

Abstract
Myocardial ischemia or reperfusion increases the generation of reactive oxygen species (ROS) from damaged mitochondria, NADPH oxidases, xanthine oxidase, and inflammation. ROS can be removed by eight endogenous antioxidant and redox systems, many components of which are expressed under the influence of the activated Nrf2 transcription factor. Transcriptomic profiling, sequencing of Nrf2-bound DNA, and Nrf2 gene knockout studies have revealed the power of Nrf2 beyond the antioxidant and detoxification response, from tissue recovery, repair, and remodeling, mitochondrial turnover, and metabolic reprogramming to the suppression of proinflammatory cytokines. Multifaceted regulatory mechanisms for Nrf2 protein levels or activity have been mapped to its functional domains, Nrf2-ECH homology (Neh)1–7. Oxidative stress activates Nrf2 via nuclear translocation, de novo protein translation, and increased protein stability due to removal of the Kelch-like ECH-associated protein 1 (Keap1) checkpoint, or the inactivation of β-transducin repeat-containing protein (β-TrCP), or Hmg-CoA reductase degradation protein 1 (Hrd1). The promise of small-molecule Nrf2 inducers from natural products or derivatives is discussed here. Experimental evidence is presented to support Nrf2 as a lead target for drug development to further improve the treatment outcome for myocardial infarction (MI).
Ischemia and reperfusion injury in the myocardium
Dramatic themes of heart attacks in movies and popular culture have illustrated the potential outcome of heart disease as deadly. Annually, coronary heart disease claims over 400 000 lives and accounts for one in every seven deaths in the USA. These numbers support the importance of developing novel therapies to improve the outcome for the prevention or treatment of MI (see Glossary). While clinical presentations of MI can be diverse, the classical definition of MI is based on pathological evidence of myocardial cell death due to prolonged ischemia related to coronary artery occlusion. Time-dependent cell death occurs following ischemia, in the form of necrosis, apoptosis, and necroptosis (Box 1). Myocardial cell death may result from non-ischemic conditions, promoting the establishment of myocardial injury as a generic term for medical conditions involving myocardial cell death [1]. With MI, timely implementation of medical procedures for cardiac protection and opening of the coronary artery are essential for patients’ survival and the minimization of adverse events.
Box 1. Time-dependent cell death and hypertrophy with MI.
Elevation of cTn in the circulation serves as a diagnostic measure of cardiac muscle cell death or cardiac injury. Whereas significant elevation of cTn is considered a hallmark of MI, reperfusion as an essential medical treatment causes elevation of cTn, with nearly all CABG patients and about 30% of PCI patients showing post-procedural cTn elevation.
Necrotic cell death first appears within 15–20 min of coronary artery occlusion in the subendocardium and spreads to the epicardial area after 24 h, peaking at 1 day. Morphologically, coagulation necrosis appears as pale, ‘empty’ cells or contraction-band necrosis shows thick, intense bands spanning across the myocytes, believed to result from hypercontraction during reperfusion. Necrosis mainly occurs in the infarct zone, whereas the border zone contains both necrotic and apoptotic cells.
Apoptosis can be detected in patients via imaging techniques employing Tc-99m-labeled annexin V, which showed apoptosis in the infarct area 5–20 h after reperfusion. Residual apoptosis remained detectable 6–8 weeks after the patients had been discharged from the hospital.
Necroptosis is a consequence of programmed events from molecular distortion involving receptor-interacting protein kinases (RIPs). RIP1 interacts with RIP3 to form necrosomes following the binding of a cell-death-inducing cytokine to its receptor. Necrosomes perturb mitochondria to generate ROS and initiate necrosis.
A fraction of myocardial cells surviving ischemia and reperfusion develop hypertrophy, with uneven loss of mitochondria, imbalance of biochemical metabolism, or myofibrillar remodeling. Extensive cardiomyocyte hypertrophy contributes to eventual heart failure.
Cell death along with hypertrophy of surviving cells, a prolonged inflammatory response, and activation of the neuroendocrine system ultimately leads to functional impairment and the development of heart failure.
To salvage myocardial tissue from ischemic injury, reperfusion is achieved through cardiothoracic surgery with coronary artery bypass grafting (CABG) or a non-surgical percutaneous coronary intervention (PCI) procedure. Early reperfusion reduces cell death and increases the survival rate. Bypass via CABG or opening of the coronary artery by PCI is crucial to relieve symptoms, limit infarct size, and preserve ventricular function. Despite its benefits, reperfusion elicits further myocardial cell death, contributing to an expansion of infarct size [2]. A number of molecular entities have been tested over the past 40 years to reduce the symptoms and curb the progression or consequences of MI, among which many are current MI treatment medications, including vasodilators, anticoagulants, thrombolytic agents, and β-blockers. The advances in reperfusion techniques combined with effective pharmacotherapies have led to today’s low mortality rate, providing that the patient can receive medical care immediately.
Patients who survive an acute MI are at a higher risk for the development of heart failure over subsequent years. Prolonged oxidative stress, continuous evolvement of cell death or degeneration, and inflammatory reactions are among the culprits in clinical complications. ROS cause cell death or tissue degeneration and contribute to inflammation over time, presenting a window of opportunity for novel pharmacological intervention.
Nrf2, a transcription factor activated by oxidative stress, increases the expression of antioxidant and detoxification enzymes in return for combating ROS and toxic metabolites. Transcriptomic profiling and sequencing of Nrf2-bound DNA have revealed that Nrf2 also regulates genes that are important for tissue repair or remodeling and the suppression of inflammation, illustrating a role for Nrf2 beyond the antioxidant and detoxification response. Understanding the characteristics of the Nrf2 protein and its mechanism of activation provides a molecular basis to hijack the regulatory pathway using pharmacological agents for cardiac protection.
The paradox of oxidative stress: activation of Nrf2
The human Nrf2 gene encodes a 605-amino-acid protein with seven functional domains, Neh1–7 (Figure 1). The conserved nature of the Nrf2 protein sequence across mammalian species agrees with its universal function and regulation. The Neh1 domain harbors a basic leucine zipper (bZIP) motif, which heterodimerizes with a small musculoaponeurotic fibrosarcoma homolog protein (MafF, MafG, or MafK) for DNA binding and transcriptional activation.
Figure 1. Structure of the Nrf2 protein depicting its function and regulation.
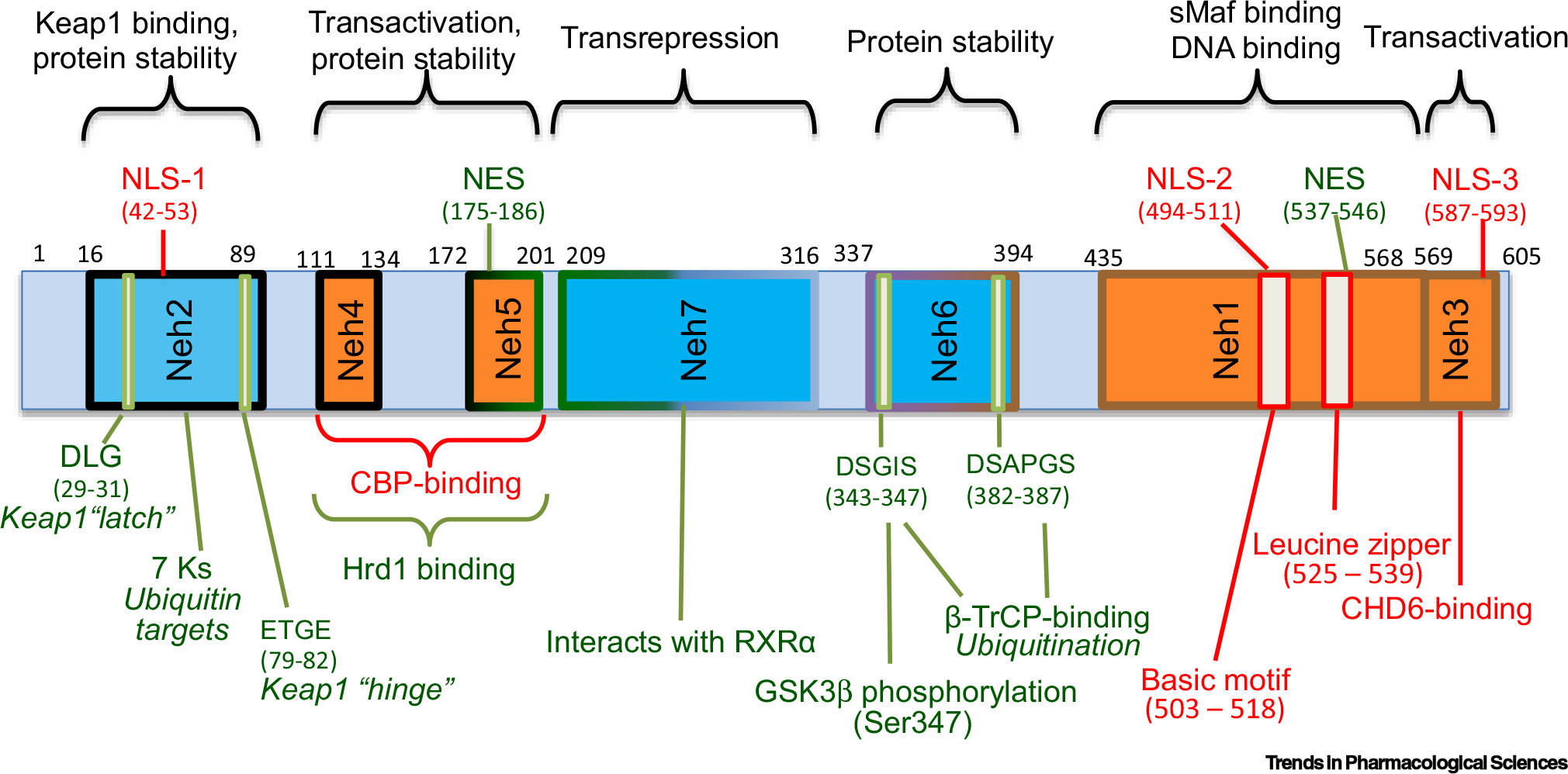
Green line or font indicates negative regulation of Nrf2 activity as a transcription factor. Red line or font indicates factors governing activation of the Nrf2 transcription factor. Orange boxes show the domains important for the activity of the Nrf2 transcription factor, whereas blue boxes indicate the domains containing negative regulators. The border colors reflect five exons encoded by the Nrf2 gene. Abbreviations: β-TrCP, β-transducin repeat-containing protein; CHD6, chromo-ATPase/helicase DNA binding protein; GSK3β, glycogen synthase kinase 3β; NES, nuclear export sequence; NLS, nuclear localization sequence; RXRα, retinoid X receptor alpha.
The Neh2 domain contains the binding sites of Keap1. Keap1 binds to the Neh2 domain at the DLG motif with low affinity and at the ETGE motif with high affinity. As a redox sensor and checkpoint partner of Nrf2, Keap1 protein bears 27 cysteine residues and normally binds to Nrf2 in a form of dimer, directing a Cullin 3 ubiquitin E3 ligase complex to conjugate ubiquitin to one of seven lysine residues in the Neh2 domain. This explains the short half-life of Nrf2 protein (10–15 mins) under physiological conditions. Keap1 oxidation at Cys151 was first reported as a key event in abolishing the Cullin 3 interaction [3]. Others have shown that redundant cysteine residues are involved in regulating Nrf2 induction. For example, Cys226/613/622/624, but not Cys151, participate in sensing H2O2, whereas either one or all of the Cys151/273/288 residues are important for Nrf2 induction by electrophiles such as dimethyl-fumarate or CDDO-Im [4]. Loss of Keap1 binding at the DLG motif has been assumed to be a main route for Nrf2 protein stabilization and level increase during oxidative stress (Figure 2). A recent study on protein–protein interaction revealed that electrophiles do not affect Keap1 binding to the DLG or the ETGE motif of Nrf2 [5]. Whether oxidation of Keap1 causes the release of DLG binding remains to be confirmed experimentally. Nevertheless, alkylation or oxidation of cysteine residues disables Keap1 from recruiting the E3 ligase for Nrf2 ubiquitination, resulting in an extension of the Nrf2 half-life to nearly 60 mins.
Figure 2. Mechanisms activating the Nrf2 transcription factor due to oxidative stress in cardiomyocytes.
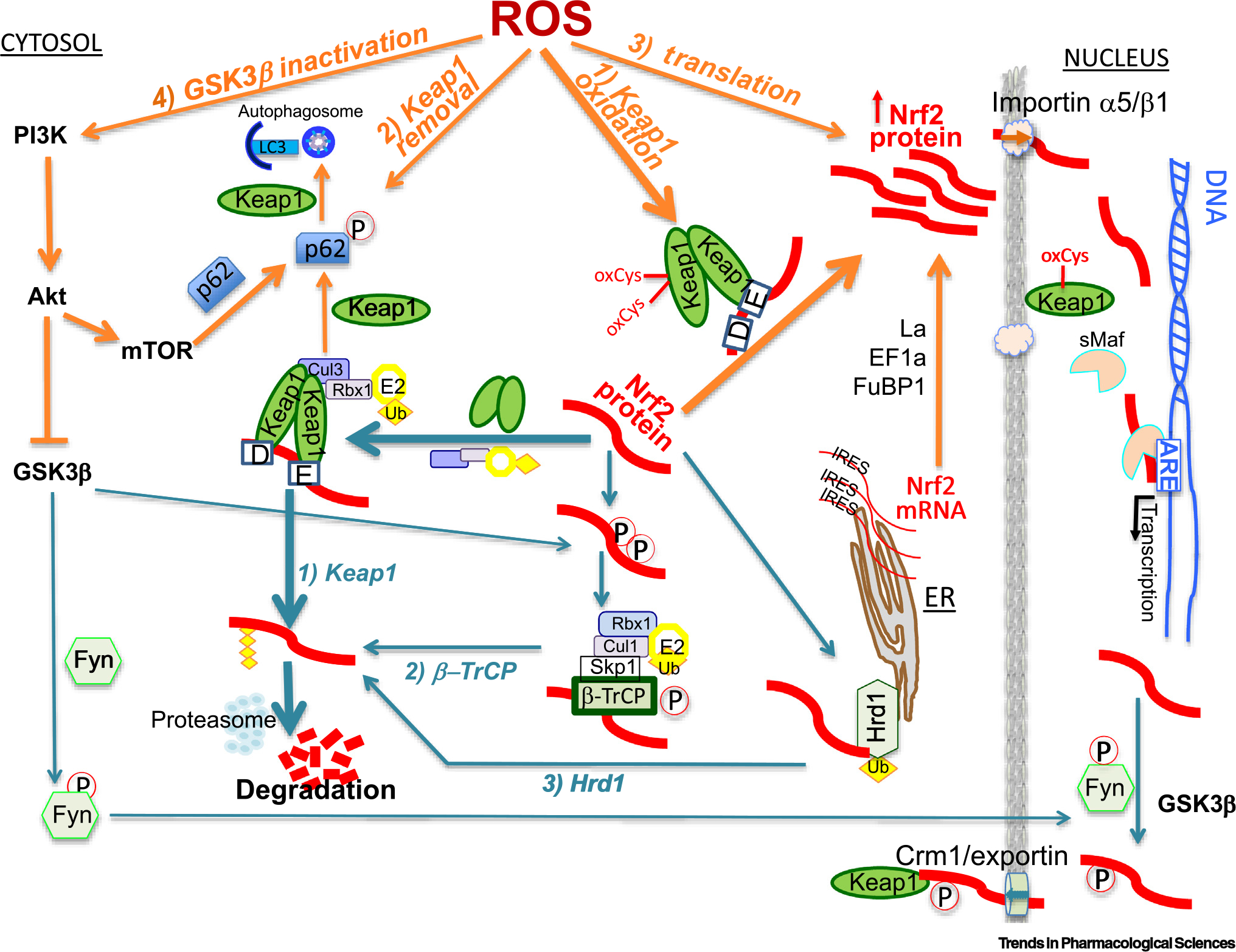
Orange font or line shows four pathways leading to Nrf2 activation, whereas turquoise-colored lines and font indicate three mechanisms of Nrf2 protein degradation plus Nrf2 inactivation via nuclear export. The weight of the lines reflects the amount of literature support and the relative importance compared with parallel pathways. Abbreviations: β-TrCP, β-transducin repeat-containing protein; FuBP1, far upstream binding protein 1; GSK3β, glycogen synthase kinase 3β; IRES, internal ribosomal entry site; Keap1, Kelch-like ECH-associated protein 1.
A second mechanism disabling Keap1 control over Nrf2 protein stability concerns the autophagy protein p62 (Figure 2). p62 normally tags damaged proteins or organelles for recognition by autophagosomes, followed by mass degradation in lysosomes. An STGE motif in p62 protein resembles the ETGE motif of Nrf2, allowing Keap1 to bind [6]. The affinity of Keap1 to p62 is enhanced when Ser349 in STGE becomes phosphorylated downstream of PI3K–Akt–mTOR signaling, escalating Keap1 degradation via autophagy [7]. As a result, the process of autophagy is accompanied by Nrf2 protein accumulation.
The Neh3, Neh4, and Neh5 domains act in synergy for Nrf2 to perform transcriptional regulation (Figure 1). Whereas Neh3 permits Nrf2 interaction with a transcriptional coactivator, chromo-ATPase/helicase DNA binding protein (CHD6), the Neh4 and Neh5 domains cooperate to interact with another transcriptional coactivator, CREB binding protein (CBP) [8,9]. The Neh4 and Neh5 domains also shelter a binding site for an E3 ubiquitin ligase, Hrd1 (also known as Synoviolin). Hrd1/Synoviolin responds to ER stress by binding to Nrf2 for ubiquitination [10]. Therefore, Hrd1/Synoviolin delivers a second line in parallel to Keap1 for Nrf2 ubiquitination and degradation.
A third line of Nrf2 ubiquitination requires Nrf2 interaction with β-TrCP at the Neh6 domain. β-TrCP recognizes the DSGIS and DSAPGS motifs and recruits the Skp1-Cult1-Rbx1/Rock ubiquitin ligase for Nrf2 ubiquitination. When Nrf2 is primed for phosphorylation by glycogen synthase kinase 3β (GSK3β) to form a phosphodegron, β-TrCP binds and recruits the ubiquitin ligase for Nrf2 ubiquitination [11,12]. Since GSK3β is a constitutively active kinase and Keap1 is a dominant regulator of Nrf2, the relationship between β-TrCP- and Keap1-mediated Nrf2 degradation remains to be elucidated. GSK3β contains a nuclear localization sequence (NLS) and may phosphorylate Nrf2 in the nucleus and facilitate its nuclear export for β-TrCP-mediated ubiquitination. It remains to be proved that GSK3β and β-TrCP may remove excess Nrf2 from the nucleus while Keap1 checks Nrf2 protein in the cytosol under physiological conditions. Nevertheless, during oxidative stress, activation of the PI3K–Ark pathway leads to inhibition of GSK3β, thereby defeating Nrf2 protein degradation via β-TrCP (Figure 2).
A negative regulatory element, the retinoid X receptor alpha (RXRα) binding site, resides in the Neh7 domain [13,14]. Retinoid binding to RXRα leads to blockage of Nrf2 activity without affecting its half-life or nuclear translocation. Interestingly, RXRα forms a heterodimer with Nrf2 in the nucleus to prohibit the transcription of Nrf2 downstream genes [13–16]. Other nuclear receptors, specifically retinoid acid receptor (RAR)α, RARγ, peroxisome proliferator-activated receptor gamma (PPARγ), estrogen receptor (ER)α, ER-related β, and glucocorticoid receptor α also exhibit Nrf2 inhibitory activity, presumably through a mechanism similar to RXRα [15]. RXRα binding-mediated negative regulation of Nrf2 provides one explanation for the failure of clinical trials employing β-carotene for the prevention of coronary artery disease and MI [17,18] (Table 1). This is related to the fact that β-carotene can be metabolized to retinoids in the body therefore inhibiting Nrf2.
Table 1.
| Trial name | Type and location | Patient population | Outcome/endpoint | Regimen and dose | Statistical method | Outcome | Adverse event | Refs |
|---|---|---|---|---|---|---|---|---|
| ATBC, N01-CN-45165 | Prevention, Helsinki | 1862 male smokers 50–69 years old, previous MI | First major coronary event (MCE) | VitE 50 mg/day, βCA 20 mg/day, 5.3 years | Kaplan-Meier method, log-rank test, Cox’s proportional-hazards regression | No difference in MCEs | Significant MI-related death in βCA (74/461) and VitE + βCA group (67/497) compared with PB (39/438) | [17] |
| ATBC | Prevention, Helsinki | 23 144 male smokers at risk, 1255 with pretrial MI | First-ever MCE | VitE 50 mg/day, βCA 20 mg/day, 5–8 years | Likelihood ratio test for homogeneity, Poisson regression for relative risk, generalized additive model for calendar time-specific rates | Increased MCE risk in βCA (1.13) and VitE + βCA groups (1.08) | [18] | |
| BEAM, NCT00811889 | Phase II, USA | 227 moderate to severe CKD adults and type 2 diabetes, 1:1:1:1 ratio for PB, 25, 75, 150 mg/day BDXL | 1st: GFR improvement in 24 weeks 2nd: 52 weeks, biomarkers of kidney function | BDXL 25, 75, 150 mg/day, 52 weeks (first 8–20 weeks dose adjust), 4 weeks follow up | Repeated-measures model for 1st and 2nd outcomes, Toeplitz structure for within-patient correlation, Kaplan-Meier method for proportion of event free | Improved GFR at 24 weeks, persisted to 52 weeks | 42–61 % calf muscle spasm (18% for PB), resolved over time, 21–32% hypomagnesemia (5% for PB), 71% transient mild elevation of ALT |
[72] |
| BEACON, NCT01351675 | Phase III, international (71% USA) | 2185 (1088 drug + 1097 PB) type 2 diabetes with stage 4 CKD, diverse age, sex, race, ethnicity, region of origin | 1st: ESRD or death 2nd: Change in GFR; heart failure hospitalization or CV death; nonfatal MI, nonfatal stroke, hospitalization for heart failure, or death from CV causes | BDXL 20 mg/day, 3–11 months (median 7 months), PB median months, median months follow up | Kaplan-Meier product limit for cumulative incidence, hazard ratios and 95% CI with Cox proportional- hazards regression models, mixed-effects regression analyses for longitudinal analysis | 27 and 19 CV deaths, 43 and 51 ESRD in BDXL and PB groups | 96 and 55 had heart-failure events in BDXL and PB, 139 and 86 in BDXL and PB had nonfatal MI, stroke, hospitalization, or death from CVD | [73] |
| BEACON | Phase III, post-hoc data analyses | See above | See above | See above | Classification and regression tree analysis to identify baseline factors predictive of adverse events | Baseline high risk of heart failure is a compound factor of heart failure by BDXL | 60% increase in heart failure with previous heart failure by BDXL, similar risk for heart failure without previous heart failure between BDXL and PB groups | [75] |
| MIRACL | Drug purposing expansion, international | 3086 unstable angina or non-Q wave acute MI | Reduction of 1 ° end-point events: death, nonfatal acute MI, cardiac arrest, recurrent myocardial ischemia | AT, 80 mg/day or PB, 24–96 h after hospital admission, for 16 weeks | Cox proportional-hazards model for primary combined end points, Cochran-Mantel-Haenszel method for each end point | Reduction of end-point events (14.8% in AT, 17.4% in PB group), reduced cholesterol | Abnormal liver transaminases more common in AT group (2.5%) than PB group (0.6%) | [76] |
| PROVE IT-TIMI 22 | Compare standard vs intensive dose, USA and Canada | 4162 acute coronary syndrome | Death from any cause, MI, unstable angina requiring hospitalization, revascularization, stroke | PV 40 mg/day or AT 80 mg/day, 2 years | Kaplan-Meier estimates of primary end point, Cox proportional-hazards model for estimates of hazard ratios and 95% CI | Reduced primary end point: 26.3% in PV, 22.4% in AT group; hazard ratio reduction of death from any cause and MI, more LDL lowering with AT than PV | Slight increase in stroke hazard ratio | [77] |
All trials were randomized and double blinded with placebo control except the PRPVE IT-TIMI 22 trial, which is randomized and double blinded with double-dummy control. All trials were completed except BEACON (NCT01351675), which was terminated early, with a median 9-month follow-up, due to safety concerns.
Abbreviations: VitE, α-tocopherol; βCA, β-carotene; PB, placebo; BDXL, bardoxolone methyl; AT, atorvastatin; PV, pravastatin; CV, cardiovascular disease; CKD, chronic kidney disease; ESRD, end-stage renal disease; GFP, glomerular filtration rate; ALT, alanine aminotransferase.
There are three NLSs, located in the Neh1, Neh2, and Neh3 domains, and two nuclear export sequences (NESs) in the Neh1 and Neh5 domains (Figure 1). Cytoplasmic to nuclear translocation through importins α5/β1 represents another layer of regulation for the activity of the Nrf2 transcription factor [19]. Sulforaphane (SFN), a well-established Nrf2 inducer, was reported to activate Akt, inhibit GSKβ, and reduce the nuclear localization of the tyrosine kinase Fyn, suggesting that Fyn phosphorylation by GSK3β mediates the nuclear export of Nrf2 [20]. Nuclear export via Crm1/exportin serves as a mechanism to inactivate Nrf2 signaling [21]. Interestingly, Keap1 also contains a consensus NES sequence in a region containing four cysteine residues for nuclear export via Crm1/exportin. While the unoxidized Keap1 may be important for Nrf2 nuclear export and keeping Nrf2 in the cytoplasm under physiological conditions, ROS inactivate Keap1’s NES, allowing Nrf2 and Keap1 to accumulate in the nucleus [22]. Therefore, Nrf2 protein undergoes active shuttling between the nucleus and cytoplasm, arbitrating its function as a transcription factor.
Besides protein stabilization and nuclear translocation, the Nrf2 protein is rapidly translated during acute oxidative stress and ischemic preconditioning [23–27]. Normally, the process of protein translation is initiated by eIF4E binding to the 7-methylguanosine cap at the 5′ end of an mRNA strand. During stress, this cap-dependent translation is inhibited. Instead, selective protein translation occurs via internal ribosomal entry sites (IRESs) [28]. Nrf2 mRNA contains an IRES in the 5′ untranslated region [24,29]. Oxidative stress in cultured cardiomyocytes or mild ischemic reperfusion in mice causes rapid onset of Nrf2 protein translation, within 10 min [23,24,26,27]. The complex theme underlying this de novo protein translation involves the recognition of Nrf2 mRNA by a set of RNA binding proteins, including the La autoantigen, EF1a, and far upstream binding protein 1 (FuBP1) [25–27]. The coordination of these RNA binding proteins with the translational machinery enables Nrf2 protein to be newly synthesized. The quick onset of Nrf2 protein translation during oxidative stress adds another framework for mobilization of the Nrf2 transcriptional cascade to limit tissue injury.
Nrf2 for endogenous antioxidant defense and beyond
Biological evolution has created multiple antioxidant and redox systems for the removal of ROS inside cells (Figure 3), including: (i) superoxide dismutase (SOD) and catalase (Cat); (ii) glutathione (GSH) peroxidase (GPx) eliminates H2O2 and lipid peroxide using GSH as a substrate – glutamate-cysteine ligase (GCL) (catalytic subunit Gclc and regulatory subunit Gclm) acts as a rate-limiting step for GSH synthesis, whereas GSH reductase (GSH-R) replenishes GSH from its oxidized form, GSSG; (iii) GSSG can also be converted back to GSH by metallothionein (MT), at the expense of the oxidation of MT cysteine residues; (iv) thioredoxin (Trx) and Trx reductase (TrxR) reduce oxidized proteins; (v) peroxiredoxin (Pdx) serves as an acceptor of peroxides at its cysteine residues, which can be reduced back by Trx and sulfiredoxin (Srxn); (vi) NAD(P)H:quinone oxidoreductase 1 (NQO1) catalyzes the reduction of quinone to hydroquinone – this obligatory two-electron reduction averts semiquinone formation, preventing the generation of superoxide or H2O2 [30,31]; (vii) tissue injury promotes hemolysis and the breakdown of hemoproteins, causing free heme to accumulate – by degrading heme, heme oxygenase 1 (HO-1) disables the redox cycling of iron in heme; and (viii) ferritin, a complex containing 24 subunits of heavy-chain FTH versus light-chain FTL, chelates iron and prevents free iron from undergoing redox cycling to generate ROS. Although some genes can be regulated by factors other than Nrf2, sufficient evidence indicates that Nrf2 plays an important role in influencing the expression of most genes in these antioxidant and redox systems (Figure 3).
Figure 3. Antioxidant and redox systems for the removal of oxidants, oxidized proteins, and lipid peroxides.
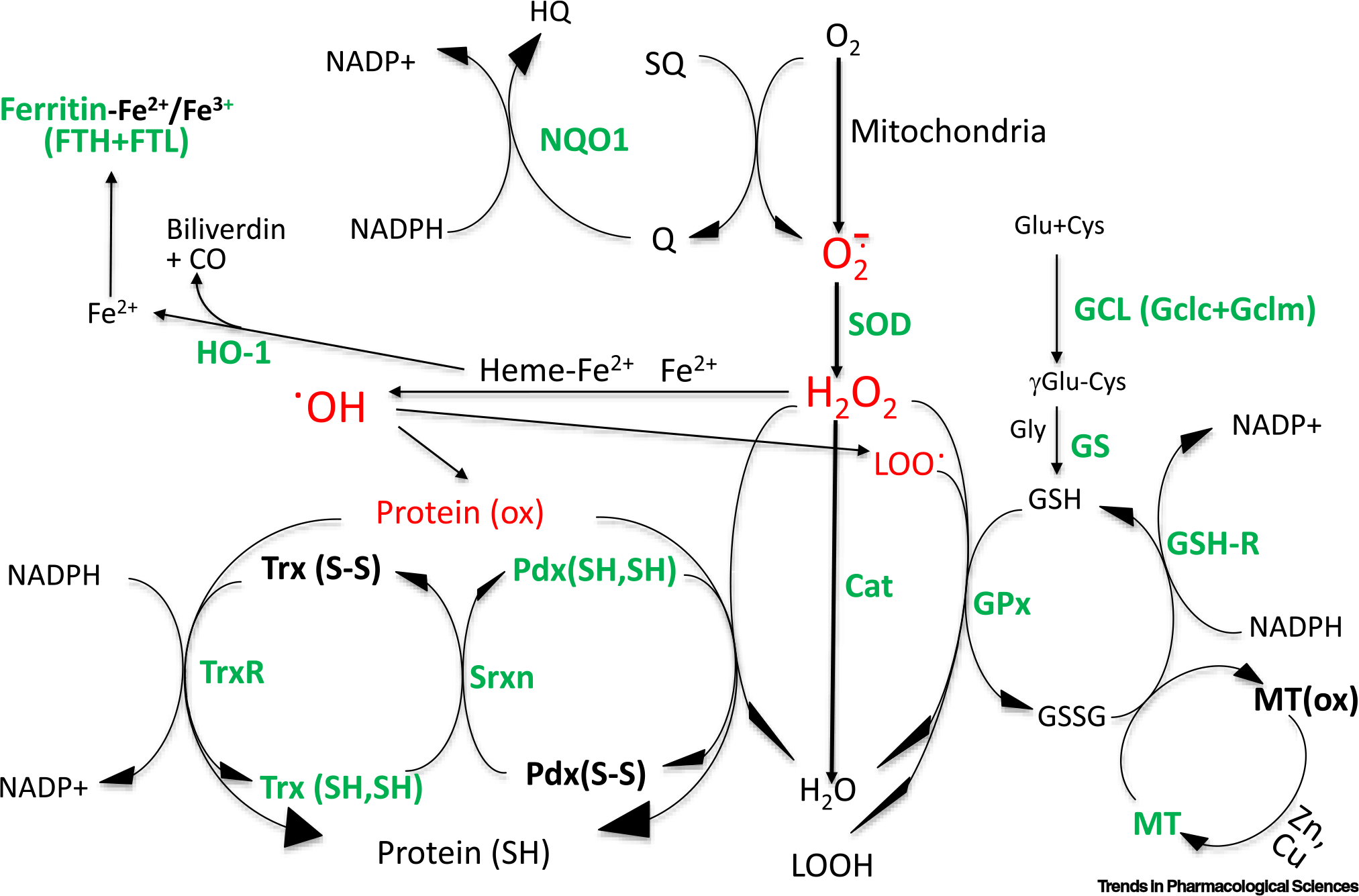
Intracellular enzyme systems, metal-binding proteins, and redox proteins for the elimination of superoxide (), hydrogen peroxide (H2O2), lipid peroxide (LOO•), and oxidized protein (ox). Red font indicates oxidative species. Green color indicates that the genes encoding the proteins have been reported to be under the influence of the Nrf2 transcription factor. Abbreviations: Cat, catalase; GCL, glutamate-cysteine ligase; GPx, glutathione peroxidase; GSH, glutathione; GSH-R, glutathione reductase; HO-1, heme oxygenase 1; MT, metallothionein; NQO1, NAD(P)H:quinone oxidoreductase 1; Prx, peroxiredoxin; SOD, superoxide dismutase; Srxn, sulfiredoxin; Trx, thioredoxin; TrxR, thioredoxin reductase.
The main mechanism of Nrf2-mediated gene expression requires its binding to the antioxidant response element (ARE). Many antioxidant and detoxification genes contain such elements in their promoters. The ARE was first characterized as a consensus DNA sequence of 5′-TMAnnRTGAYnnnGCRwww-3′ (M = A or C; R = A or G; Y = C or T; W = A or T) [32]. A number of genes (e.g., HO-1, Gclc) have polymorphic ARE sequences [33]. Considering all possible SNPs in the most common ARE sequence, TGACTCAGCA, computational analyses predicted 35.6% or 10 068 human genes containing AREs [34]. Among these, 2388 are shared with mouse and rat species, suggesting that Nrf2 is situated on the top of a hierarchy of transcriptional events ruling cellular responses to physical or chemical stress.
Typical Nrf2 downstream genes with well-characterized AREs in their promoters include NQO1, HO-1, Gclc, Gclm, and GSH S transferases (Gsts) (e.g., Gsta1/2/3/5, the Gstp1 gene), which are often used as biomarkers of Nrf2 activation. Additional antioxidant and detoxification genes reported to be under the control of Nrf2, including Gpx2, Trx, TrxR1, Pdx1/6, Srxn, MT1, MT2, FTH, and FTL [33,35–38]. Whereas ARE sequences have been defined for some genes, a long list of those affected by Nrf2 status presumably contain AREs in their promoters. As shown in the functional map of the Nrf2 protein (Figure 1), the interaction with CBP or CHD6, as well as being a bZip protein, suggests that Nrf2 may perform additional transcriptional duties besides ARE binding.
Transcriptomic profiling plus the revelation of Nrf2-bound DNA sequences led to the discovery of Nrf2 control beyond antioxidant and detoxification genes [35–37,39]. Figure 4 lists the genes with indications that those found in cardiomyocytes or myocardial tissues are effectors of changing Nrf2 status. Notably, Nrf2 upregulates a number of transcription factors, including heat shock factor 1 (Hsf1), CCAAT-enhancer-binding protein beta (c/EBPβ), activating transcription factor (ATF)3, ATF4, nuclear factor interleukin (IL)-3 regulated (NFIL3), myocyte enhancer factor 2 (MEF2A), and MafG [36,37]. Hsf1 governs a plethora of heat shock proteins (Hsps), many of which function as molecular chaperons. Deficiency of Nrf2 is coupled to decreases of six Hsp transcripts in the mouse myocardium [37]. c/EBPβ signaling during cardiac injury is essential for neutrophil infiltration, the reactivation of developmental genes, and recovery of cardiac function [40]. ATF3 or ATF4 induction is common among a variety of types of stress and can form heterodimers with c/EBP or AP-1 family members to regulate the expression of stress relief and cell survival genes [41,42]. NFIL3, an IL-3-regulated bZip transcription factor, operates as an antiapoptotic factor, likely via anti-inflammatory cytokine production and the expression of the insulin-like growth factor 2 receptor [43]. MEF2A coordinates cardiac muscle gene expression, cardiac remodeling, and hypertrophy [44]. Notch signaling determines cell fate during organ development and cardiogenesis [45]. During cardiac injury, Notch participates in the cell survival response, tissue repair, and the limitation of adverse remodeling [46,47]. In addition, the crosstalk between Nrf2 and Notch signaling appears to reduce ROS production in cultured cardiomyocytes [48].
Figure 4. Predicted functions of Nrf2 in the myocardium based on its downstream genes.

The downstream genes were revealed by microarray or RNA-seq as transcripts increased or reduced due to Nrf2 activation or deficiency or are targets of Nrf2 binding as determined by DNA sequencing following ChIP. Underlines indicate the genes, expression of which has been found in cardiomyocytes in culture or in mouse myocardium related to Nrf2 status. Color shade reflects the ratio of genes in the functional group under the control of Nrf2 in the myocardium and the amount of literature validating the function by Nrf2 inducers or Nrf2 knockout in experimental animals. Abbreviations: Cat, catalase; c/EBP, CCAAT-enhancer-binding protein; GPx, glutathione peroxidase; GSH-R, glutathione reductase; GST, glutathione S transferase; HbEGF, heparin-binding epidermal growth factor-like growth factor; HO-1, heme oxygenase 1; MT, metallothionein; NF-κB, nuclear factor kappa B; NQO1, NAD(P)H: quinone oxidoreductase 1; Pdx, peroxiredoxin; PGC1α, peroxisome proliferator-activated receptor gamma activator 1 alpha; PINK1, PTEN-induced kinase 1; SOD, superoxide dismutase; Srxn, sulfiredoxin; TGF, transforming growth factor; Trx, thioredoxin; TrxR, thioredoxin reductase; VEGF, vascular endothelial growth factor.
Induction of MafG gene expression during Nrf2 activation suggests a positive feedback mechanism for the Nrf2 transcriptional force. This is because MafG interacts with Nrf2 for transcriptional activation (Figure 1). The promoter of the Nrf2 gene contains an ARE, bolstering another layer of positive feedback [49]. Therefore, initial Nrf2 activation can lead to the amplification of Nrf2-mediated events that orchestrate cytoprotection and tissue repair or remodeling via a diverse array of transcription factors and their targets.
Nrf2 has also been linked to the expression of multitudes of growth factors, signaling molecules, proteases, and extracellular matrix proteins (Figure 4). Confirmation of the gene expression of mitogen-activated protein kinase 6 (Map3k6), connective tissue growth factor (CtGF), heparin-binding epidermal growth factor (EGF)-like growth factor (HbEGF), vascular endothelial growth factor D (VEGF-D), transforming growth factor (TGF)α and TGFβ, for their relevance to Nrf2 status in the myocardium, supports the involvement of Nrf2 in vascular growth, connective tissue proliferation, and matrix deposition [35,37]. Several proteases and extracellular matrix proteins illustrate another aspect of Nrf2 in the removal of cellular debris, matrix remodeling, and tissue repair. Inducers of Nrf2 or transgenics overexpressing the Nrf2 gene can ameliorate adverse cardiac remodeling and preserve hemodynamic function [50,51]. These reports are in alignment with a benefit of Nrf2 in protection against myocardial deterioration following an initial injury.
Mitochondria have been a focal point in understanding the mechanism and metabolic alterations of cardiac injury. Mitophagy removes damaged mitochondria with PTEN-induced kinase 1 (PINK1) as a sensor. Nrf2 regulates the expression of the PINK1 gene and facilitates mitophagy [52]. Mitochondrial biogenesis compensates for the loss of mitochondria, requiring PPARγ activator 1 alpha (PGC-1α) and nuclear respiratory factor 1 (NResF1), two transcription factors essential for the expression of nucleus-encoded mitochondrial genes. Apparently, the activation of Nrf2 is coupled to PGC1α or NResF1 induction as well as HO-1 elevation in the myocardium, contributing to mitochondrial biogenesis [53,54]. In addition, mitochondrial localization of Nrf2 has been reported [55,56]. Such localization corresponds to protection against mitochondrial decay following oxidative stress [56]. This suggests a possible transcription-independent mechanism in concert with transcriptional events for mitochondrial protection.
Nrf2 orchestrates an anti-inflammatory act by suppressing the nuclear factor kappa B (NF-κB) transcription factor. Absence of Nrf2 results in higher NF-κB basal activity and upregulation of proinflammatory genes [37,57]. Oxidants trigger a cascade of kinase signaling, leading to NF-κB activation due to IκB phosphorylation and its dissociation from the p50/p65 heterodimer. Nrf2 deficiency results in enhanced IκB phosphorylation [58]. In addition, there is evidence that Nrf2 induces HO-1 to suppress NF-κB activity [59]. HO-1 prohibits NF-κB from inducing proinflammatory cytokines, instead facilitating the expression of anti-inflammatory cytokines; for example, IL-10 [60]. In parallel to suppressing NF-κB, Nrf2 blocks the expression of the proinflammatory cytokines IL-6 and IL-1β, by binding to these genes and preventing the recruitment of RNA polymerase for transcription in macrophages [61]. Profound elevations of proinflammatory cytokines have been observed in Nrf2-deficient mice challenged with disease inducers [58,62]. Given the fact that inflammation is a culprit in the progression of many forms of cardiovascular disease, including atherosclerosis, myocardial ischemia, and heart failure, the activation of Nrf2 is expected to reduce inflammation and abate disease development.
The diversity of Nrf2 downstream events supports the complexity of Nrf2 in the etiology of MI involving atherosclerosis. The pathogenic elements of atherosclerosis include plaque formation, oxidative injury of vascular endothelial cells, infiltration by inflammatory cells, and the proliferation of smooth muscle cells. Whereas Nrf2 activity affects genes in both pro- and antiatherogenic directions, knocking out Nrf2 suppressed atherosclerotic lesions in hypercholesterolemic ApoE−/− mice and young low-density lipoprotein (LDL) receptor (LDLR)-knockout mice or LDLR−/− ApoB100/100 mice. This unexpected benefit of Nrf2 deficiency may be explained by Nrf2 effects on lipid metabolism and smooth muscle cell proliferation. Nevertheless, in older LDLR−/− ApoB100/100 mice, which develop hypercholesterolemia without a high-fat diet, Nrf2 deficiency enhanced plaque instability and inflammation and caused an increased rate of spontaneous MI [63,64]. These results point to the possibility of confounding factors (e.g. genetics, environment, aging) in Nrf2-mediated protection against atherosclerosis.
Chemical entities to activate Nrf2: proof of concept
The ability of Nrf2 to elicit myriad cytoprotective responses delineates the concept of empowering Nrf2’s transcriptional force for cardioprotection. Nrf2 deficiency in young mice results in increased cardiac injury and accelerated heart failure with induced myocardial ischemia [24,65]. Erkens et al. [66] reported baseline cardiac left ventricular hypertrophy and diastolic dysfunction in adult Nrf2 knockout mice, suggesting that Nrf2 deficiency may increase detrimental myocardial remodeling during aging. Indeed, the level of Nrf2 protein or the activity of Nrf2 transcription factor decreases in the myocardium with age in experimental animals [67–69]. This correlates with the fact obtained from clinical observations that aging is the greatest risk factor for increased incidence of MI, post-infarction heart failure, and a high mortality rate.
A fraction of Nrf2 inducers have been tested for protection against ischemic reperfusion injury in experimental models in mice or rats. Table 2 summarizes the compounds that induce the Nrf2 protein or its downstream gene NQO1, HO-1, SOD1/2, Cat, GPx, or Gclc in the myocardium. An overwhelming theme shows that Nrf2 inducers inhibited apoptosis, lowered the serum biomarkers of myocardial cell death creatine kinase myocardial band (CK-MB) and cardiac troponin (cTn), reduced infarct size, and improved contractile function. The cardioprotective effect of Nrf2 has been validated using transgenic mice with cardiac overexpression of Nrf2 [51] or miRNA inactivating Keap1 [70].
Table 2.
Nrf2 inducers capable of cardiac protection in animal models of ischemic reperfusiona
| Model | Compound | Source | Dose and schedule | Benefit | Refs |
|---|---|---|---|---|---|
| LADb ligation (rats) | SFN | Organosulfurfrom broccoli | 500 g/kg, LV cavity injection, before ischemia | ↓, ω, ⇑ | [82] |
| Bardoxolone methyl (CDDO-Me) | Synthetic triterpenoid derived from oleanolic acid | i.p. 5 mg/kg/day, 12–14 weeks post-MI | ω, ⇑ | [83] | |
| DH404 | Synthetic triterpenoid | p.o. 10 mg/kg/d for 26 d post MI | ↓, ⇑ | [84] | |
| Resveratrol | Flavonoid from grapes, red wine | i.v. 100 mol/l, 5 min before reperfusion | ↓, ϕ | [85] | |
| α-Lipoic acid | Organosulfur derived from octanoic acid | 5, 10, 15, 25, 50 mg/kg before I/R | ↓, √, ϕ, ω, ⇑ | [86] | |
| L-Carnitine | Amino acid, nutritional supplement | i.p. 150 mg/kg/day for 1 month before I/R | ϕ | [87] | |
| Urolithin B | Polyphenol metabolite from gut microbiota | i.p. 0.7 mg/kg, 0, 24, or 48 h before I/R | ↓, √, ⇑ | [88] | |
| n-Propargyl caffeamide | Caffeic acid derivative | i.p. 15 mg/kg/day for 3, 5, 7 days before I/R | ↓, ω | [89] | |
| Triptolide | Diterpenoid epoxide from Chinese herb | i.p. 0.025, 0.05, 0.1 mg/kg before I/R | ↓, ϕ, ω, ⇑ | [90] | |
| Atorvastatinc | HMG-CoA reductase inhibitor | i.v. 10 mg/kg 5 min before reperfusion | ↓, ϕ, ω | [79] | |
| Ginsenoside Rd | Ginsenoside from Panax ginseng | i.p. 50 mg/kg, 30 min before reperfusion | ↓, ϕ, ⇑ | [91] | |
| LAD ligation (diabetic rats) | Resveratrolc | See above | i.p. 20 mg/kg for 7 days | ↓, ϕ | [92] |
| LAD ligation | Dimethyl fumarate (Tecfidera)c | Citric acid cycle metabolite | p.o. 15 mg/kg 2×/day for 5 days | ↓, ⇑ | [93] |
| (mice) | Hydrogen sulfidec | Gas from rotten eggs | Tail vein i.v. Na2S 0.1 mg/kg 24 h before or daily for 7 days before surgery | ↓, √, ⇑ | [94] |
| Hydrogen sulfidec | See above | Femoral vein i.v. Na2S 100 pg/kg 24 h before I/R | ↓, ϕ, √, ⇑ | [95] | |
| Sodium sulfidec | H2S generator | 1 mg/kg LV intracardial injection at reperfusion, v. 0.1 mg/kg/day for first week after I/R |
⇑ | [96] | |
| 14p | Curcumin analog diarylheptanoid | p.o. 10 mg/kg/day | ↓, √, ϕ | [97] | |
| Sodium nitritec | Precursor of nitric oxide, H2S | 0.165 mg/kg intracardiac injection at reperfusion, 50 or 100 mg/l in drinking water for 4 weeks | ⇑, ♥ | [98] | |
| AM1241c | Aminoalkylindole, cannabinoid receptor II agonist | i.p. 20 mg/kg/day for 7 days after surgery | ⇑, ζ | [99] | |
| Butylphthalide | Celery oil | 80 mg/kg/day for 4 weeks | √, ω | [100] | |
| LAD ligation (diabetic mice) | Butin | Flavonoid from dietary plant | p.o. 10, 20, 40 mg/kg/2 days for 15 days after 4 weeks STZ injection | ↓, ⇑ | [101] |
| Langendorff (rats) | Danshensu | Hydroxypropanoic acid from Chinese herb | 1 μM perfusion for 10 min | ↓, ϕ, ⇑ | [102] |
| Sappanone | Isoflavanone from heartwood | i.p. 10, 20, or 40 mg/kg 1 h before heart isolation | ↓, √, ϕ, ⇑ | [103] | |
| Polyunsaturated fatty acidc | Dietary omega 3or6 | p.o. 0.6 g/kg/day for 8 weeks | ↓, ω, ⇑ | [104] | |
| Langendorff (diabetic rats) | Isofluranec | Anesthetic agent | 2% for 10 min after reperfusion | ↓, √, ϕ | [105] |
| Luteolin | Flavonoid from Reseda plants | ϕ, ⇑ | [106] | ||
| Langendorff (mice) | 4-Hydroxyl-2 nonenal | Lipid peroxidation product | Retro-orbital vein i.v. 4 mg/kg 24 h before heart excision | ϕ, ⇑ | [107] |
| EGFc | Peptide growth factor | i.p. 1 mg/kg/day for 7 day before surgery | ↓, √, ϕ | [108] | |
| Langendorff (diabetic mice) | bFGFc | Peptide growth factor | i.p. 2 mg/kg/day for 4th to 8th week after diabetes onset | ↓, ⇑ | [109] |
Reduce infarct size; √, reduce apoptosis; ϕ, reducethe release oftroponin, CK-MB, or LDH; ω, reduce inflammation; ⇑, preserve or improve cardiac contractile function, including ejection fraction, LV function, or coronary flow; o, reduce or suppress hypertrophy; ζ, reduce fibrosis; ♥, prevent heart failure.
Abbreviation: LAD, left anterior descending coronary artery.
Drug whose primary target or medical use is not Nrf2 inducing.
Many Nrf2 inducers are small molecules derived from natural products; namely, dietary plants, spices, or herbs. SFN, a commonly used Nrf2 inducer, alkylates Keap1 at cysteine-151 by thiocarbamylation, thereby preventing Nrf2 ubiquitination and degradation [71]. CDDO-Me and RTA408 have been registered as Nrf2 inducers for clinical trials. CDDO-Me, showed a benefit in improving renal function in patients with chronic kidney disease in a Phase II randomized clinical trial (Table 1), but failed a Phase III clinical trial in reversal of late-stage renal failure [72,73]. This Phase III trial showed increased mortality mainly due to heart failure in the CDDO-Me group [73,74]. Since renal failure patients often have heart failure, when the baseline heart failure patients were removed from the calculation there was no significant increase in the incidence of heart failure or mortality with CDDO-Me treatment [75] (Table 1). RTA408 has 13 clinical trials, mainly for safety, pharmacokinetics, and the treatment of mitochondrial myopathy. Resveratrol and L-carnitine are over-the-counter nutritional supplements, whereas dimethyl fumarate is a prescription drug for multiple sclerosis and severe psoriasis. Despite multiple ongoing clinical trials utilizing these Nrf2 inducers, none has been tested for protection against myocardial ischemia, reperfusion injury, or MI.
One category of drugs proved effective for cardiac protection is statins. Clinical trials have presented convincing evidence that high-dose statins reduce adverse events when given to acute MI patients [76–78] (Table 1). Increasing evidence points to suppression of inflammation and activation of Nrf2 signaling with statins. In a rat model of MI, atorvastatin given during reperfusion increased Nrf2 activity, elevated the level of HO-1 protein, and reduced infarct size by 20% [79]. The link between statins and Nrf2 provides one explanation for the observed cardiac protection in the clinic.
In patients with MI, activation of the neuroendocrine system is a major contributor to adverse events and the development of heart failure, as evidenced by the effectiveness of β-blockers. In mice, overexpression of Nrf2 in the rostral ventrolateral medulla ameliorated sympatho-excitation due to coronary artery occlusion, resulting in a significant reduction of heart failure [80]. This suggests that elevating Nrf2 in a specific tissue is sufficient to provide a considerable safeguard.
Concluding remarks and future perspectives
The establishment of pharmacological agents and revascularization procedures as effective therapies for MI between the 1970s and 1990s has saved millions of lives worldwide. Since then, there has not been a major breakthrough for further improvement in therapeutic outcomes. Reperfusion injury following CABG or PCI remains a major concern clinically. Heart failure in patients who have survived MI is increasingly prevalent. The concept of reducing cell death and controlling inflammation remains valid to improve the outcome of pharmacotherapy as shown in experimental animals.
The timing of when to modulate the molecular target represents a factor for consideration. Revascularization is controlled at the hands of surgeons or interventional cardiologists. The time dependency of reperfusion injury provides a golden opportunity to avert such misfortune if an effective pharmacological agent is available. It is well established that disruption of the Keap1–Nrf2 interaction is sufficient to activate the Nrf2 transcription factor. Thus, most of the small-molecule Nrf2 inducers share a common feature: they are electrophiles, either oxidizing or alkylating Keap1 to release the checkpoint on Nrf2. This creates a problem by assumption: a broad range of off-target effects or a narrowed therapeutic dose range. This conflict between the ‘beauty and beast’ may be solved by hijacking the molecular mechanism for Nrf2 protein activation using modern, advanced technology. Peptide-based inhibitors disrupting Keap1 and Nrf2 protein–protein interactions have been developed [81], providing hope to bypass the issue of toxicity. Current trends in drug development utilizing siRNA is another innovation to induce Nrf2 without introducing protein oxidation or alkylation.
This review is intended to provide answers to many aspects of the outstanding questions in basic science research on oxidative stress in myocardial ischemia and infarction (see Outstanding questions). The ultimate test relies on the outcome of clinical trials with Nrf2 inducers for cardiac protection. Multiple ongoing clinical trials targeting Nrf2 for the treatment of non-cardiovascular diseases will pave the foundation for future clinical testing in cardiac disease by providing pharmacokinetic and toxicology profiles. Unlike the diseases currently undergoing clinical trials with Nrf2 inducers, which require long-term treatment, Nrf2 induction for the treatment of MI can be a short-term affair, perhaps during revascularization and immediately after. Nrf2 elevation in a timely fashion, rather than a constitutive manner, is expected to improve therapeutic outcomes related to cardiac injury.
Outstanding questions.
How do cardiac cells deal with the sudden onset of oxidative stress due to ischemia or reperfusion?
Does cell death occur in the myocardium even when patients appear to have recovered from MI?
How does oxidative stress activate the Nrf2 transcription factor?
What does the Nrf2 transcription factor do to the myocardium when cardiac injury occurs?
How many genes does the Nrf2 transcription factor control?
Can the Nrf2 transcription factor be activated without alkylating or oxidizing cellular components?
Is the timing of when to activate Nrf2 important?
Will ongoing clinical trials of Nrf2 inducers provide useful information to test Nrf2 inducers for the prevention of reperfusion injury?
Highlights.
Myocardial ischemic reperfusion leads to increased oxidative stress and cell death by necrosis, apoptosis, and necroptosis.
During oxidative stress, the activity of Nrf2 as a transcription factor is regulated by protein stability, translation, nuclear localization, and protein–protein interactions.
The Nrf2 transcription factor controls the expression of key components in eight antioxidant and redox systems for the removal of reactive oxygen species.
The genes under the influence of Nrf2 status suggest its involvement in mitochondrial turnover, tissue recovery, repair, or remodeling, metabolic reprogramming, and the limitation of proinflammatory cytokines.
Small-molecule Nrf2 inducers have shown promise in eliciting cardiac protection and inhibiting inflammation in experimental animals, suggesting a future direction for the development of nontoxic Nrf2 inducers using modern technologies.
Acknowledgments
The author thanks Dr Douglas Holsclaw for the support through the Holsclaw Endowment. The research programs under the direction of the author have been supported by National Institutes of Health R01 HL089958, R01 GM111337, R01 GM125212, and R01 GM126165 and the University of Arizona College of Pharmacy start-up fund. The author is thankful for in-depth discussion, input of medical knowledge, and manuscript editing from Dr Joseph S. Alpert.
Glossary
- Cardiac troponin (cTn)
elevated in the blood of MI patients. Two isoforms, cTnI and cTnT, are commonly used for blood tests to determine whether chestpain patients are undergoing a MI. Recently, high-sensitivity cTn tests have been implemented in hospitals across Europe, the USA, and many other countries, providing a useful tool for the early diagnosis of MI.
- CDDO-Me, CDDO-Im, and RTA408
CDDO-Me is 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid, also known as bardoxolone methyl or RTA402, a synthetic triterpenoid modified from oleanolic acid. CDDO-Im is its imidazolide form, 1[2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oyl] imidazole, also known as RTA403. RTA408 (i.e., omaveloxolone) is a second-generation synthetic triterpenoid derivative of oleanolic acid. These compounds have been developed for clinical applications by Reata Pharmaceuticals and are therefore named after the abbreviation of the company.
- Coronary artery bypass grafting (CABG)
open heart surgery to restore or improve blood flow downstream of one or more obstructed coronary arteries.
- Creatine kinase myocardial band (CK-MB)
a biomarker for cardiac injury. The value of CK-MB can vary and assay sensitivity is low with potential nonspecificity. Muscle injury or kidney injury also causes elevation of CK-MB in the blood. This biomarker is viewed as a reference instead of a diagnostic tool for cardiac injury in patients. Blood levels of CK-MB are often measured in animal experiments with induced myocardial injury.
- Left anterior descending coronary artery (LAD)
interventricular artery; supplies blood to the muscle of the left ventricle and interventricular septum. Surgical occlusion of the LAD in a beating heart is a routine method to induce myocardial ischemia in experimental animals for studies of MI and its long-term effects such as heart failure.
- Myocardial infarction (MI)
commonly known as a heart attack. Blockage of a coronary artery causes ischemia of the downstream tissue and is the prevalent cause of MI. Often, blood clots obstruct the coronary artery as a result of the erosion or rupture of an atherosclerotic plaque. Among the typical symptoms are chest pain, upper-body discomfort, abnormal heartbeat, shortness of breath, and nausea. MI has a high mortality rate if not treated immediately. Patients who have survived MI are at a high risk of developing heart failure over time.
- Percutaneous coronary intervention (PCI)
also known as angioplasty; a non-surgical procedure for opening of the coronary artery to resume blood flow using a catheter to implant a stent.
- Sulforaphane (SFN)
a well-established Nrf2 inducer, known to alkylate Keap1 protein at cysteine residues. Cruciferous vegetables (e.g., broccoli, arugula, brussels sprouts, cabbage) are rich in glucosinolates. Hydrolysis of glucosinolates in plant cells or by microflora of the human gastrointestinal track produces isothiocyanates. Young broccoli plants contain glucoraphanin, which can be converted to SFN, an isothiocyanate organosulfur compound.
Footnotes
Declaration of interests
No interests are declared.
References
- 1.Thygesen K et al. (2018) Fourth universal definition of myocardial infarction (2018). J. Am. Coll. Cardiol. 72, 2231–2264 [DOI] [PubMed] [Google Scholar]
- 2.Yellon DM and Hausenloy DJ (2007) Myocardial reperfusion injury. N. Engl. J. Med. 357, 1121–1135 [DOI] [PubMed] [Google Scholar]
- 3.Zhang DD et al. (2004) Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 24, 10941–10953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suzuki T et al. (2019) Molecular mechanism of cellular oxidative stress sensing by Keap1. Cell Rep. 28, 746–758.e4 [DOI] [PubMed] [Google Scholar]
- 5.Horie Y et al. (2021) Molecular basis for the disruption of Keap1–Nrf2 interaction via hinge & latch mechanism. Commun. Biol. 4, 576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang T et al. (2015) p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 88, 199–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ichimura Y et al. (2013) Phosphorylation of p62 activates the Keap1–Nrf2 pathway during selective autophagy. Mol. Cell 51, 618–631 [DOI] [PubMed] [Google Scholar]
- 8.Nioi P et al. (2005) The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol. Cell. Biol. 25, 10895–10906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Katoh Y et al. (2001) Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 6, 857–868 [DOI] [PubMed] [Google Scholar]
- 10.Wu T et al. (2014) Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 28, 708–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chowdhry S et al. (2013) Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 32, 3765–3781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robertson H et al. (2018) A partnership with the proteasome; the destructive nature of GSK3. Biochem. Pharmacol. 147, 77–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang H et al. (2013) RXRα inhibits the NRF2–ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 73, 3097–3108 [DOI] [PubMed] [Google Scholar]
- 14.Wu J et al. (2014) Rexinoid inhibits Nrf2-mediated transcription through retinoid X receptor alpha. Biochem. Biophys. Res. Commun. 452, 554–559 [DOI] [PubMed] [Google Scholar]
- 15.Namani A et al. (2014) Modulation of NRF2 signaling pathway by nuclear receptors: implications for cancer. Biochim. Biophys. Acta 1843, 1875–1885 [DOI] [PubMed] [Google Scholar]
- 16.Wang XJ et al. (2007) Identification of retinoic acid as an inhibitor of transcription factor Nrf2 through activation of retinoic acid receptor alpha. Proc. Natl. Acad. Sci. U. S. A. 104, 19589–19594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rapola JM et al. (1997) Randomised trial of alpha-tocopherol and beta-carotene supplements on incidence of major coronary events in men with previous myocardial infarction. Lancet 349, 1715–1720 [DOI] [PubMed] [Google Scholar]
- 18.Tornwall ME et al. (2004) Effect of alpha-tocopherol and beta-carotene supplementation on coronary heart disease during the 6-year post-trial follow-up in the ATBC study. Eur. Heart J. 25, 1171–1178 [DOI] [PubMed] [Google Scholar]
- 19.Theodore M et al. (2008) Multiple nuclear localization signals function in the nuclear import of the transcription factor Nrf2. J. Biol. Chem. 283, 8984–8994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xin Y et al. (2018) Sulforaphane prevents angiotensin II-induced cardiomyopathy by activation of Nrf2 via stimulating the Akt/GSK-3ss/Fyn pathway. Redox Biol. 15, 405–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li W et al. (2005) Nrf2 Possesses a redox-insensitive nuclear export signal overlapping with the leucine zipper motif. J. Biol. Chem. 280, 28430–28438 [DOI] [PubMed] [Google Scholar]
- 22.Velichkova M and Hasson T (2005) Keap1 regulates the oxidation-sensitive shuttling of Nrf2 into and out of the nucleus via a Crm1-dependent nuclear export mechanism. Mol. Cell. Biol. 25, 4501–4513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Purdom-Dickinson SE et al. (2007) Translational control of Nrf2 protein in activation of antioxidant response element by oxidants. Mol. Pharm. 72, 1074–1081 [DOI] [PubMed] [Google Scholar]
- 24.Xu B et al. (2014) Myocardial ischemic reperfusion induces de novo Nrf2 protein translation. Biochim. Biophys. Acta 1842, 1638–1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang J et al. (2012) La autoantigen mediates oxidant induced de novo Nrf2 protein translation. Mol. Cell. Proteomics 11, M111.015032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee SC et al. (2017) G-Quadruplex in the NRF2 mRNA 5′ untranslated region regulates de novo NRF2 protein translation under oxidative stress. Mol. Cell. Biol. 37, e00122–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dai W et al. (2021) Far upstream binding protein 1 (FUBP1) participates in translational regulation of Nrf2 protein under oxidative stress. Redox Biol. 41, 101906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Costa-Mattioli M and Walter P (2020) The integrated stress response: from mechanism to disease. Science 368, eaat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li W et al. (2010) An internal ribosomal entry site mediates redox-sensitive translation of Nrf2. Nucleic Acids Res. 38, 778–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dinkova-Kostova AT and Talalay P (2010) NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch. Biochem. Biophys. 501, 116–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Siegel D et al. (2012) NAD(P)H:quinone oxidoreductase 1 (NQO1) in the sensitivity and resistance to antitumor quinones. Biochem. Pharmacol. 83, 1033–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nguyen T et al. (2003) Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu. Rev. Pharmacol. Toxicol. 43, 233–260 [DOI] [PubMed] [Google Scholar]
- 33.Raghunath A et al. (2018) Antioxidant response elements: discovery, classes, regulation and potential applications. Redox Biol. 17, 297–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X et al. (2007) Identification of polymorphic antioxidant response elements in the human genome. Hum. Mol. Genet. 16, 1188–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Purdom-Dickinson S et al. (2007) Induction of antioxidant and detoxification response by oxidants in cardiomyocytes: evidence from gene expression profiling and activation of the Nrf2 transcription factor. J. Mol. Cell. Cardiol. 42, 159–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malhotra D et al. (2010) Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 38, 5718–5734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quiles JM et al. (2017) Differential regulation of miRNA and mRNA expression in the myocardium of Nrf2 knockout mice. BMC Genomics 18, 509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou S et al. (2017) Intermittent hypoxia-induced cardiomyopathy and its prevention by Nrf2 and metallothionein. Free Radic. Biol. Med. 112, 224–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee JM et al. (2003) Identification of the NF-E2-related factor2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J. Biol. Chem. 278 12029–10238 [DOI] [PubMed] [Google Scholar]
- 40.Huang GN et al. (2012) C/EBP transcription factors mediate epicardial activation during heart development and injury. Science 338, 1599–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou H et al. (2014) ATF3 regulates multiple targets and may play a dual role in cardiac hypertrophy and injury. Int. J. Cardiol. 174, 838–839 [DOI] [PubMed] [Google Scholar]
- 42.Wortel IMN et al. (2017) Surviving stress: modulation of ATF4-mediated stress responses in normal and malignant cells. Trends Endocrinol. Metab. 28, 794–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Velmurugan BK et al. (2018) A minireview of E4BP4/NFIL3 in heart failure. J. Cell. Physiol. 233, 8458–8466 [DOI] [PubMed] [Google Scholar]
- 44.Potthoff MJ and Olson EN (2007) MEF2: a central regulator of diverse developmental programs. Development 134, 4131–4140 [DOI] [PubMed] [Google Scholar]
- 45.MacGrogan D et al. (2018) Notch and interacting signalling pathways in cardiac development, disease, and regeneration. Nat. Rev. Cardiol 15, 685–704 [DOI] [PubMed] [Google Scholar]
- 46.Gude NA et al. (2008) Activation of Notch-mediated protective signaling in the myocardium. Circ. Res. 102, 1025–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ferrari R and Rizzo P (2014) The Notch pathway: a novel target for myocardial remodelling therapy? Eur. Heart J. 35, 2140–2145 [DOI] [PubMed] [Google Scholar]
- 48.Zhou XL et al. (2020) Notch1–Nrf2 signaling crosstalk provides myocardial protection by reducing ROS formation. Biochem. Cell Biol. 98, 106–111 [DOI] [PubMed] [Google Scholar]
- 49.Kwak MK et al. (2002) Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: role of antioxidant response element-like sequences in the nrf2 promoter. Mol. Cell. Biol. 22, 2883–2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xing Y et al. (2012) Triterpenoid dihydro-CDDO-trifluoroethyl amide protects against maladaptive cardiac remodeling and dysfunction in mice: a critical role of Nrf2. PLoS One 7, e44899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shanmugam G et al. (2019) Enhanced Keap1–Nrf2 signaling protects the myocardium from isoproterenol-induced patho-logical remodeling in mice. Redox Biol. 27, 101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murata H et al. (2015) NRF2 regulates PINK1 expression under oxidative stress conditions. PLoS One 10, e0142438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Piantadosi CA et al. (2008) Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ. Res. 103, 1232–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liang D et al. (2018) Protective effects of exogenous NaHS against sepsis-induced myocardial mitochondrial injury by enhancing the PGC-1α/NRF2 pathway and mitochondrial biosynthesis in mice. Am. J. Transl. Res. 10, 1422–1430 [PMC free article] [PubMed] [Google Scholar]
- 55.Lo SC and Hannink M (2008) PGAM5 tethers a ternary complex containing Keap1 and Nrf2 to mitochondria. Exp. Cell Res. 314, 1789–1803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Strom J et al. (2016) Nrf2 protects mitochondrial decay by oxidative stress. FASEB J. 30, 66–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fukunaga N et al. (2020) Protective role of Nrf2 against ischemia reperfusion injury and cardiac allograft vasculopathy. Am. J. Transplant. 20, 1262–1271 [DOI] [PubMed] [Google Scholar]
- 58.Thimmulappa RK et al. (2006) Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Invest. 116, 984–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Banning A and Brigelius-Flohe R (2005) NF-κB, Nrf2, and HO-1 interplay in redox-regulated VCAM-1 expression. Antioxid. Redox Signal. 7, 889–899 [DOI] [PubMed] [Google Scholar]
- 60.Ahmed SM et al. (2017) Nrf2 signaling pathway: pivotal roles in inflammation. Biochim. Biophys. Acta 1863, 585–597 [DOI] [PubMed] [Google Scholar]
- 61.Kobayashi EH et al. (2016) Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 7, 11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rangasamy T et al. (2005) Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J. Exp. Med. 202, 47–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mimura J and Itoh K (2015) Role of Nrf2 in the pathogenesis of atherosclerosis. Free Radic. Biol. Med. 88, 221–232 [DOI] [PubMed] [Google Scholar]
- 64.Ruotsalainen AK et al. (2019) Nuclear factor E2-related factor 2 deficiency impairs atherosclerotic lesion development but promotes features of plaque instability in hypercholesterolaemic mice. Cardiovasc. Res. 115, 243–254 [DOI] [PubMed] [Google Scholar]
- 65.Strom J and Chen QM (2017) Loss of Nrf2 promotes rapid progression to heart failure following myocardial infarction. Toxicol. Appl. Pharmacol. 327, 52–58 [DOI] [PubMed] [Google Scholar]
- 66.Erkens R et al. (2015) Left ventricular diastolic dysfunction in Nrf2 knock out mice is associated with cardiac hypertrophy, decreased expression of SERCA2a, and preserved endothelial function. Free Radic. Biol. Med. 89, 906–917 [DOI] [PubMed] [Google Scholar]
- 67.Suh JH et al. (2004) Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl. Acad. Sci. U. S. A. 101, 3381–3386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Collins AR et al. (2009) Age-accelerated atherosclerosis correlates with failure to upregulate antioxidant genes. Circ. Res. 104, e42–e54 [DOI] [PubMed] [Google Scholar]
- 69.Gounder SS et al. (2012) Impaired transcriptional activity of Nrf2 in age-related myocardial oxidative stress is reversible by moderate exercise training. PLoS One 7, e45697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xiao X et al. (2018) MicroRNA miR-24–3p reduces apoptosis and regulates Keap1–Nrf2 pathway in mouse cardiomyocytes responding to ischemia/reperfusion injury. Oxidative Med. Cell. Longev. 2018, 7042105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kensler TW et al. (2013) Keap1–nrf2 signaling: a target for cancer prevention by sulforaphane. Top. Curr. Chem. 329, 163–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pergola PE et al. (2011) Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N. Engl. J. Med. 365, 327–336 [DOI] [PubMed] [Google Scholar]
- 73.de Zeeuw D et al. (2013) Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N. Engl. J. Med. 369, 2492–2503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Himmelfarb J and Tuttle KR (2014) Bardoxolone methyl in type 2 diabetes and advanced chronic kidney disease. N. Engl. J. Med. 370, 1768–1769 [DOI] [PubMed] [Google Scholar]
- 75.Chin MP et al. (2014) Mechanisms contributing to adverse cardiovascular events in patients with type 2 diabetes mellitus and stage 4 chronic kidney disease treated with bardoxolone methyl. Am. J. Nephrol. 39, 499–508 [DOI] [PubMed] [Google Scholar]
- 76.Schwartz GG et al. (2001) Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA 285, 1711–1718 [DOI] [PubMed] [Google Scholar]
- 77.Cannon CP et al. (2004) Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N. Engl. J. Med. 350, 1495–1504 [DOI] [PubMed] [Google Scholar]
- 78.Papageorgiou N et al. (2016) Statins and myocardial infarction: type, dose, and administration time: does it matter? Trends Cardiovasc. Med. 26, 433–441 [DOI] [PubMed] [Google Scholar]
- 79.Sun G et al. (2015) Atorvastatin attenuates inflammation and oxidative stress induced by ischemia/reperfusion in rat heart via the Nrf2 transcription factor. Int. J. Clin. Exp. Med. 8, 14837–14845 [PMC free article] [PubMed] [Google Scholar]
- 80.Ma A et al. (2019) Upregulating Nrf2 in the RVLM ameliorates sympatho-excitation in mice with chronic heart failure. Free Radic. Biol. Med. 141, 84–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wells G (2015) Peptide and small molecule inhibitors of the Keap1–Nrf2 protein-protein interaction. Biochem. Soc. Trans. 43, 674–679 [DOI] [PubMed] [Google Scholar]
- 82.Silva-Palacios A et al. (2019) Sulforaphane protects from myocardial ischemia-reperfusion damage through the balanced activation of Nrf2/AhR. Free Radic. Biol. Med. 143, 331–340 [DOI] [PubMed] [Google Scholar]
- 83.Tian C et al. (2019) Therapeutic effects of Nrf2 activation by bardoxolone methyl in chronic heart failure. J. Pharmacol. Exp. Ther. 371, 642–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bubb KJ et al. (2017) The NRF2 activator DH404 attenuates adverse ventricular remodeling post-myocardial infarction by modifying redox signalling. Free Radic. Biol. Med. 108, 585–594 [DOI] [PubMed] [Google Scholar]
- 85.Cheng L et al. (2015) Resveratrol attenuates inflammation and oxidative stress induced by myocardial ischemia–reperfusion injury: role of Nrf2/ARE pathway. Int. J. Clin. Exp. Med. 8, 10420–10428 [PMC free article] [PubMed] [Google Scholar]
- 86.Deng C et al. (2013) α-Lipoic acid reduces infarct size and preserves cardiac function in rat myocardial ischemia/reperfusion injury through activation of PI3K/Akt/Nrf2 pathway. PLoS One 8, e58371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhao T et al. (2020) L-Carnitine reduces myocardial oxidative stress and alleviates myocardial ischemia–reperfusion injury by activating nuclear transcription-related factor 2 (Nrf2)/heme oxygenase-1 (HO-1) signaling pathway. Med. Sci. Monit. 26, e923251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zheng D et al. (2020) Urolithin B, a gut microbiota metabolite, protects against myocardial ischemia/reperfusion injury via p62/Keap1/Nrf2 signaling pathway. Pharmacol. Res. 153, 104655. [DOI] [PubMed] [Google Scholar]
- 89.Cheng Y et al. (2018) N-Propargyl caffeamide skews macrophages towards a resolving M2-like phenotype against myocardial ischemic injury via activating Nrf2/HO-1 pathway and inhibiting NF-κB Pathway. Cell. Physiol. Biochem. 47, 2544–2557 [DOI] [PubMed] [Google Scholar]
- 90.Yu H et al. (2016) Triptolide attenuates myocardial ischemia/reperfusion injuries in rats by inducing the activation of Nrf2/HO-1 defense pathway. Cardiovasc. Toxicol. 16, 325–335 [DOI] [PubMed] [Google Scholar]
- 91.Zeng X et al. (2015) Ginsenoside Rd mitigates myocardial ischemia–reperfusion injury via Nrf2/HO-1 signaling pathway. Int. J. Clin. Exp. Med. 8, 14497–14504 [PMC free article] [PubMed] [Google Scholar]
- 92.Xu G et al. (2019) Resveratrol increase myocardial Nrf2 expression in type 2 diabetic rats and alleviate myocardial ischemia/reperfusion injury (MIRI). Ann. Palliat. Med. 8, 565–575 [DOI] [PubMed] [Google Scholar]
- 93.Ashrafian H et al. (2012) Fumarate is cardioprotective via activation of the Nrf2 antioxidant pathway. Cell Metab. 15, 361–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Peake BF et al. (2013) Hydrogen sulfide preconditions the db/db diabetic mouse heart against ischemia–reperfusion injury by activating Nrf2 signaling in an Erk-dependent manner. Am. J. Physiol. Heart Circ. Physiol. 304, H1215–H1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Calvert JW et al. (2009) Hydrogen sulfide mediates cardio-protection through Nrf2 signaling. Circ. Res. 105, 365–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shimizu Y et al. (2016) Sodium sulfide attenuates ischemic-induced heart failure by enhancing proteasomal function in an Nrf2-dependent manner. Circ. Heart Fail. 9, e002368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Li W et al. (2015) Novel curcumin analogue 14p protects against myocardial ischemia reperfusion injury through Nrf2-activating anti-oxidative activity. Toxicol. Appl. Pharmacol. 282, 175–183 [DOI] [PubMed] [Google Scholar]
- 98.Donnarumma E et al. (2016) Nitrite therapy ameliorates myocardial dysfunction via H2S and nuclear factor-erythroid 2-related factor 2 (Nrf2)-dependent signaling in chronic heart failure. J. Am. Heart Assoc. 5, e003551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li X et al. (2016) Activation of cannabinoid receptor type II by AM1241 ameliorates myocardial fibrosis via Nrf2-mediated inhibition of TGF-β1/Smad3 pathway in myocardial infarction mice. Cell. Physiol. Biochem. 39, 1521–1536 [DOI] [PubMed] [Google Scholar]
- 100.Bai M et al. (2019) Effects of butylphthalide on oxidative stress and inflammatory response in rats with myocardial infarction through Akt/Nrf2 signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 23, 9642–9650 [DOI] [PubMed] [Google Scholar]
- 101.Duan J et al. (2017) Protective effect of butin against ischemia/reperfusion-induced myocardial injury in diabetic mice: involvement of the AMPK/GSK-3β/Nrf2 signaling pathway. Sci. Rep. 7, 41491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yu J et al. (2015) Danshensu protects isolated heart against ischemia reperfusion injury through activation of Akt/ERK1/2/Nrf2 signaling. Int. J. Clin. Exp. Med 8, 14793–14804 [PMC free article] [PubMed] [Google Scholar]
- 103.Shi X et al. (2020) Sappanone A protects against myocardial ischemia reperfusion injury by modulation of Nrf2. Drug Des. Devel. Ther. 14, 61–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Farias JG et al. (2017) Polyunsaturated fatty acid induces cardioprotection against ischemia–reperfusion through the inhibition of NF-κB and induction of Nrf2. Exp. Biol. Med. (Maywood) 242, 1104–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang Y et al. (2016) Cardioprotection from emulsified isoflurane postconditioning is lost in rats with streptozotocin-induced diabetes due to the impairment of Brg1/Nrf2/STAT3 signalling. Clin. Sci. (Lond.) 130, 801–812 [DOI] [PubMed] [Google Scholar]
- 106.Xiao C et al. (2019) Luteolin attenuates cardiac ischemia/reperfusion injury in diabetic rats by modulating Nrf2 antioxidative function. Oxidative Med. Cell. Longev. 2019, 2719252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang Y et al. (2010) 4-Hydroxy-2-nonenal protects against cardiac ischemia–reperfusion injury via the Nrf2-dependent pathway. J. Mol. Cell. Cardiol. 49, 576–586 [DOI] [PubMed] [Google Scholar]
- 108.Ma J and Jin G (2019) Epidermal growth factor protects against myocardial ischaemia reperfusion injury through activating Nrf2 signalling pathway. Free Radic. Res. 53, 313–323 [DOI] [PubMed] [Google Scholar]
- 109.Tong G et al. (2020) The protective role of bFGF in myocardial infarction and hypoxia cardiomyocytes by reducing oxidative stress via Nrf2. Biochem. Biophys. Res. Commun. 527, 15–21 [DOI] [PubMed] [Google Scholar]