

Abstract
In this report, we investigate the physical and chemical properties of monocopper Cu(I) superoxo and Cu(II) peroxo and hydroperoxo complexes. These are prepared by cryoreduction/annealing of the parent [LCuI(O2)]+ Cu(I) dioxygen adducts with the tripodal, N4-coordinating, tetradentate ligands L = PVtmpa, DMMtmpa, TMG3tren and are best described as [LCuII(O2•−)]+ Cu(II) complexes that possess end-on (η1-O2•−) superoxo coordination. Cryogenic γ-irradiation (77 K) of the EPR-silent parent complexes generates mobile electrons from the solvent that reduce the [LCuII(O2•−)]+ within the frozen matrix, trapping the reduced form fixed in the structure of the parent complex. Cryoannealing, namely progressively raising the temperature of a frozen sample in stages and then cooling back to low temperature at each stage for examination, tracks the reduced product as it relaxes its structure and undergoes chemical transformations. We employ EPR and ENDOR (electron–nuclear double resonance) as powerful spectroscopic tools for examining the properties of the states that form. Surprisingly, the primary products of reduction of the Cu(II) superoxo species are metastable cuprous superoxo [LCuI(O2•−)]+ complexes. During annealing to higher temperatures this state first undergoes internal electron transfer (IET) to form the end-on Cu(II) peroxo state, which is then protonated to form Cu(II)–OOH species. This is the first time these methods, which have been used to determine key details of metalloenzyme catalytic cycles and are a powerful tools for tracking PCET reactions, have been applied to copper coordination compounds.
Graphical Abstract

INTRODUCTION
Iron- and copper-containing metalloenzymes capable of the reductive transformation of molecular oxygen (O2, dioxygen) to generate powerful oxidizing intermediates are ubiquitous in biological systems. During catalysis these intermediates are capable of efficient substrate oxidation (dehydrogenation) or O atom insertion reactions, which produce critically needed biomolecules and/or metabolic precursors. Heme (iron porphrinate)-containing enzymes are perhaps the most recognized, including the widespread cytochrome P450 (CYP) enzyme family of monooxygenases, which perform reactions such as the synthesis of hormones and cholesterol, as well as the transformation of many molecules and toxins.1,2 Nonheme iron enzymes also activate dioxygen for oxidative processes. These include the diiron-containing soluble methane monooxygenase (sMMO) and related desaturases,3 as well as monoiron enzymes, such as catechol dioxygenases and α-ketoglutarate (αKG)-dependent monooxygenases.4
Copper proteins likewise exhibit a diversity of active-site structures and reactivities. Enzymes with dicopper active sites include monooxygenases/oxidases such as tyrosinases, NspF (converts aminophenol natural product to a N-nitroso derivative), and catechol oxidases.5 However, the reaction mechanisms of peptidyl glycine α-hydroxylating monooxygenase (PHM) and dopamine-β-monooxygenase (DβM) are most often described by a mechanism in which substrate prebinding leads to a [CuII(O2•−)]+ (cupric superoxide)-mediated hydrogen atom abstraction (HAA) and net O atom transfer, all occurring at a single-copper ion active site that possesses an unusual Sthioether Met ligand.5,6,7 A recent theoretical-computational study7 suggests an alternative reaction mechanism involving a protein open-to-closed conformational change and substrate HAA chemistry carried out by a Cu2–O2-derived binuclear intermediate. Finally, lytic polysaccharide monooxygenases (LPMOs)4e,8,9 and particulate methane monooxygenases (pMMO’s)10 use a mono-Cu center to oxygenate their respective substrates (both with C–H bond dissociation energies >100 kcal/mol).
Our interest here is in the chemistry of mono-Cu centers, in particular ligand–copper(I)–dioxygen complexes relevant to practical copper-ion-mediated substrate oxidations,11 and modeling O2 activation in copper biochemistry.5,12 In this report, we study the reduction/protonation of several previously characterized cupric superoxo complexes, [CuII(O2•−)]+, formed by low-temperature oxygenation of LCuI complexes. The three complexes studied employ the family of tripodal tetradentate ligands DMMtmpa, PVtmpa, and TMG3tren, as shown in Figure 1.
Figure 1.

Parent ligand TMPA and cationic copper(II) superoxo complexes employing derivatized TMPA ligands, as formed from Cu(I) complexes on reaction with dioxygen, used in this study.
The CuI/O2 chemistry of the two ligands that are derivatives of TMPA (Figure 1) has been extensively characterized. On the basis of physical-spectroscopic and structural studies, these ligand–Cu(I)–dioxygen adducts (designated below as [LCuI(O2)]+ species) are best described as CuII superoxo complexes possessing an end-on (η1-O2•−)13 superoxo coordination (νO–O = 1119 cm−1 for the TMPA parent ligand)19.13 The third complex, [(TMG3tren)CuII(O2•−)]+, has been well established as an end-on cupric superoxo complex (Figure 1).14 These pentacoordinate [LCuII(O2•−)]+ solution species possess S = 1 ground states, having ferromagnetically coupled copper(II) (i.e., d9) and O2•− lone electrons, and do not exhibit X-band EPR.15 The [(tmpa)CuII(X)]+,2+ complexes (X = Cl−, N3−, H2O, MeCN) possess trigonal-bipyramidal (TBP) coordination geometries,16 as do [{(tmpa)CuII}2(μ-1,2-O22−)]2+ 17 and [(TMG3tren)CuII(O2•−)]+.14b TBP geometries are maintained in solution, as shown by distinctive UV–vis d–d band patterns. This geometry yields a d-orbital manifold with the Cu(II) odd electron in a dz2-based orbital, as signaled by a diagnostic “reverse-axial” EPR spectra seen for [LCuII(X)]+,2+ (g⊥ > g‖ ≅ 2.00; A⊥ ≈ 200–300 MHz, A∥ ≈ 300–400 MHz), as opposed to the familiar spectrum of a tetragonal Cu(II) with an odd electron in a dx2–y2-based orbital (g∥ > g⊥ ≅ 2.00; A∥ ≈ 500 MHz, A⊥ ≈ 50 MHz).16a,d,18
The complex [(tmpa)CuII(O2•−)]+ was first detected in UV–vis monitored stopped-flow studies under cryogenic conditions, as a very rapidly formed kinetic intermediate, with a rate constant for O2 binding of kon = 1.8 × 104 M−1 s−1 at −90 °C, in EtCN as solvent.20 Subsequent studies by flash- and-trap laser spectroscopy demonstrated that kon = 1.3 × 109 M−1 s−1 (RT) in the noncoordinating solvent tetrahydrofuran (THF), a value greater than those found for reactions of ferrous hemes (synthetic or biological) with O2.21
At −80 °C, [(tmpa)CuII(O2•−)]+ is metastable and transforms rapidly to a thermodynamic product, the binuclear complex [{(tmpa)CuII}2(μ-1,2-O22−)]2+ (Scheme 1).22 However, all of the superoxo complexes depicted in Figure 1 are stable in solution at T < −80 °C. Strong donor ligands and/or H-bonding substituents such as the pivalamido group in [(PVtmpa)CuII(O2•−)]+ (Figure 1) (i) considerably enhance both the [LCuII(O2•−)]+ complex thermal stability and reactivity with substrates—H-atom abstraction (HAA) from phenols or weak C–H bond substrates,13,23 (ii) while they suppress diversion to binuclear peroxo dicopper(II) analogues.19,24 In addition to the direct reactivity of peroxo and hydroperoxo Cu(II) complexes in H-atom abstraction and possibly in nucleophilic reactions, they may be important intermediates in Cu-based monooxygenase catalytic cycles (vide supra). For these, oxidative chemistries are initiated by the formation of an O2 adduct, such as a cupric superoxo (CuII–O–O•−) species with end-on (η1) or side-on (η2) O2•− ligation25 or a binuclear peroxo dicopper(II) complex, and proceed through peroxo, hydroperoxo, or even other highly oxidizing intermediates (see Figure S1). Thus, as part of the effort to understand Cu-catalyzed oxidations in general, it is imperative to elucidate the structures and reactivity of [LCuII(O2•−)]+, [LCuII(O22−)], and [LCuII(−OOH)]+ intermediates.
Scheme 1.

Formation of the Binuclear Complex [{(tmpa)CuII}2(μ-1,2-O22−)]2+
In this report, we extend earlier investiations of the physical and chemical properties of CuII superoxo and CuII hydroperoxo compounds25b–d,26 and present a detailed examination of all three products of reduction–protonation of the [LCuI(O2)]+ adducts given in the title, for the complexes with L = PVtmpa, DMMtmpa, TMG3tren (Figure 1), in MeTHF (2-methyltetrahydrofuran) as solvent. We employ EPR and ENDOR (electron–nuclear double resonance) as powerful spectroscopic tools to examine the properties of the states that form. ENDOR, a method that monitors the NMR spectra of nuclei associated with an EPR-active center, allows for the observation of nuclear hyperfine couplings that are unresolved in the EPR spectra of those centers.27 From the g values of such a center and the hyperfine couplings to its nuclei, its metal-ion valence, ligand covalency parameters, and coordination geometries can be identified.
The investigations are initiated by cryoreduction of the EPR-silent parent complexes. In this procedure, γ-irradiation at 77 K, using a 60Co source, ionizes solvent molecules, generating energetic mobile electrons. These “hot electrons” proceed to reduce the [LCuII(O2•−)]+ centers within the frozen matrix, typically creating a reduced form trapped in a structure comparable to that of the parent complex for study by EPR/ENDOR methods.27a,28 Reactions of the reduced species can then be examined by a procedure denoted cryoannealing: namely, progressively raising the temperature of a frozen sample in stages and then cooling back to low temperature at each stage for spectroscopic characterization. This process allows a reduced complex to be tracked as it relaxes its structure and possibly undergoes chemical transformations at higher temperatures. In the present case the final step in cryoannealing the MeTHF frozen matrix occurs upon warming to T > 116 K, at which temperature the solvent melts, with further changes possible upon warming of the fluid phase. This is the first time these methods, which have been used to determine key details of metalloenzyme catalytic cycles27a–i and are powerful tools for tracking PCET reactions,27b have been applied to copper coordination compounds.
MATERIALS AND METHODS
General Experimental Details.
All solvents and chemicals were purchased from commercial sources and used as received unless stated otherwise. Tetrahydrofuran (THF) and 2-methyltetrahydrofuran (MeTHF) were distilled over sodium metal with benzophenone under argon, deoxygenated with argon, and then stored over 4 Å sieves. Pentane was distilled 4× over calcium hydride under Ar and deoxygenated with Ar purging.
1-Hydroxy-2,2,6,6-tetramethyl-piperidine (TEMPO-H) and 1-Deuteroxy-2,2,6,6-tetramethylpiperidine (TEMPO-D) Synthesis.
TEMPO-H was synthesized by following a literature procedure.29 TEMPO–D was synthesized by following the same procedure, but with D2O instead of H2O.
Ligand Syntheses.
DMMtmpa, PVtmpa, and TMG3tren were all prepared according to literature procedures.30 Deuterated (d6) DMMtmpa (D-DMMtmpa, with the three ligand backbone methylene groups deuterated) was prepared by refluxing 200 mg of DMMtmpa in MeOD for 48 h. MeOD was removed by rotary evaporation, and the solid thus obtained was dissolved in 20 mL of hot diethyl ether and filtered through a cotton plug. The solution was then layered with 100 mL of n-hexanes and placed in a −30 °C freezer. After a solid product was obtained, the solvent was decanted and the precipitate was dried. An NMR spectrum of the product indicated 75% incorporation of deuterium into the DMMtmpa methylene positions.
Syntheses of LCuI.
[(PVtmpa)CuI(MeCN)]B(C6F5)4, [(TMG3tren)CuI(MeCN)]B(C6F5)4, and both [(DMMtmpa)CuICO]-B(C6F5)4 and [(D-DMMtmpa)CuICO]B(C6F5)4 were prepared according to literature methods.23,31
Syntheses of LCuII(Cl)2.
[(DMMtmpa)CuIICl]Cl and [(PVtmpa)-CuIICl]Cl were synthesized according to a literature procedure.32 Solutions (1 mM) for EPR samples of [(DMMtmpa)CuIICl]Cl were prepared in butyronitrile, while 1 mM EPR samples of [(PVtmpa)-CuIICl]Cl were prepared in ethanol.
EPR/ENDOR Sample Preparation.
All stock solutions for EPR/ENDOR spectroscopy samples were prepared in a Vacuum Atmospheres glovebox. Samples (0.25 mM) were prepared in volumetric flasks by dissolving the appropriate amount of LCuI solid in MeTHF, and 2.5 equiv trifluoroacetic acid or deuterated trifluoroacetic were added to the stock solutions as appropriate. Stock solutions were then placed in X- or Q-band EPR tubes that were then capped with septa and sealed with Parafilm. All samples were cooled using a cold bath, and the temperature was monitored using an Omega Engineering thermocouple. Cupric superoxide samples in MeTHF were prepared at −135 °C using a 4/1 MeTHF/THF bath cooled with liquid nitrogen, while samples of the peroxide dicopper(II) complex [{(L)CuII)2(μ-1,2-O22−)](B(C6F5)4)2 were prepared at −80 °C with an acetone bath cooled with liquid nitrogen. Oxygenated samples using 16O2 were prepared by passing O2 through a Drierite column and needle into the solution. 17O2 samples were prepared with 37% 17O2 purchased from Cambridge Isotope Laboratories by using a breakflask attached to an evacuated Schlenk flask; the 17O2 was then added to samples using a three-way syringe and the solution purged (to remove excess dioxygen) by bubbling with argon for 15 s. Product solutions were then immediately frozen in liquid nitrogen.
Synthesis of [(DMMtmpa)CuII(−OOH)]B(C6F5)4 and [(DMMtmpa)CuII(−OOD)]B(C6F5)4.
A stock solution (0.25 mM) of [(DMMtmpa)CuICO]B(C6F5)4 in MeTHF was prepared in the glovebox, and 10 equiv of TEMPO-H or TEMPO-D were added. The solution was then transferred to the EPR tube, which was capped with a septum and sealed with Parafilm. The samples were cooled to −135 °C using a 4/1 MeTHF/THF bath cooled with liquid nitrogen; the temperature was monitored using an Omega Engineering thermocouple. Samples were oxygenated by passing O2 through a Drierite column and syringe needle into the solution, and the solution was purged (to remove excess dioxygen) by bubbling with argon for 15 s. The cupric superoxide sample thus formed was allowed to react with the TEMPO-H or TEMPO-D already present for 1 h. Product solutions were then immediately frozen in liquid nitrogen.
To increase the relative population of monocopper [LCuII(O2•−)]+ species (relative to binuclear peroxo complexes that could form; vide supra), all EPR and ENDOR spectroscopy experiments were conducted at low concentrations (e.g., 0.25 mM [(DMMtmpa)CuI]+). Under these conditions, the concentration of the peroxo-bridged binuclear complex was less 0.04 mM.19
EPR and ENDOR Spectroscopy.
X-band continuous wave (CW) EPR spectra were recorded on a Bruker ESP 300 spectrometer equipped with an Oxford Instruments ESR 900 continuous He flow cryostat. All CW Q-band EPR and ENDOR spectra were recorded at 2 K in dispersion mode, under “rapid passage” conditions, which give an absorption-display line shape.10a 2H Mims Pulsed ENDOR measurements were collected at 2 K on a spectrometer described previously,33 with a SpinCore PulseBlaster ESR_PRO 400 MHz digital word generator and an Agilent Technologies Acquiris DP235 500 MS/s digitizer using SpecMan4EPR software.34 The Mims pulse sequence (π/2–τ–π/2–TRF–π/2–τ–echo) was employed.35 Simulations of EPR spectra were performed using the program Easyspin with the pepper function for powdered samples.36
RESULTS
In this report we first describe the experiments that characterize the primary product trapped upon cryoreduction of the suite of CuIO2-derived complexes. We then focus on the behavior of cryoreduced [(DMMtmpa)CuI(O2)]+, characterizing the conversion of its primary cryoreduction product to relaxed states upon successive stages of cryoannealing.
Primary Product of Cryoreduction.
1n the case of the end-on LCuI(O2) complexes being examined, given their actual electronic structure, [LCuII(O2•−)]+, one must consider multiple conceivable cryoreduction products (Scheme 2), where species shown in red are EPR-active. First and foremost, the common result in previous cryoreduction studies of FeII(O2) complexes is localization of the added electron on the O2 moiety.27j,37 In the present case such a result would yield a trapped copper(II) peroxo complex. However, in addition, there is precedent27j,37 for localization of the added electron on the metal ion, which in this case would generate O2•− coordinated to copper(I). Moreover, as it is extraordinarily difficult to completely dry a solvent, either one of these products could pick up a proton during reduction to directly yield the protonated form of the O2-derived moiety. This possibility is enhanced by adding trifluoroacetic acid (TFA) to the solution, with the prospect that it would H-bond to the [CuI(O2)]+ reactant, thus offering an easily abstracted proton to the immediate product of reduction. Measurements now described identify the actual cryoreduction product.
Scheme 2.

Conceivable Products of [LCuII(O2•−)]+ Cryoreduction
Radiolytic reduction of each of the diamagnetic [LCuI(O2)]+ complexes (≡[LCuII(O2•−)]+) in MeTHF at 77 K results in the generation of a new paramagnetic species (A) that shows the same axial EPR spectra in Q-band and X-band EPR for all of the complexes studied (Figure 2 for L = DMMtmpa; see Figure S2 for L = PVtmpa, TMG3tren). The simulation shown gives g∥ = 2.52 ≫ g⊥ ≈ 1.95; the strong broad signal at g ≈ 2 is due to trapped radiolytically generated free radicals. Multiple characteristics of this spectrum immediately indicate that it is not associated with a copper(II) center. First, although the “symmetry” of the g tensor, namely g∥ > g⊥, is reminiscent of a tetragonally coordinated copper(II) center with a dx2–y2 odd electron, the large value of g∥ argues against this assignment. Even more telling is the absence of the large 63,65Cu hyperfine splittings of the g∥ feature of the X-band spectrum invariably seen for such a Cu(II) center (such splittings commonly are washed out in Q-band spectra). Furthermore, rapid electron-spin relaxation causes the loss of the X-band EPR spectrum for species A at temperatures above 50 K, whereas within our experience such copper(II) complexes never exhibit this characteristic.
Figure 2.

(top) Q-band absorption (black) CW EPR spectrum for the 0.25 mM [(DMMtmpa)CuI(O2)]+ adduct (≡[(DMMtmpa)-CuII(O2 •−)]+) in MeTHF exposed to γ-irradiation at 77 K with a dose of 3 Mrad and a simulation (red). Instrument settings: modulation amplitude 1 G, microwave power 0.7 mW, microwave frequency 35.09 GHz, T = 2 K. (bottom) X-band derivative CW EPR spectra for 0.25 mM [(DMMtmpa)CuI(16O2)]+ and [(DMMtmpa)-CuI(17O2)]+ (37% 17O enrichment) adducts in MeTHF γ-irradiated at 77 K with a dose of 3 Mrad. The doublet of sharp peaks marked by asterisks is due to hydrogen atoms generated by γ-irradiation of the quartz tube, and the strong signal at g = 2 belong to radicals in the solvent matrix created by γ-irradiation. Instrument settings: modulation amplitude 8 G; microwave power 2 mW, microwave frequency 9.373, measurement temperature 10 K.
A more detailed examination confirms this conclusion. The coordination sphere of a metal center that undergoes radiolytic cryoreduction while it is trapped in a frozen matrix at 77 K cannot rearrange, and thus the cryoreduced product is expected to retain the geometry of the parent complex.28 If species A trapped upon cryoreduction of [LCuII(O2•−)]+ at 77 K were the end-on peroxo [LCuII(O22−)] or hydroperoxo [LCuII(−O2H)]+ species that retains the trigonal-bipyramidal structure of the precursor, it would show a copper(II) EPR spectrum characteristic of this symmetry, with g⊥ > g∥16a,b,18a accompanied by a well-resolved 63,65Cu hyperfine structure at both g∥ and g⊥, as seen for other [LCuII(X)]n+ compounds (e.g., X = Cl−; Figure S3) and in particular for [LCuII(−O2H)]+ (see below). However, this is not true for A. In addition, details of the EPR spectra of [LCuII(X)]n+ complexes depend on the nature of the ligands L, whereas that of A does not vary with L (Figure S2).
Finally, 14N and 1H ENDOR spectra of A, formed from the [(DMMtmpa)CuII(O2•−)]+ complex (Figure 3), confirm that it does not have a copper(II) center. First, if A were a copper(II) species, the presence of nitrogens from DMMtmpa in its coordination sphere would generate well-resolved 14N ENDOR signals, such as those exhibited by the chemically prepared [(DMMtmpa)CuII(−OOH)]+ complex, but species A does not show such signals (Figure 3; see further discussion in the Supporting Information). The absence of such signals is not merely the result of “detectability” issues. As further discussed in the Supporting Information, if A were a Cu(II) complex with a dx2–y2 odd electron, the ligand pyridyl 14N signals would be at a frequency (greater hyperfine coupling) even higher than those for the amine 14N of [(DMMtmpa)-CuII(−OOH)]+ and their ENDOR signal would be far more intense. Thus, such a complex would give 14N signals that are far easier to see than the quite strong signals from [(DMMtmpa)CuII(−OOH)]+, in contrast to the experimental absence of any 14N signals. Second, the 1H ENDOR spectra of A formed by the cryoreduced L = DMMtmpa, PVtmpa, and TMG3tren CuI(O2) adducts (Figure S4) are all very similar and do not show the relatively strongly coupled nonexchangeable 1H signals from the ligand methylene protons seen in LCuII(X) centers, including the [(DMMtmpa)CuII(−OOH)]+ complex (discussed below and shown in the Supporting Information). When they are taken together, these spectroscopic data indicate that the species A generated by cryoreduction of the [LCuII(O2•−)]+ parent complexes are not copper(II) centers formed by electron addition to the superoxo moiety. The cryoreductive conversion of [LCuII(O2•−)]+ to A must instead involve electron addition to the Cu(II) ion to generate end-on copper(I)-superoxo complexes, [LCuI(O2•−)]+. As we now show in detail, the properties of A confirm this conclusion.
Figure 3.
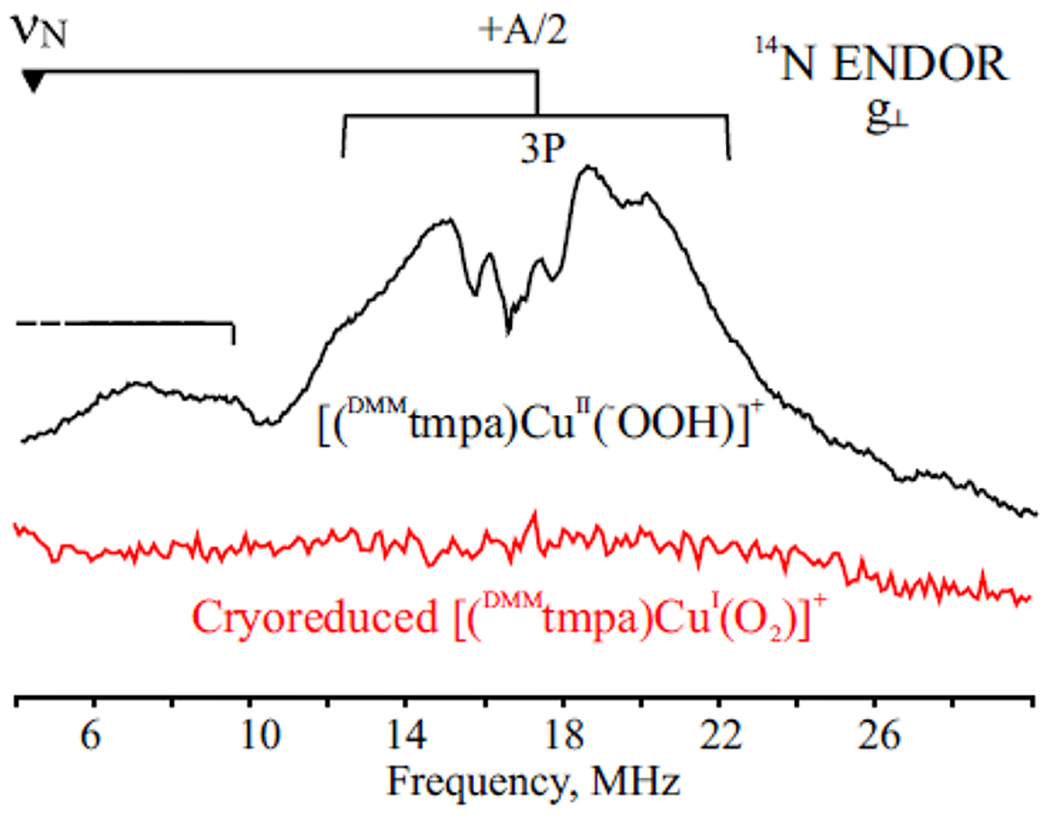
35 GHz, 2 K CW 14N ENDOR spectra, (black) Spectrum for [(DMMtmpa)CuII(−OOH)]+ in MeTHF taken at g⊥ = 2.2, with responses from two types of 14N. As discussed with additional detail in the Supporting Information, the more intense pattern centered at ~18 MHz (“goalpost” centered at the 14N Larmor frequency (∇)) is assignable to the amine 14N; the low-frequency, and therefore less intense, pattern (which extends to still lower frequencies as indicated) is assigned to the three pyridyl nitrogens. (red) Spectrum of A, the product of cryoreduction of [(DMMtmpa)CuII(O2 •−)]+ in MeTHF, taken at g⊥ = 1.95. Instrument settings: microwave power 25 db, modulation amplitude 2 G, RF sweep rate, 0.5 MHz/s, 35 scans.
This conclusion is supported by the observation of 17O hyperfine broadening of the A EPR spectrum even upon use of O2 with a low level of 17O enrichment (37%) to prepare the parent adduct [(DMMtmpa)CuI(16,17O2•−)]+ (Figure 2, bottom). This assignment is further supported by the g tensor for species A, which is typical for the 2Π3/2 state of O2•−. The influence of bonding interactions in a nonlinear Cu(I)–(O–O)•− linkage can be described as creating a local C2ν crystal field that splits the π-orbital degeneracy of the O2•− ion, placing the unpaired electron in the π*(2py) antibonding orbital normal to the Cu–O–O plane (Figure 4). As described by eq 1, this gives rise to a near-axial tensor with g∥ ≫ g⊥ ≳ ge, with the g∥ direction being along the internuclear O2•− axis (Figure 4).38 In these equations, λ is the spin–orbit coupling constant, Δ is the πy*–πx* splitting, and l represents a correction to the angular momentum about the internuclear axis.38
| (1) |
Figure 4.

(left) Schematic representation of the O2•− π* orbitals split by an energy Δ through the influence of progressively stronger bonding in a nonlinear Cu–O–O complex, (right) Plot of the superoxide g values as a function of Δ /λ, with λ being the spin–orbit coupling constant (eq 1).
As seen in Figure 4, these equations fully describe the g tensor for species A when Δ /λ ≈ 4.3. It is well-known that the dependence of the EPR spectra of superoxide ions (O2•−) on their surroundings can be described as reflecting variations in the crystal-field parameter that splits the π* orbitals (Δ), which are degenerate in an idealized axially symmetric environment.38,39 In particular, superoxide ions in alkali halide hosts show a strongly anisotropic EPR signal, with g‖ ≈ 2.46 and g⊥ ≈ 1.95 for the KCl host, values remarkably similar to these for species A.38,40 In short, the question posed by Scheme 2 has been answered by EPR and ENDOR measurements: cryoreduction of the [LCuI(O2)]+ = [LCuII(O2•−)]+ complexes generates an end-on superoxide radical coordinated to a diamagnetic copper(I) ion.
The implicit assumption in the above discussion is that the superoxo ligand in [LCuI(O2•−)]+ (A) has not abstracted a proton (i.e., from solvent or other source) upon cryoreduction to generate the hydroperoxo species, [LCuI(•O2H)]. This is confirmed by a comparison of the g tensor and 1H ENDOR of A with the properties of the well-known •O2H radical.41 The π* orbitals of the nonlinear •O–O–H are split by such a large value of Δ /λ that g‖ < 2.1, in sharp contrast with g‖ ≈ 2.46 for A, and the •O2H proton exhibits hyperfine couplings, A ≈ 40 MHz, not present for A (Figure S4). Indeed, the isolation of the parent freeze-quenched CuII(O2•−)+ moiety from interactions with its environment in the 77 K frozen solution is illustrated by cryoreduction of [(TMG3tren)CuI(O2)]+ with added trifluoroacetic acid. A previous study indicated that TFA forms an H-bond with the superoxo ligand of the [(TMG3tren)CuII(O2•−)]+ complex.42 Nonetheless, we find here that adding TFA to [(TMG3tren)CuII(O2•−)]+ in MeTHF neither alters the EPR signal of the cryoreduced sample (Figure S5) nor introduces an exchangeable 1H ENDOR response from an interacting proton (Figure S6), further demonstrating that this state has not abstracted a proton to generate the hydroperoxo species [LCuI(•O2H)]. Thus, A, the primary product of cryoreduction of the dioxygen adduct [(TMG3tren)CuI(O2)]+, indeed is “simply” the end-on superoxo complex, [(TMG3tren)CuI(O2•−)]+ (A; Scheme 3).
Scheme 3.

Primary Product of [(TMG3tren)CuI(O2)]+ Cryoreduction A
Cryoannealing.
Overview.
For clarity, we first summarize here the multistep cryoannealing process and then proceed to describe the individual stages; additional data are presented in the Supporting Information. We progressively annealed A = [(DMMtmpa)CuI(O2•−)]+ at multiple temperatures up to 260 K, at each stage recooling the sample to collect EPR spectra (Figure 5 and Figure S7). Cryoannealing the frozen solid containing the complex A at T = 90 K for 5 min allowed its relaxation and transformation to a new species with a dramatically different EPR spectrum, denoted B (Figure 5 and Figure S8), which is stable at this temperature. In turn B transforms to a third species, C, at T ≈ 116/117 K, where MeTHF becomes fluid. Species C has an EPR spectrum very similar to that of B but has a definitively different 1H ENDOR spectrum (Figures 6 and 7). Complex C is stable in fluid solution to T ≈ 135 K, but attempts to warm it further were compromised by its instability along with autoxidation of unreduced [(DMMtmpa)CuI(O2)]+ (Figure S7). We now describe the characterization of complexes B and C.
Figure 5.

X-band CW EPR spectra: (black) radiolytically cryoreduced 0.25 mM [(DMMtmpa)CuI(O2)]+ adduct, A, in MeTHF; (red) after annealing at 90 K for 5 min to generate B; (blue) after further annealing at 117 K for 2 min, creating C; (pink) 0.25 mM [(DMMtmpa)CuII(−OOH)]+ adduct in MeTHF prepared chemically as described in Materials and Methods. The 117 K annealed spectrum and the [(DMMtmpa)CuII(−OOH)]+ spectrum are both well-simulated with the parameters g = [2.21, 2.20, 1.96], A⊥ = 220 MHz, and A‖ = 330 MHz. Instrument settings: microwave frequency 9.37 GHz; modulation amplitude = 10 G; microwave power = 2 mW; T = 10 K (black) and 20 K for others.
Figure 6.

(top) 2 K Q-band, CW 1H ENDOR spectra (g = 2.23) for [(DMMtmpa)CuII(−OOH)]+ (red) and [(DMMtmpa)CuII(−OOD)]+ prepared chemically (black). (bottom) 2 K Mims pulsed 2H ENDOR spectrum for [(DMMtmpa)CuII(−OOD)]+, with the frequency axis stretched to correspond to the 1H axis. CW instrument settings: modulation amplitude 2 G, microwave power 25 db; RF scan rate 1 MHz/s (1H CW) and 0.1 MHz/s (Mims), 30 scans, T = 2 K, microwave frequency 35.2 GHz. Mims instrument settings: π = 90 ns, τ = 500 ns, repetition time 100 ms, tRF = 60 μs, RF tail 10 μs, 125 scans, microwave frequency 34.648 GHz.
Figure 7.

35 GHz 2 K 1H ENDOR spectra collected near g⊥, in the field region where there is no overlap of the EPR signal of B with radiation-produced solvent radicals (Figure S8). Spectra: complex B (black), the radiolytically cryoreduced 0.25 mM [(DMMtmpa)-CuI(O2)]+ adduct in MeTHF after annealing at 90 K for 5 min; complex C (blue), generated by further annealing at 117 K for 2 min; [(DMMtmpa)CuII(−OOH)]+ (red) prepared chemically. Instrument settings: T = 2 K, modulation amplitude 2 G, radio frequency bandwidth broadened to 60 kHz, 1 MHz/s RF scan speed, 30 scans.
Annealing at 90 K.
Complex A = [(DMMtmpa)CuI(O2•−)]+, formed directly by cyroreduction of the [(DMMtmpa)CuI(O2)]+ trapped at 77 K, relaxes upon annealing for 5 min at 90 K to the new species B showing a reverse-axial EPR signal with g⊥ ≈ 2.23 > g‖, characteristic of a trigonal-bipyramidal copper(II) dz2 ground state (Figure 5 (X-band) and Figure S8 (Q-band)). As is typical for TBP Cu(II) complexes, this signal exhibits a resolved 63,65Cu hyperfine structure at g⊥; there is clearly 63,65Cu hyperfine structure at g‖ as well, but details of the g‖ ≈ 2 region are obscured by a signal from free radicals generated during cryoreduction (Figure 5 and Figure S8). The EPR data thus show that the annealing/relaxation at 90 K of the superoxo radical primary cryoreduction product A has led to internal electron transfer (IET) in which the copper(I) of [(DMMtmpa)CuI(O2•−)] (A) is oxidized to the EPR-active copper(II) state while the superoxo radical is reduced to the closed-shell peroxo state (Scheme 4). In the next subsection we show that comparisons of proton ENDOR spectra of this species with those of the state obtained upon annealing B to higher temperature establish that the peroxo moiety of species B remains unprotonated.
Scheme 4.

Conversion of A to B by IET during Cryoannealing
Annealing to T ≈ 116 K.
The solvent-matrix radical signal almost completely disappears upon annealing B to T ≥ 116 K, the melting point of MeTHF, revealing the complete EPR signal for a further-relaxed copper(II) species, C (Figure 5 and Figure S8). The EPR signal of C has g⊥ ≈ 2.23 and g‖ ≈ 2.06, with well-resolved 63,65Cu hyperfine splittings associated with both g values, as expected for a TBP geometry (legend of Figure 5). The spectra of B and C are quite similar, but g⊥ and the 63,65Cu hyperfine structure of C differ subtly from those in the spectrum of B. Instead, the g values and 63,65Cu hyperfine splittings of C are those of the mononuclear end-on hydroperoxo [(DMMtmpa)CuII(−OOH)]+ complex state prepared by HAA from TEMPO-H by the [(DMMtmpa)CuI/O2]+ adduct (Figure 5 and Figures S7 and S8).19
Identity of C.
To test the resulting expectation that C is indeed the hydroperoxo state, we first characterized the 1H ENDOR of the Cu(II)–OOH hydroperoxo proton by comparing the properties of the [(DMMtmpa)CuII(−OOH/D)]+ isotopologue complexes, as separately prepared by the chemical reaction of [(DMMtmpa)CuII(O2•−)]+ with TEMPO-H/D: their EPR spectra are identical, but their ENDOR spectra are not. The 1H ENDOR response of [(DMMtmpa)-CuII(−OOD)]+ shows a loss of intensity and subtle changes in shape in comparison to those of [(DMMtmpa)CuII(−OOH)]+ in spectra taken across their EPR envelopes (Figure S9). In particular, at fields in the g⊥ region (Figure 6 and Figure S9) the spectra of [(DMMtmpa)CuII(−OOH)]+contain a doublet feature with a splitting of A ≈ 7 MHz, which upon closer inspection is a superposition of two signals. As a result of H/D substitution, those peaks in the 1H spectra of [(DMMtmpa)-CuII(−OOD)]+ with A ≈ 6–7 MHz lose intensity and are no longer composite, while in the 1H spectra of [(DMMtmpa)-CuII(−OOH)]+ there are broader “wings” as the field approaches g⊥, and these are suppressed upon H/D substitution. The association of these differences in the 1H ENDOR spectra with contributions from the hydroperoxo proton is confirmed by the appearance of 2H ENDOR signals for [(DMMtmpa)CuII(−OOD)]+ that correspond to the loss of 1H intensity in the 1H ENDOR response of [(DMMtmpa)-CuII(−OOH)]+ (Figure 6).
This exchangeable 1H coupling of A ≈ 7 MHz for [(DMMtmpa)CuII(−OOH)]+ is similar to that of the hydroperoxo proton of low-spin Fe(III)–OOH centers, A ≈ 10 MHz.37b The nonexchangeable 1H signals for [(DMMtmpa)-CuII(−OOH/D)]+ (Figure 6) are attributable to protons of the tripodal ligand. For example, as seen in Figure S10, deuteration of the hydrogen atoms of the three backbone methylene groups causes the loss of 1H intensity with a broad range range of couplings, with maximum values of A ≈ 10–12 MHz.
Having thus characterized the chemically formed [(DMMtmpa)CuII(−OOH)]+, we then compared its properties to those of C. Not only do their EPR spectra agree (Figure 5) but also the 1H ENDOR spectra of the cryoannealed species C correspond well, both showing the 1H intensity from the exchangeable hydroperoxo proton (Figure 7). Together, these EPR/ENDOR measurements establish that annealing B at T ≥ 116 K leads to the formation of C = [(DMMtmpa)-CuII(−OOH)]+. A plausible source of the hydroperoxo proton acquired during cryoannealing is a trace of water in the MeTHF that becomes available in the fluid solution.
Identity of B.
With these measurements and conclusions regarding C, we can now complete the assignment of B. Despite the similarity between the EPR spectra of species B, generated by annealing A at 90 K, with that of C = [(DMMtmpa)CuII(−OOH)]+, formed by annealing of B at T > 116 K, their 1H ENDOR spectra taken near g⊥ differ tellingly: the response from the exchangeable hydroperoxide proton that appears in the 1H spectrum of chemically prepared [(DMMtmpa)CuII(−OOH)]+ and of C is absent in the 1H spectrum of B (Figure 7), indicating that B lacks a hydroperoxo proton. It is important to note that the absence of this proton and its ENDOR signal for B is supported by an examination of the spectrum of the chemically prepared [(D-DMMtmpa)CuII(−OOH)]+, in which the methylene hydrogen atoms of the DMMtmpa ligand are deuterated (Figure S9). Comparing the signal of B with that of the latter shows that the residual, low-intensity features in the 1H spectrum of B that extend to frequencies associated with the hydroperoxo proton of C are in fact instead attributable to signals from the ligand methylene protons, as described above (Figure S10).
This absence of intensity from a hydroperoxo 1H in the spectrum of species B thus indicates that B is indeed the unprotonated mononuclear peroxo intermediate [(DMMtmpa)-CuII(O22−)] (Scheme 4). The further conclusion that B exhibits end-on peroxo binding (CuII–O–O configuration), rather than side-on binding, is supported by its EPR spectrum with g⊥ > g‖ ≈ 2, which is characteristic of trigonal-bipyramidal copper(II) coordination enforced by the tetradentate ligand, and thus of the peroxo ligand bound axially to copper by only a single oxygen atom.
DISCUSSION
In this report, we have applied the cryoreduction/annealing protocol to frozen solutions of [LCuI(O2)]+ Cu(I) dioxygen adducts, utilizing a set of related tripodal tetradentate N4 ligands to copper, DMMtmpa, PVtmpa, and TMG3tren (Figure 1), whose O2 adducts had been characterized as cupric superoxide ([LCuII(O2•−)]+) coordination complexes. Scheme 5 summarizes the overall outcome of the cryoreduction/annealing of these species.
Scheme 5.

Overall Outcome of Cryoreduction/Cryoannealing of [LCuII(O2•−)]+ Complexes
γ-irradiative cryoreduction of frozen solutions of the [LCuII(O2•−)]+ species produces the reduced species (A) trapped in the geometry of the parent complex at the 77 K temperature of irradiation. Progressive annealing (warming) of the frozen solution to higher temperatures, first below and then above the solvent (MeTHF) freezing point, causes successive transformation of the initial product to the new species B and C through electron-transfer and/or protonation processes (Scheme 5). These intermediates have been characterized by EPR/ENDOR spectroscopy and correspond to those formed prior to O–O cleavage in P-450 monooxygenases and in the proposed mono-Cu monooxygenase cycles of Figure S1. Thus, their physical properties, reactivity, and an understanding of their chemical interrelationships are of central importance in chemical and biochemical copper-ion-mediated O2 activation through reductive transformation to highly reactive oxidizing intermediates.
Species A and Superoxo Complexes.
Athough it was perhaps to be expected that the initial reduction of a [LCuII(O2•−)]+ complex (Figure 1) would convert the superoxide radical anion to the peroxide dianion, creating the EPR-active Cu(II) peroxide, instead it is the copper(II) ion that is reduced, yielding A = [LCuI(O2•−)], with a superoxide radical anion bound to a diamagnetic cuprous ion (Scheme 3). This assignment of A is based on multiple observations, as detailed in Results.
An early analysis of the EPR signal of the superoxide radical anion showed that the components of the g tensor vary with the splitting of the two mutually perpendicular π* superoxide anion orbitals (Figure 4). The properties of the [LCuI(O2•−)]+ complexes can be usefully compared to those of the analogous dioxygen adducts of cobalt(II) macrocycles, which have long been recognized as exhibiting an end-on superoxide coordinated to the diamagnetic cobalt(III) ion,43 [CoIII(O2•−)]2+. In those cases, g‖ ≈ 2.09 is markedly less than that for A, and the description by eq 1 thus implies a far larger π-orbital splitting, Δ / λ > 15, with the precise value depending on assumptions regarding spin delocalization. In those cobalt complexes, the large π-splitting is induced by strong bonding in the bent Co–O–O linkage (angle of ~120°). The far larger value of g‖ and thus smaller splitting for A implies that the nonlinear CuI–O–O•− moiety exhibits a substantially weaker interaction between the Cu(I) and the superoxide.
While we are unaware of any examples of previously reported copper(I) superoxo complexes, analogous heme–iron(II)–superoxo species have been generated. Davydov, Hoffman, and co-workers37a first established that cryoreduction of a monomeric oxy-ferrous hemoglobin FeII(O2) center (FeIII(O2•−)), an oxy-ferrous octaethylpoprhyrin, and an oxy-ferrous cyclen complex all produced a superoxo-ferrous state FeII(O2•−) state, where the macrocycle-iron(II) is low-spin and thus diamagnetic.37a Upon annealing/warming to T > 150 K, all of these complexes transform by internal electron transfer (IET) to FeIII–(hydro)peroxo species, driven at least in part by H-bonding or H+-donation to the superoxo moiety.
In synthetic systems, superoxide radical anion binding to heme iron(II) has also been observed. Ohta, Naruta, and co-workers44 demonstrated that γ-ray irradiation could be used to cryoreduce a heme–FeIII-superoxo, giving a porphyrinate–iron(II)–superoxide species. Ivanović-Burmazović and co-workers45 showed that an iron(II) porphyrinate with a K+ crown ether appendage undergoes reversible O2•− binding to generate an iron(II) superoxo complex shown to be in equilibrium with an iron(III) peroxo complex. Such interconversions suggest that an FeII(O2•−) moiety may function as a superoxide H atom abstractor and/or a peroxide anion nucleophilic oxidant.
Thus, on the basis of this iron(II)-superoxo complex literature, it would be of future interest to further probe the formation and reactivity of [LCuI(O2•−)] (A) species, if they could be isolated in proteins or synthetic complexes. What are the chemical and redox properties of the superoxo binding in the parent [LCuII(O2•−)]+ complexes and likewise for the initial product of cryoreduction, [LCuI(O2•−)] (A)?
Species B and Peroxo Complexes.
As shown here, cryoannealing A = [(DMMtmpa)CuI(O2•−)] complexes in the frozen solid leads to IET, giving the end-on B = [(DMMtmpa)-CuII(O22−)] CuII peroxo complex (Scheme 5), with a dramatically altered EPR spectrum (see Results). While one O atom of the CuII–O–O− center is ligated to copper(II), the other oxygen apparently is “naked”, as its properties were not changed by incorporation of TFA in the frozen solution and it does not show the 1H ENDOR signal from an H-bond to the terminal O atom. Further verification that B is not a hydroperoxo species comes from the contrasting result that C indeed does possess an exchangeable proton, with spectroscopic properties (EPR and 1H ENDOR) which match those of authentically generated Cu–OOH complex(es) (see Results).
Some time ago, the Karlin group characterized a peroxo dicopper(II) complex, [(XYLO−)CuII2(O22−)]+ (XYLO− is a binucleating ligand with phenolato bridging group), whose spectroscopic properties indicated a terminal end-on peroxo, CuII–O–O− coordination.46 The peroxo group is exceedingly basic; the conjugate acid of this complex has a pKa value of 24 in MeTHF at −125 °C.47
To our knowledge, other copper coordination complexes with a terminal end-on peroxo group, such as those we find for intermediate B, are unknown. However, two LPMO protein derivative X-ray structures are suggested to possess such entities.48 In these, the active-site copper(II) exhibits tridentate nitrogen coordination: a histidine imidazole and an imidazole + amino −NH2 group from the terminal histidine.8,9,49 In both structures the CuII–O–O− group is severely bent (~120°); the O–O moiety is assigned as a peroxo group on the basis of O–O distances calculated to be dO–O = 1.44 or 1.46 Å. One does not know unambiguously if these peroxo groups might be protonated. Otherwise, side-on binding of a peroxo group to a monocopper(II) center has been demonstrated in both a protein50 and a synthetic chemistry environment.51 In both of these cases overall highly favorable pentacoordination for Cu(II)18b is achieved by peroxo side-on coordination, whereas for the [LCuII(O22−)] studied here, with L as a tetradentate ligand, end-on peroxo binding achieves pentacoordination.
The relative reactivity toward substrates of an end-on bound copper or iron metal peroxo versus a metal hydroperoxo is of considerable interest. The expectation would be that the former possesses a nuclophilic oxidative behavior while the latter acts as an electrophile. While there are particular examples known for nucleophilic non-heme iron Fe–O–O−52 or Cu–O–O− (vide supra)46,47 species, there are also documented cases of non-heme iron53 and copper hydroperoxo54 complexes with nucleophilic or amphoteric55 reactivity. A future comparative study of the reactivity of complexes such as B and C should thus be highly informative.
Examples of synthetic hemes56,57 with end-on peroxo binding are known. This coordination is also seen as the primary product of cryoreduction of FeII(O2) complexes in heme proteins,27j,37 as confirmed via X-ray crystal structure determinations. For example, a ferric peroxo myoglobin (Mb) complex was generated by in situ synchrotron radiation of the oxy FeII(O2) species at 100 K.58 Another case comes from the cryoradiolytic reduction of a Mb Compound II.59 In both, the end-on-bound peroxo group terminal O atom (with formal negative charge) is stabilized by protein H-bonding. Most recently, an end-on heme-peroxo moiety was generated from cryoreduction of an oxy-heme in an engineered protein studied by Lu and co-workers.60
Sligar and co-workers61 recently reported on the chemistry of a steroidogenic P450, CYP17A1, which effects a biosynthetic aldehyde oxidation involving C–C bond cleavage (lyase activity); their spectroscopic interrogation points to a nucleophilic end-on peroxo heme-FeIII(OO)2− intermediate that adds to the substrate carbonyl group, leading to products. The possibility that P450 end-on bound ferric peroxo anion intermediates effect substrate transformation via nuclophillic attack had been discussed extensively,2b,62 but it is now clear that this is not usually the case (i.e., in NO synthases or aromatase). Rather, the peroxo intermediates in general simply protonate to give heme-FeIII(−OOH) intermediates that transform to the Compounds I which effect the substrate oxidative (net hydroxylation) reactions.27j,37b
Species C and Hydroperoxo Complexes.
As has already been described, annealing the end-on peroxo complex [(DMMtmpa)CuII(O22−)] (B) causes protonation and relaxation to the terminal hydroperoxo complex possessing TBP coordination, C = [(DMMtmpa)CuII(−OOH)]+. Complex C is an analogue of heme-FeIII(−OOH) hydroperoxo species, which are the reactive states in heme oxygenases and which form in P450s prior to further protonation and O–O heterolytic cleavage. Most recently, there has been an intense interest in the O–O cleavage, both homolytic and heterolytic, of synthetic ligand–CuII(−OOR) (R ≠ H) compounds, studied as a function of the ligand environment, presence of H-bonding groups, etc.63 However, a deep understanding of O–O cleavage chemistries, which likely occur in synthetic hydroperoxo–copper(II) complexes64 or in copper monooxygenases, remains to be achieved.
CONCLUSIONS
We have for the first time performed γ-irradiation cryoreduction of mononuclear copper(I) dioxygen complexes and have characterized reduced and/or protonated intermediates important in O2-activation chemistry, analogues of species observed in the early intermediates observed for the cytochrome P450 enzyme and Cu monooxygenase (Figure S1) catalytic cycles. Surprisingly, the primary product of reduction of the Cu(II) superoxo species is a set of metastable cuprous superoxo [LCuI(O2•−)]+ species. During annealing to higher temperatures this state first undergoes IET to form the Cu(II) peroxo state, which then protonates to form the Cu(II)–OOH species, according to Scheme 5.
Future studies suggested by these findings would include (a) probing for synthetic systems exhibiting a CuI superoxo/CuII peroxo (end-on) equilibrium, (b) the design of complexes that generate an analogue to complexes B, with an end-on peroxo group bound to Cu(II), (c) introduction of oxidizable organic substrates into studies such as those described here, and (d) cryoreduction of Cu(II) hydroperoxo complexes C, to study O–O reductive cleavage, a key step in all O2-derived biological and abiological chemistries.
Supplementary Material
ACKNOWLEDGMENTS
The authors are grateful for financial support of this research from the National Science Foundation (MCB-1908587 to B.M.H.) and the National Institutes of Health (T32GM008382 to R.J.J.; R01111097 to B.M.H.; R01GM28962 and R35GM139536 to K.D.K.).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c10252.
Discussion of 14N ENDOR (Figure 3) and additional EPR and ENDOR figures (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.1c10252
The authors declare no competing financial interest.
Contributor Information
Roman Davydov, Department of Chemistry, Northwestern University, Evanston, Illinois 60201, United States.
Austin E. Herzog, Department of Chemistry, Johns Hopkins University, Baltimore, Maryland 21218, United States.
Richard J. Jodts, Department of Chemistry, Northwestern University, Evanston, Illinois 60201, United States.
Kenneth D. Karlin, Department of Chemistry, Johns Hopkins University, Baltimore, Maryland 21218, United States.
Brian M. Hoffman, Department of Chemistry, Northwestern University, Evanston, Illinois 60201, United States.
REFERENCES
- (1).(a) Huang X; Groves JT Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins. Chem. Rev 2018, 118, 2491–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Poulos TL Heme Enzyme Structure and Function. Chem. Rev 2014, 114, 3919–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ortiz de Montellano PR Hydrocarbon Hydroxylation by Cytochrome P450 Enzymes. Chem. Rev 2010, 110, 932–948. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Denisov IG; Makris TM; Sligar SG; Schlichting I Structure and Chemistry of Cytochrome P450. Chem. Rev 2005, 105, 2253–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Sono M; Roach MP; Coulter ED; Dawson JH Heme Containing Oxygenases. Chem. Rev 1996, 96, 2841–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).(a) Others include heme oxygenases, NO synthases, and aromatases: [DOI] [PubMed]; (b) Zhu Y; Silverman RB Revisiting Heme Mechanisms. A Perspective on the Mechanisms of Nitric Oxide Synthase (NOS), Heme Oxygenase (HO), and Cytochrome P450s (CYP450s). Biochemistry 2008, 47, 2231–2243. [DOI] [PubMed] [Google Scholar]; (c) Davydov R; Kofman V; Fujii H; Yoshida T; Ikeda-Saito M; Hoffman BM Catalytic Mechanism of Heme Oxygenase through EPR and ENDOR of Cryoreduced Oxy-Heme Oxygenase and Its Asp 140 Mutants. J. Am. Chem. Soc 2002, 124, 1798–1808. [DOI] [PubMed] [Google Scholar]; (d) Davydov RM; Yoshida T; Ikeda-Saito M; Hoffman BM Hydroperoxy-Heme Oxygenase Generated by Cryoreduction Catalyzes the Formation of α-meso-Hydroxyheme as Detected by EPR and ENDOR. J. Am. Chem. Soc 1999, 121, 10656–10657. [DOI] [PubMed] [Google Scholar]; (e) Garcia-Bosch I; Sharma SK; Karlin KD A Selective Stepwise Heme Oxygenase Model System: An Iron(IV)-Oxo Porphyrin π-Cation Radical Leads to a Verdoheme-Type Compound via an Isoporphyrin Intermediate. J. Am. Chem. Soc 2013, 135, 16248–16251. [DOI] [PubMed] [Google Scholar]; (f) Matsui T; Unno M; Ikeda-Saito M Heme Oxygenase Reveals Its Strategy for Catalyzing Three Successive Oxygenation Reactions. Acc. Chem. Res 2010, 43, 240–247. [DOI] [PubMed] [Google Scholar]; (g) Poulos TL; Schuller DJ; Wilks A; Ortiz de Montellano PR Crystal Structure of Human Heme Oxygenase-l. Nat. Struct. Biol 1999, 6, 860–867. [DOI] [PubMed] [Google Scholar]; (h) Unno M; Matsui T; Ikeda-Saito M Structure and Catalytic Mechanism of Heme Oxygenase. Nat. Prod. Rep 2007, 24, 553–570. [DOI] [PubMed] [Google Scholar]; (i) Akhtar M; Njar VCO; Wright JN Mechanistic Studies on Aromatase and Related C-C Bond Cleaving P-450 Enzymes. J. Steoid Biochem. Mol. Biol 1993, 44, 375–387. [DOI] [PubMed] [Google Scholar]; (j) Cole PA; Bean JM; Robinson CH Conversion of a 3-Desoxysteroid to 3-Desoxyestrogen by Human Placental Aromatase. Proc. Natl. Acad. Sci. U. S. A 1990, 87, 2999–3003. [DOI] [PubMed] [Google Scholar]; (k) Yoshimoto FK; Guengerich FP Mechanism of the Third Oxidative Step in the Conversion of Androgens to Estrogens by Cytochrome P450 19A1 Steroid Aromatase. J. Am. Chem. Soc 2014, 136, 15016–15025. [DOI] [PubMed] [Google Scholar]; (l) Makris TM; Davydov R; Denisov IG; Hoffman BM; Sligar SG Mechanistic Enzymology of Oxygen Activation by the Cytochromes P450. Drug Metab. Rev 2002, 34, 691–708. [DOI] [PubMed] [Google Scholar]; (m) Griffith OW; Stuehr DJ Annu. Rev. Physiol 1995, 57, 707–736. [DOI] [PubMed] [Google Scholar]; (n) Marletta MA Nitric Oxide Synthase Structure and Mechanism. J. Biol. Chem 1993, 268, 12231–12234. [DOI] [PubMed] [Google Scholar]
- (3).Jasniewski AJ; Que L Jr. Dioxygen Activation by Nonheme Diiron Enzymes: Diverse Dioxygen Adducts, High-Valent Intermediates, and Related Model Complexes. Chem. Rev 2018, 118, 2554–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Krebs C; Fujimori DG; Walsh CT; Bollinger JM Jr. Non-Heme Fe(IV)-Oxo Intermediates. Acc. Chem. Res 2007, 40, 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chakrabarty S; Austin RN; Deng D; Groves JT; Lipscomb JD Radical Intermediates in Monooxygenase Reactions of Rieske Dioxygenases. J. Am. Chem. Soc 2007, 129, 3514–3515. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kovaleva EG; Lipscomb JD Versatility of Biological Nonheme Fe(II) Centers in Oxygen Activation Reactions. Nat. Chem. Biol 2008, 4, 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Solomon EI; DeWeese DE; Babicz JT Mechanisms of O2 Activation by Mononuclear Non-Heme Iron Enzymes. Biochemistry 2021, 60, 3497–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Lee JL; Ross DL; Barman SK; Ziller JW; Borovik AS C–H Bond Cleavage by Bioinspired Nonheme Metal Complexes. Inorg. Chem 2021, 60, 13759–13783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Solomon EI; Heppner DE; Johnston EM; Ginsbach JW; Cirera J; Qayyum M; Kieber-Emmons MT; Kjaergaard CH; Hadt RG; Tian L Copper Active Sites in Biology. Chem. Rev 2014, 114, 3659–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Klinman JP The Copper-Enzyme Family of Dopamine β-Monooxygenase and Peptidylglycine α-Hydroxylating Monooxygenase: Resolving the Chemical Pathway for Substrate Hydroxylation. J. Biol. Chem 2006, 281, 3013–3016. [DOI] [PubMed] [Google Scholar]; (b) Cowley RE; Tian L; Solomon EI Mechanism of O2 Activation and Substrate Hydroxylation in Noncoupled Binuclear Copper Monooxygenases. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 12035–12040. [DOI] [PubMed] [Google Scholar]; (c) Bhadra M; Transue WJ; Lim H; Cowley RE; Lee JYC; Siegler MA; Josephs P; Henkel G; Lerch M; Schindler S; Neuba A; Hodgson KO; Hedman B; Solomon EI; Karlin KD A Thioether-Ligated Cupric Superoxide Model with Hydrogen Atom Abstraction Reactivity. J. Am. Chem. Soc 2021, 143, 3707–3713. [DOI] [PubMed] [Google Scholar]
- (7).Wu P; Fan F; Song J; Peng W; Liu J; Li C; Cao Z; Wang B Theory Demonstrated a “Coupled” Mechanism for O2 Activation and Substrate Hydroxylation by Binuclear Copper Monooxygenases. J. Am. Chem. Soc. 2019, 141, 19776–19789. [DOI] [PubMed] [Google Scholar]
- (8).Meier KK; Jones SM; Kaper T; Hansson H; Koetsier MJ; Karkehabadi S; Solomon EI; Sandgren M; Kelemen B Oxygen Activation by Cu LPMOs in Recalcitrant Carbohydrate Polysaccharide Conversion to Monomer Sugars. Chem. Rev 2018, 118, 2593–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wang B; Walton PH; Rovira C Molecular Mechanisms of Oxygen Activation and Hydrogen Peroxide Formation in Lytic Polysaccharide Monooxygenases. ACS Catal. 2019, 9, 4958–4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) Cutsail GE; Ross MO; Rosenzweig AC; DeBeer S Towards a Unified Understanding of the Copper sites in Particulate Methane Monooxygenase: an X-ray Absorption Spectroscopic Investigation. Chem. Sci 2021, 12, 6194–6209. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Peng W; Qu X; Shaik S; Wang B Deciphering the Oxygen Activation Mechanism at the CuC site of Particulate Methane Monooxygenase. Nature Catalysis 2021, 4, 266–273. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ross MO; MacMillan F; Wang J; Nisthal A; Lawton TJ; Olafson BD; Mayo SL; Rosenzweig AC; Hoffman BM Particulate Methane Monooxygenase Contains only Mononuclear Copper Centers. Science 2019, 364, 566–570. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Jodts RJ; Ross MO; Koo CW; Doan PE; Rosenzweig AC; Hoffman BM Coordination of the Copper Centers in Particulate Methane Monooxygenase: Comparison between Methanotrophs and Characterization of the CuC Site by EPR and ENDOR Spectroscopies. J. Am. Chem. Soc 2021, 143, 15358–15368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) Wendlandt AE; Suess AM; Stahl SS Copper-Catalyzed Aerobic Oxidative C–H Functionalizations: Trends and Mechanistic Insights. Angew. Chem., Int. Ed 2011, 50, 11062–11087. [DOI] [PubMed] [Google Scholar]; (b) Allen SE; Walvoord RR; Padilla-Salinas R; Kozlowski MC Aerobic Copper-Catalyzed Organic Reactions. Chem. Rev 2013, 113, 6234–6458. [DOI] [PubMed] [Google Scholar]; (c) Esguerra KVN; Lumb J-P Cu(III)-Mediated Aerobic Oxidations. Synthesis 2019, 51, 334–358. [DOI] [PubMed] [Google Scholar]; (d) Trammell R; Rajabimoghadam K; Garcia-Bosch I Copper-Promoted Functionalization of Organic Molecules: from Biologically Relevant Cu/O2 Model Systems to Organometallic Transformations. Chem. Rev 2019, 119, 2954–3031. [DOI] [PubMed] [Google Scholar]
- (12).(a) Solomon EI; Sundaram UM; Machonkin TE Multicopper Oxidases and Oxygenases. Chem. Rev 1996, 96, 2563–2605. [DOI] [PubMed] [Google Scholar]; (b) Klinman JP Mechanisms Whereby Mononuclear Copper Proteins Functionalize Organic Substrates. Chem. Rev 1996, 96, 2541–2561. [DOI] [PubMed] [Google Scholar]
- (13).Maiti D; Fry HC; Woertink JS; Vance MA; Solomon EI; Karlin KDA 1:1 Copper-Dioxygen Adduct is an End-on Bound Superoxo Copper(II) Complex which Undergoes Oxygenation Reactions with Phenols. J. Am. Chem. Soc 2007, 129, 264–265. [DOI] [PubMed] [Google Scholar]
- (14).(a) Schatz M; Raab V; Foxon SP; Brehm G; Schneider S; Reiher M; Holthausen MC; Sundermeyer J; Schindler S Combined spectroscopic and theoretical evidence for a persistent end-on copper superoxo complex. Angew. Chem., Int. Ed 2004, 43, 4360–4363. [DOI] [PubMed] [Google Scholar]; (b) Würtele C; Gaoutchenova E; Harms K; Holthausen MC; Sundermeyer J; Schindler S Crystallographic Characterization of a Synthetic 1:1 End-On Copper Dioxygen Adduct Complex. Angew. Chem. Int. Ed 2006, 45, 3867–3869. [DOI] [PubMed] [Google Scholar]
- (15).(a) Lanci MP; Smirnov VV; Cramer CJ; Gauchenova EV; Sundermeyer J; Roth JP Isotopic Probing of Molecular Oxygen Activation at Copper(I) Sites. J. Am. Chem. Soc 2007, 129, 14697–14709. [DOI] [PubMed] [Google Scholar]; (b) Woertink JS; Tian L; Maiti D; Lucas HR; Himes RA; Karlin KD; Neese F; Würtele C; Holthausen MC; Bill E; Sundermeyer J; Schindler S; Solomon EI Spectroscopic and Computational Studies of an End-on Bound Superoxo-Cu(II) Complex: Geometric and Electronic Factors That Determine the Ground State. Inorg. Chem 2010, 49, 9450–9459. [DOI] [PubMed] [Google Scholar]; (c) Ginsbach JW; Peterson RL; Cowley RE; Karlin KD; Solomon EI Correlation of the Electronic and Geometric Structures in Mononuclear Copper(II) Superoxide Complexes. Inorg. Chem 2013, 52, 12872–12874. [DOI] [PubMed] [Google Scholar]
- (16).(a) Karlin KD; Hayes JC; Shi J; Hutchinson JP; Zubieta J Tetragonal vs. Trigonal Coordination in Copper(II) Complexes with Tripod Ligands: Structures and Properties of (Cu(C21H24N4)Cl)PF6 and (Cu(C18H18N4)Cl)PF6. Inorg. Chem 1982, 21, 4106–4108. [Google Scholar]; (b) Zubieta J; Karlin KD; Hayes JC “Structural Systematics of Cu(I) and Cu(II) Derivatives of Tripodal Ligands. In Copper Coordination Chemistry: Biochemical and Inorganic Perspectives; Karlin KD, Zubieta J, Eds.; Adenine Press: 1983; pp 97–108. [Google Scholar]; (c) Lim BS; Holm RH Molecular Heme-Cyanide-Copper Bridged Assemblies: Linkage Isomerism, Trends in nCN values, and Relation to the Heme a3/CuB Site in Cyanide-Inhibited Heme-Copper Oxidases. Inorg. Chem 1998, 37, 4898–4908.11670655 [Google Scholar]; (d) Lucchese B; Humphreys KJ; Lee D-H; Incarvito CD; Sommer RD; Rheingold AL; Karlin KD Mono- Bi- and Trinuclear CuII-Cl Contaninig Products Based on the Tris(2-pyridylmethyl)amine Chelate, Derived from Copper(I) Complex Dechlorinations of Chloroform. Inorg. Chem 2004, 43, 5987–5998.15360248 [Google Scholar]
- (17).Tyeklár Z; Jacobson RR; Wei N; Murthy NN; Zubieta J; Karlin KD Reversible Reaction of O2 (and CO) with a Copper(I) Complex: X-ray Structures of Relevant Mononuclear Cu(I) Precursor Adducts and the trans-(μ-1,2-Peroxo)- Dicopper(II) Product. J. Am. Chem. Soc 1993, 115, 2677–2689. [Google Scholar]
- (18).(a) Barbucci R; Bencini A; Gatteschi D Electron-Spin Resonance-Spectra and Spin-Hamiltonian Parameters for Trigonal-Bipyramidal Nickel(I) and Copper(II) Complexes. Inorg. Chem 1977, 16, 2117–2120. [Google Scholar]; (b) Hathaway BJ Copper. In Comprehensive Coordination Chemistry; Wilkinson G, Ed.; Pergamon: 1987; Vol. 5, pp 533–774. [Google Scholar]
- (19).Bhadra M; Lee JYC; Cowley RE; Kim S; Siegler MA; Solomon EI; Karlin KD Intramolecular Hydrogen Bonding Enhances Stability and Reactivity of Mononuclear Cupric Superoxide Complexes. J. Am. Chem. Soc 2018, 140, 9042–9045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).(a) Karlin KD; Wei N; Jung B; Kaderli S; Zuberbühler AD Kinetic, Thermodynamic and Spectral Characterization of the Primary Cu-O2 Adduct in a Reversibly Formed and Structurally Characterized Peroxo-Dicopper(II) Complex. J. Am. Chem. Soc 1991, 113, 5868–5870. [Google Scholar]; (b) Karlin KD; Wei N; Jung B; Kaderli S; Niklaus P; Zuberbühler AD Kinetics and Thermodynamics of Formation of Copper-Dioxygen Adducts: Oxygenation of Mononuclear Cu(I) Complexes Containing Tripodal Tetradentate Ligands. J. Am. Chem. Soc 1993, 115, 9506–9514. [Google Scholar]
- (21).Fry HC; Scaltrito DV; Karlin KD; Meyer GJ The Rate of O2 and CO Binding to a Copper Complex, Determined by a “Flash-and-Trap” Technique, Exceeds that for Hemes. J. Am. Chem. Soc 2003, 125, 11866–11871. [DOI] [PubMed] [Google Scholar]
- (22).(a) Jacobson RR; Tyeklár Z; Karlin KD; Liu S; Zubieta J A Cu2-O2 Complex. Crystal Structure and Characterization of a Reversible Dioxygen Binding System. J. Am. Chem. Soc 1988, 110, 3690–3692. [Google Scholar]; (b) Baldwin MJ; Ross PK; Pate JE; Tyeklár Z; Karlin KD; Solomon EI Spectroscopic and Theoretical Studies of an End-On Peroxide-Bridged Coupled Binuclear Copper(II) Model Complex of Relevance to the Active Sites in Hemocyanin and Tyrosinase. J. Am. Chem. Soc 1991, 113, 8671–8679. [Google Scholar]
- (23).(a) Maiti D; Lee D-H; Gaoutchenova K; Würtele C; Holthausen MC; Sarjeant AAN; Sundermeyer J; Schindler S; Karlin KD Reactions of a Copper(II) Superoxo Complex Lead to C–H and O–H Substrate Oxygenation: Modeling Copper-Monooxygenase C–H Hydroxylation. Angew. Chem. Int. Ed 2008, 47, 82–85. [DOI] [PubMed] [Google Scholar]; (b) Lee JY; Peterson RL; Ohkubo K; Garcia-Bosch I; Himes RA; Woertink J; Moore CD; Solomon EI; Fukuzumi S; Karlin KD Mechanistic Insights into the Oxidation of Substituted Phenols via Hydrogen Atom Abstraction by a Cupric–Superoxo Complex. J. Am. Chem. Soc 2014, 136, 9925–9937. [DOI] [PubMed] [Google Scholar]
- (24).Diaz DE; Quist DA; Herzog AE; Schaefer AW; Kipouros I; Bhadra M; Solomon EI; Karlin KD Impact of Intramolecular Hydrogen Bonding on the Reactivity of Cupric Superoxide Complexes with O–H and C–H Substrates. Angew. Chem., Int. Ed 2019, 58, 17572–17576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).(a) Itoh S Mononuclear Copper Active-Oxygen Complexes. Curr. Opin. Chem. Biol 2006, 10, 115–122. [DOI] [PubMed] [Google Scholar]; (b) Liu JJ; Diaz DE; Quist DA; Karlin KD Copper(I)-Dioxygen Adducts and Copper Enzyme Mechanisms. Isr. J. Chem 2016, 56, 738–755. [DOI] [PubMed] [Google Scholar]; (c) Elwell CE; Gagnon NL; Neisen BD; Dhar D; Spaeth AD; Yee GM; Tolman WB Copper–Oxygen Complexes Revisited: Structures, Spectroscopy, and Reactivity. Chem. Rev 2017, 117, 2059–2107. [DOI] [PubMed] [Google Scholar]; (d) Quist DA; Diaz DE; Liu JJ; Karlin KD Activation of dioxygen by copper metalloproteins and insights from model complexes. JBIC, J. Biol. Inorg. Chem 2017, 22, 253–288. [DOI] [PubMed] [Google Scholar]
- (26).Itoh S Developing Mononuclear Copper-Active-Oxygen Complexes Relevant to Reactive Intermediates of Biological Oxidation Reaction. Acc. Chem. Res 2015, 48, 2066–2074. [DOI] [PubMed] [Google Scholar]
- (27).(a) Davydov R; Laryukhin M; Ledbetter-Rogers A; Sono M; Dawson JH; Hoffman BM Electron Paramagnetic Resonance and Electron-Nuclear Double Resonance Studies of the Reactions of Cryogenerated Hydroperoxoferric–Hemoprotein Intermediates. Biochemistry 2014, 53, 4894–4903. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hoffman BM ENDOR of Metalloenzymes. Acc. Chem. Res 2003, 36, 522–529. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Huyett JE; Doan PE; Gurbiel R; Houseman ALP; Sivaraja M; Goodin DB; Hoffman BM Compound ES of Cytochrome-c Peroxidase Contains a Trp π-Cation Radical - Characterization by CW and Pulsed Q-Band ENDOR Spectroscopy. J. Am. Chem. Soc 1995, 117, 9033–9041. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Sturgeon BE; Burdi D; Chen SX; Huynh BH; Edmondson DE; Stubbe J; Hoffman BM Reconsideration of X, the Diiron Intermediate Formed during Cofactor Assembly in E-Coli Ribonucleotide Reductase. J. Am. Chem. Soc 1996, 118, 7551–7557. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Willems JP; Lee HI; Burdi D; Doan PE; Stubbe J; Hoffman BM Identification of the Protonated Oxygenic Ligands of Ribonucleotide Reductase Intermediate X by Q-band 1,2H CW and Pulsed ENDOR. J. Am. Chem. Soc 1997, 119, 9816–9824. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Burdi D; Willems JP; Riggs-Gelasco P; Antholine WE; Stubbe J; Hoffman BM The Core Structure of X Generated in the Assembly of the Diiron Cluster of Ribonucleotide Reductase: 17O2 and H2 17O ENDOR. J. Am. Chem. Soc 1998, 120, 12910–12919. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Lee HI; Dexter AF; Fann YC; Lakner FJ; Hager LP; Hoffman BM Structure of the Modified Heme in Allylbenzene-Inactivated Chloroperoxidase Determined by Q-Band CW and Pulsed ENDOR. J. Am. Chem. Soc 1997, 119, 4059–4069. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Walsby CJ; Hong W; Broderick WE; Cheek J; Ortillo D; Broderick JB; Hoffman BM Electron-Nuclear Double Resonance Spectroscopic Evidence that S-Adenosylmethionine Binds in Contact with the Catalytically Active [4Fe-4S]+ Cluster of Pyruvate Formate-Lyase Activating Enzyme. J. Am. Chem. Soc 2002, 124, 3143–3151. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Walsby CJ; Ortillo D; Broderick WE; Broderick JB; Hoffman BM An Anchoring Role for FeS Clusters: Chelation of the Amino Acid Moiety of S-Adenosylmethionine to the Unique Iron Site of the [4Fe-4S]+ Cluster of Pyruvate Formate-Lyase Activating Enzyme. J. Am. Chem. Soc 2002, 124, 11270–11271. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Davydov R; Strushkevich N; Smil D; Yantsevich A; Gilep A; Usanov S; Hoffman BM Evidence That Compound I Is the Active Species in Both the Hydroxylase and Lyase Steps by Which P450scc Converts Cholesterol to Pregnenolone: EPR/ENDOR/Cryoreduction/Annealing Studies. Biochemistry 2015, 54, 7089–7097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Della Longa S; Arcovito A; Benfatto M; Congiu-Castellano A; Girasole M; Hazemann JL; Lo Bosco A Redox-Induced Structural Dynamics of Fe-Heme Ligand in Myoglobin by X-ray Absorption Spectroscopy. Biophys. J 2003, 85, 549–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Mader EA; Davidson ER; Mayer JM Large Ground-State Entropy Changes for Hydrogen Atom Transfer Reactions of Iron Complexes. J. Am. Chem. Soc 2007, 129, 5153–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).(a) Zhang CX; Kaderli S; Costas M; Kim E.-i.; Neuhold Y-M; Karlin KD; Zuberbühler AD Copper(I)-Dioxygen Reactivity of [(L)CuI]+ (L = tris(2-pyridymethyl)amine): Kinetic/Thermodynamic and Spectroscopic Studies Concerning the Formation of Cu-O2 and Cu2-O2 Adducts as a Function of Solvent Medium and 4-Pyridyl Ligand Substituent Variations. Inorg. Chem 2003, 42, 1807–1824. [DOI] [PubMed] [Google Scholar]; (b) Wada A; Harata M; Hasegawa K; Jitsukawa K; Masuda H; Mukai M; Kitagawa T; Einaga H Structural and Spectroscopic Characterization of a Mononuclear Hydroperoxo-Copper(II) Complex with Tripodal Pyridylamine Ligands. Angew. Chem., Int. Ed 1998, 37, 798–799. [DOI] [PubMed] [Google Scholar]; (c) Wittmann H; Raab V; Schorm A; Plackmeyer J; Sundermeyer J Complexes of Manganese, Iron, Zinc, and Molybdenum with a Superbasic Tris(guanidine) Derivative of Tris(2-ethylamino)amine (tren) as a Tripod Ligand. Eur. J. Inorg. Chem 2001, 2001, 1937–1948. [DOI] [PubMed] [Google Scholar]
- (31).Peterson RL; Himes RA; Kotani H; Suenobu T; Tian L; Siegler MA; Solomon EI; Fukuzumi S; Karlin KD Cupric Superoxo-Mediated Intermolecular C–H Activation Chemistry. J. Am. Chem. Soc 2011, 133, 1702–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Eckenhoff WT; Pintauer T Atom Transfer Radical Addition in the Presence of Catalytic Amounts of Copper(I/II) Complexes with Tris(2-pyridylmethyl)amine. Inorg. Chem 2007, 46, 5844–5846. [DOI] [PubMed] [Google Scholar]
- (33).Davoust CE; Doan PE; Hoffman BM Q-band Pulsed Electron Spin-Echo Spectrometer and its Application to ENDOR and ESEEM. J. Magn. Reson. Ser. A 1996, 119, 38–44. [Google Scholar]
- (34).Epel B; Gromov I; Stoll S; Schweiger A; Goldfarb D Spectrometer Manager: A Versatile Control Software for Pulse EPR Spectrometers. Concepts Magn. Reson., Part B 2005, 26b, 36–45. [Google Scholar]
- (35).Doan PE; Hoffman BM Making Hyperfine Selection in Mims ENDOR Independent of Deadtime. Chem. Phys. Lett 1997, 269, 208–214. [Google Scholar]
- (36).Stoll S; Schweiger A EasySpin, a Comprehensive Software Package for Spectral Simulation and Analysis in EPR. J. Magn. Reson 2006, 178, 42–55. [DOI] [PubMed] [Google Scholar]
- (37).(a) Davydov R; Satterlee JD; Fujii H; Sauer-Masarwa A; Busch DH; Hoffman BM A Superoxo-Ferrous State in a Reduced Oxy-Ferrous Hemoprotein and Model Compounds. J. Am. Chem. Soc 2003, 125, 16340–16346. [DOI] [PubMed] [Google Scholar]; (b) Davydov R; Hoffman BM Active Intermediates in Heme Monooxygenase Reactions as Revealed by Cryoreduction/Annealing, EPR/ENDOR Studies. Arch. Biochem. Biophys 2011, 507, 36–43. [DOI] [PubMed] [Google Scholar]
- (38).Känzig W; Cohen MH Paramagnetic Resonance of Oxygen in Alkali Halides. Phys. Rev. Lett 1959, 3, 509–510. [Google Scholar]
- (39).(a) Petr A; Kataev V; Buchner B First direct in situ EPR spectroelectrochemical evidence of the superoxide anion radical. J. Phys. Chem. B 2011, 115, 12036–9. [DOI] [PubMed] [Google Scholar]; (b) Maricle DL; Hodgson WG Reducion of Oxygen to Superoxide Anion in Aprotic Solvents. Anal. Chem 1965, 37, 1562–1565. [DOI] [PubMed] [Google Scholar]
- (40).Callens R; Callens F; Matthys P; Boesman E EPR of a New O2− Centre in KCl. Phys. Stat. Solid B 1988, 148, 683–688. [Google Scholar]
- (41).Norizawa K; Hirai M; Kanosue K; Ikeya M Trapping of Atmospheric HO2 in Solid CO2 on Icy Satellites: Simulation Using Electron Spin Resonance and Thermoluminescence Analyses. Jpn. J. Appl. Phys. Part 2000, 39, 6759–6762. [Google Scholar]
- (42).Peterson RL; Ginsbach JW; Cowley RE; Qayyum MF; Himes RA; Siegler MA; Moore CD; Hedman B; Hodgson KO; Fukuzumi S; Solomon EI; Karlin KD Stepwise Protonation and Electron-Transfer Reduction of a Primary Copper–Dioxygen Adduct. J. Am. Chem. Soc 2013, 135, 16454–16467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).(a) Hoffman BM; Diemente DL; Basolo F Electron Paramagnetic Resonance Studies of Some Cobalt(II) Schiff Base Compounds and their Monomeric Oxygen Adducts. J. Am. Chem. Soc 1970, 92, 61–65. [Google Scholar]; (b) Hoffman BM; Weschler CJ; Basolo F The Dioxygen Adduct of meso-TetraphenylporphyrinManganese(II), a Synthetic Oxygen Carrier. J. Am. Chem. Soc 1976, 98, 5473–5482.956568 [Google Scholar]
- (44).(a) Ohta T; Liu JG; Nagaraju P; Ogura T; Naruta Y A Cryo-Generated Ferrous-Superoxo Porphyrin: EPR, Resonance Raman and DFT Studies. Chem. Commun 2015, 51, 12407–12410. [DOI] [PubMed] [Google Scholar]; (b) Ohta T; Nagaraju P; Liu J-G; Ogura T; Naruta Y The Secondary Coordination Sphere and Axial LIgand Effects on Oxygen Reduction Reaction by Iron Porphyrins: a DFT Computational Study. JBIC, J. Biol. Inorg. Chem 2016, 21, 745–755. [DOI] [PubMed] [Google Scholar]
- (45).(a) Dürr K; Macpherson BP; Warratz R; Hampel F; Tuczek F; Helmreich M; Jux N; Ivanovic-Burmazovic I Iron(III) Complex of a Crown Ether-Porphyrin Conjugate and Reversible Binding of Superoxide to Its Iron(II) Form. J. Am. Chem. Soc 2007, 129, 4217–4228. [DOI] [PubMed] [Google Scholar]; (b) Dürr K; Jux N; Zahl A; van Eldik R; Ivanovíc-Burmazovíc I Volume Profile Analysis for the Reversible Binding of Superoxide to Form Iron(II)-Superoxo/Iron(III)-Peroxo Porphyrin Complexes. Inorg. Chem 2010, 49, 11254–11260. [DOI] [PubMed] [Google Scholar]
- (46).(a) Karlin KD; Cruse RW; Gultneh Y; Hayes JC; Zubieta J Peroxide Coordination to a Dicopper(II) Center. Dioxygen Binding to a Structurally Characterized Phenoxide Bridged Binuclear Copper(I) Complex. J. Am. Chem. Soc 1984, 106, 3372–3374. [Google Scholar]; (b) Pate JE; Cruse RW; Karlin KD; Solomon EI Vibrational, Electronic and Resonance Raman Spectral Studies of [Cu2(XYL-O−)O2]+, a Copper(II) Peroxide Model Complex of Oxyhemocyanin. J. Am. Chem. Soc 1987, 109, 2624–2630. [Google Scholar]
- (47).Quist DA; Ehudin MA; Schaefer AW; Schneider GL; Solomon EI; Karlin KD Ligand Identity-Induced Generation of Enhanced Oxidative Hydrogen Atom Transfer Reactivity for a CuII2(O2 •−) Complex Driven by Formation of a CuII2(−OOH) Compound with a Strong O–H Bond. J. Am. Chem. Soc 2019, 141, 12682–12696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).(a) O’Dell WB; Agarwal PK; Meilleur F Oxygen Activation at the Active Site of a Fungal Lytic Polysaccharide Monooxygenase. Angew. Chem. Int. Ed 2017, 56, 767–770. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bacik J-P; Mekasha S; Forsberg Z; Kovalevsky AY; Vaaje-Kolstad G; Eijsink VGH; Nix JC; Coates L; Cuneo MJ; Unkefer CJ; Chen JCH Neutron and Atomic Resolution X-ray Structures of a Lytic Polysaccharide Monooxygenase Reveal Copper-Mediated Dioxygen Binding and Evidence for N-Terminal Deprotonation. Biochemistry 2017, 56, 2529–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Hangasky JA; Detomasi TC; Marletta MA Glycosidic Bond Hydroxylation by Polysaccharide Monooxygenases. Trends in Chemistry 2019, 1, 198–209. [Google Scholar]
- (50).Rudzka K; Moreno DM; Eipper B; Mains R; Estrin DA; Amzel LM Coordination of Peroxide to the CuM center of Peptidylglycine Alpha-Hydroxylating Monooxygenase (PHM): Structural and Computational Study. JBIC, J. Biol. Inorg. Chem 2013, 18, 223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Osako T; Nagatomo S; Tachi Y; Kitagawa T; Itoh S Low-Temperature Stopped-Flow Studies on the reactions of Copper(II) Complexes and H2O2: The First Detection of a Mononuclear Copper(II)-Peroxo Intermediate. Angew. Chem., Int. Ed 2002, 41, 4325–4328. [DOI] [PubMed] [Google Scholar]
- (52).McDonald AR; Van Heuvelen KM; Guo Y; Li F; Bominaar EL; Münck E; Que L Characterization of a Thiolato Iron(III) Peroxy Dianion Complex. Angew. Chem 2012, 124, 9266–9270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Cho J; Jeon S; Wilson SA; Liu LV; Kang EA; Braymer JJ; Lim MH; Hedman B; Hodgson KO; Valentine JS; Solomon EI; Nam W Structure and reactivity of a mononuclear non-haem iron(III)-peroxo complex. Nature 2011, 478, 502–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Kim B; Jeong D; Ohta T; Cho J Nucleophilic reactivity of a copper(II)-hydroperoxo complex. Communications Chemistry 2019, 2, 81. [Google Scholar]
- (55).Sankaralingam M; Lee YM; Nam W; Fukuzumi S Amphoteric reactivity of metal-oxygen complexes in oxidation reactions. Coord. Chem. Rev 2018, 365, 41–59. [Google Scholar]
- (56).Liu J-G; Shimizu Y; Ohta T; Naruta Y Formation of an End-On Ferric Peroxo Intermediate upon One-Electron Reduction of a Ferric Superoxo Heme. J. Am. Chem. Soc 2010, 132, 3672–3673. [DOI] [PubMed] [Google Scholar]
- (57).Kim H; Rogler PJ; Sharma SK; Schaefer AW; Solomon EI; Karlin KD Ferric Heme Superoxide Reductive Transformations to Ferric Heme (Hydro)Peroxide Species: Spectroscopic Characterization and Thermodynamic Implications for H-Atom Transfer (HAT). Angew. Chem., Int. Ed 2021, 60, 5907–5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Unno M; Chen H; Kusama S; Shaik S; Ikeda-Saito M Structural Characterization of the Fleeting Ferric Peroxo Species in Myoglobin: Experiment and Theory. J. Am. Chem. Soc 2007, 129, 13394–13395. [DOI] [PubMed] [Google Scholar]
- (59).Hersleth HP; Hsiao YW; Ryde U; Gorbitz CH; Andersson KK The Crystal Structure of Peroxymyoglobin Generated through Cryoradiolytic Reduction of Myoglobin Compound III during Data Collection. Biochem. J 2008, 412, 257–264. [DOI] [PubMed] [Google Scholar]
- (60).Petrik ID; Davydov R; Kahle M; Sandoval B; Dwaraknath S; Ädelroth P; Hoffman B; Lu Y An Engineered Glutamate in Biosynthetic Models of Heme-Copper Oxidases Drives Complete Product Selectivity by Tuning the Hydrogen-Bonding Network. Biochemistry 2021, 60, 346–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Mak PJ; Duggal R; Denisov IG; Gregory MC; Sligar SG; Kincaid JR Human Cytochrome CYP17A1: The Structural Basis for Compromised Lyase Activity with 17-Hydroxyprogesterone. J. Am. Chem. Soc 2018, 140, 7324–7331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Sivaramakrishnan S; Ouellet H; Matsumura H; Guan S; Moënne-Loccoz P; Burlingame AL; Ortiz de Montellano PR Proximal Ligand Electron Donation and Reactivity of the Cytochrome P450 Ferric–Peroxo Anion. J. Am. Chem. Soc 2012, 134, 6673–6684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).(a) Tano T; Ertem MZ; Yamaguchi S; Kunishita A; Sugimoto H; Fujieda N; Ogura T; Cramer CJ; Itoh S Reactivity of Copper(II)-Alkylperoxo Complexes. Dalton Trans. 2011, 40, 10326–10336. [DOI] [PubMed] [Google Scholar]; (b) Tano T; Mieda K; Sugimoto H; Ogura T; Itoh S A Copper Complex Supported by an N2S-Tridentate Ligand Inducing Efficient Heterolytic O-O Bond Cleavage of Alkylhydroper-oxide. Dalton Trans. 2014, 43, 4871–4877. [DOI] [PubMed] [Google Scholar]; (c) Abe T; Morimoto Y; Mieda K; Sugimoto H; Fujieda N; Ogura T; Itoh S Geometric Effects on O–O Bond Scission of Copper(II)-Alkylperoxide Complexes. J. Inorg. Biochem 2017, 177, 375–383. [DOI] [PubMed] [Google Scholar]; (d) Trammell R; See YY; Herrmann AT; Xie N; Díaz DE; Siegler MA; Baran PS; Garcia-Bosch I Decoding the Mechanism of Intramolecular Cu-Directed Hydroxylation of sp3 C–H Bonds. J. Org. Chem 2017, 82, 7887–7904. [DOI] [PubMed] [Google Scholar]; (e) Shimizu I; Morimoto Y; Velmurugan G; Gupta T; Paria S; Ohta T; Sugimoto H; Ogura T; Comba P; Itoh S Characterization and Reactivity of a Tetrahedral Copper(II) Alkylperoxido Complex. Chem. - Eur. J 2019, 25, 11157–11165. [DOI] [PubMed] [Google Scholar]; (f) Oh H; Ching W-M; Kim J; Lee W-Z; Hong S Hydrogen Bond-Enabled Heterolytic and Homolytic Peroxide Activation within Nonheme Copper(II)-Alkylperoxo Complexes. Inorg. Chem 2019, 58, 12964–12974. [DOI] [PubMed] [Google Scholar]
- (64).Kunishita A; Scanlon JD; Ishimaru H; Honda K; Ogura T; Suzuki M; Cramer CJ; Itoh S Reactions of Copper(II)-H2O2 Adducts Supported by Tridentate bis(2-pyridylmethyl)amine Ligands: Sensitivity to Solvent and Variations in Ligand Substitution. Inorg. Chem 2008, 47, 8222–8232. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.