

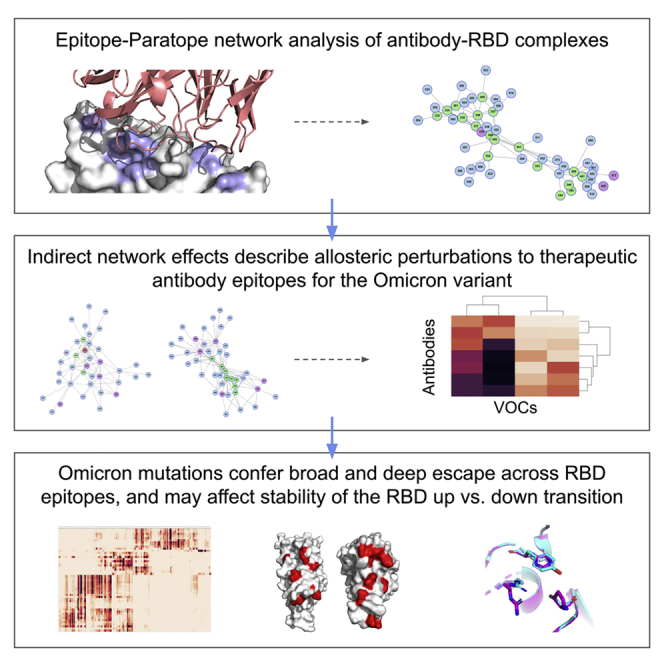
Summary
The Omicron variant features enhanced transmissibility and antibody escape. Here, we describe the Omicron receptor-binding domain (RBD) mutational landscape using amino acid interaction (AAI) networks, which are well suited for interrogating constellations of mutations that function in an epistatic manner. Using AAI, we map Omicron mutations directly and indirectly driving increased escape breadth and depth in class 1–4 antibody epitopes. Further, we present epitope networks for authorized therapeutic antibodies and assess perturbations to each antibody’s epitope. Since our initial modeling following the identification of Omicron, these predictions have been realized by experimental findings of Omicron neutralization escape from therapeutic antibodies ADG20, AZD8895, and AZD1061. Importantly, the AAI predicted escape resulting from indirect epitope perturbations was not captured by previous sequence or point mutation analyses. Finally, for several Omicron RBD mutations, we find evidence for a plausible role in enhanced transmissibility via disruption of RBD-down conformational stability at the RBDdown-RBDdown interface.
Keywords: SARS-CoV-2, variant of concern, Omicron, mutation, antigenic drift, antibody escape, network analysis, epistasis, RBD interface stability, epitope-paratope
Graphical abstract
Highlights
-
•
Network analyses offer distinct insights into RBD-neutralizing Ab interactions
-
•
Omicron mutations broadly and deeply perturb networks across RBD epitope classes
-
•
Networks capture indirect effects of Omicron mutations on Ab escape potential
-
•
Omicron mutations provide plausible structural rationale for enhanced transmission
The Omicron variant possesses 15 RBD mutations that together produce enhanced functions as compared with these mutations in isolation. Here, Miller et al. present a protein network analysis of the mutational landscape of the Omicron RBD. Their analysis can guide investigations of future variants and design of variant-proof therapeutic antibodies.
Introduction
The severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) Omicron (B.1.1.529) variant of concern (VOC) has spread globally due to an apparent fitness advantage over the Delta variant.1 Several of Omicron’s spike mutations have been observed in other VOCs and are known to enhance transmissibility and confer varying degrees of escape from neutralizing antibodies.2, 3, 4, 5, 6 However, numerous Omicron mutations have not been observed on previous VOCs nor characterized rigorously in terms of their functional effects. The position of many uncharacterized Omicron mutations within dominant antibody epitopes therefore prompted concerns that the efficacy of vaccines and therapeutic antibodies could be significantly reduced against Omicron, leading to policy decisions and research prioritizations with far-reaching consequences.
In this study, we analyze the receptor-binding domain (RBD) mutational landscape of Omicron using amino acid interaction (AAI) networks.7,8 AAI network analysis is particularly well suited for understanding the impact of constellations of mutations residing within and adjacent to an antibody epitope as occurs on the Omicron variant. For example, AAI network analysis considers how mutation of a residue that does not directly interact with a given antibody paratope (e.g., via a hydrogen bond between side chain and antibody complementarity determining region [CDR]) may still perturb antibody binding if the residue plays a significant structural role in supporting other sites that interact directly with the antibody.8,9 Such indirect effects have been conceptualized as mutations outside of the direct antibody epitope that alter higher-order protein structure or perturb protein “breathing.”10, 11, 12 AAI networks quantitate these indirect relationships through which Omicron’s mutation constellation may substantially perturb the chemical and physical properties of RBD epitope surfaces. We apply the AAI network lens to map the potential impacts of Omicron RBD mutations on polyclonal antibody responses and therapeutic monoclonal antibodies. We further discuss the limitations in predicting efficacy of therapeutic antibodies against emerging variants based on existing in vitro data for isolated mutations. Finally, we present possible functional roles for Omicron RBD mutations that are not predicted to substantially enhance antibody evasion. Our analysis using a fixed-backbone Omicron homology model and a recently released Omicron spike structure suggests Omicron mutations may modulate RBD-up versus RBD-down conformational dynamics toward enhanced infectivity.
Results
The impact of Omicron mutations on polyclonal antibody evasion
Toward investigating the antigenic impact of the Omicron RBD mutations, we first mapped direct and indirect effects of Omicron mutations on RBD antibody epitopes using AAI networks. Our RBD epitope map included at least 10 antibodies from each of the four structural classes of anti-RBD antibodies13 and thus represents the dominant functional components of population-level polyclonal antibody responses.14 First examining Omicron escape breadth, we found that Omicron mutations occur at RBD sites that interact with all antibodies examined and span the four antibody classes (Figure 1). In contrast, our analysis showed that the RBD mutations of the Beta and Delta variants are confined to sites within class 1 and 2 antibody epitopes, except for Beta N501Y, which interacts indirectly with certain class 3 antibodies. This is consistent with experimental evidence documenting Beta escape from class 1 and 2 antibodies15,16 and Delta escape primarily from class 2 antibodies.17 Our analysis therefore suggests that Omicron’s increased antibody escape breadth as compared with previous variants is driven by mutations in class 3 and 4 antibody epitopes.
Figure 1.

RBD epitopes and variant mutational constellations
(A) AAI networking between a panel of antibodies and nanobodies across the four anti-RBD antibody classes (x axis) and RBD sites (y axis) is shown, with total networking strength annotated as heatmap intensity (colorbar). Only RBD sites that interact with at least three antibodies in the panel are shown for clarity. RBD sites mutated on the Omicron, Beta, Delta, and PMS20 RBDs are highlighted by red, blue, purple, and green arrows along the y axis, respectively. The Beta and Delta variant mutations primarily reside at sites corresponding to class 1 and 2 antibodies, the PMS20 mutations occur at sites residing within class 1–3 epitopes, and the Omicron mutations cover the epitopes of all four antibody classes.
(B) VOC mutations affecting class 1–4 antibody epitopes. Two surface representations of RBD are shown for each variant, with the top view displaying surfaces targeted by class 1 and 2 antibodies and the bottom view displaying surfaces targeted by class 3 and 4 antibodies. The surface view highlights the extent of Omicron’s mutational breadth (across all four antibody classes) as well as depth (extent of accumulated mutations within a given epitope surface).
Class 3 antibodies are potent neutralizers that are immunodominant for certain individuals,5 while class 4 antibodies tend to be weakly neutralizing.13 Our network analysis associated Omicron mutations N440K, G446S, G496S, and Q498R most strongly with enhanced class 3 antibody escape based on these sites having the strongest network interactions with the antibodies surveyed. The RBD of PMS20, a research variant that escapes neutralization from most convalescent and polyclonal sera, features similar class 3 mutations to Omicron at sites 440 and 445, yet also features an R346K mutation that Omicron lacks.18 Our analysis found that R346 is the most strongly networked residue for certain class 3 antibodies, including C135. This suggests that PMS20 may escape more effectively from the class 3 antibody component of sera than Omicron, highlighting a key caveat in comparisons between Omicron and PMS20.
The AAI analysis suggested an additional set of Omicron mutations (G339D, S371L, S373P, S375F) may contribute to class 3 and 4 antibody epitopes via predominantly indirect mechanisms, with mutations at these sites appearing to affect nearly all class 4 antibodies in our panel, as well as select class 3 antibodies, including S309, S2M11, C110, CV38-142, Ab 812, and PDI-96. While mutational scanning of point mutations at these sites suggested these mutations are unlikely to confer significant class 3 and 4 antibody escape in isolation,5 it is plausible that the combined indirect effects of these four non-conservative mutations could meaningfully alter the local structure in this RBD region and thus perturb class 3 and 4 antibodies. In contrast to escape from class 3 antibodies, however, Omicron lacked mutation at a site that is strongly networked to multiple class 4 antibodies such as 368-9, 377-8, 384, or 434.
Further, we observed that Omicron mutations may enhance escape depth from class 1 antibodies beyond that observed for the Beta variant due to accumulation of additional mutations in class 1 antibody epitopes. The polyclonal antibody response to infection and vaccination has been shown to broaden over the months following exposure due to persistent somatic mutation of antibody CDRs.19 This broadening process can increase an antibody’s tolerance of escape mutations within its epitope,20 resulting in polyclonal responses with enhanced neutralizing capacity against the Beta variant over time. We found that Omicron has accumulated multiple tightly clustered mutations and therefore may have enhanced escape from matured polyclonal responses that are tolerant of certain class 1 antibody escape mutations such as K417N and N501Y (see Figure 1). That is, Omicron’s accumulated class 1 mutations may reduce Omicron’s susceptibility to class 1 antibodies tolerant of canonical class 1 escape mutations such as K417N. Specifically, Omicron mutations at residues Q493, G496, Q498, and Y505 clustered closely with the Beta and Omicron escape mutations at residues K417 and N501 indicating class 1 escape depth. Such escape depth could have contributed to PMS20’s escape from convalescent and vaccinee sera,18 as it can be seen in Figure 1 that PMS20 also features multiple class 1 mutations. Notably, however, PMS20 was shown not to fully escape from polyclonal responses generated from infection followed by vaccination,18 suggesting that Omicron’s class 1 escape depth and class 3 escape breadth alone may not confer this ability.
Omicron mutations and their effects on therapeutic antibodies
We subsequently applied AAI networks to examine the Omicron mutations in the context of their ability to evade neutralization by therapeutic antibodies of current clinical relevance (Figure 2). An important perspective offered by our network approach is the impact of mutations that are not directly located at the interface of antibody-antigen complexes, yet may still disrupt the antibody interaction. Specifically, it is important to consider the allosteric effects of the many Omicron mutations and how they may cooperatively affect antibody binding to the Omicron RBD by modulating the structural and chemical features of the epitope surface. Our results present an accessible and concise visualization of the network interactions between RBD sites mutated on Omicron and authorized therapeutic antibodies.
Figure 2.

VOCs versus therapeutic mAbs
Cumulative AAI networking between mutated sites on the Beta, Delta, Omicron, and PMS20 variant RBDs and the currently authorized therapeutic antibodies REGN10933, REGN10987, LY-CoV16, LY-CoV555, AZD1061, AZD8895, and S309 is shown and broken down into direct, indirect, and total components. Networking strength is annotated as heatmap intensity (colorbar). Network diagrams describing connectivity between specific sites are provided in the supplement as Figures S1–S3. Direct networking results are consistent with point mutation analyses and do not identify certain antibody epitope perturbations resulting from the Omicron mutations such as occurs for AZD1061. Introducing an indirect networking metric that accounts for indirect interactions successfully identifies the Omicron-AZD1061 perturbation. The combination of both direct and indirect features in total networking is highly consistent with experimental observations
We found that Omicron mutations occur at sites that are strongly directly networked to REGN10987 + REGN10933 (casirivimab + imdevimab; sites 417, 440, 446, 484, 493, 496, 498) and LY-CoV016 + LY-CoV555 (bamlanivimab + etesevimab; sites 417, 484, 493, 501, 505), suggesting the binding of these antibodies will be directly perturbed by Omicron mutations. In contrast, AAI networking revealed Omicron mutations do not appear to strongly directly interact with S309 (sotrovimab) or AZD8895 + AZD1061 (tixagevimab + cilgavimab). Our findings on the basis of direct networking between epitope residues in Omicron variant and paratope residues in the neutralizing antibodies align with existing analyses and commentary for these therapeutic antibodies derived from mutagenesis screens based on the binding perturbation induced by the Omicron mutations individually and in isolation.5,21, 22, 23
However, point mutation analyses and AAI direct networking do not explain interactions observed between these antibodies and the Omicron variant in the full context of the set of Omicron RBD mutations. Specifically, analysis of the AZD1061 network found that Omicron mutated sites are highly networked to sites directly networked to AZD1061, resulting in significant indirect networking (Figures 2 and S1A). Meanwhile, we observed that the Omicron mutated sites result in a large number of moderate direct and indirect interactions for AZD8895 (Figure S1B). Since the submission of this analysis, multiple studies reported significant reductions in pseudovirus neutralization for AZD8895 and AZD1061,23,24 which is consistent with the cumulative Omicron mutations resulting in perturbation of these epitopes.
Examination of the S309 AAI network showed indirect interactions between S309 and the Omicron mutated sites 339, 373, 440, and 446 (Figure S2). Since the submission of this manuscript, multiple studies have reported an approximately 3-fold reduction in neutralization for S309 against Omicron.21,23, 24, 25 This reduction is consistent with the AAI model prediction that the cumulative indirect effects of the Omicron mutations moderately perturb the S309 binding surface. Building on this observation, we noted that the AAI network indicated R346 is important to the epitope network of S309, and R346 K/S mutations had been detected in Omicron subclades.23 Further AAI analysis showed that R346 is networked both directly and indirectly to the S309 paratope in the context of the other Omicron mutations. While isolated mutations at R346 only minorly affect S309 binding and neutralization,21,26 the AAI network suggests R346 mutations are likely to have a greater impact in the context of the other Omicron mutations. Liu et al. and Cameroni et al. further tested Omicron + R346K and found no significant additional reduction in neutralization.23,25 Importantly, R→K is a conservative mutation, highlighting how the physiochemical features of a mutation are critical to understanding whether the mutation will result in a significant perturbation of the AAI network with effect on antibody function. Therefore, it is important to monitor whether there is a further reduction in the susceptibility (>5-fold change as defined in the sotrovimab emergency use authorization [EUA] fact sheet27) of subclades with other R346 mutations to neutralization by S309, as R→S has also been observed on Omicron subclades and could result in more substantial perturbation of the epitope network.
Similarly, our analysis suggested that such indirect effects may affect the recently identified anti-RBD antibodies such as ADG-2 that target the conserved epitope shared across clade 1a and 1b coronaviruses.28 Using the AAI network model, we established that Omicron mutations at sites 496, 498, 501, and 505 are networked to the critical ADG-2 epitope residues D405, G502, G504, and Y505 (Figure S3). Rappazzo et al. further reported that Y505 C/N/S knocked down ADG-2 binding potency.28 Single point mutation analysis on an epitope does not consider the extent to which mutations at proximal residues may affect the epitope-paratope binding interaction. For example, ADG-2 does not bind the RaTG13 RBD, and RatG13 shares sequence identity with Omicron at the four critical ADG-2 binding sites 405, 502, 504, and 505, demonstrating the importance of the network context of these four critical sites. Since the submission of this manuscript, Adagio reported a 300-fold knockdown of ADG-2 neutralization potency against Omicron in both pseudovirus and live virus experiments,29 consistent with AAI network observations and highlighting the importance of indirect effects in assessing monoclonal antibody function when multiple mutations occur within or adjacent to an epitope.
Potential additional functional roles of Omicron RBD mutations
Given the substantial transmission advantage of Omicron over Delta, it is plausible that Omicron RBD mutations contribute to enhanced transmission by mechanisms other than or in addition to antibody escape. Above (Figure 1), we identified several Omicron mutations that do not appear to play a major role as polyclonal epitope constituents or whose predicted contribution to antibody evasion is unlikely to confer a substantial fitness advantage due to the mutations occurring in class 4 antibody epitopes. Here, we highlight two mechanisms by which these RBD mutations may contribute to Omicron’s fitness via enhanced transmissibility.
Enhanced ACE-2 binding
There is existing evidence that certain Omicron mutations enhance ACE-2 binding, and this is a leading hypothesis to explain Omicron’s enhanced transmission. In particular, Zahradník et al. identified enhanced ACE2 binding by RBD’s bearing Q498R and S477N when combined with the N501Y mutation.30 Importantly, these mutation combinations resulted in a synergistic effect extending beyond the increased ACE-2 binding predicted when the effect of these mutations assayed individually was summed. It is therefore likely that a similar synergy effect occurs on the Omicron variant at these three sites and may also include additional Omicron mutations. Using AAI networks to examine the local residue dependencies in this vicinity, we observed a network extending from the known synergistic pair 498 + 501 to position 505, suggesting Omicron H505 may modulate the synergistic ACE-2 binding effect for Q498R + N501Y.
RBD-RBD interface stability
While Omicron’s mutations at sites 371, 373, and 375 may contribute to escape from class 3 and 4 antibodies, evolution of such a triplet mutation toward this escape function is not parsimonious given that other single mutations have previously been identified as conferring greater class 3 and 4 antibody escape.5 We therefore hypothesized that mutations S371L, S373P, and S375F might provide a fitness advantage other than or in addition to escape. To test this hypothesis, we sought to analyze the mutations in the context of different conformational states of the RBD in the trimeric spike protein. In a literature review, we found that Wrobel et al. had previously identified a role for sites S371 and S373 in stabilizing the RBDdown-RBDdown interface in the spike three RBD-down conformation for SARS-CoV-2 as compared with the related RaTG13 CoV.31 Further, Wrobel et al., identified interactions between sites 369, 371, 373, 403, 440, 493, and 505 as plausibly driving differences between SARS-CoV-2 and RaTG13 in RBDdown-RBDdown interface stability with implications for the RBD-up transition that enables ACE-2 binding and subsequent infection. Remarkably, Omicron mutations occur in five of these seven sites. The potential for a significant perturbation at this interface was also captured by the AAI network analysis, which identified numerous direct and indirect interactions from sites 371, 373, 375, 440, 493, and 505 extending across the RBDdown-RBDdown interface (data not shown). Additionally, mutations such as D614G have previously been linked to increased infectivity via destabilization of the RBD-down spike protein conformation (thus promoting the active RBD-up conformation) offering a plausible structure-function relationship.32
Given that no structure of the Omicron spike in the three RBD-down state was available in the days following the first identification of the Omicron variant, we built a fixed-backbone homology model of the Omicron RBDdown-RBDdown interface in the three RBD-down state to investigate the potential influence of the Omicron mutations on interface stability (Figure 3A). Our homology model suggested Omicron mutations disrupt the closed-state RBDdown-RBDdown interface. Specifically for the sites highlighted by Wrobel et al.,31 the modeled Omicron mutations disrupted hydrogen bonds from Y505 to S373 and Y505 to R403. The loss of these bonds was reflected in positive ΔΔG for Y505H and S373P, yet partially compensated for by a more energetically favorable R403 environment. A positive ΔΔG was also observed upon mutation at site 375. Further, interface analysis of our Omicron model revealed that the RBDdown-RBDdown interface buried surface area decreased by 40 Å2, as compared with a 65 Å2 change for SARS-CoV-2 versus RaTG13, and the interface surface complementarity decreased from 0.60 to 0.54.
Figure 3.

The effects of Omicron mutations on the RBDdown-RBDdown interface
(A) The RBDdown-RBDdown interface in the three RBD-down conformation, with the repacked WT structure (PDB: 6ZGI)31 and our Omicron fixed-backbone model shown in light blue and pink, respectively. Omicron mutations S373P, S375F, and Y505H reduced the energetic complementarity of the RBDdown-RBDdown interface at these sites, but enabled a more energetically favorable conformation for R403. Interface buried surface area and surface complementary were also reduced for Omicron by 40 Å2 and from 0.60 to 0.54, respectively.
(B) Structural model of the RBDdown-RBDdown interface in the one RBD-up conformation, with the WT (PDB: 6XM3)33 and Omicron (PDB: 7TB4)34 structures shown in light blue and pink, respectively. Alignment of the protomer 1 of WT and Omicron RBDdown showed a 7 to 10 Å movement of the Omicron protomer two RBDdown toward the distal RBDup (not shown), which is annotated as red arrows, resulting in loss of all three interface bonds and 190 Å2 of buried surface area per RBD.
Since our initial modeling effort, a structure of Omicron spike in the one RBD-up conformation was released (PDB: 7TB4).34 When compared with wild-type RBD in the same conformation (PDB: 6XM3),33 the Omicron RBDdown-RBDdown interface showed a dramatic physical separation, loss of all three RBDdown-RBDdown interface bonds, and an accompanying 190 Å2 reduction (−62%) in buried surface area (Figure 3B). The separation was similarly pronounced when the Omicron one RBD-up was compared with the same conformation for other wild-type structures and the Alpha, Gamma, Kappa, and Delta variants (data not shown; PDB: 6ZOW,35 7EDG,36 7M8K,37 7SBR,38 7SBL38). Our model indicating a decrease in chemical and physical complementarity for the RBDdown-RBDdown interface is therefore consistent with these structural finding for the one RBD-up conformation. Further, Zhou et al. highlight a new bond between RBDdown F486 and the distal RBDup F375, which they hypothesize to stabilize the one RBD-up conformation.34 Together, these data suggest the Omicron mutations may destabilize the RBDdown-RBDdown interface and stabilize the RBDdown-RBDup interface. In particular, S375F may function simultaneously at both the RBDdown-RBDdown and RBDdown-RBDup interfaces to stabilize the one RBD-up Omicron conformation, although other Omicron mutations such as S373P likely alter the local structure to facilitate this function. Interestingly, such a functional change may also bear a fitness tradeoff due to increased surface exposure of the RBD-neutralizing epitopes. These predictions can be validated experimentally and explained in more mechanistic detail once an Omicron spike structure in the three RBD-down conformation is solved. Indeed, early work by Yin et al. to solve a three RBD-down structure with accompanying thermal shift and hydrogen-deuterium exchange mass spectrometry experiments observed significant shifts for Omicron as compared with wild-type (WT) spike,39 suggestive of decreased three RBD-down state stability.
Discussion
The network-based analysis of the Omicron mutational landscape presented here describes Omicron’s escape breadth and depth. We find evidence for enhanced escape breadth of Omicron as compared with the Beta and Delta variants due primarily to class 3 escape mutations as well as increased escape depth within class 1 epitopes. We further identify plausible class 4 antibody escape via the mutations at sites 371, 373, and 375, which may alter class 4 epitope surfaces indirectly, but note that escape from class 4 antibodies is unlikely to confer a meaningful escape fraction from sera and that these sites are not the dominant class 4 escape sites. Additionally, clades of Omicron bearing R346 K/S should be monitored closely23 as this mutation is likely to further enhance Omicron antibody evasion.
Importantly, although neutralizing antibody titers are correlated with vaccine protection against infection,40 vaccine efficacy against severe disease is likely to be preserved even in the case of complete antibody escape due to non-neutralizing antibodies and T cell responses.41 The high degree of major histocompatibility complex (MHC) class I polymorphism at the population level means that, while CD8 T cell escape can occur via MHC class I escape within an individual during a chronic infection such as HIV, T cell escape at the population level would require mutation of nearly all T cell epitopes and is therefore unlikely to occur for SARS-CoV-2.11 Analysis by Tarke et al. predicts that Omicron CD8 epitopes are 86% conserved and CD4 epitopes are 72% conserved across the Omicron spike protein.42 A second analysis of 52 CD8 T cell epitopes spanning six human leukocyte antigen (HLA) haplotypes found just two of these epitopes contained a single Omicron mutation.43 Together these data bode well for preservation of protection from severe disease provided by vaccination and convalescence. However, early evidence suggests that 30% (CD8) and 45% (CD4) of immunocompromised individuals (those receiving anti-CD20 therapy in the study) do not maintain detectable T cell response against Omicron even following a booster vaccine dose.44 This finding therefore highlights the critical need to extend additional resources to protect vulnerable populations from Omicron and future VOCs. These results from immunocompromised individuals also provide evidence that vaccine boosters may enhance the frequency and potency of T cell responses against Omicron, wherein T cell breadth has previously been associated with mild disease courses.45
Further, it will be important to determine if the accumulation of class 1 escape mutations, which provide escape depth, reads through to a meaningful escape impact or reduction in vaccine efficacy. Thus far, such depth seems to provide little additional escape benefit given that the combination of K417 and N501 mutations appears sufficient to escape from class 1 antibodies in most convalescent and vaccine sera.46 However, it is notable that the PMS20 variant was unable to fully escape from polyclonal responses generated by infection followed by vaccination.18 Such “hybrid” immunity provided exceptionally potent and broad polyclonal responses.42 It is plausible that Omicron’s additional class 1 antibody escape mutations provide a benefit against such responses, which could suggest these Omicron mutations represent multiple rounds of virus-host evolution. Teasing out these specifics of Omicron’s escape breadth and depth will be particularly informative for designing vaccine boosters resilient to constellations of escape mutations both across antibody classes as well as within immunodominant class 1 and 2 epitopes.
Previous variants presented with mutations on largely orthogonal epitope surfaces targeted by distinct antibody classes, yet Omicron’s RBD mutational landscape is clustered within overlapping antibody epitopes. Throughout this study we therefore highlight the importance of considering the complex structural and chemical relationships between combinations of mutations within or adjacent to antigenic surfaces. While point mutation escape studies are unlikely to provide a complete mapping of how mutation constellations may perturb antibody binding, our AAI network approach maps these complex relationships between proximal residues and interface structure. The direct component of our network analysis was consistent with predictions on therapeutic antibody escape provided by traditional point mutation predictions. In contrast, the indirect component identified allosteric interactions between Omicron mutation sites and the AZD1061, S309, and ADG-2 epitopes.
Finally, we find evidence for mechanisms other than immune escape through which Omicron mutations may enhance fitness. While mutation combinations such as Q498S and N501Y have previously been shown to epistatically enhance ACE-2 binding, our analysis suggests that Omicron mutations such as Y505H may also indirectly influence the ACE-2 interaction. Further, based on the previous work of Wrobel et al.31 and our Omicron three RBD-down fixed model, we present evidence of a potential role for Omicron mutations in destabilizing the RBDdown-RBDdown interface. This finding is complementary with recent work showing stabilization of the one RBD-up conformation via a new bond at the RBDdown-RBDup interface,34 as RBDdown-RBDdown destabilization could promote both transition from three RBD-down to one RBD-up as well as maintenance of the one RBD-up state. These effects may synergize to increase the propensity for the RBD-up conformation. Increased occupancy of the RBD-up state may also present a fitness tradeoff resulting from greater surface exposure of the RBD-up antibody epitopes. Omicron’s antibody evasion mutations may largely negate this penalty, and it is plausible that such RBDdown-RBDdown interface mutations are only enabled in the context of Omicron’s unique degree of immune escape. We also note that the Omicron BA.2 subvariant, which appears to have a transmission advantage,47 features distinct RBD-RBD interface mutation S371F and additonal interface mutations T376A, D405N, and R408S. This observation further supports a relationship between mutations in this region and enhanced transmissibility. Importantly, AAI analysis of the BA.2 RBD mutations showed reduced antibody escape breadth relative to BA.1 due to the lack of the G446S mutation. This suggests BA.2's growth advantage is unlikely to be driven by superior immune escape.
In light of probable continued Omicron dominance, determining the various functional roles of the Omicron mutations is critical. In particular, it is important to understand the complex relationships between Omicron mutations, and the contribution of these indirect effects to Omicron’s antibody escape and enhanced transmissibility. Network-based approaches such as those presented in this study are particularly well suited for modeling such indirect interactions.
Limitations of the study
Limitations of our study include (1) sampling a representative polyclonal response from a set of individual antibodies and nanobodies at sufficient resolution, (2) performing Omicron analyses based on antibody interactions with WT SARS-CoV-2, and (3) the lack of a structural model of the Omicron spike in the three RBD-down conformation. We limit the impact of (1) by sampling at least 10 antibodies from each of the four major anti-RBD antibody classes, and observe a high degree of similarity between antibodies, suggesting these samples span the dominant epitope-paratope interactions for each class. Further, AAI network analysis is somewhat less sensitive to structural resolution than traditional structural analyses that rely heavily on interpretation of specific bonds. Limitation (2) exists for all predictive approaches to assessing antibody function against a recently identified variant based on existing data describing interactions between the given antibody and a prior spike structure. However, as shown in this manuscript, protein network analyses appear more robust for predicting the impact of combined mutations than other analyses relying on aggregating the cumulative effects of multiple point mutations. Due to (3), we are limited in our ability to predict whether the Omicron RBDdown-RBDdown interface in the three RBD-down conformation is sufficiently perturbed to result in a phenotypic effect. However, the recently released two RBD-down one RBD-up model (PDB: 7TB4)34 offers additional evidence, which we incorporate into our analysis in addition to our fixed model. Further early evidence of decreased three RBD-down conformational stability is provided by thermal shift assay and hydrogen deuterium exchange mass spectrometry in Yin et al.39 Together, these data and the data on RaTG13 versus SARS-CoV-2 from Wrobel et al.31 support our hypothesis and warrant further investigation of Omicron mutations at the RBDdown-RBDdown interface.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Antibody-RBD AAI Networks | This paper | https://doi.org/10.5281/zenodo.5865881 |
| Antibody-RBD structures | seeTable S1 | N/A |
| Software and algorithms | ||
| PDBePISA | Krissinel 200748 | https://www.ebi.ac.uk/pdbe/pisa/ |
| SWISS-MODEL | Waterhouse, 201849 | https://swissmodel.expasy.org |
| Custom code – Homology modeling, higher-order network analyses, and visualizations | This paper | https://doi.org/10.5281/zenodo.5865881 |
| Significant Interaction Network | Soundararajan, 20117 | https://doi.org/10.1038/srep00200 |
| PyMOL 2.3.4 | Schrödinger, LLC50 | https://pymol.org/2/ |
| Python/matplotlib | Hunter 200751 | https://matplotlib.org |
| Python/networkx | Hagberg 200852 | https://networkx.org |
| Python/numpy | Harris 202050 | https://numpy.org |
| Python/pandas | McKinney 201053 | https://pandas.pydata.org |
| Python/seaborn | Waskom 202154 | https://seaborn.pydata.org |
| Python/PyRosetta | Chaudhury 201055 | https://www.pyrosetta.org |
| Google Slides | https://www.google.com/slides | |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ram Sasisekharan (rams@mit.edu).
Materials availability
-
•
This study did not generate new unique reagents.
Experimental model and subject details
Structural data generated experimentally by others describing epitope-paratope interactions between antibodies/nanobodies and recombinant spike protein or the spike RBD was analyzed. The antibodies/nanobodies analyzed are listed in Table S1. Table S1 additionally includes Protein Data Bank accession codes for the structural model corresponding to each antibody/nanobody—where further experimental details for each structure determination are available—and DOIs for the accompanying publications describing biological experimental details such as production and purification of each antibody.
Method details
Amino acid interaction (AAI) network
AAI networks were quantified as described in the quantification and statistical analysis section. The quantification includes definition of Direct, Indirect, and Total networking metrics which are referenced in the below STAR Methods subsections.
Amino acid interaction (AAI) matrix and clustering
Total networking scores between each RBD site and the entirety of the antibody or nanobody paratope and framework regions were computed and normalized as described above. Subsequently, these data were aggregated into a single matrix describing the network interactions between all antibodies (Table S1) and all RBD sites. This matrix was then plotted as a heatmap and clustered using clustermap from the Seaborn statistical data visualization package.54 Dendrograms were generated for RBD sites (rows) and antibodies (columns) using the ‘canberra’ distance metric. For quantifying potential networking perturbations between individual therapeutic antibodies and variants of concern such as Omicron, the total networking between the given antibody and all RBD sites mutated on the given variant was summed to obtain a single value. Repeating this process for a panel of antibodies and a panel of variants results in computation of a refined AAI network matrix, which was subsequently visualized as a clustered heatmap using clustermap as described above. Note that the heatmap in Figure 1 was manually annotated with vertical dotted lines and labels for each antibody class designation, as well as arrows indicating the mutations for each variant shown on the right-side. The surface representations of RBD were constructed in PyMOL. Note that Figure 2 is a composite of three heatmaps, in which the only modification of the output is that two of the three identical color maps (legends) are removed for clarity.
AAI Network visualization for therapeutic antibodies
AAI networks were visualized (Figures S1–S4) for certain therapeutic antibodies as second- and third-order networks as follows. First, the set of all RBD sites that are networked to the given antibody paratope or framework regions was computed. These sites, which interact directly with the given antibody, form the core of the interaction network between the antibody and RBD. Next, all RBD sites that are networked to first layer of sites was computed. Both sets of RBD sites were combined and used to build a network using NetworkX,52 wherein RBD sites were defined as nodes and edges were constructed between all pairs of RBD sites with non-zero networking and weighted according to AAI networking strength. Node positions were computed from these edge weights using the Fruchterman-Reingold force-directed algorithm as implemented in NetworkX spring_layout with the optimal node distance defined as 10 times the inverse square root of the network order, such that pairs of RBD sites that are strongly networked appear as nodes that are tightly-clustered and separated by a short edge. Finally, all nodes and edges were plotted, with nodes assigned colors based on whether they are directly or indirectly networked to the antibody paratope/framework, and whether or not they are mutated on the Omicron variant. In the case of antibody ADG2, in which no structure is publicly available and so RBD sites directly networked to the antibody cannot be determined algorithmically, the four “critical” epitope sites 405, 502, 504, and 505 reported in Rappazzo et al.28 were chosen as the first directly-networked set or the network “core”. Four directly-networked sites is fewer than is typically observed for antibodies suggesting that additional RBD residues are likely involved in direct interactions with the antibody. To ensure that we captured all RBD sites that may be indirectly networked to antibody ADG2 in our network visualization, we therefore added an additional network layer to the epitope network for ADG2. That is, we followed the same workflow described above for the other antibody networks, but included an additional degree of indirect networking such that the ADG2 network is represented as: four critical sites → first layer of indirectly networked sites → second layer of indirectly networked sites. Note that legends were manually annotated on top of the raw network figures for all four supplemental figures.
Omicron RBDdown-RBDdown interface analysis
A fixed-backbone model was used to analyze the Omicron RBDdown-RBDdown interface mutations in the 3-RBD down conformation, as homology modeling is limited in its ability to accurately predict RBD structure in the context of 15 Omicron RBD mutations, particularly for probable backbone perturbations conferred by mutations such as S373P that reside within the specific interface in question. The fixed-backbone approximation offers initial insight into whether or not mutations in the RBDdown-RBDdown interface could result in structural alterations and divergent conformational preferences in the Omicron spike trimer. First, a homology model of the Omicron RBDdown-RBDdown interface was constructed via SWISS-Model49 using the 6ZGI31 template. Subsequently, with the backbone fixed, all side chains within 30 Å heavy atom distance from site 505 were repacked on the two RBD protomers of the homology model in PyRosetta.55 This repacking process was also performed for the SARS-CoV-2 wild-type (WT) template (6ZGI) as a control. Finally, the Omicron and WT RBDdown-RBDdown interfaces were analyzed using PDBePISA,48 which reports dG values at each site for the WT and Omicron RBDdown-RBDdown interfaces as well as buried surface area for the interface. Surface complementarity was computed using the InterfaceAnalzyerMoveer in PyRosetta according to the sc_value metric. Interfaces visualizations were generated using PyMOL.56 For analysis of the 1 RBD-up Omicron spike structure, the RBDdown-RBDdown interface was first obtained from PDBs 6XM333 and 7TB434 For interface analysis, both WT and Omicron RBDdown-RBDdown interfaces were analyzed using PDBePISA as above to report buried surface area. For visualization in Figure 3B, protomer 1 was assigned to the RBD monomer most distal from the RBDup for both WT and Omicron, and both promoter 1 were aligned in PyMOL. For Figure 3B, the three hydrogen bonds shown for the WT interface were consistently identified by both PDBePISA and PyMOL. Note that the red arrows indicating the 7-10 Å shift, the mutation labels and ΔΔG values, and the promoter labels were manually annotated.
Quantification and statistical analysis
Amino acid interaction (AAI) network quantification
For each antibody-RBD complex, AAI network scores between every pair of residues within the complex were computed.7,8 Network scores quantify all contacts for both side-chain and backbone atoms between residues, and weight these contacts according to the energetics of the expected interaction based on experimental energetic measurements for each interaction type. The following interactions are quantified: hydrogen bonds, pi bonds, disulfide bonds, polar interactions, salt bridges, and Van der Waals interactions. The following metrics were defined and computed for all surveyed structures from the resulting interaction network. Direct Networking: For a given residue on RBD, the sum of all interactions between the RBD residue and all residues on the complexed antibody/nanobody including both the paratope and framework regions. Indirect Networking: For a given residue on RBD, the sum of all interactions between the RBD residue and all other residues on RBD which have non-zero direct networking to the given antibody/nanobody’s paratope or framework region. Given that the AAI network is weighted more heavily toward interactions involving at least one side-chain as compared to backbone interactions, an additional computation was performed for the indirect networking scores for glycine residues. Specifically, glycine indirect networking was interpolated from nearby residues (within 5Å of the glycine alpha carbon) based on averaging the network connections from these adjacent residues to their partners. Total Networking: For a given residue on RBD, the sum of direct and indirect networking scores for the residue. Note that for all networking metrics, scores are normalized to the highest networking score within each RBD-mAb complex. Structures analyzed and their PDB identifiers are listed in Table S1. The raw networking matrices from which direct, indirect, and total networking are computed are available in the key resources table, along with additional software packages50,51,53 required for the described computation.
Acknowledgments
N.L.M. was supported in part by T32 ES007020/ES/NIEHS NIH and by SMART, Singapore.
Author contributions
N.L.M. and T.C. conducted the analyses. All authors designed the analyses and wrote the paper.
Declaration of interests
R.S. is a board member of Tychan, Singapore, which focuses on infectious diseases.
Published: January 24, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2022.100527.
Supplemental information
Data and code availability
-
•
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
-
•
All original code has been deposited at GitHub and is publicly available as of the date of publication. DOIs are listed in the key resources table. Additional Supplemental Items are available from Github/Zenodo at https://doi.org/10.5281/zenodo.5865881
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Pulliam J.R.C., Schalkwyk C. van, Govender N., Gottberg A. von, Cohen C., Groome M.J., Dushoff J., Mlisana K., Moultrie H. Increased risk of SARS-CoV-2 reinfection associated with emergence of the Omicron variant in South Africa. medRxiv. 2021 doi: 10.1101/2021.11.11.21266068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu Y., Liu J., Johnson B.A., Xia H., Ku Z., Schindewolf C., Widen S.G., An Z., Weaver S.C., Menachery V.D., et al. Delta spike P681R mutation enhances SARS-CoV-2 fitness over Alpha variant. bioRxiv. 2021 doi: 10.1101/2021.08.12.456173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu Y., Liu J., Plante K.S., Plante J.A., Xie X., Zhang X., Ku Z., An Z., Scharton D., Schindewolf C., et al. The N501Y spike substitution enhances SARS-CoV-2 transmission. Nature. 2021 doi: 10.1038/s41586-021-04245-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou D., Dejnirattisai W., Supasa P., Liu C., Mentzer A.J., Ginn H.M., Zhao Y., Duyvesteyn H.M.E., Tuekprakhon A., Nutalai R., et al. Evidence of escape of SARS-CoV-2 variant B.1.351 from natural and vaccine-induced sera. Cell. 2021;184:2348–2361.e6. doi: 10.1016/j.cell.2021.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greaney A.J., Starr T.N., Barnes C.O., Weisblum Y., Schmidt F., Caskey M., Gaebler C., Cho A., Agudelo M., Finkin S., et al. Mapping mutations to the SARS-CoV-2 RBD that escape binding by different classes of antibodies. Nat. Commun. 2021;12:4196. doi: 10.1038/s41467-021-24435-8. 2021 12:1 12, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ku Z., Xie X., Davidson E., Ye X., Su H., Menachery V.D., Li Y., Yuan Z., Zhang X., Muruato A.E., et al. Molecular determinants and mechanism for antibody cocktail preventing SARS-CoV-2 escape. Nat. Commun. 2021;12:469. doi: 10.1038/s41467-020-20789-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soundararajan V., Zheng S., Patel N., Warnock K., Raman R., Wilson I.A., Raguram S., Sasisekharan V., Sasisekharan R. Networks link antigenic and receptor-binding sites of influenza hemagglutinin: mechanistic insight into fitter strain propagation. Scientific Rep. 2011;1:200. doi: 10.1038/srep00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller N.L., Clark T., Raman R., Sasisekharan R. An antigenic space framework for understanding antibody escape of SARS-CoV-2 variants. Viruses. 2021;13:2009. doi: 10.3390/v13102009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sethi A., Tian J., Derdeyn C.A., Korber B., Gnanakaran S. A mechanistic understanding of allosteric immune escape pathways in the HIV-1 envelope glycoprotein. PLoS Comput. Biol. 2013;9:1003046. doi: 10.1371/journal.pcbi.1003046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalia V., Sarkar S., Gupta P., Montelaro R.C. Antibody neutralization escape mediated by point mutations in the intracytoplasmic tail of human immunodeficiency virus type 1 gp41. J. Virol. 2005;79:2097–2107. doi: 10.1128/JVI.79.4.2097-2107.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yewdell J.W. Antigenic drift: understanding COVID-19. Immunity. 2021;54:2681–2687. doi: 10.1016/j.immuni.2021.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kolawole A.O., Smith H.Q., Svoboda S.A., Lewis M.S., Sherman M.B., Lynch G.C., et al. Norovirus escape from broadly neutralizing antibodies is limited to allostery-like mechanisms. mSphere. 2017;2 doi: 10.1128/mSphere.00334-17. e00334–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barnes C.O., Jette C.A., Abernathy M.E., Dam K.M.A., Esswein S.R., Gristick H.B., Malyutin A.G., Sharaf N.G., Huey-Tubman K.E., Lee Y.E., et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature. 2020;588:682–687. doi: 10.1038/s41586-020-2852-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hastie K.M., Li H., Bedinger D., Schendel S.L., Moses Dennison S., Li K., Rayaprolu V., Yu X., Mann C., Zandonatti M., et al. Defining variant-resistant epitopes targeted by SARS-CoV-2 antibodies: a global consortium study. Science. 2021;374:472–478. doi: 10.1126/science.abh2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wibmer C.K., Ayres F., Hermanus T., Madzivhandila M., Kgagudi P., Oosthuysen B., Lambson B.E., de Oliveira T., Vermeulen M., van der Berg K., et al. SARS-CoV-2 501Y.V2 escapes neutralization by South African COVID-19 donor plasma. Nat. Med. 2021;27:622–625. doi: 10.1038/s41591-021-01285-x. [DOI] [PubMed] [Google Scholar]
- 16.Yuan M., Liu H., Wu N.C., Lee C.C.D., Zhu X., Zhao F., Huang D., Yu W., Hua Y., Tien H., et al. Structural basis of a shared antibody response to SARS-CoV-2. Science. 2020;369:1119–1123. doi: 10.1126/science.abd2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng L., Song S., Fan Q., Shen S., Wang H., Zhou B., Ge X., Ju B., Zhang Z. Cross-neutralization of SARS-CoV-2 Kappa and Delta variants by inactivated vaccine-elicited serum and monoclonal antibodies. Cell Discov. 2021;7:112. doi: 10.1038/s41421-021-00347-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmidt F., Weisblum Y., Rutkowska M., Poston D., da Silva J., Zhang F., Bednarski E., Cho A., Schaefer-Babajew D.J., Gaebler C., et al. High genetic barrier to SARS-CoV-2 polyclonal neutralizing antibody escape. Nature. 2021;600:512–516. doi: 10.1038/s41586-021-04005-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Z., Muecksch F., Schaefer-Babajew D., Finkin S., Viant C., Gaebler C., Hoffmann H.H., Barnes C.O., Cipolla M., Ramos V., et al. Naturally enhanced neutralizing breadth against SARS-CoV-2 one year after infection. Nature. 2021;595:426–431. doi: 10.1038/s41586-021-03696-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muecksch F., Weisblum Y., Barnes C.O., Schmidt F., Schaefer-Babajew D., Wang Z., Julio J.C., Flyak A.I., DeLaitsch A.T., Huey-Tubman K.E., et al. Affinity maturation of SARS-CoV-2 neutralizing antibodies confers potency, breadth, and resilience to viral escape mutations. Immunity. 2021;54:1853–1868.e7. doi: 10.1016/j.immuni.2021.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cathcart A.L., Havenar-Daughton C., Lempp F.A., Ma D., Schmid M.A., Agostini M.L., Guarino B., iulio J. di, Rosen L.E., Tucker H., et al. The dual function monoclonal antibodies VIR-7831 and VIR-7832 demonstrate potent in vitro and in vivo activity against SARS-CoV-2. bioRxiv. 2021 doi: 10.1101/2021.03.09.434607. [DOI] [Google Scholar]
- 22.REGENERON Regeneron evaluating REGEN-COV® and next generation antibodies against new Omicron COVID-19 variant. 1–8. 2021. https://investor.regeneron.com/static-files/969bdb0b-53f5-46c7-94fb-7473ee7f5be3
- 23.Liu L., Iketani S., Guo Y., Chan J.F.-W., Wang M., Liu L., Luo Y., Chu H., Huang Y., Nair M.S., et al. Striking antibody evasion manifested by the Omicron variant of SARS-CoV-2. Nature. 2021 doi: 10.1038/s41586-021-04388-0. [DOI] [PubMed] [Google Scholar]
- 24.Cao Y., Wang J., Jian F., Xiao T., Song W., Yisimayi A., Huang W., Li Q., Wang P., An R., et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature. 2021:1–9. doi: 10.1038/s41586-021-04385-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cameroni E., Bowen J.E., Rosen L.E., Saliba C., Zepeda S.K., Culap K., Pinto D., VanBlargan L.A., De Marco A., di Iulio J., et al. Broadly neutralizing antibodies overcome SARS-CoV-2 Omicron antigenic shift. Nature. 2021 doi: 10.1038/s41586-021-04386-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Starr T.N., Czudnochowski N., Liu Z., Zatta F., Park Y.J., Addetia A., Pinto D., Beltramello M., Hernandez P., Greaney A.J., et al. SARS-CoV-2 RBD antibodies that maximize breadth and resistance to escape. Nature. 2021;597:97–102. doi: 10.1038/s41586-021-03807-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.GSK. FDA Fact sheet for healthcare providers emergency use authorization (EUA) OF sotrovimab authorized use. 2021. https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-
- 28.Garrett Rappazzo C., Tse L.v., Kaku C.I., Wrapp D., Sakharkar M., Huang D., Deveau L.M., Yockachonis T.J., Herbert A.S., Battles M.B., et al. Broad and potent activity against SARS-like viruses by an engineered human monoclonal antibody. Science. 2021;371:823–829. doi: 10.1126/science.abf4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adagio Therapeutics Adagio therapeutics reports reduction in in vitro neutralizing activity of ADG20 against Omicron SARS-CoV-2 variant. 2021. https://investors.adagiotx.com/news-releases/news-release-details/adagio-therapeutics-reports-reduction-vitro-neutralizing Press Release.
- 30.Zahradník J., Marciano S., Shemesh M., Zoler E., Harari D., Chiaravalli J., Meyer B., Rudich Y., Li C., Marton I., et al. SARS-CoV-2 variant prediction and antiviral drug design are enabled by RBD in vitro evolution. Nat. Microbiol. 2021;6:1188–1198. doi: 10.1038/s41564-021-00954-4. [DOI] [PubMed] [Google Scholar]
- 31.Wrobel A.G., Benton D.J., Xu P., Roustan C., Martin S.R., Rosenthal P.B., Skehel J.J., Gamblin S.J. SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat. Struct. Mol. Biol. 2020;27:763–767. doi: 10.1038/s41594-020-0468-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berger I., Schaffitzel C. The SARS-CoV-2 spike protein: balancing stability and infectivity. Cell Res. 2020;30:1059–1060. doi: 10.1038/s41422-020-00430-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou T., Tsybovsky Y., Gorman J., Rapp M., Cerutti G., Chuang G.Y., Katsamba P.S., Sampson J.M., Schön A., Bimela J., et al. Cryo-EM structures of SARS-CoV-2 spike without and with ACE2 reveal a pH-dependent switch to mediate endosomal positioning of receptor-binding domains. Cell Host Microbe. 2020;28:867–879.e5. doi: 10.1016/j.chom.2020.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou T., Tsybovsky T., Kwong P.D. 2021. RCSB PDB - 7TB4: Cryo-EM Structure of the Spike of SARS-CoV-2 Omicron Variant of Concern. [DOI] [Google Scholar]
- 35.Melero R., Sorzano C.O.S., Foster B., Vilas J.L., Martínez M., Marabini R., Ramiacute, rez-Aportela E., Sanchez-Garcia R., Herreros D., et al. Continuous flexibility analysis of SARS-CoV-2 spike prefusion structures. IUCrJ. 2020;7:1059–1069. doi: 10.1107/S2052252520012725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang T.J., Yu P.Y., Chang Y.C., Liang K.H., Tso H.C., Ho M.R., Chen W.Y., Lin H.T., Wu H.C., Hsu S.T.D. Effect of SARS-CoV-2 B.1.1.7 mutations on spike protein structure and function. Nat. Struct. Mol. Biol. 2021;28:731–739. doi: 10.1038/s41594-021-00652-z. [DOI] [PubMed] [Google Scholar]
- 37.Wang P., Casner R.G., Nair M.S., Wang M., Yu J., Cerutti G., Liu L., Kwong P.D., Huang Y., Shapiro L., et al. Increased resistance of SARS-CoV-2 variant P.1 to antibody neutralization. Cell Host Microbe. 2021;29:747. doi: 10.1016/j.chom.2021.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang J., Xiao T., Cai Y., Lavine C.L., Peng H., Zhu H., Anand K., Tong P., Gautam A., Mayer M.L., et al. Membrane fusion and immune evasion by the spike protein of SARS-CoV-2 Delta variant. Science. 2021;374:1353–1360. doi: 10.1126/science.abl9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yin W., Xu Y., Xu P., Cao X., Wu C., Gu C., He X., Wang X., Huang S., Yuan Q., et al. Structures of the Omicron spike trimer with ACE2 and an anti-Omicron antibody: mechanisms for the high infectivity, immune evasion and antibody drug discovery. bioRxiv. 2021 doi: 10.1101/2021.12.27.474273. [DOI] [Google Scholar]
- 40.Khoury D.S., Cromer D., Reynaldi A., Schlub T.E., Wheatley A.K., Juno J.A., Subbarao K., Kent S.J., Triccas J.A., Davenport M.P. Neutralizing antibody levels are highly predictive of immune protection from symptomatic SARS-CoV-2 infection. Nat. Med. 2021;27:1205–1211. doi: 10.1038/s41591-021-01377-8. [DOI] [PubMed] [Google Scholar]
- 41.Cevik M., Grubaugh N.D., Iwasaki A., Openshaw P. COVID-19 vaccines: keeping pace with SARS-CoV-2 variants. Cell. 2021;184:5077–5081. doi: 10.1016/j.cell.2021.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tarke A., Coelho C.H., Zhang Z., Dan J.M., Yu E.D., Methot N., Bloom N.I., Goodwin B., Phillips E., Mallal S., et al. SARS-CoV-2 vaccination induces immunological memory able to cross-recognize variants from Alpha to Omicron. bioRxiv. 2021 doi: 10.1101/2021.12.28.474333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Redd A.D., Nardin A., Kared H., Bloch E.M., Abel B., Pekosz A., Laeyendecker O., Fehlings M., Quinn T.C., Tobian A.A. Minimal cross-over between mutations associated with Omicron variant of SARS-CoV-2 and CD8+ T cell epitopes identified in COVID-19 convalescent individuals. bioRxiv. 2021 doi: 10.1101/2021.12.06.471446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Madelon N., Heikkilä N., Royo I.S., Fontannaz P., Breville G., Lauper K., Goldstein R., Grifoni A., Sette A., Siegrist C.-A., et al. Omicron-specific cytotoxic T-cell responses are boosted following a third dose of mRNA COVID-19 vaccine in anti-CD20-treated multiple sclerosis patients. medRxiv. 2021 doi: 10.1101/2021.12.20.21268128. [DOI] [Google Scholar]
- 45.Nelde A., Bilich T., Heitmann J.S., Maringer Y., Salih H.R., Roerden M., Lübke M., Bauer J., Rieth J., Wacker M., et al. SARS-CoV-2-derived peptides define heterologous and COVID-19-induced T cell recognition. Nat. Immunol. 2021;22:74–85. doi: 10.1038/s41590-020-00808-x. [DOI] [PubMed] [Google Scholar]
- 46.Yuan M., Huang D., Lee C.C.D., Wu N.C., Jackson A.M., Zhu X., Liu H., Peng L., van Gils M.J., Sanders R.W., et al. Structural and functional ramifications of antigenic drift in recent SARS-CoV-2 variants. Science. 2021;373:818–823. doi: 10.1126/science.abh1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lyngse F.P., Kirkeby C.T., Denwood M., Christiansen L.E., Mølbak K., Møller C.H., et al. Transmission of SARS-CoV-2 Omicron VOC subvariants BA.1 and BA.2: Evidence from Danish Households. medRxiv. 2022 doi: 10.1101/2022.01.28.22270044. [DOI] [Google Scholar]
- 48.Krissinel E., Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 49.Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F.T., de Beer T.A.P., Rempfer C., Bordoli L., et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harris C.R., Millman K.J., van der Walt S.J., Gommers R., Virtanen P., Cournapeau D., Wieser E., Taylor J., Berg S., Smith N.J., et al. Array programming with NumPy. Nature. 2020;585:357–362. doi: 10.1038/s41586-020-2649-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hunter J.D. Matplotlib: a 2D graphics environment. Comput. Sci. Eng. 2007;9:90–95. [Google Scholar]
- 52.Hagberg A., Schult D., Swart P. Proceedings of the 7th Python in Science Conference. 2008. Exploring network structure, dynamics, and function using NetworkX | BibSonomy; pp. 11–15. [Google Scholar]
- 53.Mckinney W. (2010). Data structures for statistical computing in Python. PROC. OF THE 9th PYTHON IN SCIENCE CONF., 445. (SCIPY 2010). p. 51-56.
- 54.Waskom M. seaborn: statistical data visualization. J. Open Source Softw. 2021;6:3021. [Google Scholar]
- 55.Chaudhury S., Lyskov S., Gray J.J. PyRosetta: a script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics (Oxford, England) 2010;26:689–691. doi: 10.1093/bioinformatics/btq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.The PyMOL Molecular Graphics System. Version 2.0 Schrödinger, LLC.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
-
•
All original code has been deposited at GitHub and is publicly available as of the date of publication. DOIs are listed in the key resources table. Additional Supplemental Items are available from Github/Zenodo at https://doi.org/10.5281/zenodo.5865881
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.