, increasing [Mg2+]p from 0.2 mM to 1.8 mM decreased the peak ICa amplitude from 16.4 ± 2.3 pA/pF (n = 6) to 4.4 ± 0.4 pA/pF (n = 6), producing a marked reduction of peak ICa (69 ± 2.6%, n = 6) (Fig. 5, A and B), comparable to that under basal and high phosphorylation conditions (see Ref. 37). These data, therefore, suggest that Ca2+ channel phosphorylation need not change for intracellular Mg2+ to strongly modulate gating kinetics in myocytes.
Fig. 5.
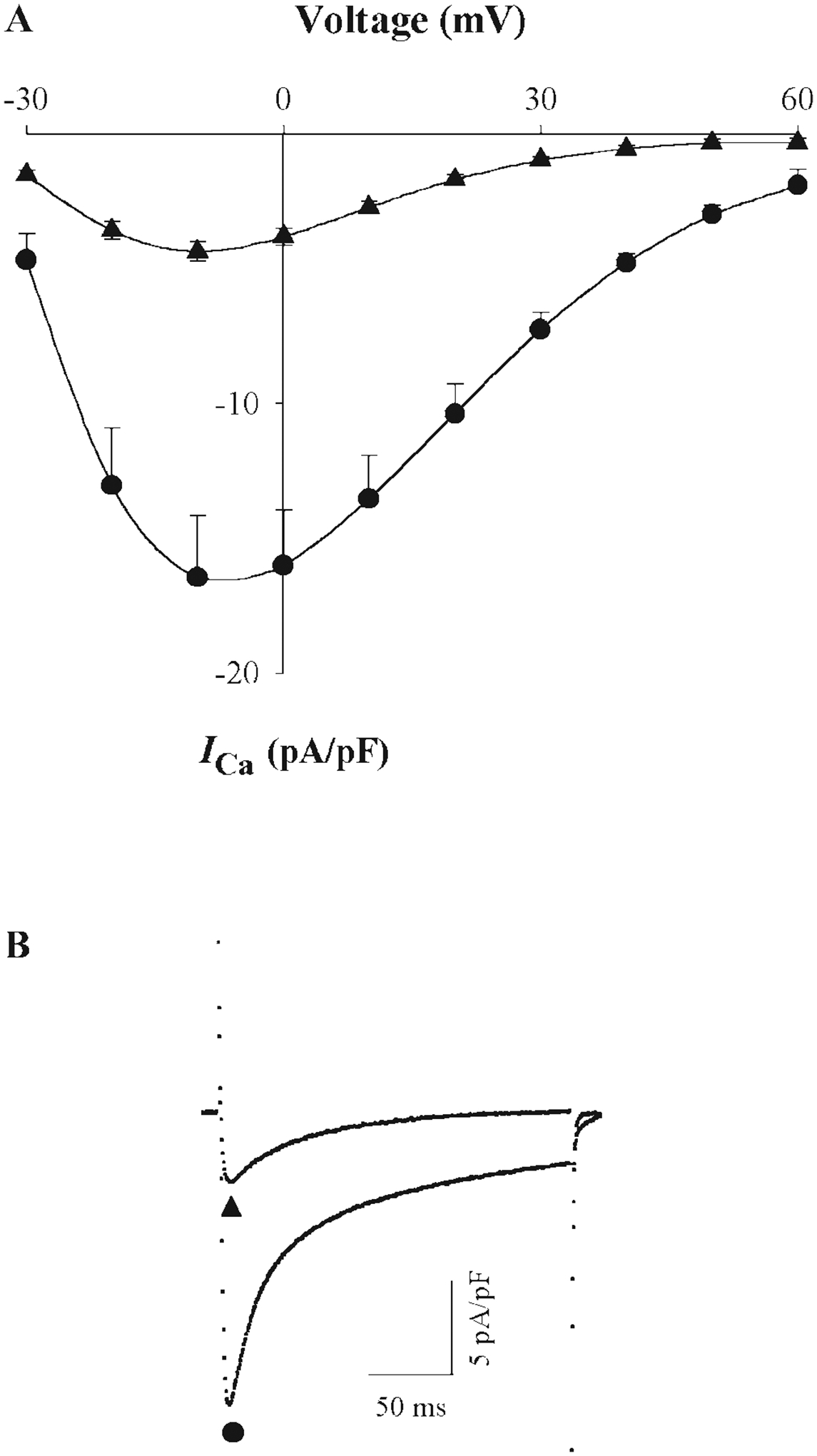
Effect of [Mg2+]p on ICa in rat cardiac ventricular myocytes under thiophosphorylation conditions. A: I-V relationships for ICa in myocytes dialyzed with 0.2 mM (●) and 1.8 mM (▲) [Mg2+]p with 4 mM ATP-γ-S in the pipette solution and inclusion of 10 μM forskolin and 300 μM IBMX in the 0.5 mM Ca2+-containing superfusion solution. External Ca2+ was 0.5 mM to avoid voltage errors due to high current density induced by forskolin and IBMX (and by BAY K8644 in Table 2). Our published data (37) showed that [Mg2+]i effects are the same with 0.5 and 2.0 mM Ca2+-containing superfusion solutions. Currents were recorded at 5 min after breakthrough into the whole cell patch-clamp configuration. Six experiments were conducted for each [Mg2+]p. B: typical ICa records at a test potential of 0 mV with 0.2 mM [Mg2+]p (●) and 1.8 mM [Mg2+]p (▲).
Data from tsA201 cells suggest that the channel phosphorylation is not necessary for [Mg2+]i-dependent modulation of gating, if channel opening is promoted by other mechanisms. To test this idea, myocytes were exposed to 0.1 μM BAY K8644 to increase channel opening, independent of PKA phosphorylation. As expected, the addition of BAY K8644 to the superfusion solution significantly increased ICa compared with control conditions in cells voltage-clamped with 0.2 mM [Mg2+]p (Table 2). In experiments with 0.2 and 1.8 mM [Mg2+]p, peak ICa amplitude was 16.7 ± 0.9 pA/pF (n = 4) and 3.9 ± 0.6 pA/pF (n = 4), respectively, i.e., increasing [Mg2+]p produced a 76 ± 2.9% decrease. To ensure that channel phosphorylation state did not affect this result, experiments were repeated in myocytes voltage-clamped with patch electrodes containing PP2A. For purposes of comparison, Table 2 includes our previously published data (37) showing that, with PP2A (1 U/ml) alone, increasing [Mg2+]p from 0.2 mM to 1.8 mM produces a relatively small decrease in ICa. In the presence of PP2A, 0.1 μM BAY K8644 still produced a significant increase in ICa to 11.6 ± 1.4 pA/pF (n = 5), but with 1.8 mM [Mg2+]p, peak ICa density was reduced by 75 ± 2.9% (Table 2). This reduction in current was significantly larger than the effect of [Mg2+]i in the presence of PP2A alone, but similar to the reduction of ICa observed under control conditions in the absence of PP2A (Table 2), with BAY K8644 alone and in high phosphorylation conditions produced with forskolin/IBMX/okadaic acid (see Ref. 37). Thus these results suggest that [Mg2+]i effects are not uniquely dependent on channel phosphorylation state but gating kinetics that promote channel opening, consistent with the results in tsA201 cells.
Table 2.
Effect of [Mg2+]i on ICa in cardiac myocytes with PP2A and BAY K8644
| Conditions | ICa, pA/pF | %Reduction of ICa | ||
|---|---|---|---|---|
| 0.2 mM [Mg2+]p | 1.8 mM [Mg2+]p | |||
| [Ca2+]o (2.0 mM) | Control | 17.0±2.0 (12) | 5.6±1.0*(10) | 64±2.8 |
| PP2A | 9.8±1.3† (12) | 6.7±0.4*(16) | 25±3.4† | |
| [Ca2+]o (0.5 mM) | Control | 8.0±0.7 (6) | 1.9±0.3*(8) | 75±2.4 |
| BAY K8644 | 16.7±0.9‡ (4) | 3.9±0.6*(4) | 76±2.9 | |
| BAY K8644 + PP2A | 11.6±1.4† (5) | 2.5±0.3*(5) | 75±2.9 | |
Values are means ± SE. The numbers in parentheses indicate replicates for each experimental condition. ICa density in rat ventricular myocytes voltage-clamped with patch electrodes containing the indicated [Mg2+]p. [Ca2+]o (2.0 mM) values were from Ref. 37. Currents were recorded at 5 min after breakthrough into the whole cell patch-clamp configuration. PP2A was included in patch electrode solutions at a concentration of 5 U/ml. BAY K8644 (0.1 μM) was added to superfusion solutions with the indicated [Ca2+]o.
P < 0.05, statistically significant changes of ICa between 0.2 and 1.8 mM [Mg2+]p.
P < 0.05, a reduction of ICa by [Mg2+]i that is significantly different than that produced under control conditions.
P < 0.05, statistically significant difference between control and experimental condition at the same [Mg2+]p.
DISCUSSION
[Mg2+]i Inhibition of L-type Ca2+ Channels
Data in this study demonstrated that increasing [Mg2+]p from 0.2 to 1.8 mM had a marked inhibitory effect on wild-type L-type Ca2+ channels expressed in HEK-tsA201 cells, decreasing peak amplitude of ICa and shifting the peak of the I-V relationship 5–10 mV in the negative direction. These effects were in general similar to [Mg2+]i modulation of ICa observed in our previous study with cardiac myocytes (37). Some previous studies (31, 42–45) have shown that cytosolic Mg2+ inhibition of ICa was manifested in the micromolar concentration range, whereas under defined experimental conditions that favor high phosphorylation, inhibition of ICa was only observed at [Mg2+]i well >1 mM (31, 44, 45). In addition to the present data, some reports also show that changes of [Mg2+]i around physiological levels can regulate L-type Ca2+ channels (1, 41); however, the degree of current inhibition is considerably smaller than that observed here and by the study of Wang et al. (37). Taken together, our data make the case that intracellular Mg2+ can be a potent modulator of ICa at physiological concentrations.
Channel Phosphorylation Affects [Mg2+]i Inhibition of L-Type Ca2+ Channels
Under conditions that antagonized channel phosphorylation in tsA201 cells (with PP2A in the patch pipette solution), an increase in [Mg2+]p from 0.2 to 1.8 mM had a less pronounced effect on ICa than in basal conditions or those promoting channel phosphorylation (with PKA in the pipette solution), consistent with our previous studies in cardiac myocytes (37). These findings are also consistent with reports that cytosolic Mg2+ inhibits ICa with high affinity (in micromolar range) under basal, presumably low, phosphorylation conditions (43) but with low apparent affinity, i.e., >1 mM, under high phosphorylation conditions (31, 44, 45). In light of these findings, we would conclude that 0.2 mM [Mg2+]i produces substantial inhibition of ICa under conditions antagonizing channel phosphorylation, so that increasing [Mg2+]i to 1.8 mM would have only modest effects on ICa.
The importance of channel phosphorylation in [Mg2+]i effects on ICa was further supported by data derived from Ca2+ channels, in which phosphorylation sites were mutated to Ala residues. Many potential phosphorylation sites are found in the α1 (23, 26, 27, 35, 46) and β-subunits of L-type Ca2+ channels (12, 16); however, the major sites of PKA-dependent protein phosphorylation in heart muscle appear to be located at Ser1928 of the α1C-subunit (8, 11, 29, 32) and two serine residues (Ser478 and Ser479) of the β2A-subunit (3). Consistent with previous reports, our data demonstrated that these three serine residues are functional targets for phosphorylation because mutation of these three amino acids to Ala produced PKA-insensitive currents. In channels with all three Ser residues mutated to Ala (α1C/S1928A/β2A/S478A/S479A/α2δ) increasing [Mg2+]p from 0.2 to 1.8 mM produced a much smaller effect on the peak ICa amplitude than that observed in wild-type channels. [Mg2+]i effects on these channels were, however, comparable to those observed in the presence of PP2A. Thus, from these data, we conclude that cytosolic Mg2+ in the concentration range tested has only marginal (~20–25%) inhibitory effects in unphosphorylated channels.
Interestingly, with 0.2 mM [Mg2+]p, current density in cells expressing α1C/S1928A/β2A/S478A/S479A/α2δ Ca2+ channels was less than that observed when wild-type channels were expressed. If these mutant channels mimic the condition of low phosphorylation in wild-type channels produced by PP2A, which reduces channel opening (15), this lower current density makes sense. However, an alternative explanation is that expression level of this mutant channel protein is lower. We have no evidence pertaining to expression levels, but irrespective of the reasons for the lower current density, these data, and the effects of PP2A in cells expressing wild-type Ca2+ channels, suggest that current density is maintained by some level of channel phosphorylation that probably accounts for [Mg2+]i actions under basal conditions.
Approximately 20–25% inhibition of peak ICa by high [Mg2+]p was still observed on increasing [Mg2+]p from 0.2 to 1.8 mM with Ca2+ channels containing the triple Ser to Ala mutation. One explanation for this result is that channel phosphorylation by other kinases such as PKC (24) and PKG (22) could also be affected by [Mg2+]i. Another explanation may be that [Mg2+]i blocks ion permeation through the channels. Although previous studies (25, 42, 44) with L-type Ca2+ channels suggest that cytosolic Mg2+, at the concentrations used in this study, would not affect divalent cation permeation rate, [Mg2+]i in the 1 mM range has been reported to produce extensive block in voltage-dependent Na+ and K+ channels (4, 17). Thus the possibility that physiologically relevant [Mg2+]i blocks ion permeation cannot be entirely neglected.
Does Channel Phosphorylation Need to Change for [Mg2+]I Effects on ICa?
Two mechanisms have been proposed to explain the phosphorylation dependence of [Mg2+]i effects on ICa. First, [Mg2+]i might change the level of channel phosphorylation by regulating the activity of the many Mg2+-dependent kinases, phosphatases, and phosphodiesterases present in the cell (31, 41). Second, Mg2+ might change the effect of phosphorylation on channel gating kinetics (44). To distinguish between these two possible mechanisms, [Mg2+]i effects on ICa were investigated under thiophosphorylation conditions induced by inclusion of ATP-γ-S in the pipette solutions used for experiments with tsA201 cells and myocytes. The addition of ATP-γ-S is reported to produce phosphatase-resistant thiophosphorylated channels (14) that would be insensitive to [Mg2+]i-induced changes in enzyme activities. Under these conditions, increasing [Mg2+]p from 0.2 mM to 1.8 mM produced a similar reduction of ICa amplitude to that observed in basal conditions and high phosphorylation conditions in both tsA201 cells and cardiac myocytes. Thus these data suggest that the level of Ca2+ channel phosphorylation need not change for intracellular Mg2+ to regulate Ca2+ channels. Thus we must consider the hypothesis that Mg2+ might change the effect of phosphorylation on channel-gating kinetics.
The data might also suggest that these Mg2+-dependent enzymes are not necessarily involved in [Mg2+]i regulation on the Ca2+ channels, consistent with our previous result that Ca2+-calmodulin-dependent kinase II is not involved in [Mg2+]i regulation of ICa (37); however, the data do not exclude the possibility that, under the appropriate conditions, such kinases, phosphatases and phosphodiesterases could participate in [Mg2+]i modulation of L-type Ca2+ channels.
Is Channel Phosphorylation Absolutely Necessary for [Mg2+]i Effects on ICa?
Previously published studies (1, 31, 37, 41, 44, 45) and our present data clearly demonstrate that channel phosphorylation affects [Mg2+]i regulation of ICa. Nonetheless, one motivation for this study was to determine whether channel phosphorylation is necessary for [Mg2+]i regulation of ICa. To address this question, we made a truncated α1C subunit (α1CΔ1905) that lacked 265 COOH-terminal amino acids including Ser1928. Channels containing this truncated α1C-subunit have been reported to display increased current amplitude due to increased channel opening probability rather than by elevated channel expression (10, 39). We took advantage of this characteristic of α1CΔ1905 and coexpressed the subunit along with a β-subunit containing the double Ser to Ala substitution (β2A/S478A/S479A) to obtain a PKA-insensitive channel that displayed high channel opening. With this channel, increasing [Mg2+]p from 0.2 to 1.8 mM produced a marked reduction of peak ICa amplitude similar to that observed under high phosphorylation conditions. These data suggested that channel phosphorylation is not absolutely necessary for [Mg2+]i regulation on ICa.
Considering that truncation of α1C-subunit is, in fact, functionally very similar to cAMP-dependent phosphorylation, i.e., it promotes channel openings (47), we supposed that [Mg2+]i regulation on ICa might simply be dependent on the functional state of the channels. To test this idea, we used the dihydropyridine agonist BAY K8644 to induce channel opening (20) and PP2A to inhibit channel phosphorylation at the same time in cardiac myocytes. Under these conditions, increasing [Mg2+]p from 0.2 to 1.8 mM produced a marked inhibitory effect on ICa, which was much bigger than that under low phosphorylation conditions, i.e., PP2A alone, but very similar to that under basal and high phosphorylation conditions produced by forskolin/IBMX/okadaic acid (37). Together with the data discussed above, these results suggest that [Mg2+]i modulation of ICa channels is promoted by conditions that favor channel opening, including channel phosphorylation and pharmacological agonists.
[Mg2+]i Effect on ICa and Channel Gating Modes
Three patterns of gating kinetics displayed by L-type Ca2+ channels have been classified as “gating modes” (20). In mode 0, channels fail to open during depolarization. In mode 1, channels display brief openings with a low frequency, whereas mode 2 gating is characterized by long-lasting and frequent channel openings (20). Transitions among these three gating modes are thought to be modulated by, among other factors, channel phosphorylation and dihydropyridines. The β-adrenergic receptor-PKA signaling pathways enhance transitions to mode 1 or mode 2 gating (2, 47). Long-lived, mode 2 gating is also promoted by pharmacological agents, such as BAY K8644 (9, 20) and structural modification of channels, such as the truncation of α1C-subunit (10, 39). Conversely, transitions from mode 2 to mode 1 or mode 0 gating are stimulated by channel dephosphorylation induced by maneuvers such as cytosolic application of PP2A (15).
Our data show that [Mg2+]i-dependent effects were enhanced by seemingly any maneuver, including channel phosphorylation, pharmacological agents (BAY K8644), α1C-subunit truncation (α1CΔ1905) that increased channel availability, and/or favored mode 2 gating but were lessened by maneuvers, such as channel dephosphorylation (PP2A), that decreased mode 2 gating. Thus one explanation for intracellular [Mg2+]i modulation of channel gating is that Mg2+ shifts channel gating to low open probability states (mode 1) and decreases channel availability (mode 0). This hypothesis is quite plausible because, in addition to our data, Yamaoka and Seyama (42) reported that channel availability increased on lowering cytosolic Mg2+ from millimolar to micomolar concentrations, reflecting the shift of channels from silent (mode 0) to active modal states (mode 1).
The three experimental maneuvers that promote [Mg2+]i inhibition of ICa, i.e., channel phosphorylation, BAY K8644 and truncation of α1C-subunits, also promote channel opening. The degree to which these maneuvers share a common mechanism in altering channel gating kinetics is unknown. Nonetheless, it is parsimonious to assume that cytosolic Mg2+ acts via a common mechanism to decrease ICa. In this regard, much attention has been focused on intracellular structures of the Ca2+ channel that might be involved in the inactivation process (34). Several domains of the protein have been shown to be critical in current inactivation and facilitation, including the COOH-terminal tail of the α-subunit. In the COOH-terminal tail, one of those domains, an EF-hand motif, has recently been suggested as a cytosolic binding site at which Mg2+ can regulate channel gating (5). Given the proximity of the EF-hand motif to the other protein domains that are also critical for channel gating (34), such a conclusion does not at all seem at odds with our finding that Mg2+ can modulate ICa under a wide variety of conditions that promote channel opening. At the same time, considerable caution must be exercised in defining the structural basis for cytosolic Mg2+ actions given the apparent complexity of channel gating properties and the reported contributions by many protein domains in the gating process.
ACKNOWLEDGMENTS
The authors thank Drs. M. M. Hosey, R. Shirokov, and D. T. Yue for supplying reagents for heterogeneous expression Ca2+ channels in tsA201 cells and helpful discussions, and Sarbjit Sandhu for participating in construction of the truncated α-subunit.
GRANTS
This project was supported by National Heart, Lung and Blood Institute Grant P01-HL-69020.
REFERENCES
- 1.Agus ZS, Kelepouris E, Dukes I, and Morad M. Cytosolic magnesium modulates calcium channel activity in mammalian ventricular cells. Am J Physiol Cell Physiol 256: C452–C455, 1989. [DOI] [PubMed] [Google Scholar]
- 2.Bean BP, Nowycky MC, and Tsien RW. β-Adrenergic modulation of calcium channels in frog ventricular heart cells. Nature 307: 371–375, 1984. [DOI] [PubMed] [Google Scholar]
- 3.Bünemann M, Gerhardstein BL, Gao T, and Hosey MM. Functional regulation of L-type calcium channels via protein kinase A-mediated phosphorylation of the β2 subunit. J Biol Chem 274: 33851–33854, 1999. [DOI] [PubMed] [Google Scholar]
- 4.Bara M, Cuiet-Bara A, and Durlach J. Regulation of sodium and potassium pathways by magnesium in cell membranes. Magnes Res 6: 167–177, 1993. [PubMed] [Google Scholar]
- 5.Brunet S, Scheuer T, Klevit R, and Catterall WA. Modulation of CaV1.2 channels by Mg2+ acting at an EF-hand motif in the COOH-terminal domain. J Gen Physiol 126: 311–323, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buri A and McGuigan JA. Intracellular free magnesium and its regulation, studied in isolated ferret ventricular muscle with ion-selective microelectrodes. Exp Physiol 75: 751–761, 1990. [DOI] [PubMed] [Google Scholar]
- 7.Chien A, Zhao X, Shirokov R, Puri T, Chang CF, Sun D, Ríos E, and Hosey M. Roles of membrance-localized β-subunit in the formation and targeting of functional L-type Ca2+ channels. J Biol Chem 270: 30036–30044, 1995. [DOI] [PubMed] [Google Scholar]
- 8.De Jongh KS, Murphy BJ, Colvin AA, Hell JW, Takahashi M, and Catterall WA. Specific phosphorylation of a site in the full-length form of the α1C subunit of the cardiac L-type calcium channel by adenosine 3′,5′-cyclic monophosphate-dependent protein kinase. Biochemistry 35: 10392–10402, 1996. [DOI] [PubMed] [Google Scholar]
- 9.Erxleben C, Gomez-Alegria C, Darden T, Mori Y, Birnbaumer L, and Armstrong DL. Modulation of cardiac CaV1.2 channels by dihydropyridine and phosphatase inhibitor requires Ser-1142 in the domain III pore loop. Proc Natl Acad Sci USA 100: 2929–2934, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao TY, Cuadra AE, Ma H, Bünemann M, Gerhardstein BL, Cheng T, Eick RT, and Hosey MM. C-terminal fragments of the α1C (Cav1.2) subunit associate with and regulate L-type calcium channels containing C-terminal-truncated α1C subunits. J Biol Chem 276: 21089–21097, 2001. [DOI] [PubMed] [Google Scholar]
- 11.Gao TY, Yatani A, Dell’Acqua ML, Sako H, Green SA, Dascal N, Scott JD, and Hosey MM. cAMP-dependent regulation of cardiac L-type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Neuron 19: 185–196, 1997. [DOI] [PubMed] [Google Scholar]
- 12.Gerhardstein BL, Puri TS, Chien AJ, and Hosey MM. Identification of the sites phosphorylated by cyclic AMP-dependent protein kinase on the β2 subunit of L-type voltage-dependent calcium channels. Biochemistry 38: 10361–10370, 1999. [DOI] [PubMed] [Google Scholar]
- 13.Goldstein A Biostatistics: An Introductory Text. New York: Macmillan, 1964, p. 187–189. [Google Scholar]
- 14.Gratecos D and Fischer EH. Adenosine-O-(3-thiotriphosphate) in the control of phosphorylase activity. Biochem Biophys Res Commun 58: 960–967, 1974. [DOI] [PubMed] [Google Scholar]
- 15.Groschner K, Schuhmann K, Mieskes G, Baumgartner W, and Romanin C. A type 2A phosphatase-sensitive phosphorylation site controls modal gating of L-type Ca2+ channels in human vascular smooth-muscle cells. Biochem J 318: 513–517, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haase H, Bartel S, Karczewski P, Morano I, and Krause EG. In-vivo phosphorylation of the cardiac L-type calcium channel β-subunit in response to catecholamines. Mol Cell Biochem 163–164: 99–106, 1996. [DOI] [PubMed] [Google Scholar]
- 17.Harris RE and Isacoff EY. Hydrophobic mutations alter the movement of Mg2+ in the pore of voltage-gated potassium channels. Biophys J 71: 209–219, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hartzell HC and White RE. Effects of magnesium on inactivation of the voltage-gated calcium current in cardiac myocytes. J Gen Physiol 94: 745–767, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herzig S and Neumann J. Effects of serine/threonine protein phosphatases on ion channels in excitable membranes. Physiol Rev 80: 173–210, 2000. [DOI] [PubMed] [Google Scholar]
- 20.Hess P, Lansman JB, and Tsien RW. Different modes of Ca channel gating behavior favored by dihydropyridine Ca agonists and antagonists. Nature 311: 538–544, 1984. [DOI] [PubMed] [Google Scholar]
- 21.Hongo K, Konishi M, and Kurihara S. Cytoplasmic free Mg2+ in rat ventricular myocytes studied with the fluorescent indicator furaptra. Jpn J Physiol 44: 357–378, 1994. [DOI] [PubMed] [Google Scholar]
- 22.Jiang LH, Gawler DJ, Hodson N, Milligan CJ, Pearson HA, Porter V, and Wray D. Regulation of cloned cardiac L-type calcium channels by cGMP-dependent protein kinase. J Biol Chem 275: 6135–6143, 2000. [DOI] [PubMed] [Google Scholar]
- 23.Kameyama A, Shearman MS, Sekiguchi K, and Kameyama M. Cyclic AMP-dependent protein kinase but not protein kinase C regulates the cardiac Ca2+ channel through phosphorylation of its α1 subunit. J Biochem (Tokyo) 120: 170–176, 1996. [DOI] [PubMed] [Google Scholar]
- 24.Kamp TJ and Hell JW. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ Res 87: 1095–1102, 2000. [DOI] [PubMed] [Google Scholar]
- 25.Kuo C and Hess P. Block of the L-type Ca2+ channel pore by external and internal Mg2+ in rat phaeochromocytoma cells. J Physiol 466: 683–706, 1993. [PMC free article] [PubMed] [Google Scholar]
- 26.Leach RN, Brickley K, and Norman RI. Cyclic AMP-dependent protein kinase phosphorylates residues in the C-terminal domain of the cardiac L-type calcium channel α subunit. Biochim Biophys Acta 128: 205–212, 1996. [DOI] [PubMed] [Google Scholar]
- 27.Mikami A, Imoto K, Tanabe T, Niidome T, Mori Y, Takeshima H, Narumiya S, and Numa S. Primary structure and functional expression of the cardiac dihydropyridine-sensitive calcium channel. Nature 340: 230–233, 1989. [DOI] [PubMed] [Google Scholar]
- 28.Mitra R and Morad M. A uniform enzymatic method of dissociation of myocytes from heart and stomachs of vertebrates. Am J Physiol Heart Circ Physiol 249: H1056–H1060, 1985. [DOI] [PubMed] [Google Scholar]
- 29.Mitterdorfer J, Froschmayr M, Grabner M, Moebius FF, Glossmann H, and Striessnig J. Identification of PKA phosphorylation sites in the carboxyl terminus of L-type calcium channel α1 subunits. Biochemistry 35: 9400–9406, 1996. [DOI] [PubMed] [Google Scholar]
- 30.Nilius B, Hess P, Lansman JB, and Tsien RW. A novel type of cardiac calcium channel in ventricular cells. Nature 316: 443–446, 1985. [DOI] [PubMed] [Google Scholar]
- 31.Pelzer S, La CC, and Pelzer DJ. Phosphorylation-dependent modulation of cardiac calcium current by intracellular free magnesium. Am J Physiol Heart Circ Physiol 281: H1532–H1544, 2001. [DOI] [PubMed] [Google Scholar]
- 32.Perets T, Blumenstein Y, Shistik E, Lotan I, and Dascal N. A potential site of functional modulation by protein kinase A in the cardiac Ca2+ channel α1C subunit. FEBS Lett 384: 189–192, 1996. [DOI] [PubMed] [Google Scholar]
- 33.Perez-Reyes E, Castellano A, Kim HS, Bertrand P, Baggstrom E, Lacerda AE, Wei XY, and Birnbaumer L. Cloning and expression of a cardiac/brain beta subunit of the L-type calcium channel. J Biol Chem 267: 1792–1797, 1992. [PubMed] [Google Scholar]
- 34.Stotz SC, Jarvis SE, and Zamponi GW. Functional roles of cytoplasmic loops and pore lining transmembrane helices in the voltage-dependent inactivation of HVA calcium channels. J Physiol 554: 263–273, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sculptoreanu A, Rotman E, Takahashi M, Scheuer T, and Catterall WA. Voltage-dependent potentiation of the activity of cardiac L-type calcium channel α1 subunits due to phosphorylation by cAMP-dependent protein kinase. Proc Natl Acad Sci USA 90: 10135–10139, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tomlinson WJ, Stea A, Bourinet E, Charnet P, Nargeot J, and Snutch TP. Functional properties of a neuronal class C L-type calcium channel. Neuropharmacology 32: 1117–1126, 1993. [DOI] [PubMed] [Google Scholar]
- 37.Wang M, Tashiro M, and Berlin JR. Regulation of L-type calcium current by intracellular magnesium in rat cardiac myocytes. J Physiol 555: 383–396, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watanabe M and Konishi M. Intracellular calibration of the fluorescent Mg2+ indicator furaptra in rat ventricular myocytes. Pflügers Arch 442: 35–40, 2001. [DOI] [PubMed] [Google Scholar]
- 39.Wei X, Neely A, Lacerda AE, Olcese R, Stefani E, Perez-Reyes E, and Birnbaumer L. Modification of Ca2+ channel activity by deletions at the carboxyl terminus of the cardiac alpha 1 subunit. J Biol Chem 269: 1635–1640, 1994. [PubMed] [Google Scholar]
- 40.Wei XY, Perez-Reyes E, Lacerda AE, Schuster G, Brown AM, and Birnbaumer L. Heterologous regulation of the cardiac Ca2+ channel α1 subunit by skeletal muscle β and γ subunits. J Biol Chem 266: 21943–21947, 1991. [PubMed] [Google Scholar]
- 41.White RE and Hartzell HC. Effects of intracellular free magnesium on calcium current in isolated cardiac myocytes. Science 239: 778–780, 1988. [DOI] [PubMed] [Google Scholar]
- 42.Yamaoka K and Seyama I. Regulation of Ca2+ channel by intracellular Ca2+ and Mg2+ in frog ventricular cells. Pflügers Arch 431: 305–317, 1996. [DOI] [PubMed] [Google Scholar]
- 43.Yamaoka K and Seyama I. Modulation of Ca2+ channels by intracellular Mg2+ ions and GTP in frog ventricular myocytes. Pflügers Arch 432: 433–438, 1996. [DOI] [PubMed] [Google Scholar]
- 44.Yamaoka K and Seyama I. Phosphorylation modulates L-type Ca channels in frog ventricular myocytes by changes in sensitivity to Mg2+ block. Pflügers Arch 435: 329–337, 1998. [DOI] [PubMed] [Google Scholar]
- 45.Yamaoka K, Yuki T, Kawase K, Munemori M, and Seyama I. Temperature-sensitive intracellular Mg2+ block of L-type Ca2+ channels in cardiac myocytes. Am J Physiol Heart Circ Physiol 282: H1092–H1101, 2002. [DOI] [PubMed] [Google Scholar]
- 46.Yoshida A, Takahashi M, Nishimura S, Takeshima H, and Kokubun S. Cyclic AMP-dependent phosphorylation and regulation of the cardiac dihydropyridine-sensitive Ca channel. FEBS Lett 309: 343–349, 1992. [DOI] [PubMed] [Google Scholar]
- 47.Yue DT, Herzig S, and Marban E. β-Adrenergic stimulation of calcium channels occurs by potentiation of high-activity gating modes. Proc Natl Acad Sci USA 87: 753–757, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]