

Abstract
Background
Imputation of untyped markers is a standard tool in genome-wide association studies to close the gap between directly genotyped and other known DNA variants. However, high accuracy with which genotypes are imputed is fundamental. Several accuracy measures have been proposed and some are implemented in imputation software, unfortunately diversely across platforms. In the present paper, we introduce Iam hiQ, an independent pair of accuracy measures that can be applied to dosage files, the output of all imputation software. Iam (imputation accuracy measure) quantifies the average amount of individual-specific versus population-specific genotype information in a linear manner. hiQ (heterogeneity in quantities of dosages) addresses the inter-individual heterogeneity between dosages of a marker across the sample at hand.
Results
Applying both measures to a large case–control sample of the International Lung Cancer Consortium (ILCCO), comprising 27,065 individuals, we found meaningful thresholds for Iam and hiQ suitable to classify markers of poor accuracy. We demonstrate how Manhattan-like plots and moving averages of Iam and hiQ can be useful to identify regions enriched with less accurate imputed markers, whereas these regions would by missed when applying the accuracy measure info (implemented in IMPUTE2).
Conclusion
We recommend using Iam hiQ additional to other accuracy scores for variant filtering before stepping into the analysis of imputed GWAS data.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12859-022-04568-3.
Keywords: GWAS, High-throughput genotyping, Genotype imputation, Accuracy measures
Background
To date information of more than 660 million reference single nucleotide polymorphisms (refSNPs) and 5.9 million regions with structural variation (SV) on the human DNA are known and stored in the publicly available databases, like dbSNP [1]. To identify those genetic variants, that are associated with common human diseases, genome-wide association studies (GWAS) can be conducted. Usually, commercial single nucleotide polymorphism (SNP) microarrays are used to carry out genotyping of DNA samples for these studies. There are two predominant companies for high throughput genotyping arrays, Thermo Fisher Scientific Inc., Santa Clara, CA (Affymetrix™) and Illumina Inc., San Diego, CA. The underlying chemistry differs but both array types can be used to ascertain genotypes in a similar fashion [2]. In contrast to the more expensive and error prone new generation sequencing technologies, the number of genotyped variants ranges from 300,000 to 4 million. Array-based markers are supposed to tag the genomic region in their vicinity, but represent only a small proportion of all known DNA variants. Furthermore, these variants are not a random selection but have been chosen according to criteria such as minor allele frequency (MAF), location in exons or blocks of linkage disequilibrium or putative associations with certain disease.
Imputation methods and strategies have been developed and are now a standard tool in GWAS to close the gap between genotyped and existing DNA variants [3–5]. These methods transfer information of DNA structure from one or several reference panels with high marker density (e.g. 1000 Genomes Project phase 3 [6] or Haplotype Reference Consortium (HRC) [7]) to the genotyped study samples [4]. Most imputation methods estimate a-posteriori genotype probabilities (referred to as dosages, ranging from 0 to 1) for each untyped variant and each individual in the sample of interest. The resulting increase of variant density in the study sample improves the genomic coverage and can increase the power to identify genomic variants associated with a trait [8]. Imputation further has the potential that an identified associated marker is located closer to a true risk locus; it facilitates fine mapping of causal variants and is essential for meta-analyses of GWAS, particularly when different genotyping arrays have been used for multiple studies [9]. However, imputation requires advanced statistical methods for data analysis and may introduce extra uncertainty in interpreting findings. Further, only DNA variants that have previously been genotyped in the used reference panel can be imputed [4, 10].
Imputation methods based on linkage disequilibrium (LD) information (e.g. fastPHASE [11]; MaCH [12, 13]; Beagle [14]; IMPUTE2 [15]) and are suitable for samples of independent individuals, as in case–control studies. Other methods use pedigree and linkage information (e.g. F-Impute [16]; α-Impute [3, 17]), and are therefore suitable for related individuals.
Known accuracy measures
It is important to evaluate the quality of imputation, e.g. to exclude poorly imputed variants from statistical analysis. Several quality indices have been developed and are routinely applied [4, 5, 18]. These comprise inter alia the squared correlation r2 between the true and imputed dose of an allele across all imputed samples (MaCH r2, Minimac or Beagle r2) or IMPUTE2’s info.
All r2 measures can be derived from a-posteriori allele probabilities without knowledge of the true allele dose, but only if the allele probabilities are well calibrated and MAF is not too low. The power of an allelic test with N samples and imputed alleles is approximately equal to the power of the same test with r2N samples and known alleles, in case of a binary trait. Differences among the known r2 measures are discussed elsewhere [4]. The commonly used info is defined as the proportion of statistical information on the population allele frequency in the imputed genotypes, relative to “known” genotypes [5]. If the Hardy–Weinberg disequilibrium (HWE) holds, info equalizes to Minimacs r2. Hence, r2-based measures and info are directly related to the power of statistical test of a marker x trait association.
In general, both metrics have preferable characteristics if the a-posteriori genotype probabilities (dosages) are accurately calculated [18]. However, multiple factors can affect imputation accuracy, e.g. sample size, sequencing coverage and haplotype accuracy of the references panel(s), density of the genotyping array, allele frequency and poor LD between genotyped and imputed variants [4]. One can calculate these accuracy measures from dosage files. However, the standard outputs of common imputation programs (e.g. Beagle or IMPUTE2) contain different metrics. Hence, choosing an imputation program binds the user to the metrics provided, although the SNPTEST program offers the option of calculating a measure similar to that of info [19].
We propose a new pair of metrics to depict additional aspects of imputation accuracy also calculable from dosage files. First, we aim to quantify the amount of individual-specific versus population-specific genotype information in the imputed genotypes. Second, we aim to assess the heterogeneity between dosages of a marker across the sample at hand. Both measures can be used to identify markers or regions in which population-specific genetic information conceal individual-specific information and are therefore less informative for e.g. association testing. These new metrics are not intended as a competitor to established scores, but are intended to support the making of well-founded decisions in SNP filtering of imputed markers prior to an analysis or in interpretation of results after an analysis.
We calculated this pair of accuracy measures on a series of 27,065 cases and controls gathered by the International Lung Cancer Consortium (ILCCO) to find meaningful thresholds for marker exclusion and compared it with info, because all of the ILCCO samples had previously been imputed with IMPUTE2 applied to a standard 1000 Genomes referent panel. Further, we contrasted the usability of the new measures to info in some simulated data.
Results
Comparison of Iam and hiQ
When applying the novel indices Iam hiQ (as defined in the section Novel accuracy measures) to 517,482 SNPs types with the OncoArray, only a small portion (n = 40,678, 4‰) can be considered as imputed without doubt (Iam = 1 and hiQ = 1). For the majority of SNPs a value between 0.95 and < 1 was assigned for hiQ (9,760,392, ~ 94%), while only 30% (n = 3,243,272 markers) achieved such a large value with respect to Iam. It is worth to mention, that we assigned a reduced value for Iam (from 0.4 to 0.75) to about as many SNPs (n = 3,491,596, 33%). More details are given in Additional file 1: Table S1.
Both components of Iam hiQ, are contrasted in a bubble plot (Fig. 1). The oversized grey bubble in the top right corner represents the vast majority of almost fully-informative markers with Iam ≥ 0.99 and hiQ ≥ 0.99. It can easily be seen that the remaining small minority of not fully accurately imputed markers take advantage of the whole theoretical range for Iam (even negative values). In contrast, hiQ always exceeds 0.4 in the sample at hand, but seems to be sensitive in markers with low values for Iam, whereas lower values of hiQ are only assigned to common markers.
Fig. 1.
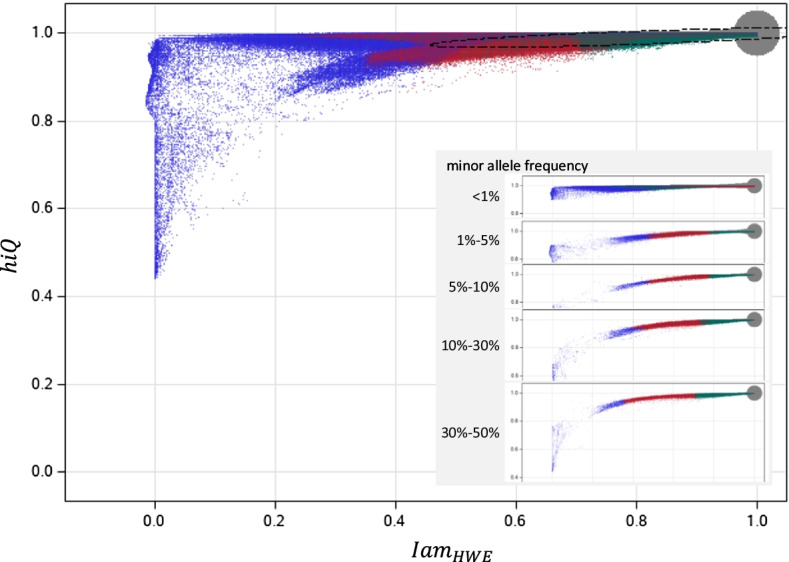
Iam by hiQ. Main panel: all markers by Iam vs. hiQ; blue dots: variants with info < 0.5; red dots: variants with 0.5 ≤ info < 0.8; 8; green dots: variants with info ≥ 0.8; dotted line: robust 99.9999999% bivariate normal random interval (assuming a two-dimensional normal distribution). The oversized grey bubble in the top right corner represents the vast majority of almost fully-informative markers with Iam ≥ 0.99 and hiQ ≥ 0.99; inserted panel: like main panel, but marker are divided according to the minor allele frequency
Defining thresholds for marker filtering
In order to exclude less accurately imputed variants from further analysis, one needs to define a meaningful and applicable threshold for any accuracy index. For the measure info threshold values like 0.8 or 0.3 have been proposed, but without sound justification [5, 20, 21]. It was even proposed to lower the threshold for info in very large samples, as those of the UK Biobank, and still maintain a good ability to detect associations [22].
We applied robust regression (PROG ROBUSTREG of SAS 9.4; cut-off-α = 10–6 for leverage points, cut-off-multiplier = 5 for outliers[23]) to estimate the expected value of Iam and hiQ and their variance–covariance matrix, assuming a hidden two-dimensional normal distribution (ignoring the upper bounds of the indices). Based on this we derived the 99.9999999% (1–10−9) random region (dashed line in Fig. 1) to define, very conservative and data driven, lower bounds for the two indices, limiting the probability of a false-exclusion to ~ 1/(100∙10,439,017) (one hundredth under Bonferroni correction assuming independent markers). The robust mean for Iam was 0.7409 that for hiQ was 0.9885. Restricted to common markers (MAF ≥ 0.1) we achieved similar mean values (Iam: 0.8101, hiQ: 0.9894). The lower bounds of the random region of hiQ were 0.9627 for all markers and 0.9673 for common markers, which is almost identical. In contrast, the lower bounds of the random region of Iam were 0.2553 for all markers and 0.4657 for common markers, demonstrating the influence of MAF, via HWE, on Iam. We decided to use the study specific thresholds of 0.47 for Iam and 0.97 for hiQ to further classify markers of poor accuracy. Because Iam ranges linearly from population informative dosages to fully individual informative dosages, the threshold of ~ 0.5 indicates markers with less than ~ 50% individual-specific genotype information (in average across all samples). Such an intuitive interpretation cannot be given for hiQ.
For the majority of 9,094,772 (87.2%) variants, sufficient imputation accuracy was achieved, according to our defined thresholds (see Table 1). Limiting the markers to those with 0.5 < info ≤ 0.8 and info ≥ 0.8, this fraction increases to 95.1% or 99.9%, respectively. In very rare genetic variants (MAF < 1%) the fraction drops to 76.5%. In contrast, only 0.6% of variants meet neither the Iam nor the hiQ criteria. Interestingly, 1,214,620 variants (11.7%) missed only the Iam criteria. The fraction was larger in very rare variants (23.2%) and in variants with info < 0.5 (58.5%), while it was moderate in variants with 0.5 < info ≤ 0.8 (2.5%).
Table 1.
Classification of markers by IamHWE and hiQ
| hiQ | IamHWE | ||||
|---|---|---|---|---|---|
| < 0.47 | ≥ 0.47 | ||||
| N | %a | N | %a | ||
| All markers | |||||
| < 0.97 | 59,077 | 0.6% | 59,130 | 0.6% | |
| ≥ 0.97 | 1,214,620 | 11.7% | 9,094,772 | 87.2% | |
| Quality defined by info | |||||
| Low quality: info < 0.8 | < 0.97 | 59,077 | 1.2% | 58,592 | 1.1% |
| ≥ 0.97 | 1,214,612 | 23.8% | 3,777,566 | 73.9% | |
| High quality: info ≥ 0.8 | < 0.97 | – | 538 | < 0.1% | |
| ≥ 0.97 | 8 | < 0.1% | 5,317,206 | 99.9% | |
| Minor allele frequency (MAF) | |||||
| < 1% | < 0.97 | 15,366 | 0.3% | 136 | < 0.1% |
| ≥ 0.97 | 1,210,505 | 23.2% | 4,000,616 | 76.5% | |
| 1% to < 5% | < 0.97 | 13,448 | 0.9% | 12,742 | 0.8% |
| ≥ 0.97 | 2,317 | 0.2% | 1,472,328 | 98.1% | |
| 5% to < 10% | < 0.97 | 4,007 | 0.6% | 12,117 | 1.8% |
| ≥ 0.97 | 7 | < 0.1% | 638,931 | 97.5% | |
| 10% to < 30% | < 0.97 | 8,441 | 0.6% | 18,576 | 1.4% |
| ≥ 0.97 | 10 | < 0.1% | 1,288,714 | 97.9% | |
| 30% to 50% | < 0.97 | 9,283 | 1.3% | 4,188 | 0.6% |
| ≥ 0.97 | 261 | < 0.1% | 721,679 | 98.1% | |
| > 50% | < 0.97 | 8,532 | 0.9% | 11,371 | 1.1% |
| ≥ 0.97 | 1,520 | 0.2% | 972,504 | 97.8% | |
Thresholds for IamHWE (0.47) and hiQ (0.97) were defined according to a robust 99.9999999% bivariate normal random interval (assuming a two-dimensional normal distribution)
aProportion within tabulated subgroup of markers
Identifying markers and regions of low accuracy
Figure 2 presents the accuracy of imputed markers according to Iam hiQ in a Manhattan-like plot, with Iam given in the lower part (blue) and hiQ given in the upper part (red). This plot contains all 10,427,599 SNPs. Regions with massively less accurate imputation can easily be identified, especially by hiQ (red needles). This is for instant the case close to the centromere of chromosomes 1, 2 and 9 (accuracy by chromosome 1 is presented in Additional file 1: Figure S1). However, variants with Iam or hiQ below the defined thresholds can be found in many regions across the whole genome. Massively less accurate imputation can be found upstream the centromere, less distinct downstream the centromere and close to the telomeres, as well as around position 50K (blue icicle). Nevertheless, it is still hard to visually find regions that are enriched with less accurately imputed markers.
Fig. 2.

Manhattan-like-plot: Iam hiQ. Upper panel: (low Q.: = 0; high Q.: = 1; Thresholds hiQ (cutoff = 0.97); lower panel: (low Q.: = 0; high Q.: = 1; Thresholds Iam cutoff = 0.47): Thresholds were defined according a robust 99.9999999% bivariate normal random interval (assuming a two-dimensional normal distribution)
To identify more genomic regions prone to host inaccurate markers we calculated the exponentially weighted moving averages (ewma) of Iam and hiQ (PROC EXPAND of SAS 9.4; smoothing factor 0.1) [23]. We consider variants with an ewma < threshold (0.47 for Iam and 0.97 for hiQ) as belonging to a “hot region” and variants with an ewma < threshold/2 (0.23 for Iam and 0.48 for hiQ) as belonging to a “very hot region”. Across the whole genome, we were able to identify 4,603 “hot regions” and 171 “very hot regions” according to IamHWE, as well as 2,899 “hot regions” according to hiQ. These regions partially overlap or are interconnected. “Hot” and “very hot” Iam-regions contain in total 85,790 variants, only about 8‰ of all variants. “Hot” hiQ-regions contain in total 53,590 variants, only about 5‰ of all variants. However, about 1 out of 3 “hot” or “very hot” regions is very small and contains only one variant. In contrast, 10% of the “hot” Iam-regions and about 20% of either the “very hot” Iam- or the “hot” hiQ-regions contain more than 20 variants (see Additional file 1: Tables S2–S4). Some of these regions on chromosome 1 are indicated by flames in Fig. 3.
Fig. 3.

Manhattan-like-plot: Iam hiQ: chromosome 1. Upper panel: (low Q.: = 0; high Q.: = 1; Thresholds hiQ (cutoff = 0.97); lower panel: (low Q.: = 0; high Q.: = 1; Thresholds Iam cutoff = 0.47): Thresholds were defined according a robust 99.9999999% bivariate normal random interval (assuming a two-dimensional normal distribution); red flames indicate “very hot” regions; orange flames indicate “hot” regions
Comparing Iam hiQ with info and certainty
The missing rate for info was about 18% and 0.6% for certainty, in the data set at hand. Iam and hiQ could be determined for all markers (see Additional file 1: Table S5). The correlation between Iam and info was largest (r2 = 0.944), indicating that both represent comparable information on accuracy. hiQ and certainty correlate only moderate among themselves as do the other measures (r2 < 0.5) (see Table 2). However, only every second fully-informative SNP (Iam = 1 and hiQ = 1) was assigned a value ≥ 0.8 for info (see Additional file 1: Table S1, red shaded points in Fig. 1), whereas this was the case for less than 2‰ of variants with reduced Iam (0.4 to 0.75). This means that Iam and info nevertheless carry different information on imputation accuracy.
Table 2.
Correlation between accuracy measures
| IamHWE | hiQ | info | certainty | |
|---|---|---|---|---|
| IamHWE | – | 0.684 | 0.944 | 0.484 |
| hiQ | 0.405 | – | 0.367 | 0.156 |
| info | 0.976 | 0.686 | – | 0.050 |
| certainty | 0.305 | 0.335 | 0.051 | – |
Right upper triangle: Pearson’s correlation coefficient, left lower triangle: Spearman’ rank correlation coefficient
Figures for a visual comparison of Iam and info are included in the Additional file 1: Figures S2 and S3. These clearly show that info is less suitable for mapping regions enriched with less accurately imputed genotypes, genome-wide and chromosome-wide.
Usability
We also investigated the usability of the proposed indices in contrast to info by simulation. Usability was considered in terms of discrimination between sufficient and insufficient imputation, rather than in terms of validity of imputation because validity is a characteristic of the imputation routine (e.g. IMPUTE2).
Eight scenarios consisting of two common genotyped tagSNPs flanking one intermediate marker for imputation were defined, differing from each other by the underlying haplotype structure, MAF and LD-patterns were defined. Two scenarios each form a pair (a scene), consisting of a scenario in which the missing marker can be imputed sufficiently/better and one scenario in which the missing marker can be imputed insufficiently/worse. Imputation was performed on 100 randomly drawn samples for each scenario, and accuracy measures were calculated. The ability of an index to discriminate a sufficient from an insufficient scenario (usability) was visually inspected plotting comparative receiver operation curves (one ROC per index) for each scene, and quantified as area under der curve (AUCs) of ROCs. Details on the simulation and the results are given in the Additional file 1.
Info and Iam appear to be comparable usable in terms of discrimination between sufficient and insufficient imputation for common SNPs. However, hiQ seems to be superior when MAF of the imputed marker is low.
Suggestion on how to use Iam hiQ
For general use, a threshold of 0.5 for Iam and 0.9 for hiQ seems reasonable to identify markers with low accuracy. However, we do not claim that this recommendation to generally optimal. All variants with values for Iam and e.g. info below the threshold value, as well as all variants with MAF < 1% and a hiQ below the threshold value should be excluded from a data analysis in order to ensure that all aspects of the imputation quality are met. This pre-analysis marker filtering can be extended to all variants in “hot” or “very hot” regions. If association results cannot be replicated across several studies, a low value of Iam indicates a reduced individual-specific information content, even if, for example, info and hiQ imply sufficient power and genotype heterogeneity.
Discussion
Imputation is a cost-effective tool for GWAS to fill gaps of non-genotyped variants instead of whole-genome sequencing for all recruited individuals, since global coverage in genomic information of available arrays with less than 1 million SNPs not exceeds 25% [10, 24]. However, imputation accuracy matters. Several accuracy measures have been proposed and implemented in imputation software, unfortunately diverse across platforms. Das et al. [4] favour r2, the squared correlation between true and imputed allele dose, because it is tightly related to the power of an allelic test. However, they also emphasized the importance of adequate imputed samples for the r2accuracy.
We introduce Iam hiQ, an independent and complementary pair of accuracy measures. Other than e.g. r2, Iam quantifies the amount of individual-specific versus population-specific genotype information in a linear manner for each individual before averaging, while hiQ addresses the inter-individual heterogeneity of dosages for a marker across the sample at hand. These new measures are not intended to compete with established scores, but should complement them. We derived meaningful, but study specific thresholds for variant filtering applying Iam hiQ to a large case–control sample at hand. We showed how regions enriched with less accurately imputed genotypes can be identified (computationally and visually), and finally compared Iam hiQ to info, as provided by IMPUTE2. Iam hiQ is simple to interpret: of 0 indicates a complete loss of genomic information for a variant. of 0 indicates a reduction to solely population-specific (not individual-specific) genomic information for a variant. of 1 indicates variants for which complete individual-specific genomic information is available. of 0 indicates complete inter-individual homogeneity of dosages across the sample. of 1 indicates that statistical tests can derive all their power from heterogeneity between dosages.
However, it has been discussed that any imputation accuracy measures assuming HWE to calculate "expected" genotype counts can be confounded. This was demonstrated for MaCH r2 [25]. For the proposed method, HWE is solely chosen as anchor point to define pure population informative dosages. One should keep in mind that Iam hiQ is just a tool for quality assurance and not a data analysis module. Thus, slight violations from HWE do not compromise their use, but in case of family data caution is advised. In such cases, one can either apply IamHWE to founders only, or use Iamchance.
Finally, accuracy measures with non-justified thresholds, as e.g. info, should be applied with caution. This in mind, we derived thresholds for Iam hiQ, in contrast to other measures, from observation on a large sample and follow a traceable logic. Because its direct and linear relationship to the average amount of individual-specific genomic information contained in the dosages of a marker, Iam is easy to interpret. By this, it differs from r2, which is approximately equal to the power of the same test with r2N samples.
For the presented quality assurance, we calculated Iam hiQ for autosomes only. Extending this to the X and Y chromosome is possible, but the sex of genotyped individual and the position of the variant on the chromosome must be taken into account when calculating a correct HWE distribution. Even an ex post application of Iam hiQ can be useful, particular to explain whether missed replication of an observed marker-phenotype association is due to inaccurate imputation. Since the imputation accuracy of particularly rare markers tend to be low, an improved imputation of the ILCCO samples is planned on newer panels that contain more SNPS with low MAF.
Conclusion
In summary, Iam hiQ is a newly proposed pair of accuracy measures for imputed genotypes. In contrast to others, it addresses directly the contents of individual-specific genotype information and the heterogeneity between dosages. It is independent of the imputation platform and can be computed for all imputed variants. We recommend using Iam hiQ additional to other accuracy scores for variation filtering before stepping into the analysis of imputed GWAS data.
Availability of data and materials
A macro for SAS® 9.4 to calculate the measures IamHWE, Iamchance and hiQ for autosomal markers based on the dosage-file as output of IMPUTE2 is provided with the Additional file 1.
Materials and methods
Novel accuracy measure
In the following we will consider markers with two alleles ( and ) and a MAF fA in the source population of the study sample consisting of N individuals. The three possible genotypes aa, aA, and AA are indicated by allele doses 0, 1 and 2 (equal to the number of minor alleles A of a genotype). Imputation will result in triplets of a-posteriori genotype probabilities , referred to as dosages, with . We assume the whole uncertainty related to genotype imputation is contained in these triplets. The allele dose of an individual i for an imputed marker will then be and can take any value between 0 and 2. Multi-allelic markers are assumed to be split into pseudo-two-allele variants.
Index of individual-specific versus population-specific genotype information: Iam
To quantify the amount of individual-specific versus population-specific genotype information in the dosages of the imputed single marker m for a single person i, we first consider the following three marginal situations:
The triplet of dosages takes on the values , or in a different order, if imputation is fully sufficient, when the missing genotype is unambiguously derived from the reference panel. Thus, the dosages contain fully individual-specific genotype information.
In contrast, if all genotypes are equally likely the dosages take on the values and imputing of the missing genotype failed (choosing a best guess genotype would be completely due to chance). The dosages contain no individual-specific genotype information at all.
If the dosages take on the values and hence follow HWE, imputation used solely MAF in the reference population and thus the dosages contain solely population-specific information.
To construct an index to distinguish dosages from , or respectively, we were guided by the well-established Herfindahl–Hirschman Index (HHI) [26]. HHI is a concentration measure for distributions of discrete random variables with k possible realisations, defined as . HHI ranges from 1 (if and ; alike (i)) to 1/k (if all ; alike [ii]). Because we are interested in anti-concentration, the opposite of HHI, we first define the quantity.
for each marker m and each individual i. takes the value 0 in case of [i]: and the value 2/3 in case of [ii]: . To achieve an imputation accuracy measure (Iam) for each marker m, we then rescaled the average across all individuals to
ranges from 0 (in case of [ii]: non-informative dosages) to 1 (in case of [i]: fully individual genotype information).
Similarly, can be rescaled to represent situation [iii]: by the index value 0. In this case takes the value
This alternative of the imputation accuracy measure (Iam) can be straightforwardly calculated by
Figure 4 visually presents these definitions of Iam.
Fig. 4.

From dosages to Iam-indices. MAF/fA minor allele frequency; HWE Hardy–Weinberg equilibrium; Iam imputation accuracy measure
indicates that the 3 genotypes are equally likely, averaged over all individuals. Therefore, imputation did not contribute any information at all.
indicates that the genotypes are just as likely as under the HWE, averaged over all individuals. Therefore, imputation contributes only information of MAF in the population (respectively the reference sample), but not for further individual-specific information.
The computation of both Iam indices requires only the dosages provided by the imputation program used. For case–control or cross-sectional studies MAF can by estimated by averaging the allele doses across all individuals, using the same data:
will be calculated fair enough for the outlined purpose even for markers that are associated to a trait and therefore have different MAFs between affected and unaffected individuals. The same applies in the presence of low grade hidden relationships. However, if the study sample consists of relatives, it is advisable to consider only unrelated founders for the estimation of .
depends on MAF. For rare markers is much closer to 0 than for common markers (see Table 3). In case all dosages correspond to HWE (as in situation [iii]) for common markers is close to 0 (indicating inaccurate imputation), whereas for rare markers it is close to 1 (misleadingly indicating accurate imputation). This shows that it is fairly hard to determine the content of individual-specific information in the triplet of dosages of rare markers.
Table 3.
Q and Iamchance by MAF
| MAF | Qchance | QHWE |
Iamchance based on QHWE |
|---|---|---|---|
| 50% | 0.667 | 0.625 | 0.0625 |
| 40% | 0.667 | 0.614 | 0.0784 |
| 30% | 0.667 | 0.575 | 0.1369 |
| 20% | 0.667 | 0.486 | 0.2704 |
| 10% | 0.667 | 0.311 | 0.5329 |
| 5% | 0.667 | 0.176 | 0.7353 |
| 1% | 0.667 | 0.039 | 0.9415 |
| 0.1% | 0.667 | 0.0040 | 0.9940 |
| 0.01% | 0.667 | 0.0004 | 0.9994 |
| 0.001% | 0.667 | 0.00004 | 0.9999 |
| 0.0001% | 0.667 | 0.000004 | 1.0000 |
MAF: minor allele frequency (), Qchance refers to a dosage of ; QHWE refers to a dosage of
It is possible that takes negative values, if the majority of triplets of dosages can be located between the non-informative and population-informative case. This might be caused by genotyping errors as well as by small deviations between sample and population MAF or locally increased inbreeding coefficients in the source population. Some values of will then be between 2/3 and . Due to the upper mentioned shift of by MAF, this is more likely for rare than for common markers. However, small negative values should be regarded as occurred by pure chance.
Index of heterogeneity in quantities: hiQ
Inter-individual heterogeneity of dosages for a marker m is a second concern with respect to the usability of imputed genotypes. Consider the following example: Table 4 gives two markers with average dosages across 10 individuals. Marker 1 is not suitable for any data analysis, because all dosages are identical. The best guess for all individuals is genotype aa. In contrast, marker 2 consists of three different dosages, leading to different best guess genotypes for the individuals. This heterogeneity serves power for statistical testing.
Table 4.
Inter-individual heterogeneity of dosages: example
| ID | Marker 1 | Marker 2 | ||||
|---|---|---|---|---|---|---|
| aa | aA | AA | aa | aA | AA | |
| 1 | 0.6 | 0.3 | 0.1 | 1 | 0 | 0 |
| 2 | 0.6 | 0.3 | 0.1 | 1 | 0 | 0 |
| 3 | 0.6 | 0.3 | 0.1 | 1 | 0 | 0 |
| 4 | 0.6 | 0.3 | 0.1 | 1 | 0 | 0 |
| 5 | 0.6 | 0.3 | 0.1 | 1 | 0 | 0 |
| 6 | 0.6 | 0.3 | 0.1 | 1 | 0 | 0 |
| 7 | 0.6 | 0.3 | 0.1 | 0 | 1 | 0 |
| 8 | 0.6 | 0.3 | 0.1 | 0 | 1 | 0 |
| 9 | 0.6 | 0.3 | 0.1 | 0 | 1 | 0 |
| 10 | 0.6 | 0.3 | 0.1 | 0 | 0 | 1 |
| Avg | 0.6 | 0.3 | 0.1 | 0.6 | 0.3 | 0.1 |
To construct an index of heterogeneity in quantities of dosages (hiQ) we compare “average dosages”(ad) across all individuals with “average of best guess dosages” (ab) applying the Hellinger H-distance. The H-distance quantifies the distance between two (trinomial) probability distributions, taking value H = 0 in case of coincident probability distributions and H = 1 if the probability vectors are perpendicular [27, 28]. Therefore, we defined
In Table 4 the “average of best guess dosages” for marker 1 compared to the average dosages () yields an . This indicates a loss of heterogeneity between dosages and reduced power of a statistical test. For marker 2, where , takes on the value 1, indicating fully achievable inter-individual heterogeneity and no reduced power of a statistical test.
A SAS® macro far calculating IamHWE, Iamchance and hiQ based on the dosage-file as output of IMPUTE2 is included in the Additional file 1.
Application to a sample of lung cancer patients and controls
We applied the novel indices to a dataset of the Integrative Analysis of Lung Cancer Etiology and Risk program of the International Lung Cancer Consortium (INTEGRAL-ILCCO) to examine the behaviour of Iam hiQ, to find appropriate thresholds for marker filtering and for comparison with an established accuracy measure. The sample comprises 14,803 lung cancer cases and 12,262 controls of European descent. They were genotyped on the OncoArray, which queried 517,482 SNPs. The array is designed to cover the whole genome (with GWAS backbone) and for fine mapping of susceptibility to common cancers as well as for de novo discovery, and hence is enriched with low frequent and rare variants [29]. About 50% of markers are considered as GWAS backbone. Details of the sample, the genotyping and the quality control are given elsewhere [30]. The OncoArray whole-genome data were imputed in a two-stage procedure, using SHAPEIT to derive phased genotypes and IMPUTEv2 to infer additional genotypes for genetic variants included in the 1000 Genomes Project (phase 3 panel) [6, 15]. We restricted calculations and comparisons to markers of the autosomes. A total of n = 10,427,599 SNPs were finally included in this quality assessment. Presumably difficult to impute, due to their MAF, are 5,226,623 of these SNPs (50%) with a MAF lower than 1% and 1,500,835 SNPs with a MAF between 1 and 5% (Additional file 2).
Supplementary Information
Additional file 1. A macro for SAS® 9.4 to calculate the measures IamHWE, Iamchance and hiQ for autosomal markers based on the dosagefile. Finally, tables and figures are given with markers and regions of low accuracy.
Additional file 2. Other members (not co-authors) of the International Lung Cancer Consortium (ILCCO).
Acknowledgements
We acknowledge support by the Open Access Publication Funds of the Göttingen University.
Abbreviations
- Iam:
Imputation accuracy measure
- hiQ:
Heterogeneity in quantities of dosages
- ILCCO:
International Lung Cancer Consortium
- SNP:
Single nucleotide polymorphism
- SV:
Structural variation
- GWAS:
Genome-wide association studies
- MAF:
Minor allele frequency
- HWE:
Hardy–Weinberg disequilibrium
Authors' contributions
AR and VT developed the new method and wrote the manuscript. VT implemented the analysis. AT and all other authors planned, designed, and/or conducted the data contributing studies and contributed to the manuscript. RH and CIA coordinate consortium activities. RH, CIA, DC, NC, GL, SB, LM, DA, MA, AT, GFT, GR, JF, MD, TL, LK, PL, AH, SZ, SL, MS, AA, ED, SA, HB, OM, GG, CC, JD, MT, AC, PW, ARi, TM, MJ, PB, MTL, SSS, HB are involved in managing contributing studies, including data collection an preparation and funding acquisition. All authors revised and approved the final manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL. The National Institutes of Health (7U19CA203654-02/ 397 114564-5111078 Integrative Analysis of Lung Cancer Etiology and Risk) supported this work. CARET is funded by the National Cancer Institute, National Institutes of Health through grants U01 CA063673, UM1 CA167462, R01 CA 111703, RO1 CA 151989, U01 CA167462 and funds from the Fred Hutchinson Cancer Research Center. Other individual funding for participating studies and members of INTEGRAL-ILCCO are listed elsewhere [10, 30]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
The data that support the findings of this study are available from ILCCO/INTEGRAL but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are however available from the authors upon reasonable request and with permission of ILCCO/INTEGRAL.
Declarations
Ethics approval and consent to participate
This work was conducted on a collaborative basis within the International Lung Cancer Consortium (ILCCO)/Integrative Analysis of Lung Cancer Etiology and Risk (INTEGRAL)). All consortium research, including the work presented, received approval from the Dartmouth Committee for Protection of Human Subjects on 7/30/2014 with id STUDY00023602. Informed consent was obtained from all participants or, if participants are under 18, from a parent and/or legal guardian. All experimental protocols and other methods used comply with institutional, national, or international guidelines.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Albert Rosenberger, Email: arosenb@gwdg.de.
the INTEGRAL-ILCCO consortium:
Rayjean J. Hung, David C. Christiani, Neil E. Caporaso, Geoffrey Liu, Stig E. Bojesen, Loic Le Marchand, Demetrios Albanes, Melinda C. Aldrich, Adonina Tardon, Guillermo Fernández-Tardón, Gad Rennert, John K. Field, Mike Davies, Triantafillos Liloglou, Lambertus A. Kiemeney, Philip Lazarus, Aage Haugen, Shanbeh Zienolddiny, Stephen Lam, Matthew B. Schabath, Angeline S. Andrew, Eric J. Duell, Susanne M. Arnold, Hans Brunnström, Olle Melander, Gary E. Goodman, Chu Chen, Jennifer A. Doherty, Marion Dawn Teare, Angela Cox, Penella J. Woll, Angela Risch, Thomas R. Muley, Mikael Johansson, Paul Brennan, Maria Teresa Landi, Sanjay S. Shete, and Christopher I. Amos
References
- 1.NCBI Variation Summary. https://www.ncbi.nlm.nih.gov/dbvar/content/org_summary/
- 2.Lindgren D, Hoglund M, Vallon-Christersson J. Genotyping techniques to address diversity in tumors. Adv Cancer Res. 2011;112:151–182. doi: 10.1016/B978-0-12-387688-1.00006-5. [DOI] [PubMed] [Google Scholar]
- 3.Hickey JM, Cleveland MA, Maltecca C, Gorjanc G, Gredler B, Kranis A. Genotype imputation to increase sample size in pedigreed populations. Methods Mol Biol. 2013;1019:395–410. doi: 10.1007/978-1-62703-447-0_17. [DOI] [PubMed] [Google Scholar]
- 4.Das S, Abecasis GR, Browning BL. Genotype imputation from large reference panels. Annu Rev Genomics Hum Genet. 2018;19:73–96. doi: 10.1146/annurev-genom-083117-021602. [DOI] [PubMed] [Google Scholar]
- 5.Marchini J, Howie B. Genotype imputation for genome-wide association studies. Nat Rev Genet. 2010;11(7):499–511. doi: 10.1038/nrg2796. [DOI] [PubMed] [Google Scholar]
- 6.Genomes Project C. Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, Kang HM, Fuchsberger C, Danecek P, Sharp K, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48(10):1279–1283. doi: 10.1038/ng.3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Winkler TW, Day FR, Croteau-Chonka DC, Wood AR, Locke AE, Magi R, Ferreira T, Fall T, Graff M, Justice AE, et al. Quality control and conduct of genome-wide association meta-analyses. Nat Protoc. 2014;9(5):1192–1212. doi: 10.1038/nprot.2014.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng J, Li Y, Abecasis GR, Scheet P. A comparison of approaches to account for uncertainty in analysis of imputed genotypes. Genet Epidemiol. 2011;35(2):102–110. doi: 10.1002/gepi.20552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tozzi V, Rosenberger A, Kube D, Bickeboller H. Global, pathway and gene coverage of three Illumina arrays with respect to inflammatory and immune-related pathways. Eur J Hum Genet. 2019;27(11):1716–1723. doi: 10.1038/s41431-019-0441-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scheet P, Stephens M. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am J Hum Genet. 2006;78(4):629–644. doi: 10.1086/502802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34(8):816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu EY, Li M, Wang W, Li Y. MaCH-admix: genotype imputation for admixed populations. Genet Epidemiol. 2013;37(1):25–37. doi: 10.1002/gepi.21690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007;81(5):1084–1097. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5(6):e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sargolzaei M, Chesnais JP, Schenkel FS. A new approach for efficient genotype imputation using information from relatives. BMC Genomics. 2014;15:478. doi: 10.1186/1471-2164-15-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hickey JM, Kinghorn BP, Tier B, van der Werf JH, Cleveland MA. A phasing and imputation method for pedigreed populations that results in a single-stage genomic evaluation. Genet Sel Evol. 2012;44:9. doi: 10.1186/1297-9686-44-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Browning BL, Browning SR. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet. 2009;84(2):210–223. doi: 10.1016/j.ajhg.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39(7):906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 20.Mitt M, Kals M, Parn K, Gabriel SB, Lander ES, Palotie A, Ripatti S, Morris AP, Metspalu A, Esko T, et al. Improved imputation accuracy of rare and low-frequency variants using population-specific high-coverage WGS-based imputation reference panel. Eur J Hum Genet. 2017;25(7):869–876. doi: 10.1038/ejhg.2017.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krithika S, Valladares-Salgado A, Peralta J, EscobedodeLaPena J, Kumate-Rodriguez J, Cruz M, Parra EJ. Evaluation of the imputation performance of the program IMPUTE in an admixed sample from Mexico City using several model designs. BMC Med Genomics. 2012;5:12. doi: 10.1186/1755-8794-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O'Connell J, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562(7726):203–209. doi: 10.1038/s41586-018-0579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.SAS/STAT 14.1 User's Guide, vol. Second Edition; 2015.
- 24.Ha NT, Freytag S, Bickeboeller H. Coverage and efficiency in current SNP chips. Eur J Hum Genet. 2014;22(9):1124–1130. doi: 10.1038/ejhg.2013.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shriner D. Impact of Hardy–Weinberg disequilibrium on post-imputation quality control. Hum Genet. 2013;132(9):1073–1075. doi: 10.1007/s00439-013-1336-x. [DOI] [PubMed] [Google Scholar]
- 26.Bleymüller J, Gehlert G, Gülicher H: Statistik für Wirtschaftswissenschaftler, 15., überarb. Aufl. edn. München: Vahlen; 2008.
- 27.Roshyara NR, Kirsten H, Horn K, Ahnert P, Scholz M. Impact of pre-imputation SNP-filtering on genotype imputation results. BMC Genet. 2014;15:88. doi: 10.1186/s12863-014-0088-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chung JK, Kannappan PL, Ng CT, Sahoo PK. Measures of distance between probability-distributions. J Math Anal Appl. 1989;138(1):280–292. doi: 10.1016/0022-247X(89)90335-1. [DOI] [Google Scholar]
- 29.Amos CI, Dennis J, Wang Z, Byun J, Schumacher FR, Gayther SA, Casey G, Hunter DJ, Sellers TA, Gruber SB, et al. The OncoArray Consortium: a network for understanding the genetic architecture of common cancers. Cancer Epidemiol Biomarkers Prev. 2017;26(1):126–135. doi: 10.1158/1055-9965.EPI-16-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McKay JD, Hung RJ, Han Y, Zong X, Carreras-Torres R, Christiani DC, Caporaso NE, Johansson M, Xiao X, Li Y, et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat Genet. 2017;49(7):1126–1132. doi: 10.1038/ng.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. A macro for SAS® 9.4 to calculate the measures IamHWE, Iamchance and hiQ for autosomal markers based on the dosagefile. Finally, tables and figures are given with markers and regions of low accuracy.
Additional file 2. Other members (not co-authors) of the International Lung Cancer Consortium (ILCCO).
Data Availability Statement
A macro for SAS® 9.4 to calculate the measures IamHWE, Iamchance and hiQ for autosomal markers based on the dosage-file as output of IMPUTE2 is provided with the Additional file 1.
The data that support the findings of this study are available from ILCCO/INTEGRAL but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are however available from the authors upon reasonable request and with permission of ILCCO/INTEGRAL.