

Abstract
Presented is an economical means of removing fluorine from various highly fluorinated arenes using NaBH4. The procedure was adapted for different classes of perfluoroarenes. A novel isomer of an emerging class of organic dyes based on the carbazole phthalonitrile motif was succinctly synthesized in two steps from tetrafluorophthalonitrile demonstrating the utility of the hydrodefluorination procedure. Initial exploration of the dye shows it to be photoactive and capable of facilitating contrathermodynamic styrenoid E/Z isomerization.
Graphical Abstract
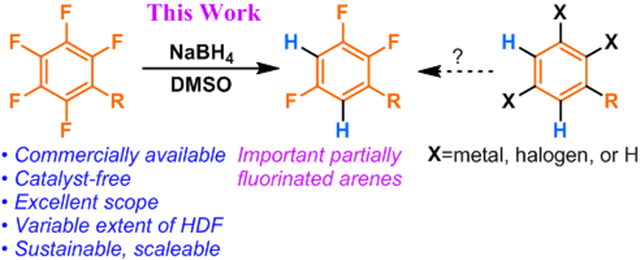
A meaningful portion of drugs, bioactive molecules, and agrochemicals bear fluorine atoms as important components. In that context, fluorine boasts an important role as a means of modulating lipophilicity, metabolic breakdown, and overall potency. There is a clear interest in having diverse and efficient means of incorporating fluorine into small molecules for probing structure-activity relationships. Obtaining specific fluorination patterns in complex small molecules can often represent a challenge, necessarily leading to inefficient multistep syntheses to add fluorine to a scaffold with the proper regioselectivity. Without regioselectivity issues, perfluorinated arenes represent the cheapest class of commercially available molecules that have all the desired fluorine atoms in their proper place – and several other superfluous ones. Selective aromatic hydrodefluorination (HDF), has the potential to be a useful alternative to the stepwise installation of individual fluorine atoms, especially when targeting higher degrees of fluorination. Starting from perfluorinated or polyfluorinated arenes; a well-defined repertoire of conditions to remove specific fluorine atoms presents a convenient path to select fluorine configurations.
Polyfluoro aromatic C─F bonds are well known handles for transition-metal-catalyzed functionalization1-4 and nucleophilic functionalization. Nucleophilic aromatic HDF of perfluoroarenes with hydride has been reported before via LiAlH4,5-8, DIBAL,9 and other transition metal-mediated aluminum hydrides.10 These procedures, however, represent harsh conditions either by virtue of the high, unselective reactivity of the aluminum hydride or by the temperatures necessary to facilitate conversion – both of which greatly limit the scope of target perfluoroarenes. Ligand-bound boranes,11 precious metal hydride catalysts,12,13 and catalytic hydrosilicate14 have also been reported featuring milder conditions, but also non-trivial catalyst and/or hydride complex preparations. Sodium borohydride is generally known to be a less reactive, more selective hydride source that is cost-effective, easily handled, and found in almost all synthetic labs. So, it was surprising to find only sparse literature reference to its use for aromatic HDF with limited scope. Perhaps this is because common conditions for NaBH4 reductions are not conducive to selective nucleophilic aromatic substitution. NaBH4 has, in an isolated example, been reported to perform hydrodefluorination at the 4-position of 3-chloro-2,5,6-trifluoropyridine in cooled ethanolic solution,15 which, in our hands, proved not to be generally applicable to other substrates owing to their tendency to undergo solvolysis, vide infra. It has also been reported to remove fluorine from the 4-position of exactly pentafluorobenzamide and pentafluorobenzonitrile with respective yields of 65% and 82%.16,17 The demonstrated scope in these patents, however, is devoid of sensitive functional groups or more reactive substrates and lack sufficient detail to be of any real use to the practitioner. Given the limited nature of literature documentation of functional group sensitivity under conditions conducive to aromatic HDF, we opted to investigate protocols for NaBH4 HDF of perfluoroarenes.
Results and Discussion
Methyl pentafluorobenzoate (MPFB), a simple and inexpensive perfluoroarenes with a moderately vulnerable electron-withdrawing functional group, was selected as the initial substrate for optimization of reaction conditions. The first parameter we examined was solvent. Given the literature precedent of polar protic solvents modulating borohydride reductions,18-20 we first examined methanol and ethanol (entries 1 and 2, Scheme 1 and Table 1). While both entries consumed MPFB, only ethanol led to product formation, albeit very little. In both alcoholic solvents the reactivity towards the ester was observed, an important functional group sensitivity absent from the substrates studied by Schlosser.15 The 19FNMR spectra confirmed the retention of fluorine at the 4-position of developing side products. Next, we examined ethereal solvents tetrahydrofuran and 1,2-dimethoxyethane (entries 3 and 4) neither of which were homogenous. Both solvent-NaBH4 suspensions resulted in the formation of significantly fewer side products, as anticipated based on the observations of Brown et al.18,21 However, the rates were variable with THF resulting in very low conversion, and DME giving 46% conversion within 10 h. The poor coordination of the sodium ion and resulting partial solubility of the reagent in the ethereal solvents likely contributed to the poor to moderate performance. Finally, we examined strongly polar aprotic solvents DMF and DMSO (entries 5 and 6), which were expected to enhance the reactivity of the borohydride due to their strong coordination of the sodium ion.22,23 Indeed, we observed that both DMF and DMSO gave clean conversion. Specifically, DMF gave 70% yield while DMSO gave 98% yield (entries 5 and 6). Of the selected solvents, DMSO proved to be the most promising, leading to the fastest and most selective conversion to product; further optimization was carried out in DMSO.
Scheme 1.

Optimization Template
Table 1.
Initial Optimizations.
| entry | Eq. NaBH4 |
Solvent | Time (hours) |
% NMR Yield |
% Conversion MPFBb |
|---|---|---|---|---|---|
| 1 | 0.50 | MeOH | 10 | 0 | ~100 |
| 2 | 0.50 | EtOH | 10 | 6 | 72 |
| 3 a | 0.50 | THF | 10 | <2 | <2 |
| 4 a | 0.50 | DME | 10 | 46 | 46 |
| 5 | 0.50 | DMF | 10 | 70 | 72 |
| 6 | 0.50 | DMSO | 10 | 97 | 98 |
| 7 | 0.33 | DMSO | 2 | 23 | 23 |
| 8 | 0.66 | DMSO | 2 | 43 | 43 |
| 9 | 1.00 | DMSO | 2 | 99 | 99 |
| 10 | 2.00 | DMSO | 2 | 99 | 99 |
Reactions were run as suspensions.
Determined using 19F NMR.
Next, we examined NaBH4 loading with the goal of exploring the reaction rate as a function of NaBH4 loading. Entries 5 and 6 (Table 1) indicated that when DMSO and DMF were used, at least 2 hydrides per BH4− were capable of being delivered to the MPFB, and that an earlier timepoint would be needed to distinguish relative rates. Thus, the loading of NaBH4 was varied (entries 7-9); and 19FNMR spectra collected after 2 hours. The results indicated that there was a significant difference in the rates of HDF due to the in situ generated reductants, and that of stoichiometric NaBH4 which gave complete conversion within 2 hours. An experiment was conducted to ascertain the stability of MPFB over extended periods in DMSO solution with NaBH4. (Suppl. S13) Minimal degradation was observed. Finally, excess NaBH4 neither removed further fluorine nor reduced the methyl ester within the same timeframe and conditions (entry 10). Increased hydrogen atom content on the aromatic ring discourages further HDF. From these experiments, we anticipated HDF rates could easily be controlled by equivalents of NaBH4 used and using ethereal solvents to attenuate reactivity by degrees, with DME lowering the reactivity moderately and THF reducing reactivity further.
With these controls in mind, we next examined the scope of the reaction using the generalized reaction conditions, specifically 1.0 equiv of NaBH4 in DMSO at room temperature (Figure 1). We examined other polyfluorinated aromatic compounds which exhibited varying degrees of activation and functional group vulnerability. As expected, the use of NaBH4 rather than more potent reductants such as LiAlH4, allowed for the incorporation of esters (1b, 2b), and amides (3b-5b). Octafluorotoluene also undergoes perfectly regioselective HDF at the 4-position with no trace of the benzylic defluorination characteristic of several HDF procedures that proceed through radical anion fragmentations.24 Complementary to Braun’s boryl-Rh-catalyzed HDF25 of pentafluoropyridine which displays C2 selectivity, we observed that pentafluoropyridine also underwent clean HDF at C4.
Figure 1. Scope of NaBH4 mediated hydrodefluorination.

Adaptations of standard conditions were as follows: a1:1 v:v THF:DMSO used as solvent, bheated to 45 °C, c2.2 equiv NaBH4 used, dpure THF used as solvent, e3 equiv NaBH4 used, fheated to 80 °C, gDME used as sole solvent, h19F NMR yield determined on crude reaction mixture using fluorobenzene as internal standard. iA 90% yield was afforded for a subsequent 5 g scale synthesis of 1b.
Owing to their volatility, no attempt was made to isolate either product 6b or 7b which could be obtained in similar yield to the photocatalyzed HDF without the need for iridium based photocatalyst.26 Both alkenyl (8b) and alkynyl (9b) substituents were found to be sufficiently activated to facilitate HDF. Notably, this took place with negligible alkene or alkyne reduction when moderately attenuating solvents were used. Alkenes would likely have been problematic under recently developed photocatalytic HDF conditions which chance contrathermodynamic styrenoid isomerization.27-31 Furthermore, use of traditional transition metal hydride chemistry32-35 would risk undesired alkenyl or alkynyl metalation.36 Attempts to perform the HDF on pentafluorotoluene (22b) revealed that mildly electron donating alkyl substituents are also viable, though they do require more elevated temperatures (80 °C) and increased NaBH4 loading. Currently, photocatalytic-HDF on this type of substrate is sluggish at best, making this an enabling advancement. Similar conditions also facilitated di-HDF in hexafluorobenzene (21b). 19FNMR spectra taken periodically revealed the sequential formation of mono- and then di-HDF (21b), but no attempt was made to obtain a yield for the mono-HDF product. Attempts to perform selective HDF on the corresponding fluorinated benzaldehyde and acetophenone failed, which instead engaged in carbonyl reduction. However, by elevating the reaction temperature to 45 °C, acetals (11b), ketals (12b) also underwent selective mono-HDF at the C4 position, formally allowing access to the aldehyde and ketone HDF products. Anilines and are synthetically important and readily converted to versatile diazoniums.37 Unfortunately, pentafluoroaniline showed no change to the fluorine configuration even under our most forcing conditions. Conveniently, di-Boc-aniline (10b) can serve as a masked aniline following gentle heating in the presence of trifluoroacetic acid.(Suppl. S12) Other substrates bearing electron donating groups attached to the ring generally exhibited no HDF under standard conditions or even when exposed to 3 equivalents of NaBH4 in DMSO at 80 °C over 12 hours. In the case of pentafluorophenol and pentafluoroaniline this may be due to the acidic OH/NH. While Zhang38 and Ogoshi14 both reported successful, albeit attenuated nucleophilic HDF of perfluoroaryl ethers with catalytic copper hydrides and catalytic hydrosilicate respectively, we did not observe any HDF in the case of the ethereal substrate, which remained unchanged. Some nitro and nitrile substituted fluoroarenes were more prone to subsequent HDF reactions. Typically, defluorination occurred para and then subsequently ortho to the functional group. Thus, it was necessary to modify the standard conditions to avoid generating mixtures of mono- and poly-defluorinated products. Moreover, these functional groups exhibited a greater tendency than others to become reduced by the solution of borohydride in DMSO, an observation previously noted by Hutchins when treating various nitrobenzenes with NaBH4 in DMSO.39 We were able to attenuate the rate by adding, or switching, the solvent to THF. The attenuated reactivity, presumably due to decreased concentration of borohydride in solution (see Table 1), allowed for more selective HDF, stopping at single HDF for pentafluorobenzonitrile (16b). Further, we were able to use the loading of NaBH4 (1 or 2 equiv) to convert tetrafluorophthalonitrile to either 3,4,6-trifluorophthalonitrile (mono-HDF, 18b) or to 3,6-difluorophthalonitrile (di-HDF, 19b) in good yield for uncatalyzed HDF.12 Even with the reduced reactivity in THF, tetrafluorophthalonitrile exhibited partial nitrile reduction. However, we found that the yield was improved by the addition of stoichiometric water which played a key role in preventing functional group reduction, analogous to Hutchins’ observation of water protecting allylic borohydride dehalogenations.23 For some substrates, there was insufficient difference in rates of defluorination of the initial and product fluoroarenes leaving only the product of multiple HDF accessible. For instance, pentafluoronitrobenzene underwent rapid triple-HDF without denitration40 to yield di-F nitrobenezene (17b) in good yield. We were able to cleanly isolate di-HDF products (14b, 15b, and 15c) by increasing the NaBH4 loading, affording complete conversion to the doubly defluorinated products. Octafluoronaphthalene is the only substrate in which less than perfect regioselectivity (determined by 19F NMR of the crude reaction mixtures) was observed which still gave a synthetically useful 8:1 ratio of products. The regioselectivity was assigned by matching the literature.10,14,41
Aromatic hydrodehalogenation which takes place via radical anion fragmentation42 displays a stronsg tendency for the heavier halogen to undergo preferential fragmentation, regardless of the aromatic regiochemistry. In a revealing experiment, 3,5-dichloro-2,4,6-trifluoropyridine (13b) underwent single C4 HDF under standard conditions. The preservation of the chlorine substituents at the meta positions, which is characteristic of nucleophilic displacements, contrasts with the trend of electron transfer- radical anion fragmentation hydrodehalogenations.26 Moreover, we observed HDF can be completely blocked at a position by substitution with a heavier halogen, which is possible via-the retrohalex reaction.26,43,44 4-Cl-tetra-F-nitrobenzene, in which the site of preferential fluoride fragmentation (4-F) has been replaced with 4-Cl, undergoes smooth double-HDF with no evidence of chlorine cleavage to yield 20b, indicating that chlorine substitution will effectively block the innate regiochemical preference of the ring.
Finally, to demonstrate the scalability of the process, 1b was synthesized on a 5 g scale, with an isolated yield of 90% and a slight improvement compared to the smaller scale.
The ability to access complex multi-fluorinated compounds by use of only commercially available reagents (perfluoroarenes and NaBH4) and solvents common to most synthetic labs (DMSO, THF), using quick operationally simple (open flasks) procedures is expected to be broadly enabling. To demonstrate the utility of this method, we used it to synthesize a novel analog of a class of carbazole-phthalonitrile organodyes (Scheme 2). These dyes are an exciting class of visible light absorbing complexes that display great triplet state lifetimes, energy transfer quantum efficiencies, and redox capabilities allowing for recent facilitation of many chemical transformations; including dearomative cycloaddition,45 cross-dehydrogenative-coupling type Minisci reactions,46 and even aromatic dehalogenation.47 Conspicuously absent among these dyes, is the isomer with the carbazole groups ortho to the nitriles. Presumably this absence is more of a reflection of a lack of commercial availability for the respective di-F-phthalonitrile (19b), which does not store well, rather than a lack of photochemical activity. Thus, we attempted to synthesize this dye by subjecting 200 mg of commercially available tetrafluorophthalonitrile to the HDF conditions along with 2 equiv of water, to generate 58% yield of the di-F nitrile after isolation. Using conditions adapted from Adachi et al.,48 we were able to smoothly synthesize 3,6-di(9H-carbazol-9-yl)phthalonitrile (19c) in 79% yield.
Scheme 2.

Synthesis of Phthalonitrile Organodye
As anticipated, 19c exhibited strong fluorescence with an emission maximum of 472 nm in toluene and 533 nm in acetonitrile. The solvatochromism is visualized in Scheme 3A and C with 5 solvents of varying polarity. The excited state was partially quenched by oxygen, demonstrated by diminished fluorescence in aerobic atmosphere. To verify the dye’s utility as an efficient photocatalyst, a styrenoid (E-23a, Scheme 3D) E-to-Z contrathermodynamic isomerization was set up in CH2Cl2 with 0.25 mol% of 19c present as photocatalyst. An isolated yield of Z-23b was obtained at 62%, with 8% of the starting E-23a remaining. In conclusion, we have reported operationally simple reactions conditions that can facilitate HDF with excellent substrate scope and good functional group tolerance. The reaction appears to proceed through a nucleophilic displacement mechanism, giving it complementary selectivity to other catalytic HDF processes. In addition to highly practical reaction conditions, it also facilitates the use of fully fluorinated arenes as precursors to less storable fluoroarenes which can enable synthesis, as demonstrated by the synthesis of a new member of the carbazole-phthalonitrile organodyes.
Scheme 3.

19c-Photocatalyzed E/Z Isomerization.
Supplementary Material
ACKNOWLEDGMENT
The authors would like to thank the National Institutes of Health NIH NIGMS (5R01GM115697) and the Herman Frasch Fund for Chemical Research for financial support of this work. Additionally, we thank Prof. Spencer Pitre (OSU) for helpful discussions.
Footnotes
ASSOCIATED CONTENT
Supporting Information (Experimental procedures, characterization data) is available free of charge at http://pubs.acs.org. FAIR Data is available as Supporting Information for Publication and includes the primary NMR FID files for compounds 1b-23b.
REFERENCES
- (1).Weaver J; Senaweera S C-F Activation and Functionalization of Perfluoro- and Polyfluoroarenes. Tetrahedron 2014, 70 (41), 7413–7428. 10.1016/j.tet.2014.06.004. [DOI] [Google Scholar]
- (2).Li J; Zheng T; Sun H; Li X Selectively Catalytic Hydrodefluorination of Perfluoroarenes by Co(PMe3)4 with Sodium Formate as Reducing Agent and Mechanism Study. J. Chem. Soc. Dalt. Trans 2013, 42 (36), 13048–13053. 10.1039/c3dt50409c. [DOI] [PubMed] [Google Scholar]
- (3).Podolan G; Lentz D; Reissig HU Selective Catalytic Hydrodefluorination as a Key Step for the Synthesis of Hitherto Inaccessible Aminopyridine Derivatives. Angew. Chemie - Int. Ed 2013, 52 (36), 9491–9494. 10.1002/anie.201301927. [DOI] [PubMed] [Google Scholar]
- (4).Vela J; Smith JM; Yu Y; Ketterer NA; Flaschenriem CJ; Lachicotte RJ; Holland PL Synthesis and Reactivity of Low-Coordinate Iron(II) Fluoride Complexes and Their Use in the Catalytic Hydrodefluorination of Fluorocarbons. J. Am. Chem. Soc 2005, 127 (21), 7857–7870. 10.1021/ja042672l. [DOI] [PubMed] [Google Scholar]
- (5).Chambers RD; Seabury MJ; Lyn D; Williams H; Hughes N Mechanisms for Reactions of Halogenated Compounds. Part 5.’ Orientating Effects of Fluorine Substituents on Nucleophilic Substitution in Naphthalene and Other Polycyclic Systems. J. Chem. Soc. Perkin Trans 1988, 1, 251. 10.1039/P19880000251. [DOI] [Google Scholar]
- (6).Banks RE; Burgess JE; Cheng WM; Haszeldine RN 93. Heterocyclic Polyfluoro-Compounds. Part IV. Nucleophilic Substitution in Pentafluoropyridine: The Preparation and Properties of Some 4-Substituted 2,3,5,6-Tetrafluoropyridines. J. Chem. Soc 1965, 67, 575. 10.1039/jr9650000575. [DOI] [Google Scholar]
- (7).Payne DT; Zhao Y; Fossey JS Ethylenation of Aldehydes to 3-Propanal, Propanol and Propanoic Acid Derivatives. Sci. Rep 2017, 7 (1), 1–8. 10.1038/s41598-017-01950-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Naumann K; Behrenz W; Hammann I; Klauke E; Marhold A COMBATING ARTHROPODS WITH 2,2-DIMETHYL-3-VINYL-CYCLOPROPANE CARBOXYLIC ACID ESTERS OF HALOGENATED BENZYL ALCOHOLS. US 4183950, 1980.
- (9).Chambers RD; Hall CW; Hutchinson J; Millar RW Polyhalogenated Heterocyclic Compounds. Part 42. Fluorinated Nitrogen Heterocycles with Unusual Substitution Patterns. J. Chem. Soc. - Perkin Trans 1 1998, No. 10, 1705–1713. 10.1039/a709291a. [DOI] [Google Scholar]
- (10).Yow S; Gates SJ; White AJP; Crimmin MR Zirconocene Dichloride Catalyzed Hydrodefluorination of CSp2─F Bonds. Angew. Chemie Int. Ed 2012, 51 (50), 12559–12563. 10.1002/anie.201207036. [DOI] [PubMed] [Google Scholar]
- (11).Phillips NA; O’Hanlon J; Hooper TN; White AJP; Crimmin MR Dihydridoboranes: Selective Reagents for Hydroboration and Hydrodefluorination. Org. Lett 2019, 21 (18), 7289–7293. 10.1021/acs.orglett.9b02515. [DOI] [PubMed] [Google Scholar]
- (12).Matsunami A; Kuwata S; Kayaki Y Hydrodefluorination of Fluoroarenes Using Hydrogen Transfer Catalysts with a Bifunctional Iridium/NH Moiety. ACS Catal. 2016, 6 (8), 5181–5185. 10.1021/acscatal.6b01590. [DOI] [Google Scholar]
- (13).Schwartsburd L; Mahon MF; Poulten RC; Warren MR; Whittlesey MK Mechanistic Studies of the Rhodium NHC Catalyzed Hydrodefluorination of Polyfluorotoluenes. Organometallics 2014, 33 (21), 6165–6170. 10.1021/om500827d. [DOI] [Google Scholar]
- (14).Kikushima K; Grellier M; Ohashi M; Ogoshi S Transition-Metal-Free Catalytic Hydrodefluorination of Polyfluoroarenes by Concerted Nucleophilic Aromatic Substitution with a Hydrosilicate. Angew. Chemie - Int. Ed 2017, 56 (51), 16191–16196. 10.1002/anie.201708003. [DOI] [PubMed] [Google Scholar]
- (15).Bobbio C; Rausis T; Schlosser M Removal of Fluorine from and Introduction of Fluorine into Polyhalopyridines: An Exercise in Nucleophilic Hetarenic Substitution. Chem. A Eur. J 2005, 11, 1903–1910. 10.1002/chem.200400837. [DOI] [PubMed] [Google Scholar]
- (16).Seisaku K; Koji S; Takahashi S PRODUCTION OF 2,3,5,6-TETRAFLUOROBENZENE DERIVATIVE. JPH06321858A, 1994.
- (17).Seisaku K; Koji S; Takahashi S PRODUCTION OF 3,5-DIFLUOROANILINE. JPH06239810A, 1994.
- (18).Brown HC; Mead EJ; Subba Rao BC A Study of Solvents for Sodium Borohydride and the Effect of Solvent and the Metal Ion on Borohydride Reductions 1. J. Am. Chem. Soc 1955, 77 (23), 6209–6213. 10.1021/ja01628a044. [DOI] [Google Scholar]
- (19).Da Costa JCS; Pais KC; Fernandes EL; De Oliveira PSM; Mendonça JS; De Souza MVN; Peralta MA; Vasconcelos TRA Simple Reduction of Ethyl, Isopropyl and Benzyl Aromatic Esters to Alcohols Using Sodium Borohydride-Methanol System. Gen. Pap. Ark 2006, No. i, 128–133. [Google Scholar]
- (20).C. P. P; Ebbin J; D S N; Ibnusaud I; Raskatov J; Singaram B Stabilization of NaBH 4 in Methanol Using a Catalytic Amount of NaOMe. Reduction of Esters and Lactones at Room Temperature without Solvent-Induced Loss of Hydride. J. Org. Chem 2018, 83, 1431–1440. 10.1021/acs.joc.7b02993. [DOI] [PubMed] [Google Scholar]
- (21).Brown HC; Ichikawa K The Influence of Solvent and Metal Ion on the Rate of Reaction of Alkali Metal Borohydrides with Acetone. J. Am. Chem. Soc 1961, 83 (21), 4372–4374. 10.1021/ja01482a018. [DOI] [Google Scholar]
- (22).Hutchins RO; Hoke D; Keogh J; Koharski D Sodium Borohydride in Dimethyl Sulfoxide or Sulfolane. Convenient Systems for Selective Reductions of Primary, Secondary and Certain Tertiary Halides and Tosylates. Tetrahedron Lett. 1969, 10 (40), 3495–3498. 10.1016/S0040-4039(01)88430-6. [DOI] [Google Scholar]
- (23).Hutchins RO; Kandasamy D; Dux F; Maryanoff CA; Rotstein D; Goldsmith B; Burgoyne W; Cistone F; Dalessandro J; Puglis J Nucleophilic Borohydride: Selective Reductive Displacement of Halides, Sulfonate Esters, Tertiary Amines, and N,N-Disulfonimides with Borohydride Reagents in Polar Aprotic Solvents. J. Org. Chem 1978, 43 (11), 2259–2267. 10.1021/jo00405a036. [DOI] [Google Scholar]
- (24).Luo C; Bandar JS Selective Defluoroallylation of Trifluoromethylarenes. J. Am. Chem. Soc 2019, 141, 14120–14125. 10.1021/jacs.9b07766, and the references therein. [DOI] [PubMed] [Google Scholar]
- (25).Teltewskoi M; Panetier JA; Macgregor SA; Braun T A Highly Reactive Rhodium(I)-Boryl Complex as a Useful Tool for C-H Bond Activation and Catalytic C-F Bond Borylation. Angew. Chemie - Int. Ed 2010, 49 (23), 3947–3951. 10.1002/anie.201001070. [DOI] [PubMed] [Google Scholar]
- (26).Senaweera SM; Singh A; Weaver JD Photocatalytic Hydrodefluorination: Facile Access to Partially Fluorinated Aromatics. J. Am. Chem. Soc 2014, 136, 3002. 10.1021/ja500031m. [DOI] [PubMed] [Google Scholar]
- (27).Day JI; Singh K; Trinh W; Weaver JD Visible Light Mediated Generation of Trans-Arylcyclohexenes and Their Utilization in the Synthesis of Cyclic Bridged Ethers. J. Am. Chem. Soc 2018, 140, 9934–9941. 10.1021/jacs.8b04642. [DOI] [PubMed] [Google Scholar]
- (28).Singh K; Staig SJ; Weaver JD Facile Synthesis of Z-Alkenes via Uphill Catalysis. J. Am. Chem. Soc 2014, 136, 5275–5278. 10.1021/ja5019749. [DOI] [PubMed] [Google Scholar]
- (29).Wei X-J; Boon W; Hessel V; Noeël T Visible-Light Photocatalytic Decarboxylation of α,β-Unsaturated Carboxylic Acids: Facile Access to Stereoselective Difluoromethylated Styrenes in Batch and Flow. ACS Catal. 2017, 7, 7136–7140. 10.1021/acscatal.7b03019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Lin QY; Xu XH; Qing FL Chemo-, Regio-, and Stereoselective Trifluoromethylation of Styrenes via Visible Light-Driven Single-Electron Transfer (SET) and Triplet-Triplet Energy Transfer (TTET) Processes. J. Org. Chem 2014, 79 (21), 10434–10446. 10.1021/jo502040t. [DOI] [PubMed] [Google Scholar]
- (31).Metternich JB; Gilmour R A Bio-Inspired, Catalytic E → Z Isomerization of Activated Olefins. J. Am. Chem. Soc 2015, 137, 11254–11257. 10.1021/jacs.5b07136. [DOI] [PubMed] [Google Scholar]
- (32).Aizenberg M; Milstein D Catalytic Activation of Carbon-Fluorine Bonds by a Soluble Transition Metal Complex. Science (80-. ) 1994, 265 (5170), 359–361. 10.1126/science.265.5170.359. [DOI] [PubMed] [Google Scholar]
- (33).Braun T; Noveski D; Ahijado M; Wehmeier F Hydrodefluorination of Pentafluoropyridine at Rhodium Using Dihydrogen: Detection of Unusual Rhodium Hydrido Complexes †. Dalt. Trans 2007, 3820–3825. 10.1039/b706846h. [DOI] [PubMed] [Google Scholar]
- (34).Kuehnel MF; Lentz D; Braun T Synthesis of Fluorinated Building Blocks by Transition-Metal-Mediated Hydrodefluorination Reactions. Angew. Chemie - Int. Ed 2013, 52 (12), 3328–3348. 10.1002/anie.201205260. [DOI] [PubMed] [Google Scholar]
- (35).Gair JJ; Grey RL; Giroux S; Brodney MA Palladium Catalyzed Hydrodefluorination of Fluoro-(Hetero)Arenes. Org. Lett 2019, 21, 2482–2487. 10.1021/acs.orglett.9b00889. [DOI] [PubMed] [Google Scholar]
- (36).Roşca DA; Radkowski K; Wolf LM; Wagh M; Goddard R; Thiel W; Fürstner A Ruthenium-Catalyzed Alkyne Trans-Hydrometalation: Mechanistic Insights and Preparative Implications. J. Am. Chem. Soc 2017, 139 (6), 2443–2455. 10.1021/jacs.6b12517. [DOI] [PubMed] [Google Scholar]
- (37).Filimonov VD; Trusova M; Postnikov P; Krasnokutskaya EA; Min Lee Y; Yun Hwang H; Kim H; Chi K-W Diazo Chemistry I; VCH: Weinheim. Org. Lett 2008, 10 (18), 3961–3964. 10.1021/ol8013528. [DOI] [PubMed] [Google Scholar]
- (38).Lv H; Cai YB; Zhang JL Copper-Catalyzed Hydrodefluorination of Fluoroarenes by Copper Hydride Intermediates. Angew. Chemie - Int. Ed 2013, 52 (11), 3203–3207. 10.1002/anie.201208364. [DOI] [PubMed] [Google Scholar]
- (39).Hutchins RO; Lamson DW; Rua L; Milewski C; Maryanoff B Reduction of Aromatic Nitro Compounds with Sodium Borohydride in Dimethyl Sulfoxide or Sulfolane. Synthesis of Azo or Azoxy Derivatives. J. Org. Chem 1971, 36 (6), 803–806. [Google Scholar]
- (40).Lamson DW; Ulrich P; Hutchins RO Aromatic Denitration with Borohydride. Nucleophilic Displacement of Nitrite by Hydride; 1973; Vol. 38. [Google Scholar]
- (41).Facundo AA; Aré A; Fundora-Galano G; Flores-A ĺamo M; Orgaz E; Garcí JJ Hydrodefluorination of Functionalized Fluoroaromatics with Triethylphosphine: A Theoretical and Experimental Study †. New J. Chem 2019, 43, 6897. 10.1039/c9nj00721k. [DOI] [Google Scholar]
- (42).Beregovaya IV; Shchegoleva LN Potential Energy Surface and Dissociative Cleavage of Chlorobenzene Radical Anion. Chem. Phys. Lett 2001, 348 (5–6), 501–506. 10.1016/S0009-2614(01)01171-X. [DOI] [Google Scholar]
- (43).Mulryan D; White AJP; Crimmin MR Organocatalyzed Fluoride Metathesis. Org. Lett 2020, 0–4. 10.1021/acs.orglett.0c03593. [DOI] [PubMed] [Google Scholar]
- (44).Whiteker GT; Froese RDJ; Arndt KE; Renga JM; Zhu Y; Roth GA; Yang Q; Canturk B; Klosin J Synthesis of 6-Aryl-5-Fluoropicolinate Herbicides via Halex Reaction of Tetrachloropicolinonitrile. Org. Process Res. Dev 2019, 23 (10), 2166–2174. 10.1021/acs.oprd.9b00197. [DOI] [Google Scholar]
- (45).Rolka AB; Koenig B Dearomative Cycloadditions Utilizing an Organic Photosensitizer: An Alternative to Iridium Catalysis. Org. Lett 2020, 22, 5040. 10.1021/acs.orglett.0c01622. [DOI] [PubMed] [Google Scholar]
- (46).Tian H; Yang H; Tian C; An G; Li G Cross-Dehydrogenative Coupling of Strong C(Sp 3 )−H with N-Heteroarenes through Visible-Light-Induced Energy Transfer Scheme 1. Visible-Light-Mediated CDC Minisci Reactions. Org. Lett 2020, 22, 7709–7715. 10.1021/acs.orglett.0c02912. [DOI] [PubMed] [Google Scholar]
- (47).Ou W; Zou R; Han M; Yu L; Su C Tailorable Carbazolyl Cyanobenzene-Based Photocatalysts for Visible Light-Induced Reduction of Aryl Halides. Chinese Chem. Lett 2020, 31 (7), 1899–1902. 10.1016/j.cclet.2019.12.017. [DOI] [Google Scholar]
- (48).Uoyama H; Goushi K; Shizu K; Nomura H; Adachi C Highly Efficient Organic Light-Emitting Diodes from Delayed Fluorescence. Nature 2012, 492 (7428), 234–238. 10.1038/nature11687. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.