

SUMMARY
Macrophages undergoing M1- versus M2-type polarization differ significantly in their cell metabolism and cellular functions. Here, global quantitative time-course proteomics and phosphoproteomics paired with transcriptomics provide a comprehensive characterization of temporal changes in cell metabolism, cellular functions, and signaling pathways that occur during the induction phase of M1- versus M2-type polarization. Significant differences in, especially, metabolic pathways are observed, including changes in glucose metabolism, glycosaminoglycan metabolism, and retinoic acid signaling. Kinase-enrichment analysis shows activation patterns of specific kinases that are distinct in M1- versus M2-type polarization. M2-type polarization inhibitor drug screens identify drugs that selectively block M2- but not M1-type polarization, including mitogen-activated protein kinase kinase (MEK) and histone deacetylase (HDAC) inhibitors. These datasets provide a comprehensive resource to identify specific signaling and metabolic pathways that are critical for macrophage polarization. In a proof-of-principle approach, we use these datasets to show that MEK signaling is required for M2-type polarization by promoting peroxisome proliferator-activated receptor-γ (PPARγ)-induced retinoic acid signaling.
In brief
He et al. provide a detailed characterization of dynamic temporal changes in cell signaling and metabolism during macrophage polarization by using quantitative time-course proteomics and phosphoproteomics and identify pharmacologic inhibitors of M2-type macrophage polarization. These data uncover a critical role of MEK/ERK signaling for PPARγ/retinoic acid-induced M2-type macrophage polarization.
Graphical Abstract
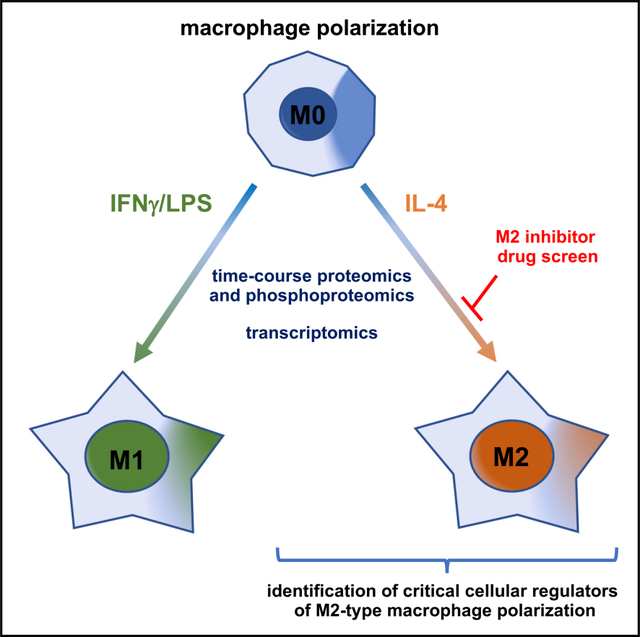
INTRODUCTION
Specific cues from the tissue microenvironment can result in macrophage activation and polarization into functionally distinct subpopulations, a process associated with extensive epigenetic modifications, transcriptional reprogramming, and metabolic changes (Covarrubias et al., 2016; Martinez and Gordon, 2014; Sica and Mantovani, 2012; Viola et al., 2019; Xue et al., 2014). Activated macrophages can be found along a gradient of polarization states, with polar opposites having partly antagonistic functions (Mantovani et al., 2013; Martinez and Gordon, 2014; Pelegrin and Surprenant, 2009; Xue et al., 2014). Macrophages activated by interferon-γ (IFN-γ) or lipopolysaccharides (LPS) are referred to as M1-type macrophages, whereas interleukin-4 (IL-4) leads to M2-type macrophages (Martinez and Gordon, 2014; Murray et al., 2014). M1-type macrophages are anti-angiogenic and promote chronic inflammatory conditions but inhibit tumor growth. M2-type macrophages can promote pathologic angiogenesis, organ fibrosis, tumor growth, or allergic and parasitic diseases (Hughes et al., 2015; Lavin et al., 2017; Mantovani et al., 2013; Mantovani and Sica, 2010; Pyonteck et al., 2013; Rodell et al., 2018; Sica and Mantovani, 2012; Zilionis et al., 2019). Even fully polarized macrophages can be reprogrammed when transferred into a new microenvironment, suggesting that they can be “reeducated” into their polar opposites to influence their effects in specific disease conditions (Gosselin et al., 2014; Hagemann et al., 2008; Hobson-Gutierrez and Carmona-Fontaine, 2018; Lavin et al., 2014). Thus, therapeutic strategies to shift the balance from M2- to M1-type macrophages may inhibit pathologic angiogenesis, fibrosis, or tumor growth (Wynn and Vannella, 2016). For example, M2-type macrophages are increased in neovascular age-related macular degeneration (NV-AMD), and they promote choroidal neovascularization (CNV) and fibrosis in a laser-induced model of NV-AMD, whereas ablating macrophages in this model strongly inhibits angiogenesis and fibrosis (He and Marneros, 2013, 2014; Lad et al., 2015; Marneros, 2013; Strittmatter et al., 2016; Yang et al., 2016; Zandi et al., 2015).
M1-type polarization has been linked to metabolic reprogramming toward glycolysis, the pentose phosphate pathway, and fatty acid synthesis, whereas M2-type polarization shows a preferential metabolic switch to oxidative phosphorylation and fatty acid oxidation (Viola et al., 2019). However, a comprehensive understanding of the dynamic temporal changes in metabolic pathways and associated kinase activation patterns that occur during the induction phase of M1- versus M2-type polarization is lacking. This is, in part, explained by the lack of comparative, global quantitative time-course proteomic and phosphoproteomic analyses of the changes that occur during the induction phase of M1- versus M2-type macrophage polarization. Previous phosphoproteomic studies were limited in scale and temporal resolution and only assessed LPS-induced, but not IL-4-induced, macrophage polarization and mainly examined only the very early phase of M1-type polarization (Daniels et al., 2020; Sharma et al., 2010; Sjoelund et al., 2014; Weintz et al., 2010). Similarly, prior proteomic studies of fully polarized macrophages have been limited in depth and scale (Court et al., 2017; Meijer et al., 2015; Wiktorowicz et al., 2019). Thus, a detailed global quantitative characterization of the dynamic changes in metabolic and signaling pathways that are linked to the induction of M1- versus M2-type macrophage polarization is currently lacking.
Here, global quantitative time-course proteomics and phosphoproteomics during the first 24 h of M1- versus M2-type macrophage polarization, combined with transcriptomics, provide a detailed picture of changes in cellular functions and metabolic pathways that occur with each polarization process. In addition, quantitative proteomics provided a comprehensive characterization of the protein landscape of fully polarized macrophages. Kinase enrichment analysis (KEA) of the time-course phosphoproteomic data identified multiple kinases whose temporal activation differs significantly during M1- versus M2-type polarization. The KEA data suggest that specific kinase inhibitors may selectively block M2-type polarization without inhibiting M1-type polarization. Indeed, we identified, in M2-type polarization-inhibitor drug screens, multiple kinase inhibitors that block kinases found to be differentially activated during M2-type polarization in the KEA and that selectively inhibit M2- but not M1-type polarization. For example, we found that a spike in mitogen-activated protein kinase kinase (MEK) signaling occurs during the induction of M2-type polarization and that MEK inhibitors selectively blocked M2-, but not M1-type, polarization. A similar effect was observed with various histone deacetylase (HDAC) inhibitors. The identified MEK and HDAC inhibitors also potently blocked M2-type macrophage polarization and angiogenesis in mouse models of NV-AMD and wound healing. Our datasets provide a comprehensive resource to explore the relevance of specific metabolic pathways and signaling axes for macrophage polarization. In a proof-of-principle approach, we use these datasets to show that IL-4-induced MEK/ERK signaling is critical for the induction of peroxisome proliferator-activated receptor-γ (PPARγ) expression that subsequently promotes M2-type polarization via activation of retinoic acid (RA) signaling.
RESULTS
Global quantitative proteomics of fully polarized M1- versus M2-type macrophages
Macrophages derived from the human monocytic cell line THP-1 are a well-established system to assess macrophage polarization mechanisms. Treatment of THP-1 cells with phorbol myristate acetate (PMA) for 24 h results in differentiation into macrophages (M0) that polarize with subsequent treatment with IL-4 into M2-type macrophages, whereas treatment with IFN-γ/LPS leads to M1-type macrophages (Figure S1A), which allows us to examine the opposite polar ends of this spectrum with uniform cell populations. We aimed to identify mechanisms that drive these opposite polarization states in these macrophages and subsequently validate key findings also in primary macrophages. We chose 4 days of treatment with IL-4 or IFN-γ/LPS as a duration to achieve full polarization because we observed, at that time point, established polarization markers were present at high levels and at a further increase compared with earlier time points. For example, protein levels of the prototypical M2-type marker TGM2 progressively increased during the first 24 h and showed a further increase at 4 days when compared with 24 h (Figure S1B). Thus, we defined the first 24 h of treatment with IL-4 or IFN-γ/LPS as the phase of polarization induction, and 4 days of treatment as a time point when full polarization is achieved.
First, by using multiplexed mass spectrometry (Kreuzer et al., 2019; McAlister et al., 2014; Ting et al., 2011), we performed quantitative proteomics of fully polarized THP-1-derived M1- and M2-type macrophages to define the global landscape of cellular markers that characterize these polarized macrophages (Figures S1C–S1E). This approach led to the quantification of ~7,900 proteins (Figures S1F; Table S1). Among the most upregulated proteins identified were well-established M2-type markers (e.g., MRC1, TGM2, FABP4, CCL24, and CCL26) in IL-4-treated groups and M1-type markers in IFN-γ/LPS-treated cells (e.g., IDO1, FAM26F, CXCL9, and CXCL10) (Figure S1C). Thus, findings in these THP-1-derived macrophages likely closely resemble molecular changes that occur in polarized primary macrophages as well, which upregulate the expression of these markers with either M1- or M2-type polarization (Martinez et al., 2013; Murray et al., 2014). This global proteomics approach also identified additional proteins to be highly upregulated with either polarization state that have previously not been linked to M1- or M2-type macrophage polarization (Figures S1D and S1E; Table S1). Based on these data, as well as published findings (Martinez et al., 2006, 2013; Murray et al., 2014), we chose MRC1 and TGM2 as key selective M2-type markers to monitor the effects of pharmacological interventions on M2-type polarization in human macrophages (Arg1 serves as a prototypical M2-type marker in mouse macrophages).
Time-course global quantitative proteomics during the induction phase of M1- versus M2-type macrophage polarization
Next, we performed a quantitative time-course analysis of both proteomic and phosphoproteomic changes that occur during the first 24 h of IL-4- versus IFNγ/LPS-induced polarization of macrophages in order to identify mechanisms that drive M1- versus M2-type polarization (Figure S2 shows an overview of the different experimental groups and OMICs approaches used in this study). Proteomic and phosphoproteomic data were obtained by multiplexed mass spectrometry from THP-1-derived macrophages that were treated with IL-4 or IFN-γ/LPS for 10 min, 30 min, 1 h, 2 h, 4 h, 8 h, or 24 h (Table S2). This approach provided quantitative time-course data on more than 9,400 proteins and more than 38,000 phosphopeptides. These datasets are validated by the observation of temporal changes in specific phosphorylation events that are known to be associated with M1- (e.g., STAT1 [Y701]) or M2-type (e.g., STAT6 [Y641]) macrophage polarization, which we confirmed by western blotting (Figures S1B and S1G). Moreover, we also confirmed some of the key proteomic/phosphoproteomic data subsequently in western blots, using not only THP-1-derived macrophages but also primary mouse and/or human macrophages to establish the physiological relevance of our findings. The time-course proteomics data also showed a progressive increase of well-established polarization markers during the first 24 h, validating that time period as the induction phase of polarization (Figure S1H).
Gene set enrichment analysis (GSEA) and functional pathway analysis of the proteomics datasets show which pathways are distinctly activated or downregulated during the different temporal phases of M1- versus M2-type polarization. Among the most significant KEGG pathways that were predominantly upregulated during the early and mid-phases of M1-type polarization were nucleotide-binding oligomerization domain (NOD)-like receptor signaling, chemokine signaling, Toll-like receptor (TLR) signaling, cytosolic DNA sensing pathway, retinoic acid-inducible gene 1 (RIG-I)-like receptor signaling, antigen processing and presentation, tryptophan metabolism, cytokine/cytokine receptor interaction, and adipocytokine signaling (Figure 1A). An upregulation predominantly during the early phase of M1-type polarization was linked to KEGG terms glycolysis/gluconeogenesis, pentose phosphate pathway, endocytosis, mTOR signaling, and ubiquitin-mediated proteolysis (Figure 1A). Activation of KEGG pathways during the early stage of M2-type polarization included oxidative phosphorylation and insulin signaling (Figures 1A and 1B). Upregulation of pathways during the mid- to late phases of M2-type polarization included PPAR signaling, arginine and proline metabolism, oxidative phosphorylation, ECM/receptor interaction, lysosome, VEGF signaling, galactose metabolism, and pentose phosphate pathway (Figure 1A).
Figure 1. Time-course global quantitative proteomics identify dynamic temporal changes in cellular and metabolic pathways during M1- versus M2-type macrophage polarization.

(A) Heatmap shows KEGG pathways upregulated in at least one time point, based on the time-course proteomics data of macrophages undergoing M1- (treated with IFN-γ/LPS) or M2-type polarization (treated with IL-4). Color coding indicates −log10 p values when comparing the M1 or M2 group at the indicated time point with the M0 group (after 24 h PMA treatment only) at the same corresponding time point. *p < 0.05. Select pathways with distinct activation pattern during M1- versus M2-type polarization are indicated in bold.
(B) Insulin signaling pathway protein changes and KEGG enrichment plot shown in macrophages after 4 h of treatment with IL-4 (compared with M0 macrophages). Heatmap shows Z scores.
See also Figures S1–S4; Tables S1 and S2.
Similarly, Gene Ontology (GO) pathway analyses showed particularly striking differences in cell metabolism between M1- and M2-type macrophage polarization processes (Figures S3 and S4). A strong upregulation of GO terms with M2- but not M1-type polarization also included terms associated with RA signaling, PPAR signaling, and glycosaminoglycan metabolism (Figures S3 and S4). The association of increased RA signaling with M2- but not M1-type polarization is reflected by a high increase in the retinal dehydrogenase ALDH1A2, a rate-limiting enzyme in the generation of RA, at 24 h after IL-4 treatment, whereas no upregulation was observed in IFN-γ/LPS-treated macrophages (Figure S1H). Notably, we observed a relative upregulation of GO terms associated with histone deacetylase activity particularly during the mid-phase of IL-4-induced M2-type polarization compared with the same time points in IFN-γ/LPS-treated macrophages (Figure S3F).
Collectively, these proteomics data provide a comprehensive resource to link a specific temporal activation pattern of distinct metabolic and cellular signaling pathways to the induction of M1- versus M2-type macrophage polarization.
Time-course global quantitative phosphoproteomics identify distinct kinase activation patterns during the induction phase of M1- versus M2-type macrophage polarization
To determine which signaling mechanisms lead to the distinct activation of these metabolic and cellular pathways and regulate the induction of M1- versus M2-type polarization, we performed a time-course analysis of the phosphoproteomic changes that occur during the first 24 h of IL-4- versus IFN-γ/LPS-induced polarization of macrophages. Kinase enrichment analyses (KEAs) of these data showed that induction of M2-type polarization is associated with temporal activation patterns of specific kinases that are largely distinct from those activated during M1-type polarization (Figure 2A; Table S2). For example, we found that strong JAK2 activation occurs during M1- but not M2-type polarization (Figure 2A), providing validation of the data because JAK2 activation is known to occur during IFN-γ-induced macrophage polarization (Martinez and Gordon, 2014). Activation of the mTOR pathway occurred early during M1- but not M2-type polarization, consistent with the KEEG pathway analysis findings of the proteomics dataset (Figures 1A and 2A). AKT2 activation occurred preferentially with M1-type polarization (Figure 2A), which is also supported by previous findings (Vergadi et al., 2017). Additional signaling pathways whose activation was mainly associated with M1-type polarization but whose role for M1-type polarization has not been well established so far included signaling via PRKCH, RET, PRKCQ, PRKG1 (and isoform 2), TAK1, RPS6KA1, JNK1, PRKD1, MAPKAPK2, and MERTK (Figure 2A).
Figure 2. Time-course global quantitative phosphoproteomics identify distinct kinase activation patterns during the induction phase of M1- versus M2-type macrophage polarization.

(A) Heatmap based on KEA of time-course phosphoproteomics data. The activities of kinases are displayed as the mean relative levels of the phosphosites that they phosphorylate (log2 ratios relative to its mean activity). Kinases with a false discovery rate (FDR) < 1% are shown.
(B) Visualization of the KEA data for MEK1. Red lines show individual target phosphosites of MEK1, and black lines show averaged values.
(C) ERK1 (T202/Y204) phosphorylation peaks between 2 and 8 h (*) after IL-4 treatment in primary human macrophages, which is blocked by trametinib. This inhibition of ERK1 (T202/Y204) phosphorylation by trametinib is associated with inhibition of induction of MRC1 protein by IL-4.
(D) Western blotting with cell lysates (THP-1-derived macrophages after 24 h of treatment with IL-4 or IFN-γ/LPS) used in the phosphoproteomics and proteomics experiments in (E) and (F) confirm that GDC-0879 increases ERK (T202/Y204) phosphorylation and TGM2 in IL-4 treated cells, whereas trametinib blocks ERK activation and reduces TGM2.
(E) M1- or M2-type marker protein levels based on quantitative proteomics of THP-1-derived macrophages after 24 h of treatment with either IL-4 or IFN-γ/LPS in the presence or absence of trametinib or GDC-0879. The data for each protein are shown as log2 ratios relative to its mean level.
(F) Quantitative phosphoproteomics of THP-1-derived macrophages treated for 24 h with IL-4 (performed in duplicate); cell lysates were also used in the proteomics experiments in (E). GDC-0879 induces phosphorylation events associated with activation of the MEK/ERK pathway and downstream targets. The y axis shows mass spectrometry (MS) intensity values (arbitrary unit). KEA heatmap shows an increase in signaling activities of MEK1 and its downstream targets ERK1/ERK2 and RSK2 with GDC-0879 treatment. The activities of each kinase are shown as log2 ratios relative to its mean activity.
(G) Top: the MEK inhibitor trametinib and the pan-Raf inhibitor TAK-632 potently reduce IL-4-induced MRC1 levels in THP-1-derived macrophages, whereas GDC-0879 increases MRC1. Bottom: trametinib inhibits IL-4-induced Arg1, whereas GDC-0879 increases Arg1 in murine BMDMs. Cytokine treatment for 4 days.
(H) Western blotting shows effects of chemicals on MRC1 and phospho-STAT1(Y701) levels in primary human macrophages treated for 24 h with the chemicals in the presence of either IL-4 or IFN-γ/LPS.
Western blot values indicate normalization to β-actin or β-tubulin loading control and DMSO control sample (no IL-4) in (C) and (D) or IL-4 DMSO control sample (IL-4 with DMSO vehicle but no inhibitors) in (G) and (H). Phospho-STAT1 (Y701) normalized to IFN-γ/LPS control (H). See also Figures S1 and S2; Tables S1 and S2.
The KEA showed clearly distinct temporal activation patterns of specific mitogen-activated protein (MAP) kinases with M1- versus M2-type polarization, also supported by the KEGG pathway analysis findings of the proteomic datasets (Figures 1A and 2A). For example, MEK1 showed an activation peak at 4–8 h in IL-4-treated macrophages undergoing M2-type polarization, whereas MEK1 activity was strongly downregulated at 24 h once M2-type polarization had progressed. In contrast, MEK1 activity peaked at 24 h in IFN-γ/LPS-treated M1-type macrophages, which we also confirmed by western blotting (Figures 2A, 2B, and S1B). Thus, a brief increase in MEK1/ERK signaling during the induction phase of IL-4-induced polarization may promote M2-type polarization. A distinct temporal activation pattern between M1- and M2-type polarization was also observed for multiple other kinases, including p38α and its downstream target MAPKAPK2, JNK1, PRKD1, AMPKA1, AMPKA2, FYN, CAMK1, PDK1, PDK2, LYN, and JNK3 (Figure 2A). Overall, the KEA provides a comprehensive resource to characterize the contributions of specific signaling pathways for M1- versus M2-type macrophage polarization.
MEK/ERK signaling promotes M2-type macrophage polarization
We observed the temporal spike in ERK1 activation at 2–8 h with IL-4 treatment not only in THP-1-derived (Figure S1B) but also in primary human macrophages (Figures 2C). Blocking MEK/ERK signaling with the selective MEK inhibitor trametinib effectively inhibited IL-4-induced M2-type polarization of both THP-1-derived and primary human macrophages (Figures 2C–2E). Thus, the temporal spike in MEK/ERK activity is required for macrophages to undergo IL-4-induced M2-type polarization.
Because inhibition of MEK signaling blocks M2-type polarization, increased activation of this pathway may stimulate M2-type polarization. To test this hypothesis, we treated macrophages with the highly selective B-Raf inhibitor GDC-0879 (Hoeflich et al., 2009), because B-Raf inhibition with GDC-0879 in the presence of activated Ras induces B-Raf binding to c-Raf, leading to c-Raf hyperactivation and thereby elevated MEK/ERK signaling (Hatzivassiliou et al., 2010; Heidorn et al., 2010; Sieber et al., 2018). First, we confirmed that GDC-0879 indeed promotes paradoxical MEK/ERK signaling in IL-4-treated macrophages by quantitative phosphoproteomics (Figure 2F; Table S1). The increase in MEK/ERK signaling activity with GDC-0879 treatment in macrophages undergoing IL-4-induced polarization was associated with a strong increase in M2-type marker expression not only in polarized THP-1-derived macrophages (Figures 2D and 2G) but also in murine bone-marrow-derived macrophages (BMDMs) (Figure 2G) and primary human macrophages (Figures 2H). Even at early stages of IL-4-induced macrophage polarization, we found increased protein levels of M2-type markers with GDC-0879 treatment but a decrease in those markers with trametinib treatment (Figure 2E). In contrast, trametinib did not decrease M1-type markers in IFN-γ/LPS-treated cells, and GDC-0879 even increased M2-type markers in IFN-γ/LPS-treated cells (Figure 2E). That GDC-0879 promotes M2-type polarization through paradoxical activation of MEK/ERK signaling via c-Raf is supported by the observation that M2-type polarization is inhibited also by pan-Raf inhibitors that block both B-Raf and c-Raf and, therefore, prevent paradoxical activation of MEK/ERK via c-Raf (Figures 2G, 3A, and 3B). Thus, our data show that IL-4-induced MEK/ERK signaling activity is a critical regulator of M2-type macrophage polarization. These findings serve as proof-of-principle validation that the KEA data provide a framework to systematically interrogate the functions of different kinases and signaling pathways for M1- versus M2-type macrophage polarization.
Figure 3. Chemical screens identify pharmacologic blockers of M2-type macrophage polarization.

(A) Scatterplot shows results of small-molecule chemical screen performed in duplicate (plates 1 and 2) with MRC1 promoter-driven luciferase activity as a readout. Axes show log2 FC (fold change) of luciferase activity. Select HDAC inhibitors, pan-Raf inhibitors, MEK inhibitors, proteasome/immunoproteasome inhibitors, or HSP90 inhibitors are highlighted in the plots.
(B) Luciferase assays show a dose-dependent inhibition of MRC1 promoter-driven luciferase activity by the MEK inhibitor trametinib, the pan-Raf inhibitor AZ628, the HDAC inhibitor panobinostat, and the HSP90 inhibitor NMS-E973 at concentrations that do not affect cell viability (CTG). Concentrations of chemicals used indicated in nM on the x axis; values indicated on the y axis normalized to the control carrier (DMSO). Red: luciferase activity; green: cell viability assay (CTG). Graphs represent data as means ± SD. N = 4/group.
(C) Inhibitors of IL-4-induced MRC1 expression in the chemical screens were tested for their effects on expression of M2-type markers MRC1 and CD209 and M1-type markers CXCL9, CCR7, and iNOS in THP-1-derived macrophages that were treated with either IL-4 or IFN-γ/LPS for 4 days in the presence or absence of these inhibitors. Values were normalized to CD68 expression levels and the IL-4-treated or IFN-γ/LPS-treated control group. Expression fold changes based on semiquantitative RT-PCR shown in y axes. Graphs represent data as means ± SEM. N = 3/group (each in triplicate).
(D) HDAC inhibitors attenuate M2-type polarization of IL-4-treated THP-1-derived macrophages with diminished MRC1 and TGM2 without affecting phospho-STAT1 (Y701) levels in IFN-γ/LPS-treated macrophages. Cytokine treatment for 4 days.
(E) Effects of inhibitors on Arg1 and phospho-STAT1 (Y701) protein levels in mouse BMDMs treated with IL-4. Panobinostat and the JAK½ inhibitor S-ruxolitinib potently block Arg1 expression, and MEK inhibitors (trametinib, PD0325901) reduce Arg1 protein. GW9662 has only a moderate inhibitory effect at the concentration tested. The Src inhibitor KX2–391 and the B-Raf inhibitor GDC-0879 increased Arg1 protein. Cytokine treatment was for 24 h.
(F) Semiquantitative RT-PCR shows that four different MEK inhibitors strongly reduce MRC1 expression in IL-4-treated THP-1-derived macrophages, whereas they increase expression of CXCL9 in IFN-γ/LPS-treated macrophages (normalized to CD68 and to IL-4 or IFN-γ/LPS value). Cytokine treatment for 4 days. Expression fold changes shown in y axes. Graphs represent data as means ± SEM. N = 3/group (each in triplicate). p values are shown (t test).
Western blot values indicate normalization to β-actin loading control and DMSO control sample with no inhibitors (D) or IL-4 DMSO control sample (IL-4 with DMSO vehicle but no inhibitors) (E). See also Figure S5; Table S3.
Chemical screens identify pharmacologic blockers of M2-type macrophage polarization
Based on the KEA findings, we hypothesized that pharmacologic targeting of specific signaling pathways or downstream transcriptional regulators that are linked to M2-type polarization may allow for selective inhibition of M2-type polarization without inhibiting M1-type polarization. To determine which of the signaling events identified in the KEA are not only associated with but also required for IL-4-induced M2-type polarization and could, thus, be targeted pharmacologically to block M2-type polarization, we conducted small-molecule screens for inhibitors of M2-type polarization that included a large number of kinase inhibitors. For those screens, we used human macrophages derived from a clonal cell population of THP-1 cells that stably express the promoter of the prototypical M2-type marker MRC1 driving luciferase (He and Marneros, 2014). Our proteomic data of fully polarized macrophages showed that MRC1 is a suitable selection marker for M2-type polarization because it is upregulated with IL-4 treatment but downregulated with IFN-γ/LPS treatment in these macrophages (Figure S1A). We used these macrophages for these screens instead of primary macrophages to ensure uniform cell populations with little cell heterogeneity and a stable readout of luciferase activity. However, key hits of these screens were subsequently confirmed in primary macrophages to establish the physiological relevance of the findings. We identified specific classes of small molecules as potent inhibitors of IL-4-induced M2-type macrophage polarization that target the same kinase or cellular protein, suggesting that these are class-specific inhibitory effects and not off-target observations of a particular small molecule (Figures 3A and S5A; Table S3). Among the most potent inhibitors of IL-4-induced macrophage polarization in these screens were multiple highly selective MEK inhibitors (e.g., trametinib, selumetinib, MEK162, pimasertib, or PD0325901) as well as pan-Raf inhibitors (e.g., AZ628 or TAK632), which showed a dose-dependent inhibition of M2-type polarization at concentrations that did not impair cell viability (Figures 3A, 3B, S5A, and S5B). These findings are consistent with the KEA data that linked a spike in MEK activation to M2-type polarization and our data showing that trametinib effectively blocks M2-type macrophage polarization.
In addition, various HDAC inhibitors (such as panobinostat, trichostatin A, dacinostat, pracinostat, or givinostat) also strongly blocked M2-type polarization of THP-1-derived macrophages (Figures 3A–3C, 3D, and S5C) and primary mouse (Figures 3E and S5D) and primary human macrophages (Figure 2H) at concentrations that did not affect cell viability. This finding is supported by the GSEA of our time-course proteomics data, which showed a relatively greater increase in HDAC activity with M2- than with M1-type polarization (Figure S3F). Multiple, additional drugs were identified in this screen to potently block M2-type macrophage polarization, including heat shock protein 90 (HSP90) inhibitors, bromodomain and extra-terminal domain (BET) domain inhibitors, the immunoproteasome inhibitor ONX-0914, and dihydroartemisinin (Figures 3A and S5A).
Particularly those inhibitors that can either shift macrophage populations from a M2- to a M1-type or that can selectively block M2-type polarization without affecting M1-type polarization would have therapeutic relevance for diseases promoted by M2-type macrophages. Thus, we tested whether the identified M2-type polarization inhibitors showed selective effects in inhibiting M2- but not M1-type polarization. For that purpose, we assessed the effects of those inhibitors on the expression of well-established M2-type markers (MRC1 and CD209) or M1-type markers (iNOS, CXCL9, and CCR7) in both THP-1-derived macrophages and murine BMDMs (normalized to a macrophage differentiation marker [CD68]); these markers were chosen based on our proteomic datasets and published work (Martinez and Gordon, 2014; Martinez et al., 2013; Murray et al., 2014). We also confirmed these findings by western blotting in polarized THP-1-derived macrophages and in primary human macrophages (M2 markers: MRC1 and TGM2; M1 marker: phospho-STAT1 [Y701], induced with M1-type polarization [Figure S1G]), as well as in polarized murine BMDMs (M2-marker: Arg1; M1 marker: phospho-STAT1 [Y701]). We found that MEK inhibitors potently blocked M2- but not M1-type macrophage polarization, and they even induced expression of some key M1-type markers (Figures 3C and 3F). This selectivity of preferentially inhibiting M2- but not M1-type polarization was also observed for multiple HDAC inhibitors (Figures 3C and 3D). For example, panobinostat strongly inhibited IL-4-induced expression of M2-type markers MRC1 or CD209, but markedly increased expression of M1-type markers iNOS or CXCL9 (Figure 3C). Panobinostat treatment also resulted in an almost complete loss of Arg1 protein in IL-4-treated mouse BMDMs (Figure 3E) and of MRC1 or TGM2 in IL-4-treated THP-1-derived human macrophages (Figure 3D) or primary human macrophages (Figure 2H), whereas STAT1 (Y701) phosphorylation was not blocked in IFN-γ/LPS-treated macrophages. Similar results were observed with other HDAC inhibitors as well (Figures 3C and 3D). HSP90 inhibitors, the immunoproteasome inhibitor ONX-0914, and dihydroartemisinin also inhibited expression of M2-type markers (but not expression of the M1-type markers CCR7 or iNOS) (Figure 3C). In contrast, BET bromodomain inhibitors or IκB kinase (IKK) inhibitors blocked expression of M1- as well as M2-type polarization markers (Figure 3C).
Increased PPARγ and RA signaling and a switch to oxidative phosphorylation during M2-type macrophage polarization
To determine how MEK and HDAC inhibitors block M2-type macrophage polarization, we assessed transcriptional programs associated with M1- versus M2-type macrophage polarization in both THP-1-derived macrophages and primary human macrophages by RNA sequencing (RNA-seq) and H3K27Ac chromatin immunoprecipitation sequencing (ChIP-seq) in the presence or absence of trametinib or panobinostat (polarized with either IFN-γ/LPS or IL-4 for 24 h) (Table S4). This allowed us not only to determine which gene programs are associated with M1- versus M2-type polarization but also to identify the subset of genes whose inhibition by trametinib or panobinostat correlates with blockade of M2-type polarization.
Several key pathways associated with M1- versus M2-type polarization that were identified in the GSEA of the proteomics data were also identified in the GSEA of the RNA-seq data (Figures 4A, 4B, S6, and S7; Table S5). We also observed similarities in key transcriptional programs associated with M1- or M2-type polarization when comparing THP-1 derived macrophages with primary human macrophages (Figures 4A and S8). Resembling the results from the proteomic analyses, among the most significantly enriched functional annotations based on the RNA-seq data of IL-4-treated THP-1-derived or primary human macrophages were oxidative phosphorylation, PPAR signaling, retinol metabolism, carboxylic acid biosynthetic process (a term that includes synthesis of fatty acids, RA, glycosaminoglycans, amino acids and leukotrienes), or protein kinase B (AKT) signaling (a pathway known to regulate M2-type polarization) (Covarrubias et al., 2016; Vergadi et al., 2017) (Figures 4A–4C). Both THP-1-derived and primary human macrophages that were polarized with IFN-γ/LPS to the M1-type also showed activation of similar pathways as observed in the proteomic data of M1-type macrophages (Figures S8).
Figure 4. HDAC and MEK activities promote PPARγ/RA signaling to drive M2-type macrophage polarization.

(A) GO enrichment analysis of highly upregulated genes (lg FC > 2) found in RNA-seq data after 24 h of treatment with IL-4 in THP-1-derived macrophages or in primary human macrophages. In both macrophage groups, RA signaling is significantly associated with M2-type polarization (underlined).
(B) GSEA enrichment plots show a high correlation of oxidative phosphorylation and PPAR signaling with M2-type polarization both in the RNA-seq as well as in the proteomics datasets (both at 24 h of treatment with IL-4 [M2] versus vehicle-treated control [M0]).
(C) Heatmaps show upregulated genes associated with oxidative phosphorylation, retinol metabolism, or PPAR signaling in M2-type macrophages (lg FC is shown), including ALDH1A2, PPARG, FABP4, and ANGPTL4 (arrows). GSEA-KEGG pathway analysis in THP-1-derived M2-type macrophages shows that among the most significant terms associated with M2-type polarization are oxidative phosphorylation and retinol metabolism. GSEA enrichment plot shown for retinal metabolism comparing M2-type macrophages with macrophages not treated with IL-4 (M0).
(D) Top-ranking GSEA KEGG terms in THP-1-derived and primary human macrophages show that panobinostat inhibits IL-4-induced transcriptional programs associated with retinol metabolism, PPAR signaling, and oxidative phosphorylation.
(E) Left: Venn diagram shows overlap of IL-4-induced genes in THP-1-derived macrophages inhibited by trametinib and panobinostat greater 2-fold (lg FC > 1). Among those genes are key regulators of PPARγ and RA signaling. Right: heatmap shows effects of trametinib or panobinostat on expression of some of these genes (lg FC, normalized to DMSO). N.D. = not detected.
(F) KEGG and GO pathway analyses of M2-type genes inhibited by both trametinib as well as panobinostat identify key pathways associated with M2-type polarization, including PPAR signaling, retinol metabolism, ERK signaling, and carboxylic acid biosynthetic process (same pathways identified in both analyses underlined in same color).
(G) Extent of inhibition of expression of PPAR signaling genes inhibited by panobinostat or trametinib (lg FC). MMP1, ANGPTL4, and PPARG are among those genes with the greatest inhibition.
GeneRatio shows percentage of differentially expressed genes (DEGs) in the given GO term. “Count” shows number of DEGs in the given term. Unless otherwise indicated, data refers to THP-1-derived macrophages. lg = log2. See also Figures S2 and S6–S11; Tables S2, S3, S4, and S5.
Among the highest upregulated transcripts in M2-type macrophages were those from genes that regulate oxidative phosphorylation or are involved in PPARγ or RA signaling (e.g., ALDH1A2, DHRS9, CYP26B1, PPARG, FABP4, and ANGPTL4) (Figures 4B and 4C). In particular, ALDH1A2 was highly upregulated in response to IL-4 (Figure 4C). Thus, both transcriptomic and proteomic datasets show that IL-4-induced M2-type macrophage polarization is associated with a switch to oxidative phosphorylation and changes in cell metabolism that included increased PPARγ and RA signaling.
Next, we assessed genes associated with IL-4-induced M2-type polarization whose expression is inhibited by trametinib and/or panobinostat. The expression of a subset of those IL-4-induced genes inhibited by these small molecules is required for M2-type polarization because their inhibition by trametinib and panobinostat was linked to a block in M2-type polarization. Panobinostat strongly inhibited IL-4-induced transcriptional programs associated with retinol metabolism, PPAR signaling, oxidative phosphorylation, carboxylic acid biosynthesis, and MAPK/ERK signaling (Figures 4D and S9–S11; Table S5). Trametinib inhibited similar M2-type polarization-associated cellular pathways in THP-1-derived and primary human macrophages, including MAP kinase signaling, phosphatidylinositol 3-kinase (PI3K)/AKT signaling, focal adhesion, and extracellular matrix-receptor interactions as well as retinol metabolism (Figures S9–S11). Notably, trametinib also inhibited genes of the histone deacetylase complex or genes that regulate histone binding in both THP-1-derived and primary human macrophages (Figures S9A, S9B, and S11). The highly significant inhibition of ERK signaling by panobinostat in IL-4-treated macrophages was associated with downregulation of the downstream target MYC and of expression of proangiogenic growth factors (e.g., IL-1β and platelet-derived growth factor subunit B [PDGFB]) or adhesion molecules (e.g., ICAM1) (Figure S10A). Similarly, inhibition of MAP kinase signaling by trametinib was associated with downregulation of downstream targets MYC and AP-1 transcription factors FOS and JUN and proangiogenic growth factors (e.g., IL-1β and FGF2) (Figure S11). These findings likely explain, in part, the overlap of effects of panobinostat and trametinib on transcriptional programs that are associated with IL-4-induced M2-type polarization. Genes inhibited by both panobinostat and trametinib >2-fold in IL-4-treated THP-1-derived macrophages included MYC, MMP1, MRC1, the adhesion molecules ICAM1 and VCAM1, and key genes of PPARγ/RA signaling, including PPARγ, ALDH1A2, RDH10, DHRS9, and ANGPTL4 (Figure 4E). GSEA and pathway analyses of IL-4-induced genes that are inhibited by both trametinib and panobinostat showed among the highest-ranking terms carboxylic acid biosynthetic process, ERK signaling, retinol metabolic process, PPAR signaling, and oxidative phosphorylation (Figures 4F and S11).
The inhibition of PPAR signaling and retinol metabolism by both trametinib and panobinostat was associated with a strong reduction in transcript levels of PPARγ and key enzymes of RA signaling that are normally induced by IL-4 (ALDH1A2, RDH10, and DHRS9) (Figures 4E, 4G, S9C, and S9D). The inhibition of IL-4-induced ALDH1A2 expression by trametinib is also supported by our proteomic data at the same time point (24 h of IL-4 treatment): trametinib inhibited the strong increase in ALDH1A2 induced by IL-4, whereas GDC-0879 showed an increase in ALDH1A2 (Figure 2E). Analysis of enhancer regions by H3K27Ac ChIP-seq showed that IL-4 strongly increased ALDH1A2 enhancer occupancy, which was markedly reduced by both trametinib and panobinostat (Figure S9E). Similar reductions in enhancer marks by trametinib or panobinostat were also observed for PPARγ, MRC1, and TGM2 (Figure S9E). Collectively, these data demonstrate that, in the presence of IL-4, increased MEK activity promotes PPARγ expression, which is associated with increased RA signaling, oxidative phosphorylation, and M2-type polarization, and all of that can be blocked by trametinib or panobinostat. This suggests a critical role for MEK/ERK activity in promoting the PPARγ/RA signaling axis to drive M2-type macrophage polarization.
MEK/ERK links IL-4 signaling with PPARγ-induced RA signaling during M2-type polarization
Which upstream signaling events regulate PPARγ/RA-induced M2-type polarization is not well understood. Moreover, a comprehensive understanding of the successive signaling events and temporal interactions that occur between different signaling pathways to drive M2-type polarization is currently lacking. Thus, we analyzed the time-course proteomics and phosphoproteomics data to establish a model for the sequence of temporal signaling events that are induced by IL-4 and lead to activation of signaling via MEK, PPARγ, and RA and which result in M2-type polarization. We found that IL-4 treatment leads to phosphorylation of the IL-4Rα and to activating phosphorylation events of STAT6 (Y641) and JAK1 (S574) within 10–30 min after adding IL-4 (Figures 5A and 5B). Thereafter, a steady decrease in total IL-4Rα, phosphorylated IL-4Rα (S387; T476; T487), JAK1 (S574), and STAT6 (Y641) is observed. JAK1 activates IRS2 with a peak at ~30 min by increasing tyrosine phosphorylation (Y823) and decreasing serine phosphorylation (S365, S388, S391, and S679) (Figure S12A), consistent with data showing that IL-4-induced tyrosine phosphorylation of IRS2 increases its activity and serine phosphorylation inhibits its activity (Warren et al., 2016). IRS2 (Y823) and total IRS2 protein levels decrease subsequently, and there is a concomitant relative increase in serine phosphorylation (Figure S12A). IL-4Rα/JAK1-induced activation of IRS2 serves as a signaling node that leads to activation of PI3K/AKT as well as Ras/Raf signaling, which occur in a parallel pathway to IL-4Rα/STAT6 signaling (Covarrubias et al., 2015, 2016; Heller et al., 2008; Warren et al., 2016). Our pathway analyses of the transcriptomic studies and previous reports show that PI3K/AKT signaling promotes M2-type polarization (Figure 4A) (Vergadi et al., 2017), and we found that IL-4 induces activation of the p85α subunit of PI3K (Y467 and Y580) and AKT1 (S124, S126, and S129) with a peak at ~30 min to 1 h of IL-4 treatment (Figures 5B and S12B). The adaptor protein GRB2 links IRS2 to the Ras/Raf signaling cascade, and activating phosphorylating events in BRAF (e.g., at S446) occurred at ~1 h of IL-4 treatment, whereas inactivating phosphorylation events (e.g., T440) were downregulated at that time (Figure S12C). Following this BRAF activation, a steady increase of downstream MEK-mediated activating ERK phosphorylation events (ERK2 [T185; Y187]) occurred at 2–8 h of IL-4 treatment (Figure 5B). Thus, we observed a sustained ERK activation pattern with IL-4 treatment and not a short-lived immediate response. Notably, p38α (Y128) activation occurred at a similar time point during the induction phase of M2-type polarization as ERK activation did (Figure S12D).
Figure 5. MEK signaling links IL-4 signaling with PPARγ and RA signaling during M2-type polarization.

Quantitative time-course proteomics and phosphoproteomics show temporal activation events after initiating treatment with either IL-4 or IFN-γ/LPS in THP-1-derived macrophages.
(A) Left: temporal dynamics of levels of IL-4Rα protein and phosphorylated IL-4Rα (S387, T476, and T487) in response to IL-4. Right: correlation matrix for IL-4-treated groups.
(B) Left: graph shows temporally distinct and successive peaks (arrows) of activating phosphorylating events for JAK1 and STAT6 at 10 min after the addition of IL-4, for AKT1 at 1 h, for ERK2 at 4–8 h (and diminished activity at 24 h), and for MYC at ~4 h, whereas PPARγ (S112) peaks at 24 h. Right: correlation matrix for IL-4-treated groups shows that JAK1 and STAT6 activations strongly correlate temporally (high r value), whereas the delayed activation of ERK2, MYC, or the S112 phosphorylation of PPARγ is reflected in a low r value or an adverse correlation in the comparison with JAK1 or STAT6.
(C) Left: a strong increase in downstream targets of PPARγ is observed at 24 h, including FABP4, LPL, and rate-limiting enzymes of RA signaling (RDH10 and ALDH1A2). TGM2 and CD209 are markedly increased at 24 h. Right: correlation matrix for all treatment groups shows a high correlation for all these proteins.
(D) Proposed model of temporal sequence of activation events during IL-4-induced M2-type macrophage polarization based on time-course proteomic and phosphoproteomic data and functional studies. Inhibitors of pan-Raf, MEK, HDACs, and PPARγ inhibit M2-type polarization. BRAF-inhibitor-mediated activation of MEK/ERK signaling, PPARγ agonist treatment, or direct stimulation of RA signaling with the RAR agonist AM580 or with ATRA promotes M2-type polarization. “P” indicates phosphorylation. Time when phosphorylation peaks is shown. TF: transcription factor.
For correlation matrices: Pearson correlation coefficients (r) shown in heatmap with p values in parenthesis. See also Figures S2 and S12; Table S2.
ERK signaling can stabilize the AP-1 transcription factor family member FOS, and our motif analyses of enhancers based on H3K27Ac ChIP-seq data in IL-4-treated THP-1-derived and primary human macrophages showed that IL-4 treatment induced an enrichment of AP-1 transcription factor binding at enhancers (Figure S12E) (Chen et al., 1996; Fontana et al., 2015a, 2015b). Phosphorylation of MYC at S62 (which is induced by ERK2 and promotes transcription) increased at 2–8 h (Figure 5B). This observation, together with our finding that IL-4 induces MYC expression, an effect that was inhibited by both trametinib and panobinostat (Figure 4E), suggest a co-regulatory role of MYC for IL-4-induced transcription of M2-type polarization genes. Thus, IL-4-induced activation of STAT6, MYC, and AP-1 transcription factors is associated with the early phase of M2-type macrophage polarization.
PPARγ protein levels peak at ~8 h of IL-4 treatment and are markedly reduced at 24 h (Figure S12F). Together with our findings that IL-4 induces PPARγ expression and that trametinib inhibits PPARγ expression, this demonstrates that MEK signaling is a critical inducer of PPARγ expression, which leads to a peak of PPARγ protein levels at ~8 h, following the peak of ERK signaling activity (2–8 h). PPARγ activity can be switched off through S112 phosphorylation by ERK, which leads to degradation of PPARγ (Hu et al., 1996). Indeed, we observe that PPARγ (S112) phosphorylation peaks at 24 h of IL-4 treatment when PPARγ protein levels are also reduced (Figures 5B and S12F). This suggests that IL-4-induced MEK/ERK signaling promotes PPARγ activity during the early induction phase of M2-type polarization by increasing its protein levels (at ~8 h), and once M2-type polarization has progressed, PPARγ activity is switched off by ERK-mediated S112 phosphorylation, which leads to its degradation.
PPARγ induces M2-type polarization by promoting a metabolic switch to oxidative phosphorylation and by induction of RA signaling via increasing expression of rate-limiting enzymes of RA signaling, including ALDH1A2 (Gyöngyösi et al., 2013; Szatmari et al., 2006; Zhu et al., 2013). Indeed, we observed that PPARγ targets involved in RA signaling or lipid metabolism (including RDH10, ALDH1A2, FABP4, and lipoprotein lipase [LPL]) and M2-type polarization markers induced by RA signaling (e.g., TGM2) all peaked after 8 h (24-h time point) (Figure 5C, 5D, and S12G). In addition to its role of inducing fatty acid oxidation, IL-4-induced PPARγ also promotes lipolysis by inducing expression of LPL (Odegaard et al., 2007), which we found to be highly upregulated at 24 h after IL-4 treatment (Figure 5C).
IL-4-induced and MEK/ERK-mediated PPARγ and RA signaling are required for M2-type macrophage polarization
We found that MEK/ERK-induced PPARγ and RA signaling not only is associated with M2-type polarization but also is required for that polarization process. The PPARγ antagonist GW9662 dose-dependently inhibited MRC1 expression in macrophages at concentrations that do not affect cell viability (Figure 6A). Our proteomic and transcriptomic data show that inhibition of M2-type polarization by panobinostat or trametinib is linked to inhibiting the expression of PPARγ and its downstream target ALDH1A2. We confirmed by western blotting that panobinostat and trametinib completely blocked the IL-4-mediated strong increase in ALDH1A2 protein levels (Figure 6B). Moreover, GW9662 reduced IL-4-induced ALDH1A2 protein levels, whereas the PPARγ agonist rosiglitazone increased ALDH1A2 protein levels (Figure 6B). ALDH1A2 protein levels correlated to protein levels of the RA-dependent downstream target and M2-type marker TGM2 (Figure 6B). We confirmed in both primary mouse and human macrophages that GW9662 reduced protein levels of the M2-type markers Arg1 (mouse) and TGM2 (human), whereas the PPARγ agonist rosiglitazone increased their protein levels (Figures 6C and 6D). Activation of RA signaling directly with either the retinoic acid receptor (RAR) agonist AM580 or all-trans-RA (ATRA) also strongly increased Arg1 or TGM2 protein levels in mouse and human macrophages, respectively (Figures 6B–6D).
Figure 6. IL-4-induced and MEK/ERK-mediated PPARγ and RA signaling are required for M2-type macrophage polarization.

(A) GW9662 reduces IL-4-induced MRC1 promoter-driven luciferase activity in a dose-dependent manner at concentrations that do not affect cell viability in THP-1-derived macrophages. Concentrations of chemicals used indicated in nM on the x axis; light units on y axis are normalized to the control carrier (DMSO). Red: luciferase activity; green: CTG. Graphs represent data as means ± SD. N = 4/group.
(B) THP-1-derived macrophages treated with IL-4 at the indicated time points (in hours). Western blotting for ALDH1A2, TGM2, and as a loading control β-actin or β-tubulin. High ALDH1A2 protein levels are detected at 24 h, but they are completely blocked by trametinib and panobinostat (upper band is the specific ALDH1A2 band; lower band is an unspecific background band). GW9662 reduces and rosiglitazone increases ALDH1A2. Panobinostat, trametinib, and GW9662 inhibit TGM2 levels (peaking at 24 h), whereas rosiglitazone, ATRA, or AM580 increase TGM2.
(C) GW9662 reduces M2-type polarization in primary mouse macrophages with reduction of Tgm2 and Arg1, whereas rosiglitazone, AM580, and ATRA strongly promote M2-type polarization. AM580 and ATRA even induce Arg1 and Tgm2 in macrophages treated with IFN-γ/LPS. 4 days of chemokine treatment.
(D) IL-4-induced TGM2 is reduced with GW9662 but increased with rosiglitazone, AM580, or ATRA. Primary human macrophages after 4 days of treatment with IL-4.
(E) Primary human macrophages treated with IL-4 at the indicated time points (in hours) in the presence or absence of GDC-0879. IL-4 increases ERK1 (T202/Y204) phosphorylation at 2–8 h with a concomitant increase in PPARγ, which then both decline at 24 h. GDC-0879 increases ERK1(T202/Y204) phosphorylation and PPARγ levels, which leads to an increase in MRC1 and TGM2.
(F) Primary human macrophages treated with IL-4 for either 8 h or for 24 h in the presence or absence of trametinib or GDC-0879 (DMSO as controls). The increase in ERK1 (T202/Y204) phosphorylation and PPARγ with GDC-0879 is associated with increased MRC1 and TGM2, whereas the block of ERK1 (T202/Y204) phosphorylation and the low levels of PPARγ with trametinib treatment lead to reduced MRC1 and TGM2.
Western blots with β-actin or β-tubulin loading control. Western blot values indicate normalization to loading control and IL-4 DMSO control sample at 24 h (B), IL-4 DMSO control sample (C) and (D), IL-4 DMSO control sample at 0 h (for MRC1 at 24 h) (E), or DMSO control sample with no IL-4 (F).
Our proteomic and phosphoproteomic data showed that the increase in ERK phosphorylation at 2–8 h of IL-4 treatment was associated with an increase in PPARγ protein levels, which then both decreased at 24 h (Figures 5B and S12F). We confirmed these findings also in primary human macrophages by western blotting (Figures 6E and 6F). To provide further evidence that ERK signaling activity is a critical determinant of PPARγ protein levels during IL-4-induced M2-type polarization, we treated primary human macrophages with IL-4 in the presence or absence of GDC-0879 (to activate ERK signaling) or trametinib (to inhibit ERK signaling). GDC-0879 increased ERK activation and concomitantly led to increased protein levels of PPARγ, MRC1, and TGM2, whereas trametinib had the opposite effects (Figures 6E and 6F). The increase in ERK1 (T202/Y204) phosphorylation and in PPARγ observed with IL-4 treatment at 8 h was further increased with GDC-0879 but completely blocked with trametinib. At 24 h of IL-4 treatment, PPARγ levels again decreased but remained increased with GDC-0879 treatment (Figures 6E and 6F). Thus, M2-type macrophage polarization is dependent on IL-4-induced and MEK/ERK-mediated PPARγ and RA signaling.
Trametinib and panobinostat block M2-type macrophage polarization in vivo and inhibit inflammatory angiogenesis and fibrosis
The inhibition of M2-type polarization of macrophages by trametinib or panobinostat also resulted in reduced expression of the proangiogenic growth factor VEGF-A in these macrophages (Figure 7A). Thus, we tested whether trametinib and panobinostat also inhibit M2-type polarization and aberrant angiogenesis in vivo. We have shown that laser-induced CNV is an animal model for wound healing and NV-AMD, in which Arg1+ M2-type macrophages strongly accumulate within the first 48–72 h at the site of the laser-induced injury and promote CNV (He and Marneros, 2013, 2014; Marneros, 2013; Strittmatter et al., 2016). Here, we find that both panobinostat and trametinib potently inhibited M2-type macrophage polarization in these CNV lesions without blocking macrophage infiltration (Arg1−F4/80+), which was associated with strong inhibition of CNV (Figures 7B and 7C). Similarly, panobinostat and trametinib diminished Arg1 expression in wound macrophages after skin wounding (Figures 7D and 7E).
Figure 7. Panobinostat and trametinib block M2-type macrophage polarization in vivo and inhibit CNV.

(A) MEK inhibitors and HDAC inhibitors attenuate VEGF-A expression in IL-4-treated THP-1-derived macrophages. Normalized to housekeeping gene 36B4 (semiquantitative RT-PCR). Cytokine treatment for 4 days. N = 3/group (each in triplicate).
(B) Semiquantitative RT-PCR of choroid/RPE lysates of mice treated with either panobinostat, trametinib, or DMSO (control) for 3 days after laser-induced CNV induction. Graphs show Arg1 (M2), COX2 (M1), and CXCL9 (M1) expression normalized to CD68. N = 8–10 mice/group (each in triplicate).
(C) Confocal microscopy images of CNV lesions 3 days after laser-induced injury and treatment with either DMSO, trametinib, or panobinostat. Arg1+ macrophages (white; yellow arrows), SMA+ cells (red), and CD31+ neovessels (green) in DMSO-treated mice, but no Arg1 or CD31 and only reduced SMA staining is observed in trametinib or panobinostat treated mice (top row). Panobinostat or trametinib treatment does not prevent macrophage infiltration (F4/80+ cells [white]; bottom row). Scale bars, 100 μm.
(D) In skin wounds infiltrating F4/80+ (white) macrophages are strongly Arg1+ (green). Panobinostat-treated mice show infiltration of F4/80+ macrophages, which do not stain for Arg1. Scale bars, 20 μm.
(E) Semiquantitative RT-PCR shows inhibition of Arg1 expression in skin wounds after trametinib or panobinostat treatment (normalized to 36b4). N = 8–10 mice/group (each in triplicate).
Graphs represent data as means ± SEM. p values are shown (t test). See also Table S3.
DISCUSSION
Our datasets provide a comprehensive resource to define which temporal changes in cell signaling and metabolism occur during specific phases of the macrophage-polarization process and have critical roles for M1- versus M2-type polarization. They allow an in-depth, direct comparison of IFN-γ/LPS- versus IL-4-induced quantitative changes of the proteome and phosphoproteome at a temporal resolution and scale that has not been available thus far. Moreover, our quantitative proteomics of fully polarized M1- and M2-type macrophages resulted in the quantification of many more proteins than in previous proteomic studies of polarized macrophages (Court et al., 2017; Meijer et al., 2015; Wiktorowicz et al., 2019).
Macrophage polarization has been linked to metabolic reprogramming. M1-type macrophages rely mainly on glycolysis and synthesize fatty acids from acetyl-coenzyme A (acetyl-CoA), whereas IL-4-induced PPARγ is required for M2-type macrophage polarization and stimulates oxidative phosphorylation (Bouhlel et al., 2007; Chawla, 2010; Lavin et al., 2017; Nelson et al., 2018; Odegaard et al., 2007). Notably, our findings not only validated previously known distinct changes in cell metabolism that occur with M1- versus M2-type polarization but also defined the precise temporal activation pattern for these metabolic pathways during M1- versus M2-type polarization. Moreover, we also detected metabolic changes that were previously not well recognized, such as the upregulation of glycosaminoglycan metabolism with M2-type polarization. Our data show that IL-4-induced M2-type polarization is associated with increases in key genes and proteins involved in PPARγ and RA signaling, oxidative phosphorylation, and carbohydrate, glycosaminoglycan, and lipid metabolism at specific phases of the polarization process.
These datasets can be used to identify critical signaling and metabolic pathways required for macrophage polarization. As such, we examined the role of IL-4-induced MEK signaling for M2-type macrophage polarization. Although it has previously been reported that PPARγ-induced RA signaling promotes M2-type polarization, our finding that MEK signaling is a critical regulator of this PPARγ/RA axis was not previously established. The demonstration that pharmacologic blockade of MEK signaling can inhibit the PPARγ/RA axis and thereby block M2-type polarization, whereas pharmacologic activation of MEK signaling has the opposite effect, shows the therapeutic relevance of these findings and demonstrates the value of combing information from the proteomic/phosphoproteomic datasets with those from chemical screens. The integration of these data provided a model for the temporal sequence of signaling events that are required for M2-type polarization and that can be regulated by small molecules. Based on our data, we propose a model in which IL-4 induces JAK1/STAT6 activation and, via parallel pathways, PI3K/AKT and MEK/ERK signaling, which cooperatively lead to the activation of transcription that is regulated, at least in part, by the activities of STAT6, HDACs, AP-1, and MYC. This early phase of M2-type polarization results in increased PPARγ, which then functions in a second subsequent phase via activation of RA signaling to further promote M2-type polarization (Figure 5D). We show that this MEK/PPARγ/RA signaling axis can be targeted by small molecules to block M2-type polarization, thereby providing therapeutic opportunities for conditions that are exacerbated by M2-type macrophages. For example, MEK inhibitors potently inhibited expression of M2-type polarization markers not only in vitro but also in vivo in assays of wound healing and NV-AMD.
The increase in PPARγ with IL-4 treatment preceded the increase in rate-limiting enzymes required for RA signaling (e.g., ALDH1A2), consistent with data showing that ALDH1A2 expression is upregulated by IL-4 through the cooperative actions of PPARγ and STAT6 and promotes M2-type macrophage polarization (Chiba et al., 2016; Daniel et al., 2018; Gyöngyösi et al., 2013; Ho et al., 2016; Huang et al., 1999; Lee et al., 2016; Nelson et al., 2018; Szatmari et al., 2006; Vellozo et al., 2017; Zhu et al., 2013). We show that stimulation of PPARγ activity with an agonist in IL-4-treated macrophages increases ALDH1A2 levels and M2-type polarization, whereas a PPARγ inhibitor had the opposite effect. IL-4-induced MEK/ERK signaling is required for the increase in PPARγ expression and PPARγ-induced RA signaling that promotes M2-type polarization. Moreover, panobinostat had a similar effect as trametinib, implicating HDAC activity as a key regulator of PPARγ-induced M2-type polarization.
IL-4 induces MYC expression that promotes expression of PPARγ and a subset of M2-genes (Pello et al., 2012). We also observed an upregulation of MYC expression by IL-4 and an induction of activating phosphorylating events of MYC by ERK at the time when ERK activity peaked. Moreover, trametinib and panobinostat both inhibited IL-4-induced expression not only of PPARγ but also of MYC. In addition, AP-1 transcription factors can be activated by MEK/ERK signaling and have been shown to promote macrophage polarization (Fontana et al., 2015a, 2015b), which is supported by our enhancer analysis data as well. This suggests that functional cooperation between STAT6, AP-1, MYC, and HDACs induces genes that promote M2-type polarization, including PPARγ, during the early phase of M2-type polarization and that MEK/ERK signaling promotes that process by activating AP-1 and MYC. ERK-induced phosphorylation of PPARγ at S112 inhibits its transcriptional activity and leads to its degradation (Burns and Vanden Heuvel, 2007; Hu et al., 1996). Consistent with a phase-dependent activating versus inhibitory function of MEK/ERK signaling for PPARγ activity and M2-type polarization, we find that the MEK/ERK-induced increased PPARγ activity in the early phase of M2-type polarization is subsequently switched off in the second phase (at 24 h) by ERK-induced PPARγ (S112) phosphorylation, which leads to its degradation at a time when M2-type polarization has progressed. In fully polarized M2-type macrophages, MEK/ERK signaling activity is diminished. Thus, MEK/ERK activity controls PPARγ levels that determine induction of IL-4-dependent M2-type polarization.
Whether and how MEK/ERK signaling affects macrophage polarization has previously been unclear. Differences in the reported effects of MEK inhibitors on M2-type macrophage polarization in earlier studies may, in part, be explained by differences in the time points when MEK/ERK phosphorylation was examined (Heller et al., 2008; Long et al., 2017). We show in human macrophages that IL-4 leads to an increase in ERK phosphorylation between 2–8 h but not at 10–30 min, explaining why a previous study that assessed ERK phosphorylation only at 10–30 min after adding IL-4 did not observe an increase in ERK phosphorylation (Heller et al., 2008). The duration of ERK activation can be either transient and short-lived or sustained, lasting several hours (Muta et al., 2019). Sustained ERK signaling can occur because of activated receptors that are translocated upon ligand stimulation to long-lived signaling endosomes, where they avoid degradation (Bergeron et al., 2016; Valdez et al., 2007; Villaseñor et al., 2015). Notably, ligand-induced IL-4 receptor dimers become enriched in cortical endosomes after IL-4 treatment to stimulate downstream signaling (Kurgonaite et al., 2015), which may explain our observation that M2-type polarization of macrophages is associated with sustained ERK activation between 2 and 8 h after IL-4 treatment. Collectively, our data show that IL-4-induced sustained MEK/ERK signaling is required for M2-type macrophage polarization.
The contributions of HDACs for macrophage polarization are not well understood. Panobinostat was a particularly potent inhibitor of M2-type marker expression and could block Arg1 expression in activated macrophages also in vivo, which was associated with an inhibition of CNV. Panobinostat inhibited expression of genes induced by MEK/ERK signaling (e.g., MYC) as well as of PPARγ and ALDH1A2, suggesting that HDAC inhibition acts upstream of RA signaling in blocking M2-type polarization. This is also supported by the observation that panobinostat and trametinib showed a significant overlap of inhibited genes and pathways associated with M2-type polarization. This overlap may, in part, be explained by our finding that panobinostat inhibited expression of genes linked to MEK/ERK signaling, as well as by our observation that trametinib inhibited expression of genes associated with histone deacetylase activity. Notably, pan-HDAC inhibitor treatment only affects expression of a subset of genes in a cell-type-dependent manner and only a subset of HDAC-bound genomic locations are targeted by HDAC inhibitors (Hanigan et al., 2018; Peart et al., 2005; Richon et al., 2000; Van Lint et al., 1996). Consistent with such a context-dependent, selective effect of HDAC inhibitors on transcriptional programs, panobinostat inhibited M2-type marker gene expression but increased expression of various M1-type markers. Varying findings regarding the roles of HDAC inhibitors on macrophage polarization may be explained not only by different activities of various HDAC inhibitors on specific HDACs, different origins of macrophages, or different in vivo assays used but also by different HDAC concentrations used in these experimental assays (Lohman et al., 2016). This may explain why some previous studies also implicated some HDAC inhibitors as potential inducers of M2-type macrophage polarization (Mohammadi et al., 2018). However, our observation that multiple, different HDAC inhibitors could block M2-type marker expression in macrophages and that panobinostat could prevent M2-type polarization of macrophages also in vivo demonstrates their therapeutic potential as M2-type polarization inhibitors.
Among the diseases for which M2-type macrophage polarization inhibitors may offer therapeutic benefits would be conditions in which increased M2-type macrophages are associated with pathologic angiogenesis, such as in NV-AMD (Yang et al., 2016; Zandi et al., 2015). Our previous observation that CNV-infiltrating macrophages/microglia in the laser-induced NV-AMD model predominantly express M2-type markers and that ablation of CNV macrophages/microglia prevented CNV formation suggests that the potent inhibition of CNV and M2-type polarization with panobinostat and trametinib is, at least to a significant extent, due to the block of M2-type polarization (He and Marneros, 2013; Marneros, 2013). However, MEK and HDAC inhibitors can also inhibit angiogenesis via direct effects on endothelial cells (Bullard et al., 2003; Deroanne et al., 2002; Zhu et al., 2002). Thus, it is likely that they inhibit CNV not only by blocking M2-type macrophage polarization but also to some extent through direct effects on endothelial cells. This dual inhibitory activity of MEK and HDAC inhibitors on both M2-type macrophage polarization and endothelial cells makes these drugs particularly promising as therapeutic options in diseases with pathologic angiogenesis.
In summary, our data provide a comprehensive resource to define signaling mechanisms and changes in cell metabolism that regulate macrophage polarization. We used these data to show that a MEK/PPARγ/RA signaling axis is required for IL-4-induced M2-type macrophage polarization, which can be effectively blocked by pharmacologic inhibitors of MEK, PPARγ, or HDACs. Thus, these small-molecule inhibitors are likely to have therapeutic relevance for conditions exacerbated by M2-type macrophages.
Limitations of study
A limitation of studying macrophage polarization in vitro is that this approach only partially captures the tissue microenvironment context in which many different factors affect macrophage polarization. However, it is likely that the identified signaling mechanisms that promote polarization in vitro are also critical for polarization mechanisms that occur in vivo. This is supported by our observation that trametinib and panobinostat inhibited M2-type macrophage polarization not only in vitro but also in skin wounds and laser-induced CNV lesions.
Our proteomic studies and chemical screens used a well-characterized human macrophage cell line (THP-1 cells). This allowed us to conduct these experiments with a sufficiently large number of cells that had less heterogeneity than primary macrophages. In contrast, RNA-seq experiments were conducted in both THP-1-derived and primary human macrophages and showed a significant overlap in activation of specific cellular pathways that occur with either M1- or M2-type polarization between both macrophage populations. Thus, it is likely that many of the findings from the proteomics and chemical screen experiments in THP-1-derived macrophages also apply to primary human macrophages. However, differences exist between these cells and, therefore, findings in THP-1-derived macrophages cannot be directly extrapolated to cellular changes that occur in primary macrophages. This highlights the importance of functional validation experiments to assess whether a particular pathway activation or drug effect observed in THP-1-derived macrophages also occurs in primary human macrophages, as has been demonstrated for key findings in this study.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact, Alexander G. Marneros (amarneros@mgh.harvard.edu).
Materials availability
Materials described in this study are commercially available. The MRC1-promoter luciferase THP-1 macrophage cell line is available from the lead contact.
Data and code availability
All RNA-Seq and ChiP-Seq sequencing data were deposited on the National Cancer for Biotechnology Information Gene Expression Omnibus (GEO): GSE154347. All proteomics data were deposited into the MassIVE repository (MSV000084672). Accession numbers are also listed in the key resources table. All other data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
| Rabbit anti-MRC1 | Sigma Aldrich | HPA004114, RRID: AB_1846270 |
| Rabbit anti-CD68 antibody | Santa Cruz Biotechnology | SC-9139, RRID:AB_2275738 |
| Rabbit anti-F4/80 | Santa Cruz Biotechnology | SC-25830, RRID:AB_2246477 |
| Goat anti-Arg-1 | Santa Cruz Biotechnology | SC-18354, RRID:AB_2227469 |
| Rabbit anti-TGM2 | Cell Signaling Technologies | #3557, RRID:AB_2202883 |
| Rabbit anti-phospho-Stat1 (Y701) | Cell Signaling Technologies | #9167, RRID:AB_561284 |
| Rabbit anti-phospho-ERK1, Thr202/Tyr204 | Cell Signaling Technologies | #9101, RRID:AB_331646 |
| Rabbit anti-ERK1/2 | Cell Signaling Technologies | #4695, RRID:AB_390779 |
| Rabbit anti-β-actin | Lab Vision | Rb-9421-P1, RRID:AB_720056 |
| Mouse anti-β-actin | Sigma Aldrich | A5316, RRID:AB_476743 |
| Rabbit anti-PPARγ | Cell Signaling Technologies | # 2443, RRID:AB_823598 |
| Rabbit anti-phospho-STAT6 (Y641) | Cell Signaling Technologies | # 56554, RRID:AB_2799514 |
| Rabbit anti-β-tubulin | Cell Signaling Technologies | #2128, RRID:AB_823664 |
| Mouse anti-ALDH1A2 | Santa Cruz Biotechnology | sc-393204 |
| Rabbit IgG HRP Linked Whole Ab antibody | GE Healthcare | GENA934, RRID:AB_2722659 |
| Sheep Anti-Mouse IgG - Horseradish Peroxidase antibody | GE Healthcare | NA931, RRID:AB_772210 |
| Rabbit anti-H3K27ac | Abcam | ab4729, RRID:AB_2118291 |
| Rat anti-CD31 | BD Biosciences | # 550274, RRID:AB_393571 |
| Mouse anti-SMA-Cy3 conjugate (clone 1A4) | Sigma Aldrich | C6198, RRID:AB_476856 |
| Rat anti-F4/80 (conjugated with Alexa647) | BioLegend | # 123121, RRID:AB_893492 |
|
Chemicals, peptides, and recombinant proteins | ||
| PMA | Sigma Aldrich | P8139 |
| Murine M-CSF | Peprotech | #315–02 |
| Murine IL-4 | Peprotech | #214–14 |
| LPS | Sigma Aldrich | L4391 |
| Human M-CSF | Invitrogen | 14–8789-80 |
| Human IL-4 | Peprotech | #200–04 |
| Human IFNγ | Peprotech | #300–02 |
| MNase | New England Biolabs | M0247S |
| small molecule chemicals | Selleckchem or Cayman Chemicals | N/A |
|
Critical commercial assays | ||
| CellTiter-Glo luminescent cell viability assay (CTG) | Promega | G7573 |
| Steady-Glo Luciferase Assay System | Promega | E2520 |
| mRNeasy Mini Kit | QIAGEN | No. 217004 |
| TruSeq RNA Library Prep Kit V2 | Illumina | RS-122–2001 |
|
Deposited data | ||
| Proteomics | MassIVE repository | MSV000084672 |
| RNA-Seq and ChIP-Seq | GEO | GSE154347 |
|
Experimental models: cell lines and primary cells | ||
| THP-1 cell line | ATCC | Cat# TIB-202, RRID:CVCL_0006 |
| mouse BMDMs (primary cells) | JAX Labs | C57BL/6J mice |
| human PBMCs (primary cells) | iXCells Biotechnologies | 10HU-003CR100M |
| THP-1 cell line with MRC1 promoter-driven luciferase, 1.0k, clone#4 | He and Marneros, 2014 | N/A |
|
Experimental models: mice | ||
| C57BL/6J mice | JAX labs | JAX:000664 |
|
Oligonucleotides | ||
| semiquantitative RT-PCR primers | Life Technologies | N/A |
|
Software and algorithms | ||
| GelQuantNET, western blot band quantifications | Biochemlabsolutions | http://biochemlabsolutions.com/GelQuantNET.html |
| In-house developed software suite | Huttlin et al., 2010 | N/A |
| Sequest algorithm | Eng et al., 1994 | N/A |
| Ascore algorithm | Beausoleil et al., 2006 | N/A |
| GSEA | UCSD and Broad Institute | http://www.gsea-msigdb.org/gsea/login.jsp;jsessionid=CF17CD01EADE3916721F5DA4F6FEFFAA; version 4.0.2) |
| nf-core/rnaseq | Ewels et al., 2020 | https://github.com/nf-core/rnaseq |
| nf-core/chipseq | Ewels et al., 2020 | https://github.com/nf-core/chipseq |
|
Other | ||
| Chemical Libraries, 143 natural products | Selleckchem | Library L1400–01 and -02 |
| Chemical Libraries, 1836 bioactive compounds | Selleckchem | Library L1700–01 and -02 |
| Xvivo10 medium | Lonza | 04–380Q |
| Human serum AB plasma | Sigma | H4522 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and reagents
THP-1 cells were obtained from ATCC (ATCC Cat# TIB-202, RRID:CVCL_0006) and cultured in RPMI1640+10% FBS+1% Anti-Anti (Life Technologies, 15240) on plastic cell culture dishes (Nunclon Delta, Thermo Fisher). BMDMs were isolated from both femurs and tibias of 8-week-old C57BL/6J mice. Briefly, mice were euthanized and immersed in 70% ethanol for 2 minutes. After removing attached tissues from femurs and tibias, bones were immersed in 70% ethanol for 30 s and subsequently bone marrow was flushed out by injecting DMEM+10% FBS into bones using 10 mL syringes with 27G needles. The bone marrow suspension was spun down. Pellets were resuspended in 25 mL of DMEM + 10% FBS+1% Anti-Anti+ murine 10 ng/ml M-CSF. Cells were cultured in 140 mm Petri dishes for 7 days. One day before use, attached macrophages were washed with PBS for 3 times and split into 12 well cell culture dishes for the following experiments. One day after splitting, BMDMs were treated with mouse IL-4 (20 ng/ml) or IFNγ (20 ng/ml) + LPS (10 ng/ml) for either 24 hr or 4 days. Cryopreserved human PBMCs (100 million cells/vial) were purchased from iXCells Biotechnologies (10HU-003CR100M). PBMCs were suspended in cell culture medium (Xvivo10 [Lonza, 04–380Q, with gentamicin and phenol red], 1.8% human serum [AB plasma, Sigma, H4522], 100 ng/ml human M-CSF [Invitrogen, 14–8789-80]). Medium was changed on day 1 and day 3 and cells floating in media were span down and re-seeded to cell culture dishes. On day 6, PBMCs were treated with human IL-4 (20ng/ml) or human IFNγ (20ng/ml) + LPS (10ng/ml). For small molecule treatments, PBMC-derived macrophages were treated together with indicated chemicals for 24 hours or 4 days. Recombinant human IL-4 (#200–04), human IFNγ (#300–02), murine IL-4 (#214–14) and murine M-CSF (#315–02) were purchased from Peprotech, (Peprotech, NJ); and LPS (L4391) and PMA (P8139) were purchased from Sigma.
Study approval
All animal studies were approved by the institutional animal review board (IACUC) of Massachusetts General Hospital.
METHOD DETAILS
Cell-based small molecule inhibitor screen
We described the generation of the MRC1 promoter luciferase construct and the establishment of a clonal THP-1 cell line stably expressing this construct previously (He and Marneros, 2014). The THP-1 MRC1 promoter luciferase reporter cells (1.0kb, clone #4) were seeded into 384-well plates at a density of 4×104 cells/well by a Wellmate machine (Matrix Technologies Corp) and treated with 40 nM PMA for 24 hours and followed by the treatments of IL-4 (20 ng/ml) + 2.5 μM chemicals each for additional 3 days. The chemical libraries used were obtained from Selleckchem and included 143 natural products (library L1400–01 and L1400–02) and 1836 bioactive compounds (library L1700–1 to 21). Luciferase activity was determined using the Steady-Glo Luciferase Assay System (Promega, E2520) as per manufacturer’s protocol. All treatments in this library screen were performed in duplicate.
Verification of candidate chemicals using a luciferase reporter assay in THP-1 cells
THP-1 MRC1 promoter luciferase reporter cells (1.0k, clone #4) were seeded into 384 well plates, 4×104 cells/well in 30 μL RPMI1640+10% FBS+1% Anti-Anti with PMA (40 nM) by a Wellmate machine. 24 hours later, 50 μL RPMI1640+10% FBS+1% Anti-Anti with human IL-4 (20ng/ml) were added to primed cells. Candidate compounds were transferred to 384 well plates by pin plates. 4 days later, 10 μL Steady Glo was added. Plates were read with a luminescence plate reader. These experiments were performed in quadruplicate. To test for dose-dependent effects, chemicals were used at concentrations between 0 and 22,500nM (concentrations indicated in graphs). Results were normalized to the control carrier (DMSO). Chemicals were obtained from Selleckchem or Cayman Chemical.
Cytotoxicity assay
Concentration-dependent chemical cytotoxicity on THP-1-derived macrophages was measured with a CellTiter-Glo luminescent cell viability assay (CTG assay, Promega, G7573). Briefly, chemical-treated cells were split into two groups, one for the luciferase reporter assay and another for the cytotoxicity assay. Four days later, 5 μL 1:1 diluted CTG were added to the cells in 384 well plates by a Wellmate machine. All treatments in this assay were in quadruplicate.
Semiquantitative RT-PCR
Concentrations of chemicals that inhibited MRC1-driven luciferase activity without affecting cell viability were determine in dose-dependent luciferase assays. These concentrations were used in experiments to determine the effects of these chemicals on polarization of THP-1-derived macrophages or BMDMs (Table S3). THP-1 cells were primed with 40nM PMA for 24 hours. Cells were treated with candidate chemicals + human IL-4 (20 ng/ml) or IFNγ (20 ng/ml) + LPS (10 ng/ml) for 4 days before harvest. BMDMs from 10 C57BL/6J mice were pooled and split once before use. One day after splitting, BMDMs were pretreated with candidate chemicals plus mouse IL-4 (20 ng/ml) or IFNγ (20 ng/ml) + LPS (10 ng/ml) for 4 days.
RNA was isolated from cells with Trizol reagent (Life Technologies). For gene expression studies, cDNA was obtained using 0.5 μg RNA and the Transcriptor First strand synthesis kit utilizing hexamer primers (Roche). Semiquantitative RT-PCR was performed using a LightCycler 480 system with the LightCycler 480 SYBR Green I master mix according to standard procedures (45 amplification cycles) (Roche Applied Science, Indianapolis, IN). Primers for CD68 or 36b4 were used as a normalization control. Concentrations were determined using a standard dilution curve. Experiments for all samples were performed in triplicate with n = 3 wells/experimental group. An unpaired two-tailed Student’s t test was used for statistical analyses. P values < 0.05 were considered to be statistically significant.
Western blotting
Cells were harvested and proteins were extracted using NP-40 lysis buffer (Life Technologies, FNN0021) + 1mM PMSF and protease inhibitor cocktail (cOmplete, Roche). 20–30 μg protein was loaded onto NuPage 4%–12% Bis-Tris gradient gels (Life Technologies) and transferred to nitrocellulose membranes (GE Lifesciences). Primary antibodies used were against MRC1 (Sigma, HPA004114, 1:300; RRID: AB_1846270), CD68 (SCBT, SC-9139, 1:1000; RRID: AB_2275738), F4/80 (SCBT, SC-25830, 1:200, RRID: AB_2246477), Arg-1 (SCBT, SC-18354, 1:1000, RRID: AB_2227469), TGM2 (Cell Signaling, #3557, 1:1000; RRID: AB_2202883), phospho-Stat1 (Y701) (Cell Signaling, #9167, 1:1000; RRID: AB_561284), phospho-ERK1 (Thr202/Tyr204, Cell Signaling, #9101, 1:1500, RRID: AB_331646), ERK1/2 (Cell Signaling, #4695, 1:1500, RRID: AB_390779), β-actin (Lab Vision, Rb-9421-P1, 1:1000; RRID: AB_720056; and A5316 from Sigma, RRID: AB_476743), PPARγ (Cell Signaling Technology Cat# 2443, RRID: AB_823598), phospho-STAT6 (Y641) (Cell Signaling Technology Cat# 56554, RRID: AB_2799514), ALDH1A2 (Santa Cruz Biotechnology, sc-393204), and β-tubulin (Cell Signaling Technology Cat# 2128, RRID: AB_823664). HRP-conjugated secondary antibodies were purchased from GE Healthcare (ECL rabbit or mouse whole molecule, NA934 and NA931). Chemiluminescence signal was determined with the SuperSignal WestPico chemiluminescent substrate or the ECL western blotting substrate (Pierce).
Proteomics and phosphoproteomics samples
For proteomics of fully polarized THP-1-derived macrophages, THP-1 cells (2.5×106 cells/treatment) were treated with PMA (50nM) for 24 hours in 60 mm cell culture dishes. After PMA treatment, cells were incubated for 4 additional days with either (1) fresh medium (RPMI1640+10% FBS+1% Anti-Anti), (2) fresh medium + IL-4 (20 ng/ml) or (3) fresh medium + IFNγ (20 ng/ml) and LPS (10 ng/ml). Cells were then washed with cold PBS three times, scraped down and flash frozen in liquid nitrogen for proteomic experiments. Additional experimental groups included cells treated with IL-4 and (+)-JQ1 (0.093 μM). All experiments were performed in duplicate (n = 2/experimental group).
For time-course experiments, THP-1 cells (3×106 cells in 60 mm dish per time point for proteomics and 3.5×108 cells in 9×150 mm dishes per time point for phosphoproteomics) were treated with PMA for 24 hours to induce differentiation of THP-1 cells into M0 macrophages. Thereafter, these cells were harvested at 0 minutes (baseline comparison), and at 10 minutes, 30 minutes, 1 hour, 2 hours, 4 hours, 8 hours and 24 hours after initiation of treatment with either IL-4 (20 ng/ml) or IFNγ (20 ng/ml) and LPS (10 ng/ml). As controls, we utilized THP-1 cells (3×106 cells in 60 mm dish per time point) that were treated for 24 hours with PMA and harvested at the same time points as the treatment groups. We performed a multiplexed quantitative proteomics and phosphoproteomics approach based on the use of tandem mass tag reagents according to experimental protocols previously reported (Edwards and Haas, 2016).
To test the effects of trametinib or GDC-0879, THP-1 cells were first primed with PMA for 24 hours, then THP-1 cells were subjected to GDC-0879 (2.5 μM) or DMSO (control) pre-treatments for 1 hour and subsequently either IL-4 (20 ng/ml) or IFNγ (20 ng/ml)/ LPS (10 ng/ml) was added for M1 or M2 polarization as above for 24 hours. After this, cells were harvested for proteomics and phosphoproteomics profiling. These treatments were performed in duplicate (except for M1 cells treated with GDC-0879 for one sample).
Proteomics and phosphoproteomics methods
Proteome profiling was done by using a multiplexed quantitative proteomics approach based on the use of tandem mass tag (TMT) reagents (McAlister et al., 2012, 2014; Thompson et al., 2003; Ting et al., 2011). Cells were lysed by addition of 500 μL lysis buffer (75mM NaCl, 50mM HEPES pH 8.5, 10mM Sodium pyrophosphate, 10mM Sodium Fluoride, 10mM B-Glycerophosphate, 10mM Sodium Orthovanadate, Roche complete mini EDTA free protease inhibitors, 3% SDS, 10mM PMSF) (Edwards and Haas, 2016). The disulfide bonds were reduced by adding dithiothreitol (DTT) to a final concentration of 5 mM and incubation at 56°C for 30 min followed by adding iodoacetamide to adjust a final concentration of 15 mM and an incubation in the dark at room temperature for 20 min. The reaction was stopped by adding DTT to a final concentration of 5 mM and an incubation in the dark at room temperature for 15 minutes. Protein was isolated by adding one part of trichloroacetic acid to 4 parts of protein solution and incubation for 10 min on ice. The precipitated protein was pelleted by centrifugation (15,000 g, 10 min, 5°C) and washed twice with prechilled acetone (−20°C, 300 μL, 15,000 g, 10 min, 5°C). The remaining protein pellets were resuspended in 500μL 1 M urea, 50 mM HEPES (pH 8.5) and digested overnight at room temperature with 1 μg/μL endoproteinase Lys-C (Wako) followed by digestion with sequencing-grade trypsin (Promega) at a final concentration of 1 ng/μL 6 h at 37°C. The digestion was quenched with 1% trifluoroacetic acid (TFA), and peptides were desalted using Sep-Pak C18 solid-phase extraction (SPE) cartridges (Waters). The peptide concentration of each sample was determined using a BCA assay (Thermo Scientific).
For labeling with TMT10 or 11-plexed reagents (Thermo Scientific), 50 μg of peptides were dried and resuspended in 50 μL of 200 mM HEPES (pH 8.5), 30% acetonitrile (ACN). Labeling was performed by adding 150 μg TMT reagent in anhydrous ACN and incubating at room temperature for 1 h. The reaction was stopped by the addition of 5% (w/v) hydroxylamine in 200 mM HEPES (pH 8.5) to a final concentration of 0.5% hydroxylamine and incubation at room temperature for 15 min. Samples were acidified with 1% TFA, and samples were combined. The pooled samples were desalted using Oasis HLB cartridges (Waters) (Kreuzer et al., 2019). The combined multiplexed samples underwent a prefractionation on an HPLC under basic pH (basic pH reversed-phase liquid chromatography, bRPLC), as previously described, and fractions were dried (Edwards and Haas, 2016). A total of two TMT sets were analyzed, with twelve fractions assayed for each set. To enable a quantitative comparison of all analyzed samples, we used a common “bridge” sample in both TMT sets comprising all studied samples (Lapek et al., 2017). The proteome mapping experiments were done using an Orbitrap Fusion or an Orbitrap Fusion Lumos mass spectrometer. Peptides were resuspended in 5% ACN/5% formic acid and subjected to LC-MS2/MS3 analysis on an Orbitrap Lumos mass spectrometer. Peptides were separated on an in-house pulled, in-house packed microcapillary column (inner diameter, 100 μm; outer diameter, 360 μm). Columns were packed to a final length of 30 cm with GP-C18 (1.8 μm, 120 Å, Sepax Technologies). Peptides were eluted with a linear gradient of increasing ACN in 0.125% formic acid over 165 minutes at a flow rate of 300 nl/minute while the column was heated to 60°C. The Orbitrap Fusion was operated in data-dependent mode, with a survey scan performed over an m/z range of 500–1,200 in the Orbitrap with a resolution of 6×104, automatic gain control (AGC) of 5 × 105, and a maximum injection time of 100 ms. The most abundant ions detected in the survey scan were subjected to MS2 and MS3 experiments to be acquired in a 5 s experimental cycle. For MS2 analysis, doubly charged ions were selected from an m/z range of 600–1200, and triply and quadruply charged ions from an m/z range of 500–1200. The ion intensity threshold was set to 5×104 and the isolation window to 0.5 m/z. Peptides were isolated using the quadrupole and fragmented using CID at 30% normalized collision energy at the rapid scan rate using an AGC target of 1 × 104 and a maximum ion injection time of 35 ms. MS3 analysis was performed using synchronous precursor selection (SPS) (Ting et al., 2011; McAlister et al., 2014). Up to 10 MS2 fragment ions precursors were simultaneously isolated and further fragmented for MS3 analysis with an isolation window of 2.5 m/z and HCD fragmentation at 55% normalized collision energy. MS3 spectra were acquired at a resolution of 6×104 with an AGC target of 1 × 105 and a maximum ion injection time of 100 ms. A complete set of method parameters are listed in the RAW files provided through the MassIVE repository (MSV000084672).
Sample preparation for phosphoproteome mapping was done as described previously (Kreuzer et al., 2019; Lyons et al., 2018). Peptides from the Lys-C/trypsin digest were enriched for phosphopeptides using a 4: 1 ratio of titanium dioxide beads to peptide (w/w). Peptides were resuspended in 2 M lactic acid in 50% ACN and added to 1.8 mg of titanium dioxide beads. The mixture was shaken gently for 1 hour. Beads were collected by centrifugation and washed 3 times with 2 M lactic acid in 50% ACN and 3 times with 50% ACN / 0.1% TFA. Phospshopeptides were eluted with 2 × 200 μL of 50 mM KH2PO4, pH 10, and acidified with 1% TFA. Eluted phosphopeptides were desalted, lyophilized, and labeled with TMT reagents as described above. The combined sample was enriched for phosphotyrosine-containing peptides using phosphotyrosine antibody-conjugated beads (Cell Signaling Technology) following the protocol provided by the manufacturer. Unbound peptides (phosphoserine and phosphothreonine peptides) were desalted, lyophilized, and fractionated by bRPLC. A total of 96 fractions were collected, and fractions were combined into 24 fractions. Bound peptides (phosphotyrosine peptides) were eluted and desalted. All 25 fractions were re-suspended in 5% ACN / 5% formic acid and analyzed on an Orbitrap Fusion or an Orbitrap Lumos mass spectrometer using LC-MS2/MS3 for identification and quantification of the phosphopeptides. Analyses were done using both HCD fragmentation with Orbitrap fragment ion detection and CID fragmentation with ion trap fragment ion detection (Kreuzer et al., 2019; Lyons et al., 2018). The following key settings were used for the mass spectrometry methods: MS1 m/z, 500–1500; MS1 resolution, 6×105; MS1 AGC, 1×106; MS1 maximum ion injection, 100 ms; data acquisition cycle time, 5 s; MS2 precursor m/z selection range, 500–1500; MS2 ion intensity threshold, 3×106; MS2 isolation width, 0.5 m/z; MS2 AGC target, 2×104 (Fusion), 4×104 (Lumos); HCD-MS2 resolution, 1.5×104; HCD-MS2 collision energy, 40%; CID-MS2 collision energy, 30%; number of MS2 fragment ions collected for MS3, 3; MS3 isolation width, 2.5 m/z (Fusion), 2 m/z (Lumos); MS3 collision energy, 55%; MS3 resolution, 6×104 (Fusion), 5×104 (Lumos); MS3 AGC target, 1×105 (Fusion), 3×105 (Lumos). If multiple TMT sets were required to analyze all samples from an experiment two bridge channels pooled from all samples were used to connect the data from the individual TMT sets (Lapek et al., 2017).
Data were processed using an in-house developed software suite (Huttlin et al., 2010). MS2 data were annotated using the Sequest algorithm (Eng et al., 1994) to search the Uniprot database of human protein sequences (02/2014) including known contaminants such as trypsin, and a target-decoy database strategy was applied to measure false-discovery rates of peptide and protein identifications (Elias and Gygi, 2007). Searches were performed with a 50 ppm precursor mass tolerance; 10-plex TMT tags on lysine residues and peptide n-termini (+229.162932 Da) and carbamidomethylation of cysteines (+57.02146 Da) were set as static modifications and oxidation of methionine (+15.99492 Da) as a variable modification for proteome data searches and additionally phosphorylation of serine, threonine, and tyrosine (+79.966331 Da) as variable modification for phosphoproteome data searches. Based on the target-decoy database search strategy (Elias and Gygi, 2007) and employing linear discriminant analysis and posterior error histogram sorting, peptide and protein assignments were filtered to false discovery rate (FDR) of < 1% (Huttlin et al., 2010). Peptides that matched to more than one protein were assigned to that protein containing the largest number of matched redundant peptide sequences following the law of parsimony (Huttlin et al., 2010). The Ascore algorithm was used to evaluate the correct assignment of phosphorylation within the peptide sequence (Beausoleil et al., 2006). TMT reporter ion intensities were extracted from the MS3 spectra selecting the most intense ion within a 0.003 m/z window centered at the predicted m/z value for each reporter ion and spectra were used for quantification if the average signal-to-noise ratio per channel was 40 and 20 for proteomics and phosphoproteomics experiments, respectively, and the isolation specificity (Ting et al., 2011) for the precursor ion was ≥ 0.75. Protein intensities were calculated by summing the TMT reporter ions for all peptides assigned to a protein. Intensities were first normalized by the average intensity across all samples relative to the median average across all proteins (Lapek et al., 2017). In a second normalization step protein intensities measured for each sample were normalized by the average of the median protein intensities measured across the samples (Lapek et al., 2017).
Five proteomics experiments are described in the paper:
-
Quantitative proteomics of fully polarized macrophages after 4 days of treatment with IL4 or IFNγ/LPS to determine global proteome landscape of fully polarized THP-1-derived M1-type versus M2-type macrophages. In addition, the effects of the BET bromodomain inhibitor (+)-JQ1 on IL4-induced M2-type polarization was included (IL4 + JQ1).
Proteome mapping on Orbitrap Fusion (one TMT set)
RAW files: Marneros_4DayTreatment_proteome_fraction01 to …fraction12
8 samples
Labeling: PMA.1 (126), PMA.2 (127n), IL4.1 (127c), IL4.2 (128n), IL4+JQ1.1 (128c), IL4+JQ1.2 (129n), IFNgamma+LPS.1 (129c), IFNgamma+LPS.2 (130n)
-
Time-course quantitative proteomics during the first 24 hours after induction of macrophage polarization in THP-1-derived macrophages to identify protein changes associated with M1-type (IFNγ/LPS-induced) versus M2-type (IL-4-induced) polarization:
Proteome mapping on Orbitrap Fusion (three TMT sets: set01, set02, set03)
RAW files:
Marneros_time course_proteome_TMTset01_fraction01 to …fraction12
Marneros_time course_proteome_TMTset02_fraction01 to …fraction12
Marneros_time course_proteome_TMTset03_fraction01 to …fraction12
22 samples
Labeling:
set01; bridge 1 (126), PMA 0min (127n), PMA 10min (127c), PMA 30min (128n), PMA 1h (128c), PMA 2h (129n), PMA 4h (129c), PMA 8h (130n), PMA 24h (130c), bridge 2 (131n)
set02; bridge 1 (126), IL4 10min (127n), IL4 30min (127c), IL4 1h (128n), IL4 2h (128c), IL4 4h (129n), IL4 8h (129c), IL4 24h (130n), IFNgamma+LPS 10min (130c), bridge 2 (131n)
set03; bridge 1 (126), IFNgamma+LPS 30min (127n), IFNgamma+LPS 1h (127c), IFNgamma+LPS 2h (128n), IFNgamma+LPS 4h (128c), IFNgamma+LPS 8h (129n), IFNgamma+LPS 24h (129c), bridge 2 (131n)
-
Time-course quantitative phosphoproteomics during the first 24 hours after induction of macrophage polarization in THP-1-derived macrophages to identify phosphorylation events associated with M1-type (IFNγ/LPS-induced) versus M2-type (IL-4-induced) polarization (same time points as in group 2):
Phosphoproteome mapping on Orbitrap Fusion (three TMT sets: set01, set02, set03)
RAW files:
Marneros_time course_phospho_TMTset01_fraction01 to …fraction06, …fraction08 to …fraction24, …pY
Marneros_time course_phospho_TMTset02_fraction01 to …fraction12, …fraction16 to …fraction24, …pY
Marneros_time course_phospho_TMTset03_fraction01 to …fraction24, …pY
The following RAW files gave no phosphopeptide quantifications (they are not included in the group of provided RAW files): set01_fraction07, set02_fraction13, set02_fraction14, set02_fraction15.
22 samples
Labeling: as in (ii)
-
Quantitative proteomics to assess the effects of the B-Raf inhibitor GDC-0879 or of the MEK inhibitor trametinib on IL-4-induced M2-type polarization and IFNγ/LPS-induced M1-type polarization on THP-1-derived macrophages after 24 hours of treatment:
Proteome mapping on Orbitrap Fusion (one TMT set)
RAW files: Marneros_BRAF_inhibitor_proteine_fraction01 to …fraction12
11 samples
Labeling: IL4 1 (131c), IL4 2 (127n), IL4+GDC 1 (131n), IL4+GDC 2 (128n), IL4+TRAM 1 (130c), IL4+TRAM 2 (129n), IFNgamma+LPS 1 (130n), IFNgamma+LPS 2 (127c), IFNgamma+LPS+TRAM 1 (126), IFNgamma+LPS+TRAM 2(129c), IFNgamma+LPS+GDC (128c)
-
Quantitative phosphoproteomics to assess the effects of the B-Raf inhibitor GDC-0879 on IL-4-induced M2-type polarization and IFNgamma/LPS-induced M1-type polarization on THP-1-derived macrophages after 24 hours of treatment:
Phosphoproteome mapping on Orbitrap Fusion Lumos (one TMT set)
RAW files: Marneros_BRAF_inhibitor_phospho_fraction01 to …fraction24, …pY
7 samples
Labeling: IL4 1 (131c), IL4 2 (127n), IL4+GDC 1 (130n), IL4+GDC 2 (128n), IFNgamma+LPS 1 (128c), IFNgamma+LPS 2 (127c), IFNgamma+LPS+GDC (130c).
RNA-Seq experiments
THP-1 cells were primed with PMA (100 nM) for 24 hours and then panobinostat (0.0034 μM), trametinib (0.011 μM) or DMSO (vehicle) were added one hour prior to starting treatment with either BSA (control), IL-4 (20 ng/ml) or IFNγ (20 ng/ml)/LPS (10 ng/ml) for 24 hours. PBMCs were cultured as shown in the section of “Cell lines and reagents.” On day 6, PBMCs were treated with either BSA (control), IL-4 (20 ng/ml) or IFNγ (20 ng/ml)/LPS (10 ng/ml) in the presence of either panobinostat (0.0034 μM), trametinib (0.011 μM) or DMSO (vehicle) for 24 hours. All treatments were performed as replicates (n = 2/group) for both THP-1-derived and PBMC-derived macrophages. 2×106 cells per sample from 12 well dishes were harvested to extract RNA using mRNeasy Mini Kit (QIAGEN, No. 217004). We used 0.4 μg total RNA for RNA-Seq library preparation with the TruSeq RNA Library Prep Kit V2 (Illumina, RS-122–2001). Concentrations of RNA-Seq libraries were determined by a Bioanalyzer. RNA-Seq libraries were sequenced using a NextSeq 500/550 High Output V2.5 75 cycles kit from Illumina on a NextSeq 550 sequencer.
ChIP-Seq experiments
THP-1 cells and PBMCs were treated as described for the RNA-Seq experiments. We used 4×106 THP-1-derived macrophages and 8×106 PBMC-derived macrophages per sample from 6 well dishes for H3K27ac ChIP-Seq (n = 2/group). During cell harvesting, cell culture medium was removed and cells were treated with PBS+1% PFA (Amresco, M134) at RT for 15 minutes and then quenched with 0.125 M glycine at RT for 5 minutes. After washing 6 times with cold PBS, the nuclei were extracted by incubating cells in RLB buffer (50 mM HEPES pH7.9, 140 mM NaCl, 2 mM EDTA, 2 mM EGTA, 1% Triton X-100, and cOmplete ULTRA Protease Inhibitor Cocktail, Sigma 582988001) on ice for 1 hour. After washing with MNase buffer (50 mM HEPES pH 7.9, 140 mM NaCl, 1% Triton X-100, 1 mM CaCl2, 1 mM DTT), chromatins were digested with 66 units MNase (New England Biolabs, M0247S) at 37°C for 10 minutes and the reactions were stopped using EGTA (5 μM) on ice for 5 minutes. Chromatins were sonicated with a Branson Microtip Sonifier 450 (4×15 s at output 4.5 and duty cycle 60%). The sizes of the chromatins after these treatments were ~150–500 bp in length. For ChIP, we incubated the chromatins with 20 μl Dynabead protein G (Thermo Fisher Scientific, 10004D) and 1.5 μg anti-H3K27ac antibodies (Abcam Cat# ab4729, RRID:AB_2118291) in IP dilution buffer (20 mM Tris HCl pH8.0, 150 mM NaCl, 2 mM EDTA, 2 mM EGTA) at 4°C overnight. IPed chromatins were sequentially washed with IP dilution buffer+0.1% Triton, ChIP wash I (20 mM Tris HCl pH8.0, 150 mM NaCl, 0.5% Triton, 2 mM EDTA, 2 mM EGTA), ChIP wash II (20 mM Tris HCl pH8.0, 500 mM NaCl, 1% Triton, 2 mM EDTA, 2 mM EGTA), ChIP wash III (20 mM Tris HCl pH8.0, 250 mM LiCl, 1% Triton, 2 mM EDTA, 2 mM EGTA) and TE buffer. Chromatins were eluted using elution buffer (20 mM Tris HCl pH8.0, 5 mM EDTA, 5 mM EGTA, 1% SDS) at 65°C for 2 × 15 minutes. Chromatins were digested using RNase ONE Ribonuclease (Promega, PAM4261, 1:10 dilution) at 37°C for 1 hour and Pronase (Roche, 10165921001, 1.5 μg/μl) at 42°C for 2 hours and 65°C for 6 hours. Digested chromatins were purified using QIAGEN PCR purification kits. ChIP-Seq libraries were prepared using a standard protocol, including End-Repair, dA-Tailing, Single-Index Adaptor Ligation and final PCR Amplification (17 cycles) using Phusion DNA polymerase. ChIP-Seq libraries were purified from 2% agarose gels. Concentrations of ChIP-Seq libraries were determined using a Bioanalyzer. ChIP-Seq libraries were sequenced using a NextSeq 500/550 High Output V2.5 75 cycles kit from Illumina on a Nextseq 550 platform and all data were deposited to GEO. For normalization controls, 1% of the chromatin that was used for ChIP was used to make input libraries.
Experimental laser-induced CNV model and skin wound healing model
Experimental induction of CNV lesions and analysis was performed as previously described (He and Marneros, 2013; Strittmatter et al., 2016). Briefly, eyes of age- and gender-matched mice were exposed to laser photocoagulation for induction of experimental CNV after eyes had been dilated with 1% tropicamide. Laser photocoagulation was performed using a 532-nm laser (Visulas 532S; Carl Zeiss Meditec, Dublin, Ireland). Lesions were induced using a power of 120mW, a spot size of 100 μm, and a duration of 100ms. At the same time, four 6mm full thickness skin punch biopsies were induced on the back of the mice for skin wound healing analysis.
Eight-week-old male C57BL/6J mice (n = 10 mice per group) were treated with (1) trametinib at 1mg/kg daily per intraperitoneal injection, (2) with panobinostat at 20mg/kg daily per intraperitoneal injection, or with (3) DMSO (vehicle controls) on days 0, 1, 2, and 3 after laser injury. Eyes were harvested and fixed in 4% PFA 3 days after laser treatment. The size of CNV lesions was measured in choroidal flat mounts from these mice. The eyes were enucleated and fixed in 4% paraformaldehyde. The RPE/choroid tissue (posterior eye) was used for immunolabeling with anti-CD31 antibodies to detect blood vessels (rat anti-mouse CD31, BD Biosciences Cat# 550274, RRID:AB_393571; Alexa Fluor-488 secondary anti-rat antibody), SMA antibodies to detect myofibroblasts (mouse monoclonal SMA-Cy3 conjugate (clone 1A4), C6198, Sigma; RRID:AB_476856) and anti-Arg1 antibodies (goat anti-mouse arginase-1; sc-18354, Santa Cruz, 1:100; RRID:AB_2227469). In a subset of eyes co-labeling for F4/80 (rat anti-mouse F4/80 conjugated with Alexa647, BioLegend Cat# 123121, RRID:AB_893492), and Arg1 was performed. Choroidal flat mounts were analyzed by epifluorescence microscopy using a Zeiss microscope (Carl Zeiss Microscopy, Jena, Germany). Images were obtained with a 10x objective. CNV lesions were measured using Zeiss AxioVision software version 4.8.2. Average CNV size was determined for each mouse, and differences between treatment groups were assessed by a Student’s t test. For semiquantitative RT-PCR experiments 14 laser spots were induced in each eye and RPE/choroids were used for RNA isolation. An unpaired two-tailed Student’s t test was used for statistical analyses. P values < 0.05 were considered to be statistically significant.
QUANTIFICATION AND STATISTICAL ANALYSIS
Bioinformatics analysis of proteomic and phosphoproteomic data
To control for slight differences in the input protein amount and the efficiency of sample preparation, the protein levels in each sample were normalized to a median level of 1. Then, the relative level of each protein was divided by its mean level across all samples and these ratios (relative protein levels) displayed on a log2 scale. Proteins were clustered using hierarchical clustering with cosine distance as a similarity measure.
To identify biological functions with differential regulation across samples, we performed gene set enrichment analysis similar to Franks et al. (2017). We selected the subset of proteins associated with each GO term and used ANOVA analysis to compute the probability (p value) that these proteins are not changing across the samples (macrophage treatments). Thus, low p values correspond to biological functions changing across the macrophages treated with PMA, INFγ/LPS and IL-4. The p values were corrected for multiple hypotheses testing, and all reported results have a false discovery rate (FDR) < 1%. To quantify the magnitude of change, for each GO term we computed the mean of the relative levels of its associated proteins. The kinase enrichment analysis was performed analogously, except that instead of defining protein groups by GO terms, protein groups were defined based on a curated database of protein phospho-sites known to be phosphorylated by a particular kinase (Krug et al., 2019). Then, the activity of the kinase is displayed as the mean relative levels of the phosphosites that it phosphorylates. For increased specificity, each kinase was associated with specific phospho-sites rather than with a target protein, and the phospho-sites supporting the kinase activities reported in Figure 2A are shown in Table S2.
In a second set of analyses, to explore protein functions differentially activated during macrophage polarization, GSEA was applied on all proteins ranked according to their normalized intensity values (http://www.gsea-msigdb.org/gsea/login.jsp; version 4.0.2) (Subramanian et al., 2005). Two databases of gene functional sets, namely Gene Ontology (GO, c5.all.v7.1.symbols.gmt) and Kyoto Encyclopedia of Genes and Genomes (KEGG, c2.cp.kegg.v7.1.symbols.gmt), were selected from the Molecular Signature Database (MSigDB) (Liberzon et al., 2015). According to a permutation test, the biological functions with sufficiently low p value and FDR were identified when comparing M1 (INFγ/LPS+PMA) or M2 (IL-4+PMA) with M0 (PMA) at N min (10min, 30min, 1h, 2h, 4h, 8h, 24h, 4d). After identification of significant GO terms or KEGG pathways at each time point, to quantify the magnitude of functional change across time points, all significant pathways obtained from different time points (10min-4d) were combined into a single heatmap with hierarchical clustering. KEGG pathways with p value < 0.05 in at least one time point were considered significantly changed during macrophage polarization treated with INFγ/LPS or IL-4. Then, these significant pathways differentially expressed between INFγ/LPS and IL-4 treatments were visualized using R package pheatmap (version 1.0.12) based on their normalized enrichment scores (NES). Analysis of proteome data treated with GDC-0879 and Trametinib was performed analogously.
Correlation analyses matrices (Figure 5) were obtained with Graphpad Prism 9.2.0.
Western blot band quantifications were performed with GelQuantNET (http://biochemlabsolutions.com/GelQuantNET.html).
RNA-Seq bioinformatics analysis
Analyses of RNA-Seq data were conducted by using the framework of nf-core/rnaseq (https://github.com/nf-core/rnaseq) (Ewels et al., 2020). Based on parameters of the workflow version 1.3 used for RNA-Seq analysis, a total of 24 samples of THP-1-derived macrophages and 12 samples of primary human macrophages (PBMC-derived) with an average of 15.9 million raw reads in FastQ format were processed. In the nfcore/rnaseq pipeline, FastQC (Andrews, 2010) and TrimGalore (Krueger, 2015) were used to perform the removal of adaptor contamination and the trimming of low quality regions. MultiQC (Ewels et al., 2016) was adopted for sequence quality assessment. Over 98.5% of raw reads were identified as high-quality sequences for subsequent analyses. The retained reads were aligned against the iGenome Ensembl human reference genome GRCh37 (https://support.illumina.com/sequencing/sequencing_software/igenome.html) using STAR (Dobin et al., 2013). The average length of total reads aligned to the human genome is 14.47 million (91.4%), while the unaligned sequences were discarded. Next, the aligned reads were used to determine the count of reads for each gene by using the feature Counts (Liao et al., 2014). Additionally, the FPKM metrics for genes and transcripts as well as the transcript features were determined by StringTie (Pertea et al., 2015). Finally, the FPKM values of two replicates from the same experimental condition were combined into a single value for all genes. This analytical pipeline is available on the Nextflow platform (Di Tommaso et al., 2017). After determining gene expression values by the FPKM metric, comparisons of gene expression changes between control groups (DMSO condition) and experimental groups (panobinostat or trametinib conditions) were conducted to identify differentially expressed genes. In addition, an analysis was performed to assess M2-type polarization-associated genes whose expression was inhibited by trametinib and/or panobinostat. Expression changes for genes with a log2 ratio of DMSO_M2 over DMSO_M0 larger than one (log2 [DMSO_M2/DMSO_M0] > 1) were regarded as significantly upregulated in the M2-type group. To identify genes whose expression was inhibited by trametinib and/or panobinostat > 2-fold compared to DMSO groups, the log2 ratio of DMSO_M2 over Trametinib_M2 or Panobinostat_M2 > 1 were assessed.
Functional enrichment analysis: To explore gene functions significantly involved in macrophage polarization, gene set enrichment analysis (GSEA) was applied on all genes ranked by their FPKM values in the RNA-Seq data (http://www.gsea-msigdb.org/gsea/login.jsp; version 4.0.2) (Subramanian et al., 2005). Two databases of gene sets, namely Gene Ontology (GO, c5.all.v7.1.symbols.gmt) and Kyoto Encyclopedia of Genes and Genomes (KEGG, c2.cp.kegg.v7.1.symbols.gmt), were selected from the Molecular Signature Database (MSigDB) (Liberzon et al., 2015). In the MSigDB collection, the Array Annotations database (Human_ENSEMBL_Gene_ID_MSigDB.v7.0.chip) was employed to map Ensembl IDs to Gene Symbols for all genes. According to a permutation test, the biological functions with low p value and FDR were identified. To investigate biological functions enriched in a set of differentially expressed genes (DEGs) among the different experimental conditions, GO/KEGG enrichment analysis was performed by using Clusterprofile (Yu et al., 2012) (http://www.bioconductor.org/packages/release/bioc/html/clusterProfiler.html). GO/KEGG enrichment analysis with the p value < 0.05 was performed on these selected genes.
Principal component analysis (PCA) used transcripts that have 10 or more reads in at least 2 samples. The data for these genes were log2 transformed, normalized to mean zero across the conditions, and the vector for each condition (treatment) converted to z-scores. The right singular vectors of these matrices were computed as principal components of the data.
ChIP-Seq bioinformatics analysis, detection of enhancers and motif analyses
The nf-core/chipseq (https://github.com/nf-core/chipseq) is a bioinformatics pipeline specialized for ChIP-Seq data analysis. The pipeline is built upon Nextflow, which is a workflow management platform for arranging multiple tasks across various computational infrastructures in a portable manner. According to parameters of the version 1.0.0 of the nf-core/chipseq workflow, samples of experimental groups from either THP-1-derived macrophages or primary human macrophages (PBMC-derived) with an average of 25.2 million raw reads in FastQ format were processed. In the first stage of the ChIP-Seq workflow, after data preprocessing, which involves quality assessment and sequence alignment of raw reads to the genome, low-quality reads were eliminated using FastQC and TrimGalore software (Andrews, 2010; Krueger, 2015). The remaining high-quality reads were aligned against the iGenome Ensembl human reference genome GRCh37 by BWA and SAMtools (Li, 2011; Li, 2013; Li et al., 2009). Then, the obtained BAM files (alignment result) were converted into bigWig files for subsequent analyses. In the second stage of ChIP-Seq workflow, the normalized bigWig files were scaled to one million mapped reads through BEDTools (Quinlan and Hall, 2010) and bedGraphToBigWig (Kent et al., 2010). To identify detailed sequences captured by ChIP-Seq, we then used a peak calling method to explore areas of a genome that have been enriched with aligned reads, indicating the potential regulatory elements, such as enhancers, superenhancers, and transcription factor binding sites. MACS2 (Zhang et al., 2008) is a model-based method dedicated to identification of read-rich areas (peak calling) from ChIP-Seq data. In order to identify the peaks specifically occurring in an experimental condition, input control samples from both PBMC-derived and THP-1-derived macrophage experimental groups were included in the analysis for the detection of significant peaks in the various experimental groups. In comparison to the control samples, peaks with a q-value (minimum FDR) < 0.05 were defined as significant regions. Finally, HOMER (Heinz et al., 2010) was employed to link the selected peaks to genes and IGV (Robinson et al., 2017) or DNANexus software was used to visualize the distribution of peaks with their intensity values throughout the whole genome.
ROSE (rank ordering of superenhancers) (Lovén et al., 2013; Whyte et al., 2013) is a tool dedicated to finding enhancers and their downstream genes in ChIP-Seq BAM files and GFF files. A genome-wide occupancy analysis for the different macrophage states (M0_PMA, M1_INFγ/LPS and M2_IL4) with the various treatment conditions (DMSO, panobinostat or trametinib) was conducted utilizing the ChIP-Seq data. Genes located within the downstream regions (50kb) of the enhancers were annotated as the proximal related transcriptionally active genes, based on the definition of ROSE (Lovén et al., 2013).
To identify sequence motifs enriched in enhancers in macrophages, based on the enriched regions defined by MACS2 (Zhang et al., 2008), HOMER software (version 4.11.1) with the motif size ranging from 8 to 13 bps was adopted to scan across the entire genomic sequence. Without specifying a background sequence set in HOMER, the genomic regions with the same GC content were selected randomly as the background sequence set, which can be used to identify the over-represented motifs in enhancers.
Supplementary Material
Highlights.
Time-course quantitative proteomics/phosphoproteomics of polarizing macrophages
Distinct changes in cell metabolism and signaling in M1- and M2-type macrophages
Chemical screens identify pharmacologic M2-type macrophage polarization inhibitors
MEK induces PPARγ that promotes M2-type polarization by activating RA signaling
ACKNOWLEDGMENTS
This study was supported by funds from Massachusetts General Hospital and from the NIH (R21AI131044, R01DK118134, and R01DK121178) to A.G.M. W.H. was supported by NIH (NCI U01 CA215798). We would like to thank Drs. Jin Mo Park and Katia Georgopoulos for critical reading of the manuscript.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109955.
REFERENCES
- Andrews S (2010). FastQC: A quality control tool for high throughput sequence data (Babraham Bioinformatics). https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- Beausoleil SA, Villén J, Gerber SA, Rush J, and Gygi SP (2006). A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24, 1285–1292. [DOI] [PubMed] [Google Scholar]
- Bergeron JJ, Di Guglielmo GM, Dahan S, Dominguez M, and Posner BI (2016). Spatial and temporal regulation of receptor tyrosine kinase activation and intracellular signal transduction. Annu. Rev. Biochem. 85, 573–597. [DOI] [PubMed] [Google Scholar]
- Bouhlel MA, Derudas B, Rigamonti E, Dièvart R, Brozek J, Haulon S, Zawadzki C, Jude B, Torpier G, Marx N, et al. (2007). PPARγ activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 6, 137–143. [DOI] [PubMed] [Google Scholar]
- Bullard LE, Qi X, and Penn JS (2003). Role for extracellular signal-responsive kinase-1 and −2 in retinal angiogenesis. Invest. Ophthalmol. Vis. Sci. 44, 1722–1731. [DOI] [PubMed] [Google Scholar]
- Burns KA, and Vanden Heuvel JP (2007). Modulation of PPAR activity via phosphorylation. Biochim. Biophys. Acta 1771, 952–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla A (2010). Control of macrophage activation and function by PPARs. Circ. Res. 106, 1559–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RH, Juo PC, Curran T, and Blenis J (1996). Phosphorylation of c-Fos at the C-terminus enhances its transforming activity. Oncogene 12, 1493–1502. [PubMed] [Google Scholar]
- Chiba T, Skrypnyk NI, Skvarca LB, Penchev R, Zhang KX, Rochon ER, Fall JL, Paueksakon P, Yang H, Alford CE, et al. (2016). Retinoic acid signaling coordinates macrophage-dependent injury and repair after AKI. J. Am. Soc. Nephrol. 27, 495–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court M, Petre G, Atifi ME, and Millet A (2017). Proteomic signature reveals modulation of human macrophage polarization and functions under differing environmental oxygen conditions. Mol. Cell. Proteomics 16, 2153–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covarrubias AJ, Aksoylar HI, and Horng T (2015). Control of macrophage metabolism and activation by mTOR and Akt signaling. Semin. Immunol. 27, 286–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covarrubias AJ, Aksoylar HI, Yu J, Snyder NW, Worth AJ, Iyer SS, Wang J, Ben-Sahra I, Byles V, Polynne-Stapornkul T, et al. (2016). AktmTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. eLife 5, e11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel B, Nagy G, Horvath A, Czimmerer Z, Cuaranta-Monroy I, Poliska S, Hays TT, Sauer S, Francois-Deleuze J, and Nagy L (2018). The IL-4/STAT6/PPARγ signaling axis is driving the expansion of the RXR heterodimer cistrome, providing complex ligand responsiveness in macrophages. Nucleic Acids Res. 46, 4425–4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels CM, Kaplan PR, Bishof I, Bradfield C, Tucholski T, Nuccio AG, Manes NP, Katz S, Fraser IDC, and Nita-Lazar A (2020). Dynamic ADP-ribosylome, phosphoproteome, and interactome in LPS-activated macrophages. J. Proteome Res. 19, 3716–3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deroanne CF, Bonjean K, Servotte S, Devy L, Colige A, Clausse N, Blacher S, Verdin E, Foidart JM, Nusgens BV, and Castronovo V (2002). Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene 21, 427–436. [DOI] [PubMed] [Google Scholar]
- Di Tommaso P, Chatzou M, Floden EW, Barja PP, Palumbo E, and Notredame C (2017). Nextflow enables reproducible computational workflows. Nat. Biotechnol. 35, 316–319. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards A, and Haas W (2016). Multiplexed quantitative proteomics for high-throughput comprehensive proteome comparisons of human cell lines. Methods Mol. Biol. 1394, 1–13. [DOI] [PubMed] [Google Scholar]
- Elias JE, and Gygi SP (2007). Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214. [DOI] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, and Yates JR (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989. [DOI] [PubMed] [Google Scholar]
- Ewels P, Magnusson M, Lundin S, and Käller M (2016). MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewels PA, Peltzer A, Fillinger S, Alneberg J, Patel H, Wilm A, Garcia MU, Di Tommaso P, and Nahnsen S (2020). The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 38, 276–278. [DOI] [PubMed] [Google Scholar]
- Fontana MF, Baccarella A, Kellar D, Oniskey TK, Terinate P, Rosenberg SD, Huang EJ, Herbert DR, and Kim CC (2015a). Myeloid expression of the AP-1 transcription factor JUNB modulates outcomes of type 1 and type 2 parasitic infections. Parasite Immunol. 37, 470–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana MF, Baccarella A, Pancholi N, Pufall MA, Herbert DR, and Kim CC (2015b). JUNB is a key transcriptional modulator of macrophage activation. J. Immunol. 194, 177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks A, Airoldi E, and Slavov N (2017). Post-transcriptional regulation across human tissues. PLoS Comput. Biol. 13, e1005535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, Stender JD, Chun HB, Garner H, Geissmann F, and Glass CK (2014). Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 159, 1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyöngyösi A, Szatmari I, Pap A, Dezso B, Pos Z, Széles L, Varga T, and Nagy L (2013). RDH10, RALDH2, and CRABP2 are required components of PPARγ-directed ATRA synthesis and signaling in human dendritic cells. J. Lipid Res. 54, 2458–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, Robinson SC, and Balkwill FR (2008). “Re-educating” tumor-associated macrophages by targeting NF-kB. J. Exp. Med. 205, 1261–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanigan TW, Danes JM, Taha TY, Frasor J, and Petukhov PA (2018). Histone deacetylase inhibitor-based chromatin precipitation for identification of targeted genomic loci. J. Biol. Methods 5, e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJ, Stokoe D, Gloor SL, Vigers G, et al. (2010). RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 464, 431–435. [DOI] [PubMed] [Google Scholar]
- He L, and Marneros AG (2013). Macrophages are essential for the early wound healing response and the formation of a fibrovascular scar. Am. J. Pathol. 182, 2407–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, and Marneros AG (2014). Doxycycline inhibits polarization of macrophages to the proangiogenic M2-type and subsequent neovascularization. J. Biol. Chem. 289, 8019–8028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, Hussain J, Reis-Filho JS, Springer CJ, Pritchard C, and Marais R (2010). Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 140, 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller NM, Qi X, Junttila IS, Shirey KA, Vogel SN, Paul WE, and Keegan AD (2008). Type I IL-4Rs selectively activate IRS-2 to induce target gene expression in macrophages. Sci. Signal. 1, ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho VW, Hofs E, Elisia I, Lam V, Hsu BE, Lai J, Luk B, Samudio I, and Krystal G (2016). All trans retinoic acid, transforming growth factor β and prostaglandin E2 in mouse plasma synergize with basophil-secreted interleukin-4 to M2 polarize murine macrophages. PLoS ONE 11, e0168072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobson-Gutierrez SA, and Carmona-Fontaine C (2018). The metabolic axis of macrophage and immune cell polarization. Dis. Model. Mech. 11, dmm034462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeflich KP, Herter S, Tien J, Wong L, Berry L, Chan J, O’Brien C, Modrusan Z, Seshagiri S, Lackner M, et al. (2009). Antitumor efficacy of the novel RAF inhibitor GDC-0879 is predicted by BRAFV600E mutational status and sustained extracellular signal-regulated kinase/mitogen-activated protein kinase pathway suppression. Cancer Res. 69, 3042–3051. [DOI] [PubMed] [Google Scholar]
- Hu E, Kim JB, Sarraf P, and Spiegelman BM (1996). Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARgamma. Science 274, 2100–2103. [DOI] [PubMed] [Google Scholar]
- Huang JT, Welch JS, Ricote M, Binder CJ, Willson TM, Kelly C, Witztum JL, Funk CD, Conrad D, and Glass CK (1999). Interleukin-4-dependent production of PPAR-gamma ligands in macrophages by 12/15-lipoxygenase. Nature 400, 378–382. [DOI] [PubMed] [Google Scholar]
- Hughes R, Qian BZ, Rowan C, Muthana M, Keklikoglou I, Olson OC, Tazzyman S, Danson S, Addison C, Clemons M, et al. (2015). Perivascular M2 macrophages stimulate tumor relapse after chemotherapy. Cancer Res. 75, 3479–3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villén J, Haas W, Sowa ME, and Gygi SP (2010). A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Zweig AS, Barber G, Hinrichs AS, and Karolchik D (2010). BigWig and BigBed: Enabling browsing of large distributed datasets. Bioinformatics 26, 2204–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuzer J, Edwards A, and Haas W (2019). Multiplexed quantitative phosphoproteomics of cell line and tissue samples. Methods Enzymol. 626, 41–65. [DOI] [PubMed] [Google Scholar]
- Krueger F (2015). Trim galore: A wrapper tool around Cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files (Babraham Bioinformatics). https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/.
- Krug K, Mertins P, Zhang B, Hornbeck P, Raju R, Ahmad R, Szucs M, Mundt F, Forestier D, Jane-Valbuena J, et al. (2019). A curated resource for phosphosite-specific signature analysis. Mol. Cell. Proteomics 18, 576–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurgonaite K, Gandhi H, Kurth T, Pautot S, Schwille P, Weidemann T, and Bökel C (2015). Essential role of endocytosis for interleukin-4-receptormediated JAK/STAT signalling. J. Cell Sci. 128, 3781–3795. [DOI] [PubMed] [Google Scholar]
- Lad EM, Cousins SW, Van Arnam JS, and Proia AD (2015). Abundance of infiltrating CD163+ cells in the retina of postmortem eyes with dry and neovascular age-related macular degeneration. Graefes Arch. Clin. Exp. Ophthalmol. 253, 1941–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapek JD Jr., Greninger P, Morris R, Amzallag A, Pruteanu-Malinici I, Benes CH, and Haas W (2017). Detection of dysregulated protein-association networks by high-throughput proteomics predicts cancer vulnerabilities. Nat. Biotechnol. 35, 983–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, and Amit I (2014). Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159, 1312–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin Y, Kobayashi S, Leader A, Amir ED, Elefant N, Bigenwald C, Remark R, Sweeney R, Becker CD, Levine JH, et al. (2017). Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell 169, 750–765.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B, Wu CY, Lin YW, Park SW, and Wei LN (2016). Synergistic activation of Arg1 gene by retinoic acid and IL-4 involves chromatin remodeling for transcription initiation and elongation coupling. Nucleic Acids Res. 44, 7568–7579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv, 1303.3997v2. [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohman RJ, Iyer A, Fairlie TJ, Cotterell A, Gupta P, Reid RC, Vesey DA, Sweet MJ, and Fairlie DP (2016). Differential anti-inflammatory activity of HDAC inhibitors in human macrophages and rat arthritis. J. Pharmacol. Exp. Ther. 356, 387–396. [DOI] [PubMed] [Google Scholar]
- Long ME, Eddy WE, Gong KQ, Lovelace-Macon LL, McMahan RS, Charron J, Liles WC, and Manicone AM (2017). MEK1/2 inhibition promotes macrophage reparative properties. J. Immunol. 198, 862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, and Young RA (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons J, Brubaker DK, Ghazi PC, Baldwin KR, Edwards A, Boukhali M, Strasser SD, Suarez-Lopez L, Lin YJ, Yajnik V, et al. (2018). Integrated in vivo multiomics analysis identifies p21-activated kinase signaling as a driver of colitis. Sci. Signal 11, eaan3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, and Sica A (2010). Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Curr. Opin. Immunol. 22, 231–237. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Biswas SK, Galdiero MR, Sica A, and Locati M (2013). Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 229, 176–185. [DOI] [PubMed] [Google Scholar]
- Marneros AG (2013). NLRP3 inflammasome blockade inhibits VEGF-A-induced age-related macular degeneration. Cell Rep. 4, 945–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FO, and Gordon S (2014). The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 6, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FO, Gordon S, Locati M, and Mantovani A (2006). Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: New molecules and patterns of gene expression. J. Immunol. 177, 7303–7311. [DOI] [PubMed] [Google Scholar]
- Martinez FO, Helming L, Milde R, Varin A, Melgert BN, Draijer C, Thomas B, Fabbri M, Crawshaw A, Ho LP, et al. (2013). Genetic programs expressed in resting and IL-4 alternatively activated mouse and human macrophages: similarities and differences. Blood 121, e57–e69. [DOI] [PubMed] [Google Scholar]
- McAlister GC, Huttlin EL, Haas W, Ting L, Jedrychowski MP, Rogers JC, Kuhn K, Pike I, Grothe RA, Blethrow JD, and Gygi SP (2012). Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal. Chem. 84, 7469–7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, Nusinow DP, Jedrychowski MP, Wühr M, Huttlin EL, Erickson BK, Rad R, Haas W, and Gygi SP (2014). MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 86, 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer K, Weening D, de Vries MP, Priebe MG, Vonk RJ, and Roelofsen H (2015). Quantitative proteomics analyses of activation states of human THP-1 macrophages. J. Proteomics 128, 164–172. [DOI] [PubMed] [Google Scholar]
- Mohammadi A, Sharifi A, Pourpaknia R, Mohammadian S, and Sahebkar A (2018). Manipulating macrophage polarization and function using classical HDAC inhibitors: Implications for autoimmunity and inflammation. Crit. Rev. Oncol. Hematol. 128, 1–18. [DOI] [PubMed] [Google Scholar]
- Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, et al. (2014). Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 41, 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muta Y, Matsuda M, and Imajo M (2019). Divergent dynamics and functions of ERK MAP kinase signaling in development, homeostasis and cancer: Lessons from fluorescent bioimaging. Cancers (Basel) 11, 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson VL, Nguyen HCB, Garcìa-Cañaveras JC, Briggs ER, Ho WY, DiSpirito JR, Marinis JM, Hill DA, and Lazar MA (2018). PPARγ is a nexus controlling alternative activation of macrophages via glutamine metabolism. Genes Dev. 32, 1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Red Eagle A, Vats D, Brombacher F, Ferrante AW, and Chawla A (2007). Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature 447, 1116–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peart MJ, Smyth GK, van Laar RK, Bowtell DD, Richon VM, Marks PA, Holloway AJ, and Johnstone RW (2005). Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 102, 3697–3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelegrin P, and Surprenant A (2009). Dynamics of macrophage polarization reveal new mechanism to inhibit IL-1β release through pyrophosphates. EMBO J. 28, 2114–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pello OM, De Pizzol M, Mirolo M, Soucek L, Zammataro L, Amabile A, Doni A, Nebuloni M, Swigart LB, Evan GI, et al. (2012). Role of c-MYC in alternative activation of human macrophages and tumor-associated macrophage biology. Blood 119, 411–421. [DOI] [PubMed] [Google Scholar]
- Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT, and Salzberg SL (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, et al. (2013). CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 19, 1264–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richon VM, Sandhoff TW, Rifkind RA, and Marks PA (2000). Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc. Natl. Acad. Sci. USA 97, 10014–10019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Wenger AM, Zehir A, and Mesirov JP (2017). Variant review with the integrative genomics viewer. Cancer Res. 77, e31–e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodell CB, Arlauckas SP, Cuccarese MF, Garris CS, Li R, Ahmed MS, Kohler RH, Pittet MJ, and Weissleder R (2018). TLR7/8-agonistloaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat. Biomed. Eng. 2, 578–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K, Kumar C, Kéri G, Breitkopf SB, Oppermann FS, and Daub H (2010). Quantitative analysis of kinase-proximal signaling in lipopolysaccharide-induced innate immune response. J. Proteome Res. 9, 2539–2549. [DOI] [PubMed] [Google Scholar]
- Sica A, and Mantovani A (2012). Macrophage plasticity and polarization: In vivo veritas. J. Clin. Invest. 122, 787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieber J, Wieder N, Clark A, Reitberger M, Matan S, Schoenfelder J, Zhang J, Mandinova A, Bittker JA, Gutierrez J, et al. (2018). GDC-0879, a BRAFV600E inhibitor, protects kidney podocytes from death. Cell Chem. Biol. 25, 175–184.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoelund V, Smelkinson M, and Nita-Lazar A (2014). Phosphoproteome profiling of the macrophage response to different toll-like receptor ligands identifies differences in global phosphorylation dynamics. J. Proteome Res. 13, 5185–5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter K, Pomeroy H, and Marneros AG (2016). Targeting platelet-derived growth factor receptor β+ scaffold formation inhibits choroidal neovascularization. Am. J. Pathol. 186, 1890–1899. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szatmari I, Pap A, Rühl R, Ma JX, Illarionov PA, Besra GS, Rajnavolgyi E, Dezso B, and Nagy L (2006). PPARγ controls CD1d expression by turning on retinoic acid synthesis in developing human dendritic cells. J. Exp. Med. 203, 2351–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A, Schäfer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Johnstone R, Mohammed AK, and Hamon C (2003). Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 75, 1895–1904. [DOI] [PubMed] [Google Scholar]
- Ting L, Rad R, Gygi SP, and Haas W (2011). MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 8, 937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez G, Philippidou P, Rosenbaum J, Akmentin W, Shao Y, and Halegoua S (2007). Trk-signaling endosomes are generated by Rac-dependent macroendocytosis. Proc. Natl. Acad. Sci. USA 104, 12270–12275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lint C, Emiliani S, and Verdin E (1996). The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Expr. 5, 245–253. [PMC free article] [PubMed] [Google Scholar]
- Vellozo NS, Pereira-Marques ST, Cabral-Piccin MP, Filardy AA, Ribeiro-Gomes FL, Rigoni TS, DosReis GA, and Lopes MF (2017). All-trans retinoic acid promotes an M1- to M2-phenotype shift and inhibits macrophage-mediated immunity to Leishmania major. Front. Immunol. 8, 1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, and Tsatsanis C (2017). Akt signaling pathway in macrophage activation and M1/M2 polarization. J. Immunol. 198, 1006–1014. [DOI] [PubMed] [Google Scholar]
- Villaseñor R, Nonaka H, Del Conte-Zerial P, Kalaidzidis Y, and Zerial M (2015). Regulation of EGFR signal transduction by analogue-to-digital conversion in endosomes. eLife 4, e06156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T, and Castegna A (2019). The metabolic signature of macrophage responses. Front. Immunol. 10, 1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren KJ, Fang X, Gowda NM, Thompson JJ, and Heller NM (2016). The TORC1-activated proteins, p70S6K and GRB10, regulate IL-4 signaling and M2 macrophage polarization by modulating phosphorylation of insulin receptor substrate-2. J. Biol. Chem. 291, 24922–24930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintz G, Olsen JV, Frühauf K, Niedzielska M, Amit I, Jantsch J, Mages J, Frech C, Dölken L, Mann M, and Lang R (2010). The phosphoproteome of toll-like receptor-activated macrophages. Mol. Syst. Biol. 6, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiktorowicz JE, Chowdhury IH, Stafford S, Choudhuri S, Dey N, and Garg NJ (2019). Integrated functional analysis of the nuclear proteome of classically and alternatively activated macrophages. Mediators Inflamm. 2019, 3481430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA, and Vannella KM (2016). Macrophages in tissue repair, regeneration, and fibrosis. Immunity 44, 450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, De Nardo D, Gohel TD, Emde M, Schmidleithner L, et al. (2014). Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 40, 274–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Liu F, Tang M, Yuan M, Hu A, Zhan Z, Li Z, Li J, Ding X, and Lu L (2016). Macrophage polarization in experimental and clinical choroidal neovascularization. Sci. Rep 6, 30933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang L-G, Han Y, and He Q-Y (2012). clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandi S, Nakao S, Chun KH, Fiorina P, Sun D, Arita R, Zhao M, Kim E, Schueller O, Campbell S, et al. (2015). ROCK-isoform-specific polarization of macrophages associated with age-related macular degeneration. Cell Rep. 10, 1173–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu WH, MacIntyre A, and Nicosia RF (2002). Regulation of angiogenesis by vascular endothelial growth factor and angiopoietin-1 in the rat aorta model: distinct temporal patterns of intracellular signaling correlate with induction of angiogenic sprouting. Am. J. Pathol. 161, 823–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B, Buttrick T, Bassil R, Zhu C, Olah M, Wu C, Xiao S, Orent W, Elyaman W, and Khoury SJ (2013). IL-4 and retinoic acid synergistically induce regulatory dendritic cells expressing Aldh1a2. J. Immunol. 191, 3139–3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, Krishnan I, Maroni G, Meyerovitz CV, Kerwin CM, et al. (2019). Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity 50, 1317–1334.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All RNA-Seq and ChiP-Seq sequencing data were deposited on the National Cancer for Biotechnology Information Gene Expression Omnibus (GEO): GSE154347. All proteomics data were deposited into the MassIVE repository (MSV000084672). Accession numbers are also listed in the key resources table. All other data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
| Rabbit anti-MRC1 | Sigma Aldrich | HPA004114, RRID: AB_1846270 |
| Rabbit anti-CD68 antibody | Santa Cruz Biotechnology | SC-9139, RRID:AB_2275738 |
| Rabbit anti-F4/80 | Santa Cruz Biotechnology | SC-25830, RRID:AB_2246477 |
| Goat anti-Arg-1 | Santa Cruz Biotechnology | SC-18354, RRID:AB_2227469 |
| Rabbit anti-TGM2 | Cell Signaling Technologies | #3557, RRID:AB_2202883 |
| Rabbit anti-phospho-Stat1 (Y701) | Cell Signaling Technologies | #9167, RRID:AB_561284 |
| Rabbit anti-phospho-ERK1, Thr202/Tyr204 | Cell Signaling Technologies | #9101, RRID:AB_331646 |
| Rabbit anti-ERK1/2 | Cell Signaling Technologies | #4695, RRID:AB_390779 |
| Rabbit anti-β-actin | Lab Vision | Rb-9421-P1, RRID:AB_720056 |
| Mouse anti-β-actin | Sigma Aldrich | A5316, RRID:AB_476743 |
| Rabbit anti-PPARγ | Cell Signaling Technologies | # 2443, RRID:AB_823598 |
| Rabbit anti-phospho-STAT6 (Y641) | Cell Signaling Technologies | # 56554, RRID:AB_2799514 |
| Rabbit anti-β-tubulin | Cell Signaling Technologies | #2128, RRID:AB_823664 |
| Mouse anti-ALDH1A2 | Santa Cruz Biotechnology | sc-393204 |
| Rabbit IgG HRP Linked Whole Ab antibody | GE Healthcare | GENA934, RRID:AB_2722659 |
| Sheep Anti-Mouse IgG - Horseradish Peroxidase antibody | GE Healthcare | NA931, RRID:AB_772210 |
| Rabbit anti-H3K27ac | Abcam | ab4729, RRID:AB_2118291 |
| Rat anti-CD31 | BD Biosciences | # 550274, RRID:AB_393571 |
| Mouse anti-SMA-Cy3 conjugate (clone 1A4) | Sigma Aldrich | C6198, RRID:AB_476856 |
| Rat anti-F4/80 (conjugated with Alexa647) | BioLegend | # 123121, RRID:AB_893492 |
|
Chemicals, peptides, and recombinant proteins | ||
| PMA | Sigma Aldrich | P8139 |
| Murine M-CSF | Peprotech | #315–02 |
| Murine IL-4 | Peprotech | #214–14 |
| LPS | Sigma Aldrich | L4391 |
| Human M-CSF | Invitrogen | 14–8789-80 |
| Human IL-4 | Peprotech | #200–04 |
| Human IFNγ | Peprotech | #300–02 |
| MNase | New England Biolabs | M0247S |
| small molecule chemicals | Selleckchem or Cayman Chemicals | N/A |
|
Critical commercial assays | ||
| CellTiter-Glo luminescent cell viability assay (CTG) | Promega | G7573 |
| Steady-Glo Luciferase Assay System | Promega | E2520 |
| mRNeasy Mini Kit | QIAGEN | No. 217004 |
| TruSeq RNA Library Prep Kit V2 | Illumina | RS-122–2001 |
|
Deposited data | ||
| Proteomics | MassIVE repository | MSV000084672 |
| RNA-Seq and ChIP-Seq | GEO | GSE154347 |
|
Experimental models: cell lines and primary cells | ||
| THP-1 cell line | ATCC | Cat# TIB-202, RRID:CVCL_0006 |
| mouse BMDMs (primary cells) | JAX Labs | C57BL/6J mice |
| human PBMCs (primary cells) | iXCells Biotechnologies | 10HU-003CR100M |
| THP-1 cell line with MRC1 promoter-driven luciferase, 1.0k, clone#4 | He and Marneros, 2014 | N/A |
|
Experimental models: mice | ||
| C57BL/6J mice | JAX labs | JAX:000664 |
|
Oligonucleotides | ||
| semiquantitative RT-PCR primers | Life Technologies | N/A |
|
Software and algorithms | ||
| GelQuantNET, western blot band quantifications | Biochemlabsolutions | http://biochemlabsolutions.com/GelQuantNET.html |
| In-house developed software suite | Huttlin et al., 2010 | N/A |
| Sequest algorithm | Eng et al., 1994 | N/A |
| Ascore algorithm | Beausoleil et al., 2006 | N/A |
| GSEA | UCSD and Broad Institute | http://www.gsea-msigdb.org/gsea/login.jsp;jsessionid=CF17CD01EADE3916721F5DA4F6FEFFAA; version 4.0.2) |
| nf-core/rnaseq | Ewels et al., 2020 | https://github.com/nf-core/rnaseq |
| nf-core/chipseq | Ewels et al., 2020 | https://github.com/nf-core/chipseq |
|
Other | ||
| Chemical Libraries, 143 natural products | Selleckchem | Library L1400–01 and -02 |
| Chemical Libraries, 1836 bioactive compounds | Selleckchem | Library L1700–01 and -02 |
| Xvivo10 medium | Lonza | 04–380Q |
| Human serum AB plasma | Sigma | H4522 |