

Abstract
Background
Despite the development of new treatment options, the prognosis of high‐risk neuroblastoma patients is still poor; more than half of patients experience disease recurrence. High‐dose chemotherapy and haematopoietic stem cell rescue (i.e. myeloablative therapy) might improve survival. This review is the second update of a previously published Cochrane review.
Objectives
Primary objective
To compare the efficacy, that is event‐free and overall survival, of high‐dose chemotherapy and autologous bone marrow or stem cell rescue with conventional therapy in children with high‐risk neuroblastoma.
Secondary objectives
To determine adverse effects (e.g. veno‐occlusive disease of the liver) and late effects (e.g. endocrine disorders or secondary malignancies) related to the procedure and possible effects of these procedures on quality of life.
Search methods
We searched the electronic databases The Cochrane Central Register of Controlled Trials (CENTRAL) (The Cochrane Library 2014, issue 11), MEDLINE/PubMed (1966 to December 2014) and EMBASE/Ovid (1980 to December 2014). In addition, we searched reference lists of relevant articles and the conference proceedings of the International Society for Paediatric Oncology (SIOP) (from 2002 to 2014), American Society for Pediatric Hematology and Oncology (ASPHO) (from 2002 to 2014), Advances in Neuroblastoma Research (ANR) (from 2002 to 2014) and American Society for Clinical Oncology (ASCO) (from 2008 to 2014). We searched for ongoing trials by scanning the ISRCTN register (www.isrct.com) and the National Institute of Health Register (www.clinicaltrials.gov). Both registers were screened in April 2015.
Selection criteria
Randomised controlled trials (RCTs) comparing the efficacy of myeloablative therapy with conventional therapy in high‐risk neuroblastoma patients.
Data collection and analysis
Two authors independently performed study selection, data extraction and risk of bias assessment. If appropriate, we pooled studies. The risk ratio (RR) and 95% confidence interval (CI) was calculated for dichotomous outcomes. For the assessment of survival data, we calculated the hazard ratio (HR) and 95% CI. We used Parmar's method if hazard ratios were not reported in the study. We used a random‐effects model.
Main results
We identified three RCTs including 739 children. They all used an age of one year as the cut‐off point for pre‐treatment risk stratification. The first updated search identified a manuscript reporting additional follow‐up data for one of these RCTs, while the second update identified an erratum of this study. There was a significant statistical difference in event‐free survival in favour of myeloablative therapy over conventional chemotherapy or no further treatment (three studies, 739 patients; HR 0.78, 95% CI 0.67 to 0.90). There was a significant statistical difference in overall survival in favour of myeloablative therapy over conventional chemotherapy or no further treatment (two studies, 360 patients; HR 0.74, 95% CI 0.57 to 0.98). However, when additional follow‐up data were included in the analyses the difference in event‐free survival remained statistically significant (three studies, 739 patients; HR 0.79, 95% CI 0.70 to 0.90), but the difference in overall survival was no longer statistically significant (two studies, 360 patients; HR 0.86, 95% CI 0.73 to 1.01). The meta‐analysis of secondary malignant disease and treatment‐related death did not show any significant statistical differences between the treatment groups. Data from one study (379 patients) showed a significantly higher incidence of renal effects, interstitial pneumonitis and veno‐occlusive disease in the myeloablative group compared to conventional chemotherapy, whereas for serious infections and sepsis no significant difference between the treatment groups was identified. No information on quality of life was reported. In the individual studies we evaluated different subgroups, but the results were not univocal in all studies. All studies had some methodological limitations.
Authors' conclusions
Based on the currently available evidence, myeloablative therapy seems to work in terms of event‐free survival. For overall survival there is currently no evidence of effect when additional follow‐up data are included. No definitive conclusions can be made regarding adverse effects and quality of life, although possible higher levels of adverse effects should be kept in mind. A definitive conclusion regarding the effect of myeloablative therapy in different subgroups is not possible. This systematic review only allows a conclusion on the concept of myeloablative therapy; no conclusions can be made regarding the best treatment strategy. Future trials on the use of myeloablative therapy for high‐risk neuroblastoma should focus on identifying the most optimal induction and/or myeloablative regimen. The best study design to answer these questions is a RCT. These RCTs should be performed in homogeneous study populations (e.g. stage of disease and patient age) and have a long‐term follow‐up. Different risk groups, using the most recent definitions, should be taken into account.
It should be kept in mind that recently the age cut‐off for high risk disease was changed from one year to 18 months. As a result it is possible that patients with what is now classified as intermediate‐risk disease have been included in the high‐risk groups. Consequently the relevance of the results of these studies to the current practice can be questioned. Survival rates may be overestimated due to the inclusion of patients with intermediate‐risk disease.
Plain language summary
High‐dose chemotherapy and stem cell transplant compared to conventional therapy for children with high‐risk neuroblastoma
Despite the development of new treatment options, the prognosis of high‐risk neuroblastoma patients still remains poor; in more than half of patients the disease returns. Stem cell rescue replaces blood‐forming stem cells that were destroyed by high‐dose chemotherapy in order to recover the bone marrow. It is also known as myeloablative therapy and might improve the survival of these patients. A well‐informed decision on the use of myeloablative therapy in the treatment of children with high‐risk neuroblastoma should be based on high quality evidence of effectiveness for treating tumours and side effects.
This systematic review focused on randomised studies comparing the effectiveness of myeloablative therapy with conventional therapy in children with high‐risk neuroblastoma. The authors found three studies including 739 patients. These studies provide evidence that myeloablative therapy improves event‐free survival (that is, the time until a certain event, for example tumour progression, the development of a second tumour, or death from any cause occurs). For overall survival (that is, the time until a patient dies from any cause, so not only from the tumour or its treatment, but for example, also from a car accident) there is no evidence of a better outcome in patients treated with myeloablative therapy. Side effects such as renal (kidney) effects, interstitial pneumonitis (a type of lung disease) and veno‐occlusive disease (a condition in which some of the small veins in the liver are obstructed) were more common in patients treated with myeloablative therapy than conventional chemotherapy. It should be noted that this systematic review only allows a conclusion on the concept of myeloablative therapy; no conclusions regarding the best treatment strategy with regard to, for example, types of chemotherapeutic agents and the use of radiation therapy, could be made. More high quality research is needed.
It should be noted that recently the age cut‐off for high‐risk disease was changed from one year to 18 months. As a result it is possible that patients with what is now classified as intermediate‐risk disease were included in the high‐risk groups. Consequently the relevance of the results of these studies to the current practice can be questioned. Survival rates may be overestimated due to the inclusion of patients with intermediate‐risk disease.
Background
Description of the condition
Neuroblastoma is the most common extracranial solid tumour in children comprising 8% to 10% of all childhood cancers. The incidence is nearly 10 per 1,000,000 children under the age of 15 years and 90% of cases are diagnosed in the first 10 years of life. Children with neuroblastoma mostly present with abdominal disease and more than half of the cases have advanced disease (defined as stage III or IV disease) (Aydin 2009; Brodeur 2006; Goldsby 2004).
Neuroblastoma is one of the most challenging and enigmatic neoplasms of childhood because of its biological heterogeneity and contrasting patterns of clinical behaviour. Some young infants with favourable disease may experience complete spontaneous regression while older children with metastatic disease mostly relapse despite initial response to chemotherapy.
The prognosis and management of children with neuroblastoma is highly dependent on clinical, histopathological and biological characteristics, and they are stratified into risk groups based on prognostic factors (Goldsby 2004; Maris 2005; Maris 2007; Weinstein 2003). For pre‐treatment risk stratification traditionally an age of one year was used as a cut‐off point. The low‐risk disease group included cases younger than one year who had stage I, II or IV‐S disease with favourable histopathology and no MYCN oncogene amplification (Brodeur 2006; Goldsby 2004; Maris 2005; Maris 2007; Weinstein 2003).
High‐risk neuroblastoma cases were traditionally characterised by an age older than one year, disseminated disease, MYCN oncogene amplification and unfavourable histopathologic findings (Brodeur 2006; Goldsby 2004; Maris 2005; Weinstein 2003). However, in recent years a new neuroblastoma risk classification system has been developed by the International Neuroblastoma Risk Group (INRG) Task Force resulting in standardised approaches for the initial evaluation and treatment stratification of neuroblastoma patients (Cohn 2009). In this INRG classification system an age cut‐off of 18 months is used for pre‐treatment risk stratification, as opposed to the traditional cut‐off of one year. In many of the currently published studies patients with what is now understood to be intermediate‐risk disease were classified as high‐risk patients. Consequently the relevance of the results of these studies to current practice can be questioned.
Description of the intervention
Substantial improvement has been achieved in the cure of patients with low‐risk neuroblastoma resulting in survival rates up to 90% (Brodeur 2006; Goldsby 2004). In high‐risk cases, despite intensified combination chemotherapies, surgery, radiotherapy and the use of differentiation agents, prognosis improved only modestly with long‐term survival in less than one‐third of patients (Brodeur 2006; De Bernardi 2003; Goldsby 2004; Weinstein 2003; Peinemann 2015a). In the last two decades higher remission rates have been achieved with intensive induction chemotherapy regimens combined with surgical resection, external irradiation or both (Brodeur 2006; Castel 1995; Goldsby 2004; Kaneko 2002; Kushner 2004; Laprie 2004; Sawaguchi 1990). The effects of retinoic acid post consolidation therapy for high‐risk neuroblastoma patients treated with autologous hematopoietic stem cell transplantation are currently not clear (Peinemann 2015b).
The challenge is to maintain remission since more than half of patients with high‐risk disease develop systemic disease recurrence with or without a relapse at the primary tumour site (Brodeur 2006; Goldsby 2004; Matthay 1993; Weinstein 2003). AntiGD2 antibody‐based immunotherapy has been shown to improve event‐free and overall survival in high‐risk neuroblastoma patients in remission after multimodality treatment (Yu 2010). Therapy failures are mostly attributed to development of resistance to chemotherapy and minimal residual disease is considered an important cause of recurrence (Burchill 2004; Keshelava 1998; Reynolds 2001; Reynolds 2004).
The idea that further increasing dose intensity may overcome chemotherapy resistance has provided a rationale for aggressive high‐dose chemotherapy consolidation protocols (Cheung 1991; Pritchard 1995). Such myeloablative chemotherapy regimens utilise effective high‐dose drug combinations which can be safely escalated to levels above those causing bone marrow ablation. Rapid bone marrow reconstitution can be achieved by autologous stem cell rescue. Autologous peripheral blood stem cells (PBSC) are the preferred source for rescue as this has several advantages over autologous bone marrow grafts. For example, the procedure of PBSC collection is easier, the incidence of tumour cell contamination is lower, and the yield of stem cells is higher (Brodeur 2006; Cohn 1997; Ladenstein 1994). A possible limitation of using autologous products is the risk of tumour cell contamination in the graft, which has been shown to contribute to relapse. Considerable efforts have been made to detect and remove tumour (negative purging) or select progenitor cells (positive purging) before reinfusion (Ladenstein 2004). Disease status prior to stem cell rescue has a crucial influence on final outcome. Patients in complete, very good partial or partial remission have a better prognosis, while those with stable disease or no response have a poor outcome (Ladenstein 2004).
Patients undergoing stem cell rescue may experience toxicities related to conditioning regimens like veno‐occlusive disease of the liver or haemorrhagic cystitis (Bollard 2006). Growth failure, endocrine disorders such as gonadal or thyroid dysfunction, hearing impairment, renal impairment, orthopedic complications as well as the occurrence of secondary malignancies are among the late complications following high‐dose chemotherapy and stem cell rescue (Bollard 2006; Trahair 2007).
Why it is important to do this review
Many retrospective and prospective non‐randomised studies support the use of a myeloablative consolidation in neuroblastoma. Retrospective studies mostly suggest that intensification of consolidation therapy with autologous stem cell rescue following high‐dose chemotherapy improves survival (Castel 1995; Di Caro 1994; Matthay 1995; Philip 1997; Stram 1996; Verdeguer 2004). The results of non‐randomised pilot studies by the Children's Cancer Group also suggest a modest prolongation of event‐free survival for children with high‐risk neuroblastoma (Matthay 1995).
This is the second update of the first systematic review evaluating the current state of evidence on the efficacy of high‐dose chemotherapy and autologous haematopoietic stem cell rescue compared with conventional therapy in patients diagnosed with high‐risk neuroblastoma (Yalçin 2010; Yalçin 2013).
Objectives
Primary objective
To compare the efficacy, that is event‐free and overall survival, of high‐dose chemotherapy and autologous bone marrow or stem cell rescue with conventional therapy in children with high‐risk neuroblastoma.
Secondary objectives
To determine adverse effects (e.g. veno‐occlusive disease of the liver) and late effects (e.g. endocrine disorders or secondary malignancies) related to the procedure and possible effects of these procedures on quality of life.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials (RCTs) that compared the efficacy of high‐dose chemotherapy and autologous bone marrow or stem cell rescue with conventional therapy were considered for inclusion.
Types of participants
Participants included children (aged < 21 years) with high‐risk neuroblastoma. High‐risk neuroblastoma is defined as a combination of the following characteristics: unfavourable age (as defined by the authors of the included studies), advanced stage disease (as defined by the authors of the included studies), presence of MYCN oncogene amplification, or unfavourable histopathologic findings (as defined by the authors of the included studies) or both. Recently the age cut‐off for high‐risk disease was changed from one year to 18 months. As a result it is possible that patients with what is now classified as intermediate‐risk disease were included in the high‐risk group.
Types of interventions
Interventions included high‐dose chemotherapy with autologous bone marrow or stem cell rescue versus conventional therapy (i.e. either conventional chemotherapy or no further treatment). High‐dose chemotherapy is defined as chemotherapy sufficient to require stem cell rescue. Conventional chemotherapy is defined as chemotherapy at a lower dose than the high‐dose chemotherapy without the need for stem cell rescue.
Types of outcome measures
Primary outcomes
Event‐free survival (usually defined as the time to recurrence or progression of disease or death from any cause or varying definitions, as used by the authors).
Overall survival (defined as the time to death from any cause).
Secondary outcomes
Adverse events (e.g. stomatitis, diarrhoea, fatigue, anaemia, leukopenia, thrombocytopenia or veno‐occlusive disease of the liver) and late effects (e.g. endocrine disorders or secondary malignancies).
Quality of life.
Search methods for identification of studies
We searched the following electronic databases: the Cochrane Central Register of Controlled Trials (CENTRAL) (The Cochrane Library 2014, issue 11), MEDLINE/PubMed (from 1966 to December 2014) and EMBASE/Ovid (from 1980 to December 2014). The search strategies for the different electronic databases (using a combination of controlled vocabulary and text word terms) are shown in Appendix 1, Appendix 2 and Appendix 3.
We located information about trials not registered in CENTRAL, MEDLINE or EMBASE, either published or unpublished, by searching the reference lists of relevant articles and review articles. We also scanned the conference proceedings of the International Society for Paediatric Oncology (SIOP) (from 2002 to 2014), the American Society for Pediatric Hematology and Oncology (ASPHO) (from 2002 to 2014), Advances in Neuroblastoma Research (ANR) (from 2002 to 2014) and the American Society for Clinical Oncology (ASCO) (from 2008 to 2014), if available electronically and otherwise by handsearching. We searched for ongoing trials by scanning the ISRCTN register (www.isrct.com) and the National Institute of Health Register (www.clinicaltrials.gov). Both registers were screened in April 2015. We imposed no language restriction.
Data collection and analysis
Selection of studies
Two authors independently screened the search results to identify studies meeting the inclusion criteria. We obtained full manuscripts for any study that appeared to meet the inclusion criteria based on title, or abstract or both for closer inspection. We clearly reported reasons for exclusion for any study considered for the review. Discrepancies between authors were resolved by consensus. No third party arbitration was needed.
Data extraction and management
Two authors independently extracted data using standardised forms. We extracted the following data:
the characteristics of the participants at the time of randomisation (such as age, sex, stage of disease, histology, MYCN oncogene amplification, received induction treatment, received external irradiation, remission status at myeloablative treatment and stem cell rescue);
the characteristics of interventions (details of myeloablative therapy: drugs used, routes of delivery, dose, timing, use of total body irradiation; details of the stem cell rescue: timing and methods of cell harvest, timing of stem cell rescue, number of cells infused, contamination with tumour cells);
details of supportive care (such as the use of prophylactic antibiotics, growth factors, granulocyte infusions);
details of outcome measures as described above; and
length of follow up.
In case of disagreement between authors, we re‐examined the abstracts and articles and discussed these until consensus was achieved. No third party arbitration was needed.
Assessment of risk of bias in included studies
Two authors independently performed assessment of risk of bias in the included studies. For this second update we used the most recent recommendations of the Childhood Cancer Group (that is selection bias, performance bias, detection bias (for each outcome separately), attrition bias (for each outcome separately), reporting bias and other bias). We used the 'risk of bias' items and definitions of low risk, unclear risk and high risk as described in the module of the Cochrane Childhood Cancer Group (Kremer 2014), which is based on the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). All RCTs were scored using the new 'risk of bias' items. Discrepancies between authors were resolved by consensus. No third party arbitration was needed. The risk of bias in the included studies was taken into account in the interpretation of the review's results.
Measures of treatment effect
The risk ratio (RR) was calculated for dichotomous outcomes along with the corresponding 95% confidence interval (CI). For the assessment of survival, we used the generic inverse variance function of RevMan to combine logs of the hazard ratios (HR) and estimate corresponding 95% CI. We used Parmar's method if hazard ratios were not reported in the study (Parmar 1998).
Assessment of heterogeneity
We assessed heterogeneity both by visual inspection of the forest plots and by a formal statistical test for heterogeneity, i.e. the I² statistic (Higgins 2003). If there was evidence of substantial heterogeneity (I² > 50%), this was reported.
Assessment of reporting biases
In addition to the evaluation of reporting bias, as described in the Assessment of risk of bias in included studies section, we planned to construct a funnel plot to graphically ascertain the existence of publication bias. However, as a rule of thumb, tests for funnel plot asymmetry should be used only when there are at least 10 studies included in the meta‐analysis, because when there are fewer studies the power of the tests is too low to distinguish chance from real asymmetry (Sterne 2011). Since only three trials could be included in the review, we did not construct funnel plots.
Data synthesis
We entered data into Review Manager (RevMan 2014) and analysed data according to the guidelines of the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2011). We pooled studies for meta‐analysis when appropriate. We extracted data by allocation intervention, irrespective of compliance with the allocated intervention, in order to allow for an 'intention‐to‐treat' (ITT) analysis. We used a random‐effects model for the estimation of treatment effects throughout the review. Where possible, we presented data separately for different prognostic factors such as disease status at the time of stem cell rescue, MYCN amplification status and serum lactate dehydrogenase levels. For outcomes where only one study was available, we were unable to calculate a RR if one of the treatment groups experienced no events and the Fischer’s exact test was used instead; this option is not available in RevMan and therefore we used http://graphpad.com/quickcalcs/contingency2/.
Sensitivity analysis
For all outcomes for which pooling was possible we performed sensitivity analyses for all quality criteria separately. We excluded low quality studies and studies for which the quality was unclear, and compared the results of the good quality studies with the results of all available studies.
Results
Description of studies
Results of the search
We ran searches of the electronic databases CENTRAL, MEDLINE/PubMed and EMBASE/Ovid in January 2009 for the original version of this review. This search yielded a total of 1463 references. Initial screening excluded 1427 references which clearly did not meet all the criteria for considering studies for this review. We obtained 36 references in full for closer inspection. Three studies fulfilled all the criteria for considering studies for this review and were included in the review (Berthold 2005; Matthay 1999; Pritchard 2005). We excluded the remaining 33 articles for reasons described in the Characteristics of excluded studies table.
For the first update we ran searches of CENTRAL, MEDLINE/PubMed and EMBASE/Ovid in June 2012 yielding a total of 266 references which were added to the search results from January 2009. Initial screening excluded 265 references which clearly did not meet the inclusion criteria. We obtained one full‐text study for closer inspection. It fulfilled the inclusion criteria and was thus included. This study provides additional follow‐up data for the Matthay 1999 study.
For the second update we ran searches of CENTRAL, MEDLINE/PubMed and EMBASE/Ovid in December 2014 yielding a total of 313 references. Initial screening excluded all 313 references; no studies needed to be seen in full text.
We scanned the reference lists of relevant articles and reviews and conference proceedings (i.e. SIOP, ASPHO and ASCO) and did not identify any additional eligible studies. One trial was added to the Characteristics of excluded studies table. By scanning the ANR conference proceedings we identified one study that has not yet been published in full and is awaiting further assessment (see Characteristics of studies awaiting classification table). This study provides additional follow‐up for the Berthold 2005 study. We identified an erratum on one of the included studies (Matthay 1999). No eligible ongoing trials were identified.
In summary, the total number of included RCTs was three (described in four publications and one erratum). Characteristics of included studies are summarised below (for further details see the Characteristics of included studies table). We also identified one study awaiting assessment.
Included studies
All three eligible RCTs were multi‐centre studies, two of which were based in Europe (Berthold 2005; Pritchard 2005) and one in North America (Matthay 1999). Patients were recruited between January 1982 (Pritchard 2005) and November 2002 (Berthold 2005). The total number of patients included in the three studies was 739; 370 children received high‐dose chemotherapy and stem cell rescue, whereas 369 children received either conventional chemotherapy (Berthold 2005; Matthay 1999) or no further treatment (Pritchard 2005). All trials used different induction regimens (i.e. different chemotherapeutic agents and different (cumulative) dosages) and different myeloablative treatments. In Berthold 2005 and Matthay 1999 there were also differences in treatment patients received in the study itself (for example, external radiotherapy, therapeutic MIBG‐I131, immunotherapy and retinoic acid treatment). See the Characteristics of included studies table for further details. All patients were diagnosed with high‐risk neuroblastoma. The definitions of high‐risk neuroblastoma differed between studies (See Characteristics of included studies table). Matthay 1999 included newly diagnosed patients. It was unclear if the patients in the other studies were newly diagnosed. None of the studies reported the exact age of the participants; only the number of cases above and below one year of age was reported.
Risk of bias in included studies
See the risk of bias section of the Characteristics of included studies table and Figure 1 for the exact scores per included study.
1.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study (it should be noted that in the study describing additional follow‐up the associated risk of detection bias was thus unclear for all outcomes except overall survival)
Allocation
For evaluating selection bias we have assessed the random sequence generation and the allocation concealment. The risk of selection bias was low in two studies (Berthold 2005; Pritchard 2005), while in one study it was unclear (Matthay 1999).
Blinding
For evaluating performance bias we have assessed the blinding of participants and personnel. In two studies there was a high risk of bias (Berthold 2005; Matthay 1999), while in one study it was unclear (Pritchard 2005). However, it should be noted that due to the nature of the interventions blinding of care providers and patients was virtually impossible.
For evaluating detection bias we have evaluated the blinding of outcome assessors for all separate outcomes, with the exception of overall survival since for that outcome blinding is not relevant and the risk of detection bias was thus automatically judged as low for all three studies evaluating this outcome. All three studies evaluated event‐free survival; in two studies the risk of detection bias was unclear (Berthold 2005; Pritchard 2005), while in the other study it was low (Matthay 1999). However, for the additional follow‐up data of this study it became unclear. Two studies evaluated adverse effects; in one study the risk of detection bias was unclear (Berthold 2005), while in the other study it was low (Matthay 1999). However, again, for the additional follow‐up data of this study it was unclear if the outcome assessor evaluating adverse events was blinded.
Incomplete outcome data
For evaluating attrition bias we have assessed incomplete outcome data for all separate outcomes. Three studies evaluated both event‐free and overall survival; in all studies the risk of attrition bias was unclear. Two studies evaluated adverse effects other than treatment‐related death; in both studies the risk of attrition bias was unclear (Berthold 2005; Matthay 1999). Two studies evaluated treatment‐related death; in one study the risk of attrition bias was low (Berthold 2005), while in the other study it was high (Matthay 1999).
Selective reporting
For evaluating reporting bias we have assessed selective reporting. We defined 'all expected outcomes' as reporting on event‐free survival, overall survival and early and late adverse effects (irrespective of the number of evaluated adverse effects; for studies presenting additional follow‐up only late adverse effects were considered appropriate). In one study we judged the risk of reporting bias to be low (Matthay 1999), while in two studies it was judged to be high (Berthold 2005; Pritchard 2005).
Other potential sources of bias
For evaluating other potential sources of bias we have assessed the difference in length of follow‐up between treatment arms and inappropriate influence of funders. In all three studies there was an unclear risk of other bias. For a more detailed description of the different items see the risk of bias section of the Characteristics of included studies table.
Effects of interventions
Event‐free survival
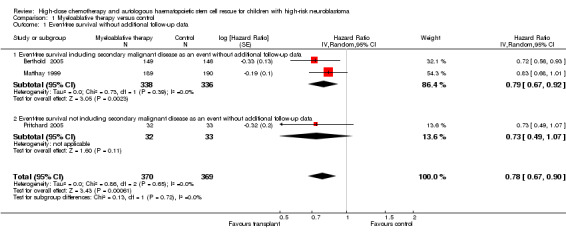
Data on event‐free survival were extracted from three trials (Berthold 2005; Matthay 1999; Pritchard 2005) with a total of 739 patients (see Figure 2). Two of these trials included secondary malignant disease as an event (Berthold 2005; Matthay 1999), whereas the other did not (Pritchard 2005; see the Characteristics of included studies table for the exact definitions). However, since only a very small percentage of patients developed secondary malignant disease (see the description of adverse effects later on), we were able to pool the results of these three trials. There was a significant statistical difference in event‐free survival in favour of myeloablative therapy over conventional chemotherapy or no further treatment (HR 0.78, 95% CI 0.67 to 0.90, P = 0.0006). No heterogeneity was detected for this comparison (I2 = 0%).
2.
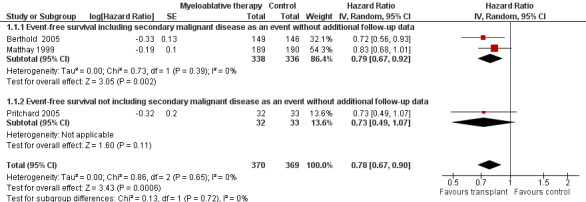
Forest plot of comparison: 1 Myeloablative therapy versus control, outcome: 1.1 Event‐free survival without additional follow‐up data.
The meta‐analysis of the two trials including secondary malignant disease as an event also showed a significant statistical difference in event‐free survival in favour of myeloablative therapy over conventional chemotherapy (HR 0.79; 95% CI 0.67 to 0.92, P = 0.002, 674 patients). No heterogeneity was detected for this comparison (I2 = 0%). The study not including secondary malignant disease as an event showed no significant statistical difference between myeloablative therapy and no further treatment (HR 0.73, 95% CI 0.49 to 1.07, P = 0.11, 65 patients).
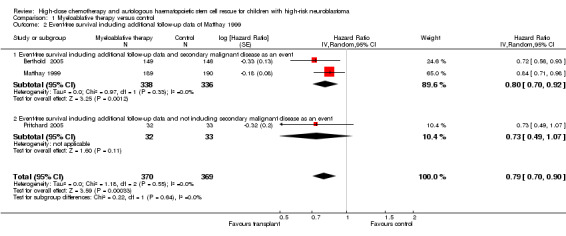
Additional follow‐up data from the Matthay 1999 study have been published. The length of follow‐up was not reported, but the median follow‐up of patients alive without an event was 7.7 years (range 130 days to 12.8 years). Relapse was included as an event in the follow‐up study, as opposed to the original publication. The results of this meta‐analysis of three trials including additional follow‐up data were in line with the earlier data. There was a significant statistical difference in event‐free survival in favour of the myeloablative therapy group over conventional chemotherapy or no further treatment (HR 0.79, 95% CI 0.70 to 0.90, P = 0.0003, 739 patients). No heterogeneity was detected for this comparison (I2 = 0%). When additional follow‐up data were included the meta‐analysis of the two trials including secondary malignant disease as an event again showed a significant statistical difference in event‐free survival in favour of the myeloablative therapy group over conventional chemotherapy (HR 0.80, 95% CI 0.70 to 0.92, P = 0.001, 674 patients). No heterogeneity was detected for this comparison (I2 = 0%). The results of the analysis of the study not including secondary malignant disease as an event did not change, since no new data were available (i.e. no significant difference between the treatment groups). See Figure 3.
3.
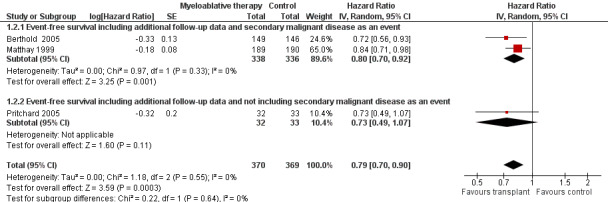
Forest plot of comparison: 1 Myeloablative therapy versus control, outcome: 1.2 Event‐free survival including additional follow‐up data of Matthay 1999.
The above mentioned HRs were calculated using the complete follow‐up period of the trials. The individual studies also reported three or five‐year event‐free survival rates. Berthold 2005 and Matthay 1999 reported significantly better three‐year event‐free survival rates in the myeloablative therapy group compared to conventional chemotherapy (P = 0.0221 and P = 0.034 respectively). Pritchard 2005 found no significant statistical difference in five‐year event‐free survival between the treatment groups (P = 0.08). Additional follow‐up data from Matthay 1999 showed a significant statistical difference in five‐year event‐free survival in favour of the myeloablative therapy group (P = 0.0434).
It should be noted that the individual studies all used different starting points for event‐free survival, either years since randomisation or years from diagnosis. In the Matthay 1999 study randomisation was performed a median of 60 days after diagnosis. Pritchard 2005 did not report the time of randomisation but the decision to randomise was made five to nine months after diagnosis. In the Berthold 2005 study randomisation was performed a median of 39 days after diagnosis.
Subgroups
Subgroups are summarised descriptively (i.e. as reported in the individual studies).
Berthold 2005 performed subgroup analyses based on commonly accepted risk factors. Three‐year event‐free survival was significantly better in the myeloablative therapy group compared to conventional chemotherapy: (1) in patients who had a complete remission or very good partial remission to induction chemotherapy/before randomisation (P = 0.0083); (2) in patients with raised serum concentration of lactate dehydrogenase (P = 0.0357; cut‐off value not reported); and (3) in patients receiving antibody treatment (P = 0.0061). By contrast, no significant statistical difference in three‐year event‐free survival was identified between the treatment groups: (1) in patients who had partial remission, mixed remission or stable disease before randomisation (P = 0.1809); (2) in patients treated with retinoic acid (P = 0.9732); (3) in patients with MYCN amplification with stage I, II, III, or IV‐S disease or with stage IV disease aged younger than one year (P = 0.2183); (4) in patients with MYCN amplification (P = 0.0669); (5) in patients without MYCN amplification (P = 0.0643); (6) in patients with normal concentrations of serum lactate dehydrogenase (P = 0.3132; cut‐off value not mentioned); and (7) in patients with stage IV disease and age more than one year (P = 0.0549).
Matthay 1999 performed subgroup analyses based on stage IV disease, MYCN amplification, unfavourable histopathological findings, a serum ferritin level of at least 143 ng per millilitre, a partial response to initial chemotherapy (as compared to complete response or nearly complete response), age at diagnosis more than two years and, among stage IV patients, bone metastases at diagnosis, and the presence at diagnosis of more than 100 tumour cells per 105 normal nucleated bone marrow cells. Matthay 1999 reported that event‐free survival among patients assigned to myeloablative therapy was longer in each subgroup (univariate analysis) than that among patients assigned to conventional chemotherapy (no P values reported). This difference in event‐free survival was most pronounced among the subgroups of patients who were older than two years at diagnosis (P = 0.01) and among patients with MYCN amplification (P = 0.03). Additional follow‐up data showed no significant statistical difference between treatment groups in event‐free survival in patients with stage IV disease.
Pritchard 2005 reported significantly better five‐year event‐free survival in patients aged more than one year with stage IV disease assigned to the myeloablative therapy group (P = 0.01).
Overall survival
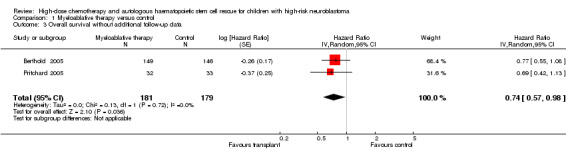
Data on overall survival were reported in three trials (Berthold 2005; Matthay 1999; Pritchard 2005). The results from two of these trials could be pooled (Berthold 2005; Pritchard 2005). There was a significant statistical difference in overall survival in favour of the myeloablative therapy group over conventional chemotherapy or no further treatment (HR 0.74, 95% CI 0.57 to 0.98, P = 0.04, 360 patients). No heterogeneity was detected for this comparison (I2 = 0%). See Figure 4. The other study (Matthay 1999) only provided descriptive results: overall survival was similar for both regimens (379 patients).
4.
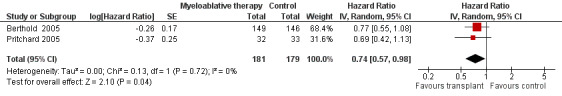
Forest plot of comparison: 1 Myeloablative therapy versus control, outcome: 1.3 Overall survival without additional follow‐up data.
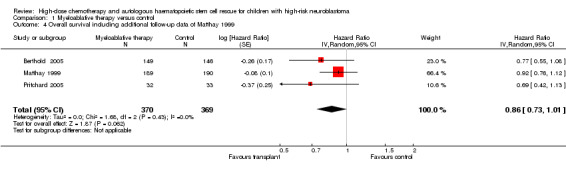
As opposed to the original publication, the additional follow‐up data from the Matthay 1999 study included information on overall survival. The length of follow‐up was not mentioned, but the median follow‐up of patients alive without an event was 7.7 years (range 130 days to 12.8 years). In contrast to the earlier results, the meta‐analysis including the additional follow‐up data showed no significant statistical difference in overall survival between treatment groups (HR = 0.86, 95% CI 0.73 to 1.01, P = 0.06, 739 patients). No heterogeneity was detected (I2 = 0%) (Figure 5).
5.
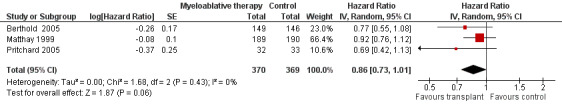
Forest plot of comparison: 1 Myeloablative therapy versus control, outcome: 1.4 Overall survival including additional follow‐up data of Matthay 1999.
The above mentioned HR is calculated using the complete follow‐up period of the trials. The individual studies also reported three‐ or five‐year overall survival rates. Berthold 2005 and Matthay 1999 found no significant statistical difference in three‐year overall survival between the treatment groups (P = 0.0875 and P = 0.87 respectively). Pritchard 2005 found no significant statistical difference in five‐year overall survival between the treatment groups (P = 0.1). Additional follow‐up data from Matthay 1999 showed no significant statistical difference in five‐year overall survival between the treatment groups (P = 0.3917). As opposed to the data provided in the additional follow‐up publication (at five years overall survival was significantly better in the myeloablative therapy group compared to the conventional chemotherapy group), in the erratum it was stated that at five years the overall survival curves were not significantly different (P = 0.08).
It should be noted that the individual studies all used different starting points for overall survival, either years since randomisation or years from diagnosis. In the Matthay 1999 study randomisation was performed a median of 60 days after diagnosis. Pritchard 2005 did not report the time of randomisation but the decision to randomise was made five to nine months after diagnosis. In the Berthold 2005 study randomisation was performed a median of 39 days after diagnosis.
Subgroups
Subgroups are summarised descriptively (i.e. as reported in the individual studies).
Berthold 2005 performed subgroup analyses based on commonly accepted risk factors. Three‐year overall survival was significantly better in the myeloablative therapy group compared to conventional chemotherapy: (1) in patients who had a complete remission or very good partial remission before randomisation (P = 0.0436), or (2) in patients receiving antibody treatment (P = 0.0221). By contrast, no significant difference in three‐year event‐free survival was identified between the treatment groups in the following subgroups: (1) in patients who had partial remission, mixed remission or stable disease before randomisation (P = 0.1311); (2) in patients treated with retinoic acid (P = 0.8918); (3) in patients with MYCN amplification with stage I, II, III or IV‐S disease or with stage IV disease aged younger than one year (P = 0.2287); (4) in patients with MYCN amplification (P = 0.1658); (5) in patients without MYCN amplification (P = 0.1921); (6) in patients with normal concentrations of serum lactate dehydrogenase (P = 0.1985; cut‐off value not reported); (7) in patients with raised serum concentration of lactate dehydrogenase (P = 0.1247; cut‐off value not reported); and (8) in patients with stage IV disease and aged more than one year (P = 0.1820).
Matthay 1999 did not report any subgroup analyses regarding overall survival in the original manuscript. Using additional follow‐up data a subgroup analysis of patients with stage IV disease found no significant statistical difference in overall survival between the treatment groups.
Pritchard 2005 reported that five‐year overall survival in patients aged more than one year with stage IV disease was significantly better in the myeloablative therapy group than in the no further treatment group (P = 0.03).
Adverse effects
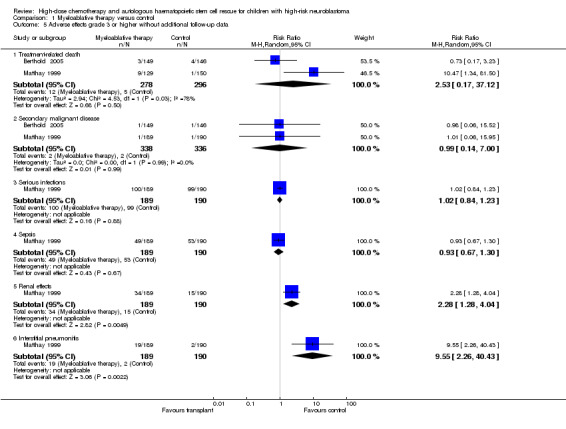
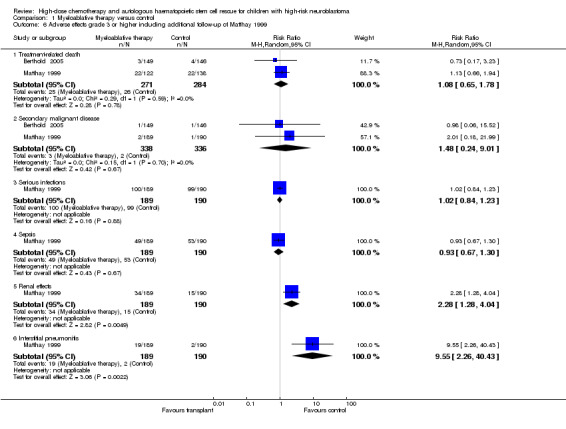
Since all patients receiving antineoplastic treatment will suffer from adverse events, we decided to analyse only severe and life‐threatening effects. We defined this as toxicity grade three or higher. Only adverse effects occurring from the start of the randomised treatment (i.e. myeloablative therapy or control) were eligible for inclusion in this review. See Figure 6 and Figure 7.
6.
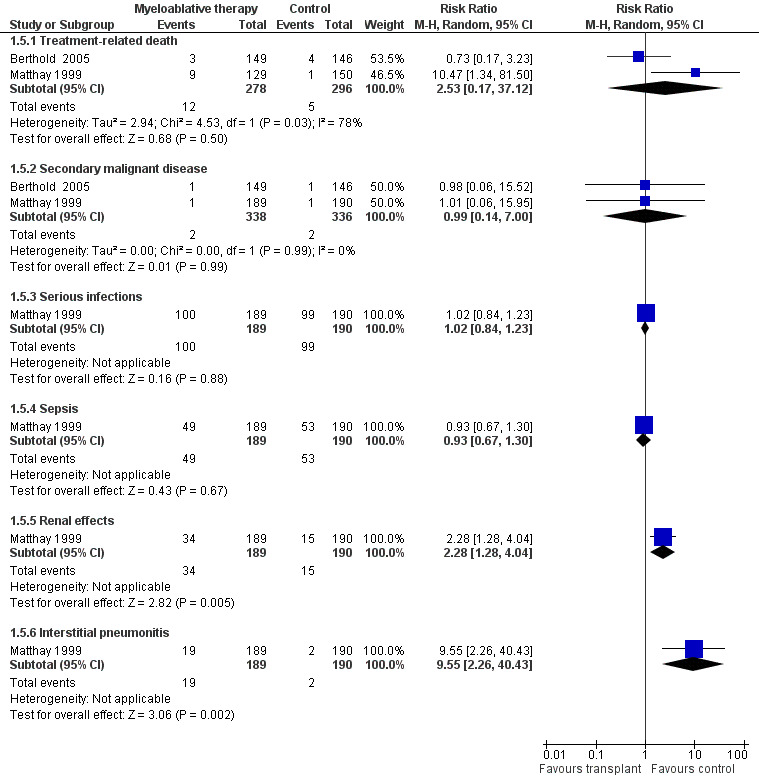
Forest plot of comparison: 1 Myeloablative therapy versus control, outcome: 1.5 Adverse effects grade 3 or higher without additional follow‐up data.
7.
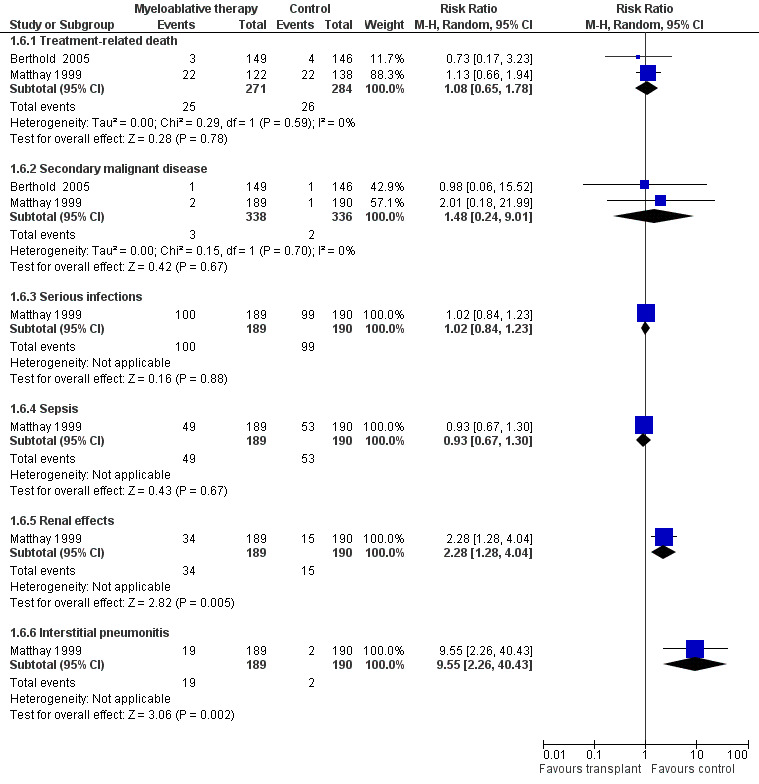
Forest plot of comparison: 1 Myeloablative therapy versus control, outcome: 1.6 Adverse effects grade 3 or higher including additional follow‐up of Matthay 1999.
Treatment‐related death
Data on treatment‐related death could be extracted from two trials with a total of 574 patients (Berthold 2005; Matthay 1999). There were 12 deaths among 278 patients randomised to the myeloablative therapy group and five among 296 in the control group. There was no significant statistical difference in treatment‐related death between the treatment groups (RR 2.53, 95% CI 0.17 to 37.12, P = 0.50). However, unexplained heterogeneity was detected (I2 = 78%). It should be noted that it was not possible to perform an ITT analysis using the Matthay 1999 data, therefore we performed an as‐treated analysis.
When additional follow‐up data from Matthay 1999 were included in the meta‐analysis there was still no significant statistical difference in treatment‐related death between the treatment groups (RR 1.08, 95% CI 0.65 to 1.78, P = 0.78). There were 25 deaths among 271 patients in the myeloablative group compared to 26 among 284 patients in the control group (RR 1.08, 95% CI 0.65 to 1.78, P = 0.78). No unexplained heterogeneity was detected for this comparison (I2 = 0%). Again, it was not possible to perform an ITT analysis for the study of Matthay 1999, so an as‐treated analysis was performed.
Secondary malignant disease
Data on secondary malignant disease could be extracted from two trials with a total of 674 patients (Berthold 2005; Matthay 1999). There were two cases of secondary malignant disease among 338 patients in the myeloablative group (i.e. one acute myeloid leukaemia and one unknown malignancy) compared to two cases among 336 conventional chemotherapy patients (i.e. one acute lymphoblastic leukaemia and one unknown malignancy). The meta‐analysis showed no significant statistical difference between the treatment groups (RR 0.99; 95% CI 0.14 to 7.00, P = 0.99). No heterogeneity was detected for this comparison (I2 = 0%).
When additional follow‐up data from the Matthay 1999 study were included in the meta‐analysis there was still no significant statistical difference between the treatment groups (RR 1.48, 95% CI 0.24 to 9.01, P = 0.67). There were three cases of secondary malignant disease among 338 patients in the myeloablative therapy group (i.e. two cases of acute myeloid leukaemia and one follicular carcinoma of the thyroid) compared to two among 336 patients in the conventional chemotherapy group (i.e. both acute lymphoblastic leukaemia). No unexplained heterogeneity was detected for this comparison (I2 = 0%).
Other adverse effects
Data on serious infections, sepsis, renal effects, interstitial pneumonitis and veno‐occlusive disease could be extracted from one trial with a total of 379 patients (Matthay 1999). There was a significantly higher incidence of renal effects, interstitial pneumonitis and veno‐occlusive disease in the myeloablative group compared to conventional chemotherapy, whereas for serious infections and sepsis no significant difference between the treatment groups was identified (see Figure 6 and Figure 7 for more information; since for veno‐occlusive disease none of the 190 patients in the control group experienced an event (while 17 out of 189 patients in the myeloablative therapy group did), we used the Fischer's exact test; P = 0.0001). These toxicity data did not change with additional follow‐up.
Quality of life
None of the studies evaluated quality of life.
Sensitivity analyses
The results of the sensitivity analyses for the risk of bias criteria were consistent among the trials and did not differ from the overall analyses.
Discussion
Neuroblastoma is the most common extracranial solid tumour in children. Unfortunately, despite the development of new treatment options, the prognosis of high‐risk patients is poor; and disease recurs in more than half of patients (Brodeur 2006; Goldsby 2004; Matthay 1993; Weinstein 2003). High‐dose chemotherapy and stem cell transplantation might increase survival in these patients. This systematic review evaluated the current state of evidence on the efficacy of high‐dose chemotherapy and autologous haematopoietic stem cell rescue compared with conventional therapy in patients diagnosed with high‐risk neuroblastoma. Only randomised controlled trials (RCTs) were included since it is widely recognised that a RCT is the only study design which can be used to obtain unbiased evidence on the use of different treatment options, provided that the design and execution of the RCTs are adequate.
We identified three RCTs that compared high‐dose chemotherapy and autologous haematopoietic stem cell rescue (i.e. myeloablative therapy) with conventional chemotherapy or no further treatment (Berthold 2005; Matthay 1999; Pritchard 2005). The first updated literature search identified a manuscript reporting additional follow‐up data from the Matthay 1999 study and the second one an erratum for this study. The exact length of follow‐up was not reported, but the median follow‐up of patients alive without an event was 7.7 years (range 130 days to 12.8 years). Even though all patients were diagnosed with high‐risk neuroblastoma, definitions of high‐risk patients differed between the studies, as did the required response to induction therapy to be eligible for randomisation. The three included RCTs used an age of one year as the cut‐off point for pre‐treatment risk stratification. Recently the age cut‐off for high‐risk disease was changed from one year to 18 months. As a result it is possible that patients with what is now classified as intermediate‐risk disease were included in the high‐risk groups. Consequently the relevance of the results of these studies to the current practice can be questioned. Survival rates may be overestimated due to the inclusion of patients with intermediate‐risk disease. Randomisation was performed at different time intervals since diagnosis. All trials used different induction treatments, myeloablative treatments and control treatments. Also, in two trials the received treatment slightly differed between patients, but we assumed that as a result of randomisation these differences in treatment were evenly distributed in both treatment groups. Two studies used bone marrow cells (Matthay 1999; Pritchard 2005) and one study used peripheral blood cells (Berthold 2005) for stem cell transplant (See the Characteristics of included studies table for further information).
The individual studies reported three or five‐year event‐free survival. In the Berthold 2005 and Matthay 1999 studies (including the additional follow‐up data from Matthay 1999) a significant statistical difference in favour of myeloablative therapy was identified. In the Pritchard 2005 study this difference was not statistically significant. Our meta‐analysis using their complete follow‐up period also showed a significant statistical difference in event‐free survival in favour of the myeloablative therapy group (HR 0.78, 95% CI 0.67 to 0.90, P = 0.0006). When the two studies in which secondary malignant disease was included as an event were analysed separately (Berthold 2005; Matthay 1999), this finding was confirmed. In the study which did not include secondary malignant disease as an event, no significant statistical difference in event‐free survival between the treatment groups was identified (Pritchard 2005). However, the direction of the result of this study was the same as the overall result of all trials. The results of the meta‐analysis of event‐free survival did not change when additional follow‐up data from Matthay 1999 were included in the analysis.
The individual studies also reported three‐ or five‐year overall survival. In all three studies (including the additional follow‐up data from Matthay 1999) no significant statistical difference between the treatment groups was identified. Our original meta‐analysis using the complete follow‐up period showed a significant statistical difference in overall survival in favour of the myeloablative therapy group (HR 0.74, 95% CI 0.57 to 0.98, P = 0.04; 2 studies, 360 patients). However, when additional follow‐up data from Matthay 1999 was included in the meta‐analysis the overall result was no longer statistically significant (HR 0.86, 95% CI 0.73 to 1.01, P = 0.43; 3 studies, 739 patients). A possible explanation could be the mortality caused by different late effects (e.g. as described by Mertens 2001; Reulen 2010). Another possible explanation could be that treatment options for progressive disease or relapse in patients who received myeloablative therapy are more limited than in patients who received conventional therapy or no further treatment. However, more data are needed for a definitive conclusion.
For two evaluated adverse effects (toxicity grade three or higher) it was possible to perform a meta‐analysis. No significant statistical differences in secondary malignant disease or treatment‐related death were identified between the treatment groups (both with the original data and with the inclusion of additional follow‐up data from Matthay 1999). While unexplained heterogeneity was detected in the original analysis of treatment‐related death, this was not the case when additional follow‐up data were included. Serious infections, sepsis, renal effects, interstitial pneumonitis and veno‐occlusive disease were only evaluated in the Matthay 1999 study. There was a significantly higher incidence of renal effects, interstitial pneumonitis and veno‐occlusive disease in the myeloablative therapy group, whereas for serious infections and sepsis no significant statistical differences between the treatment groups were identified. These toxicity data did not change with additional follow‐up. None of the included studies evaluated quality of life.
In the individual studies different subgroups were evaluated, but the results were not univocal in all studies. As a result, no definitive conclusions can be made regarding the effect of myeloablative therapy or conventional treatment in the different subgroups.
In this review we performed intention‐to‐treat (ITT) analyses, since they provide the most realistic and unbiased answer to the question of clinical effectiveness (Lachin 2000; Lee 1991). However, for treatment‐related death it was not possible to perform an ITT analysis using the Matthay 1999 data and we therefore performed an as‐treated analysis.
'No evidence of effect', as identified in this review, is not the same as 'evidence of no effect'. The reason that some studies did not identify a significant statistical difference between study groups could be the fact that the number of patients included in these studies was too small to detect a difference between the treatment groups (i.e. low power). Furthermore, the length of follow‐up could have been too short to detect a significant difference between the treatment groups.
The risk of bias in the included studies was difficult to assess due to a lack of reporting. As a result, the presence of selection bias, performance bias, detection bias, attrition bias, reporting bias and other bias could not be ruled out. However, at the moment this is the best available evidence from RCTs comparing the efficacy of high‐dose chemotherapy and autologous haematopoietic stem cell rescue with conventional therapy in patients diagnosed with high‐risk neuroblastoma. With regard to performance bias, it should be noted that due to the nature of the interventions, blinding of care providers and patients was virtually impossible.
The results of myeloablative therapy must be viewed in the context of the different treatment regimens used in the included studies. The included studies differed in terms of the preceding therapy (e.g. induction chemotherapy, surgery and radiotherapy), the myeloablative regimen with or without total body irradiation, the stem cells (e.g. whether purging was used or not, and the origin of the stem cells, i.e. bone marrow or peripheral blood) and the presence and type of consolidation chemotherapy. As a result, in this systematic review we can only provide conclusions on the concept of myeloablative treatment versus conventional or no further treatment. We cannot make conclusions with regard to specific versions of these treatments. However, the concept seems to work in terms of event‐free survival, both for all randomised patients treated with myeloablative therapy and for specific subgroups. For overall survival there is currently no evidence of effect. No definitive conclusions can be made regarding adverse effects and quality of life.
We are awaiting the full text publication of the study published as a conference abstract (Hero 2010), which presented additional follow‐up data from the Berthold 2005 study. Hero 2010 reported that myeloablative therapy proved to be effective with respect to long‐term outcome, but is complicated by acute and long‐term toxicity. However, as it is known that information provided in conference abstracts is often substantially different from information provided in subsequent full text publications (Yoon 2012), these results were not included in the analyses of this systematic review.
Authors' conclusions
Implications for practice.
Our original meta‐analyses of both event‐free and overall survival showed a significant statistical difference in favour of myeloablative therapy. However, when additional follow‐up data from Matthay 1999 were included in the meta‐analyses the difference in event‐free survival remained statistically significant, but the difference in overall survival was no longer statistically significant. A possible explanation could be that treatment options for progressive disease or relapse in patients who received myeloablative therapy are more limited than in patients who received conventional therapy or no further treatment. Another possible explanation could be the mortality caused by different late effects. Nonetheless, more data are needed for a definitive conclusion. Data on adverse effects were very limited, but significantly more events in the myeloablative therapy group were identified for renal effects, interstitial pneumonitis and veno‐occlusive disease. The fact that no significant differences in the occurrence of other adverse effects between treatment groups were identified could be the result of low power or too short a follow‐up period. No information on quality of life was provided. No definitive conclusions regarding the effect of myeloablative therapy or conventional treatment in different subgroups can be made.
It should be kept in mind that recently the age cut‐off for high‐risk disease was changed from one year to 18 months. As a result it is possible that patients with what is now classified as intermediate‐risk disease were included in the high‐risk groups. Consequently the relevance of the results of these studies to the current practice can be questioned. Survival rates may be over estimated due to the inclusion of patients with intermediate‐risk disease.
Based on the currently available evidence, we cannot make recommendations for the use of high‐dose chemotherapy and autologous haematopoietic stem cell rescue for children with high‐risk neuroblastoma in clinical practice. This systematic review only allows a conclusion on the concept of myeloablative treatment; no conclusions regarding the best treatment strategy can be made.
Implications for research.
Future trials on the use of high‐dose chemotherapy and autologous haematopoietic stem cell rescue for children with high‐risk neuroblastoma should focus on identifying the optimal induction therapy and myeloablative regimen, with regard to survival, (late) adverse effects and quality of life. The best study design to answer these questions is a randomised controlled trial (RCT). RCTs should be performed in homogeneous study populations (e.g. stage of disease) and have a long‐term follow up. Different risk groups, using the most recent definitions, should be taken into account. The number of included patients should be sufficient to obtain the power needed for the results to be reliable.
Also, it will be very interesting to examine additional data from the RCTs already performed. We are awaiting the full text publication of additional follow‐up data from the Berthold 2005 study. The performance of an individual patient data analysis including only patients that are classified as high‐risk according to the latest definition is another possibility to assess the efficacy of high‐dose chemotherapy and autologous haematopoietic stem cell rescue for children with high‐risk neuroblastoma.
What's new
| Date | Event | Description |
|---|---|---|
| 24 June 2015 | New citation required but conclusions have not changed | Summary of most important changes in the update: The search for eligible studies was updated to December 2014. No new RCTs were identified, but we did identify an erratum for one of the already included studies. Based on data provided in the erratum a descriptive result of overall survival at 5 years changed from significantly different in favour of the myeloablative therapy group to not significantly different between treatment groups, which is in accordance with our overall analysis. For the risk of bias assessment we now used the most recent recommendations of the Childhood Cancer Group. All studies were re‐assessed using the new criteria. |
| 24 June 2015 | New search has been performed | The search for eligible studies was updated to December 2014. |
History
Protocol first published: Issue 4, 2006 Review first published: Issue 5, 2010
| Date | Event | Description |
|---|---|---|
| 28 November 2012 | New citation required and conclusions have changed | Summary of most important changes in the update: The search for eligible studies was updated to June 2012. Additional follow‐up data of one of the three already included RCTs were available. For overall survival the results changed from a significant difference in favour of myeloablative therapy into no significant difference between the treatment groups. It should be noted that all included studies used an age of one year as the cut‐off point for pre‐treatment risk stratification. Recently the age cut‐off for high risk disease was changed from one year to 18 months. As a result it is possible that patients with what is now classified as intermediate risk disease have been included in the high risk groups. Consequently the relevance of the results of these studies to the current practice can be questioned. |
| 28 November 2012 | New search has been performed | The search for eligible studies was updated to June 2012. |
Acknowledgements
Leontien Kremer and Elvira van Dalen, the Co‐ordinating Editors of the Childhood Cancer Group, are both co‐authors of this review and therefore they could not act as Co‐ordinating Editor for this review. Marianne van de Wetering (Department of Paediatric Oncology, Emma Children's Hospital/Academic Medical Center, Amsterdam, the Netherlands) was willing to take on this task, for which we would like to thank her. Huib Caron was a co‐author of the protocol for this systematic review, the original version and the first update and we thank him for his valuable input. Also, we would like to thank Edith Leclercq, the Trials Search Co‐ordinator of the Childhood Cancer Group, for running the search strategy in the different databases and providing us with the titles and abstracts of the searches, Marcello DiNisio for translating an Italian article and, the Foundation of Paediatric Cancer Research (SKK), the Netherlands and Stichting Kinderen Kankervrij (KIKA), the Netherlands, for the financial support which made it possible to perform this systematic review. The editorial base of the Cochrane Childhood Cancer Group is funded by Kinderen Kankervrij (KIKA). For survival analyses, the hazard ratio and associated statistics were calculated using an Excel spreadsheet developed by Matthew Sydes and Jayne Tierney of the MRC Clinical Trials Unit, London, the United Kingdom.
Appendices
Appendix 1. Search strategy for Central Register of Controlled Trials (CENTRAL)
(1) For neuroblastoma we have used the following subject headings and text words (both in the original version of the review and the updates):
(neuroblastoma OR neuroblastomas OR ganglioneuroblastoma OR ganglioneuroblastomas OR neuroepithelioma OR olfactory esthesioneuroblastoma OR neuroblast*) in Clinical Trials
(2) For bone marrow and stem cell rescue the following subject headings and text words were used (both in the original version of the review and the updates):
(stem cell rescue OR bone marrow rescue OR bone marrow transplantation OR bone marrow grafting OR bone marrow cell transplantation OR stem cell transplantation OR stem cell transplantations OR hematopoietic stem cell transplantation OR peripheral blood stem cell transplantation OR peripheral stem cell transplantation OR cord blood stem cell transplantation OR placental blood stem cell transplantation OR umbilical cord stem cell transplantation OR autograft OR autografts OR autologous transplantation OR autotransplant OR autotransplants OR ABMT OR transplant* OR autolog* OR BMT OR myeloablative therapy OR myeloablative agonist OR myeloablative agonists OR myeloablativ* OR mega therapy OR high‐dose therapy OR high dose therapy) in Clinical Trials
(3) For children the following subject headings and text words were used (both in the original version of the review and the updates):
(infant OR infan* OR child OR child* OR schoolchild* OR schoolchild OR school child OR school child* OR kid OR kids OR toddler* OR adolescent OR adoles* OR teen* OR boy* OR girl* OR minors OR minors* OR underag* OR under ag* OR juvenil* OR youth* OR kindergar* OR puberty OR puber* OR pubescen* OR prepubescen* OR prepuberty* OR pediatrics OR pediatric* OR paediatric* OR peadiatric* OR schools OR nursery school* OR preschool* OR pre school* OR primary school* OR secondary school* OR elementary school* OR elementary school OR high school* OR highschool* OR school age OR schoolage OR school age* OR schoolage* OR infancy) in Clinical Trials
Finally, searches were combined as (1) AND (2) AND (3).
The search was performed in title, abstract or keywords.
[* = zero or more characters]
Appendix 2. Search strategy for MEDLINE/PubMed
(1) For neuroblastoma we have used the following subject headings and text words (both in the original version of the review and the updates):
neuroblastoma OR neuroblastomas OR ganglioneuroblastoma OR ganglioneuroblastomas OR neuroepithelioma OR esthesioneuroblastoma, olfactory OR neuroblast*
(2) For bone marrow and stem cell rescue the following subject headings and text words were used (both in the original version of the review and the updates) :
stem cell rescue OR bone marrow rescue OR bone marrow transplantation OR bone marrow grafting OR bone marrow cell transplantation OR stem cell transplantation OR stem cell transplantations OR hematopoietic stem cell transplantation OR peripheral blood stem cell transplantation OR peripheral stem cell transplantation OR cord blood stem cell transplantation OR placental blood stem cell transplantation OR umbilical cord stem cell transplantation OR autograft OR autografts OR transplantation, autologous OR autotransplant OR autotransplants OR ABMT OR transplant* OR autolog* OR BMT OR myeloablative therapy OR myeloablative agonist OR myeloablative agonists OR myeloablativ* OR mega therapy OR high‐dose therapy OR high dose therapy
(3) For children the following subject headings and text words were used (both in the original version of the review and the updates):
infant OR infan* OR child OR child* OR schoolchild* OR schoolchild OR school child OR school child* OR kid OR kids OR toddler* OR adolescent OR adoles* OR teen* OR boy* OR girl* OR minors OR minors* OR underag* OR under ag* OR juvenil* OR youth* OR kindergar* OR puberty OR puber* OR pubescen* OR prepubescen* OR prepuberty* OR pediatrics OR pediatric* OR paediatric* OR peadiatric* OR schools OR nursery school* OR preschool* OR pre school* OR primary school* OR secondary school* OR elementary school* OR elementary school OR high school* OR highschool* OR school age OR schoolage OR school age* OR schoolage* OR infancy OR schools, nursery
(4) For identifying RCTs and CCTs we used the highly sensitive search strategy as described in the Cochrane Handbook for Systematic Reviews of Interventons (Higgins 2005 for the original version, and Higgins 2008 and Lefebvre 2011 for the updates).
Finally, searches were combined as (1) AND (2) AND (3) AND (4).
[pt = publication type; tiab = title, abstract; sh = subject heading; mh = MeSH term; *=zero or more characters; RCT = randomized controlled trial; CCT = controlled clinical trial]
Appendix 3. Search strategy for EMBASE/Ovid
(1) For neuroblastoma we have used the following subject headings and text words (both in the original version of the review and the updates):
(neuroblastoma or neuroblastomas or ganglioneuroblastoma or ganglioneuroblastomas or neuroepithelioma or olfactory esthesioneuroblastoma or neuroblast$ or neuroganglioblastoma).mp or neuroblastoma/ or olfactory neuroepithelioma/ or neuroepithelioma/ or esthesioneuroblastoma/ or neuroblastoma cell/
(2) For bone marrow and stem cell rescue the following subject headings and text words were used (both in the original version of the review and the updates):
(stem cell rescue or bone marrow rescue).mp or (bone marrow transplantation or bone marrow grafting).mp or (bone marrow cell transplantation or stem cell transplantation or stem cell transplantations).mp or (hematopoietic stem cell transplantation or peripheral blood stem cell transplantation).mp or (peripheral stem cell transplantation or cord blood stem cell transplantation).mp or (placental blood stem cell transplantation or umbilical cord stem cell transplantation).mp or (autograft or autografts).mp or (autologous transplantation or autotransplant or autotransplants).mp or (BMT or ABMT or PBSCT).mp or transplant$.mp or autolog$.mp or (bone marrow transplant or bone marrow transfusion or bone marrow graft).mp or myeloablative therapy.mp or (myeloablative agonist or myeloablative agonists).mp or myeloablativ$.mp or mega therapy.mp or (high‐dose therapy or high dose therapy).mp or autologous bone marrow transplantation/ or allogenic bone marrow transplantation/ or bone marrow transplantation/ or bone marrow rescue/ or myeloablative agent/ or stem cell transplantation/ or allogenic bone marrow transplantation/ or allogeneic stem cell transplantation/ or autologous stem cell transplantation/ or peripheral blood stem cell transplantation/ or cord blood stem cell transplantation/ or hematopoietic stem cell transplantation/ or autograft/ or autotransplantation/ or allotransplantation/
(3) For children the following subject headings and text words were used (both in the original version of the review and the updates):
Infant/ or infancy/ or newborn/ or baby/ or child/ or preschool child/ or school child/ or adolescent/ or juvenile/ or boy/ or girl/ or puberty/ or prepuberty/ or pediatrics/ or primary school/ or high school/ or kindergarten/ or nursery school/ or school/ or infant$.mp or newborn$.mp or new born$.mp or (baby or baby$ or babies).mp or neonate$.mp or child$.mp or (school child$ or schoolchild$).mp or (school age$ or schoolage$).mp or (pre school$ or preschool$).mp or (kid or kids).mp or toddler$.mp or adoles$.mp or teen$.mp or boy$.mp or girl$.mp or minors$.mp or (under ag$ or underage$).mp or juvenil$.mp or youth$.mp or puber$.mp or pubescen$.mp or prepubescen$.mp or prepubert$.mp or (pediatric$ or paediatric$ or peadiatric$).mp or (school or schools).mp or (high school$ or highschool$).mp or primary school$.mp or nursery school$.mp or elementary school.mp or secondary school$.mp or kindergar$.mp
(4) For RCTs and CCTs the following subject headings and text words were used (in the original version of the review):
(a) clinical trial/ or controlled study/ or randomized controlled trial/ or double blind procedure/ or single blind procedure/ or comparative study/ or randomization/ or prospective study/ or placebo/ or phase 2 clinical trial/ or phase 3 clinical study.mp or phase 4 clinical study.mp or phase 3 clinical trial/ or phase 4 clinical trial/ or allocat$.mp or blind$.mp or control$.mp or placebo$.mp or prospectiv$.mp or random$.mp or ((singl$ or doubl$ or trebl$ or tripl$) and (blind$ or mask$)).mp or (versus or vs).mp or (randomized controlled trial$ or randomised controlled trial$).mp or controlled clinical trial$.mp or clinical trial$.mp
(b) human/ or nonhuman/ or animal/ or animal experiment/
(c) (b) not human/
(d) (a) not (c)
For the updates the following subject headings and text words were used:
1. Randomized Controlled Trial/ 2. Controlled Clinical Trial/ 3. randomized.ti,ab. 4. placebo.ti,ab. 5. randomly.ti,ab. 6. trial.ti,ab. 7. groups.ti,ab. 8. drug therapy.sh. 9. or/1‐8 10. Human/ 11. 9 and 10
Finally, searches were combined as (1) AND (2) AND (3) AND (4).
[mp = title, abstract, subject headings, drug trade name, original title, device manufacturer, drug manufacturer name; sh = subject heading; ti,ab = title, abstract; / = Emtree term; $=zero or more characters; RCT = randomised controlled trial; CCT = controlled clinical trial].
Data and analyses
Comparison 1. Myeloablative therapy versus control.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Event‐free survival without additional follow‐up data | 3 | 739 | Hazard Ratio (Random, 95% CI) | 0.78 [0.67, 0.90] |
| 1.1 Event‐free survival including secondary malignant disease as an event without additional follow‐up data | 2 | 674 | Hazard Ratio (Random, 95% CI) | 0.79 [0.67, 0.92] |
| 1.2 Event‐free survival not including secondary malignant disease as an event without additional follow‐up data | 1 | 65 | Hazard Ratio (Random, 95% CI) | 0.73 [0.49, 1.07] |
| 2 Event‐free survival including additional follow‐up data of Matthay 1999 | 3 | 739 | Hazard Ratio (Random, 95% CI) | 0.79 [0.70, 0.90] |
| 2.1 Event‐free survival including additional follow‐up data and secondary malignant disease as an event | 2 | 674 | Hazard Ratio (Random, 95% CI) | 0.80 [0.70, 0.92] |
| 2.2 Event‐free survival including additional follow‐up data and not including secondary malignant disease as an event | 1 | 65 | Hazard Ratio (Random, 95% CI) | 0.73 [0.49, 1.07] |
| 3 Overall survival without additional follow‐up data | 2 | 360 | Hazard Ratio (Random, 95% CI) | 0.74 [0.57, 0.98] |
| 4 Overall survival including additional follow‐up data of Matthay 1999 | 3 | 739 | Hazard Ratio (Random, 95% CI) | 0.86 [0.73, 1.01] |
| 5 Adverse effects grade 3 or higher without additional follow‐up data | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Treatment‐related death | 2 | 574 | Risk Ratio (M‐H, Random, 95% CI) | 2.53 [0.17, 37.12] |
| 5.2 Secondary malignant disease | 2 | 674 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.14, 7.00] |
| 5.3 Serious infections | 1 | 379 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.84, 1.23] |
| 5.4 Sepsis | 1 | 379 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.67, 1.30] |
| 5.5 Renal effects | 1 | 379 | Risk Ratio (M‐H, Random, 95% CI) | 2.28 [1.28, 4.04] |
| 5.6 Interstitial pneumonitis | 1 | 379 | Risk Ratio (M‐H, Random, 95% CI) | 9.55 [2.26, 40.43] |
| 6 Adverse effects grade 3 or higher including additional follow‐up of Matthay 1999 | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 Treatment‐related death | 2 | 555 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.65, 1.78] |
| 6.2 Secondary malignant disease | 2 | 674 | Risk Ratio (M‐H, Random, 95% CI) | 1.48 [0.24, 9.01] |
| 6.3 Serious infections | 1 | 379 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.84, 1.23] |
| 6.4 Sepsis | 1 | 379 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.67, 1.30] |
| 6.5 Renal effects | 1 | 379 | Risk Ratio (M‐H, Random, 95% CI) | 2.28 [1.28, 4.04] |
| 6.6 Interstitial pneumonitis | 1 | 379 | Risk Ratio (M‐H, Random, 95% CI) | 9.55 [2.26, 40.43] |
1.1. Analysis.
Comparison 1 Myeloablative therapy versus control, Outcome 1 Event‐free survival without additional follow‐up data.
1.2. Analysis.
Comparison 1 Myeloablative therapy versus control, Outcome 2 Event‐free survival including additional follow‐up data of Matthay 1999.
1.3. Analysis.
Comparison 1 Myeloablative therapy versus control, Outcome 3 Overall survival without additional follow‐up data.
1.4. Analysis.
Comparison 1 Myeloablative therapy versus control, Outcome 4 Overall survival including additional follow‐up data of Matthay 1999.
1.5. Analysis.
Comparison 1 Myeloablative therapy versus control, Outcome 5 Adverse effects grade 3 or higher without additional follow‐up data.
1.6. Analysis.
Comparison 1 Myeloablative therapy versus control, Outcome 6 Adverse effects grade 3 or higher including additional follow‐up of Matthay 1999.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Berthold 2005.
| Methods | Randomisation was performed centrally by computer‐generated sequence with a block size of 8. The stratification criteria were MYCN alone (amplified vs not amplified vs unknown), concentration of serum LDH at diagnosis (raised vs not raised vs unknown), and age (1 to < 2 years vs > 2 years at diagnosis). Randomisation was performed at a median of 39 days (range 7 to 224 days) after diagnosis. Patients were randomised to myeloablative therapy or conventional chemotherapy. |
|
| Participants | 295 children with high‐risk neuroblastoma. High‐risk neuroblastoma was defined as: stage IV disease in patients older than 1 year or as MYCN‐amplified tumours in patients with stage I, II, III or IV‐S disease or with stage IV disease and aged younger than 1 year. Staging was done in accordance with the International Neuroblastoma Staging System criteria (Brodeur 1993). Age: 22 patients < 1 year and 273 patients > 1 year Sex: not reported Stage of disease: I (n = 2), II (n = 6), III (n = 19), IV‐S (n = 5) and IV (n = 263) Primary disease or recurrence: not reported Histology: not reported Bone metastases: yes n = 179, no n = 116 (including 84 stage IV patients) Immunocytologic analysis of bone marrow at diagnosis: not reported MYCN amplification: yes n = 114, no n = 176, unclear n = 5 Serum LDH level (cut‐off values not mentioned): raised n = 262, not raised n = 29, not reported n = 4 Induction regimen: Data for all randomised patients were not available; these are data for 212 of the 295 patients. According to protocol, they received 3 N5 cycles with 40 mg/m2 cisplatin a day and 100 mg/m2 etoposide a day, both given as continuous infusion over 96 hours on days 1 to 4, and 3 mg/m2 vindesine given intravenously over 1 hour on day 1; and 3 N6 cycles with 1.5 mg/m2 vincristine a day given intravenously over 1 hour on days 1 and 8, 200 mg/m2 dacarbazine a day given intravenously over 1 hour on days 1 to 5, 1.5 g/m2 ifosfamide a day given as continuous infusion over 120 hours on days 1 to 5, and 30 mg/m2 doxorubicin a day given intravenously over 4 hours on days 6 and 7. In a pilot setting, 3 patients received cycles with slightly higher doses of etoposide (125 mg/m2 a day given as continuous infusion over 96 hours on days 1 to 4) and a different infusion time for doxorubicin (48 hours continuous infusion instead of 2 infusions of 4 hours each). Actually received cumulative doses were not reported. The N5 and N6 cycles were alternated; the total duration of the induction regimen was 5 to 7 months. External radiotherapy: Patients with contrast medium or MIBG uptake in the primary tumour at the end of induction chemotherapy had local radiotherapy to the primary residual tumour. Data for all randomised patients were not available; these are data for 212 of the 295 patients. Yes (36‐40 Gy to residual tumour) n = 24, no n = 188. Surgery: Timing of surgery varied during induction chemotherapy regimen: at diagnosis or after 2nd, 4th or 6th cycles of chemotherapy. Therapeutic MIBG‐I131: Patients with clear uptake of 123‐iodine MIBG in metastatic lesions at the end of induction chemotherapy received MIBG therapy. Data for all randomised patients were not available; these are data for 212 of the 295 patients. Yes n = 28. Immunotherapy: For 1 year was started 4 to 6 weeks after the end of the conventional chemotherapy or after recovery of the haemopoiesis after megatherapy. Six infusion cycles (one cycle every 2 months) of 20 mg/m2 ch14.18 (chimeric monoclonal antibody against GD2) a day given intravenously over 8 to 12 hours on days 1 to 5 of every cycle. Yes n = 146. Retinoic acid: After November 2002, instead of immunotherapy: 160 mg/m2/day, oral on days 1 to 14, followed by a break for 14 days, for 6 months. After 3 months’ break, retinoic acid was resumed for another 3 months. Yes n = 35. Supportive care: Prophylactic antibiotics not reported, growth factors not reported, granulocyte infusions not reported, other: drugs were given to control pain and allergic reactions during immunotherapy (n = not reported). Response before randomisation: Complete remission or very good partial remission n = 163, partial remission n = 87, stable disease (or mixed remission) n = 13, progressive disease (or death) n = 25, not reported n = 1, not applicable n = 6. Response was assessed by use of the International Neuroblastoma Remission Criteria (Brodeur 1993) |
|
| Interventions | Data for all randomised patients were not available; data were available for 212 of the 295 patients. Myeloablative therapy (n = 149): According to protocol: melphalan 45 mg/m2 a day given intravenously over 30 minutes on days 8 to 5 before transplantation, etoposide 40 mg/kg a day given intravenously over 4 hours on day 4 before transplantation, carboplatin 500 mg/m2 a day given intravenously over 1 hour on days 4 to 2 before transplantation. Actually received cumulative doses not mentioned. Note: some dose and drug adjustments were made in 6 patients because of hearing loss: 3 received cyclophosphamide instead of carboplatin and 3 received no carboplatin. In 9 children, melphalan was combined with busulfan. Use of total body irradiation was not reported. Source of stem cells: autologous peripheral blood. Timing of cell harvest: between 2nd and 5th induction regimen. Timing of stem cell rescue: after 6th induction chemotherapy cycle. Number of cells infused: 1‐2 X 106/kg CD 34(+) cells was recommended. Contamination with tumour cells: not mentioned. Purging: yes n = 96, no n = 1, not mentioned n = 13 Conventional therapy (n = 146): According to protocol: 4 N7 cycles: cyclophosphamide, 150 mg/m2 a day, orally, with mesna 50 mg/m2 3 times a day on days 1 to 8. Actually received cumulative doses not mentioned |
|
| Outcomes |
Event‐free survival (defined as time until disease progression or relapse, a second neoplastic disease, or death from any cause, whichever occurred first, or until the last examination). Overall survival (defined as death from any cause or until the last examination if the patient survived). Adverse effects, i.e. second malignancies and treatment‐related deaths (according to World Health Organization criteria) |
|
| Notes | Length of follow up not reported (median follow up for all cases alive at the censoring date was 3.57 years (range 1.01 to 7.02)) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was performed centrally by computer‐generated sequence with a block size of 8. |
| Allocation concealment (selection bias) | Low risk | Randomisation was performed centrally by computer‐generated sequence with a block size of 8. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants and personnel were not blinded. However, it should be noted that due to the nature of the interventions blinding of participants and personnel was virtually impossible. |
| Blinding of outcome assessment (detection bias) Event‐free survival | Unclear risk | No information on blinding of outcome assessors was provided. |
| Blinding of outcome assessment (detection bias) Overall survival | Low risk | No information on blinding of outcome assessors was provided, but since this is not applicable for overall survival we judged this as a low risk of bias. |
| Blinding of outcome assessment (detection bias) Adverse effects | Unclear risk | No information on blinding of outcome assessors was provided. |
| Incomplete outcome data (attrition bias) Event‐free survival | Unclear risk | It was not clear if all participants were included in the analysis. |
| Incomplete outcome data (attrition bias) Overall survival | Unclear risk | It was not clear if all participants were included in the analysis. |
| Incomplete outcome data (attrition bias) Adverse effects except treatment‐related death | Unclear risk | It was not clear if all participants were included in the analysis. |
| Incomplete outcome data (attrition bias) Treatment‐related death | Low risk | All participants were included in the analysis. |
| Selective reporting (reporting bias) | High risk | There was no protocol mentioned in the manuscript (and we did not search for it), but not all expected outcomes were reported. |
| Other bias | Unclear risk |
Difference in length of follow‐up between treatment arms: unclear (not reported). Inappropriate influence of funders: not present. |
Matthay 1999.
| Methods | Randomisation was performed by permuted‐block design using strata with and without metastatic disease. Randomisation was performed at week 8, before the 3rd cycle of chemotherapy for patients without disease progression at a median of 60 days after diagnosis. Patients were randomised to myeloablative therapy or conventional chemotherapy. | |
| Participants | 379 children with high‐risk neuroblastoma. High‐risk neuroblastoma was defined as: stage IV neuroblastoma; stage III disease with one or more of the following: amplification of the MYCN oncogene, a serum ferritin level of at least 143 ng per millilitre, and unfavourable histopathological findings; stage II disease with amplification of MYCN (age > 1 year); stage I or II disease with bone metastases before therapy other than surgery; and stage IV disease with MYCN amplification for less than 1 year. Staging was done using the Evans staging criteria (Evans 1971). Unfavourable histopathological findings were based on the Shimada classification (Shimada 1984). Age: 9 patients < 1 year and 370 patients > 1 year Sex: not reported Stage of disease: I (n = 0), II with MYCN amplification (n = 1), III (n = 44), IV‐S (n = 0) and IV (n = 334) Primary disease or recurrence: all primary disease Histology: favourable n = 15, unfavourable n = 248, unclear n = 116 Bone metastases: yes n = 230, no n = 149 (including 104 stage IV patients) Immunocytologic analysis of bone marrow at diagnosis: negative n = 66, positive n = 184, unclear n = 129 MYCN amplification: yes n = 104, no n = 179, unclear n = 96 Serum LDH level: not reported Induction regimen: According to protocol all patients received 5 courses at 28‐day intervals of cisplatin 60 mg/m2 on day 0; doxorubicin 30 mg/m2 on day 2; etoposide 100 mg/m2on days 2 and 5; cyclophosphamide 1 gr/m2 on days 3 ‐ 4. Actually received cumulative doses not reported. External radiotherapy: Following induction regimen patients with gross residual disease received surgery and radiotherapy. Number of patients not reported. Surgery: Timing of surgery: after cycle 4 of induction chemotherapy. Therapeutic MIBG‐I131: No Immunotherapy: No Retinoic acid: 50 children randomised to myeloablative therapy received 13‐cis‐retinoic acid (whereas 48 did not) and 52 children randomised to conventional chemotherapy received 13‐cis‐retinoic acid (whereas 53 did not). Treatment with 13‐cis‐retinoic acid was part of a second randomisation (see notes); not all patients included in the first randomisation were eligible for inclusion in the second one. Supportive care: Prophylactic antibiotics not mentioned, growth factors: 250 mcg/m2/day intravenously in patients undergoing myeloablative therapy and 5 mcg/kg/day s.c. in patients undergoing maintenance therapy, granulocyte infusions not reported, other not reported. Response before randomisation: Complete remission n = 117, (very good) partial remission n = 147, stable disease or mixed response n = 34, progressive disease n = 52, not mentioned n = 29. Response was assessed by use of the International Neuroblastoma Remission Criteria (Brodeur 1988; Brodeur 1993) |
|
| Interventions |
Myeloablative therapy (n = 189): According to protocol: carboplatin 1000 mg/m2, 96‐hour continuous infusion, day ‐8; etoposide 640 mg/m2, 96‐hour continuous infusion, day ‐8; melphalan 140 mg/m2 and 70 mg/m2 bolus infusion, days ‐7 and ‐6. Cumulative doses not mentioned. Total body irradiation: 333 cGy/day, days ‐3, ‐2, ‐1. Actually received cumulative doses not reported. Source of stem cells: bone marrow Timing of cell harvest: between 3rd and 4th and 4th and 5th cycles of induction regimen Timing of stem cell rescue: after 5th induction chemotherapy cycle Number of cells infused: 2 X 108 mnc/kg, day 0 (median dose) Contamination with tumour cells: no Purging: yes (performed by sedimentation, filtration, immunomagnetic separation) Conventional therapy (n = 190): According to protocol: 3 cycles of cisplatin 160 mg/m2; etoposide 500 mg/m2; doxorubicin 40 mg/m2 96 hours continuous infusion; simultaneously with bolus ifosfamide 2500 mg/m2 + mesna 1500 mg/m2 days 0 to 3. Actually received cumulative doses were not reported |
|
| Outcomes |
Event‐free survival (defined as the time from randomisation to disease progression, death from any cause and a second neoplasm, whichever occurred first). Overall survival (definition not reported). Adverse effects, i.e. treatment‐related death, secondary malignant disease, veno‐occlusive disease, interstitial pneumonitis, renal effects, sepsis, serious infections (according to the common toxicity criteria of the National Cancer Institute). |
|
| Notes | Length of follow up not reported. In this study a second randomisation was included to answer the question of whether subsequent treatment with 13‐cis‐retinoic acid (isotretinoin) could further improve event‐free survival. Additional follow‐up data of this study have been published (Matthay 2009): for response before randomisation slightly different numbers of patients were reported (i.e. complete remission n = 117, (very good) partial remission n = 148 (instead of 147), stable disease or mixed response n = 34, progressive disease n = 46 (instead of 52), not mentioned n = 34 (instead of 29). The length of follow‐up was not reported, but the median follow‐up of patients alive without an event was 7.7 years (range 130 days to 12.8 years). In this publication another definition of event‐free survival was used (relapse was included as an event): the time from randomisation to disease progression, relapse, death from any cause and a second neoplasm, whichever occurred first) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation was performed by permuted‐block design; no further information provided. |
| Allocation concealment (selection bias) | Unclear risk | Randomisation was performed by permuted‐block design; no further information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants and personnel were not blinded. However, it should be noted that due to the nature of the interventions blinding of participants and personnel was virtually impossible. |
| Blinding of outcome assessment (detection bias) Event‐free survival | Low risk | Outcome assessors were blinded*. |
| Blinding of outcome assessment (detection bias) Overall survival | Low risk | Outcome assessors were blinded. In the additional follow‐up study no information on blinding of outcome assessors was provided, but since this is not applicable for overall survival we judged this as a low risk of bias. |
| Blinding of outcome assessment (detection bias) Adverse effects | Low risk | Outcome assessors were blinded*. |
| Incomplete outcome data (attrition bias) Event‐free survival | Unclear risk | It was not clear if all participants were included in the analysis. |
| Incomplete outcome data (attrition bias) Overall survival | Unclear risk | It was not clear if all participants were included in the analysis. |
| Incomplete outcome data (attrition bias) Adverse effects except treatment‐related death | Unclear risk | It was not clear if all participants were included in the analysis. |
| Incomplete outcome data (attrition bias) Treatment‐related death | High risk | Not all participants were included in the analysis. |
| Selective reporting (reporting bias) | Low risk | There was no protocol mentioned in the manuscript (and we did not search for it), but all expected outcomes were reported. |
| Other bias | Unclear risk |
Difference in length of follow‐up between treatment arms: unclear (not reported) Inappropriate influence of funders: unclear (no information provided). |
Pritchard 2005.
| Methods | Randomisation was performed at the co‐ordinating data centre by means of a minimisation technique using stages (III or IV) and individual participating centres as stratification factors. Time of randomisation not reported (the decision to randomise was made 5 to 9 months after diagnosis). Patients were randomised to myeloablative therapy or no further treatment. |
|
| Participants | 65 children with high‐risk neuroblastoma. High‐risk neuroblastoma was defined as: age > 6 months, stage III or IV. Staging was done using the Evans staging criteria (Evans 1971). Age: 5 patients < 1 year and 60 patients > 1 year Sex: 34 males and 31 females Stage of disease: I (n = 0), II (n = 0), III (n = 13), IVS (n = 0) and IV (n = 52) Primary disease or recurrence: not reported Histology: not reported Bone metastases: yes n = 40/52 stage IV patients, unclear n = 12/52 stage IV patients Immunocytologic analysis of bone marrow at diagnosis: not reported MYCN amplification: not reported Serum LDH level: not reported Induction regimen: According to protocol all patients received vincristine 1.5 mg/m2 on day 1, cyclophosphamide 600 mg/m2 on day 1, cisplatin 60 mg/m2 on day 2, and teniposide 150 mg/m2 on day 4 repeated every 21 days or as soon as possible afterwards if recovery of neutrophil and platelet counts was slow. Cumulative doses not reported. External radiotherapy: No Surgery: Surgery was performed before randomisation took place. Therapeutic MIBG‐I131: No Immunotherapy: No Retinoic acid: No Supportive care: Prophylactic antibiotics not mentioned, growth factors no, granulocyte infusions not mentioned, other: nutritional supplementation (n = not reported). Response before randomisation: Complete remission n = 36, good partial remission n = 29. This study was carried out and completed prior to the publication of the International Neuroblastoma Remission Criteria, but the definitions used in this study were agreed unanimously by the trialists as they were considered to be "standard practice" at the time of the study. Complete remission was defined as disappearance of primary tumour as judged by abdominal imaging and of secondary deposits and normal urine catecholamine metabolite levels. Good partial remission was defined as (a) shrinkage of primary tumour by more than 50% in each of three dimensions, (b) reduction of urine catecholamine metabolite levels by more than 50% of the original values and (c) disappearance of secondary deposits including bone marrow involvement. In both types of response, however, isotope bone scan appearances need not have normalised as long as there was improvement at all sites reported as abnormal on the scan performed at diagnosis, with no new lesions |
|
| Interventions |
Myeloablative therapy (n = 32): According to protocol: melphalan 180 mg/ m2, i.v. bolus, 4 to 8 weeks after surgery or final course of chemotherapy (whichever came first). Cumulative doses not reported. Total body irradiation not reported. Source of stem cells: bone marrow Timing of cell harvest: the day before melphalan Timing of stem cell rescue: after induction regimens Contamination with tumour cells: not reported Number of cells infused: 4.6 X 108 nucleated cells of bone marrow per kg (range 1.8 to 14.2) Purging: no Conventional therapy (n = 33): No further treatment |
|
| Outcomes |
Event‐free survival (defined as the time to relapse or death prior to relapse from randomisation). Overall survival (defined as the time to death from any cause from randomisation). |
|
| Notes | Length of follow up not mentioned (median follow up for 21 surviving children was 14.3 years from randomisation (range 8.8 to 17.1 years)). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was performed at the co‐ordinating data centre by means of a minimisation technique. |
| Allocation concealment (selection bias) | Low risk | Randomisation was performed at the co‐ordinating data centre by means of a minimisation technique. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information on blinding of participants and personnel was provided. However, it should be noted that due to the nature of the interventions blinding of participants and personnel was virtually impossible. |
| Blinding of outcome assessment (detection bias) Event‐free survival | Unclear risk | No information on blinding of outcome assessors was provided. |
| Blinding of outcome assessment (detection bias) Overall survival | Low risk | No information on blinding of outcome assessors was provided, but since this is not applicable for overall survival we judged this as a low risk of bias. |
| Incomplete outcome data (attrition bias) Event‐free survival | Unclear risk | It was not clear if all participants were included in the analysis. |
| Incomplete outcome data (attrition bias) Overall survival | Unclear risk | It was not clear if all participants were included in the analysis. |
| Selective reporting (reporting bias) | High risk | There was no protocol mentioned in the manuscript (and we did not search for it), but not all expected outcomes were reported. |
| Other bias | Unclear risk |
Difference in length of follow‐up between treatment arms: unclear (not reported) Inappropriate influence of funders: unclear (no information provided). |
i.v.: intravenous; LDH: lactate dehydrogenase; s.c.: subcutaneous; vs: versus; *: it should be noted that in the study describing additional follow‐up this was not reported and the associated risk of detection bias was thus unclear for all outcomes
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Adkins 2004 | Patients also included in Matthay 1999 |
| Berthold 1990 | Not a RCT |
| Castel 2001 | Not a RCT |
| Castel 2002 | Not a RCT |
| Dini 1989 | All patients underwent BMT |
| Dini 1991a | All patients underwent BMT |
| Dini 1991b | All patients underwent BMT |
| Garaventa 1993 | Not a RCT; all patients underwent BMT |
| Haas‐Kogan 2003 | Patients also included in Matthay 1999 |
| Hartmann 1985 | All patients underwent BMT |
| Kaneko 1999 | Not a RCT |
| Klingebiel 1994 | All patients underwent transplant |
| Ladenstein 1991 | Not a RCT; all patients underwent BMT |
| Ladenstein 1996 | Not a RCT; all patients with stage IV underwent BMT |
| Madon 1985 | Randomisation between chemotherapy and chemotherapy + immunotherapy |
| Matthay 1995 | Not a RCT |
| Matthay 1996 | Review of different study protocols |
| Matthay 1998 | Not a RCT |
| Mugishima 1999 | Not a RCT; review |
| Ohnuma 1995 | Not a RCT |
| Olgun 2008 | Not a RCT (even though this was stated in the abstract) |
| Paolucci 1989 | Not a RCT |
| Philip 1991 | All patients underwent BMT |
| Pinkerton 1991 | Preliminary results of Pritchard 2005 |
| Saarinen 1996 | Not a RCT |
| Sawaguchi 1989 | Not a RCT |
| Sawaguchi 1990 | Not a RCT |
| Schmidt 2005 | Patients also included in Matthay 1999 |
| Seeger 1991 | All patients underwent BMT |
| Seeger 2000 | Patients also included in Matthay 1999 |
| Shuster 1991 | Not a RCT |
| Stram 1994 | Not a RCT |
| Stram 1996 | Not a RCT |
| Zucker 1991 | All patients underwent BMT |
RCT: randomised controlled trial; BMT: bone marrow transplant
Characteristics of studies awaiting assessment [ordered by study ID]
Hero 2010.
| Methods | No information on method of randomisation provided. |
| Participants | 295 children with high‐risk neuroblastoma (defined as stage 4 or MYCN amplified). No information was provided on age, sex, stage of disease, primary disease or recurrence, histology, presence of bone metastases, immunocytologic analysis of bone marrow at diagnosis, MYCN amplification, serum LDH level, treatment details, response before randomisation. |
| Interventions | Myeloablative therapy (n=143) versus conventional therapy, i.e. 4 cycles of oral cyclophosphamide (n=119). For 33 patients no information was provided. |
| Outcomes | Relapses occurred statistically significantly (P < 0.001) later in the transplant group (74 out of 143 (52%) patients; median 21 months (range 8 to 85 months)) than in the control group (83 out of 119 (70%) patients; median 16 months (range 7 to 60 months, median 16 months)). Patients died up to nine years after diagnosis: in the transplant group at a median of 30 months; in the control group at a median of 20 months (P < 0.001). Early complications (the abstract did not report if these were significant statistical differences): (a) pneumonia n = 10 in the transplant group versus n = 1 in the control group, (b) other severe infectious complications n = 8 in the transplant group, (c) veno‐occlusive disease n = 8 in the transplant group, (d) renal failure n = 3 in the transplant group, (e) treatment‐related death n = 5 in the transplant group versus n = 0 in the control group. Secondary malignant disease: 1 in each treatment group (both leukaemia cases; no further information provided). Major late effects in 109 patients surviving 5 years or longer, i.e. n = 68 in the transplant group versus n = 41 in the control group (it was unclear if this were all patients who survived 5 years or longer): (a) hearing loss 72% versus 51% (P = 0.04), (b) tubular damage 18% versus 12% (NS), (c) hypothyroidism 19% versus 2% (P = 0.02), (d) focal nodular hyperplasia of the liver 10% versus 0 % (P = 0.04), (e) impaired growth 10% versus 0% (P = 0.04). In conclusion: myeloablative therapy proved effective with respect to long‐term outcome, but is complicated by acute and long‐term toxicity. |
| Notes | This study has not been published in full text (April 2015), but was presented at the ANR conference 2010. It provides additional follow‐up of the Berthold 2005 study. Length of follow‐up was up to 12 years (median observation time 8.4 years). |
LDH: lactate dehydrogenase; NS: not significant
Differences between protocol and review
For the first update of this review we extended the search strategy as described in the protocol: we also included the conference proceedings of the American Society of Clinical Oncology (ASCO).
For the second update we used the most recent recommendations of the Childhood Cancer Group for the assessment of risk of bias in the included studies. All RCTs were scored using the new 'risk of bias' items. Also, since performing the original review and the first update of this review, the Childhood Cancer Group has adjusted some of its recommendations regarding analyses: when for a particular outcome only one study is available and there are no events in one of the treatment groups, it is impossible to calculate an adequate risk ratio using the RevMan software, instead the Fischer's exact test should be used. We have adjusted this where necessary. Finally, the websites for the ongoing trials registers changed since doing the original review and the first update; we used the new websites to access the registers.
Contributions of authors
Bilgehan Yalçin designed the study and wrote the protocol. He identified studies meeting the inclusion criteria. He searched for unpublished and ongoing studies. He performed the data extraction and risk of bias assessment of the included studies. He contributed to the interpretation of the results. He critically reviewed the review.
Leontien CM Kremer designed the study and critically reviewed the protocol. She contributed to the interpretation of the results. She critically reviewed the review.
Elvira C van Dalen designed the study and wrote the protocol. She developed the search strategy. She identified the studies meeting the inclusion criteria. She searched for unpublished and ongoing studies. She performed the data extraction and risk of bias assessment of the included studies. She analysed the data and interpreted the results. She wrote and revised the review.
All authors approved the final version.
Sources of support
Internal sources
No sources of support supplied
External sources
Foundation of Pediatric Cancer Research (SKK) Amsterdam, Netherlands.
Stichting Kinderen Kankervrij (KIKA), Netherlands.
Declarations of interest
Bilgehan Yalçin: none known. Leontien CM Kremer: none known, Elvira C van Dalen: none known.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Berthold 2005 {published data only}
- Berthold F, Boos J, Burdach S, Erttmann R, Henze G, Hermann J, et al. Myeloablative megatherapy with autologous stem‐cell rescue versus oral maintenance chemotherapy as consolidation treatment in patients with high‐risk neuroblastoma: a randomised controlled trial. Lancet Oncology 2005;6:649‐58. [DOI] [PubMed] [Google Scholar]
Matthay 1999 {published data only}
- Matthay KK, Reynolds CP, Seeger RC, Shimada H, Adkins ES, Haas‐Kogan D, et al. Long‐term results for children with high‐risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13‐cis‐retinoic acid: a children's oncology group study. Journal of Clinical Oncology 2009;27(7):1007‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthay KK, Villablanca JG, Seeger RC, Stram DO, Harris RE, Ramsay NK, et al. Treatment of high‐risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13‐cis‐retinoic acid. Children's Cancer Group. New England Journal of Medicine 1999;341:1165‐73. [DOI] [PubMed] [Google Scholar]
- No authors named. Erratum. Journal of Clinical Oncology 2014;32(17):1862‐3. [Google Scholar]
Pritchard 2005 {published data only}
- Pritchard J, Cotterill SJ, Germond SM, Imeson J, Kraker J, Jones DR. High dose melphalan in the treatment of advanced neuroblastoma: results of a randomised trial (ENSG‐1) by the European Neuroblastoma Study Group. Pediatric Blood and Cancer 2005;44:348‐57. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Adkins 2004 {published data only}
- Adkins ES, Sawin R, Gerbing RB, London WB, Matthay KK, Haase GM. Efficacy of complete resection for high‐risk neuroblastoma: a Children's Cancer Group study. Journal of Pediatric Surgery 2004;39:931‐6. [DOI] [PubMed] [Google Scholar]
Berthold 1990 {published data only}
- Berthold F, Burdach S, Kremens B, Lampert F, Niethammer D, Riehm H, et al. The role of chemotherapy in the treatment of children with neuroblastoma stage IV: the GPO (German Pediatric Oncology Society) experience. Klinische Pädiatrie 1990;202:262‐9. [DOI] [PubMed] [Google Scholar]
Castel 2001 {published data only}
- Castel V, Cañete A, Navarro S, García‐Miguel P, Melero C, Acha T, et al. Outcome of high‐risk neuroblastoma using a dose intensity approach: improvement in initial but not in long‐term results. Medical and Pediatric Oncology 2001;37:537‐42. [DOI] [PubMed] [Google Scholar]
Castel 2002 {published data only}
- Castel V, Tovar JA, Costa E, Cuadros J, Ruiz A, Rollan V, et al. The role of surgery in stage IV neuroblastoma. Journal of Pediatric Surgery 2002;37:1574‐8. [DOI] [PubMed] [Google Scholar]
Dini 1989 {published data only}
- Dini G, Philip T, Hartmann O, Pinkerton R, Chauvin F, Garaventa A, et al. Bone marrow transplantation for neuroblastoma: a review of 509 cases. EBMT Group. Bone Marrow Transplant 1989;4(Suppl 4):42‐6. [PubMed] [Google Scholar]
Dini 1991a {published data only}
- Dini G, Lanino E, Garaventa A, Paolucci P, Amici A, Locatelli F, et al. Unpurged ABMT for neuroblastoma: AIEOP‐BMT experience. Bone Marrow Transplant 1991;7(Suppl 2):92. [PubMed] [Google Scholar]
Dini 1991b {published data only}
- Dini G, Lanino E, Garaventa A, Paolucci P, Amici A, Locatelli F, et al. Unpurged autologous bone marrow transplantation for neuroblastoma: the AIEOP‐BMT group experience. Bone Marrow Transplant 1991;7(Suppl 3):109‐11. [PubMed] [Google Scholar]
Garaventa 1993 {published data only}
- Garaventa A, Ladenstein R, Chauvin F, Lanino E, Philip I, Corciulo P, et al. High‐dose chemotherapy with autologous bone marrow rescue in advanced stage IV neuroblastoma. European Journal of Cancer 1993;29A:487‐91. [DOI] [PubMed] [Google Scholar]
Haas‐Kogan 2003 {published data only}
- Haas‐Kogan DA, Swift PS, Selch M, Haase GM, Seeger RC, Gerbing RB, et al. Impact of radiotherapy for high‐risk neuroblastoma: a Children's Cancer Group study. International Journal of Radiation Oncology, Biology, Physics 2003;56:28‐39. [DOI] [PubMed] [Google Scholar]
Hartmann 1985 {published data only}
- Hartmann O, Zucker JM, Pinkerton R, Philip T, Beaujean F, Bernard JL, et al. Metastatic neuroblastoma in children older than one year old at diagnosis. Treatment with intensive chemo‐radiotherapy and autologous bone marrow transplant. Revue Française de Transfusion et Immuno‐Hématologie 1985;28:539‐46. [DOI] [PubMed] [Google Scholar]
Kaneko 1999 {published data only}
- Kaneko M, Tsuchida Y, Uchino J, Takeda T, Iwafuchi M, Ohnuma N, et al. Treatment results of advanced neuroblastoma with the first Japanese study group protocol. Study Group of Japan for Treatment of Advanced Neuroblastoma. Journal of Pediatric Hematology/Oncology 1999;21:190‐7. [DOI] [PubMed] [Google Scholar]
Klingebiel 1994 {published data only}
- Klingebiel T, Handgretinger R, Herter M, Lang P, Dopfer R, Scheel‐Walter H, et al. Peripheral stem cell transplantation in neuroblastoma stage 4 with the use of [131I‐m]IBG. Progress in Clinical and Biological Research 1994;385:309‐17. [PubMed] [Google Scholar]
Ladenstein 1991 {published data only}
- Ladenstein R, Lasset C, Bouffet E, Brunat‐Mentigny M, Biron P, Philip I, et al. A single center experience with bone marrow transplantation for high risk neuroblastoma. Bone Marrow Transplant 1991;7(Suppl 2):93. [PubMed] [Google Scholar]
Ladenstein 1996 {published data only}
- Ladenstein R, Ambros PF, Urban C, Ambros IM, Fink FM, Zoubek A, et al. Value of prognostic factors in the Austrian A‐NB87 Neuroblastoma Study. Klinische Pädiatrie 1996;208:210‐20. [DOI] [PubMed] [Google Scholar]
Madon 1985 {published data only}
- Madon E, Pastore G, Cordero di Montezemolo L, Brach del Prever A, Lanino E. Italian multicenter experience in the treatment of neuroblastoma and soft tissue sarcoma. Minerva Pediatrica 1985;37:603‐10. [PubMed] [Google Scholar]
Matthay 1995 {published data only}
- Matthay KK, O'Leary MC, Ramsay NK, Villablanca J, Reynolds CP, Atkinson JB, et al. Role of myeloablative therapy in improved outcome for high risk neuroblastoma: review of recent Children's Cancer Group results. European Journal of Cancer 1995;31A:572‐5. [DOI] [PubMed] [Google Scholar]
Matthay 1996 {published data only}
- Matthay KK. Impact of myeloablative therapy with bone marrow transplantation in advanced neuroblastoma. Bone Marrow Transplant 1996;18(Suppl 3):S21‐4. [PubMed] [Google Scholar]
Matthay 1998 {published data only}
- Matthay KK, Perez C, Seeger RC, Brodeur GM, Shimada H, Atkinson JB, et al. Successful treatment of stage III neuroblastoma based on prospective biologic staging: a Children's Cancer Group study. Journal of Clinical Oncology 1998;16:1256‐64. [DOI] [PubMed] [Google Scholar]
Mugishima 1999 {published data only}
- Mugishima H. High‐dose chemotherapy and hematopoietic stem cell transplant in the treatment of advanced neuroblastoma. Gan To Kagaku Ryoho 1999;26:1056‐67. [PubMed] [Google Scholar]
Ohnuma 1995 {published data only}
- Ohnuma N, Takahashi H, Kaneko M, Uchino J, Takeda T, Iwafuchi M, et al. Treatment combined with bone marrow transplantation for advanced neuroblastoma: an analysis of patients who were pretreated intensively with the protocol of the Study Group of Japan. Medical and Pediatric Oncology 1995;24:181‐7. [DOI] [PubMed] [Google Scholar]
Olgun 2008 {published data only}
- Olgun N, Gunes D, Aksoylar S, Varan A, Erbay A, Hazar V, et al. The Turkish Pediatric Oncology Group Neuroblastoma 2003 (TPOG‐NB‐2003): treatment results of the high risk group (abstract D110). Pediatric Blood and Cancer. 2008:140.
Paolucci 1989 {published data only}
- Paolucci P, Mancini AF, Rosito P, Vecchi V, Vivarelli F, Granchi D, et al. Intensification of induction prior to intensification of consolidation and bone marrow rescue in disseminated neuroblastoma. Bone Marrow Transplant 1989;4(Suppl 4):151‐2. [PubMed] [Google Scholar]
Philip 1991 {published data only}
- Philip T, Zucker JM, Bernard JL, Lutz P, Bordigoni P, Plouvier E, et al. The LMCE1 unselected group of stage IV neuroblastoma revisited with a median follow up of 59 months after ABMT. Progress in Clinical and Biological Research 1991;366:517‐25. [PubMed] [Google Scholar]
Pinkerton 1991 {published data only}
- Pinkerton CR. ENSG 1‐randomised study of high‐dose melphalan in neuroblastoma. Bone Marrow Transplant 1991;7(Suppl 3):112‐3. [PubMed] [Google Scholar]
Saarinen 1996 {published data only}
- Saarinen UM, Wikström S, Mäkipernaa A, Lanning M, Perkkiö M, Hovi L, et al. In vivo purging of bone marrow in children with poor‐risk neuroblastoma for marrow collection and autologous bone marrow transplantation. Journal of Clinical Oncology 1996;14:2791‐802. [DOI] [PubMed] [Google Scholar]
Sawaguchi 1989 {published data only}
- Sawaguchi S, Kaneko M, Nakajo T, Sawada T, Takahashi H, Tsuchida Y. A preliminary treatment report of the Study Group of Japan for Advanced Neuroblastoma. Nippon Gan Chiryo Gakkai Shi 1989;24:1020‐6. [PubMed] [Google Scholar]
Sawaguchi 1990 {published data only}
- Sawaguchi S, Kaneko M, Uchino J, Takeda T, Iwafuchi M, Matsuyama S, et al. Treatment of advanced neuroblastoma with emphasis on intensive induction chemotherapy. A report from the Study Group of Japan. Cancer 1990;66:1879‐87. [DOI] [PubMed] [Google Scholar]
Schmidt 2005 {published data only}
- Schmidt ML, Lal A, Seeger RC, Maris JM, Shimada H, O'Leary M, et al. Favorable prognosis for patients 12 to 18 months of age with stage 4 nonamplified MYCN neuroblastoma: a Children's Cancer Group Study. Journal of Clinical Oncology 2005;23:6474‐80. [DOI] [PubMed] [Google Scholar]
Seeger 1991 {published data only}
- Seeger RC, Villablanca JG, Matthay KK, Harris R, Moss TJ, Feig SA, et al. Intensive chemoradiotherapy and autologous bone marrow transplantation for poor prognosis neuroblastoma. Progress in Clinical and Biological Research 1991;366:527‐33. [PubMed] [Google Scholar]
Seeger 2000 {published data only}
- Seeger RC, Reynolds CP, Gallego R, Stram DO, Gerbing RB, Matthay KK. Quantitative tumor cell content of bone marrow and blood as a predictor of outcome in stage IV neuroblastoma: a Children's Cancer Group Study. Journal of Clinical Oncology 2000;18:4067‐76. [DOI] [PubMed] [Google Scholar]
Shuster 1991 {published data only}
- Shuster JJ, Cantor AB, McWilliams N, Pole JG, Castleberry RP, Marcus R, et al. The prognostic significance of autologous bone marrow transplant in advanced neuroblastoma. Journal of Clinical Oncology 1991;9:1045‐9. [DOI] [PubMed] [Google Scholar]
Stram 1994 {published data only}
- Stram DO, Matthay KK, O'Leary M, Reynolds CP, Seeger RC. Myeloablative chemoradiotherapy versus continued chemotherapy for high risk neuroblastoma. Progress in Clinical and Biological Research 1994;385:287‐91. [PubMed] [Google Scholar]
Stram 1996 {published data only}
- Stram DO, Matthay KK, O'Leary M, Reynolds CP, Haase GM, Atkinson JB, et al. Consolidation chemoradiotherapy and autologous bone marrow transplantation versus continued chemotherapy for metastatic neuroblastoma: a report of two concurrent Children's Cancer Group studies. Journal of Clinical Oncology 1996;14:2417‐26. [DOI] [PubMed] [Google Scholar]
Zucker 1991 {published data only}
- Zucker JM, Philip T, Bernard JL, Michon J, Bouffet E, Gentet JC, et al. Single or double consolidation treatment according to remission status after initial therapy in metastatic neuroblastoma: first results of LMCE 3 study in 40 patients. Bone Marrow Transplant 1991;7(Suppl 2):91. [PubMed] [Google Scholar]
References to studies awaiting assessment
Hero 2010 {published data only}
- Hero B, Thorsten S, Dillo D, Kremens B, Grigull L, Scheel‐Walter HG, et al. Long term outcome: the price of treatment for surviving high‐risk neuroblastoma. Advances in Neuroblastoma Research Association. 2010:105.
Additional references
Aydin 2009
- Aydin GB, Kutluk MT, Yalçin B, Büyükpamukçu M, Kale G, Varan A, et al. Neuroblastoma in Turkish children: experience of a single center. Journal of Pediatric Hematology and Oncology 2009;31:471‐80. [DOI] [PubMed] [Google Scholar]
Bollard 2006
- Bollard CM, Krance RA, Heslop HE. Hematopoietic stem cell transplantation in pediatric oncology. In: Pizzo PA, Poplack DG editor(s). Principles and practice of pediatric oncology. 5th Edition. Philadelphia: Lippincott Williams & Wilkins, 2006:476‐500. [Google Scholar]
Brodeur 1988
- Brodeur GM, Seeger RC, Barrett A, Berthold F, Castleberry RP, D'Angio G, et al. International criteria for diagnosis, staging, and response to treatment in patients with neuroblastoma. Journal of Clinical Oncology 1988;6:1874‐81. [DOI] [PubMed] [Google Scholar]
Brodeur 1993
- Brodeur GM, Pritchard J, Berthold F, Carlsen NL, Castel V, Castelberry RP, et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. Journal of Clinical Oncology 1993;11:1466‐77. [DOI] [PubMed] [Google Scholar]
Brodeur 2006
- Brodeur GM, Maris JM. Neuroblastoma. In: Pizzo PA, Poplack DG editor(s). Principles and practice of pediatric oncology. 5th Edition. Philadelphia: Lippincott Williams & Wilkins, 2006:933‐70. [Google Scholar]
Burchill 2004
- Burchill SA. Micrometastases in neuroblastoma: are they clinically important?. Journal of Clinical Pathology 2004;57:14‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Castel 1995
- Castel V, García‐Miguel P, Melero C, Navajas A, Navarro S, Molina J, et al. On behalf of the Spanish Neuroblastoma Study Group. The treatment of advanced neuroblastoma. Results of the Spanish Neuroblastoma Study Group (SNSG) Studies. European Journal of Cancer 1995;31A:642‐5. [DOI] [PubMed] [Google Scholar]
Cheung 1991
- Cheung NK, Heller G. Chemotherapy dose intensity correlates strongly with response, median survival, and median progression‐free survival in metastatic neuroblastoma. Journal of Clinical Oncology 1991;9:1050‐8. [DOI] [PubMed] [Google Scholar]
Cohn 1997
- Cohn SL, Moss TJ, Hoover M, Katzenstein HM, Haut PR, Morgan ER, et al. Treatment of poor‐risk neuroblastoma patients with high‐dose chemotherapy and autologous peripheral stem cell rescue. Bone Marrow Transplantation 1997;20:543‐51. [DOI] [PubMed] [Google Scholar]
Cohn 2009
- Cohn SL, Pearson AD, London WB, Monclair T, Ambros PF, Brodeur GM, et al. The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. Journal of Clinical Oncology 2009;27(2):289‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
De Bernardi 2003
- Bernardi B, Nicolas B, Boni L, Indolfi P, Carli M, Montezemolo LC, et al. Disseminated neuroblastoma in children older than one year at diagnosis: comparable results with three consecutive high‐dose protocols adopted by the Italian Co‐Operative Group for Neuroblastoma. Journal of Clinical Oncology 2003;21:1592‐601. [DOI] [PubMed] [Google Scholar]
Deeks 2011
- Deeks JJ, Higgins JPT, Altman DG (editors). Chapter 9: Analysing data and undertaking meta‐analyses. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Di Caro 1994
- Caro A, Bostrom B, Moss TJ, Neglia J, Ramsay NK, Smith J, et al. Autologous peripheral blood cell transplantation in the treatment of advanced neuroblastoma. American Journal of Pediatric Hematology/Oncology 1994;16:200‐6. [DOI] [PubMed] [Google Scholar]
Evans 1971
- Evans AE, D'Angio GJ, Randolph J. A proposed staging for children with neuroblastoma. Children's cancer study group A. Cancer 1971;27:374‐8. [DOI] [PubMed] [Google Scholar]
Goldsby 2004
- Goldsby RE, Matthay KK. Neuroblastoma: evolving therapies for a disease with many faces. Paediatric Drugs 2004;6:107‐22. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2005
- Higgins JPT, Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions [updated May 2005]. In: The Cochrane Library, Issue 3, 2006. John Wiley & Sons Ltd, Chichester (UK).
Higgins 2008
- Higgins JPT, Green S. Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.0 [updated February 2008]. The Cochrane Collaboration, 2008. Available from www.cochrane‐handbook.org.
Higgins 2011
- Higgins JPT, Altman DG, Sterne JAC (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Kaneko 2002
- Kaneko M, Tsuchida Y, Mugishima H, Ohnuma N, Yamamoto K, Kawa K, et al. Intensified chemotherapy increases the survival rates in patients with stage 4 neuroblastoma with MYCN amplification. Journal of Pediatric Hematology/Oncology 2002;24:613‐21. [DOI] [PubMed] [Google Scholar]
Keshelava 1998
- Keshelava N, Seeger RC, Groshen S, Reynolds CP. Drug resistance patterns of human neuroblastoma cell lines derived from patients at different phases of therapy. Cancer Research 1998;58:5396‐405. [PubMed] [Google Scholar]
Kremer 2014
- Kremer LCM, Leclercq E, Dalen EC. Cochrane Childhood Cancer Group. About The Cochrane Collaboration (Cochrane Review Groups (CRGs)) 2014, Issue 5. Art. No.: CHILDCA.
Kushner 2004
- Kushner BH, Kramer K, LaQuaglia MP, Modak S, Yataghene K, Cheung NK. Reduction from seven to five cycles of intensive induction chemotherapy in children with high‐risk neuroblastoma. Journal of Clinical Oncology 2004;22:4888‐92. [DOI] [PubMed] [Google Scholar]
Lachin 2000
- Lachin JM. Statistical considerations in the intent‐to‐treat principle. Controlled Clinical Trials 2000;21:167‐89. [DOI] [PubMed] [Google Scholar]
Ladenstein 1994
- Ladenstein R, Lasset C, Hartmann O, Klingebiel T, Bouffet E, Gadner H, et al. Comparison of auto versus allografting as consolidation of primary treatments in advanced neuroblastoma over one year of age at diagnosis: report from the European Group for Bone Marrow Transplantation. Bone Marrow Transplantation 1994;14:37‐46. [PubMed] [Google Scholar]
Ladenstein 2004
- Ladenstein R, Hartmann O, Koscielnak E, Philip T. Megatherapy with stem cell rescue in solid tumours. In: Pinkerton R, Plowman PN, Pieters R editor(s). Pediatric Oncology. 3rd Edition. London: Arnold Publishers, 2004:538‐569. [Google Scholar]
Laprie 2004
- Laprie A, Michon J, Hartmann O, Munzer C, Leclair MD, Coze C, et al. Neuroblastoma Study Group of the French Society of Pediatric Oncology. High‐dose chemotherapy followed by locoregional irradiation improves the outcome of patients with international neuroblastoma staging system Stage II and III neuroblastoma with MYCN amplification. Cancer 2004;101:1081‐9. [DOI] [PubMed] [Google Scholar]
Lee 1991
- Lee YJ, Ellenberg JH, Hirtz DG, Nelson KB. Analysis of clinical trials by treatment actually received: is it really an option?. Statistics in Medicine 1991;10:1595‐605. [DOI] [PubMed] [Google Scholar]
Lefebvre 2011
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Searching for studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org. Available from www.cochrane‐handbook.org.
Maris 2005
- Maris JM. The biologic basis for neuroblastoma heterogeneity and risk stratification. Current Opinion in Pediatrics 2005;17:7‐13. [DOI] [PubMed] [Google Scholar]
Maris 2007
- Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet 2007;369(9579):2106‐20. [DOI] [PubMed] [Google Scholar]
Matthay 1993
- Matthay KK, Atkinson JB, Stram DO, Selch M, Reynolds CP, Seeger RC. Patterns of relapse after autologous purged bone marrow transplantation for neuroblastoma: a Children's Cancer Group pilot study. Journal of Clinical Oncology 1993;11:2226‐33. [DOI] [PubMed] [Google Scholar]
Matthay 2009
- Matthay KK, Reynolds CP, Seeger RC, Shimada H, Adkins ES, Haas‐Kogan D, et al. Long‐term results for children with high‐risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13‐cis‐retinoic acid: a children's oncology group study. Journal of Clinical Oncology 2009;27:1007‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mertens 2001
- Mertens AC, Yasui Y, Neglia JP, Potter JD, Nesbit ME Jr, Ruccione K, et al. Late mortality experience in five‐year survivors of childhood and adolescent cancer: the Childhood Cancer Survivor Study. Journal of Clinical Oncology 2001;19:3163‐72. [DOI] [PubMed] [Google Scholar]
Parmar 1998
- Parmar MK, Torri V, Stewart L. Extracting summary statistics to perform meta‐analyses of the published literature for survival endpoints. Statistics in Medicine 1998;17:2815‐34. [DOI] [PubMed] [Google Scholar]
Peinemann 2015a
- Peinemann F, Tushabe DA, Dalen EC, Berthold F. Rapid COJEC versus standard induction therapies for high‐risk neuroblastoma. Cochrane Database of Systematic Reviews 2015, Issue 5. [DOI: 10.1002/14651858.CD010774.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Peinemann 2015b
- Peinemann F, Dalen EC, Tushabe DA, Berthold F. Retinoic acid post consolidation therapy for high‐risk neuroblastoma patients treated with autologous hematopoietic stem cell transplantation. Cochrane Database of Systematic Reviews 2015, Issue 1. [DOI: 10.1002/14651858.CD010685.pub2] [DOI] [PubMed] [Google Scholar]
Philip 1997
- Philip T, Ladenstein R, Lasset C, Hartmann O, Zucker JM, Pinkerton R, et al. 1070 myeloablative megatherapy procedures followed by stem cell rescue for neuroblastoma: 17 years of European experience and conclusions. European Group for Blood and Marrow Transplant Registry Solid Tumour Working Party. European Journal of Cancer 1997;33:2130‐5. [DOI] [PubMed] [Google Scholar]
Pritchard 1995
- Pritchard J. "Megatherapy" for advanced neuroblastoma ‐ rationale and role. European Journal of Cancer 1995;31A:134‐6. [DOI] [PubMed] [Google Scholar]
Reulen 2010
- Reulen RC, Winter DL, Frobisher C, Lancashire ER, Stiller CA, Jenney ME, et al. Long‐term cause‐specific mortality among survivors of childhood cancer. Journal of American Medical Association 2010;304:172‐9. [DOI] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Reynolds 2001
- Reynolds CP, Seeger RC. Detection of minimal residual disease in bone marrow during or after therapy as a prognostic marker for high‐risk neuroblastoma. Journal of Pediatric Hematology/Oncology 2001;23:150‐2. [DOI] [PubMed] [Google Scholar]
Reynolds 2004
- Reynolds CP. Detection and treatment of minimal residual disease in high‐risk neuroblastoma. Pediatric Transplantation 2004;8(Suppl 5):56‐66. [DOI] [PubMed] [Google Scholar]
Shimada 1984
- Shimada H, Chatten J, Newton WA Jr, Sachs N, Hamoudi AB, Chiba T, et al. Histopathologic prognostic factors in neuroblastic tumors: definition of subtypes of ganglioneuroblastoma and an age‐linked classification of neuroblastomas. Journal of the National Cancer Institute 1984;73:405‐16. [DOI] [PubMed] [Google Scholar]
Sterne 2011
- Sterne JAC, Egger M, Moher D (editors). Chapter 10: Addressing reporting biases. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Intervention. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Trahair 2007
- Trahair TN, Vowels MR, Johnston K, Cohn RJ, Russell SJ, Neville KA, et al. Long‐term outcomes in children with high‐risk neuroblastoma treated with autologous stem cell transplantation. Bone Marrow Transplantation 2007;40(8):741‐6. [DOI] [PubMed] [Google Scholar]
Verdeguer 2004
- Verdeguer A, Munoz A, Canete A, Pardo N, Martinez A, Donat J, et al. Long‐term results of high‐dose chemotherapy and autologous stem cell rescue for high‐risk neuroblastoma patients: a report of the Spanish working party for BMT in children (Getmon). Pediatric Hematology and Oncology 2004;21:495‐504. [DOI] [PubMed] [Google Scholar]
Weinstein 2003
- Weinstein JL, Katzenstein HM, Cohn SL. Advances in the diagnosis and treatment of neuroblastoma. The Oncologist 2003;8:278‐92. [DOI] [PubMed] [Google Scholar]
Yoon 2012
- Yoon U, Knobloch K. Assessment of reporting quality of conference abstracts in sports injury prevention according to CONSORT and STROBE criteria and their subsequent publication rate as full papers. BMC Medical Research Methodology 2012;12:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yu 2010
- Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, et al. Anti‐GD2 antibody with GM‐CSF, interleukin‐2, and isotretinoin for neuroblastoma. New England Journal of Medicine 2010;363(14):1324‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Yalçin 2006
- Yalçin B, Kremer LC, Caron HN, Dalen EC. High dose chemotherapy and autologous haematopoietic stem cell rescue for children with high‐risk neuroblastoma. Cochrane Database of Systematic Reviews 2006, Issue 4. [DOI: 10.1002/14651858.CD006301] [DOI] [Google Scholar]
Yalçin 2010
- Yalçin B, Kremer LC, Caron HN, Dalen EC. High‐dose chemotherapy and autologous haematopoietic stem cell rescue for children with high‐risk neuroblastoma. Cochrane Database of Systematic Reviews 2010, Issue 5. [DOI: 10.1002/14651858.CD006301.pub2] [DOI] [PubMed] [Google Scholar]
Yalçin 2013
- Yalçin B, Kremer LCM, Caron HN, Dalen EC. High‐dose chemotherapy and autologous haematopoietic stem cell rescue for children with high‐risk neuroblastoma. Cochrane Database of Systematic Reviews 2013, Issue 8. [DOI: 10.1002/14651858.CD006301.pub3] [DOI] [PubMed] [Google Scholar]