

Summary
The SARS-CoV-2 main protease of (Mpro) is an important target for SARS-CoV-2 related drug repurposing and development studies. Here, we describe the steps for structural characterization of SARS-CoV-2 Mpro, starting from plasmid preparation and protein purification. We detail the steps for crystallization using the sitting drop, microbatch (under oil) approach. Finally, we cover data collection and structure determination using serial femtosecond crystallography.
For complete details on the use and execution of this protocol, please refer to Durdagi et al. (2021).
Subject areas: Protein Biochemistry, Protein expression and purification, Structural Biology, X-ray Crystallography
Graphical abstract
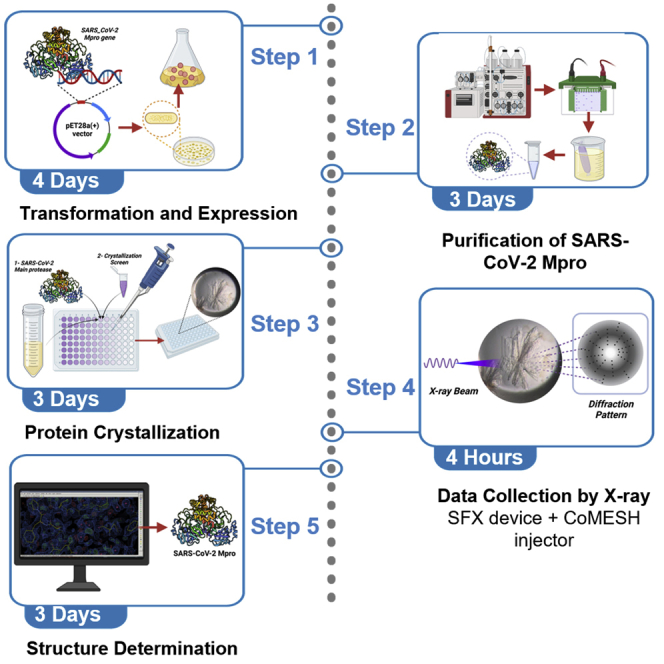
Highlights
-
•
Protocol for SARS-CoV-2 Main protease production and purification
-
•
Sitting drop, microbatch screening (under oil) approach for protein crystallization
-
•
Data collection and structure determination procedure for SARS-CoV-Mpro
The SARS-CoV-2 main protease of (Mpro) is an important target for SARS-CoV-2 related drug repurposing and development studies. Here, we describe the steps for structural characterization of SARS-CoV-2 Mpro, starting from plasmid preparation and protein purification. We detail the steps for crystallization using the sitting drop, microbatch (under oil) approach. Finally, we cover data collection and structure determination using serial femtosecond crystallography.
Before you begin
Severe acute respiratory syndrome coronavirus 2 main protease (SARS-CoV-2 Mpro) is a potential key drug target because of its essential role in this virus’ life cycle. Mpro mainly functions by processing the 1ab polyprotein. Inhibition of the Mpro activity with drugs is one of the main goals of the research community to tackle the COVID-19 pandemic (Zhang et al., 2020). In this protocol, we report the steps to obtain high-throughput SARS-CoV-2 Mpro structures with a Serial Femtosecond X-ray crystallography (SFX) technique performed at XFEL. This protocol describes step-by-step high-resolution structure determination of the native SARS-CoV-2 Mpro at ambient temperature.
Prepare the plasmids
Timing: 15 days
-
1.
Obtain the gene sequence from the UniProt database (UniProt ID: P0DTD1).
-
2.
Add a C-terminal stop codon.
-
3.
Escherichia coli (E. coli) is selected as the host expression organism.
-
4.
pET28a(+) (Addgene: Cat#69864-3) should be selected as the target vector. The plasmid encodes kanamycin resistance and 6× Histidine tag at N and C terminus which are cleavable with thrombin enzyme (Figure 1).
-
5.
Select NdeI and BamHI restriction cleavage sites at 5′ and 3′ ends.
Figure 1.

Plasmid map of Native Mpro construct and multiple cloning site of pET28a(+) vector
SnapGene Viewer (GSL, Biotech; available at snapgene.com).
Prepare the antibiotics and IPTG stocks
-
6.
Prepare kanamycin, chloramphenicol, and isopropyl-beta-d-thiogalactopyranoside (IPTG) stock solutions for E. coli BL21 (DE3) Rosetta™ 2 culture. Rosetta™ 2 contains a chloramphenicol-resistant plasmid to enhance protein expression by providing tRNAs for rare codons.
-
7.
Weigh 2.25 g of kanamycin and dissolve it in 45 mL EDI ultrapure water Stock concentration is 50 mg/mL. Store at −20°C.
-
8.
Weigh 1.575 g of chloramphenicol and dissolve it in 45 mL 100% ethanol. Stock concentration is 35 mg/mL. Store at −20°C.
-
9.
Weigh 4.20 g of IPTGand dissolve it in 45 mL of EDI ultrapure water. Stock concentration is 0.4 M. Store at −20°C.
Prepare LB agar plates
-
10.Prepare Luria-Bertani (LB) Agar plates for bacterial cell culture.
-
a.Weigh 10 g of LB Agar and dissolve it in 150 mL of EDI ultrapure water in a 500 mL volume bottle.
-
b.Add 100 mL more EDI ultrapure water to bring the final volume to 250 mL.
-
c.Close the lid loosely and autoclave it at 121°C for 20 min.
 CRITICAL: Never fill bottles more than ⅔ volume. Loosely closing the lid allows gas exchange and prevents pressure build-up that may lead to over-pressurization.
CRITICAL: Never fill bottles more than ⅔ volume. Loosely closing the lid allows gas exchange and prevents pressure build-up that may lead to over-pressurization. -
d.Take the bottle out of the autoclave and wait until it cools down to ∼60°C
-
e.Add 250 μL (1:1000) of chloramphenicol and kanamycin from stock solutions.CRITICAL: Antibiotics must be added after cooling down as they degrade at high temperatures.
-
f.Pour the agar mixture into the Petri dishes until 1:4 volume is filled by rotating the dish to spread agar equally under aseptic conditions inside the fume hood
-
g.Wait until the agar solidifies and cover the edges of the plate with parafilm.
-
h.Store it at 4°C upside down.CRITICAL: Keeping the agar plates upside down prevents humidity on the surface of the LB-Agar.
-
a.
Prepare LB Broth
-
11.
For small-scale protein production, weigh 10 g tryptone, 5 g yeast extract, and 10 g sodium chloride (NaCl) per liter and mix the powders well.
-
12.
Measure 500 mL EDI ultrapure water using a graduated cylinder.
-
13.
Add 25 g of LB medium to the graduated cylinder containing 500 mL EDI ultrapure water.
-
14.
Then, add EDI ultrapure water until the total volume reaches 1 L.
-
15.
Place the graduated cylinder on a magnetic stirrer and add a magnetic fish to mix the solution.
-
16.
Aliquot 100 mL of LB medium into 250 mL glass Erlenmeyer flasks.
-
17.
Cover the lids of the flasks with aluminum foil. Place autoclave tape on top of the foil.
-
18.
Sterilize LB media in an autoclave at liquid mode (121°C, 20 min).
-
19.
Store them in a 4°C fridge once the flasks cool down.
-
20.
For large-scale protein production, weigh 120 g tryptone, 60 g yeast extract, and 120 g NaCl and mix the powders well.
-
21.
Measure 12 L EDI ultrapure water by using a graduated cylinder.
-
22.
Add 12 L EDI ultrapure water to a demijohn.
-
23.
Add the mixed powder to 12 L EDI ultrapure water in the demijohn.
-
24.
Place the demijohn on a stirrer and add a magnetic fish to mix the solution.
-
25.
Aliquot 2 L LB medium into 2.8 L glass Erlenmeyer flasks.
-
26.
Cover the lids of the flask with aluminum foilPlace autoclave tape on top of the foil.
-
27.
Sterilize the LB media in an autoclave at liquid mode (121°C 20 min).
-
28.
Store them in a 4°C fridge once the flasks cool down.
Prepare the competent cells
-
29.
Take out a 250 mL Erlenmeyer flask containing 100 mL LB for E. coli BL21 (DE3) Rosetta™ 2 culture.
-
30.
Warm it to 22°C.
-
31.
Add 100 μL (1:1000) chloramphenicol to the LB medium.
-
32.
Inoculate LB medium with E. coli BL21 (DE3) Rosetta™ 2 strains by using the tip of the 1000 μL pipette under aseptic conditions inside the fume hood.
-
33.
Take a bit of E. coli BL21 (DE3) Rosetta™ 2 glycerol stock and inoculate it into LB medium.
-
34.
Incubate the flask in the incubator shaker at 37°C 110 rpm for 18 h.
-
35.
Take out the overnight cultures from the incubator shaker.
-
36.
Dilute the overnight cultures 1:50 in a fresh 150 mL LB containing 150 μL (1:1000) chloramphenicol.
-
37.
Place the fresh flask in the incubator shaker at 37°C 110 rpm until optical density (OD) reaches 0.5 at 600 nm (NanoDrop 2000c, Thermo Fisher Sci).
Note: The growth should take around 3 hours. Make sure to check OD600 every 30 min.
-
38.
When OD reaches 0.5, spin down diluted flask using a 50 mL centrifuge tube at 3500 rpm (2850 g) for 15 min at 4°C.
-
39.
Discard the supernatant and fill centrifuge bottles with the remaining cell culture.
-
40.
Continue to centrifuge remaining of the diluted cultures until the flasks are empty.
-
41.
Place the centrifuge tube containing the cell pellets on crushed ice.
-
42.
Prepare calcium chloride (100 mM) solution by using a graduated cylinder.
-
43.
Take 8 mL of calcium chloride (100 mM) and 32 mL of EDI ultrapure water.
-
44.
Add the mixture into a glass beaker with fish inside and mix it on the stirrer.
-
45.
Distribute 40 mL into each centrifuge tube, then gently vortex.
-
46.
Once the pellets are dissolved in the solution, place the tubes on ice.
-
47.
Incubate them on ice for 20 min.
-
48.
Prepare 25 mL 100 mM calcium chloride, 30% glycerol solution.
-
49.
After incubation, centrifuge the mixture at 4°C 3500 rpm (2850 g) for 5 min.
-
50.
Discard the supernatant.
-
51.
Dissolve the pellet in 10 mL of 100 mM calcium chloride, 30% glycerol.
-
52.
Gently vortex it.
-
53.
Aliquot 200 μL of competent cells and store them in a −80°C freezer.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, consumables and recombinant proteins, and other | ||
| Acrylamide/bisacrylamide, 30% Solution 29:1 | Sigma-Aldrich | Cat#A3574 |
| Acrylamide/bisacrylamide, 30% Solution 29:1 | Sigma-Aldrich | Cat#A3574 |
| APS | Sigma-Aldrich | Cat#A3678 |
| TEMED | Sigma-Aldrich | Cat#T9281 |
| Calcium Chloride | Sigma-Aldrich | Cat#C4901 |
| Acetic Acid | ISOLAB | Cat#901.013.1000 |
| HCl | ISOLAB | Cat#932.106.2501 |
| NaOH | ISOLAB | Cat#969.112.0500 |
| SDS | ISOLAB | Cat#915.02P.0051 |
| Beta Mercaptoethanol (BME) | Merck | Cat#8057400250 |
| SDS-PAGE casting system | Bio-Rad | Cat#1658015 |
| SDS-PAGE system | Bio-Rad | Cat#1658004 |
| Nanodrop 2000c | Thermo Scientific | Cat#ND2000C |
| Amicon Ultra 15 mL 10K centrifugal filter unit | Amicon | Cat#UFC901024 |
| Kimwipes | Kimberly-Clark | Cat#13258179 |
| Innova 44R | Eppendorf, New Brunswick | Cat#16524370 |
| Akta Go | Cytiva | Cat#29383015 |
| Regenerated Cellulose Dialysis tubing | Thermo Scientific | Cat#68100 |
| Tryptone | Sigma-Aldrich | T7293-1KG |
| Yeast Extract | Sigma-Aldrich | Y1625-1KG |
| IPTG | GOLDBIO | Cat#12481C100 |
| Chloramphenicol | GOLDBIO | Cat#C-105-100 |
| Kanamycin | GOLDBIO | Cat#K-120-100 |
| EDI ultrapure water | Milli-Q | Cat#ZR0Q008WW |
| NaCl | ISOLAB | Cat#969.036.0500 |
| Glycerol | ISOLAB | Cat#927.043.1000 |
| Dithiothreitol (DTT) | Roche | Cat#10708984001 |
| Tris | Sigma-Aldrich | Cat#93362 |
| Triton X-100 | Sigma-Aldrich | Cat#T9284 |
| Ni-NTA Agarose resin | QIAGEN | Cat#30250 |
| PreScission | GenScript | Cat#Z02799 |
| Terasaki Plate | Greiner Bio-One | Cat#654 102 |
| Pact Premier | Molecular Dimensions | Cat#MD1-29 |
| Pure Cotton | Ipek | N/A |
| Bacterial and virus strains | ||
| Rosetta™ 2(DE3) | Sigma-Aldrich | Cat#71400-M |
| Deposited data | ||
| Monoclinic structure of Mpro at ambient temperature | Durdagi et al. (2021) | PDB ID:7CWB |
| Recombinant DNA | ||
| pET28a(+) Construct | GenScript | |
| Software and algorithms | ||
| PSOCAKE | Damiani et al. (2016) | https://github.com/lcls-psana/psocake |
| CHEETAH | Barty et al. (2014) | https://github.com/biochem-fan/cheetah |
| CrystFEL | White et al. (2012) | https://www.desy.de/∼twhite/crystfel/ |
| CCP4 | Winn et al. (2011) | https://www.ccp4.ac.uk/ |
| SnapGene | GSL Biotech | https://www.snapgene.com |
| Structure Refinement: PHENIX | Adams et al. (2010) | http://www.phenix-online.org/ |
| Structure Modeling: COOT | Emsley and Cowtan (2004) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| PyMOL | Schrödinger, LLC | https://pymol.org/2/ |
| Maestro molecular modeling program | Madhavi Sastry et al. (2013) | https://www.schrodinger.com/products/maestro |
| PROPKA | Bas et al. (2008) | https://pypi.org/project/propka/ |
| R (Bio3D package) | Grant et al. (2006) and Yao and Grant (2013) | https://www.r-project.org/ |
Materials and equipment
Separating gel (4× 15% gels)
| Reagent | Final concentration | Amount |
|---|---|---|
| EDI ultrapure water | 4.4 mL | |
| 30% Acrylamide/0.8% bisacrylamide | 15% acrylamide/0.4% bisacrylamide | 10 mL |
| 1.5 M Tris-HCl (pH:8.8) | 390 mM | 5.2 mL |
| 10% SDS | 0.1% | 200 μL |
| 10% APS | 0.05% | 100 μL |
| TEMED | 0.5% | 100 μL |
| Total | 20 mL |
Store at 4°C
Stacking gel (4× 15% gels)
| Reagent | Final concentration | Amount |
|---|---|---|
| EDI ultrapure water | 5.86 mL | |
| 30% Acrylamide/0.8% bisacrylamide | 4.02% acrylamide/0.11% bisacrylamide | 1.34 mL |
| 0.5 M Tris-HCl (pH:6.8) | 130 mM | 2.6 mL |
| 10% SDS | 0.1% | 100 μL |
| 10% APS | 0.1% | 100 μL |
| TEMED | 0.1% | 10 μL |
| Total | 10 mL |
Store at 4°C
SDS-PAGE running buffer (10×)
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris base | 120 mM | 30.30 g |
| Glycine | 1.92 M | 144.10 g |
| SDS | 1% | 10 g |
| EDI ultrapure water | Up to 1 L | |
| Total | 1 L |
Store at RT
5× sample loading buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl pH:6.8 | 250 mM | 2.5 mL |
| SDS | 10% | 1 g |
| Glycerol | 30% | 3 mL (from 100%) |
| Bromophenol Blue | 0.25% | 5 mg |
| BME | 5% | 500 μL |
| EDI ultrapure water | Up to 10 mL | |
| Total | 16 mL |
Store at −20°C
Coomassie brilliant blue (CBB) staining solution
| Reagent | Final concentration | Amount |
|---|---|---|
| CBB R-250 | 0.1% | 10 g |
| Methanol | 50% | 500 mL |
| Acetic acid | 10% | 100 mL |
| EDI ultrapure water | 400 mL | |
| Total | 1 L |
Store at RT
Lysis buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| NaCl | 300 mM | 17.53 g |
| Tris | 50 mM | 6.06 g |
| Glycerol | 5% | 5 mL |
| Triton X-100 | 0.5% | 5 mL |
| EDI ultrapure water | Fill to 1 L | |
| Total | 1 L |
Adjust pH to 7.5 and Store at 4°C
Buffer A
| Reagent | Final concentration | Amount |
|---|---|---|
| NaCl | 150 mM | 8.77 g |
| Tris | 20 mM | 2.42 g |
| Imidazole | 5 mM | 0.34 g |
| EDI ultrapure water | Fill to 1 L | |
| Total | 1 L |
Adjust pH to 7.5 and Store at 4°C
Buffer B
| Reagent | Final concentration | Amount |
|---|---|---|
| NaCl | 150 mM | 8.77 g |
| Tris | 20 mM | 2.42 g |
| Imidazole | 500 mM | 34.03 g |
| EDI ultrapure water | Fill to 1 L | |
| Total | 1 L |
Adjust pH to 7.5 and Store at 4°C
Step-by-step method details
Transformation and expression of SARS-CoV-2 Mpro
Transform pET28a(+) plasmid, which contains target gene, to the E. coli BL21 (DE3) Rosetta™ 2 competent cells.
-
1.Resuspension of plasmid
-
a.Take a sterile 50 mL falcon tube and fill it with EDI ultrapure water.
-
b.Take the commercial plasmid tube.
-
c.Spin lyophilized plasmid down at 5000 rpm (2650 g) for 2 min.
-
d.Add 25 μL EDI ultrapure water directly on lyophilized plasmid (bottom of the tube).
-
e.Vortex it for 10 s.
-
f.Spin it down at 5000 rpm (2650 g) for 2 min.
-
g.Repeat steps c. and d. three times.
-
a.
-
2.
Take Rosetta™ 2 competent cells from −80°C and place them on ice.
-
3.
Take 50 μL of Rosetta™ 2 competent cells into a sterile 1.5 mL Eppendorf tube once the competent cells are fully dissolved.
-
4.
Thaw both the component cells and the plasmid on ice for 20 min.
-
5.
Add 2 μL of the plasmid into the competent cells.
-
6.
Mix the plasmid gently through the bacterial solution by pipetting.
-
7.
Incubate the cells for 20 min more on ice.
-
8.
After incubation, give them a heat shock at 42°C for 45 s.
-
9.
Put the cells back on ice for 2 min.
-
10.
Add 500 μL of room temperature (22°C) LB medium into the Eppendorf tube which contains transformed competent cells.
-
11.
Incubate the culture tube in an incubator at 37°C for 90 min.
-
12.
After incubation, centrifuge the tube for 2 min at 1500 rpm (238 g) at 22°C.
-
13.
Discard most of the supernatant by inverting the tube once and dissolve the pellet with the leftover supernatant.
-
14.
Light the Bunsen burner.
-
15.
Sterilize the spreader with alcohol (70% ethanol) and burn the tip of the spreader. Or use a commercial sterile spreader.
-
16.
Spread 50 μL of cell suspension by the sterilized spreader into kanamycin (50 μg/mL) and chloramphenicol (35 μg/mL)-containing agar plate that is preheated to 37°C.
-
17.
Put the plate upside down into a 37°C incubator.
-
18.
Incubate it for 18 h.
-
19.
Take out 150 mL LB from 4°C after 18 h incubation.
-
20.
Warm it at 22°C.
-
21.
Add kanamycin and chloramphenicol antibiotics to 150 mL LB medium in a 1:1000 ratio.
-
22.
Inoculate the cells into LB medium by making several streaks in different directions with the help of a pipette tip.
-
23.
Incubate the starter culture in the incubator shaker at 37°C 110 rpm for 18 h (Figure 2).
-
24.
Add 2 mL (1:1000) kanamycin and 2 mL (1:1000) chloramphenicol antibiotics to 6 × 2 L LB media prepared beforehand.
-
25.
Take out the starter culture from the incubator shaker.
-
26.
Split the starter culture into 2 L glass Erlenmeyer flasks containing LB equally.
-
27.
Incubate the 2 L flasks in the incubator shaker at 37°C 110 rpm until the growth OD reaches 0.8–1.2 at 600 nm.
Note: The growth should take almost 3 h. Make sure to check the optical density value every 30 min.
-
28.
The temperature of the incubator shaker is then lowered to 16°C to cool the cultures.
-
29.
Add 2 mL (1:1000) 0.4 M isopropyl-beta-d-thiogalactopyranoside (IPTG) to each flask once the cultures cool down.
-
30.
Induce the cultures for 72 h at 16°C and 110 rpm.
-
31.
After 3 days of induction, transfer the cultures into 750 mL Thermo Bottles.
-
32.
Harvest the cells by centrifugation in an Allegra X-15R Centrifuge at 4°C (precooled) 3500 rpm (2850 g) for 45 min.
-
33.
After centrifugation, discard the supernatant.
-
34.
Place four of the centrifuge tubes on crushed ice.
-
35.
Collect pellets with the help of a sterilized spoon.
-
36.
Use the back of the spoon for gently transferring the pellets into 4 × 50 mL centrifuge tubes
-
37.
Add 25 mL of supernatant to each flask (when the pellet becomes fluid and cannot be collected by spoon) with a serological pipette to dissolve the remaining pellets.
-
38.
Transfer dissolved pellets to the same falcon tubes which contain pellets.
-
39.
Collect the remaining dissolved pellets and supernatants until the centrifuge tubes are completely empty.
-
40.
Spin down 50 mL centrifuge tubes containing bacterial pellets.
-
41.
Discard the remaining supernatant.
-
42.
Store the bacterial pellets in the −80°C freezer.
Pause point: The cell pellets can be stored at −80°C until the next step is planned and started.
Figure 2.

Workflow of expression and purification steps
Purification of SARS-CoV-2 Mpro
Purify Mpro protein for crystallization screening; steps 43–71 describe extract preparation; steps 69–77 details nickel affinity purification and steps 74–97 describe protease cleavage and dialysis.
-
43.
Take out the samples from −80°C, place them on ice.
-
44.
Turn on the Beckman Optima™ XL-80K Ultracentrifuge ultracentrifuge beforehand.
-
45.
Ultracentrifuge is set to 4°C from the precool option after placing the Type 45 Ti Fixed-Angle Titanium Rotor inside.
-
46.
Attach the horn-type Titanium tip to the sonicator.
-
47.
Clean the sonicator tip with a paper towel sprayed with ethanol.
-
48.
Add 40 mL Lysis buffer to each sample tube right before sonication.
-
49.
Set the sonication time to 45 s.
-
50.
Set the sonication amplitude to 60%.
-
51.
Sonicate the cell lysate with the cycle above.
-
52.
Place the next tube in the sonicator.
-
53.
Apply step 49–51 for each tube.
-
54.
Repeat step 51 × 4 cycles (or one more cycle after pellet is dissolved).
-
55.
Clean the sonicator with ethanol and a paper towel.
-
56.
Transfer the dissolved samples to empty ultracentrifuge tubes.
-
57.
Balance the centrifuge tubes using the precision scale.
-
58.
Clean the balance and the bench with paper towels, and turn off the balance.
-
59.
Close the ultracentrifuge tube lids well, ensure that the plastic caps of the ultracentrifuge tube lids are properly placed.
-
60.
Dry the centrifuge tubes and inside of the rotor with Kimwipes and place the samples inside the rotor according to their balance.
-
61.
Close the rotor lid well and clean the outside of the rotor. Make sure to double-check if the O–rings fit well.
-
62.
Close the lid of the equipment and press the VACUUM button.
-
63.Set up the centrifuge as below.
-
a.Speed: 40,000 rpm (186,000 g)
-
b.Time: 45 min
-
c.Temperature: 4°C
-
d.The rotor type.
-
a.
-
64.
Start the centrifuge.
-
65.
Release the vacuum after centrifugation is completed.
-
66.
Take the centrifuge tubes from the rotor and transfer supernatants to a sterilized glass bottle that is placed in an ice box.
-
67.
Power off the centrifuge after cleaning with a paper towel.
-
68.Prepare AKTA GO FPLC system for Nickel affinity purification.
-
a.Turn on the AKTA GO FPLC protein purification system.
-
b.Clean all the tubes with EDI ultrapure water to discard the remains of the previous run.
-
c.Place the Nickel affinity column to the column port of AKTA GO FPLC.CRITICAL: The column should be packed with 2 mL Ni-agarose beads for each liter of bacterial culture. We expressed 12 L culture and 24 mL resin used for the Nickel affinity column.
-
d.Wash the column with 3× column volume water.
-
e.Wash the column with 2× column volume buffer B.
-
f.Equilibrate the column with 3× column volume buffer A (running buffer).
-
a.
-
69.
Load the sample to the column with 2.5 mL per minute speed.
-
70.
Take a 40 μL sample from the load and flowthrough and mix with 10 μL loading dye for SDS-PAGE gel electrophoresis.
-
71.
Wash the column with 5× column volume of buffer A after loading of the sample is completed.
-
72.
Elute the sample with 2× column volume of buffer B.
-
73.
Take a 40 μL sample from the elution and mix with 10 μL loading dye. Then, check all samples with SDS-PAGE gel electrophoresis.
-
74.
Cut an appropriate length of dialysis membrane and leave it in 22°C water for 10 min to soften.
-
75.
Close one end of the dialysis membrane with a clip and put the elution sample from the previous step inside.
-
76.
Take a 40 μL elution sample and mix with 10 μL loading dye for SDS-PAGE gel electrophoresis.
-
77.
Add 1 μL (2 units) of PreScission Protease for each 100 μg of the fusion protein in the eluate.
-
78.
Add DTT (final concentration of 1 mM).
-
79.
Close the other end of the dialysis tube with a clip and check if there is any leakage to prevent protein loss.
-
80.
Fill a 2 L beaker with 1.5 L of buffer A with 1 mM DTT.
-
81.
Put the dialysis tube in it and stir the buffer with a magnetic fish in the beaker at 4°C for 5 h on a magnetic stirrer.
-
82.
Change the dialysis buffer after 5 h with a fresh 1.5 L buffer A with 1 mM DTT.
-
83.
Stir the buffer in the beaker slowly at 4°C for 18 h.
-
84.
Transfer the protein solution in the dialysis membrane to a centrifuge tube the next morning.
-
85.
Spin it down for 20 min at 3500 rpm (2850 g).
-
86.
Take a 40 μL sample from the supernatant and pellet and mix with 10 μL loading dye for SDS-PAGE gel electrophoresis.
-
87.Prepare AKTA GO for reverse Nickel affinity purification.
-
a.Turn on AKTA GO protein purification system.
-
b.Clean all the tubes that are placed in an EDI ultrapure water bottle to flush the capillaries to discard the remains of the previous run.
-
c.Place the Nickel affinity column to the column port of AKTA GO.
-
d.Wash the column with 3 × column volume water.
-
e.Wash the column with 2 × column volume buffer B.
-
f.Equilibrate the column with 3 × column volume buffer A (running buffer).
-
a.
-
88.
Load sample to the column with 0.5 mL per minute speed. Troubleshooting 1
-
89.
Collect all flowthrough as a fraction.
-
90.
Take a 40 μL sample from load and flowthrough and mix with 10 μL loading dye for SDS-PAGE gel electrophoresis.
Note: The recommended well-loading volume for Bio-Rad SDS-PAGE gels is 15 uL The extra amount of sample taken includes a backup sample in case of a problem with the gel (i.e. power outage)
-
91.
After loading of the sample is completed, wash the column with 5× column volume buffer A.
-
92.
Elute the uncut protein and Histag from the column with 2× column volume buffer B.
-
93.
Take a 40 μL sample from elution and mix with 10 μL loading dye. Then, check all samples with SDS-PAGE gel electrophoresis. Troubleshooting 2
-
94.
Check the purity of the protein with SDS-PAGE electrophoresis, concentrate protein with a 15 mL 10K cut-off centrifugal concentrator to 10 mg per mL, and divide it into 1 mL fractions. Troubleshooting 3
-
95.
The protein concentration is measured by UV absorption spectroscopy using NanoDrop. The required extinction coefficient value and the molecular weight of the protein are obtained from ProtParam tool (ExPASy) and used to calculate the concentration.
-
96.
Store the protein aliquots at −80°C after flash freezing with liquid nitrogen.
Crystallization of SARS-CoV-2 Mpro
Crystallize Mpro for structural data collection.
-
97.
The crystallization screening is performed by using commercially available crystallization screen kits.
Note: The protocol for each screen can be reached at https://kuybim.ku.edu.tr/wp-content/uploads/2021/12/Crystallization-screens.pdf
-
98.
Sitting drop, microbatch screening (under oil), performed by mixing the protein and crystallization screens at a 1:1 ratio, is used for crystallization.
Note: Sitting drop, microbatch screening (under oil) is a cheap and robust method. However, as an alternative method for crystallization, hanging drop vapor diffusion can be applied instead of sitting drop for proper crystal growth without a direct contact with the edges of well and easy crystal harvest.
-
99.
Thaw the protein samples, which are stored at −80°C, on ice.
-
100.Prepare all required materials and tools while the protein samples are thawing (Figure 3).
-
a.Two electronic micropipettes must be ready for use.
-
i.Set one of them to 0.83 μL (12 repeats) for protein sample loading (Figure 4.1).
-
ii.Set the other one to 16.6 μL (12 repeats) for paraffin oil loading (Figure 4.2).
-
iii.Set a 0.1–2.5 μL micropipette to 0.83 μL for crystallization screen loading.
-
i.
-
b.Label 72-well Terasaki plate (15 μL volume) with protein name, date, crystallization screen name, and initials of the researcher’s name performing the experiment (Figure 4).
-
c.Fill a 50 mL falcon tube with paraffin oil and put it into a proper rack.
-
d.Take the crystallization screen sets (3 boxes, each including 48 different screening conditions).
-
a.
-
101.
Open the lid of the Terasaki plate.
-
102.
Use the electronic micropipette set to 0.83 μL, start loading the protein samples of 25 mg/mL concentration into the first two columns (12 wells) in the Terasaki plate (Figure 4.1).
-
103.
Load and mix 0.83 μL of each crystallization condition by pipetting gently in a well containing 0.83 μL protein sample (Figure 4.2). Repeat this step until each of the 12 protein samples in the wells has been mixed with one condition.
-
104.
Cover these 12 wells with paraffin oil using the electronic micropipette set to 16.6 μL to minimize evaporation (Figure 4.3).
-
105.
Repeat steps 105–107 until all wells in the Terasaki plate are loaded.
-
106.
Cover the plate with the lid and keep it at 22°C or 4°C room/fridge.
-
107.
Observe the plate under a light microscope after 1 week, 2 weeks, and 1 month of incubation to check for protein crystal formation. Troubleshooting 4
-
108.
The SARS-CoV-2 Mpro crystals are obtained in Pact PremierTM crystallization screen 1 condition #39, 100 mM MMT buffer pH 6.0 and 25% w/v PEG 1500 [MMT buffer; DL-Malic acid, 4-Morpholine Ethane Sulfonic acid (MES) monohydrate, 2-Amino-2-(hydroxymethyl)-1,3- propanediol (TRIS)-HCl) from Molecular Dimensions, UK.
-
109.
Bundle sword-shaped crystals are obtained after 2 days.
-
110.Replicate the conditions in which protein crystals are formed for large-scale crystal production. Paraffin oil is not used in large-scale crystal production.
-
a.Mix 50 μL protein sample with 50 μL crystallization condition in an Eppendorf tube.
-
b.Wait 2–3 days for crystal formation.
-
c.Crystal seeds, obtained from step a by vortexing, were added to the 100 μL protein reservoir mixture solution.
-
d.After seeding and scaling crystals to 100 μL volume, this seeded solution is used to further inoculate 1:1 1 mL protein reservoir mixture solution.
-
e.Wait 2–3 days for crystal formation.
-
f.Mix 10 mL protein sample with 10 mL crystallization condition in a falcon tube.
-
g.Similarly, the 1 mL seeded crystal solution is used to inoculate the 10 mL protein solution.
-
a.
-
111.
The crystals are grown on a large-scale within 4 days.
-
112.
Cover the 50 mL centrifuge tube containing the crystals with three layers of organic cotton Figure 5.1, 5.2.
-
113.
Place the centrifuge tube in a zip lock bag.
-
114.
Cover the centrifuge bag with cotton Figure 5.3.
-
115.
Transfer the bag into a round plastic container Figure 5 4.
-
116.
Place the plastic container into a styrofoam box Figure 5.5.
-
117.
Fill the empty spaces with cotton Figure 5.6, 5.7.
Figure 3.

Crystallization setup includes electronic pipettes and tips, terasaki plate, protein sample on ice, crystallization conditions, paraffin oil and tip box
Figure 4.

Crystallization steps of SARS-CoV-2 Mpro
4.1. Loading protein samples. 4.2. Addition of crystallization screen conditions. 4.3. Covering protein-condition mixture with paraffin oil.
Figure 5.

Packaging steps for transport of crystal samples to LCLS
Ship the styrofoam box in a UN3373 Biological Substance Category B cardboard box Figure 5.8, 5.9.
Data collection and processing of SARS-CoV-2 Mpro
Collect and process the SFX data from Mpro crystals for structure determination (Figure 6).
-
118.
Load the 1.6 mL sample reservoir with Mpro crystals in the unaltered mother liquor.
-
119.
Use Microfluidic Electrokinetic Sample Holder (MESH) (Sierra et al., 2012, 2016) to inject the samples. The measure of the long-fused silica sample capillary should be 200 μm ID × 360 μm OD × 1.0 m. Troubleshooting 5
-
120.
The voltage should be 2500–3000 V and the counter electrode should be grounded.
-
121.
Adjust sample flow rate between 2.5 and 8 μL per minute.
-
122.
SFX experiments are conducted at the ambient temperature at the SLAC National Accelerator Laboratory (Menlo Park, CA)
-
123.
Focus the LCLS X-ray beam with a vertically polarized pulse 30 femtosecond (fs) duration with refractive beryllium compound lenses.
-
124.
The beam size should be ∼6 × 6 μm full width half maximum (FWHM).
-
125.
The pulse energy and photon energy should be 0.8 mJ and 9.8 keV (1.25 Å) respectively.
-
126.
The repetition rate should be 120 Hz.
-
127.Before data collection, Psocake (www.github.com/lcls-psana/psocake) was used to determine the initial diffraction geometry of the ePix10k 2-megapixel detector (ePix10k2M). For data processing, Cheetah was used. Crystal hit rates were monitored using OM. Troubleshooting 7Note: Access the detailed visual steps of the Psocake software here:https://kuybim.ku.edu.tr/wp-content/uploads/2021/12/Psocake_SOP.pdf. A live version of the document can be found via: https://docs.google.com/document/d/180L8uuemtm9LjN_xU96APJrnSyRs4Dxe4HAmPhWRdyU/edit
-
a.Data collection:
-
i.To perform detector readout calibration into the physically meaningful layout by assembling multi-panel detector tiles (asics).
-
ii.Subtract the photon background through radial averaging/median kernel.
-
iii.Obtain the interactive histogram including Analog to Digital Units (ADUs) in region-of-interest (ROI).
-
iv.Do detector translation through lab coordinates (x, y, z).
-
i.
-
b.Data processing:
-
i.Use the peakfinder8 algorithm in Cheetah to determine the hit finding parameters. Consider a minimum pixel count above 2 and adc-threshold of 500 with a minimum signal to noise ratio of 7 as a peak. Classify an image with a minimum of 20 peaks as a crystal hit.
-
ii.Generate the small data providing each event including hits, resolution.
-
iii.The hits were calibrated (detector gain threshold applied) and converted to HDF5 file format using Cheetah, including entries for the image data, instrument data and event codes.
-
iv.Observable hot pixels and bad detector areas (such as the panel edges) were masked out during peak finding by applying a mask in HDF5 format (consisting of zeros and ones, which gets multiplied with the image data).CRITICAL: Accurate peak positions are required for correct indexing and reflection intensity determination of the diffraction pattern, hence it is critical to complete this step meticulously.
-
v.Optimize the detector geometry by updating the mask as well as optimizing the mapping of the detector panels to the lab frame. The latter can be done using geoptimiser, which is included in the CrystFEL package up to version 0. 9.1. Troubleshooting 8
-
vi.Do CrystFEL indexing of the diffraction patterns applying the correct unit cell and Bravais lattice type once these have been successfully determined.CRITICAL: Once a correct indexing solution has been found, the predicted Bragg reflection positions can be displayed overlaid onto the image. For an indexing solution to be correct, it is important for any observed peaks to align with these predictions.CRITICAL: For accurate intensity determination of the individual reflections, the integration rings should be adjusted for optimal local background subtraction. In detail, the narrowest integration radius should be set to the size of the broadest peak observed and the middle and widest integration radii set an annulus between which the local background contribution to the respective reflection is estimated and subsequently subtracted.
-
vii.Obtain CrystFEL stream file that contains a list of intensity values for every reflection predicted by the indexing solution in every indexable pattern.
-
viii.Reiterate through the detector geometry optimization, adjusting the panel positions relative to the beam ensuring the most accurate indexing results.CRITICAL: Detector geometry correction is exceedingly important for SFX experiments. Normally, the same geometry files from the previous experiment are used. However, detector geometry optimization is inevitable when the interaction point or the detector motor position has changed.
-
i.
-
a.
-
128.Index the hits by using CrystFEL version 0.9.0. CrystFEL (Github Repository: https://github.com/taw10/crystfel) is a free and open-source software package to process the data from serial crystallography (SX) experiments characterized by XFEL and with other X-ray sources. It processes the data in which “snapshot” diffraction patterns are provided in a “serial” manner. Troubleshooting 9Note: Access the detailed visual steps of the CrystFEL software here:
https://kuybim.ku.edu.tr/wp-content/uploads/2021/12/CrystFEL_SOP.pdf
A live version of the document can be found via: https://drive.google.com/drive/u/1/folders/1B8FmsopNLJzTKmIO6NCE3vksMLz5hreHCRITICAL: It can be easily operated by scientists who know crystallography techniques. For tutorial (https://www.desy.de/∼twhite/crystfel/tutorial.html) and technical details (https://journals.iucr.org/d/issues/2019/02/00/ba5291/#BB40) can be reviewed in order.-
a.If the peak locations determined with CrystFEL are not satisfactory, peakfinding of the HDF5 files obtained with Cheetah can be re-done in CrystFEL using the same peakfinder8 algorithm
-
i.Likewise, consider a minimum pixel count above 2 and adc-threshold of 500 with a minimum signal to noise ratio of 7 as a peak.
-
i.
-
b.Determine the unit cell parameters and generate unit cell file using XGANDALF, DirAx, MOSFLM, and XDS in this order via indexamajig.CRITICAL: Use cell axis tolerances of 5 Å and angle tolerances of 5° (set this from the tolerance option in CrystFEL) to index the data after finding an approximate cell.
-
c.After finding the approximate cell and optimizing the detector geometry, HDF5 data is re-indexed through the indexamajig algorithm.
-
d.Set the integration radii to 2,3,5 and switch the multi-option to index multiple crystal lattices in a single image.
-
e.If an "indexing ambiguity" imposed by the serial nature of data collection is present for the particular point group (reference) it can be resolved using the ambigator algorithm, and the stream file is produced by re-indexing.
-
f.Integrate and merge the indexed reflections by using partialator (or process_hkl) a core program of CrystFEL and get .hkl, .hkl1, .hkl2 files.
-
i.“Merged data (.hkl)” provides the calculation of I/σ, completeness etc. via check_hkl algorithm.
-
ii.“Merged half data set A (.hkl1)” and “Merged half data set B (.hkl2)” provide the calculation of Rsplit, CC∗ etc. via compare_hkl algorithm.
-
iii.The previous two steps provide data collection statistics that are used to determine the resolutions cut-offs for the data, so as to maximize signal without incorporating an abundance of noise.
-
i.
-
g.Apply the unity model over 3 iterations and set the max-ADU to 7500.
-
h.Export .mtz file from “Merged data (.hkl)” file via create_mtz algorithm using CCP4's F2MTZ algorithm.
-
i.Scale and cut the reflection intensity list from CrystFEL by using the TRUNCATE software. Refer to the Wilson plot as well as the aforementioned data collection statistics to determine the ideal high resolution cut-off.
-
a.
Figure 6.

Protein crystallization procedures consist of protein crystallization, data collection and structure determination
Structure determination of SARS-CoV-2 Mpro
Calculate the crystal structure of Mpro protein.
-
a.
Once a list of scaled and merged structure factor amplitudes has been obtained using CrystFEL and associated software as described above, export the SFX mtz file (White et al., 2012) (Methods video S1).
-
129.Scan the existing structures in the database (Protein Data Bank) using the SWISS-MODEL (Waterhouse et al., 2018) server and find the most suitable “model structure”.
-
a.The SWISS-MODEL web server can be found here: https://swissmodel.expasy.org/
-
b.Select ‘Start Modeling’.
-
c.Add the FASTA sequence of your protein into the textbox.
-
d.Select ‘Search for Templates’ and wait until a list of template results are available.
-
e.Select an appropriate model based on coverage of sequence and Identity (Note: normally top selection may be sufficient).
-
f.Select the file icon
 next to ‘Coordinates PDB Format’.
next to ‘Coordinates PDB Format’.
-
a.
Note: If the structure is already known, the structure may be searched for on PDB and downloaded by selecting Download File -> PDB format
-
130.
Determine the structure of the crystal form with known space group, using the automated molecular replacement program PHASER (McCoy et al., 2007) implemented in PHENIX (Adams et al., 2010) by following the below command in “Terminal” of macOS or Linux:
phenix.automr name.pdb Scaled_name.mtz mass = 30000 resolution=4.0 component_type = protein RMS=0.7 copies=1 space_group=C121 unit_cell = "114 53.5 45 90 102 90" build=False nproc=7 decision_making.use_all_plausible_sg = True
Note:name.pdb: model structure; name.mtz: relevant structure’s mtz reflection. All parameters can be changed according to the data of the relevant structure
Note: Do not forget to open the required file with this .pdb and .mtz files in Terminal by command of “% cd”.
Note: The PDB file obtained through molecular replacement and the .mtz file obtained through data collection is used to achieve the best possible fit.
Note: Output files will be deposited in an generated folder named ‘AutoMR_run_1’. Resulting structure and map files will by default be named MR.1.mtz and MR.1.pdb
-
131.Review all residues in the relevant structure one by one in Coot (Emsley and Cowtan, 2004) software.
-
a.Open Coot and select File -> Open Coordinates… before selecting the pdb generated in the Molecular Replacement step (i.e., MR.1.pdb).
-
b.Open Coot and select File -> Auto Open MTZ… before selecting the mtz generated in the Molecular Replacement step (i.e., MR.1.mtz).
-
c.Select ‘Go To Atom…’ (or press F6) in the top action bar, and press okay to center on residue 1 of chain A.
-
d.Select ‘Real Space Refine Zone’ (or press ‘r’ on the keyboard) to locally refine the centered atom.
-
e.Press ‘space’ on the keyboard to shift to the next residue in the chain.
-
f.Repeat steps d and e for every residue in the protein; watching for appropriate values for Bonds, Angles, Planes, Chirals, Non-bonded Interactions, and the Ramachandran Plot. An indicator next to these values will display green for acceptable, yellow and orange for moderately acceptable, and red for inacceptable.
-
g.Select Validate -> Open Ramachandran Plot -> name.pdb to view a ramachandran plot. Red dots are outliers and may be selected for further inspection. These can be minimized through iterative refinement of the residue and surrounding residues.
-
h.After the coot refinement is finished, click File -> Save Coordinates to save the resultant structure.
-
a.
-
132.
Use the model structure's coordinates for initial rigid-body refinement executing the phenix.refinecommand with the parameters given below using this new PDB file and the .mtz file in “Terminal” of macOS or Linux:
phenix.refine MR.pdb MR.mtz strategy = rigid_body + individual_sites + individual_adp + occupancies + tls tls.params write_eff_file = false write_geo_file = false wxc_scale = 0.2 main.number_of_macro_cycles = 3 ramachandran_restraints = True nproc = 7 refinement.input.xray_data.r_free_flags.generate=True
Note:MR.pdb: molecular replacement pdb file; MR.mtz: molecular replacement mtz file. All parameters can be changed according to the data of the relevant structure
Note: Do not forget to open the required file with this .pdb and .mtz files in Terminal by command of “% cd”.
-
133.
Repeat steps 133–136 to iteratively improve your structure. Ideally, an appropriate structure has a minimum number of ramachandran outliers with r-free and r-work values less than 2, if not then less than 3. Troubleshooting 10
-
134.
When you are satisfied with your structure, open the structure and map in coot again, Check the individual water molecules in the Coot program and retain positions with strong differential density. Those which do not, can be deleted by selecting ‘Delete Item…’ in the right action bar, selecting ‘Water’ in the pop-up, and clicking on the desired water molecule. Troubleshooting 11
-
135.
Strong empty differential densities can be filled with water by selecting ‘Place Atom At Pointer…’ in the right action bar, selecting ‘Water’ in the pop-up, and selecting ‘OK’.
-
136.Upload the final PDBx/mmCIF format to the PDB database.
-
a.Before submission, convert the coordinate file to mmCIF format using this link: https://pdb-extract.wwpdb.org/
-
b.After receiving the mmCIF file, start the PDB submission from the submission page: https://deposit-pdbj.wwpdb.org/deposition/new
-
c.After getting an email about the ID and password, enter the ID and password, and click to start deposition.
-
d.Make sure having all the required files:
-
i..pdb file
-
ii..mtz file
-
iii..log file
-
iv..map file
-
v..lp file
-
i
-
e.Upload the .mmCIF file to the system.
-
f.Complete the required elements specified in the requirement charts list.
-
a.
Expected outcomes
We expect to obtain 200 mg/mL of pure protein from 12 L bacterial culture. Regular LB medium and 3–4 days of induction time improves growth. After conducting reverse Nickel affinity purification, dialysis and concentration, we obtain 80 mg/mL protein. Using the commercial crystallization conditions with crystal seeding would yield more reliable results.After SARS-CoV-2 Mpro data is processed and visualized, the final aim will be achieved. Accordingly, detailed information about the interaction between Mpro and inhibitors can be obtained by examining the alternative ligand-binding pocket conformations in detail via Molecular Dynamics (MD) simulations, docking and drug repurposing studies (Figure 7).
Figure 7.

Overview of SARS-CoV-2 Main Protease crystal structure Cartoon and surface representation of Mpro in space group C121
After this study, SARS-CoV-2 Mpro crystal structure was obtained (PDB ID: 7CWB).
Limitations
After obtaining the crystals, sending them to another country for data collection poses the possibility of damaging the crystals.
Troubleshooting
Problem 1
High pressure when loading the protein into the column.
Potential solution
This problem may be due to the sample not centrifuging for enough time or because it contains too much target protein. To prevent this, the sample can be passed through a 0.22 um filter before it is loaded on the column, or the sample can be divided into 2 fractions, reducing the total load of the column, and the purification can be conducted in 2 sections.
Problem 2
Majority of the protein is not cut by PreScission Protease.
Potential solution
The concentration of the restriction enzyme and the incubation time can be increased. Since commercial PreScission Protease is very expensive, it can be produced with a cleavable His-tag to overcome enzyme availability problems and eliminate contamination after cleavage as the histidine tags will bind to the column with the cleaved tag during reverse Ni-NTA column chromatography.
Problem 3
Protein impurity is not overcome with single-step Ni-NTA column chromatography.
Potential solution
Reverse-nickel affinity chromatography after PreScission Protease cleavage is performed to get rid of non-specific bindings. Size exclusion chromatography can also be used.
Problem 4
Proteins do not form crystals.
Potential solution
Crystallization of proteins can be performed at different temperatures and humidity, or the waiting time required for crystallization can be extended.
Problem 5
The injector is clogged.
Potential solution
Changing the injector and using a micro-capillary can solve the problem.
Problem 6
The hatch door gets stuck and does not close.
Potential solution
Call the responsible department to fix the door.
Problem 7
Bragg spots are not found during peak finding/hit finding on Psocake.
Potential solution
The intensity and position of the pixel can be displayed and examined by hovering the mouse over the corresponding pixel.
Problem 8
When the interaction point or the detector motor position has changed, detector geometry optimization is necessary.
Potential solution
If you have silver behenate data, find the region-of-interest widget that is shown a green ring which is the new detector center. With a mouse, this widget can be moved or resized around x, y. Adjust the detector distance (z) in the Diffraction Geometry panel until resolution rings overlap silver behenate ring positions. Thus, the newly determined detector position is ready to be deployed in the calibration folder. In CrystFEL, indexed Bragg peaks can be used to optimize the beam center using the detector-shift script.
Problem 9
There can be assorted reasons for indexing failure including detector center offset, wrong detector distance or numerous false peaks found.
Potential solution
A convenient way is to be able to get feedback and tweak each parameter if indexing has succeeded or not.
Problem 10
PDB structure does not have appropriate r-free/r-work values or is otherwise unsatisfactory after repeated refinement.
Potential solution
Many tools exist for refinement.Further information can be sought in the COOT, PHENIX and other websites through google searches or at their respective websites.
COOT: https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/
PHENİX: https://phenix-online.org/
R-free/r-work values greater than 6 suggests total randomness. High R values indicate that MPRO models do not fit well into your map. You could therefore consider why radical changes would occur in your map - either poor data or protein denaturation.
Problem 11
The PDB structure has too many Ramachandran outliers.
Potential solution
A few Ramachandran outliers may be expected as part of enzymatic function. If you do not believe this to be the case, further rounds of refinement may be necessary to reduce the number of outliers.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, [Hasan DeMirci] (hdemirci@ku.edu.tr).
Materials availability
Any unique reagents/materials used in this study are available from the lead contact with a completed Materials Transfer Agreement.
Acknowledgments
The authors gratefully acknowledge use of the services and facilities of the Koç University IsBank Infectious Disease Center (KUIS-CID). HD acknowledges support from National Science Foundation (NSF) Science and Technology Centers grant NSF-1231306 (Biology with X-ray Lasers, BioXFEL). This publication has been produced benefiting from the 2232 International Fellowship for Outstanding Researchers Program of TUBİTAK (Project No: 118C270), 2244 Industrial PhD program of TUBITAK (Project No: 119C132), and 1001 TUBITAK Supporting Scientific and Technological Research Projects Program (Project No: 120Z520). However, the entire responsibility of the publication belongs to the owner of the publication. The financial support received from TUBİTAK does not mean that the content of the publication is approved in a scientific sense by TUBİTAK. Use of the Linac Coherent Light Source (LCLS), SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. C.D. acknowledges support from TUBİTAK (Project No: 120Z594). The authors gratefully acknowledge use of the services and facilities of the Koç University Research Center for Translational Medicine (KUTTAM), funded by the Presidency of Turkey, Presidency of Strategy and Budget. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Presidency of Strategy and Budget. Portions of this research were carried out at Stanford Synchrotron Light Source (SSRL) at the SLAC National Accelerator Laboratory. SSRL is supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences (OBES) under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (NIGMS) (including P41GM103393).
Author contributions
Conceptualization, H.D. and C.D.; Methodology and writing, C.D., H.D., O.G., F.B.E., B.C., E.A., B.Y., M.G., O.G., C.B., G.U., and J.A.J.; Supervision, H.D. and C.D.; Data collection, Z.S., F.P., C.Y., E.H.D., C.K., B.H., M.L., M.S.H., A.B.,R.G.S., G.K., and S.B.; Funding acquisition, H.D.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2022.101158.
Data and code availability
The 3D electron density map of SARS-CoV-2 Main protease has been deposited in the Protein DataBank under accession numbers 7CWB. Any additional information required to reanalyze the data reported is available from the lead contact upon request.
References
- Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barty A., Kirian R.A., Maia F.R.N.C., Hantke M., Yoon C.H., White T.A., Chapman H. Cheetah: software for high-throughput reduction and analysis of serial femtosecond X-ray diffraction data. J. Appl. Crystallogr. 2014;47:1118–1131. doi: 10.1107/S1600576714007626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bas D.C., Rogers D.M., Jensen J.H. Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins Struct. Funct. Bioinformatics. 2008;73:765–783. doi: 10.1002/prot.22102. [DOI] [PubMed] [Google Scholar]
- Damiani D., Dubrovin M., Gaponenko I., Kroeger W., Lane T.J., Mitra A., O’Grady C.P., Salnikov A., Sanchez-Gonzalez A., Schneider D., et al. Linac coherent light source data analysis using psana. J. Appl.Crystallogr. 2016;49:672–679. [Google Scholar]
- Durdagi S., Dag C., Dogan B., Yigin M., Avsar T., Buyukdag C., DeMirci H. Near-physiological-temperature serial femtosecond X-ray crystallography reveals novel conformations of SARS-CoV-2 main protease active site for improved drug repurposing. Structure. 2021;29:1–15. doi: 10.1016/j.str.2021.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. Section D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Grant B.J., Rodrigues A.P.C., ElSawy K.M., McCammon J.A., Caves L.S.D. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics. 2006;22:2695–2696. doi: 10.1093/bioinformatics/btl461. [DOI] [PubMed] [Google Scholar]
- Madhavi Sastry G., Adzhigirey M., Day T., Annabhimoju R., Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra R.G., Gati C., Laksmono H., Dao E.H., Gul S., Fuller F., Kern J., Chatterjee R., Ibrahim M., Brewster A.S., et al. Concentric-flow electrokinetic injector enables serial crystallography of ribosome and photosystem II. Nat. Methods. 2016;13:59–62. doi: 10.1038/nmeth.3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra R.G., Laksmono H., Kern J., Tran R., Hattne J., Alonso-Mori R., Lassalle-Kaiser B., Glöckner C., Hellmich J., Schafer D.W., et al. Nanoflow electrospinning serial femtosecond crystallography. Acta Crystallogr. Section D Biol. Crystallogr. 2012;68:1584–1587. doi: 10.1107/S0907444912038152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F.T., de Beer T.A.P., Rempfer C., Bordoli L., et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White T.A., Kirian R.A., Martin A.V., Aquila A., Nass K., Barty A., Chapman H.N. CrystFEL: a software suite for snapshot serial crystallography. J. Appl. Crystallogr. 2012;45:335–341. [Google Scholar]
- Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G.W., McCoy A., et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X.-Q., Grant B.J. Domain-opening and dynamic coupling in the α-subunit of heterotrimeric G proteins. Biophys. J. 2013;105:L08–L10. doi: 10.1016/j.bpj.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The 3D electron density map of SARS-CoV-2 Main protease has been deposited in the Protein DataBank under accession numbers 7CWB. Any additional information required to reanalyze the data reported is available from the lead contact upon request.