

Abstract
Purpose:
Efforts are required to improve potency and durability of CD38- and BCMA-based immunotherapies in human multiple myeloma (MM). We here delineated the molecular and cellular mechanisms underlying novel immunomodulatory effects triggered by BCMA pyrrolobenzodiazepine (PBD) antibody drug conjugate MEDI2228 which can augment efficacy of these immunotherapies.
Experimental Design:
MEDI2228-induced transcriptional and protein changes were investigated to define significantly impacted genes and signaling cascades in MM cells. Mechanisms whereby MEDI2228 combination therapies can enhance cytotoxicity or overcome drug resistance in MM cell lines and patient MM cells were defined using in vitro models of tumor in the bone marrow (BM) microenvironment, as well as in human NK-reconstituted NSG mice bearing MM1S tumors.
Results:
MEDI2228 enriched type I interferon (IFN I)-signaling and enhanced expression of IFN-stimulated genes in MM cell lines following the induction of DNA damage-ATM/ATR-CHK1/2 pathways. It activated cGAS-STING-TBK1-IRF3 and STAT1-IRF1-signaling cascades and increased CD38 expression in MM cells but did not increase CD38 expression in BCMA-negative NK effector cells. It overcame CD38 downregulation on MM cells triggered by IL6 and patient BM stromal cell culture supernatant via activation of STAT1-IRF1, even in immunomodulatory drugs (IMiDs)- and bortezomib-resistant MM cells. In vitro and in vivo upregulation of NKG2D ligands and CD38 in MEDI2228-treated MM cells was further associated with synergistic Daratumumab (Dara) CD38 MoAb triggered NK-mediated cytotoxicity of both cell lines and autologous drug resistant patient MM cells.
Conclusions:
These results provide the basis for clinical evaluation of combination MEDI2228 with Dara to further improve patient outcome in MM.
Keywords: multiple myeloma, MM; monoclonal antibody, MoAb; immunotherapy; CD38; Daratumumab, Dara; antibody-dependent cellular cytotoxicity, ADCC; B-cell maturation antigen, BCMA; antibody drug conjugate, ADC; MEDI2228; DNA damage response, DDR; interferon, IFN; IFN type I, IFN I; STAT1; cyclic GMP-AMP synthase, cGAS; stimulator of interferon genes, STING; IRF1; bone marrow, BM; BM microenvironment; BM stromal cell, BMSC; IL6; NKG2D ligands, NKG2DLs
Introduction
Daratumumab (Dara) targeting CD38 is the first monoclonal antibody (MoAb) with clinical activity as a single agent and in combination to treat relapsed and refractory multiple myeloma (RRMM) and is also approved in combination to treat newly diagnosed MM (1–3). Dara-induced MM cytotoxicity mainly depends on FcR-expressing immune cells to activate cytotoxic effector functions, including antibody-dependent cellular cytotoxicity (ADCC) via NK cells, antibody-dependent cellular phagocytosis via macrophages, and complement-dependent cytotoxicity (1,4,5). Dara is well tolerated and its use in combination with other agents, i.e., immunomodulatory drugs (IMiDs) and proteasome inhibitors (PIs), is becoming a new standard-of-care MM treatment (3). However, relapse of disease remains, highlighting the need for novel immunotherapeutic strategies to further improve potency, extend response durability, and overcome drug resistance.
CD38 expression on MM cells correlates with response to Dara therapy (6,7) and downregulation or loss of CD38 has been reported in Dara-resistant MM (6). Since NK cells express high levels of CD38, NK depletion by Dara may negatively impact Dara efficacy (8,9). Although IMiDs enhance Dara-induced cytoxicity, relapse after combination IMiDs with Dara therapies is common. All-transretinoic acid (10), histone deacetylase inhibitors (11), and ruxolitinib targeting JAK-STAT3 (12) are reported to modulate CD38 expression. However, these agents may affect normal cells with or without CD38 expression, thereby limiting their clinical application in combination with Dara. Thus, novel agents selectively upregulating CD38 in MM but not normal cells, are needed to promote Dara efficacy and prevent disease recurrence.
Compared with CD38, BCMA is more exclusively expressed at high levels in MM cell lines and patient MM cells, but not other normal tissues (13,14). It is the first validated MM antigen for both chimeric antigen receptor T cell and antibody drug conjugate (ADC) therapies for RRMM (15–17). Specifically, the first BCMA ADC belantamab mafodotin was recently approved for RRMM (17) and directly kills MM cells via a microtubule disrupting monomethyl auristatin-F (MMAF) (18,19). MEDI2228 is a novel BCMA ADC delivering a highly cytotoxic DNA minor groove interstrand crosslinking pyrrolobenzodiazepine (PBD) dimer warhead to induce DNA breaks and DNA damage response (DDR) prior to MM cell apoptosis (20,21). This mechanism is distinct from MMAF which cannot trigger DDR at the same concentration as PBD and predominantly inhibits proliferating tumor cells. Most recently, MEDI2228 showed significant clinical activity in a first-in-human trial (22). However, the potential immunomodulatory function of MEDI2228, both alone and in combination with MoAbs targeting different MM antigens, is undefined.
We here identified immnomodulatory effects of MEDI2228 and delineated the molecular and cellular mechanisms mediating these processes. We further investigated whether combination MEDI2228 with Dara enhanced efficacy of either agent alone against human MM cells both in vitro and in mice reconstituted with human NK cells. These studies provide the rationale for clinical trials of combination BCMA- and CD38-directed immunotherapies to further improve patient outcome in MM.
Materials and Methods
Reagents and compounds
MEDI2228 (M2) and its MMAF-ADC homolog M3 were previouly described (20,21). Abs for immunoblottings and all other reagents for various experiments are detailed in Supplementary Methods, including cell culture and patient samples, real-time quantitative reverse transcription-polymerase chain reaction (qRT-PCR), flow cytometry (FC), cytotoxicity assays, lentivirus transfection, ADCC assays, animal models of human MM, immunohistochemistry (IHC), and statistics. Patient MM and normal donor samples were obtained after informed consent was provided, in accordance with the Declaration of Helsinki and under the auspices of a Dana-Farber Cancer Institute (DFCI) Institutional Review Board approved protocol. Informed written consent was obtained from each subject or each subject’s guardian.
RNA-seq analysis of H929 MM cells after M2 treatment
Total RNA from M2- and control vehicle-treated H929 cells (>85% viability) was extracted using RNeasy Mini Kit (Qiagen). Library was prepared and samples were sequenced on NovaSeq 6000 PE150. RNA-seq data sets were aligned to human reference genome hg19 using SRAR followed by VIPER NGS Analysis pipeline (23,24). RSEM was used to do the transcript quantification and differential expression analysis was performed with DESeq2. The list of differentially expressed genes (DEGs) was applied to the Gene Set Enrichment Analysis (GSEA) software to identify biological pathways modulated by M2. The biologically defined gene sets were obtained from the Molecular Signatures Database (http://software.broadinstitute.org/gsea/msigdb/index.jsp). Genes used for GSEA analysis were pre-ranked based on log2 fold change of TPM (Transcripts Per Kilobase Million) between M2 and control. Significantly M2-affected genes identified in H929 cells were further assayed by real-time qRT-PCR, FC analysis, and immunoblotting in 4–8 other MM cell lines.
FC acquisition and analysis
Quantitative FC-based analysis was used to determine geometric mean fluorescence intensity (MFI) values for indicated proteins, ADCC assays under mono- or combination treatment settings, live vs dead cells, as well as CD107a degranulation and IFNγ production in NK cells (CD56+CD3-) (25–29). All data were obtained using BD FACSCanto™ II and BD LSRFortessa™ flow cytometers, and analyzed by FlowJo V8.6.6 (BD Life Sciences).
Generation of CRISPR-modified and small hairpin RNAs (shRNA) MM transfectants
STAT1 knockout- and IRF1 shRNA-H929 transfectants were generated using lentivirus transduction with two sgRNAs targeting STAT1 gene and two shRNAs targeting IRF1 gene, respectively (Horizon Discovery/Dharmacon, Lafayette, CO). Lentiviral particles were transduced into H929 cells, as previously described (30).
ADCC assays mediated by NK effector cells against MM cell lines
Various target MM cell lines treated with M2 vs control vehicle, were pre-stained with CFSE dye or calcein-AM (Invitrogen, C34554), according to manufacturer’s instructions. Human NK (CD56+CD3-, >90% purity) cells were collected from fresh buffy coat of healthy donors with the RosetteSep human NK cell enrichment cocktail (StemCell Technologies Inc. Vancouver, Canada). In brief, NK cells were incubated with CFSE-labeled MM cells (20,000 target cells/well in a U bottom 96-well plate) at an effector:target (E:T) ratio of 1:1 in the presence of Dara overnight at 37°C in RPMI1640 media supplemented with 10% FBS and 10 ng/ml IL2. MM cells were also stained with the LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Aqua L/D) (Invitrogen, L34957), according to manufacturer’s directions to define viable cells as Aqua-/Annexin V-. Viable MM cells were determined by gating on CFSE+ cells after co-incubation with NK cells. The percentage of cytotoxicity was calculated as [(total CFSE+ MM cells - viable MM cells)/total CFSE+ MM cells] × 100%. When specified, PBMCs from patient samples were also used for Dara-induced ADCC against indicated target MM cells, in the presence or absence of BMSCs. M2-induced ADCC assays were also performed using indicated calcein-AM labeled MM cells as target cells and NK cells from normal donor (n=3) as effector cells at E:T ratio of 10:1.
FC-based ADCC assays using patient bone marrow mononuclear cells (BMMCs) with paired PBMC effector cells
BMMCs derived from patients with RRMM and autologous PBMC effector cells, were used in Dara-induced ADCC assays. Sample viability start at co-incubation was > 98%, as confirmed by Annexin V-/Aqua-. BMMCs and the paired PBMCs were co-cultured in the presence of Dara in 96-well U-bottom plates for 2 days. Surviving CD138+ patient cells were enumerated by quantitative FC analysis of CD138+ cells in the presence of Flow-Count 123 beads (Thermo Fisher Scientific) and Aqua L/D. The percentage of Dara-induced ADCC was calculated using the following formula: % lysis of CD138+ cells = [1 - (number of surviving CD138+ cells in the presence of Dara/number of surviving CD138+ cells in the presence of isotype control IgG group)] × 100%.
Human NK-reconstituted murine model of human MM
All experimental procedures and protocols were approved by the Institutional Animal Care and Use Committee (Dana Farber Cancer Institute). MM1S cells were subcutaneously injected into 6- to 8-week-old NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice lacking functional NK cells (Jackson Labs; Bar Harbor, ME). Fourteen days later, mice with MM1S tumor growth were randomized into 6 groups (6 mice per group except 7 mice in M2+NK cells+Dara combination group). Mice received a single M2 (0.3 mg/kg) i.v. injection at treatment day 1 following randomization. At treatment day 2, some mice were injected intraperitoneally (i.p.) with human NK (5 × 106 cells) once per week for 3 weeks (31,32). Dara was given i.p. the following day after NK cell injection for a total of 3 administrations (8 mg/kg per injection per mouse). MM1S tumors were measured twice per week, as previously described (13,18,21). Tissue sections from tumors harvested from mice 3 days after M2 administration were subject to immunoblotting, FC analysis, and IHC staining for indicated proteins.
Statistical analysis
Statistical significance (P-value < .05 ) of no-RNA-seq data were assessed with GraphPad Prism 8.3.1 (La Jolla, CA). Data were analyzed using Student t tests for 2 group comparisons or analysis of variance (ANOVA) for multiple comparisons, unless specified. Each in vitro experiment was performed at least thrice for each condition: each drug concentration, sources of NK or PBMC effector cells, MM target cell lines sensitive and resistant to current therapies, and patient samples. All data were presented as means ± standard deviations (SDs) from ≥ 3 independent experiments except when otherwise indicated. All error bars indicate SDs.
Drug interactions were assessed by CompuSyn software using the Chou-Talalay method to determine combination index (CI) (33). CI < 1 indicates synergism, CI > 1 antagonism, and CI = 1 additive effect.
Accession code
RNA-seq data was deposited in Gene Expression Omnibus (GEO) database under accession number GSE168967.
Results
Interferon (IFN) signaling and IFN I-stimulated genes (ISGs) are significantly enriched by M2 treatment in MM cells
We performed RNA sequencing followed by GSEA to profile transcriptional changes and identified affected signaling pathways in H929 MM cells treated with a sublethal M2 concentration for 1 day. Genes related to IFN signaling pathway were significantly enriched after treatment with M2 (FDR = 0), represented by 11 of the top 51 upregulated genes in M2 vs control vehicle treatment (P < .0002, Supplementary Table S1) (Fig. 1A, in black). Type I IFN (IFN I)-associated genes were differentially impacted by M2 (FDR = 0.0013, Fig. 1B, left), with the most M2-upregulated IFN-driven genes (Fig. 1B, right, in black, P < .004, Supplementary Table S2) including chemokines/cytokines and receptors (i.e., CXCL10, CCL4L1/2, CCL22, CCL1/3/4/5, CCL3L1/3, TNFSF9/10, CCR7), CASP1, IFN-stimulated genes (ISGs, i.e., XAF1, TRIMs, IFITs, ISG15, GBP2/3, OAS1/2/L, RNASEL, MX1/2, FAS), IKBKE, IRF1/6/7/9, STAT1, TMEM173 (STING), and SOCS1/3 (Fig. 1B, right, in black).
Figure 1. Targeting BCMA by MEDI2228 (M2) significantly induces IFN signaling and IFN I-stimulated genes in MM cells.
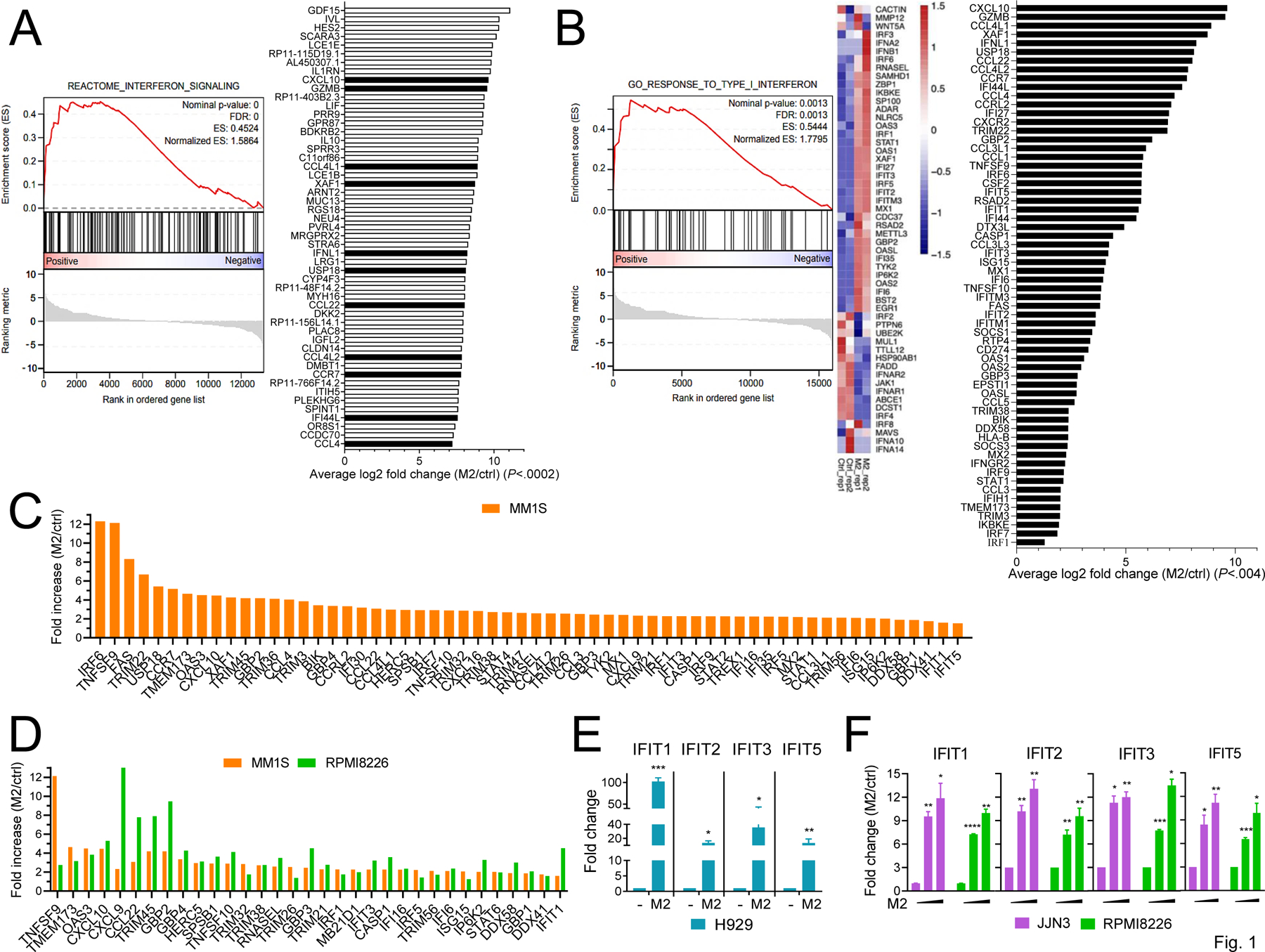
A-B The transcriptional changes resulting from MEDI2228 (M2, 0.5 μg/ml) treatment were studied in H929 MM cells following 1-day incubation (>85% viable H929 cells). RNA sequencing was performed followed by gene set enrichment analysis (GSEA), showing enrichment of interferon (IFN) pathways and Type I IFN genes. Shown are the top 51 upregulated genes by M2 vs control vehicle (ctrl) (P < .0002, Supplementary Table S1) with IFN-related genes (A) and IFN I-upregulated and/or IFN stimulated genes (ISGs) (B, n = 62, P < .004) in black. Normalized enrichment scores (NES) and false discovery rate (FDR) scores are indicated (FDR = 0.0013 for the comparison). C-F Using real-time qRT-PCR, fold increases in indicated IFN I-related genes are shown for M2- vs ctrl-treated MM cell lines sensitive (C-D, MM1S; E, H929) and relatively insensitive (D and F, RPMI8226; F, JJN3) to M2 (20,21). MM1S (C, D) and RPMI8226 (D, F) cells were treated with 1 and 5 µg/ml of M2, respectively, for 16h (> 90% viability), followed by the examination of upregulated IFN I-related and/or ISG genes (C, n = 60; D, n = 33). Indicated IFITs were further validated (E, ctrl (−) vs 0.5 µg/ml M2; F, 0, 1, 10 µg/ml of M2). Transcripts were normalized by geomean of internal controls. Two biological repeats (A, B) or three (C, F) independent experiments were done for each treatment. Each experiment performed in triplicate at each condition (C-F). Data are presented as means ± standard deviations (SDs) (error bars). *P < .05, **P < .01, ***P < .001, ****P < .0001
We next used real-time qRT-PCR to determine whether M2 altered expression of IFN 1-regulated genes in 3 other MM cell lines with various levels of sensitivity to M2 (H929>MM1S>JJN3>>RPMI8226) (20,21). Fold increases in mRNA expression of 60 tested IFN I-related genes are shown in M2- (16h at the sublethal concentration) vs control-treated MM1S cells (Fig. 1C). M2 also induced 33 IFN I-associated genes in relatively insensitive RPMI8226 cell line which expresses a lower BCMA level than MM1S and H929 cells, including TNFSF9/10, CXCL9/10, CCL22, IRF1/5, TMEM173 (STING), OAS3, TRIM45/32/38/21, GBP1/2/3/4, HERC5, RNASEL, MB21D1 (cGAS), DDX41/58, STAT6, IFIT1/3, and ISG15 (Fig. 1D). Furthermore, in agreement with the RNA-seq results, M2 significantly induced IFIT1/2/3/5 transcripts in H929 cells (Fig. 1E). The IFIT proteins confer immunity against viral infection and promote tumor cell death when overexpressed (34). The expression of IFIT1/2/3/5 was also increased by M2 in JJN3 and RPMI8226 cells which are relatively insensitive to M2 when compared with H929 and MM1S (Fig. 1F). Thus, M2 significantly induced numerous IFN I-signaling and/or ISGs in MM cell lines following DNA damage and prior to apoptosis, despite their genetic heterogeneity and drug sensitivity.
CD38 expression is increased by M2, associated with DNA damage and activation of STAT1 and cGAS-STING pathways
Using qRT-PCR, we next showed that M2, in a concentration- and time-dependent manner, induced CD38, an IFN-regulated gene, in H929 cells (Fig. 2A), supporting the RNA-seq data (P = .012, M2 vs control at d1). Using FC analysis gated on viable MM cells with various baseline CD38 levels, enhanced CD38 surface expression was seen on 8 MM cell lines triggered by M2 treatment (Fig. 2B), including IMiDs-resistant H929(R) and MM1S(R) cells derived from H929 and MM1S cells (21), respectively, and RPMI8226 cells. M2 also increased membrane CD38 expression on other MM cell lines, including MOLP8 and JJN3 with highest and undetectable baseline CD38 protein expression, respectively. Other DNA-damaging agents including gamma irradiation could induce CD38 expression in H929 cells (Supplementary Fig. 1A).
Figure 2. CD38 is upregulated by M2-induced DNA damage response associated with activation of STAT1 and cGAS-STING signaling in MM cells.
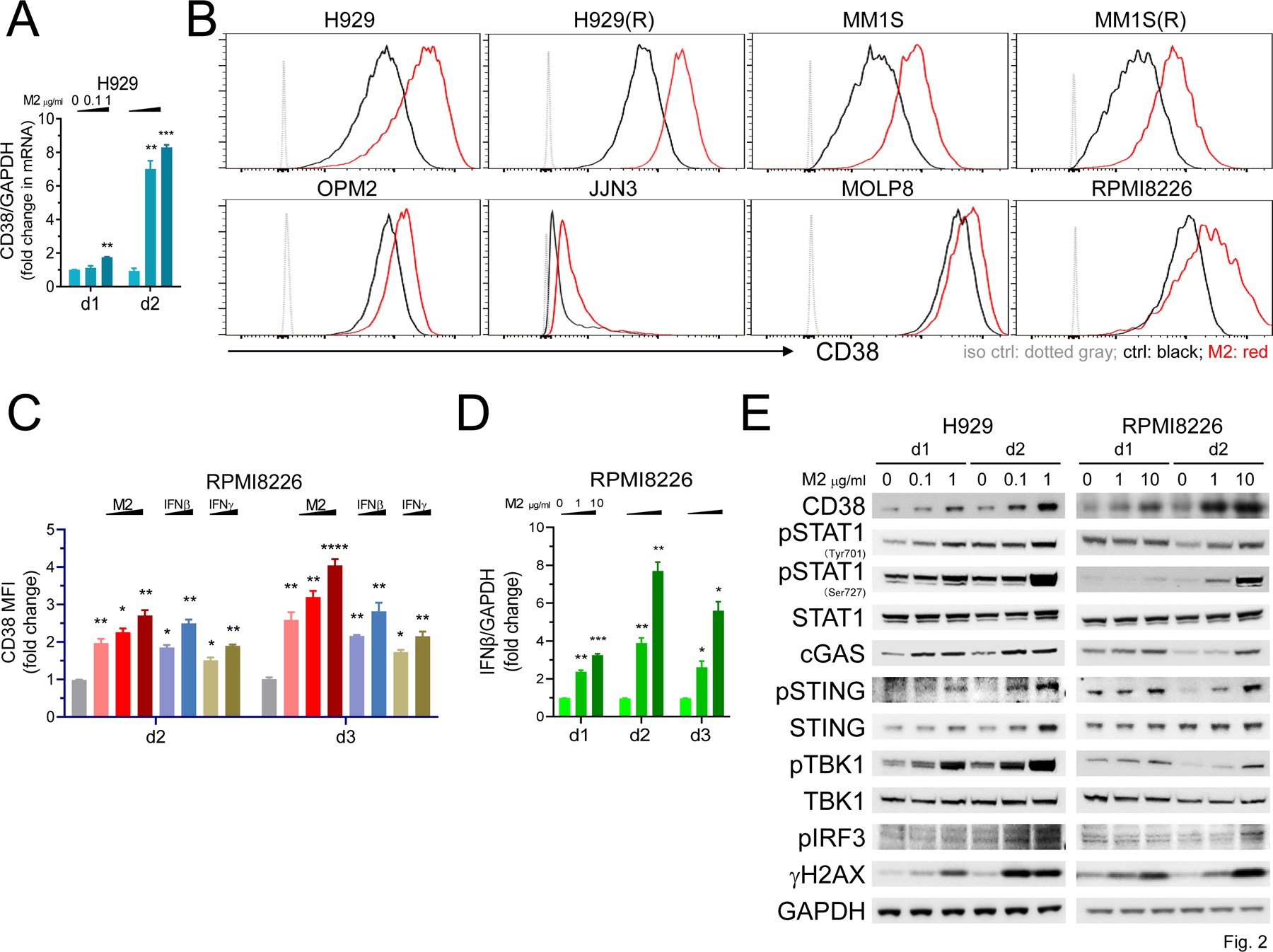
A Using real-time qRT-PCR, CD38 transcript levels were quantitated in M2-treated H929 cells at day 1 (d1) and d2. B-C Using flow cytometry (FC) analysis, CD38 membrane expression was determined on viable (Aqua-/Annexin V-) MM cells. B, Eight MM cell lines were treated with M2 for 3 days, including 4 sensitive (0.1 μg/ml, upper panel) and 4 less sensitive (1 μg/ml, lower panel) to M2. Shown are histograms (B) and fold increases in geometric mean fluorescence intensity (MFI) values of CD38 at indicated treatments vs ctrl vehicle groups (C). C RPMI8226 cells were treated with M2 (1, 5, 10 μg/ml), IFNβ or IFNγ (100, 500 U units/ml) at d2 and d3. D IFNβ expression levels were quantitated by real-time qRT-PCR analysis following M2 treatment for various time intervals. E Protein lysates were made from H929 (left) and RPMI8226 (right) cells followed by immunoblotting using antibodies specific for indicated molecules to examine activation of STAT1 and cGAS-STING signaling cascades. GAPDH served as a loading control. Each experiment was performed at least thrice in triplicate for each condition, and data are presented as means ± SDs (error bars). *P < .05, **P < .01, ***P < .001, ****P < .0001
Exogenous IFNβ and IFNγ can modulate CD38 expression in MM cells (12,35). After 2 day-treatment in RPMI8226 cells, we observed that M2-enhanced CD38 expression was comparable to IFNβ- and higher than IFNγ-induced expression (Fig. 2C). At day 3, M2 further increased CD38 membrane expression and induced higher CD38 protein levels than either IFNβ or IFNγ. M2-induced IFNβ was reduced from d2 and at later time points (Fig. 2D), suggesting that M2-induced CD38 expression via IFNβ-dependent and -independent mechanisms. Of note, compared with IFNβ induction, M2 only weakly enhanced IFNα and could not induce IFNγ (type II IFN) in H929 and RPMI8226 MM cells (data not shown).
Immunoblotting was next performed to confirm M2-induced CD38 protein levels and defined major IFN I-related signaling cascades. After 1-day treatment, M2 enhanced CD38 protein expression in a concentration-dependent fashion in H929 and RPMI8226 cells, associated with increased DNA damage shown by γH2AX (Fig. 2E) and consistent with our previous report (21). CD38 and γH2AX were further upregulated at day 2. In a similar fashion, M2 activated STAT1 signaling, evidenced by increased phosphorylation at Tyr701 and Ser727, more prominently at Ser727 which is essential for STAT1 transcriptional response mediating full-fledged IFNγ-dependent innate immunity (36,37). M2 simultaneously induced cGAS-STING signal transduction cascade, evidenced by elevated cGAS protein and phosphorylation of STING as well as downstream TBK1 and IRF3. STING protein levels were increased to a greater extent in H929 than RPMI8226 cells, consistent with higher levels of MB21D1 (cGAS) and TMEM173 (STING) transcripts triggered by M2 (Fig. 1B–D).
M2 significantly induces expression of IRF1 via STAT1 activation to sustain CD38 upregulation in MM cells
As in H929 and RPMI8226 cells, M2 treatment also consistently induced STAT1 phosphorylation, CD38 expression, and increased γH2AX in MM1S and OPM2 cells (Fig. 3A, Supplementary Fig. 1B). The transcription of IRF1, a primary IFN response gene highly induced by IFNγ, is regulated by STAT1 DNA binding element. M2 treatment enhanced IRF1 protein in all 3 MM cells in a concentration-dependent fashion (Fig. 3A), consistent with our mRNA data by RNA-seq and qRT-PCR (Fig. 1B–D). To directly demonstrate that STAT1 protein and its activation was required for M2-induced IRF1 and CD38 expression, H929 cells were transduced with lentiviruses expressing constitutive Cas9 expression vectors tagged with STAT1 sgRNAs to efficiently knock out STAT1 (Fig. 3B–C). Loss of STAT1 significantly decreased IRF1, and to a lesser extent CD38, proteins. Moreover, IRF1 and CD38 proteins were not induced by M2 treatment in STAT1-knockout vs control vector H929 transfectants. CD38 membrane expression was also decreased and M2 failed to enhance CD38 expression in sgSTAT1s- vs control vector-transduced H929 cells (Fig. 3B–C, lower graphs).
Figure 3. M2 treatment activates IRF1 and STAT1 pathways to enhance CD38 expression in MM cells.
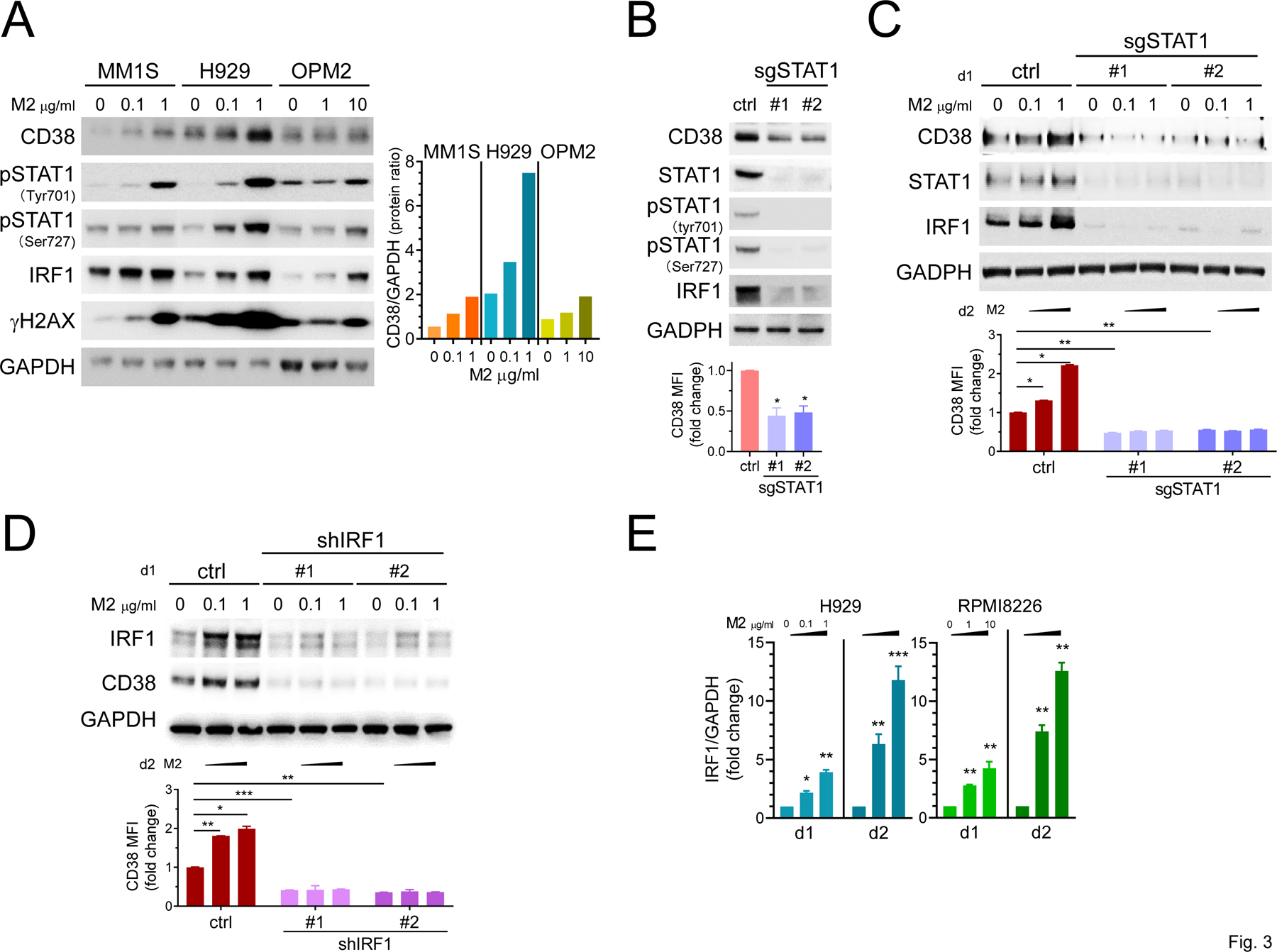
A Three representative MM cells with various CD38 levels were treated with M2 for 1d. Cell lysates were made for immunoblotting using antibodies specific for indicated proteins. GAPDH served as a loading control. Ratios of CD38 to GAPDH protein levels at indicated M2 concentrations were also graphed (right). B-E Changes in CD38, STAT1, phosphorylation of STAT1 (pSTAT1), and IRF1 were examined by immunoblotting in H929 cells with indicated gene manipulation in the presence or absence of M2 treatment for 1d. B-C STAT1 knockout and changes in indicated molecules were determined in cells treated with M2 vs ctrl vehicle following transfection with sgSTAT1#1 or #2 vs ctrl vector. Using quantitative FC analysis (B, C, D, lower panels), CD38 cell surface levels were also evaluated on Aqua-negative viable cells following 2d treatments with M2 vs ctrl vehicle. D Lentiviruses expressing shIRF1# or #2 vs ctrl vector were used followed by treatments with M2 vs ctrl vehicle for 1d (upper panel) and 2d (lower panel). E Using real-time qRT-PCR, IRF1 expression was evaluated in M2-treated MM cells. Each experiment was performed at least thrice and in triplicate for each condition. Data are presented as means ± SDs (error bars). *P < .05, **P < .01, ***P < .001
We also showed that shIRF1 knock-down significantly downregulated baseline CD38 levels (Fig. 3D, Supplementary Fig. 1C) and M2 failed to induce CD38 in lentivirus shIRF1-transduced H929 cells. Finally, concentration- and time-dependent induction of IRF1 expression by M2 treatment was further shown in RPMI8226 cells (Fig. 3E). Thus, M2 induced IRF1 expression via STAT1 phosphorylation, and both IRF1 and STAT1 are required to upregulate CD38 expression on MM cells.
M2 enhances CD38 levels in drug-resistant MM cells in the presence of CD38-downregulating factors in the bone marrow (BM) microenvironment
We next demonstrated that M2 effectively induced STAT1-IRF1-dependent CD38 protein expression, associated with increased γH2AX levels, even in MM cell lines resistant to IMiDs (H929(R)) or bortezomib (ANBL6-BR) (Fig. 4A). In addition, since M2 treatment was done in IL6 (2 ng/ml)-containing culture media for paired ANBL6 and ANBL6-BR cells, these data indicated that M2 could activate STAT1-dependent upregulation of IRF1 and CD38 in the presence of IL6.
Figure 4. M2 overcomes downregulation of CD38 in MM cells triggered by IL6 and BMSC supernatant, and further enhances Dara-induced lysis of drug-resistant patient MM cells.
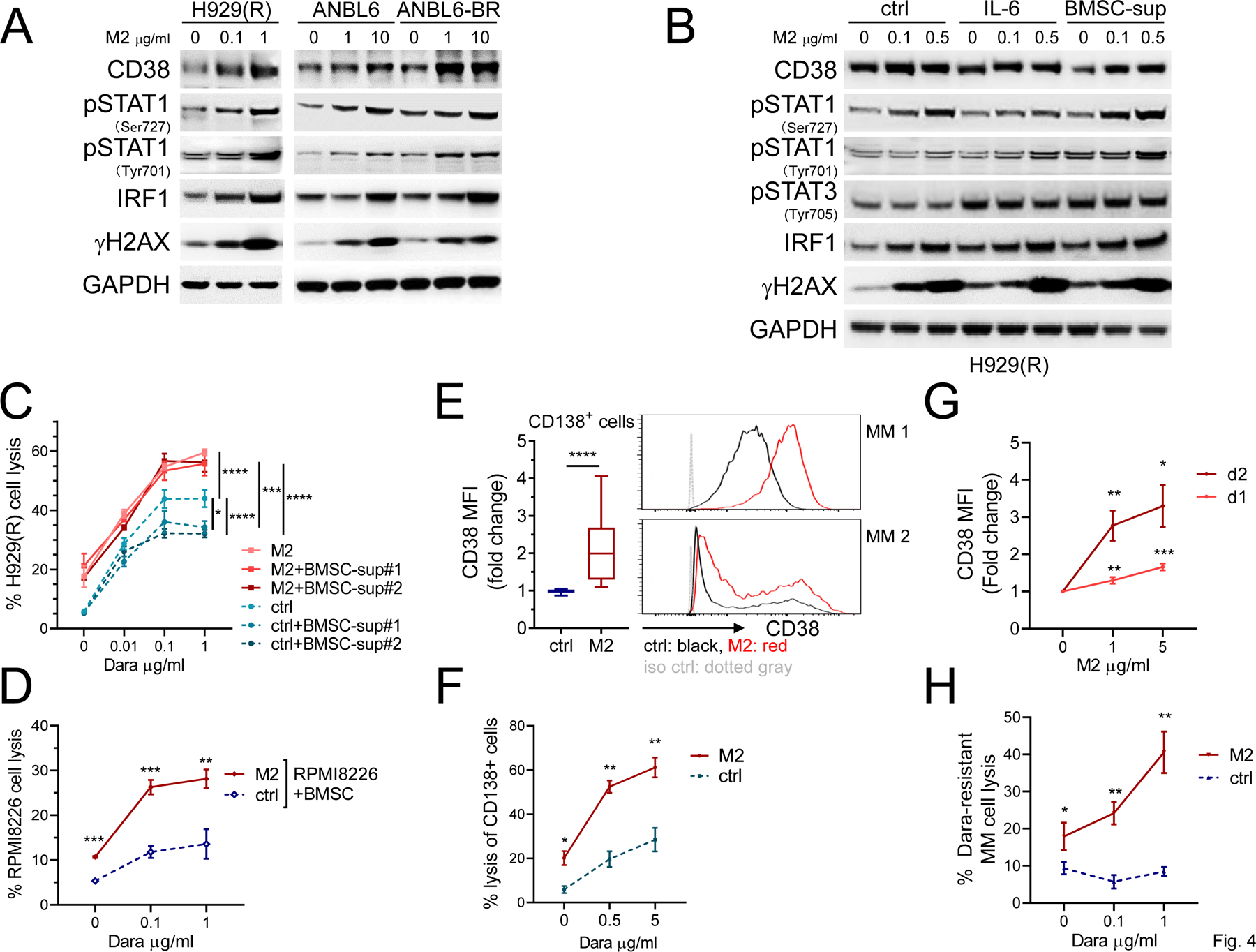
A IMiDs-resistant H929(R), ANBL6, and its paired bortezomib-resistant ANBL6-BR cells were incubated with M2 for 1d. IL6 (2 ng/ml) was also added in ANBL6 and ANBL6-BR cultures. Cell lysates were made for immunoblotting to determine levels of CD38 and indicated proteins, with GAPDH as a loading control. B BMSCs from patients were cultured for 3d in the same culture medium as MM cells. Culture supernatants were then harvested, passed through 0.2 μm filters, mixed with 50 % of control culture medium, and used as BMSC culture supernatant (BMSC-sup). H929(R) cells were pretreated with ctrl or M2 (0.1, 0.5 µg/ml) for 1d in the absence or presence of IL6 (2 ng/ml) or BMSC-sup for 2d. C H929(R) target cells, pretreated with M2 or ctrl for 1d, were co-cultured with NK cells (n = 3 normal donors) in Dara-induced ADCC assay in the absence or presence of BMSC-sup from cultures of 2 patient samples. Using quantitative FC analysis, the % H929(R) cell lysis was determined. D Dara-induced lysis of M2- vs ctrl-treated RPMI8226 cells was assayed in the presence of BMSCs (n = 3). Patient PBMCs (n = 3) were used as effector cells. E Using FC analysis, CD38 surface expression was analyzed on CD138+Aqua-/Annexin V- viable patient cells treated with 1 µg/ml M2 vs ctrl for 2d (RRMM, n = 24). Two representative samples are shown on the right. F BM mononuclear cells (BMMCs) from a RRMM patient were treated with M2 vs ctrl (1 µg/ml) for 1d, followed by co-culture with PBMC from the same patient in the presence of Dara for 2d. Using quantitative FC analysis, the % lysis of CD138+ cells was determined. BMMCs from Dara-resistant patients (n = 3) were treated with M2 followed by assessment of CD38 surface expression on viable CD138+ cells (G); samples after M2 (1 µg/ml) treatment for 1d were used to determine % Dara-induced autologous patient MM lysis in the presence of paired PBMCs (H). Three independent experiments (C-D) were done using NK cells from 3 individuals in triplicates at each condition. All results are shown as means ± SDs (error bars). *P < .05, **P < .01, ***P < .001, ****P < .0001
In IMiDs-resistant H929(R) cells, M2 induced γH2AX at the similar levels regardless of the presence of IL6 or supernatants collected from cultures of BM stromal cells (BMSC-sup) (Fig. 4B). M2 also induced STAT1 phosphorylation associated with IRF1 upregulation under all test conditions (Fig. 4B). While IL6 and BMSC-sup enhanced STAT3 phosphorylation and decreased baseline CD38 levels compared with media control, M2 overcame CD38 downregulation by IL6 and BMSC-sup. These results also complement M2-induced expression of SOCS3 (Fig. 1B), which suppresses IL6 signal (38) and modulates innate immune responses caused by viruses.
M2 improves Dara-induced cytotoxicity against drug-resistant MM cell lines and patient MM cells protected by soluble and cellular BM components
Although Dara cannot directly induce MM cell apoptosis in vitro without FcR-expressing NK effector cells (39,40), M2 directly kills MM cells without ADCC as a consequence of the site-specific conjugation technology employed by this ADC (Supplementary Fig. 2). We next showed that Dara-induced cytotoxicity against IMiDs-resistant H929(R) cells was significantly reduced in the presence of BMSC-sup, evidenced by decreased % cell lysis and maximum cell lysis (Fig. 4C). In contrast, when M2-treated H929(R) target cells were used in the NK-MM cell co-cultures, Dara-induced lysis was increased even in the presence of BMSC-sup (P < .0001). Concentration-response curves of Dara were almost superimposable for M2-treated H929(R) target cells cultured with or without BMSC-sup. Furthermore, in the presence of MM-protecting BMSCs, Dara still induced higher % lysis of RPMI8226 cells pretreated with M2 vs control (P < .01).
We next treated BM mononuclear cells (BMMCs) from MM patients with 1 μg/ml M2 vs control vehicle for 2 days, followed by FC analysis for CD38 on viable CD138+ cells. M2 significantly upregulated CD38 surface expression on patient MM cells (n=24) (P < .001, Fig. 4E). Dara-induced patient MM cell lysis was next evaluated using M2-pretreated vs control BMMC target cells from a RRMM patient co-cultured with paired autologous PBMC effector cells. Importantly, M2 increased Dara-induced autologous effector cell-mediated CD138+ MM cell lysis (Fig. 4F, P < .02). Moreover, M2 induced CD38 upregulation in a concentration- and time-dependent manner in RRMM including Dara resistant disease (P < .02, Fig. 4G). Dara-induced CD138+ autologous patient MM cell lysis was improved following M2 treatment (n=3, Fig. 4H). These data indicated that M2-triggered upregulation of CD38 expression in patient MM cells was associated with increased Dara-induced autologous patient MM cell lysis.
M2 and Dara induce synergistic NK-mediated lysis of drug-resistant MM cell lines
Under sub-optimal test conditions comprising lower M2 concentrations with lower NK:MM cell ratios (1:1) and incubation times, we next determined % of Dara-induced lysis of MM cells (n=3) using 2 concentrations of M2. Target cells including RPMI8226, MM1S, and its paired IMiDs-resistant MM1S(R), were co-cultured with NK effector cells (n=3 normal donors). Dara increased % lysis of all M2-treated vs control MM cells (Supplementary Fig. 3), with CI values (33) <1 at all concentrations of both drugs indicating synergistic cytotoxicity.
M2 simultaneously upregulates multiple NKG2D ligands (NKG2DLs) and CD38, further improving MM susceptibility to NK cells
Target MM cell lines or patient MM cells pretreated with sub-lethal concentrations of M2 consistently showed increased % NK-mediated cell lysis, even without Dara (P < .05, Fig. 4C–D, F, H, and Supplementary Fig. 3). To further examine mechanism of this enhanced NK cytotoxicity triggered by M2, H929 and RPMI8226 MM cells were next pre-treated with sub-optimal concentrations of M2 (>90% target cell viability) before co-culture with NK cells at 2 E:T ratios. NK-induced MM cell lysis was followed by quantitative FC analysis. NK cells (n=3 normal donors) killed more M2-pretreated vs control MM target cells in an effector cell number-dependent manner (Fig. 5A, P < .0004 for H929; P < .008 for RPMI8226). In contrast, M2 treatment for 3d had no impact on viability and CD38 expression in NK cells (Supplementary Fig. 4A, 3 normal donors), confirming the lack of BCMA expression on NK cells. We also showed that the % IFNγ+NK cells was significantly increased when patient effector cells (n=4) were co-cultured with M2-pretreated vs control H929 MM target cells (Fig. 5B, P < .007 for NK1; P < .004 for NK2; P < .006 for NK3; P < .008 for NK4). Moreover, M2 enhanced % CD107a+ NK cells from 4 additional patient samples co-cultured with M2-treated vs control H929 target cells (Supplementary Fig. 4B–C, P < .05). Thus, M2 increased susceptibility of MM cells to NK cell killing via enhanced CD107a and IFNγ.
Figure 5. M2 increases expression of NKG2DLs and CD38 on MM cells, further promoting lytic function of NK cells against MM cells.
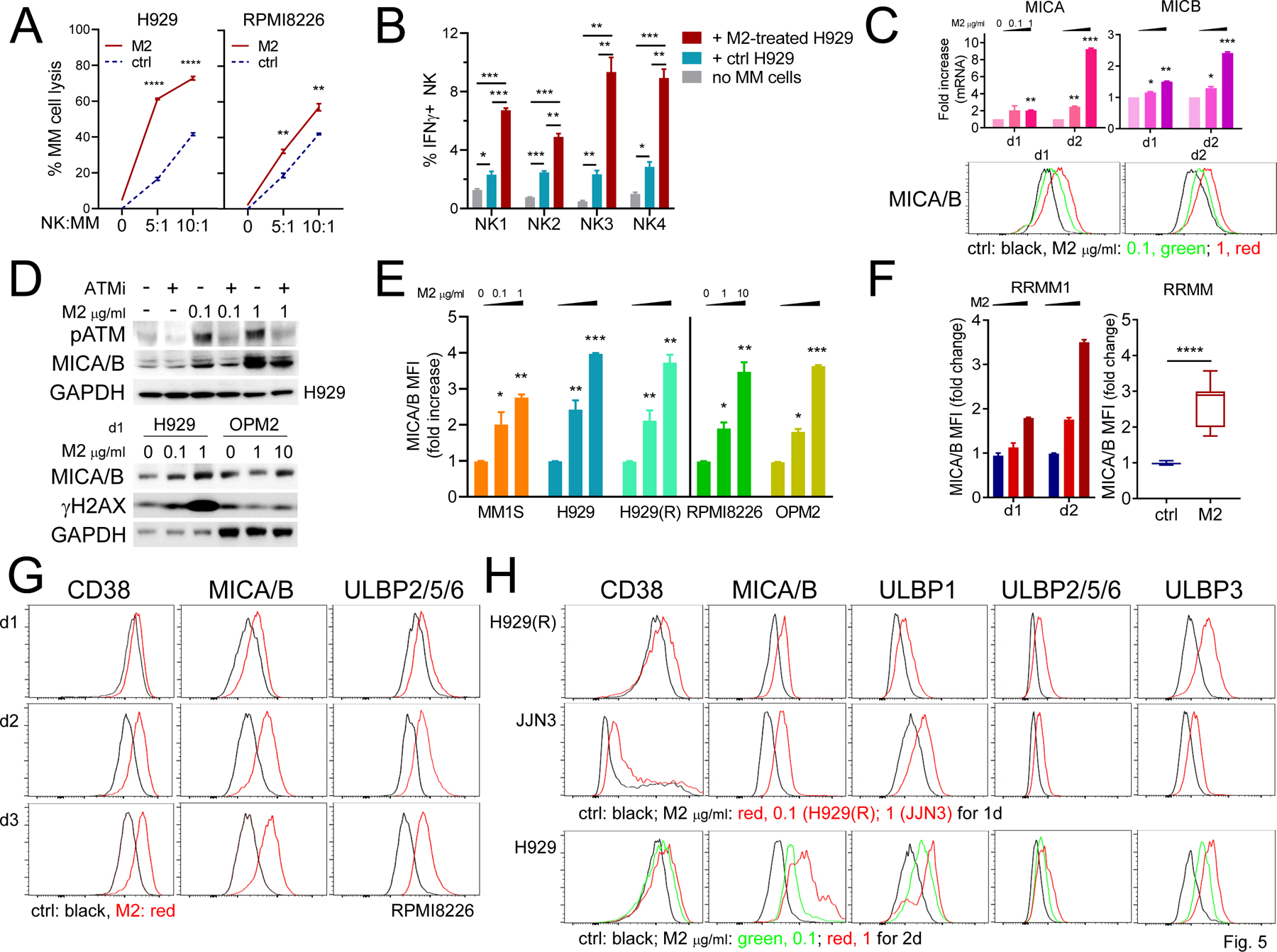
A Indicated MM target cells were pretreated with M2 for 12h (1 and 4 μg/ml for H929 and RPMI8226, respectively), washed, and then labeled with CFSE before co-culture with NK cells (n = 3) at increasing NK:MM (E:T) ratios. MM cell viability (Aqua-/AnnexinV-) was >90–95% before co-culture with NK cells. Using FC analysis, the % of lysis in CFSE+ MM cells was quantitated. B PBMCs from 4 patient samples were cultured alone or with (+) M2-treated vs control H929 cells for 6h, followed by quantitative FC analysis to determine the % of IFNγ+ NK (CD56+CD3-) cells. C Using real-time qRT-PCR (upper graph) and FC analysis (histograms, lower panel), MICA/B expression was studied at transcript and protein levels following M2 treatments for 1d and 2d in H929 cells. D Cell lysates were made from H929 cells pre-treated with or without ATM inhibitor (ATMi, Ku22933 10 μM) prior to M2 treatment (upper panel). Immunoblotting was also done to evaluate protein levels of MICA/B and γH2AX in H929 and OPM2 cells treated with M2 for 1d (lower panel). E-H Using FC analysis gated on viable MM cells, cell membrane expression levels were determined for MICA/B (E-H), other NKG2DLs (ULBP2/5/6 in G-H; ULBP1 and ULBP3 in H), and CD38 (G-H) in indicated MM cell lines (E, n = 5; G, RPMI8226; H, n = 3), and CD138+ cells of BMMCs from RRMM patients (F, n = 9). F BMMCs from a patient with RRMM were incubated with M2 at 0.5 and 2 μg/ml for 1d and 2d (left); and 9 additional RRMM samples were incubated with M2 at 1 μg/ml vs ctrl for 2d (right). H CD38 and 4 NKG2DLs were further investigated at d1 (H929(R), JJN3) and d2 (H929). Each experiment was performed at least thrice and in triplicate for each condition, and data are presented as means ± SDs (error bars). *P < .05, **P < .01, ***P < .001, ****P <.0001
Real-time qRT-PCR and immunoblotting analysis next showed that M2 enhanced expression of MICA/B, NKG2D ligands (NKG2DLs), at both mRNA and protein levels in H929 (Fig. 5C) and RPMI8226 (Supplementary Fig. 4D) cells in a concentration-and time-dependent manner. These data confirmed RNA-seq data for increased MICA/B transcripts in M2- vs control-treated H929 cells (Supplementary Fig. 4E, P < .01). Consistent with our recent report (21), M2 significantly induced phosphorylation of ATM and H2AX in H929 cells (Fig. 5D, upper panel). Importantly, M2, in a concentration-dependent manner, increased MICA/B expression which was blocked by an ATM inhibitor (ATMi, Ku22933). Upregulation of MICA/B protein associated with increased γH2AX levels was also demonstrated in M2-treated OPM2 cells (Fig. 5D, lower panel). Using FC analysis, membrane expression of MICA/B was further enhanced by M2 in all MM cell lines tested (n=5, Fig. 5E, Supplementary Fig. 4F), as well as in CD138+ tumor cells from patients with RRMM (P < .0001, n=9, Fig. 5F).
We next showed that M2, in a time-dependent manner, upregulated other NKGD2Ls including ULBP2/5/6, along with CD38 and MICA/B in RPMI8226 cells (Fig. 5G). ULBP1 and ULBP3 were also enhanced on 4 MM cells tested, associated with elevated levels of CD38, MICA/B, and ULBP2/5/6 (Fig. 5H, Supplementary Fig. 4E). Induction of NKG2DLs (i.e., MICA/B, ULBP1, ULBP3) by M2 treatment was inhibited by ATMi (Supplementary Fig. 5A), further confirming that ATM-activated DDR regulates M2-induced expression of NKG2DLs. In addition, the MMAF ADC homolog M3, even at 1-log higher concentrations than M2, failed to upregulate CD38 and MICA/B in MM cells tested (n=3, Supplementary Fig. 5B). These results indicate that multiple DNA damage-activated signaling cascades are critical for M2-induced upregulation of CD38 and NKG2DLs in MM cells.
Combined treatments with M2 and Dara completely eliminated tumors in the NK-humanized NSG mouse model
We next evaluated the in vivo efficacy of combination M2 with Dara in a human NK-cell NSG model of human MM. MM1S cells, which express relatively lower baseline CD38 levels than RPMI8226 and H929 MM cells, were injected into mice 14 days before start of treatment. Animals with palpable MM1S tumors were randomized into six groups receiving vehicle control, NK cells only, a single low M2 concentration (0.3 mg/kg), M2+NK cells, NK cells+Dara (8 mg/kg per injection per week for 3 administrations in total), or M2+NK cells+Dara (Supplementary Fig. 6A). After 7 days of treatment, mice in the M2+NK cells+Dara cohort showed decreased tumor growth vs all other 5 groups (Fig. 6A, Supplementary Fig. 6B–D, P < .01). At day 14, M2+NK cells group had decreased tumor growth, compared with NK cells alone (P < .01) or NK cells+Dara (P < .05) groups. M2+NK cells+Dara treatment most efficiently suppressed tumor growth, compared with all other groups (P < .001). Mice receiving M2+NK cells+Dara had undetectable tumor at 34 days and remained tumor-free and without weight loss (Fig. 6B) out to 150-day follow-up. A significantly prolonged median overall survival was also observed in the combination-treated group vs cohorts treated with these agents alone (control, 25 days; NK cells, 25 days; NK cells+Dara, 48 days; M2, 50 days; M2+NK cells, 62 days; M2+NK cells+Dara, all mice alive, P < .0001) (Fig. 6C–D).
Figure 6. Combination M2 with Dara treatment completely eradicates MM1S tumors and extends host survival in NK-humanized NSG mice.
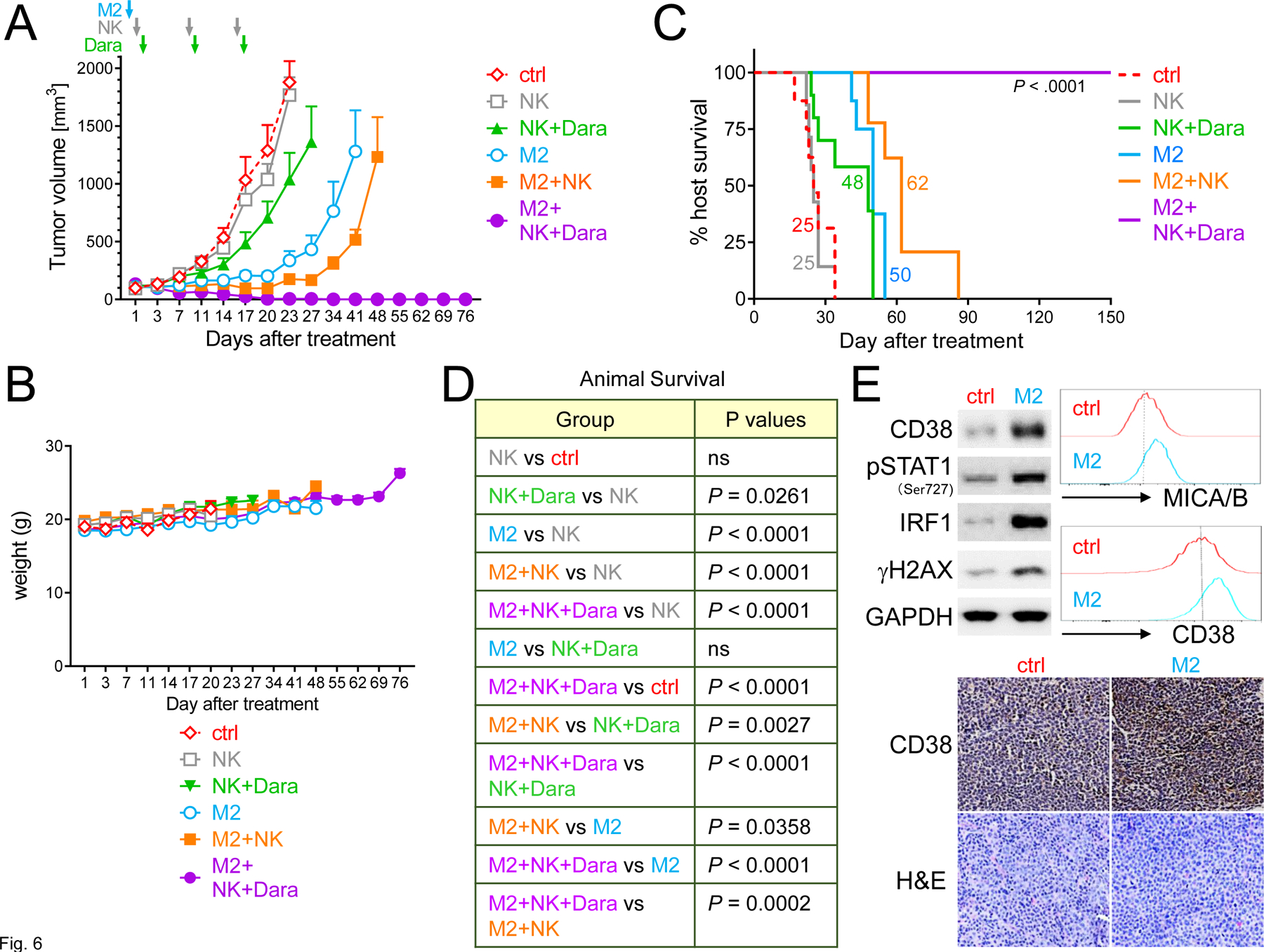
NSG mice were s.c. injected with MM1S cells 14 days before start of treatment. Mice with tumor growth were then randomized into 6 groups (6 mice per group except M2+NK+Dara): saline control (ctrl, red), NK (once weekly for 3 times, brown), NK+Dara (8 mg/kg per injection for 3 injections, green), M2 (0.3 mg/kg, only one injection, blue), M2+NK (orange), or M2+NK+Dara (n = 7, purple). A Shown are mean tumor volumes (mm3) ± SDs (error bars) at following days vs start of treatment (d1). Tumor growth was significantly inhibited in the combination-treated group compared with other groups from d7 (P < .01, Supplementary Fig. 6B). B Weights of mice were followed. C-D Using Kaplan-Meier and log-rank analysis, the median overall survival of animals was derived (ctrl, 25 days; NK, 25 days; NK+Dara, 48 days; M2, 50 days; M2+NK, 62 days). All 7 mice were tumor-free in M2+NK+Dara group at 150-day follow-up (****P < .0001). E Tumors were removed from mice 3 days after treatments with M2 vs ctrl, followed by immunoblotting analysis using indicated antibodies (upper left panel) and FC analysis for MICA/B and CD38 surface expression on viable MM1S tumor cells (Aqua-/AnnexinV-) (upper right histograms). Tumor tissue sections were also immunohistochemically analyzed for CD38 (Original magnification, x200) (lower panel).
In MM1S tumors harvested from these mice, M2 activated STAT1 signaling and upregulated protein levels of IRF1 and CD38, associated with increased γH2AX (Fig. 6E). FC and IHC analysis further confirmed enhanced MICA/B and CD38 protein levels in M2- vs control-treated tumors. These data indicate that M2 still activated STAT1/IRF1 signaling to induce CD38 and MICA/B expression in MM tumors grown in mice, resulting in enhanced CD38 targeting and increased NK cell killing in vivo when combined with Dara.
Discussion
We here characterized novel immunomodulatory effects triggered by M2, which were mediated via DDR-dependent activation of cGAS-STING-TBK1-IRF3 and STAT1-IRF1 signaling cascades and downstream IFN I-stimulated genes, even in MM cells resistant to current therapies. Since these pathways are central DNA-sensing machinery in innate immunity and viral defense (41–43), these data suggest that M2 may overcome MM cell immune evasion associated with resistance to multiple therapies. M2 selectively induced CD38 and NKG2DLs on MM cells, but not other normal cells including NK cells. It restored MM sensitivity to CD38 targeting by Dara via NK cells, even in the presence of immunosuppressive BM components. Importantly, all MM1S tumor-bearing NSG mice reconstituted with human NK cells became tumor-free following a single low concentration M2 with Dara treatment, with 100% host survival.
In our prior study of M2, we showed that DDR was induced followed by apoptosis; furthermore, combination of ADC with bortezomib or DDR (ATR/ATMWEE1) inhibitor mediated synergistic cytotoxicity (21). Here we focused on impact of M2 treatment on DDR and immunomodulation. We showed that M2 targeting BCMA on MM cells activated cGAS, an indiscriminate immune sensor for virus-derived double-strand DNA (dsDNA), as well as downstream stimulated adapter protein STING and related type I IFN signaling, thereby enhancing immune recognition and subsequent destruction of nascent tumors by immunomodulatory cells. Increased cGAS transcript and protein levels indicate that M2 induces an immunomodulatory response to promote tumor immunogenicity, analagous to those against invasion of microbial pathogens including viruses. Besides STING phosphorylation, M2 also enhanced STING trancript and protein levels in MM cells; therefore, M2 may restore STING expression which is suppressed during tumor progression (44), thereby maintaining sensing of cytoplasmic dsDNA. We also observed activation of TBK1, downstream of STING, without increasing TBK1 levels and related persistent inflammation which can be linked to cancer progression (45,46). Among IFN I-related genes downstream of cGAS-STING and STAT1-IRF1 pathways, we here focused on CD38 and showed consistent CD38 upregulation triggered by M2 treatment in MM cell lines and patient MM cells including those resistant to current therapies therapies.
In all MM cell lines tested here, we showed that M2 induced IRF family genes critical for IFN-driven immune effects, except IRF2 and IRF4. IRF2 acts as an antagonist to suppress IRF1/3-mediated transcriptional activation of IFNα/β for immune stimulation (47) and IRF4 is required for MM cell survival (48). Importantly, M2 inhibited IRF2 and IRF4 while induced expression of other IRF family members including IRF1 to mediate its immunostimulatory function. We went on to show that IRF1 was induced by M2 via STAT1 activation in MM cells, regardless of drug resistance or the presence of IL6 or BMSC-sup which downregulate CD38 levels. This M2-enhanced CD38 expression required DNA damage-induced STAT1-IRF1 signaling at both transcriptional and translational levels. Coupled with increased expression of IFN I and ISGs, upregulated cell surface CD38 expression on tumor cells further improves Dara targeting of MM cells.
Absence of NKG2DLs, cognate ligands for the major activating receptor NKG2D on NK cells, contributes to immune evasion of leukemia stem cells (49). Here we showed that M2 significantly increased NKG2DLs on the MM cell membrane to provide “kill me” signals, leading to enhanced CD107a degranulation and IFNγ secretion in patient NK cells in co-cultures. Along with increased chemokines/cytokines critical for IFN-induced MM cell apoptosis, as well as effector cell migration, homing, and function, M2 facilitated recognition and killing of MM cells by NK cells. Since M2, but not its MMAF-ADC homolog M3 even used at >1-log higher concentrations enhanced surface expression of CD38 and NKG2DLs on MM cells, the potent DDR-mediated immunomodulation triggered by M2 vs M3 is critical in rendering MM cells more susceptible to Dara-induced NK cell killing.
M2-induced CD38 expression and MM cell vulnerability to NK killing shown here could be particularly important to eliminate MM cells expressing very low BCMA and CD38 levels, including low proliferative or dormant MM-initiating cells or drug-resistant clones. The addition of Dara with M2 may also be useful to target minimal residual disease. Since M2 specifically triggers DNA damages in MM cells but not other normal cells which lack BCMA, upregulation of CD38 and NKG2DLs would be restricted to MM cells and adverse effects on CD38-expressing normal blood cells could be avoided. In this way, M2 may minimize an on-target off tumor toxicity of Dara in normal immune cells including NK and T cells, monocytes, and macrophages. Moreover, compared with other DNA damaging therapies including irradiation, melphalan, doxorubicin, or bortezomib which would affect normal cells, combining M2 with Dara might reduce nonspecific toxicity and thereby improve therapeutic index.
We next showed that a low single-concentration M2 (0.3 mg/kg) in MM1S tumor-bearing NSG mice effectively blocked tumor growth and improved host survival, compared with vehicle control or NK cells alone. In addition, MM1S tumors harvested from M2-treated mice showed activated STAT1-IRF-1 signaling and increased protein expression of CD38 and MICA/B, associated with DNA damages, mirroring our in vitro results. These results provide molecular mechanisms for underlying enhanced susceptibility of M2-treated MM1S tumors to Dara targeting and NK killing in vivo. Of note, M2+NK cells treated mice also showed prolonged survival, compared with M2 alone or NK cells+Dara treated cohorts. Most importantly, M2 with Dara triggered synergistic cytotoxicity in vivo, leading to tumor-free animals with 100% survival.
Taken together, our data showed that M2 potentiated immunogenicity of MM cells, even in the presence of immunosuppressive IL6 or BMSCs, via induction of cGAS-STING-TBK1-IRF3 and STAT1-IRF1-driven IFN I siganling, as well as ATM/ATR-CHK1/2-related DDR. M2 specifically upregulated CD38 and NKG2DLs on MM cells but not other BCMA-negative normal cells including NK cells. It enhanced Dara cytotoxicity without depleting NK cells, resulting in improved NK surveillance of MM cells resistant to current therapies. Importantly, combination M2 with Dara at low concentrations confers enhanced CD38 targeting and efficacy compared with either agent alone, which may ultimately deepen treatment responses with a more favorable therapeutic index. Ongoing studies are evaluating whether these M2-regulated immunomodulatory effects may also enhance tumor recognition and destruction by effector cells other than NK cells. Moreover, the prominent upregulation of activating NKG2DLs after M2 treatment may predict synergy with NK activating therapeutics such as elotuzumab (anti-SLAMF7 MoAb). Future studies will also further delineate the impact of ADC treatment on BCMA signaling and its sequelae.
Supplementary Material
Key points:
MEDI2228 upregulates CD38 and NKG2D ligands in MM cells via cGAS-STING-TBK1-IRF3-, STAT1-IRF1-IFN- and ATM/ATR-CHK1/2-signaling cascades.
MEDI2228 restores daratumumab sensitivity, and combination therapy eradicates MM tumors in vivo, resulting all animals tumor-free.
Translational Relevance.
Investigations are focused on promoting the efficacy of current targeted immunotherapies for patients with multiple myeloma (MM). We identified that a novel BCMA antibody drug conjugate MEDI2228 triggers IFN-regulated innate immune function via cGAS-STING-TBK1-IRF3 and STAT1-IRF1 signaling cascades in MM cells following DNA damage. Membrane expression of CD38 and NKG2D ligands MICA/B and ULBPs are increased in MEDI2228-treated MM cell lines and patient MM cells, including those resistant to standard-of-care MM therapies, without impacting normal BCMA-negative cells including NK cells. MEDI2228 treatment restores Dara sensitivity in MM cells even in the immunosuppressive bone marrrow microenvironment, and the combination synergistically enhance NK-mediated MM cell killing. Moreover, MEDI2228-Dara combination completely eradicates in vivo MM cell growth in a murine xenograft model. These results therefore provide the mechanistic rationale for clinical evaluation of combination CD38- and BCMA-directed immunotherapies to further improve patient outcome in MM.
Acknowledgements
The authors acknowledge the flow cytometry assistance from the flow cytometry facility at DFCI. We thank all other lab members, as well as the clinical research coordinators at the Jerome Lipper Multiple Myeloma Center and the LeBow Institute for Myeloma Therapeutics of the Dana-Farber Cancer Institute, for their support and help in providing primary tumor specimens for this study.
Financial support
This work was supported in part by grants from the National Institutes of Health Specialized Programs of Research Excellence (SPORE) P50 CA100707, P01 CA078378, P01CA155258, and RO1 CA 207237. This work was supported in part by Dr Miriam and Sheldon G Adelson Medical Research Foundation, as well as the Paula and Rodger Riney Family Foundation. This study was funded by AstraZeneca.
Footnotes
Conflict of Interest
K.K. is an employee of AstraZeneca and has stock and/or stock interests in AstraZeneca. N.C.M. serves on advisory boards to Millennium-Takeda, Celgene, and Novartis and is a Scientific Founder of Oncopep. K.C.A. serves on advisory boards to Pfizer, Amgen, AstraZeneca, Janssen, Sanofi-Aventis, and Precision Biosciences, and is a Scientific founder of OncoPep and C4 Therapeutics. All other authors declare no competing financial interests.
References
- 1.Palumbo A, Chanan-Khan A, Weisel K, Nooka AK, Masszi T, Beksac M, et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N Engl J Med 2016;375(8):754–66 doi 10.1056/NEJMoa1606038. [DOI] [PubMed] [Google Scholar]
- 2.Facon T, Kumar S, Plesner T, Orlowski RZ, Moreau P, Bahlis N, et al. Daratumumab plus lenalidomide and dexamethasone for untreated myeloma. N Engl J Med 2019;380(22):2104–15 doi 10.1056/NEJMoa1817249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mateos MV, Cavo M, Blade J, Dimopoulos MA, Suzuki K, Jakubowiak A, et al. Overall survival with daratumumab, bortezomib, melphalan, and prednisone in newly diagnosed multiple myeloma (ALCYONE): a randomised, open-label, phase 3 trial. Lancet 2020;395(10218):132–41 doi 10.1016/S0140-6736(19)32956-3. [DOI] [PubMed] [Google Scholar]
- 4.de Weers M, Tai YT, van der Veer MS, Bakker JM, Vink T, Jacobs DC, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol 2011;186(3):1840–8 doi 10.4049/jimmunol.1003032. [DOI] [PubMed] [Google Scholar]
- 5.Overdijk MB, Verploegen S, Bogels M, van Egmond M, Lammerts van Bueren JJ, Mutis T, et al. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. mAbs 2015;7(2):311–21 doi 10.1080/19420862.2015.1007813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nijhof IS, Casneuf T, van Velzen J, van Kessel B, Axel AE, Syed K, et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood 2016;128(7):959–70 doi 10.1182/blood-2016-03-703439. [DOI] [PubMed] [Google Scholar]
- 7.Krejcik J, Frerichs KA, Nijhof IS, van Kessel B, van Velzen JF, Bloem AC, et al. Monocytes and granulocytes reduce CD38 expression levels on myeloma cells in patients treated with daratumumab. Clin Cancer Res 2017;23(24):7498–511 doi 10.1158/1078-0432.CCR-17-2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krejcik J, Casneuf T, Nijhof IS, Verbist B, Bald J, Plesner T, et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016;128(3):384–94 doi 10.1182/blood-2015-12-687749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Casneuf T, Xu XS, Adams HC 3rd, Axel AE, Chiu C, Khan I, et al. Effects of daratumumab on natural killer cells and impact on clinical outcomes in relapsed or refractory multiple myeloma. Blood Adv 2017;1(23):2105–14 doi 10.1182/bloodadvances.2017006866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nijhof IS, Groen RW, Lokhorst HM, van Kessel B, Bloem AC, van Velzen J, et al. Upregulation of CD38 expression on multiple myeloma cells by all-trans retinoic acid improves the efficacy of daratumumab. Leukemia 2015;29(10):2039–49 doi 10.1038/leu.2015.123. [DOI] [PubMed] [Google Scholar]
- 11.Garcia-Guerrero E, Gotz R, Doose S, Sauer M, Rodriguez-Gil A, Nerreter T, et al. Upregulation of CD38 expression on multiple myeloma cells by novel HDAC6 inhibitors is a class effect and augments the efficacy of daratumumab. Leukemia 2021;35(1):201–14 doi 10.1038/s41375-020-0840-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ogiya D, Liu J, Ohguchi H, Kurata K, Samur MK, Tai YT, et al. The JAK-STAT pathway regulates CD38 on myeloma cells in the bone marrow microenvironment: therapeutic implications. Blood 2020;136(20):2334–45 doi 10.1182/blood.2019004332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tai YT, Acharya C, An G, Moschetta M, Zhong MY, Feng X, et al. APRIL and BCMA promote human multiple myeloma growth and immunosuppression in the bone marrow microenvironment. Blood 2016;127(25):3225–36 doi 10.1182/blood-2016-01-691162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cho SF, Lin L, Xing L, Li Y, Yu T, Anderson KC, et al. BCMA-targeting therapy: driving a new era of immunotherapy in multiple myeloma. Cancers (Basel) 2020;12(6) doi 10.3390/cancers12061473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med 2019;380(18):1726–37 doi 10.1056/NEJMoa1817226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trudel S, Lendvai N, Popat R, Voorhees PM, Reeves B, Libby EN, et al. Antibody-drug conjugate, GSK2857916, in relapsed/refractory multiple myeloma: an update on safety and efficacy from dose expansion phase I study. Blood Cancer J 2019;9(4):37 doi 10.1038/s41408-019-0196-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lonial S, Lee HC, Badros A, Trudel S, Nooka AK, Chari A, et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): a two-arm, randomised, open-label, phase 2 study. Lancet Oncol 2020;21(2):207–21 doi 10.1016/S1470-2045(19)30788-0. [DOI] [PubMed] [Google Scholar]
- 18.Tai YT, Mayes PA, Acharya C, Zhong MY, Cea M, Cagnetta A, et al. Novel anti-B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood 2014;123(20):3128–38 doi 10.1182/blood-2013-10-535088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tai YT, Anderson KC. Targeting B-cell maturation antigen in multiple myeloma. Immunotherapy 2015;7(11):1187–99 doi 10.2217/imt.15.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kinneer K, Flynn M, Thomas SB, Meekin J, Varkey R, Xiao X, et al. Preclinical assessment of an antibody-PBD conjugate that targets BCMA on multiple myeloma and myeloma progenitor cells. Leukemia 2019;33(3):766–71 doi 10.1038/s41375-018-0278-7. [DOI] [PubMed] [Google Scholar]
- 21.Xing L, Lin L, Yu T, Li Y, Cho SF, Liu J, et al. A novel BCMA PBD-ADC with ATM/ATR/WEE1 inhibitors or bortezomib induce synergistic lethality in multiple myeloma. Leukemia 2020;34(8):2150–62 doi 10.1038/s41375-020-0745-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar SK, Migkou M, Bhutani M, Spencer A, Ailawadhi S, Kalff A, et al. Phase 1, first-in-human study of MEDI2228, a BCMA-targeted ADC in patients with relapsed/refractory multiple myeloma. Blood 2020;Supplement_1(136):26–7 doi 10.1182/blood-2020-136375. [DOI] [Google Scholar]
- 23.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 2014;15(12):550 doi 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cornwell M, Vangala M, Taing L, Herbert Z, Koster J, Li B, et al. VIPER: Visualization Pipeline for RNA-seq, a snakemake workflow for efficient and complete RNA-seq analysis. BMC Bioinformatics 2018;19(1):135 doi 10.1186/s12859-018-2139-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng X, Zhang L, Acharya C, An G, Wen K, Qiu L, et al. Targeting CD38 suppresses induction and function of T regulatory cells to mitigate immunosuppression in multiple myeloma. Clin Cancer Res 2017;23(15):4290–300 doi 10.1158/1078-0432.CCR-16-3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang L, Tai YT, Ho M, Xing L, Chauhan D, Gang A, et al. Regulatory B cell-myeloma cell interaction confers immunosuppression and promotes their survival in the bone marrow milieu. Blood Cancer J 2017;7(3):e547 doi 10.1038/bcj.2017.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tai YT, Lin L, Xing L, Cho SF, Yu T, Acharya C, et al. APRIL signaling via TACI mediates immunosuppression by T regulatory cells in multiple myeloma: therapeutic implications. Leukemia 2019;33(2):426–38 doi 10.1038/s41375-018-0242-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cho SF, Lin L, Xing L, Li Y, Wen K, Yu T, et al. The immunomodulatory drugs lenalidomide and pomalidomide enhance the potency of AMG 701 in multiple myeloma preclinical models. Blood Adv 2020;4(17):4195–207 doi 10.1182/bloodadvances.2020002524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin L, Cho SF, Xing L, Wen K, Li Y, Yu T, et al. Preclinical evaluation of CD8+ anti-BCMA mRNA CAR T cells for treatment of multiple myeloma. Leukemia 2021;35(3):752–63 doi 10.1038/s41375-020-0951-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu T, Chaganty B, Lin L, Xing L, Ramakrishnan B, Wen K, et al. VIS832, a novel CD138-targeting monoclonal antibody, potently induces killing of human multiple myeloma and further synergizes with IMiDs or bortezomib in vitro and in vivo. Blood Cancer J 2020;10(11):110 doi 10.1038/s41408-020-00378-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu H, Blum RH, Bjordahl R, Gaidarova S, Rogers P, Lee TT, et al. Pluripotent stem cell-derived NK cells with high-affinity noncleavable CD16a mediate improved antitumor activity. Blood 2020;135(6):399–410 doi 10.1182/blood.2019000621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chan WK, Kang S, Youssef Y, Glankler EN, Barrett ER, Carter AM, et al. A CS1-NKG2D Bispecific Antibody Collectively Activates Cytolytic Immune Cells against Multiple Myeloma. Cancer immunology research 2018;6(7):776–87 doi 10.1158/2326-6066.CIR-17-0649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 2010;70(2):440–6 doi 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 34.Pidugu VK, Pidugu HB, Wu MM, Liu CJ, Lee TC. Emerging functions of human IFIT proteins in cancer. Front Mol Biosci 2019;6:148 doi 10.3389/fmolb.2019.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fedele PL, Willis SN, Liao Y, Low MS, Rautela J, Segal DH, et al. IMiDs prime myeloma cells for daratumumab-mediated cytotoxicity through loss of Ikaros and Aiolos. Blood 2018;132(20):2166–78 doi 10.1182/blood-2018-05-850727. [DOI] [PubMed] [Google Scholar]
- 36.Varinou L, Ramsauer K, Karaghiosoff M, Kolbe T, Pfeffer K, Muller M, et al. Phosphorylation of the Stat1 transactivation domain is required for full-fledged IFN-gamma-dependent innate immunity. Immunity 2003;19(6):793–802 doi 10.1016/s1074-7613(03)00322-4. [DOI] [PubMed] [Google Scholar]
- 37.Kovarik P, Mangold M, Ramsauer K, Heidari H, Steinborn R, Zotter A, et al. Specificity of signaling by STAT1 depends on SH2 and C-terminal domains that regulate Ser727 phosphorylation, differentially affecting specific target gene expression. The EMBO journal 2001;20(1–2):91–100 doi 10.1093/emboj/20.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, et al. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol 2003;4(6):540–5 doi 10.1038/ni931. [DOI] [PubMed] [Google Scholar]
- 39.Jiang H, Acharya C, An G, Zhong M, Feng X, Wang L, et al. SAR650984 directly induces multiple myeloma cell death via lysosomal-associated and apoptotic pathways, which is further enhanced by pomalidomide. Leukemia 2016;30(2):399–408 doi 10.1038/leu.2015.240. [DOI] [PubMed] [Google Scholar]
- 40.Overdijk MB, Jansen JH, Nederend M, Lammerts van Bueren JJ, Groen RW, Parren PW, et al. The Therapeutic CD38 Monoclonal Antibody Daratumumab Induces Programmed Cell Death via Fcgamma Receptor-Mediated Cross-Linking. J Immunol 2016;197(3):807–13 doi 10.4049/jimmunol.1501351. [DOI] [PubMed] [Google Scholar]
- 41.Canadas I, Thummalapalli R, Kim JW, Kitajima S, Jenkins RW, Christensen CL, et al. Tumor innate immunity primed by specific interferon-stimulated endogenous retroviruses. Nat Med 2018;24(8):1143–50 doi 10.1038/s41591-018-0116-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li T, Chen ZJ. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med 2018;215(5):1287–99 doi 10.1084/jem.20180139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kwon J, Bakhoum SF. The cytosolic DNA-sensing cGAS-STING pathway in cancer. Cancer discovery 2020;10(1):26–39 doi 10.1158/2159-8290.CD-19-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kitajima S, Ivanova E, Guo S, Yoshida R, Campisi M, Sundararaman SK, et al. Suppression of STING associated with LKB1 loss in KRAS-driven lung cancer. Cancer discovery 2019;9(1):34–45 doi 10.1158/2159-8290.CD-18-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009;462(7269):108–12 doi 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ahn J, Xia T, Konno H, Konno K, Ruiz P, Barber GN. Inflammation-driven carcinogenesis is mediated through STING. Nature communications 2014;5:5166 doi 10.1038/ncomms6166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harada H, Fujita T, Miyamoto M, Kimura Y, Maruyama M, Furia A, et al. Structurally similar but functionally distinct factors, IRF-1 and IRF-2, bind to the same regulatory elements of IFN and IFN-inducible genes. Cell 1989;58(4):729–39 doi 10.1016/0092-8674(89)90107-4. [DOI] [PubMed] [Google Scholar]
- 48.Shaffer AL, Emre NC, Lamy L, Ngo VN, Wright G, Xiao W, et al. IRF4 addiction in multiple myeloma. Nature 2008;454(7201):226–31 doi 10.1038/nature07064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paczulla AM, Rothfelder K, Raffel S, Konantz M, Steinbacher J, Wang H, et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature 2019;572(7768):254–9 doi 10.1038/s41586-019-1410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.