

Abstract
Vitamins and essential minerals are micronutrients that are essential for the normal functioning of the human body. However, they may lead to adverse health effects if consumed in excess. The concept of a tolerable upper intake level (UL) is a science‐based reference value, which was introduced to support policy‐makers and other relevant actors in managing the risks of excess nutrient intake. EFSA’s principles for establishing ULs for vitamins and minerals were originally developed by the Scientific Committee on Food in 2000. Since then, experience has been gained and the scientific field developed. This guidance from the EFSA Panel on Nutrition, Novel Foods and Food Allergens provides an updated framework to support EFSA’s UL assessments. It covers aspects related to the planning of the risk assessment (problem formulation and definition of methods) and its implementation (evidence retrieval, appraisal, synthesis, integration, uncertainty analysis). As in the previous framework, the general principles developed for the risk assessment of chemicals in food are applied (hazard identification, hazard characterisation, intake assessment, risk characterisation). Peculiar to nutrients are their biochemical and physiological roles and the specific and selective mechanisms that maintain the systemic homoeostasis and body burden of the nutrient. These must be considered when conducting a risk assessment of nutrients. This document constitutes a draft guidance that will be applied in EFSA’s assessments during a 1‐year pilot phase and be revised and complemented as necessary. Before finalisation of the guidance, a public consultation will be launched.
Keywords: dietary reference value, tolerable upper intake level, UL, vitamin, mineral
1. Background and terms of reference as provided by the European Commission
1.1. Background
Article 6 of Regulation (EC) No 1925/2006 on the addition of vitamins and minerals and of certain other substances to foods and Article 5 of Directive 2002/46/EC on the approximation of the laws of the Member States relating to food supplements provide that maximum amounts of vitamins and minerals added to foods and to food supplements respectively, shall be set.
The above‐mentioned provisions lay down the criteria to be taken into account when establishing these maximum amounts that include the upper safe levels (ULs) of vitamins and minerals established by scientific risk assessment based on “generally accepted scientific data, taking into account, as appropriate, the varying degrees of sensitivity of different groups of consumers”.
To set maximum amounts of vitamins and minerals in fortified foods and food supplements, the Commission would like to ask the European Food Safety Authority (EFSA) to review the previous opinions of the Scientific Committee on Food (SCF) or the NDA Panel on the ULs for vitamin A 1 , folic acid1/folate, vitamin D1, vitamin E1, vitamin B6, iron1, manganese1 and β‐carotene1 to take into account recent scientific developments and evidence.
In this context, EFSA should first review the guidelines of the SCF1 for the development of tolerable upper intake levels for vitamins and minerals (adopted on 19 October 2000).
Tolerable Upper Intake Levels should be presented separately for the age group from 4/6 months onwards until 3 years of age and the general population group from 3 years onwards, taking into account, as appropriate, the varying degrees of sensitivity of different consumer groups. As foods intended for the general population are also consumed by young children, young children should be considered as a potentially sensitive consumer group.
1.2. Terms of Reference
In accordance with Article 29(1)(a) of Regulation (EC) No 178/2002, the European Commission requests the European Food Safety Authority to:
Update the guidelines of the SCF for the development of Tolerable Upper Intake Levels for vitamins and minerals in the light of available recent scientific and methodological developments.
-
Review existing scientific evidence and provide advice on Tolerable Upper Intake Levels for the following vitamins and minerals including their currently authorised forms for the addition to fortified foods and food supplements for the general population and, as appropriate, for vulnerable subgroups of the population:
vitamin A
folic acid/folate
vitamin D
vitamin E
iron
manganese
β‐carotene
vitamin B6.
For nutrients for which there are no, or insufficient, data on which to base the establishment of a UL, an indication should be given on the highest level of intake where there is reasonable confidence in data on the absence of adverse effects.
2. Introduction
Vitamins and essential minerals are micronutrients that are essential for the normal functioning of the human body and need to be obtained from the diet. Like other chemical substances, micronutrients may lead to adverse health effects if consumed in excess. The concept of a UL refers to the maximum daily intake from all dietary sources (i.e. food and beverages, fortified foods and dietary supplements) above which a nutrient may cause adverse health effects. It was introduced to support policy‐makers and other relevant actors in managing the risks of ‘excess’ nutrient intake.
In 2000, the Scientific Committee on Food published guidelines for establishing ULs for vitamins and minerals (SCF, 2000a). It outlined general principles for the evaluation of the adverse effects of micronutrients in humans and for establishing ULs. In 2010, the NDA Panel published principles for deriving and applying dietary reference values (DRVs) (EFSA NDA Panel, 2010), where the concept and definition of UL were reiterated as part of DRVs for nutrients. Three DRVs (average requirement (AR), population reference intake (PRI) and lower threshold of intake (LTI)) describe the requirement distribution when average requirements can be determined (Figure 1). If not, two other values (adequate intake (AI) or reference intake range for macronutrients (RI)) can be proposed. In addition, a UL is established whenever possible for nutritional safety purposes (EFSA NDA Panel, 2010).
Figure 1.
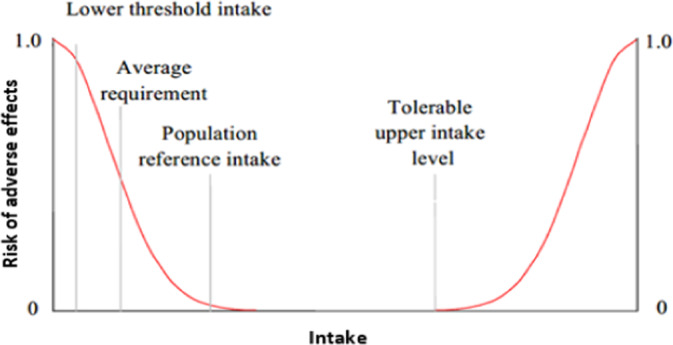
Relationship between individual intake and (cumulative) risk of adverse effects due to ‘insufficient’ or ‘excess’ intake. At intakes between the population reference intake and the tolerable upper intake level, the risk of inadequacy and the risk of excess are both close to zero. At intakes above the tolerable upper level, the risk of adverse effects increases
Examples of application of a UL include:
the setting of maximum amounts of micronutrients that can be added to foods or used in food supplements (by risk managers);
the evaluation of the safety of a new nutrient source prior to its marketing authorisation (by risk assessors);
the safety assessment of the intake of micronutrients by individuals or populations (by risk assessors, public health authorities or other health professionals).
Although the current framework for establishing ULs has proven to be useful in past evaluations, some aspects need to be reconsidered in light of the experience gained and relevant scientific developments in the field.
This guidance provides an updated framework for establishing ULs for vitamins and essential minerals (which include essential trace elements). In contrast to chemical substances which are not nutrients, nutritional requirements need to be considered when establishing ULs for essential nutrients, i.e. there is a level of intake below which the risk of deficiency or suboptimal function arises (EFSA NDA Panel, 2010). The guidance also provides further explanations on the interpretation and potential applications of ULs.
3. Data and methodologies
Several reports on ULs for nutrients have been consulted in preparing the present document (WHO/IPCS, 2002; FAO/WHO, 2006, 2009, 2020; US NASEM, 2017; Australian Government Department of Health, 2015; NTP OHAT, 2019; EFSA Scientific Committee, 2021a).
The revision of the guidance was also informed by the feedback collected through an expert workshop organised by EFSA, held on 28–29 September 2021, on data and methodologies for establishing ULs for vitamins and minerals (EFSA, 2022).
This document constitutes draft guidance that will be applied in EFSA’s assessments for a 1‐year pilot phase and be revised and complemented as necessary. Before finalisation of the document, a public consultation will be launched.
4. Definitions
Tolerable upper intake level (UL): The maximum level of total chronic daily intake of a nutrient (from all sources) which is not expected to pose a risk of adverse health effects to humans.
The previous definition of a UL from the SCF was as follows: ‘the maximum level of total chronic daily intake of a nutrient (from all sources) judged to be unlikely to pose a risk of adverse health effects to humans.’ It has been reworded to address ambiguity related to the term ‘unlikely’ which may be understood as a probability statement (EFSA Scientific Committee, 2018a,b).
A UL is a type of health‐based guidance value 2 which is applied to nutrients (EFSA Scientific Committee, 2021a).
‘Tolerable intake’ in this context connotes what is physiologically tolerable and can be established based on an assessment of risk, i.e. the probability of an adverse effect occurring at a specified level of exposure. The UL is not a recommended level of intake. As the intake increases above the UL, the risk of adverse effects increases.
The following terms are embedded in the definition of a UL:
Adverse (health) effects: An effect is considered ‘adverse’ when ‘leading to a change in the morphology, physiology, growth, development, reproduction or life span of an organism, system or (sub)population that results in an impairment of functional capacity to compensate for additional stress or an increase in susceptibility to other influences’ (FAO/WHO, 2009; EFSA Scientific Committee, 2017a). The observable effects of high nutrient intake within the causal pathway of an adverse health effect can range from biochemical changes without functional significance (e.g. certain changes in enzyme activity) to irreversible clinical outcomes (Box 1). Notably, some changes that occur before clinical manifestations could be used as surrogate or predictive markers of subsequent adverse health effects, i.e. biomarkers of effect (see below).
Biomarkers of effect: ‘A measurable biochemical, physiological, behavioural or other alteration within an organism that, depending upon the magnitude, can be recognised as associated with an established or possible health impairment or disease’ (WHO/IPCS, 1993; EFSA Scientific Committee, 2017a). Its biological relevance depends on its relation to the mode of action and the linkage with the adverse effect or the relevant adverse outcome pathway (EFSA Scientific Committee, 2017a).
Box 1. Sequence of potential effects related to high nutrient intake
Total chronic daily intake: Average daily nutrient intake over a substantial part of the lifespan, also referred to as the ‘usual’ or ‘habitual’ intake of a nutrient. It is recognised that intakes of vitamins and minerals may be characterised by considerable intra‐individual day‐to‐day and seasonal variations. ‘Total’ refers to ‘from all sources’, which is intake from all dietary sources, i.e. food (including fortified foods), beverages (including water) and food supplements.
Risk of adverse (health) effect: Probability of an adverse effect in an organism, system or (sub)population caused under specified circumstances by exposure to an agent (WHO/IPCS, 2004). In the context of a nutrient risk assessment, ‘risk’ refers to the probability of an adverse effect at a given nutrient intake level.
A theoretical representation of the risks of adverse health effects associated with the intake of a given essential micronutrient and the corresponding DRV values is depicted in Figure 1.
Target population: ULs should be protective for all members of the general population, including sensitive individuals, throughout their lifetime.
Regarding the effects of ‘excess’ nutrient intake, no risk of adverse effects is expected unless a threshold of intake is exceeded. Thresholds for any given adverse effect vary among members of the population, i.e. there is a distribution of individual thresholds within the general population (inter‐individual variation in sensitivity). In theory, ULs should be established by defining a point in the distribution of thresholds that would be protective for the whole population.
Adverse effects of excess nutrient intake may be influenced by the changes associated with growth, development and ageing that occur during an individual’s lifespan. Therefore, where necessary, and to the extent possible, ULs are derived for each separate life‐stage group, e.g. infants, children, adults, older adults and women during pregnancy or lactation. Sex‐specific values should be established where relevant.
Even within relatively homogeneous life‐stage groups, there is a range of sensitivities to adverse effects. The derivation of ULs accounts for the expected variability in sensitivity among individuals. In principle, individuals under medical care are not excluded unless: (a) There is an expected interaction between the medical condition and the occurrence of possible adverse effects of a nutrient, or (b) they are under medical treatment with the nutrient under assessment.
However, the UL may exclude subpopulations with extreme and distinct vulnerabilities to adverse effects of the nutrient due to specific genetic predisposition or other factors. Including those subpopulations would result in ULs which are significantly lower than needed to protect most people against adverse effects of high intakes. Subpopulations needing special protection are better served through the use of public health screening, healthcare providers, product labelling or other individualised strategies. The exclusion of such subpopulations must be considered on a nutrient‐by‐nutrient basis and is an area of scientific and expert judgement and of risk management. In practice, the exclusion of a subpopulation from a UL should take into consideration whether individuals from that group can be recognised (e.g. through screening, diagnostic) and handled through specific measures.
5. Problem formulation and definition of methods
In general, the principles developed for the risk assessment of chemicals in food (FAO/WHO, 2009) also apply to nutrients. However, peculiar to nutrients are their biochemical and physiological roles and the specific and selective mechanisms that maintain the systemic homoeostasis and body burden 3 of the nutrient over a range of intakes. This must be taken into account when conducting a risk assessment for nutrients. The four steps of the risk assessment process are illustrated in Figure 2.
Figure 2.
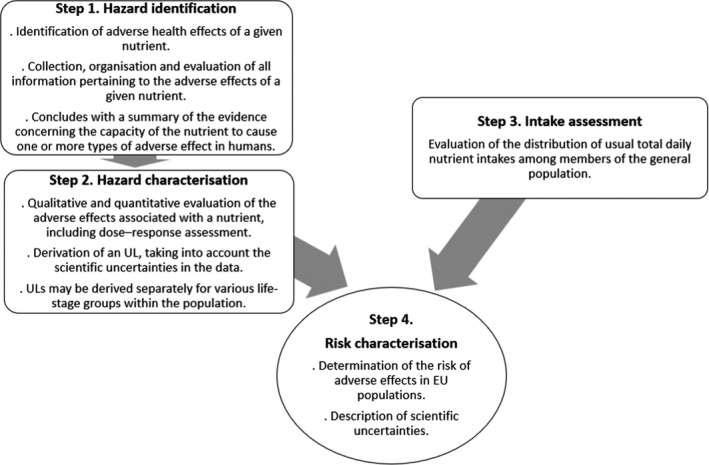
Four‐step process of nutrient risk assessment
The assessment questions underlying the UL evaluation can be formulated as follows:
What is the maximum level of total chronic daily intake of NUTRIENT X (from all sources) which is not expected to pose a risk of adverse health effects to humans? (Hazard identification and characterisation)
What is the daily intake of NUTRIENT X from all dietary sources in EU populations? (Intake assessment)
What is the risk of adverse effects related to the intake of NUTRIENT X in EU populations, including the attendant uncertainties? (Risk characterisation).
The UL evaluation follows EFSA’s scientific assessment processes (EFSA, 2020) (Figure 3). As a first step, a protocol is developed which clarifies the aim and scope of the assessment (problem formulation) and defines the methods to address the problem. For each UL evaluation, the problem formulation requires the exposure of interest to be specified (Section 5.1) and the relevant endpoints to be identified (Section 5.2) along with the subpopulations, where appropriate (Section 5.3). The assessment questions are broken down into subquestions that are specific to the nutrient under evaluation and the evidence needs and the methods used to address each subquestion are defined (Section 5.4).
Figure 3.
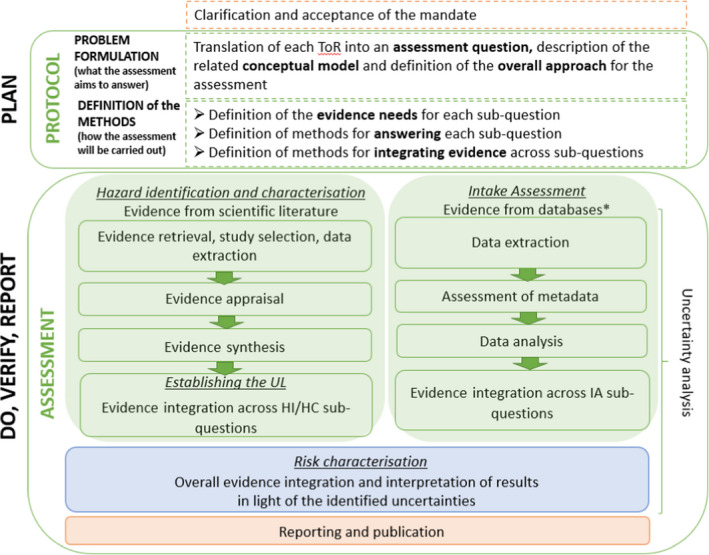
- HC: hazard characterisation; HI: hazard identification; IA: intake assessment; ToR: terms of reference; UL: tolerable upper intake level.*: Data may also be extracted from published intake assessment reports.
5.1. Definition of the exposure of interest
By definition, the UL refers to the total chronic daily intake of the nutrient from all dietary sources.
A nutrient can occur in different chemical forms in the diet, which may have different properties with regard to absorption, distribution, metabolism, elimination and biological functions in the body. Relevant information on the chemical forms of the nutrient and their sources should be considered to define the exposure of interest for the risk assessment. Specific considerations may be necessary regarding the bioavailability of the nutrient (or its specific chemical forms), as it may affect the nature and severity of adverse effects.
As a result, it may be possible to define a priori the focus of the risk assessment on a specific chemical form of the nutrient (or selected forms), or a particular source of the nutrient from which the specific chemical form originates (e.g. food supplements). At times, the need to derive a UL for specific chemical forms of the nutrient (or dietary sources thereof) may only be identified as a result of the hazard identification and characterisation.
In past evaluations, ULs have been established for specific forms of a nutrient, e.g. added folic acid (SCF, 2000b), nicotinamide and nicotinic acid (SCF, 2002) or readily dissociable magnesium salts (e.g. chloride, sulfate, aspartate, lactate) and compounds like MgO when added to foods or consumed as food supplements (i.e. in addition to Mg normally present in foods and beverages) (SCF, 2001).
Of note, the chemical forms of a nutrient that are authorised to be added to foods and for use in food supplements are those listed in Annex II of Regulation (EC) No 1925/2006, in Annex to Regulation (EC) No 609/2013 4 and in Annex II of Directive 2002/46/EC. The addition of a new form of a vitamin or a mineral to these Annexes is subject to their evaluation by EFSA as a nutrient source, in which the safety and bioavailability of the nutrient from that source are assessed (EFSA ANS Panel, 2018). Whether the established UL for a nutrient also applies to its new form(s) is considered in the evaluation of new nutrient sources.
5.2. Identification of relevant endpoints
Prior knowledge of the biological responses resulting from nutrient intake is needed to identify the relevant endpoints. The generic chain of potential events accompanying increasing intake and body burden of nutrients is illustrated in Figure 4. The nature of the endpoints relevant to establishing a UL can be diverse, ranging from the initial biochemical changes in response to ‘excess’ nutrient intake, to clinical signs or symptoms of toxicity, or (chronic) disease endpoints.
Figure 4.
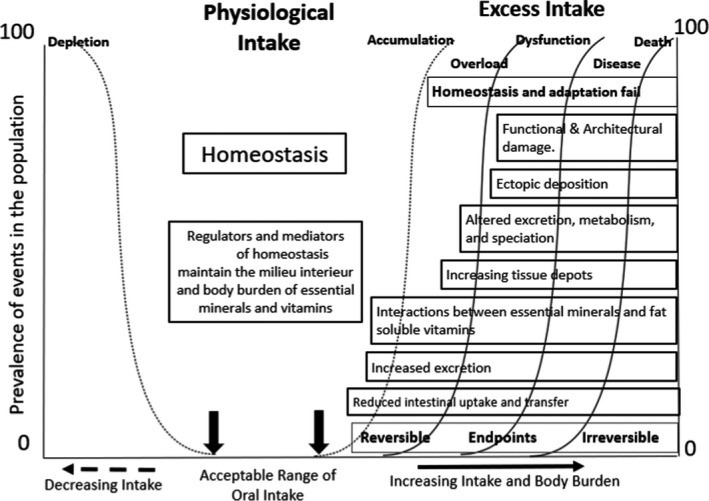
- The intakes indicated at the top progress from adequate to excessive. The boxes describe potential physiological and pathological responses to increasing intakes, and, in this context of chronic excess and subsequent toxicity, the increasing body burden of the nutrient being considered. This figure illustrates a schema for the integration of evidence on adverse health effects and pathophysiological sequelae which in turn would aid the identification of endpoints as candidate biomarkers and an appreciation of the mechanisms of adverse health effects (i.e. mode of action) for subsequent features such as disease (Figure adapted from EFSA Scientific Committee, 2021a ).
Specific to nutrients, the identification of relevant endpoints from among the homoeostatic and adaptive responses to excessive intakes has been identified as a useful approach for nutrient risk assessment (WHO/IPCS, 2002; FAO/WHO, 2006; EFSA Scientific Committee, 2021a). In this context, relevant endpoints may not necessarily be hazards or adverse effects, but early biochemical changes or biological markers for which a mechanistic pathway can be discerned, and which can be characterised and validated as predictive of adverse effects.
On the left half of Figure 4, the physiological regulators and mediators of homoeostasis refer to the mechanisms of absorption, distribution, metabolism and excretion (ADME) involved in maintaining a constant body burden (i.e. the milieu intérieur). As intakes increase, the homoeostatic mechanisms become overwhelmed and begin to fail to control the body burden. The increasing body burden elicits adaptive responses involving, amongst others, altered metabolism and speciation, increased deposition of the nutrient and/or its metabolites in tissues (in many instances, the liver is the key organ involved in both homoeostasis and adaptation), to different extents depending on the nutrient. Prolonged excessive intake leads to overload and pathological events develop. Initially these features are reversible (as per the categorisation depicted in Box 1), in that adverse biochemical and physiological changes are likely to reverse in response to a reduced intake. However, if a high intake is maintained, phenomena arising from abnormal metabolite production, excess tissue deposition and ultimately ectopic deposition, with resultant tissue and organ damage, and organ failure, will occur. The latter are associated with clinical features, the reversibility of which is uncertain, and which may contribute to overt clinical disease (Box 1). The periods over which the endpoints appear are highly variable; they can extend over decades and often the events occur concurrently.
Various sources of evidence may be used for the identification of relevant endpoints. These include both experimental and observational studies in humans. Animal data may be helpful, e.g. to identify target organs and pathologies, or to describe the sequential development of toxicological endpoints, which might enable the tracing of pathogenic events in the physio‐pathological pathway (EFSA Scientific Committee, 2021a). Also, developments in bioinformatics have fostered ‘systems biology’ approaches. These are being developed to enhance toxicological risk assessment and nutritional science (Krewski et al., 2020) and which embrace and enable the integrated use of genomics, transcriptomics, proteomics and metabolomics to explore the dynamics and systemic kinetics of compounds (EFSA Scientific Committee, 2021a).
5.3. Identification of relevant subpopulations
ULs should be protective for all members of the general population, throughout the lifetime (i.e. infants, children, adults, older adults, pregnant and lactating women). Prior knowledge should be used to identify any life‐stage groups of the population or subpopulations particularly relevant for assessment (e.g. in relation to specific endpoints).
As stated above (Section 4), a UL may exclude subpopulations with extreme and distinct vulnerabilities due to genetic predisposition or other factors. Including those subpopulations would result in ULs which are significantly lower than needed to protect most people against adverse effects of high intakes. This may be identified as part of the problem formulation for individual nutrients, based on prior knowledge. The exclusion of such subpopulations is an area of scientific and expert judgement and of risk management. Decisions on the exclusion of specific subpopulations at this stage should be documented and this should be specified in the risk characterisation (Section 8).
5.4. Definition of assessment subquestions and related methods
Based on the above, the assessment question is broken down into a series of subquestions.
Subquestions addressing the identification and characterisation of relationships between high intake of the nutrient and adverse health effects (or related markers) are typically addressed through systematic reviews (EFSA, 2010).
As conducting systematic reviews is resource intensive, the formulation of key subquestions requires the identification of priority endpoints with an expected critical role for establishing a UL. Scoping literature searches and expert judgement can be used to that end. The selection of priority endpoints should consider:
the nature of their relationship with adverse health effects (Figure 5). As mentioned above, relevant endpoints are not restricted to clinical outcomes (i.e. signs or symptoms of toxicity, clinically diagnosed disease) but may also include relevant biomarkers of effect. Biomarkers of effect that are associated with adverse health effects are preferred. However, biomarkers of exposure could also be used when they are indicative of ‘normal’ homoeostatic mechanisms being overwhelmed and body burden increasing, or of the induction of adaptive and potential subsequent pathological processes as illustrated in Figure 4. The selection of a biomarker should be based on considerations of its validity and reproducibility as a surrogate for or predictor of adverse health effects.
the availability of experimental and/or observational data (preferably in humans) that could be used to characterise a dose–response relationship between the intake of the nutrient and the selected endpoint(s). Availability of such data will be critical to the hazard characterisation step of the risk assessment and, ultimately, the derivation of a ‘value’.
Figure 5.
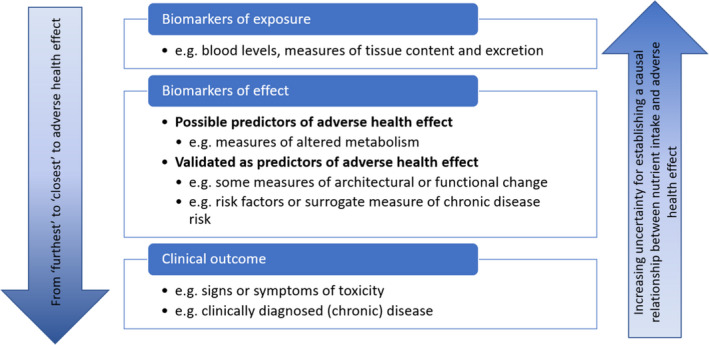
The sequential relationship between potential endpoints for adverse health effects
The rationale for the prioritisation of endpoints should be documented.
Other subquestions may aim at collecting important information to support the risk assessment (e.g. ADME, knowledge about the potential mode of action), and may be addressed through narrative reviews or other approaches.
Examples of subquestions relevant to the evaluation of a UL are provided in Table 1.
Table 1.
Examples of assessment subquestions for the evaluation of a tolerable upper level
| Risk assessment step | Subquestion | Example of method to answer the subquestion |
|---|---|---|
| HI/HC | What is the ADME of NUTRIENT X in humans? | Narrative review |
| HI/HC | What are the potential mode(s) of action underlying the relationships between NUTRIENT X intake and adverse health effects? | Narrative review |
| HI | Is there a positive and causal relationship between NUTRIENT X intake and endpoint Y in humans? | Systematic review |
| HC | What is the dose–response relationship between NUTRIENT X intake and endpoint Y in humans? | Dose–response modelling |
| IA | What is the daily NUTRIENT X intake from all dietary sources in EU populations? | Narrative review |
HI: hazard identification; HC: hazard characterisation; IA: intake assessment; RC: risk characterisation.
6. Hazard identification and characterisation
Following the planning phase, the hazard identification and characterisation steps are implemented following a structured process (Figure 3).
6.1. Evidence retrieval, study selection, data extraction
For nutrient risk assessments, human studies provide the most relevant data for hazard identification and characterisation. Other studies (e.g. animal studies) may also be used when human studies are deemed not to be feasible (e.g. to investigate specific toxicity endpoints such as genotoxicity, reproductive toxicity) or sufficient (or of sufficient quality). As stated above, animal data may be helpful, e.g. to identify target organs and pathologies, or to describe the sequential development of toxicological endpoints, which might enable the tracing of pathogenic events in the physio‐pathological pathway (EFSA Scientific Committee, 2021a).
As mentioned above, systematic reviews are favoured to address subquestions on the relationships between high intake of the nutrient and adverse health effects (or related markers). The evidence is retrieved through a predefined search strategy. Studies identified through the search are then selected based on criteria that have been defined a priori in the protocol (i.e. eligibility criteria). For each study included in a systematic review, relevant data on the study findings and characteristics are extracted in a standardised format (e.g. evidence tables). This step forms the basis for the evidence synthesis.
The body of evidence for evaluating causality (hazard identification) and the body of evidence for characterising the nature of the dose–response (hazard characterisation) will largely overlap but do not necessarily coincide (e.g. the body of evidence on dose–response may be more restricted as it requires quantitative estimates of intake). This must be taken into account in designing the literature search strategy, in screening the evidence for eligibility and also in extracting the data.
An important consideration in selecting human studies is whether the study population is representative of the target population (e.g. a study among diseased individuals). This requires case‐by‐case decisions based on expert knowledge. The rationale for such decisions should be documented.
Other experimental studies (e.g. animal studies, studies in vitro) can be useful as supporting evidence for the hazard identification and characterisation, e.g. to assess the biological plausibility of a nutrient–effect relationship or mode(s) of action.
6.2. Evidence appraisal
The internal validity or risk of bias (RoB) of eligible studies is appraised. Internal validity is the extent to which a piece of evidence provides an unbiased estimate of the causal association between exposure and outcome, i.e. the extent to which the study results reflect the ‘truth’ among the study population (EFSA Scientific Committee, 2020). For a given study, assessment of internal validity refers to the evaluation of its design and conduct, particularly in terms of the likelihood, magnitude and direction of possible biases.
The NDA Panel evaluates the internal validity of individual studies (RoB) through the use of a critical appraisal tool (CAT). CATs are structured checklists that allow the identification of potential threats to the internal validity of studies using a set of criteria (EFSA Scientific Committee, 2020). Specific tools are available to appraise RoB relevant to different study designs (e.g. NTP OHAT (NTP OHAT, 2015), Cochrane RoB‐2 (Sterne et al., 2019), NESR RoB‐NObs (NESR, 2019)). They support the formulation of RoB judgements on RoB domains identified as critical for each design. The method to appraise RoB in individual studies should be described in the protocol and the outcomes of the appraisal should be reported in the scientific opinion.
6.3. Evidence synthesis
If several studies address the same endpoint and are sufficiently comparable that they can be combined, the evidence may be synthesised through a meta‐analysis to estimate the pooled effect size and related confidence interval. Strengths of meta‐analyses include their ability to increase the statistical power and the precision of effect estimates and to provide a summary of the strength and consistency of the evidence, which are important elements in judging causal relationships and in shaping the relationship between exposure and effect. However, if heterogeneity among available studies is high, a narrative synthesis of the evidence is the most appropriate approach.
Data modelling should be used, where possible. Dose–response meta‐analyses can be valuable in describing the shape of the relationship (e.g. linear or non‐linear; monotonic or not) and for the quantification of any relationship between the nutrient intake and the occurrence/level of the endpoint of interest. Mechanistic data can help to interpret the biological plausibility of the dose–response shape. The possibility of modelling the dose–response will depend on the nature and extent of the available data. An array of modelling approaches may be used and/or adapted to address the relevant assessment questions. The choice of the approach requires technical support and expertise, and should consider methodological developments in the field (Vinceti et al., 2020).
Subgroup analyses or multivariable meta‐regression can be helpful in exploring potential sources of heterogeneity in the available data.
Sensitivity analyses should be conducted where possible to examine the influence of specific assumptions, methodological choices and studies on the results of the analysis.
6.4. Evidence integration and conclusions
6.4.1. Hazard identification: judging the level of certainty for causal relationships and biological relevance
An important element of the hazard identification step is judging the level of certainty for a causal relationship between the intake of the nutrient and adverse health effects based on the available body of evidence.
Some nutrient–effect relationships are well established in the scientific literature and the assessment will focus on the characterisation of the dose–response.
For other relationships (e.g. association with chronic diseases risk), inference about causality requires consideration of critical characteristics of the available body of evidence, such as study designs, internal validity of the available studies (RoB), consistency, magnitude and precision of the relationship, dose–response relationship (a biological gradient), mode of action. Several frameworks have been developed to support the formulation of a scientific judgement on the causality of an exposure–endpoint relationship (e.g. GRADE, NTP OHAT). It is important to apply a consistent framework to reach a common understanding of the level of certainty in the causality of a nutrient–endpoint relationship among experts and provide transparency to the risk assessment. The existing approaches are not completely consistent with EFSA’s methodological recommendations (e.g. EFSA guidance on uncertainty analysis (EFSA Scientific Committee, 2018a,b)) or fully take into account the variability of the sources of evidence and approaches to deal with evidence thus they require tailoring to EFSA’s risk assessment questions (EFSA NDA Panel, 2022). Further work is needed to adapt the existing approaches or develop new ones. This is beyond the scope of this guidance.
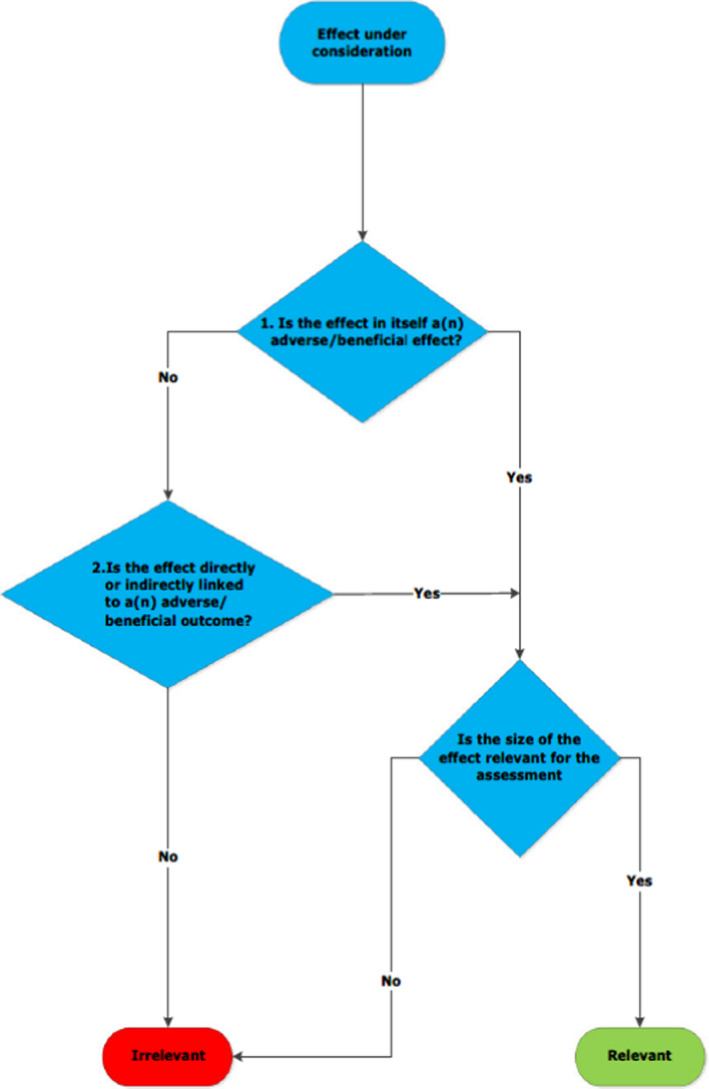
The biological relevance of observed effects should be considered. The principles outlined in the guidance on the assessment of the biological relevance of data in scientific assessments (EFSA Scientific Committee, 2017a) will be applied to assess the relevance of each effect (nature and size). This includes consideration of: (i) whether the effect is adverse in itself; (ii) whether it is directly or indirectly linked to an adverse effect; and (iii) whether the size of the effect is considered relevant for the assessment (Appendix A).
6.4.2. Hazard characterisation: establishing the UL
At this step, evidence is integrated to select the critical effect and identify a reference point (RP) for establishing the UL. ULs are derived for different life‐stage groups using relevant data for each group, where available.
The UL is derived as follows:
where UF is an uncertainty factor which accounts for the uncertainties associated with extrapolating from the observed data to the general population.
Subpopulations with particular sensitivities to the adverse effects of the nutrient may have been identified from the literature review. When applicable, a decision needs to be taken on whether to include or exclude them when establishing the UL.
6.4.2.1. Selection of a reference point
Based on the conclusions of the hazard identification step (Section 6.4.1), only endpoints biologically relevant for the assessment should be considered for hazard characterisation and identification of an RP.
Whenever the data allow, dose–response modelling should be used for that purpose.
The RP on which to base the UL would correspond to the intake level on the dose–response curve at which the risk of a specific adverse effect is minimal (Figure 6A), or to the upper bound of the intake range for which the risk is minimal (Figure 6B).
Figure 6.
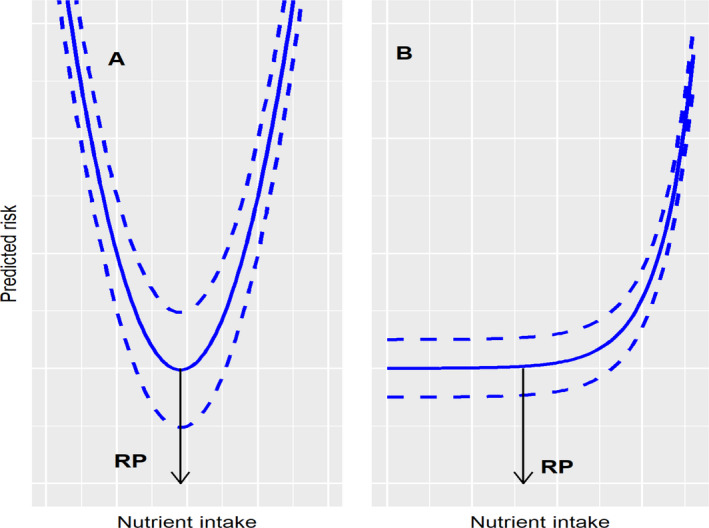
An illustration of the derivation of a reference point, depending on the shape of the relationship between nutrient intake and risk of adverse effect
If a biologically relevant change in a selected endpoint can be identified, the principles of the benchmark dose approach could be considered to identify an RP (FAO/WHO, 2009, 2020; EFSA Scientific Committee, 2017b). An illustration of the implementation of these principles for the assessment of the UL for vitamin D in infants (EFSA NDA Panel, 2018) is provided in Appendix B.
When available data are not suitable for dose–response modelling, a no‐observed‐effect level (NOEL) or a lowest‐observed‐effect level (LOEL) might be identified and used as the RP.
Where several adverse effects (or related markers) are identified for a nutrient, the critical effect to be used for the identification of the RP for establishing a UL needs to be selected (e.g. the effect occurring at the lowest dose). This needs expert judgement and integration of the totality of the evidence, considering the reliability, relevance and consistency of available pieces of evidence (EFSA Scientific Committee, 2017c).
It is recognised that challenges will arise when the nature of identified relationships varies widely (e.g. contribution of a nutrient to the risk of multifactorial chronic diseases vs. its specific toxicity). The framework for establishing a UL in this situation needs to be developed.
The rationale for the RP used as a basis for the UL should be documented, including information on the underlying assumptions and uncertainties.
6.4.2.2. Application of a UF
The application of a UF is intended to establish a UL which is protective for the general population, accounting for the expected variability in sensitivity among individuals.
Several judgements must be made in determining the UF to account for uncertainties in the body of evidence.
When the RP is identified from human data, a default UF of 10 is used in chemical risk assessment to account for inter‐individual variability in the absence of chemical‐specific data on kinetics and/or dynamics (EFSA Scientific Committee, 2012). For nutrients, the default UF of 10 may be reduced based on one or more of the following:
the amount, quality and diversity of human studies available, as they may account for part of the inter‐individual variability;
the severity and reversibility of the adverse health effects, if these are mild and reversible or are biomarkers of a potential future effect;
availability of data on the toxicokinetics and toxicodynamics of the nutrient in humans.
Applying a UF of 1.0 expresses confidence that, based on the available body of evidence, intakes of the nutrient up to the identified RP are not expected to pose a risk of adverse health effects for the general population, and the RP can thus be used as the UL.
When the RP is identified from animal data, additional uncertainties need to be considered in relation to extrapolation of data from animals to humans. This is addressed through the application of an additional UF, which accounts for inter‐species variation in toxicokinetics and toxicodynamics (EFSA Scientific Committee, 2012). If relevant nutrient‐specific data on kinetics and/or dynamics are available, this additional factor should preferably be based on real data (EFSA Scientific Committee, 2012; EFSA FAF Panel, 2019; Smeraldi et al., 2020). In the absence of such data, as with chemical risk assessment, a default factor of 10 may also be applied in nutrient risk assessment (EFSA Scientific Committee, 2012).
Importantly, nutritional needs need to be considered in determining the UF, so that the UL does not trigger a risk of deficiency in the general population.
The choice of the UF is a matter of scientific judgement, and the rationale for that choice should be explained, taking into account uncertainties in the body of evidence and the methods applied.
6.4.2.3. Scaling approaches
For a specific life‐stage group for which insufficient or no data are available (e.g. children), scaling may be made from the UL for other groups (e.g. adults). Selection of the appropriate scaling method for a nutrient should consider the relationship of its disposition to the metabolic rate or measures of body weight of the various age groups. Scaling between different population groups, such as isometric (linear with body weight) or allometric (body weight to the power of a chosen exponent) scaling, is made according to scientific judgement on a case‐by‐case basis (Appendix C). Metabolic weight has been defined as 0.75 power of body mass (weight) and is used as the preferred method to adjust for metabolic differences between age groups (EFSA NDA Panel, 2010). Reference body weights are used for scaling, which are based on observed body weights in EU populations for the relevant age groups (Appendix C).
Because of the assumptions made in scaling, due to lack of knowledge on the proportionality of nutrient requirements with parameters such as body weight or metabolic rate during growth, reference values for infants and children might have a higher uncertainty than those for adults (EFSA NDA Panel, 2010).
6.4.3. Approaches when there are insufficient data to establish a UL
For nutrients for which there are insufficient data on which to base the UL, an indication should be given on the highest level of intake for which there is reasonable confidence on the absence of adverse effects (e.g. from the highest supplemental intakes; Figure 7), drawing from the totality of the available evidence. The application of such a value for risk assessment and risk management is more limited than a UL because the proportion of people at risk of adverse effects in a population cannot be estimated, as the intake level at which the risk of adverse effects starts to increase is not defined.
Figure 7.
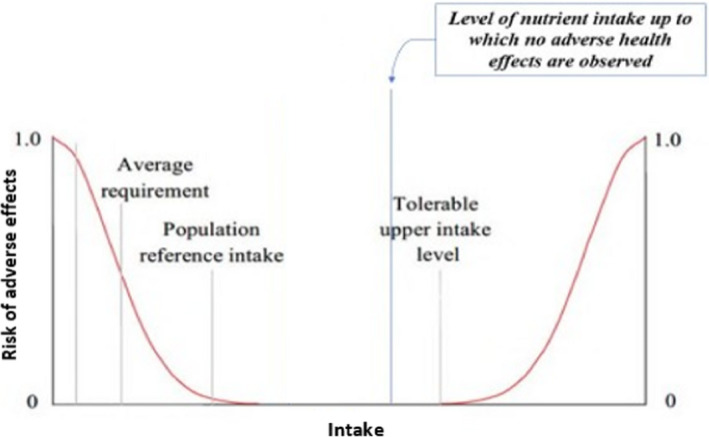
Theoretical representation of the ‘highest level of intake where there is reasonable confidence on the absence of adverse effects’. The threshold above which the actual risk of adverse effects starts to increase is unknown
For instance, it may be based on the highest supplemental intakes with no observed adverse effects in long‐term human trials, if the evidence is considered adequate and sufficient (EFSA NDA Panel, 2012).
6.4.4. Approaches when no hazard is identified
For a nutrient for which, on the basis of the available data (e.g. toxicity data, intake estimates), the Panel considers that it does not represent a hazard to health, a UL may not need to be specified.
7. Intake assessment
Intake assessment in the field of nutrients aims at characterising the dietary intake by combining data on the concentrations of the nutrient in foods and beverages with the quantity of those foods and beverages consumed. However, a comprehensive characterisation of risks associated with the dietary intake of a nutrient requires a complete intake assessment from all dietary sources, i.e. accounting for the natural nutrient content of foods as well as any additional contributions of fortified foods and food supplements. Moreover, since some nutrients find additional uses in regulated products (e.g. food additives, feed additives, pesticides), the intake resulting from these uses (direct intake for food additives, indirect intake through residues for feed additives and pesticides) should also be taken into account (EFSA Scientific Committee, 2021a).
Total dietary intake of nutrients can be estimated by combining data from food composition databases 5 and food consumption surveys. 6
When estimating the total dietary intake of nutrients, sources of uncertainties pertaining to nutrient composition data include:
The extent to which the values reported in food composition databases are representative of the foods consumed.
The contributions of nutrient content in foods due to fortification and to other uses in nutrient‐containing regulated products.
The speciation of the nutrient, i.e. distribution of its various chemical and physical forms, where applicable.
With regard to points (1) and (2) above, issues that may limit the representativeness of nutrient values in food composition databases include their update in relation to changes in production processes and in product formulations (i.e. the capacity to reflect such changes in a timely manner), the ability to reveal complex transformations in nutrient content induced by cooking methods and the extent to which they capture variability of nutrient content across seasons and geographical areas. Indeed, the use of robust databases representative of the foods and beverages consumed in each country would lead to more accurate estimates; unfortunately, such databases do not currently exist for the totality of EU Member States.
The availability of data from total diet studies (TDS) may enable the uncertainties associated with these limitations of the existing food composition databases to be reduced. TDS are specifically tailored to population chronic intake assessment, usually at the national level, and are based on representative sampling of the whole diet, with food analysed as consumed and pooled into defined food groups (EFSA/FAO/WHO, 2011). Therefore, when available, TDS data can be used for comparison with nutrient intake estimates based on the use of food composition databases or other methods and provide an indication of the attendant uncertainties.
The nature of the consumption data used also affects the overall uncertainty of intake assessment. Intake to nutrients resulting from usual (i.e. habitual) dietary intake, intended as long‐term average daily intake, is of interest in relation to UL. However, consumption data are collected via short‐term measurements of food intake and the resulting intake distributions incorporate bias from within‐person variability. Such bias especially affects the estimation of the higher percentiles and thus the percentage of the population that may be estimated to exceed the UL. Statistical modelling can be applied to improve the reliability of these estimates (Dodd et al., 2006; van Klaveren et al., 2012).
When available, biomarkers of exposure may be useful to estimate overall nutrient intake. Biomarkers of exposure reflect the internal dose and exposure from all sources. With few exceptions (e.g. vitamin D), the internal dose of nutrients (and their metabolites) reflects dietary intake. However, reliable biomarkers of exposure are only available for a limited number of nutrients. When these biomarkers are used, backcalculation to dietary intake using kinetic modelling may be explored (EFSA Scientific Committee, 2021a).
8. Risk characterisation
This step aims at estimating the probability of occurrence of potential adverse health effects in a population by integrating the results of the hazard identification, hazard characterisation and intake assessment steps, including the related uncertainties. Expressions of risk may be qualitative, quantitative or both. Associated uncertainties and data gaps should be discussed.
Typically, the UL is compared with estimates of usual intake in EU populations. If usual intakes for all individuals in a population are below the UL, no adverse effects are expected to occur. Conversely, the proportion of the population with usual intakes above the UL represents a potential at‐risk group. Factors to consider when assessing the risk of excess intakes of the nutrient include:
the accuracy of the intake data;
the percentage of the population with usual intakes above the UL and the magnitude and duration of the exceedance;
the nature and severity of the adverse effect, e.g. the extent to which the adverse effect is reversible when intakes are reduced to levels below the UL.
The risk characterisation should indicate whether subpopulations having distinct and exceptional sensitivities to the adverse health effects of the nutrient have been excluded.
When no UL is established, the identified ‘highest level of intake for which there is reasonable confidence on the absence of adverse effects’ (Section 6.4.3) can be considered a conservative value to assist risk management decisions (e.g. on the setting of maximum amounts of vitamins and minerals to be added to foods or used in food supplements). Associated uncertainties and implications for risk management should be discussed.
9. Application of ULs to assess risks for individuals or populations
9.1. Application of ULs to assess risks for individuals
If an individual’s usual nutrient intake remains below the UL, no adverse effects are expected to occur. At habitual intakes above the UL, the risk of adverse effects increases as the level of intake increases. However, the intake at which a given individual will develop adverse effects because of taking excessive amounts of a nutrient is not known with certainty. In practice, the UL can be used as an upper bound for the maximum tolerable level of usual intake for individuals. The UL is not a recommended intake.
Occasional, short‐term and limited excursions above the UL are possible without adverse effects.
9.2. Application of ULs to assess risks for populations
The UL applies to the most sensitive members of the general population. Some individuals may regularly consume nutrients at or even somewhat above the UL without experiencing adverse effects. However, because it is not known which individuals are most sensitive, it is necessary to interpret the UL as applying to all individuals.
Usual intake distributions (i.e. percentiles) allow determination of the fraction of the population exceeding the UL, i.e. at risk. The accuracy of available estimates of usual intakes will affect the reliability of the assessment of risks of adverse effects. Physiological measures can be helpful when assessing the dietary intake of individuals or groups of people and could in theory be used to complement or confirm estimates of risk based on dietary data. However, such indicators are often lacking.
Abbreviations
- ADME
Absorption, distribution, metabolism and excretion
- AI
Adequate intake
- ANS
Panel on Food Additives and Nutrient Sources added to Food
- AR
Average requirement
- CAT
Critical appraisal tool
- DRVs
Dietary reference values
- FAF
Panel on Food Additives and Flavourings
- FAO
Food and Agriculture Organization
- GRADE
Grading of Recommendations, Assessment, Development and Evaluations
- IPCS
International Programme on Chemical Safety
- LOEL
Lowest‐observed‐effect level
- LTI
Lower threshold of intake
- NASEM
National Academies of Sciences, Engineering and Medicine
- NDA
Panel on Nutrition, Novel Foods and Food Allergens
- NOEL
No‐observed‐effect level
- NTP OHAT
National Toxicology Programme, Office of Health Assessment and Translation
- PRI
Population reference intake
- RI
Reference intake range for macronutrients
- RoB
Risk of bias
- RP
Reference point
- SCF
Scientific Committee on Food
- TDS
Total diet study
- UF
Uncertainty factor
- UL
Tolerable upper intake level
- WHO
World Health Organization
Glossary
- Adequate intake (AI)
The value estimated when a population reference intake cannot be established because an average requirement cannot be determined. An adequate intake is the average observed daily level of intake by a population group (or groups) of apparently healthy people that is assumed to be adequate.
- Adverse effect
Change in the morphology, physiology, growth, development, reproduction or lifespan of an organism, system or (sub)population that results in an impairment of functional capacity to compensate for additional stress or an increase in susceptibility to other influences (FAO/WHO, 2009; EFSA Scientific Committee, 2017a).
- Adverse health effect
See adverse effect.
- Average requirement (AR)
The level of (nutrient) intake that is enough for half of the people in a healthy group, given a normal distribution of requirement.
- Bioavailability
Nutrient fraction which is absorbed and becomes available to normal metabolic and physiological processes.
- Biomarker of exposure
An exogenous substance or its metabolite or the product of an interaction between a xenobiotic agent and some target molecule or cell that is measured in a compartment within an organism (WHO/IPCS, 1993; EFSA Scientific Committee, 2017a). Urine, blood, faeces or nails are common media for the measurements of biomarkers of exposure (EFSA Scientific Committee, 2017a).
- Biomarkers of effect
A measurable biochemical, physiological, behavioural or other alteration within an organism that, depending upon the magnitude, can be recognised as associated with an established or possible health impairment or disease (WHO/IPCS, 1993; EFSA Scientific Committee, 2017a). Its biological relevance depends on its relation to the mode of action of an adverse effect or an adverse outcome pathway (EFSA Scientific Committee, 2017a).
- Body burden
The body burden is the amount of nutrient, usually a mineral, in the body. For essential nutrients this amount is maintained at a constant level by the homeostatic equilibrium of absorption, distribution, metabolism and excretion. If homeostasis fails at high and low intakes of the nutrient the body burden of the nutrient and its metabolites increases or declines, respectively.
- Critical effect
Effect selected for the derivation of a health‐based guidance value.
- Dietary reference values (DRVs)
A set of nutrient reference values that includes the average requirement, the population reference intake, the adequate intake and the reference intake range for macronutrients.
- Endpoint
Qualitative or quantitative expression of a specific factor with which a risk may be associated as determined through an appropriate risk assessment (FAO/WHO, 2009).
- Hazard
Inherent property of an agent or situation having the potential to cause adverse effects when an organism, system, or (sub)population is exposed to that agent (WHO/IPCS, 2004; FAO/WHO, 2009).
- Lower threshold intake (LTI)
The level of intake below which, on the basis of current knowledge, almost all individuals will be unlikely to maintain ‘metabolic integrity’, according to the criterion chosen for each nutrient.
- Lowest‐observed‐adverse‐effect level
The lowest concentration or amount of a substance, found by experiment or observation, that causes an adverse alteration of morphology, functional capacity, growth, development or lifespan of the target organism distinguishable from normal (control) organisms of the same species and strain under the same defined conditions of exposure (FAO/WHO, 2009).
- Lowest‐observed‐effect level (LOEL)
The lowest concentration or amount of a substance, found by experiment or observation, that causes any alteration of morphology, functional capacity, growth, development or lifespan of the target organism distinguishable from normal (control) organisms of the same species and strain under the same defined conditions of exposure (FAO/WHO, 2009).
- Mechanism of action
The specific biochemical interaction through which a substance produces an effect on a living organism or in a biochemical system (FAO/WHO, 2009).
- Mode of action
A biologically plausible sequence of key events leading to an observed effect supported by robust experimental observations and mechanistic data. A mode of action describes key cytological and biochemical events – that is, those that are both measurable and necessary to the observed effect – in a logical framework. Related term: mechanism of action (FAO/WHO, 2009).
- Monotonic relationship
The slope of the dose–response curve does not change sign at any point along the range of doses examined.
- Non‐monotonic relationship
The slope of the dose–response curve changes sign from positive to negative or vice versa at some point along the range of doses examined (EFSA Scientific Committee, 2021b). Therefore, the curve expressing the relationship switches from increasing to decreasing or vice versa at some points along the dose range.
- No‐observed‐adverse‐effect level
The greatest concentration or amount of a substance, found by experiment or observation, that causes no adverse alteration of morphology, functional capacity, growth, development or lifespan of the target organism distinguishable from those observed in normal (control) organisms of the same species and strain under the same defined conditions of exposure (FAO/WHO, 2009).
- No‐observed‐effect level (NOEL)
The greatest concentration or amount of a substance, found by experiment or observation, that causes no alteration of morphology, functional capacity, growth, development or lifespan of the target organism distinguishable from those observed in normal (control) organisms of the same species and strain under the same defined conditions of exposure (FAO/WHO, 2009).
- Population reference intakes (PRI)
The level of (nutrient) intake that is enough for virtually all healthy people in a group.
- Reference intake ranges for macronutrients (RI)
The reference intake range for macronutrients, expressed as a percentage of the daily energy intake, defined by a lower and an upper bound.
Appendix A – General decision tree for whether a biological effect is relevant or not

Source: EFSA Scientific Committee (2017a)
Appendix B – Illustration of the application of the principles of the benchmark dose approach to the evaluation of the tolerable upper intake level for vitamin D in infants
In the opinion on the tolerable upper intake level (UL) for vitamin D in infants (EFSA NDA Panel, 2018), the NDA Panel reviewed the evidence from trials and observational studies on the relationship between vitamin D intake and the risk of hypercalciuria, hypercalcaemia, nephrocalcinosis and abnormal growth patterns and concluded that the available evidence could not be used alone to establish a UL. The Panel used data on serum 25(OH)D concentration in relation to vitamin D intake, recognising that a ‘high’ concentration is not an adverse health effect per se, but can be considered as a surrogate endpoint. The Panel used a dose–response meta‐analysis of aggregated data from trials to establish the reference point for a daily intake of vitamin D in infants associated with an acceptably low prevalence of elevated serum 25(OH)D concentrations, as a basis for establishing the UL for vitamin D.
The Panel defined a maximum concentration of 25(OH)D of 200 nmol/L as unlikely to pose a risk of adverse health effects. The dose–response relationship between serum 25(OH)D and vitamin D intake was modelled to generate the predicted distribution of the 25(OH)D serum concentration of individual infants at different levels of intake (Figure B.1A). From this, the percentage of infants expected to exceed the predefined threshold for the serum concentration of 25(OH)D could be estimated. An acceptable threshold for that percentage was defined (in analogy with the benchmark response concept) and the intake of vitamin D associated with it identified as the reference point (Figure B.1B).
Figure B.1.
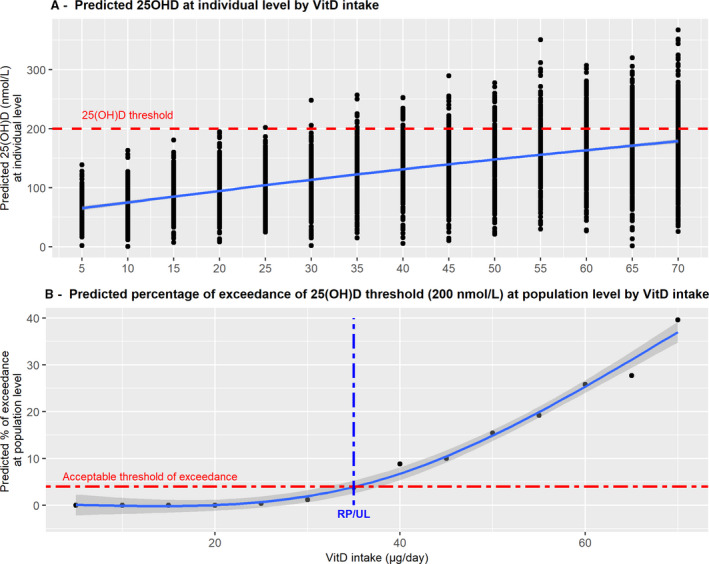
Illustration of the application of the principles of the benchmark dose approach to establish the tolerable upper intake level for vitamin D for infants aged 6–12 months
The latter was considered to be a conservative intake value that could be used as a basis for establishing the UL for vitamin D in infants, without the application of an uncertainty factor (Figure B.1B).
Several challenges were encountered when applying the ‘principles of the benchmark dose approach’, illustrative of the difficulties of using the method in the nutritional context, such as: (i) the identification of the best fitting intake–biomarker model shape; (ii) the identification of a reasonable distribution for the vitamin D intake to express the inter‐individual variability; (iii) the assessment of a threshold for the biomarker corresponding to a ‘healthy status’; (iv) the identification of an ‘acceptable’ fraction of the population which may be at risk of being in excess.
Appendix C – Scaling
In isometric scaling, the tolerable upper intake level (UL) for the population group X under consideration is derived by multiplication of the known and sex‐specific UL of group Y with the quotient between the typical weight of group X and the weight of group Y (Table C.1):
Table C.1.
Reference body weights for children and adults used for scaling
| Population group | Age taken as reference | Reference weight (kg) | References | ||
|---|---|---|---|---|---|
| Males a | Females a | Males and females b | |||
| 0–6 months | 3 months | 6.4 | 5.8 | 6.1 | WHO Multicentre Growth Reference Study Group (2006) |
| 7–11 months | 9 months | 8.9 | 8.2 | 8.6 | WHO Multicentre Growth Reference Study Group (2006) |
| 1–3 years | 2 years | 12.2 | 11.5 | 11.9 | WHO Multicentre Growth Reference Study Group (2006) |
| 4–6 years | 5 years | 19.2 | 18.7 | 19.0 | van Buuren et al. (2012) |
| 7–10 years | 8.5 years | 29.0 | 28.4 | 28.7 | van Buuren et al. (2012) |
| 11–14 years | 12.5 years | 44.0 | 45.1 | 44.6 | van Buuren et al. (2012) |
| 15–17 yrs | 16 years | 64.1 | 56.4 | 60.3 | van Buuren et al. (2012) |
| ≥ 18 years | n.a. | 82.0 c | 66.0 c | 70 d | EFSA Scientific Committee (2012) |
| Pregnant women | n.a. | – | 70.5 | – | EFSA NDA Panel (2013) |
The median weight for age at the age taken as reference was used as reference weight.
Mean of the values for males and females.
Values for adult subjects aged 18–64 years in all surveys in the EFSA Comprehensive Database.
Default body weight value for adults, as recommended by the EFSA Scientific Committee.
Isometric scaling:
Allometric scaling reflects the fact that the metabolic rate of an organism is an exponential function of body mass (weight). In allometric scaling, the UL for the population group X under consideration is derived by multiplication of the known and sex‐specific UL of group Y with the quotient between the typical weight of group X and the weight of group Y (Table C.1), to the power of 0.75:
Allometric scaling:
Suggested citation: EFSA NDA Panel (EFSA Panel on Nutrition, Novel Foods and Food Allergens) , Turck D, Bohn T, Castenmiller J, De Henauw S, Hirsch‐Ernst KI, Knutsen HK, Maciuk A, Mangelsdorf I, McArdle HJ, Peláez C, Pentieva K, Siani A, Thies F, Tsabouri S, Vinceti M, Aggett P, Crous Bou M, Cubadda F, de Sesmaisons Lecarré A, Martino L and Naska A, 2022. Guidance for establishing and applying tolerable upper intake levels for vitamins and essential minerals. Draft for internal testing. EFSA Journal 2022;20(1):e200102, 27 pp. 10.2903/j.efsa.2022.e200102
Requestor: European Commission
Question number: EFSA‐Q‐2021‐00364
Declarations of interest: The declarations of interest of all scientific experts active in EFSA’s work are available at https://ess.efsa.europa.eu/doi/doiweb/doisearch
Acknowledgements: The NDA Panel wishes to thank for the support provided to this scientific output: the hearing expert Josef Schlatter; the EFSA staff Laura Ciccollalo, Ionut Craciun, Ariane Titz and Silvia Valtuena Martinez.
Endorsed: The document was endorsed for publication and testing on 24 November 2021
Notes
SCF (2000). Scientific Committee on Food. Guidelines of the Scientific Committee on Food for the Development of Tolerable Upper Intake Levels for Vitamins and Minerals. In: Scientific Committee on Food, Scientific Panel on Dietetic Products, Nutrition and Allergies (2006). Tolerable Upper Intake Levels for Vitamins and Minerals. European Food Safety Authority. SCF (2001). Scientific Committee on Food. Opinion of the Scientific Committee on Food on the Tolerable Upper Intake Level of Magnesium. In: Scientific Committee on Food, Scientific Panel on Dietetic Products, Nutrition and Allergies (2006). Tolerable Upper Intake Levels for Vitamins and Minerals. European Food Safety Authority.
I.e. a level of intake that can be ingested daily over a lifetime without appreciable health risk to the consumer (EHC 240, FAO/WHO, 2009).
The body burden is the amount of nutrient, usually a mineral, in the body. For essential nutrients, this amount is maintained at a constant level by the homoeostatic equilibrium of absorption, distribution, metabolism and excretion. If homoeostasis fails at high and low intakes of the nutrient, the body burden of the nutrient and its metabolites increases or declines, respectively.
Regulation (EU) No 609/2013 of the European Parliament and of the Council of 12 June 2013 on food intended for infants and young children, food for special medical purposes and total diet replacement for weight control and repealing Council Directive 92/52/EEC, Commission Directives 96/8/EC, 1999/21/EC, 2006/125/EC and 2006/141/EC, Directive 2009/39/EC of the European Parliament and of the Council and Commission Regulations (EC) No 41/2009 and (EC) No 953/2009. OJ L 181, 29.6.2013, p. 35–56.
The EFSA Food composition database is available at: https://www.efsa.europa.eu/it/data‐report/food‐composition‐data
The EFSA Food consumption database is available at: https://www.efsa.europa.eu/en/data‐report/food‐consumption‐data
References
- Australian Government Department of Health and New Zealand Ministry of Health , 2015. Methodological framework for the review of Nutrient Reference Values. 70 pp. Available online: https://www.nrv.gov.au/resources
- Dodd KW, Guenther PM, Freedman LS, Subar AF, Kipnis V, Midthune D, Tooze JA and Krebs‐Smith SM, 2006. Statistical methods for estimating usual intake of nutrients and foods: a review of the theory. Journal of the American Dietetic Association, 106, 1640–1650. 10.1016/j.jada.2006.07.011 [DOI] [PubMed] [Google Scholar]
- EFSA (European Food Safety Authority) , 2010. Application of systematic review methodology to food and feed safety assessments to support decision making. EFSA Journal 2010;8(6):1637, 45 pp. 10.2903/j.efsa.2010.1637 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority) , 2022. Workshop on data and methodologies for establishing tolerable upper intake levels for vitamins and minerals. EFSA Supporting Publication 2022;EN‐6972, 86 pp. 10.2903/j.efsa.2022.EN-6972 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority) , Martino L, Aiassa E, Halldórsson TI, Koutsoumanis PK, Naegeli H, Baert K, Baldinelli F, Devos Y, Lodi F, Lostia A, Manini P, Merten C, Messens W, Rizzi V, Tarazona J, Titz A and Vos S, 2020. Draft framework for protocol development for EFSA’s scientific assessments. EFSA Supporting Publication 2020;EN‐1843, 46 pp. 10.2903/sp.efsa.2020.EN-1843 [DOI] [Google Scholar]
- EFSA ANS Panel (EFSA Panel on Food Additives and Nutrient Sources added to Food) , 2018. Guidance on safety evaluation of sources of nutrients and bioavailability of nutrient from the sources. EFSA Journal 2018;16(6):5294, 35 pp. 10.2903/j.efsa.2018.5294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA FAF Panel (EFSA Panel on Food Additives and Flavourings) , 2019. Scientific Opinion on the re‐evaluation of phosphoric acid–phosphates – di‐, tri‐ and polyphosphates (E 338–341, E 343, E 450–452) as food additives and the safety of proposed extension of use. EFSA Journal 2019;17(6):5674, 156 pp. 10.2903/j.efsa.2019.5674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies) , 2010. Scientific Opinion on principles for deriving and applying Dietary Reference Values. EFSA Journal 2010;8(3):1458, 30 pp. 10.2903/j.efsa.2010.1458 [DOI] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies) , 2012. Scientific Opinion related to the Tolerable Upper Intake Level of eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA) and docosapentaenoic acid (DPA). EFSA Journal 2012;10(7):2815, 48 pp. 10.2903/j.efsa.2012.2815 [DOI] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies) , 2013. Scientific Opinion on Dietary Reference Values for energy. EFSA Journal 2013;11(1):3005, 112 pp. 10.2903/j.efsa.2013.3005 [DOI] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies) , 2018. Scientific opinion on the update of the tolerable upper intake level for vitamin D for infants. EFSA Journal 2018;16(8):5365, 118 pp. 10.2903/j.efsa.2018.5365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Nutrition, Novel Foods and Food Allergens) , Turck D, Bohn T, Castenmiller J, De Henauw S, Hirsch‐Ernst KI, Knutsen HK, Maciuk A, Mangelsdorf I, McArdle HJ, Naska A, Peláez C, Pentieva K, Siani A, Thies F, Tsabouri S, Adan R, Emmett P, Galli C, Kersting M, Moynihan P, Tappy L, Ciccolallo L, de Sesmaisons‐Lecarré A, Fabiani L, Horvath Z, Martino L, Muñoz Guajardo I, Valtueña Martínez S and Vinceti M, 2022. Scientific opinion on the Tolerable Upper Intake Level for dietary sugars. EFSA Journal 2022;20(2):7074. 10.2903/j.efsa.2022.7074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , 2012. Guidance on selected default values to be used by the EFSA Scientific Committee, Scientific Panels and Units in the absence of actual measured data. EFSA Journal 2012;10(3):2579, 32 pp. 10.2903/j.efsa.2012.2579 [DOI] [Google Scholar]
- EFSA Scientific Committee , 2017a. Guidance on the assessment of the biological relevance of data in scientific assessments. EFSA Journal 2017;15(8):4970, 73 pp. 10.2903/j.efsa.2017.4970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , 2017b. Update: Guidance on the use of the benchmark dose approach in risk assessment. EFSA Journal 2017;15(1):4658, 41 pp. 10.2903/j.efsa.2017.4658 [DOI] [Google Scholar]
- EFSA Scientific Committee , 2017c. Scientific Opinion on the guidance on the use of the weight of evidence approach in scientific assessments. EFSA Journal 2017;15(8):4971, 69 pp. 10.2903/j.efsa.2017.4971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , 2018a. Scientific Opinion on the principles and methods behind EFSA’s Guidance on Uncertainty Analysis in Scientific Assessment. EFSA Journal 2018;16(1):5122, 235 pp. 10.2903/j.efsa.2018.5122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , 2018b. Guidance on Uncertainty Analysis in Scientific Assessments. EFSA Journal 2018;16(1):5123, 39 pp. 10.2903/j.efsa.2018.5123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , 2020. Draft for internal testing Scientific Committee guidance on appraising and integrating evidence from epidemiological studies for use in EFSA’s scientific assessments. EFSA Journal 2020;18(8):6221, 83 pp. 10.2903/j.efsa.2020.6221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , 2021a. Statement on the derivation of Health‐Based Guidance Values (HBGVs) for regulated products that are also nutrients. EFSA Journal 2021;19(3):6479, 55 pp. 10.2903/j.efsa.2021.6479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , 2021b. Opinion on the impact of non‐monotonic dose responses on EFSA’s human health risk assessments. EFSA Journal 2021;19 (10):6877, 68 pp. 10.2903/j.efsa.2021.6877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA/FAO/WHO (European Food Safety Authority, Food and Agriculture Organization, World Health Organization) , 2011. Towards a harmonised Total Diet Study approach: a guidance document. EFSA Journal 2011;9(11):2450, 66 pp. 10.2903/j.efsa.2011.2450 [DOI] [Google Scholar]
- FAO/WHO (Food and Agriculture Organization of the United Nations and World Health Organization) , 2006. A model for establishing Upper Levels of Intake for nutrients and related substances. Report of a Joint FAO/WHO Technical Workshop on Nutrient Risk Assessment WHO Headquarters, Geneva, Switzerland 2‐6 May 2005. Available at: https://apps.who.int/iris/handle/10665/43451
- FAO/WHO (Food and Agriculture Organization of the United Nations and World Health Organization) , 2009. Principles and Methods for the Risk Assessment of Chemicals in Food. Environmental Health Criteria 240. WHO, Geneva. Available online: https://www.who.int/publications/i/item/9789241572408
- FAO/WHO (Food and Agriculture Organization of the United Nations and World Health Organization) , 2020. Chapter 5: Dose‐Response Assessment and Derivation of Health‐Based Guidance Values (Updated Chapter). In: Principles and Methods for the Risk Assessment of Chemicals in Food. Environmental Health Criteria 240. WHO, Geneva. Available online: https://www.who.int/publications/i/item/9789241572408
- Krewski D, Andersen ME, Tyshenko MG, Krishan K, Hartung T, Boekelheide K, Wambaugh JF, Jones D, Whelan M, Thomas R, Yauk C, Barton‐Maclaren T and Cote I, 2020. Toxicity testing in the 21st century: progress in the past decade and future perspectives. Archives of Toxicology, 94, 1–58. 10.1007/s00204-019-02613-4 [DOI] [PubMed] [Google Scholar]
- NESR (Nutrition Evidence Systematic Review) , 2019. Risk of Bias for Nutrition Observational Studies (RoB‐NObs) Tool 2019. US Department of Agriculture. Available online: https://nesr.usda.gov/sites/default/files/2019‐07/RiskOfBiasForNutritionObservationalStudies‐RoB‐NObs.pdf [Google Scholar]
- NTP OHAT (National Toxicology Programme Health Assessment and Translation Group) , 2015. OHAT Risk of Bias Rating Tool for Human and Animal Studies. US Department of Health and Human Services, 37 pp. Available online: https://ntp.niehs.nih.gov/ntp/ohat/pubs/riskofbiastool_508.pdf
- NTP OHAT (National Toxicology Programme Health Assessment and Translation Group) , 2019. Handbook for Conducting a Literature‐Based Health Assessment Using OHAT Approach for Systematic Review and Evidence Integration. US Department of Health and Human Services. Available online: https://ntp.niehs.nih.gov/whatwestudy/assessments/noncancer/handbook/index.html [Google Scholar]
- SCF (Scientific Committee on Food) , 2000a. Guidelines of the Scientific Committee on Food for the development of tolerable upper intake levels for vitamins and minerals. Available online: https://www.efsa.europa.eu/sites/default/files/efsa_rep/blobserver_assets/ndatolerableuil.pdf [Google Scholar]
- SCF (Scientific Committee on Food) , 2000b. Opinion of the Scientific Committee on Food on the Tolerable Upper Intake Level of folate. Available online https://www.efsa.europa.eu/sites/default/files/efsa_rep/blobserver_assets/ndatolerableuil.pdf [Google Scholar]
- SCF (Scientific Committee on Food) , 2001. Opinion of the Scientific Committee on Food on the Tolerable Upper Intake Level of magnesium. Available online: https://www.efsa.europa.eu/sites/default/files/efsa_rep/blobserver_assets/ndatolerableuil.pdf [Google Scholar]
- SCF (Scientific Committee on Food) , 2002. Opinion of the Scientific Committee on Food on the Tolerable Upper Intake Level of nicotinamide and nicotinic acid (niacin). Available online: https://www.efsa.europa.eu/sites/default/files/efsa_rep/blobserver_assets/ndatolerableuil.pdf [Google Scholar]
- Smeraldi C, Giarola A, Aggett P, Moldeus P and Gundert‐Remy U, 2020. Use of mechanistic information to derive chemical‐specific adjustment factors – Refinement of risk assessment. Regulatory Toxicology and Pharmacology, 117, 104776. 10.1016/j.yrtph.2020.104776 [DOI] [PubMed] [Google Scholar]
- Sterne JAC, Savović J, Page MJ, Elbers RG, Blencowe NS, Boutron I, Cates CJ, Cheng H‐Y, Corbett MS, Eldridge SM, Hernán MA, Hopewell S, Hróbjartsson A, Junqueira DR, Jüni P, Kirkham JJ, Lasserson T, Li T, McAleenan A, Reeves BC, Shepperd S, Shrier I, Stewart LA, Tilling K, White IR, Whiting PF and Higgins JPT, 2019. RoB 2: a revised tool for assessing risk of bias in randomised trials. British Medical Journal, 366, 4898. [DOI] [PubMed] [Google Scholar]
- US NASEM (National Academies of Sciences, Engineering, and Medicine) , 2017. Guiding principles for developing Dietary Reference Intakes based on chronic disease. The National Academies Press, Washington, DC. 10.17226/24828 [DOI] [PubMed] [Google Scholar]
- van Buuren S, Schönbeck Y and van Dommelen P, 2012. Collection, collation and analysis of data in relation to reference heights and reference weights for female and male children and adolescents (0–18 years) in the EU, as well as in relation to the age of onset of puberty and the age at which different stages of puberty are reached in adolescents in the EU. Project developed on the procurement project CT/EFSA/NDA/2010/01. EFSA Supporting Publication 2012;EN‐255, 59 pp. 10.2903/sp.efsa.2012.EN-255 [DOI] [Google Scholar]
- van Klaveren JD, Goedhart PW, Wapperom D and van der Voet H, 2012. A European tool for usual intake distribution estimation in relation to data collection by EFSA. EFSA Supporting Publication 2012;EN‐300, 42 pp. Available online: https://edepot.wur.nl/222740 [Google Scholar]
- Vinceti M, Filippini T, Malavolti M, Naska A, Kasdagli M, Torres D, Lopes C, Carvalho C, Moreira P and Orsini N, 2020. Dose‐response relationships in health risk assessment of nutritional and toxicological factors in foods: development and application of novel biostatistical methods. EFSA Supporting Publication 2020;EN‐1899, 57 pp. 10.2903/sp.efsa.2020.EN-1899 [DOI] [Google Scholar]
- WHO Multicentre Growth Reference Study Group (World Health Organization) , 2006. WHO Child Growth Standards: length/height‐for‐age, weight‐for‐age, weight‐for‐length, weight‐for‐height and body mass index‐for‐age: methods and development. 312 pp.
- WHO/IPCS (World Health Organization and International Programme on Chemical Safety) , 2004. IPCS risk assessment terminology. World Health Organization. Available online: https://apps.who.int/iris/handle/10665/42908
- WHO/IPCS (World Health Organization and International Programme on. Chemical Safety) , 1993. Biomarkers and Risk Assessment: Concepts and Principles. Environmental Health Criteria 155. 86 pp. Available online: https://apps.who.int/iris/handle/10665/39037
- WHO/IPCS (World Health Organization/International Programme on Chemical Safety) , 2002. Principles and methods for the assessment of risk from essential trace elements. Environmental Health Criteria 228. Available online: https://apps.who.int/iris/handle/10665/42416