

Abstract
Neurotrophin-3 growth factor can improve cochlear neuron survival, and localized delivery of this protein to the round window membrane in the middle ear may be able to reverse sensorineural hearing loss. Thus, the goal of this work was to develop an injectable hydrogel delivery system that can allow localized release of neurotrophin-3 in a controlled and sustained manner. We identified a PEG hydrogel formulation that uses thiol-vinyl sulfone Michael addition for crosslinking. This injectable formulation provides elastic hydrogels with higher mechanical rigidity, better bio-adhesion and longer residence time than Poloxamer hydrogels currently being investigated clinically for hearing loss. In vivo, PEG hydrogels induce local immune responses comparable to biocompatible Poloxamer hydrogels, yet they released payloads at a ~5-fold slower rate in the subcutaneous area. Based on this injectable hydrogel formulation, we designed an affinity-based protein release system by modifying PEG hydrogels with affinity peptides specific to neurotrophin-3 proteins. We verified the sustained release of neurotrophin-3 from peptide-conjugated PEG hydrogels resulting from the reversible interaction between peptides and proteins. The rate of affinity-controlled release depends on the polymer concentrations, the affinity of peptides and the peptide-to-protein ratios. Collectively, we developed an injectable hydrogel formulation for localized delivery of neurotrophin-3, which provides affinity-controlled release and longer delivery time compared to Poloxamer hydrogels.
Keywords: Injectable hydrogel, Michael addition, affinity-controlled release, phage display, neurotrophin-3, round window membrane
Graphical Abstract
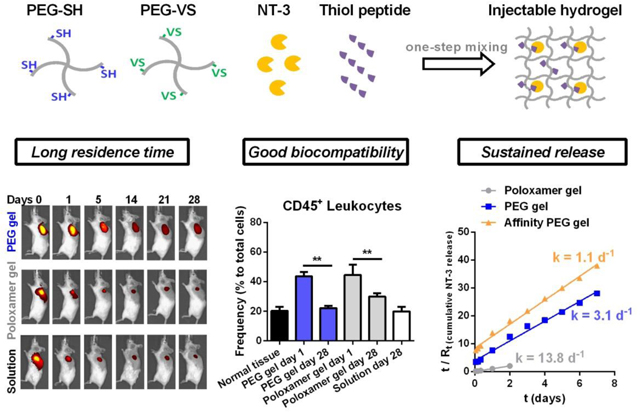
1. Introduction
Aging and injury cause synaptic loss (synapthopathy) and compromise function of the central and peripheral nervous systems.1 For example, acoustic trauma and aging lead to loss of cochlear synapses and the consequent hearing loss.2 Studies using genetically engineered mouse models demonstrated that damaged synapses can be regenerated through transgenic overproduction of the neurotrophic factor neurotrophin-3 (NT-3).3 Multiple systems have been studied to locally deliver growth factors, such as NT-3, to the inner ear by intracochlear administration or intratympanic administration.4–6 Intratympanic administration that delivers drugs to the inner ear by round window membrane (RWM) applications is much safer than directly administrating drugs to cochlea, and therefore it only needs an outpatient procedure rather than hospitalization to perform the surgery for intracochlear administration.4 Medical delivery systems for applying drugs to the RWM include implantable devices, such as microwicks and microcatheters, and injectable systems, such as hydrogels.4–6
In contrast to implantable medical devices, injectable hydrogels can deliver therapeutic growth factors by a minimally invasive intratympanic injection and match the irregular structure of the middle ear.7, 8 Several characteristics are required to design a hydrogel-based delivery system for intratympanic administration, including injectability, safety, sufficient residence time, and sustained drug release.4 Injectable hydrogels can be crosslinked by sol-gel transition induced by physical forces (i.e. electrostatic interactions and hydrophobic interactions) or chemical reactions (i.e. Michael addition and Schiff base reaction).7, 8 Physically crosslinked hydrogels require external stimuli such as temperature and pH to drive the gelation through non-covalent reversible forces, and these gels usually have low mechanical forces that result in fast gel elimination and short residence time in vivo.7, 8 In contrast, chemically crosslinked hydrogels formed by irreversible covalent bonds provide higher mechanical strength and stability, but some toxic agents such as the initiator that are needed to trigger the crosslinking and the chemical reaction itself may damage the local cells and tissues when gels are in situ formed on the RWM.9 In addition, choosing a chemical reaction that is slow and controllable to ensure the injectability of hydrogel is also important.10
In a recent report,11 the therapeutic value of NT-3 delivered by injectable and biocompatible Poloxamer 407 hydrogels through intratympanic administration was well studied, which showed that applying NT-3-loaded Poloxamer hydrogels to the RWM can reverse noise-induced hearing loss in roughly 50% of animals, suggestive of limitations in the delivery of NT-3 to the cochlea by this type of hydrogel.11 Thermo-reversible Poloxamer hydrogels are liquid at 4°C and gel at body temperature,12 and therefore a thermo-controlling device is required to maintain the liquid status of Poloxamer in the long, fine needle as the gel is injected in small volumes of viscous gels to mouse RWM (~2 μL) or human RWM (~50 μL).11 In addition, due to their water-solubility and poor mechanical rigidity, physically-crosslinked Poloxamer hydrogels are characterized by a short residence time ranging from hours to days, depending on the local environments in the biological sites (e.g. vaginal, subcutaneous, or other areas) and the size of the gel.12 These characteristics of Poloxamer hydrogels might play a role in their limited success in delivering NT-3 to the RWM for reversing hearing loss.11
In this study, we explored a chemically-crosslinked hydrogel formulation for localized delivery of NT-3 that can overcome the limitations of Poloxamer hydrogels by meeting abovementioned requirements. First, this hydrogel formulation is injectable and its gelation behavior can allow easier gel handling and injection than thermo-sensitive hydrogels. Second, the hydrogel can provide higher stability and longer residence time than Poloxamer 407 hydrogels in vivo. Third, the hydrogel is free of toxic initiators and is biocompatible in vivo, ensuing minimal damage to the delicate tissue in the RWM. Fourth, the hydrogel includes active sites that allow chemical modifications with ligands for improving release behavior and extending delivery time by different strategies, such as affinity-based release.13–18 Herein, a poly (ethylene glycol) (PEG) hydrogel formulation crosslinked via thiol (SH)-vinyl sulfone (VS) Michael addition was identified as a candidate carrier for NT-3 delivery. VS was chosen over Maleimide and other function groups because VS is moderately reactive to thiol and does not require a toxic catalyst, which can permit the generation of injectable, non-toxic hydrogels.19–21 PEG hydrogels prepared by this formulation were then characterized and compared with Poloxamer hydrogels in terms of physical properties, bio-adhesion, stability, in vivo residence time and biocompatibility to evaluate the potential of this PEG formulation for localized delivery of NT-3 to the RWM.
After identifying the carrier, we further designed an affinity-based release system based on this chemically-crosslinked hydrogel formulation by conjugating hydrogels with affinity peptides specific to NT-3. Affinity hydrogels have been reported to successfully control the protein release by leveraging the interaction between proteins and different ligands.13 These formulations include naturally derived affinity molecules that are capable of binding to multiple growth factors in vivo via electrostatic attractions,14, 15 as well as synthetic affinity molecules that can specifically bind to a target protein.16–18 In this study, we would test the hypothesis that the transient association and dissociation of polymer-anchored peptides to physically loaded NT-3 proteins delay the protein release and extend the delivery time. Affinity peptides that are specific to NT-3 proteins were identified by phage display, a well-established technology for high-throughput screening of peptide ligands,22, 23 and then their binding affinity to NT-3 were quantitatively analyzed. The release profiles of NT-3 from peptide-modified PEG hydrogels were studied at the air-liquid interface in an in vitro model that mimics the RWM. Three key factors that may control the release rate of NT-3 were then studied, including the polymer concentration, the binding affinity of peptide ligands to NT-3 proteins, and peptide-to-protein ratios. Through these studies we developed an injectable hydrogel delivery system that can address the physical limitations of traditional Poloxamer hydrogels while providing sustained, localized release of neurotrophin-3 in a controlled manner.
2. Experimental
2.1. Materials.
4-arm PEG thiol (20 kD), 4-arm PEG vinyl sulfone (20 kD) and 4-arm PEG Maleimide (20 kD) that have a pentaerythritol core structure were purchased from JenKem Technology USA. Poloxamer 407 and L-cysteine were purchased from Sigma-Aldrich. Human NT-3 protein ELISA kit was from R&D Systems. Alexa Fluor 488 (AF488) C5 Maleimide, CellTrace Far-Red dye and Ellman’s reagent were purchased from ThermoFisher Scientific. All the antibodies were from BioLegend.
2.2. Preparation of PEG hydrogels by one-step mixing and measurement of thiol conversion.
4-arm PEG thiol (PEG-4SH) and 4-arm PEG vinyl sulfone (PEG-4VS) were separately dissolved in PBS to prepare 30% (w/v) PEG stock solution. 30% PEG-4SH solution, 30% PEG-4VS solution and PBS were mixed together at 1: 1: 4, 1: 1: 2 or 1: 1: 1 ratios to prepare cargo-free 10%, 15% or 20% (w/v) PEG hydrogels, respectively. To prepare protein-loaded and peptide-conjugated 20% PEG hydrogels, 3 μM of NT-3 proteins were first mixed with thiol-functionalized peptides at the desired ratios in 10 μl of PBS, and then mixed well with 10 μl of 30% PEG-4SH solution, followed by mixing with 10 μl of 30% PEG-4VS solution. The mixed solution was then placed on the desired surface and incubated at 37 °C to allow gel formation.
To identify hydrogel crosslinking efficiency, thiol conversion was quantified by measuring the unreacted thiols in PEG hydrogels using Ellman’s reagent. Briefly, PEG-4SH solutions and PEG-4VS solutions were mixed and incubated at 37 °C to allow gel formation. 30 μl of PEG hydrogels were immersed in 300 μl of Ellman’s reagent solution and then incubated at room temperature on a shaker for 15 min. Absorbance of colored products of thiols reacting with Ellman’s reagent was measured at 412 nm on a plate reader (BioTex), and the amount of free thiols in PEG hydrogels was quantified by comparison to a standard curve composed of known concentrations of L-cysteine.
2.3. Swelling behavior of PEG hydrogels.
200 μl of 10%, 15% and 20% PEG hydrogels prepared in a mold (~10 mm in diameter and ~3 mm in height) were dried up in a 60 °C incubator for 2 days, and then immersed in water at room temperature. At the desired time points, hydrogels were removed from the water, dried with Kimwipe paper and then weighed. Swelling degree at time t (Ht) was calculated by the following equation:
| (1) |
And water content at time t (Wt) in the hydrogel was calculated by the following equation:
| (2) |
where mt is the weight of the hydrogel at time t, and m0 is the weight of the dry hydrogel, and (mt-m0) is the water weight (Mt) at t time. Winfinite is the water content when the equilibrium is reached. Three hydrogels were prepared and measured at each polymer concentration as replicates and their values were averaged and shown as mean ± SD in Figure 1.
Figure 1. Rheological properties and swelling behavior of PEG hydrogels with different polymer concentrations.

(A-D) Rheological analysis of PEG hydrogels with different polymer concentration. Gels were placed in the PBS solution and measured by a rheometer at room temperature. G’: storage modulus; G”: loss modulus; tan δ: damping factor, and tan δ = G” / G’. (E) Swelling isotherms of PEG hydrogels in water at room temperature. Ht (%): swelling ratio at time t. (F) Water content in PEG hydrogels over time. Wt (%): water content at time t. (G) Experimental data of water content and time t plotted according to Equation (5) for different PEG hydrogels. (H) Experimental data of water weight and time t plotted according to Equation (7) for different PEG hydrogels. Mt: water weight at time t; Minfinite: water weight at equilibrium. All values were shown as Mean ± SD, yet note that error bars in F, G, and H were too small to be seen.
2.4. Elasticity and rheological analysis of PEG hydrogels.
The rheological properties of pre-fabricated 10%, 15% and 20% PEG hydrogels (~10 mm in diameter and ~3 mm in height) were measured on a rheometer (Discovery HR-2, TA Instruments). The storage modulus (G′) and loss modulus (G″) were detected by keeping strain at 2% with a continuous frequency (0.1−50 rad/s). Hydrogels were placed in PBS solutions and measured at room temperature. Three hydrogels were prepared and measured at each polymer concentration as replicates and their values were averaged and shown as mean ± SD in Figure 1.
2.5. Bioadhesion and residence time analysis of hydrogels.
In vitro, bioadhesion and residence time of PEG hydrogels and Poloxamer 407 hydrogels were studied on a tissue culture treated surface. Tissue culture treated polystyrene surfaces can maximize the adhesion and proliferation of a broad range of cells in vitro. We used the tissue culture treated surface as an easy-to-use in vitro model to test the bioadhesion and the residence time of hydrogels. In contrast to fresh tissues that are hard to preserve due to dehydration and denaturation after extraction from animals, a tissue culture treated surface is stable, inexpensive, and suitable for the long-term observation of gel-tissue binding. In this study, 30 μl of freshly mixed PEG-4VS/PEG-4SH solution or ice-cold Poloxamer 407 solution was dropped to the bottom of tissue culture treated 6-well plates, and then placed at a 37 °C incubator for 20 min to allow in situ formation of 20% (w/v) hydrogels. To test the adhesion of hydrogels to the tissue culture treated surface, we scratched the hydrogels with pipette tips to see whether gels would deform or detach by mechanical forces. To identify the residence time of hydrogels in aqueous solutions, pre-warmed PBS was added to the plate wells to immerse hydrogels that had been attached to the bottom, and then the plate was placed on a shaker (20 rpm) in a 37 °C incubator. Hydrogels were imaged at the desired time points.
Ex vivo, bioadhesion of fluorescently labeled PEG hydrogels to fresh tissue with a flat tissue surface and to a fixed tissue with irregular structure (the tympanic cavity in the middle ear) was studied. 10–20 μl of 20% (w/v) of PEG-4SH/PEG-4VS mixed solution that included 5 molar% of AF488-C5 Maleimide was loaded to the hypodermis layer (subcutaneous tissue) of fresh skin, the inner side of the skull or the large intestine extracted from BALB/c mice. PEG gels were fluorescently labeled with AF488 in order to visualize the gels. These hydrogels were then incubated for 20 min to allow in situ gel formation at room temperature. Afterwards, we tested the adhesion of hydrogels to the skin by trying to separate gels from tissues with forceps. In another study, human temporal bones that were preserved in formalin solution were rinsed with PBS and dried by Kimwipe paper. Then ~5 μl of PEG-4SH/PEG-4VS mixed solution containing AF488-C5 Maleimide was applied to the round window niche of the temporal bone and incubated for 20 min to allow in situ gel formation at room temperature. We tested the attachment of hydrogels to the tympanic cavity with forceps, and then we immersed the human temporal bones in PBS and rocked on a shaker (20 rpm) at room temperature. Fluorescence of AF488-labeled PEG hydrogels was imaged by a fluorescent microscope (Nikon) at days 1, 2, 9 after gel formation.
The adhesive strength of hydrogels to the tissue culture treated surface was measured by the detachment force method. PEG or Poloxamer hydrogels (50 μL, ~4 mm in diameter and ~2 mm in height) were formed between a tissue culture treated plastic slide and the bottom of a tissue culture treated plate which was placed on and attached to a hot plate (37 °C). The plastic slide was glued to one arm of a modified balance while the other arm was attached with a plastic cup. Water was added to the cup drop by drop until the gel detaches the tissue culture treated slide. Measurements were repeated three times for each of the hydrogels. The weight of the water was recorded and the adhesive force was calculated by the following equation:
| (3) |
Where m is the water’s weight added to the balance in grams, and g is the acceleration due to gravity (980 cm/sec2), and A is the area of the gel attached to the tissue culture treated surface (A = πr2, cm2). A schematic diagram of the detachment force method used to measure the bioadhesion of hydrogels to tissue culture treated surface at 37 °C is shown in Figure S1.
2.6. In vivo imaging of hydrogels.
PEG-4SH, PEG-4VS and Poloxamer 407 solutions were sterilized by filtering ice-cold solutions through pre-cooled 0.22 μm membranes (Millipore). 5 μl of 5 mM Far-Red dye was added to 100 μl of 20% (w/v) PEG-4SH/PEG-4VS solution or 20% (w/v) ice-cold Poloxamer 407 solution or PBS solution right before injection. And then 50 μl of each ice-cold solution was subcutaneously injected to the back of BALB/c mice with 1 ml syringes with 27 G needles (the body temperature of mice is about 36 °C). Each mouse received one injection at the right side and another injection at the left side of the back. Mice bearing fluorescent hydrogels were imaged at the desired time points by an IVIS Spectrum in vivo imaging system (PerkinElmer). The first imaging was performed 3 hours after injection. The reduction in the fluorescence intensity of hydrogels in vivo was quantified and plotted over time.
2.7. Immunofluorescence staining.
Female BALB/c mouse skin tissues (from epidermis through hypodermis) surrounding hydrogels or dye solutions were collected, minced to small pieces and then digested in 2 mg/ml Liberase TM (Roche) for 30 min at 37 °C. Liberase was neutralized with FACS buffer (PBS containing 0.5% bovine serum albumin and 2 mM EDTA). Cells were then isolated by passing the tissues through 70 μm strainers (BD Bioscience). Red blood cells were lysed using 0.2% sodium chloride solution, followed by neutralization with 1.6% sodium chloride solution. For analysis of immune cell populations, 1 million cells were suspended in 100 μl of FACS buffer and blocked with 1 μg anti-mouse CD16/32 antibody for 30 min at 4 °C. Cells were then stained with an antibody cocktail solution at 4 °C for 1 h, and then fixed with 4% paraformaldehyde (PFA) and measured by Bio-Rad ZE5 cell analyzer. The 100 μl of antibody cocktail solution included 0.4 μg Brilliant Violet 510 anti-mouse/human CD11b antibody, 0.25 μg Brilliant Violet 605 anti-mouse CD11c antibody, 0.25 μg FITC anti-mouse Ly6C antibody, 0.25 μg PE antimouse Ly6G antibody, 0.25 μg PE/Cy7 anti-mouse F4/80 antibody, and 0.25 μg Alexa Fluor 700 anti-mouse CD45 antibody. In another panel, 100 μl of antibody cocktail solution included 0.25 μg Brilliant Violet 421 anti-mouse CD3 antibody and 0.25 μg Alexa Fluor 700 anti-mouse CD45 antibody.
2.8. Screening of NT-3-targeting peptides by phage display.
The Ph.D.−7 peptide library (New England Biolabs) was used to screen for NT-3 binding peptides according to the manufacturer’s instructions with slight modifications. Briefly, 96-well plates were coated with 150 μL/well of the recombinant human NT-3 protein (Peprotech) at a concentration of 30 μg/mL. Biopanning was carried out by incubating the phage display library for 1 hour at 37 °C. After washing away the unbound phage with Tris-buffered saline (TBS + 0.1% Tween 20), bound phages were eluted in TBS and titrated as described in the standard protocol, and then subjected to the next round of panning. The second, third and fourth rounds of panning were done under more stringent conditions using shorter incubation times (40 min, 25 min and 20 min in the 2, 3 and 4th round, respectively). After four rounds of panning, individual clones were randomly picked and amplified in E. coli ER2738 strain to prepare their DNA.
Individual positive phage clones were amplified and phage single-stranded DNA was extracted and purified using M13 Phage DNA Rapid Extraction Kit (Signalway Biotechnology). DNA was sequenced by the Genomics Core Facility in the Center for Genetic Medicine using the 96 gIII sequencing primer 5′-CCCTCATAGTTAGCGTAACG-3′ provided by the Ph.D.−7 peptide library kit to identify common peptide sequences among the clones and thus the extent of consensus. Amino acid sequences were deduced from phage display peptide DNA sequences by ExPASy Translate tool. Three peptide sequences were randomly picked from this pool. These peptides containing a C-terminal cysteine, were synthesized by GenScript.
2.9. Binding analysis of NT-3-targeting peptides
The binding kinetics between the selected phage display peptides and target protein NT-3 were studied using a biolayer interferometry based instrument, Octet Red (ForteBio). The biotinylation of NT-3 was prepared by incubating NT-3 with EZ-Link NHS-LC-LC-biotin (Pierce) at a 1: 1 molar ratio for 2 hours on ice, followed by dialysis to remove the excess biotin reagent. Super Streptavidin (SSA) sensors were equilibrated for 10 min in black 96-well microplates containing 200 μL/well of PBS buffer. Then, SSA-coated tips were saturated with 10 μg/mL biotinylated NT-3 at capture levels of 0.70 ± 0.15 nm. Purified binding phage display peptides at concentrations ranging from 10 nM to 200 μM were associated to protein-saturated sensors followed by dissociation in buffer. Reference binding sensors containing only the control peptide were corrected for baseline drift. Nanometer shift data were analyzed in Data Analysis 6.4 (ForteBio). To estimate a direct binding affinity via the kinetic rate constants (KD = Koff / Kon, where KD is equilibrium dissociation rate constant, kon is association rate constant, and koff is dissociation rate constant), the buffer-subtracted octet data were fitted globally to a simple 1: 1 Langmuir model.
2.10. Cumulative in vitro protein release analysis.
Transwell permeable membrane inserts placed in the Boyden chamber were used to mimic the round window membrane that separates the air-filled middle ear and liquid-filled inner ear. Boyden chambers and Transwell inserts (6.5 mm diameter, 8 μm pore size, Corning) were blocked with PBS containing 1% bovine serum albumin (BSA) overnight at room temperature. Then 30 μl of PEG hydrogel solution or Poloxamer 407 hydrogel solution including 1 μM of NT-3 proteins with or without thiol-functionalized peptides was dropped on the surface of the Transwell permeable insert to allow in situ gel formation. Afterwards, the lower chamber was filled with 0.5 ml of pre-warmed PBS containing 1% BSA that can block nonspecific binding of released proteins to the well surface. Plates were rocked at 20 rpm in a 37°C incubator with full humidity. At the desired time points, 100 μl of solution was collected from the lower chamber followed by supplementing 100 μl of fresh PBS containing 1% BSA. Released NT-3 proteins were quantified by ELISA. The accumulative amount of released NT-3 proteins was compared with the initial loading amount to obtain the drug release ratio (Rt) at time t.
2.11. Statistics.
Data was shown as mean ± SD. Statistical tests were performed with the Student’s t test and *p < 0.05 were considered to be statistically significant.
3. Results and discussion
3.1. Gelation behavior of PEG hydrogels via SH-VS Michael addition.
In this study, we prepared injectable PEG hydrogels by one-step mixing of PEG-4SH and PEG-4VS solutions at a 1: 1 molar ratio (Schematic 1A). Unreacted SH groups were quantified after gel formation, and we found that the conversion of SH in 10%, 15% and 20% (w/v) PEG hydrogels was close to 100% (Table S1), demonstrating SH-VS Michael addition reaction provides high crosslinking efficiency.
Schematic 1.

(A) Schematic representation of preparing PEG hydrogel by one-step mixing through SH-VS Michael addition. SH: thiol; VS: vinyl sulfone. (B) Schematic view of phage display used to screen peptides that specifically bind to human NT-3 proteins. (C) Schematic representation of affinity-controlled NT-3 release from PEG hydrogels conjugated with peptides that can reversibly bind to NT-3 proteins in the gel.
We then studied the gelation behavior of the PEG-4SH/PEG-4VS formulation at different temperatures. The gelation time of 60 μl of 10%−20% PEG hydrogels ranges from 5 to 15 min at body temperature (37 °C). This time range would allow the mixed solutions to fill the irregular structure in tympanic cavity before forming solid gels. The gelation time of this formulation increases to 18–46 min at room temperature (25 °C), and further extends to several hours at 4 °C (Table 1), providing sufficient time for preparation, mixing, and injection of the solution.
Table 1.
Gelation time of PEG-4SH/PEG-4VS mixed solutions under different conditions.
| PEG concentration (w/v) | 10% | 15% | 20% |
|---|---|---|---|
|
| |||
| 37°C | ~15 min | ~7 min | ~5 min |
| 25°C | ~46 min | ~27 min | ~18 min |
| 4°C | ~6.5 h | ~3.5 h | ~2 h |
VS groups are suitable for preparing injectable hydrogels crosslinked by thiol-Michael addition compared to other functional groups that have higher or far less reactivity to thiol.19–21 Maleimide (MAL) groups are highly reactive to thiol21 and the SH-MAL Michael addition reaction crosslinked PEG polymers within a few seconds. This rapid gelation provides insufficient time for mixing and injecting the PEG-4SH/PEG-4MAL solution, as well as results in hydrogels with uncontrollable gel shape and non-uniform internal structure (Figure S2) and low crosslinking efficiency (e.g. the conversion of thiol groups is 89% in 20% PEG hydrogels, Table S1). On the other hand, acrylate groups that are far less reactive to thiol than VS need catalysts to accelerate the reaction and increase the conversion rate.19, 20 Most catalysts such as triethylamine (TEA) are toxic and not suitable for in vivo applications.
3.2. Viscoelastic properties of PEG hydrogels.
With pre-fabricated hydrogels at hand, we next characterized the rheological properties of PEG hydrogels by measuring their storage (elastic) modulus (G’) and loss (viscous) modulus (G”) and plotted their values as a function of angular frequency (Figure 1A–C). Greater G’ than G” for PEG hydrogels with multiple polymer concentrations indicated the hydrogels have elastic properties. G’ values of PEG hydrogels were independent of angular frequency while G’ values of Poloxamer hydrogels were reported to increase with the frequency,24 suggesting the rheological behavior of chemically crosslinked PEG hydrogels are different from physical gels.
G’ and G” values of PEG hydrogels increased with the polymer concentration. Specifically, the mean G’ values of 10%, 15%, and 20% hydrogels were 293.89 ± 13.16, 521.14 ± 25.35, and 823.12 ± 27.94 Pa, respectively, and their mean G” values were 1.1920 ± 0.2573, 1.7153 ± 0.1989 and 2.3555 ± 0.3719 Pa, respectively (Table 2, Figure 1A–C). These results indicated 20% PEG hydrogels had a higher extent of crosslinking and a greater mechanical rigidity than 15% and 10% hydrogels.25 We then plotted the damping factor (tan δ), the ratio of G” to G’, as a function of angular frequency (Figure 1D). The tan δ values of all hydrogels were independent of frequency with a mean value ranging from 0.002 to 0.005 (Table 2), an indicator of ideally elastic solid materials (tan δ < 0.01).25
Table 2.
Characteristics of PEG hydrogels crosslinked by SH-VS Michael addition.
| Polymer concentration (w/v) | 10% | 15% | 20% |
|---|---|---|---|
|
| |||
| 1 Average G’ (Pa) | 293.89 ± 13.16 | 521.14 ± 25.35 | 823.12 ± 27.94 |
| 2 Average G” (Pa) | 1.1920 ± 0.2573 | 1.7153 ± 0.1989 | 2.3555 ± 0.3719 |
| 3 Average tan δ (×10−3) | 4.2101 ± 0.5294 | 3.2992 ± 0.6235 | 2.8738 ± 0.0195 |
| 4 Winfinite (%) | 95.79 ± 0.03 | 95.33 ± 0.40 | 94.68 ± 0.20 |
| 5 K (min−1) | 0.47 ± 0.01 | 0.38 ± 0.04 | 0.26 ± 0.02 |
| 6 n | 0.543 ± 0.004 | 0.547 ± 0.008 | 0.557 ± 0.006 |
G’: storage modulus;
G”: loss modulus;
tan δ: damping factor;
Winfinite: water content when hydrogels reach equilibrium, from equation (5);
K: kinetic rate constant, from equation (5);
n: diffusion exponent, from equation (7). Values were shown as mean ± SD.
3.3. Swelling behavior of PEG hydrogels.
We next investigated the swelling behavior of PEG hydrogels. Swelling reflects water diffusion in the matrix and is another indicator of the relative crosslinking density, with stiffer networks exhibiting lower swelling for hydrogels with the identical composition.26 This property is associated with the rate and pattern of drug release from hydrogels as well.27, 28 Herein, we measured the weight gain of 10%, 15% and 20% dried PEG hydrogels placed in water at room temperature, and plotted their swelling ratio (Ht) and water content (Wt) as a function of swelling time (t) (Figure 1E–F). All PEG hydrogels reached equilibrium at ~120 min. After 24 h, the swelling ratios of 10%, 15% and 20% PEG hydrogels were approximately 2222%, 1993% and 1718% (Figure 1E), respectively, yet their water contents were close to 95% (Figure 1F, Table 2). These results indicated that an increase in the polymer concentration decreases the swelling ratio of PEG hydrogels, yet did not significantly influence the water content at equilibrium. A decreased swelling ratio at a higher PEG concentration (20% vs. 15% and 10%) indicated a decrease in the capacity of 20% PEG hydrogels to deform themselves due to their more restricted structure resulting from a higher extent of crosslinking,25 implying a slower drug release from hydrogels with a higher polymer concentration, which was verified in the following study.
The time-programed swelling behavior of PEG hydrogels in water shows second-order kinetics (Figure 1F), which can be expressed as:29
| (4) |
where K is the kinetic rate constant, and Wt and Winfinite denote the water content at t time and at infinite time (at equilibrium), respectively. After integration between the limits (t = 0 and Wt = 0) and rearranging, the above equation can be also expressed as:29
| (5) |
We plotted the variation of t/Wt against time in Figure 1G and then calculated Winfinite and K (Table 2) by linear regression with R2 > 0.99. The polymer concentration does not significantly change Winfinite, yet the kinetics rate constant K decreased from 0.47 min−1 for 10% PEG hydrogels, to 0.38 min−1 and 0.26 min−1 for 15% and 20% PEG hydrogels (Table 2), respectively, suggesting the swelling rate is negatively correlated with the polymer concentration.
We further analyzed the rate of water transportation in PEG hydrogels using the following empirical Ritger-Peppas equation:7, 29
| (6) |
| (7) |
where Mt and Minfinite denote the water weight in the gel at time t and at infinite time, respectively. k is a constant and n is a characteristic exponent that reflects the mechanism of diffusion. We plotted the variance of log (Mt / Minfinite) as a function of log (t) (Figure 1H) and obtained n by linear regression with R2 > 0.99. The values for n range from 0.54 to 0.56 for 10–20% PEG hydrogels (Table 2), indicating non-Fickian diffusion (0.5 < n < 1) in PEG hydrogels which occurs when water diffusion and polymer relaxation rates are comparable.30 Our data was consistent with a previous report which identified that PEG hydrogels prepared by PEG-2k, 4k or 6k also showed non-Fickian diffusion during the gel swelling.31 In this report, people further reported that n values for water diffusion into PEG hydrogels decreased and approached to 0.5 (Fickian diffusion) when PEG gels became less rigid and more flexible by decreasing the molecular weight of PEG from 6k to 2k Da. Similarly, we found decreasing the polymer concentration from 20% to 10% for hydrogels prepared by PEG-20k, which also increased the flexibility of the polymer networks, slightly decreased the n value from 0.557 to 0.543, a slight trend towards Fickian diffusion. Taken together, we concluded that the water diffusion pattern in PEG hydrogels depends on the polymer relaxation, which can be affected by the molecular weight and the concentration of polymers.
3.4. Bioadhesion and residence time of PEG hydrogels.
In addition to the physical properties, the adhesion and residence time of hydrogels are also important characteristics influencing their use as local drug delivery systems in vivo. Inefficient delivery can be caused by poor hydrogel adhesion that causes discontinuity to biological tissues, or short residence time resulting from gel degradation and dissolution.7
To measure bioadhesion, we prepared in situ formed hydrogels on multiple tissue surfaces and then tested whether hydrogels would detach from surfaces upon mechanical forces. These surfaces included a tissue culture treated petri dish, fresh tissues (hypodermis, skull and large intestine) and fixed tissue with irregular structure (the tympanic cavity in the middle ear). As shown in Figure 2, using a tip or forceps failed to detach in situ formed PEG hydrogels from biological surfaces. Tight adhesion of PEG hydrogels to tissue surfaces might be attributed to the involvement of tissue-derived cysteine residues in SH-VS Michael addition reaction.32 In addition to flat surfaces, in situ formed PEG hydrogel can fill the tympanic cavity and tightly bind to the RWM (Figure 2C). In contrast, Poloxamer 407 hydrogels that bind to surfaces by reversible physical bioadhesive forces are easily deformed partly due to their low mechanical strength (Figure 2A).12
Figure 2. Bioadhesion and in vitro residence time analysis of PEG hydrogels.

(A) Images of freshly prepared PEG and Poloxamer 407 hydrogels and those scratched by a tip or incubated in PBS solutions at 37 °C for the desired times. In situ formed PEG hydrogels bind tightly to tissue culture treated surface and continue to have consistent size in aqueous solutions for over 28 days. Poloxamer 407 hydrogels dissolved in pre-warmed PBS solution within 5 min (The arrow indicates the original location of the hydrogel). Scale bar = 5 mm. (B) Adhesion of in situ formed AF488-labeled 20% PEG hydrogels (green, indicated by arrows) to different fresh tissues, including hypodermis, skull and large intestine. Gels did not detach from the tissue surface when they are lifted with a forceps (the background water level in lower panels indicates the horizontal level). Scale bar = 1 cm. (C) Residence of AF488-labeled 20% PEG hydrogels (green) in the tympanic cavity for over 9 days in PBS solutions. The arrow indicates the round window membrane in the tympanic cavity of middle ear. Scale bar = 5 mm.
Better bioadhesion of PEG hydrogels than Poloxamer hydrogels were further evaluated through the adhesive forces of hydrogels to tissues as measured by the detachment force.33–35 Our results indicate the force/area required to detach 20% PEG hydrogels from tissue culture treated plates at 37 °C was 26.18 ± 3.87 N/cm2, in contrast to 0.35 ± 0.05 N/cm2 for 20% Poloxamer gels (Table 3). We also observed that the force to detach PEG hydrogels from tissues increased with the polymer concentration (Table 3), which might be due to an increase in the number of chemical bonds formed between gels and tissues after in situ gelation.32
Table 3.
In vitro evaluation of hydrogel bioadhesion at 37 °C using detachment force measurement.
| Hydrogel | 10% PEG | 15% PEG | 20% PEG | 20% Poloxamer |
|---|---|---|---|---|
|
| ||||
| Weight required for detachment (g) | 133.3 ± 25.9 | 222.8 ± 51.5 | 335.5 ± 49.6 | 4.4 ± 0.7 |
| 1 Force (N) | 1.30 ± 0.25 | 2.18 ± 0.50 | 3.29 ± 0.49 | 0.043 ± 0.007 |
| 2 Force/Area (N/cm2) | 10.40 ± 2.02 | 17.38 ± 4.02 | 26.18 ± 3.87 | 0.35 ± 0.05 |
N = 105 dyne;
A = πr2 = 3.14 × 0.2 × 0.2 (cm2). The diameter of the hydrogel is around 4 mm.
PEG hydrogels attached to different surfaces were then immersed in aqueous solutions and rocked at 37 °C or room temperature to test their residence time in vitro. In contrast to Poloxamer 407 hydrogels that dissolved in aqueous solutions within a few minutes due to their water-solubility,12 PEG hydrogels had a consistent size and remained attached to tissue surfaces for over a month (Figure 2A, C). The residence time of PEG and Poloxamer 407 hydrogels was further compared in vivo. Freshly mixed PEG-4SH/PEG-4VS solutions or ice-cold Poloxamer 407 solutions were physically loaded with Far-Red dyes and then subcutaneously (s.c.) injected to the back of mice to allow in situ gel formation at body temperature. The uniform distribution of Far-Red fluorescence in the gel at 3 h after injection indicates in situ formed PEG hydrogels had a similar thickness across the gel while Poloxamer hydrogels had a thicker area in the center than the edge (Figures 3A and S2). The size and shape of PEG hydrogels in the subcutaneous area did not change over time despite the decay of Far-Red fluorescence, demonstrating their minimal degradation and stability in vivo. In contrast, because Poloxamer hydrogels were gradually degraded, dissolved and absorbed by tissues in vivo,12 these hydrogels, similar to Far-Red dye solutions (control), showed a significant decrease in the fluorescence intensity over time (Figure 3A and S2) and only trace amounts of Poloxamer gels were observed in the subcutaneous area at day 28 (Figure 3B).
Figure 3. in vivo residence time analysis of PEG hydrogels.

PEG-4SH/PEG-4VS or ice-cold Poloxamer 407 solutions that included Far-Red dyes were injected into the subcutaneous areas at the back of mice to allow in situ hydrogel formation. (A) Whole-animal images of dye-loaded hydrogels and dye solutions (control) on the left back of mice. Gels on the right back of mice are shown in Figure S3. (B) Images of Far-Red dye-loaded hydrogels in the subcutaneous areas at day 28 post s.c. injection; solid line distinguishes the PEG hydrogel edge or Far-Red traces at day 28 while dash line indicates the original size the gel/solution at day 0. Scale bar = 5 mm. (C) Quantitative analysis of the decay of Far-Red fluorescence over time; Ft: fluorescence reduction ratio at day t post injection, Ft = (It –I0) / I0, I: fluorescence intensity. Ft values were averaged by the two hydrogels at the mouse back. (D) Experimental data of the fluorescence reduction ratio Ft and time t plotted according to Equation (5).
The influences of residence times on in vivo drug delivery were subsequently studied by analyzing the decay of Far-Red dyes in PEG and Poloxamer hydrogels. PEG hydrogels lost ~33% of Far-Red fluorescence in the first three days, followed by a slow decrease in fluorescence (Figure 3C). Dye quenching can decrease the fluorescence signal; however, Far-Red was proven as a photo-stable fluorescent dye, as seen by their insignificant quenching in vitro over a month (Figure S4). Therefore, the decreased Far-Red signal in PEG hydrogels in vivo was likely caused by the diffusion of dyes from the gels followed by clearance within the surrounding cells while those dyes remaining within the gel maintained their fluorescence. As shown in Figure 3C, in contrast to PEG hydrogels, degradable and water-soluble Poloxamer hydrogels lost 82% of Far-Red fluorescence in the first three days, slightly below 92% loss of Far-Red fluorescence from injected solutions. Fast degradation and collapse of Poloxamer gels accelerated the reduction of Far-Red fluorescence in vivo. The decay of Far-Red fluorescence in both hydrogels shows second-order kinetics, which was then converted into plots of t/Ft vs. t according to Equation (5) where Wt was replaced with Ft. Herein, Ft is defined as the fluorescence reduction ratio at t day after s.c. injection (Ft = (It –I0) / I0, I: fluorescence intensity). After linear fitting of t/Ft vs. t with R2 > 0.99, we found the kinetics rate constants (K) for 20% PEG hydrogels and 20% Poloxamer hydrogels in vivo are 1.1 d−1 and 5.3 d−1 (Table 4), respectively, indicating a ~5-fold slower loss of Far-Red fluorescence in PEG hydrogels, which can be attributed to the higher stability and longer residence time of PEG hydrogels for sustainable payload delivery.
Table 4.
Quantitative analysis of in vivo Far-Red decay and in vitro NT-3 protein release from PEG and Poloxamer 407 hydrogels.
| Far-Red fluorescence reduction in vivo | Cumulative NT-3 release in vitro | |||||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||
| 20% PEG | 20% Poloxamer | Dye solution | 10% PEG | 15% PEG | 20% PEG | 20% PEG with peptides 2 | 20% Poloxamer | |
|
| ||||||||
| 1 K | 1.1 d−1 | 5.3 d−1 | 9.7 d−1 | 4.5 d−1 | 3.9 d−1 | 3.1 d−1 | 1.1 d−1 | 13.8 d−1 |
K: kinetic rate constant calculated from equation (5).
NLKEPYA peptides are conjugated to PEG hydrogels at a peptide: NT-3 = 50: 1 molar ratio.
3.5. Biocompatibility and local immune responses to in situ formed PEG hydrogels.
We next assessed the biocompatibility for in vivo applications of hydrogels.36 Host immune responses to biomaterial implants are inevitable, yet can be attenuated by choosing biocompatible materials and avoiding toxic and irritant reagents.36, 37 Biocompatibility of Poloxamer hydrogels has been well proven.12, 38 Herein, we studied the local immune responses to in situ formed PEG hydrogels and Poloxamer hydrogels to compare their biocompatibility.
After s.c. injection of hydrogels in mouse models, we collected the skin tissues surrounding hydrogels at the desired time, and then prepared a single cell suspension for immunofluorescence staining and flow cytometric analysis (Figure S5). Well-established immune cell markers were used to distinguish the frequency of total immune cells (leukocytes, Figure 4A) and their multiple subsets (Figure 4B–F).39 Similar to the acute immune response to Poloxamer hydrogels in skin tissues, at one day after gel injection, neutrophils (CD11b+Ly6G+) and monocytes (F4/80–Ly6C+) infiltrated tissues surrounding PEG hydrogels, resulting in a ~2-fold increase in the amount of total immune cells (CD45+) compared with normal skin tissues (Figure 4A–C). This acute inflammatory response to PEG hydrogels did not cause visible tissue redness or ulceration after injection and gel formation (Figure S6), indicating in situ crosslinking of PEG hydrogels induces minimal cytotoxicity and tissue damage in vivo.
Figure 4. Immune responses to in situ formed PEG hydrogels in subcutaneous areas.

Skin tissues that attached to 20% PEG hydrogels or 20% Poloxamer 407 hydrogels (control) or dye solutions (control) were collected at the desired times after gel injection and prepared to single cell suspension. Cells were stained by markers to distinguish leukocytes (A) and multiple immune cell subsets (B-F), and then analyzed by flow cytometry. **p < 0.01, from student’s t test.
The acute inflammatory response to PEG hydrogels and Poloxamer hydrogels was attenuated over time, as seen by the decrease in the frequency of total immune cells, neutrophils and monocytes at day 28 relative to day 1 (Figure 4A–C). Macrophages are reported to accumulate at biomaterials during the chronic inflammation phase and are involved in the formation of foreign body giant cells.36, 37 We did not observe a significant change in the frequency of macrophages over 28 days (Figure 4D). This observation might result from macrophage infiltration being limited to the interface between host tissues and hydrogels. The immune cell composition in tissues surrounding PEG hydrogels after a month is similar to that in normal tissues, and similar to tissues surrounding Poloxamer hydrogels. However, the degraded Poloxamer was shown to activate dendritic cells and accelerate their recruitment to the hydrogel (Figure 4E).40 Taken together, we concluded that in situ formed PEG hydrogels crosslinked by SH-VS Michael addition cause a low-level and resolvable inflammatory response and have in vivo biocompatibility comparable to Poloxamer hydrogels.
Collectively, we identified an injectable hydrogel formulation that provides elastic and biocompatible PEG hydrogels with higher mechanical strength, better bioadhesion, and longer residence time than Poloxamer hydrogels. Due to these advantages, the release of dye payloads from PEG hydrogels is ~5-fold slower than from Poloxamer hydrogels when gels are in situ formed in the subcutaneous area in vivo.
3.6. Screening and identification of NT-3 binding peptides.
With identification of a hydrogel carrier for in vivo applications, we next employed an affinity-controlled NT-3 release system. The injectable in situ crosslinking PEG hydrogel would be designed to minimize the initial burst release while providing sustained release for extended time periods. The SH and VS groups on PEG allow chemical conjugation to thiol-functionalized peptides that can reversibly interact with NT-3 proteins to control release.
We first screened short peptides that can selectively bind to NT-3 using a Ph.D.−7™ phage display peptide library (Schematic 1B). Following four rounds of biopanning, the amounts of output phages on NT-3 coated wells vs. control proteins (bovine serum albumin (BSA) and casein) were determined by titration (Figure 5A). The output for NT-3 coated wells was approximately 105-fold higher than BSA and casein, suggesting that the phages had a specific affinity to NT-3. After the fourth round of panning, a total of 17 phage clones were randomly selected and amplified, and the DNA of these selected clones was extracted and sequenced (Table S2). Three peptides, including NLKEPYA, ADARYKS, and SLTEPSS, were picked from the fourth-round phage pool, and all three showed strong NT-3 binding in a monoclonal phage ELISA assay compared to casein proteins (control) (Figure 5B).
Figure 5. Identification of NT-3 binding peptides.

(A) Plot of round four phage output on plates coated overnight with NT-3, BSA, or casein. (B) ELISA measurements of NLKEPYA, ADARYKS and SLTEPSS peptides or control Ph.D.™-7 peptide binding to NT-3 or casein (control). (C) Binding kinetics for NLKEPYA, ADARYKS and SLTEPSS peptides at 10, 100 and 200 μM concentrations to NT-3 proteins. kon, koff and KD are summarized in Table 5.
The affinity and binding kinetics of the three peptides were then quantified by binding analyses using Octet RED bio-layer interferometry (BLI) technology to evaluate biomaterial interactions. In this test, NT-3 proteins were immobilized to the super streptavidin coated biosensor surface, and soluble peptides were injected at concentrations ranging from 10 to 200 μM. Real-time association and dissociation of these peptides to immobilized NT-3 are shown in Figure 5C and the association rate kon, the dissociation rate koff and the binding affinity KD (KD = koff / kon) are summarized in Table 5. The binding analyses identified that NLKEPYA (KD = 3.4 μM) and SLTEPSS (KD = 10 μM) peptides have superior binding affinity to NT-3 than the ADARYKS peptide (KD = 29.6 μM) (Table 5).
Table 5.
Binding kinetics and binding affinity of NT-3 binding peptides
| Peptides | kon (×103, M−1s−1) | koff (×10−2, s−1) | KD (μM) |
|---|---|---|---|
|
| |||
| NLKEPYA | 3.99 | 1.37 | 3.4 |
| ADARYKS | 5.24 | 15.5 | 29.6 |
| SLTEPSS | 3.37 | 3.39 | 10.0 |
3.7. Peptide-conjugated PEG hydrogels show affinity-controlled NT-3 release.
Affinity-based hydrogels were prepared by mixing unmodified NT-3 proteins and thiol-functionalized peptides with the PEG-4SH/PEG-4VS solutions. We placed the hydrogels at the air-liquid interface in a Boyden chamber to study the behavior of NT-3 release and diffusion into the aqueous solutions through a porous membrane that mimics the RWM.
NT-3 release from unmodified PEG hydrogels had second-order kinetics, including the burst release in the first 24 hours followed by the slow release afterwards (Figure 6A). We defined Rt as the ratio of cumulative released NT-3 amount at t time vs. total NT-3 loaded in the gel, and then plotted t/Rt against time (Figure 6B). The kinetic rate constant (K) of NT-3 release from PEG gels was obtained according to Equation (5) where Wt was replaced to Rt. A correlation between release rates and polymer concentrations were observed with higher polymer concentration decelerating the NT-3 release. Specifically, K values for NT-3 release from 10%, 15% and 20% PEG hydrogels are 4.5 d−1, 3.9 d−1 and 3.1 d−1, respectively (Table 4). These results are consistent with the observation that water diffusion into dried 20% PEG hydrogels is slower than 10% and 15% hydrogels (Table 2) because 20% hydrogels have a more constrained structure and a relatively smaller pore size inside the hydrogels, which limit the diffusion of payloads or solutions. In contrast to stable PEG hydrogels, Poloxamer 407 hydrogels gradually dissolved and collapsed at the air-liquid interface, causing a > 95% of release of payloads within the first 2 days, while at this time point, only 16% of NT-3 proteins were released from 20% PEG hydrogels. Afterwards, NT-3 was continuously released from PEG hydrogels and 25% of NT-3 was released from 20% PEG hydrogels within the first week (Figure 6A), with 6% of NT-3 released in the next week (Figure S7). The slower NT-3 release rate over time fits the second order kinetic (Figure 6B) and implies a complete release of NT-3 on the time scale of months. In vitro NT-3 release from 20% Poloxamer hydrogels (K = 13.8 d−1) was ~4.5-fold faster than from 20% PEG hydrogels (K = 3.1 d−1, Table 4), comparable to the ~5-fold faster loss of Far-Red fluorescence from Poloxamer hydrogels than from PEG hydrogels in vivo (Figure 3, Table 4).
Figure 6. Cumulative NT-3 protein release from unmodified or peptide-conjugated PEG hydrogels in vitro.

(A) NT-3 release from 10%, 15% and 20% of unmodified PEG hydrogels and 20% Poloxamer 407 hydrogels for over 7 days. Rt: released ratio, Rt = released protein amount at t time / total protein amount. (B) Experimental data of Rt in (A) and time t plotted according to Equation (5). (C) NT-3 release from 20% PEG hydrogels conjugated with NLKEPYA, ADARYKS, or SLTEPSS at a peptide: protein = 50: 1 molar ratio within the first 2 days. (D) NT-3 release from 20% PEG hydrogels conjugated with NLKEPYA at a peptide: protein = 50: 1 molar ratio for over a week. (E) Experimental data of Rt in (C) and time t plotted according to Equation (5). (F) NT-3 release from 20% PEG hydrogels conjugated with NLKEPYA at different peptide: protein molar ratios within the first 2 days. **p < 0.01, from student’s t test.
Next, we studied the NT-3 release from the affinity-based 20% PEG hydrogels that were conjugated with the three peptides, respectively, at a peptide: protein = 50: 1 molar ratio. NLKEPYA and SLTEPSS peptides, which have high affinity to NT-3 (KD ≤ 10 μM, Table 5), significantly reduced the protein release within the first 2 days, while the low-affinity ADARYK (KD = 29.6 μM) did not change the burst release of NT-3 (Figure 6C). Protein release from NLKEPYA-conjugated PEG hydrogels exhibited second-order kinetics, which is similar to the release from unmodified PEG hydrogels except for a slower release profile (Figure 6D, S8). Quantitatively, the kinetic constant K calculated from Equation (5) for NT-3 release from 20% PEG hydrogels decreased from 3.1 d−1 to 1.1 d−1 after conjugation with NLKEPYA peptides at a peptide: protein = 50: 1 molar ratio (Figure 6E, S9, Table 4), suggesting a ~3-fold slower NT-3 release by affinity peptides and thus demonstrating that reversible binding within the gel can successfully control the release of NT-3 (Schematic 1C). In addition to peptide affinity, we also observed that a higher peptide-to-protein ratio in the gel can further decelerate the NT-3 release, yet a plateau was reached at peptide-to-protein ratios above 100 (Figure 6F).
Overall, PEG hydrogels conjugated with NLKEPYA and SLTEPSS peptides that have affinity for NT-3 with KD ≤ 10 μM successfully reduced the burst release of NT-3 from the gel and resulted in sustained release. Affinity-controlled protein release can be tuned by changing the ratio of peptides to proteins in the gel. In this study, we have demonstrated that affinity peptides can suppress the burst release of NT-3 from PEG hydrogels in addition to achieving a more sustainable release than unmodified hydrogels for over one week (Figure 6D, 6E, S8, S9). While these in vitro release studies provide a proof of concept, they provide a limited representation of the release behavior of NT-3 from affinity gels in vivo as well as the long-term drug release behavior. Future studies will be needed to clarify the affinity-controlled protein release behavior in animal models at longer times (months or longer), which would have more factors affect the drug release, including the degradation of peptides and proteins as well as the changes in the internal environment of hydrogels after in vivo implantation (e.g. cell infiltration, pH, water content).
4. Conclusions
In this study, we identified a one-step mixing formulation to prepare injectable in situ crosslinking PEG hydrogels by using SH-VS Michael addition. Tunable gelation time for this PEG-4SH/PEG-4VS formulation, depending on the temperature and polymer concentrations, provides sufficient time for gel handling at room temperature, yet efficient gelation at body temperature without the need of toxic catalysts. PEG hydrogels crosslinked by SH-VS Michael addition were elastic and stable, with higher mechanical rigidity, better bioadhesion, longer in vivo residence time and similar biocompatibility compared to physically crosslinked Poloxamer 407 hydrogels.
This injectable PEG hydrogel formulation is suitable for delivering drugs to biological sites that have irregular structure and require low cytotoxicity, such as the tympanic cavity in the middle ear. Based on this hydrogel carrier, we successfully designed an affinity-based release system for NT-3 delivery by conjugating hydrogels with high-affinity peptides specific to NT-3 proteins. Hydrogel-anchored peptides reversibly bind to physically loaded NT-3 proteins and therefore sustain their release. Polymer concentrations, affinity of peptides and the peptide-to-protein ratios were found critical for adjusting the rate of NT-3 release from PEG hydrogels. Overall, we developed an injectable protein delivery system for application in the RWM that can provide sustained release of NT-3 proteins in an affinity-controlled manner. However, additional studies will be necessary for translation, such as analysis of the host response within the inner ear and possibly developing a degradable formulation, which could be achieved by crosslinking with cleavable linkers.
Supplementary Material
Highlights.
PEG hydrogels crosslinked by thiol-vinyl sulfone Michael reaction are injectable and safe for localized delivery of growth factors to the middle ear.
Chemically crosslinked PEG hydrogels are as biocompatible as physically crosslinked Poloxamer 407 hydrogels in vivo.
In situ formed PEG hydrogels release payloads at a ~5-fold slower rate in the subcutaneous area than Poloxamer 407 hydrogels.
PEG hydrogels conjugated with affinity peptides specific to the neurotrophin-3 growth factor release proteins in vitro at a ~3-fold slower rate than unmodified PEG hydrogels.
Acknowledgements
Funding for this work was provided by NIH R01EB005678 and R01AI148076
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- (1).Mietto BS, Mostacada K, Martinez AM Neurotrauma and inflammation: CNS and PNS responses. Mediators Inflamm. (2015), 251204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kujawa SG, Liberman MC Adding insult to injury: cochlear nerve degeneration after “temporary” noise-induced hearing loss. J. Neurosci. 29 (2009), 14077–14085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wan G, Gómez-Casati ME, Gigliello AR, Liberman MC, Corfas G. Neurotrophin-3 regulates ribbon synapse density in the cochlea and induces synapse regeneration after acoustic trauma. eLife 3 (2014), e03564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Kechai NE, Agnely F, Mamelle E, Nguyen Y, Ferrary E, Bochot A. Recent advances in local drug delivery to the inner ear. Int. J. Pharm, 494 (2015), 83–101. [DOI] [PubMed] [Google Scholar]
- (5).Piu F, Bishop KM Local drug delivery for the treatment of neurotology disorders. Front. Cell. Neurosci. 13 (2019), 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Rathnam C, Chueng SD, Ying YM, Lee K, Kwan K. Developments in bio-inspired nanomaterials for therapeutic delivery to treat hearing loss. Front. Cell. Neurosci. 13 (2019), 493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Li J, Mooney DJ Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 1 (2016), 16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Dimatteo R, Darling NJ, Segura T. In situ forming injectable hydrogels for drug delivery and wound repair. Adv. Drug Deliv. Rev. 127 (2018), 167–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Mironi-Harpaz I, Wang DY, Venkatraman S, Seliktar D. Photopolymerization of cell-encapsulating hydrogels: Crosslinking efficiency versus cytotoxicity. Acta Biomater. 8 (2012), 1838–1848. [DOI] [PubMed] [Google Scholar]
- (10).Patenaude M, Campbell S, Kinio D, Hoare T. Tuning gelation time and morphology of injectable hydrogels using ketone–hydrazide cross-linking. Biomacromolecules 15 (2014), 781–790. [DOI] [PubMed] [Google Scholar]
- (11).Suzuki J, Corfas G, Liberman MC Round-window delivery of neurotrophin 3 regenerates cochlear synapses after acoustic overexposure. Sci. Rep. 6 (2016), 24907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Giuliano E, Paolino D, Fresta M, Cosco D. Mucosal applications of poloxamer 407-based hydrogels: an overview. Pharmaceutics 10 (2018), 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Zustiak SP, Wei Y, Leach JB Protein-hydrogel interactions in tissue engineering mechanisms and applications. Tissue Eng. Part B Rev. 19 (2013), 160–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Jeon O, Powell C, Solorio LD, Krebs MD, Alsberg E. Affinity-based growth factor delivery using biodegradable, photocrosslinked heparin-alginate hydrogels. J. Control. Release. 154 (2011), 258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Jaylor SJ, McDonald III JW, Sakiyama-Elbert SE Controlled release of neurotrophin-3 from fibrin gels for spinal cord injury. J. Control. Release 98 (2004), 281–294. [DOI] [PubMed] [Google Scholar]
- (16).Vulic K, Shoichet MS Tunable growth factor delivery from injectable hydrogels for tissue engineering. J. Am. Chem. Soc. 134 (2012), 882–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lin C, Anseth KS Controlling Affinity Binding with Peptide-Functionalized Poly(ethylene glycol) Hydrogels. Adv Funct Mater. 19(2009): 2325–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Soontornworajit B, Zhou J, Snipes MP, Battig MR, Wang Y. Affinity hydrogels for controlled protein release using nucleic acid aptamers andcomplementary oligonucleotides. Biomaterials, 32 (2011), 6839–6849. [DOI] [PubMed] [Google Scholar]
- (19).Chatani S, Nair DP, Bowman CN Relative reactivity and selectivity of vinyl sulfones and acrylates towards the thiol–Michael addition reaction and polymerization. Poly. Chem. 4 (2013), 1048–1055. [Google Scholar]
- (20).Nair DP, Podgorski M, Chatani S, Gong T, Xi W, Fenoli CR, Bowman CN The thiol-Michael addition click reaction: a powerful and widely used tool in materials chemistry. Chem Mater, 26 (2014), 724–744. [Google Scholar]
- (21).Northrop BH, Frayne SH, Choudhary U. Thiol–Maleimide “click” chemistry: evaluating the influence of solvent, initiator, and thiol on the reaction mechanism, kinetics, and selectivity. Poly. Chem. 6 (2015) 3415–3430. [Google Scholar]
- (22).Liu JK, Teng Q, Garrity-Moses M, Federici T, Tanase D, Imperiale MJ, Boulis NM A novel peptide defined through phage display for therapeutic protein and vector neuronal Targeting. Neurobiol. Dis. 19 (2005), 407–418. [DOI] [PubMed] [Google Scholar]
- (23).Wu J, Park JP, Dooley K, Cropek DM, West AC, Banta S. Rapid development of new protein biosensors utilizing peptides obtained via phage display. PLoS ONE 6 (2011), e24948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Chang JY, Oh YK, Choi HG, Kim YB, Kim CK Rheological evaluation of thermosensitive and mucoadhesive vaginal gels in physiological conditions. Int. J. Pharm. 241 (2002), 155–163. [DOI] [PubMed] [Google Scholar]
- (25).Yan C, Pochan DJ Rheological properties of peptide-based hydrogels for biomedical and other applications. Chem. Soc. Rev. 39 (2010), 3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Grassi M, Grassi G. Mathematical modelling and controlled drug delivery: matrix systems. Curr. Drug Deliv. 2 (2005), 97–116. [DOI] [PubMed] [Google Scholar]
- (27).Kim SW, Bae YH, Okano T. Hydrogels: swelling, drug loading, and release. Pharm. Res. 9 (1992), 283–290. [DOI] [PubMed] [Google Scholar]
- (28).Park H, Guo X, Temenoff JS, Tabata Y, Caplan AI, Kasper FK, Mikos AG Effect of swelling ratio of injectable hydrogel composites on chondrogenic differentiation of encapsulated rabbit marrow mesenchymal stem cells in vitro. Biomacromolecules 10 (2009), 541–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Katime I, Mendizábal E. Swelling properties of new hydrogels based on the dimethyl amino ethyl acrylate methyl chloride quaternary salt with acrylic acid and 2-methylene butane-1,4-dioic acid monomers in aqueous solutions. Mater. Sci. Appl. 1 (2010), 162–167. [Google Scholar]
- (30).Ritger PL, Peppas NA A simple equation for description of solute release I. Fickian and non-Fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J. Controlled Release 5 (1987), 23–36. [PubMed] [Google Scholar]
- (31).Saidi M, Dabbaghi A, Rahmani S. Swelling and drug delivery kinetics of click-synthesized hydrogels based on various combinations of PEG and star-shaped PCL: influence of network parameters on swelling and release behavior. Polymer Bulletin, 77 (2020), 3989–4010. [Google Scholar]
- (32).Requejo R, Hurd TR, Costa NJ, Murphy MP Cysteine residues exposed on protein surfaces are the dominant intramitochondrial thiol and may protect against oxidative damage. FEBS J. 277 (2020), 1465–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Sudarshan S, Sunil B B. In vivo mucoadhesive strength appraisal of gum Manilkara zapota. Braz. J. Pharm. Sci. 51 (2015), 689–698. [Google Scholar]
- (34).Kantibhai PT, Patel KS, Shah S. Test methods of bioadhesive system. World J. Pharm. Res. 7 (2018), 267–278. [Google Scholar]
- (35).Ibrahim EA, Ismail S, Fetih G, Shaaban O, Hassanein K, Abdellah NH Development and characterization of thermosensitive pluronic based metronidazole in situ gelling formulations for vaginal application. Acta Pharm, 62 (2012), 59–70. [DOI] [PubMed] [Google Scholar]
- (36).Brown BN, Badylak SF Biocompatibility and immune response to biomaterials. Regenerative Medicine Applications in Organ Transplantation. Chapter 11 (2014), 151–162. [Google Scholar]
- (37).Anderson JM, Rodriguez A, Chang DT Foreign body reaction to biomaterials. Semin. Immunol. 20 (2008), 86–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kwon JW, Han YK, Lee WJ, Cho CS, Paik SJ, Cho DI, Lee JH, Wee WR Biocompatibility of poloxamer hydrogel as an injectable intraocular lens: a pilot study. J. Cataract. Refract. Surg. 31 (2005), 607–613. [DOI] [PubMed] [Google Scholar]
- (39).Azarin SM, Yi Y, Gower RM, Aguado BA, Sullivan ME, Goodman AG, Jiang EJ, S Rao S, Ren Y, Tucker SL, Backman V, Jeruss JS, Shea L. D. LD In vivo capture and label-free detection of early metastatic cells. Nat. Commun. 6 (2015), 8094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Keselowsky BG, Lewis JS Dendritic cells in the host response to implanted materials. Semin. Immunol. 29 (2017), 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.